Transition-metal-free, room-temperature radical azidofluorination of unactivated alkenes in aqueous solution†‡
Zhaodong
Li
,
Chengwei
Zhang
,
Lin
Zhu
,
Chao
Liu
and
Chaozhong
Li
*
Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: clig@mail.sioc.ac.cn; Fax: (+) 86-21-5492-5099
First published on 20th December 2013
Abstract
We report herein the transition-metal-free azidofluorination of unactivated alkenes. Thus, the condensation of various alkenes with TMSN3 and Selectfluor in aqueous CH3CN at RT led to the efficient and regioselective synthesis of β-fluorinated alkyl azides with excellent functional group compatibility and good stereoselectivity. A single electron transfer mechanism involving the oxidative generation of azidyl radicals is proposed.
The growing importance of fluorine in agrochemicals and pharmaceuticals1 as well as the use of 18F-labeled organic compounds as contrast agents for positron emission tomography (PET)2 has spurred vigorous research for the development of new methods for C–F bond formation under mild conditions.3 In this context, the synthesis of β-fluorinated amines has received considerable attention in the past few years. Vicinal aminofluorine moieties are key building blocks of anticancer, anticholinergic and anti-inflammatory drugs4 as well as therapeutic β-peptides5 because fluorine can improve the bioavailability of amine drugs by decreasing the basicity of neighboring amine groups. Among various methods developed,6 the aminofluorination7,8 of alkenes provides rapid access to this type of molecule. For example, palladium-catalyzed intramolecular aminofluorination of N-tosyl-4-pentenyl amines with AgF led to the synthesis of 3-fluoropiperidines.7a This fluorocyclization could be carried out enantioselectively using chiral [ArIF2] reagents.8f Enantioselective intramolecular aminofluorination of indoles and conjugated dienes with Selectfluor9 under organocatalysis7b or chiral-anion phase-transfer catalysis7g was also achieved. However, only limited examples of intermolecular aminofluorination were reported8 and they were restricted to the use of glycals,8a stilbenes,8b styrenes8c,d,f and other activated alkenes such as α,β-unsaturated aldehydes.8e A general and efficient intermolecular aminofluorination of unactivated alkenes is certainly highly desirable in view of the important role of β-fluorinated amines in medicinal chemistry. Herein we report a variant of intermolecular aminofluorination, the unprecedented azidofluorination of unactivated alkenes under transition metal-free conditions.
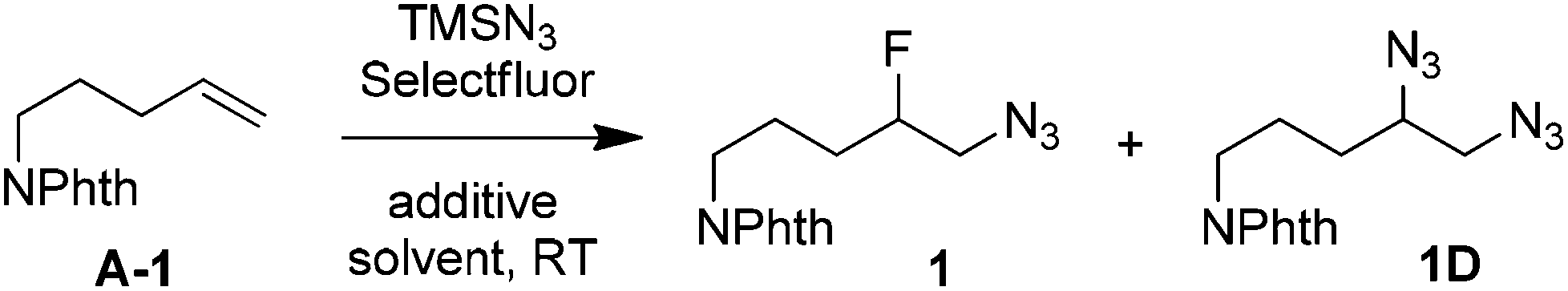
Our idea originated from our recent finding that, under the catalysis of AgNO3, the reactions of aliphatic carboxylic acids with Selectfluor resulted in oxidative fluorodecarboxylation.10a This was then successfully extended to the intramolecular radical11,12 aminofluorination of N-aryl-4-pentenamides in aqueous media.10b We envisioned that an intermolecular version of the above aminofluorination might be achieved under similar conditions. To test this idea, we chose N-(pent-4-en-1-yl)phthalimide (A-1) as the model alkene to screen a suitable nitrogen partner. Thus a number of amides or sulfonamides, including AcNHPh, BzNHPh, BzNHMe, TsNHMe and TfNHPh, were subjected to treatment with A-1, Selectfluor and a catalytic amount of AgNO3 (20 mol%) in aqueous CH3CN or CH2Cl2 solution at ambient temperature. To our disappointment, no reaction occurred in all cases. However, when TMSN3 was used as the nitrogen source, we were delighted to see that the corresponding azidofluorination product 1 was observed. We then went on to optimize the reaction conditions (Table 1). We found that AgNO3 was not required at all for the azidofluorination, and the direct treatment of A-1 with Selectfluor (2 equiv.) and TMSN3 (2 equiv.) in CH3CN–H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at room temperature for 5 h afforded β-fluoroalkyl azide 1 in 59% yield along with the bis-azidation product 1D in 29% yield (entry 2). Switching the mixed solvent to acetone–H2O or AcOH–H2O did not modulate yield or selectivity (entries 3 and 4). On the other hand, no azidofluorination could be observed in biphasic systems or in anhydrous organic solvents such as DMSO or acetonitrile (entries 5 and 6). Increasing the ratio of H2O–CH3CN from 1:1 to 2:1 slightly improved the yield of 1 (entry 7). Changing the reaction temperature did not help (not shown). In order to inhibit bis-azidation, various additives were screened. The use of NaOAc or K2CO3 as a base decreased the yield of 1 (entry 8). These phenomena urged us to test the effect of acids other than AcOH. Indeed, with an acid such as TsOH, TfOH, CF3CO2H (TFA) or even HNO3 as the additive, the yield of 1D was decreased. The use of these additives requires a longer reaction time (18 h) for complete conversion (entries 9–13). In the case of TFA (3 equiv.) as the additive, the reaction furnished the desired product 1 in 85% NMR yield (83% isolated yield) while only a small amount of byproduct 1D could be observed by 1H NMR (entry 11). To the best of our knowledge, this is the first example of azidofluorination of unsaturated carbon–carbon bonds. It is worth mentioning that no azidofluorination could be detected when the fluorine source was changed from Selectfluor to N-fluorobis(benzenesulfonyl)imide (NFSI). Note that the azidofluorination also proceeded with NaN3 as the substitute for TMSN3, but in lower (63%) efficiency.
:1) at room temperature for 5 h afforded β-fluoroalkyl azide 1 in 59% yield along with the bis-azidation product 1D in 29% yield (entry 2). Switching the mixed solvent to acetone–H2O or AcOH–H2O did not modulate yield or selectivity (entries 3 and 4). On the other hand, no azidofluorination could be observed in biphasic systems or in anhydrous organic solvents such as DMSO or acetonitrile (entries 5 and 6). Increasing the ratio of H2O–CH3CN from 1:1 to 2:1 slightly improved the yield of 1 (entry 7). Changing the reaction temperature did not help (not shown). In order to inhibit bis-azidation, various additives were screened. The use of NaOAc or K2CO3 as a base decreased the yield of 1 (entry 8). These phenomena urged us to test the effect of acids other than AcOH. Indeed, with an acid such as TsOH, TfOH, CF3CO2H (TFA) or even HNO3 as the additive, the yield of 1D was decreased. The use of these additives requires a longer reaction time (18 h) for complete conversion (entries 9–13). In the case of TFA (3 equiv.) as the additive, the reaction furnished the desired product 1 in 85% NMR yield (83% isolated yield) while only a small amount of byproduct 1D could be observed by 1H NMR (entry 11). To the best of our knowledge, this is the first example of azidofluorination of unsaturated carbon–carbon bonds. It is worth mentioning that no azidofluorination could be detected when the fluorine source was changed from Selectfluor to N-fluorobis(benzenesulfonyl)imide (NFSI). Note that the azidofluorination also proceeded with NaN3 as the substitute for TMSN3, but in lower (63%) efficiency.
|
|
||||
|---|---|---|---|---|
| Entrya | Solvent | Additive (equiv.) | Yieldb (%) | |
| 1 | 1D | |||
| a Reaction conditions: A-1 (0.3 mmol), TMSN3 (0.6 mmol), Selectfluor (0.6 mmol), solvent (3 mL), RT, 5 h. b 1H NMR yield based on A-1 with p-nitroacetophenone as the internal standard. c Reaction time: 18 h. | ||||
| 1 | CH3CN–H2O (1:1) |
AgNO3 (0.2) | 50 | 30 |
| 2 | CH3CN–H2O (1:1) |
None | 59 | 29 |
| 3 | Me2CO–H2O (1:1) |
None | 52 | 32 |
| 4 | AcOH–H2O (1:1) |
None | 58 | 29 |
| 5 | CH2Cl2–H2O (1:1) |
None | 0 | 0 |
| 6 | DMSO, CH2Cl2, CH3CN, or AcOH | None | 0 | 0 |
| 7 | CH3CN–H2O (1:2) |
None | 64 | 25 |
| 8 | CH3CN–H2O (1:2) |
NaOAc (2) | 40 | 20 |
| 9c | CH3CN–H2O (1:2) |
HNO3 (2) | 84 | 7 |
| 10c | CH3CN–H2O (1:2) |
TFA (2) | 80 | 8 |
| 11 |
CH3CN–H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :2) :2)
|
TFA (3) | 85 | <5 |
| 12c | CH3CN–H2O (1:2) |
TfOH (3) | 68 | 7 |
| 13c | CH3CN–H2O (1:2) |
TsOH (3) | 76 | 5 |
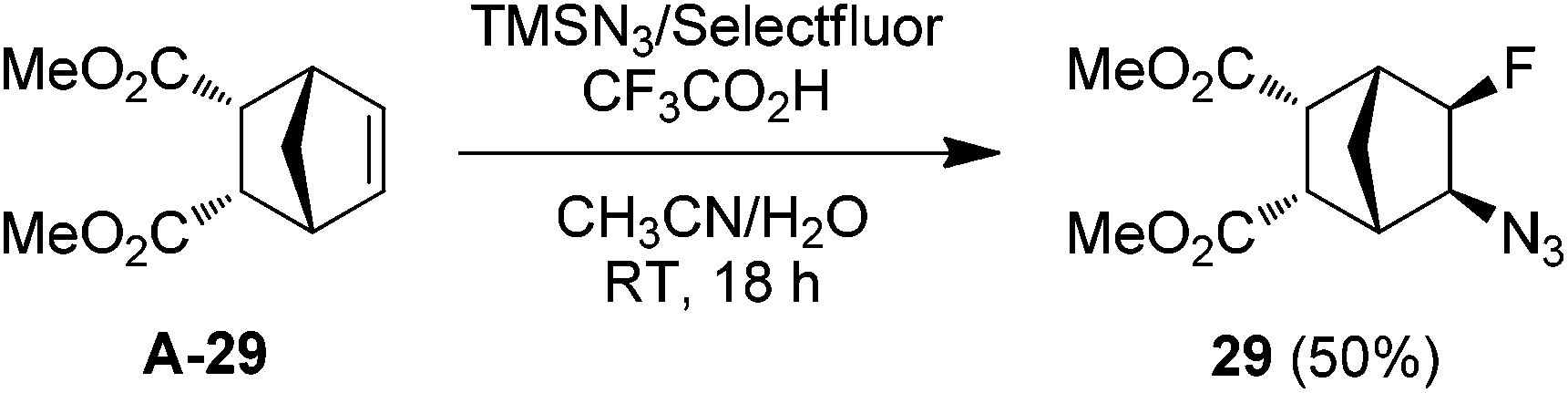
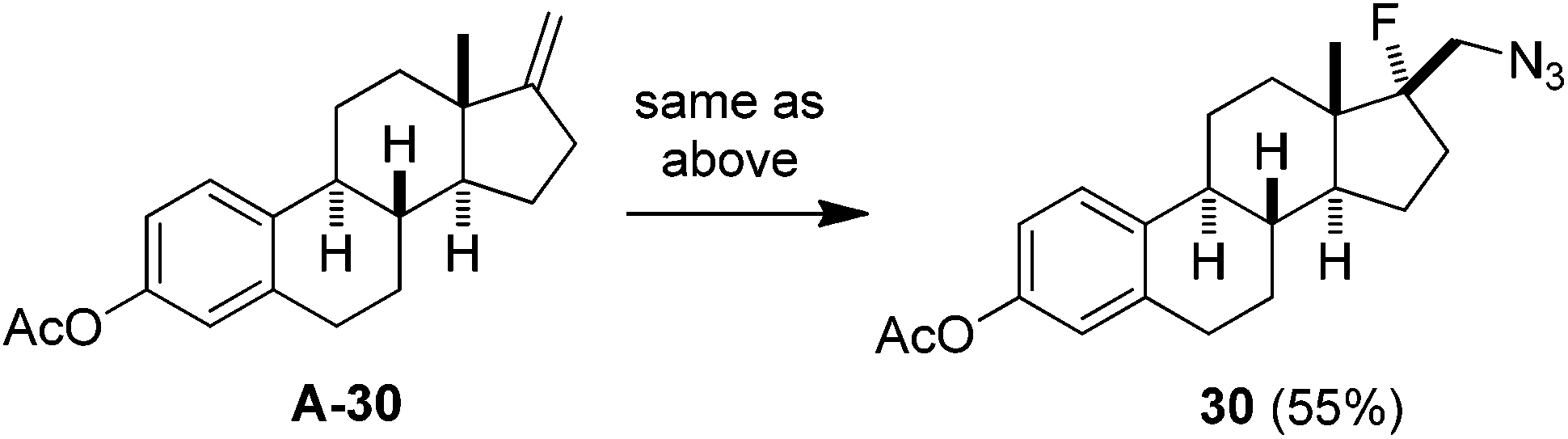
With the optimized conditions in hand (entry 11, Table 1), we set out to explore the scope and limitations of the above azidofluorination (Scheme 1). The alkene substrate scope proved quite general as not only various unactivated mono- and di-substituted alkenes but also tri-substituted alkenes participated in the azidofluorination reactions effectively. In addition, styrenes could also be used as substrates, as exemplified by the synthesis of azide 28. Nevertheless, electron-deficient alkenes such as methyl acrylate failed to give the desired product. A wide range of functional groups were well tolerated, including unprotected and protected alcohol, protected amine, alkyl chloride, ether, ketone, ester, sulfonate, amide, sulfonamide, nitrile and N-protected indole. Excellent regioselectivity was observed as the azido group reliably added to the less sterically hindered carbon of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond. Moreover, stereoselective azidofluorination could also be achieved. For example, the reaction of norbornene A-29 gave the cis-addition product 29 as the only stereoisomer isolated (eqn (1)). A high diastereoselectivity (>10:1) was also obtained in the reaction of 17-methylene steroid A-30 (eqn (2)). Thus, the above azidofluorination provides a convenient and efficient entry to β-fluorinated alkyl azides,13 which can be readily converted to the corresponding β-fluorinated amines by reduction (see also ESI‡).13 More importantly, by taking advantage of the versatility of alkyl azides in organic synthesis,14 β-fluoroalkyl azides can be elaborated into a variety of fluorinated molecules via cycloaddition, rearrangement, nitrene chemistry, etc., thus expanding the application of the above azidofluorination reactions (see also ESI‡).
C bond. Moreover, stereoselective azidofluorination could also be achieved. For example, the reaction of norbornene A-29 gave the cis-addition product 29 as the only stereoisomer isolated (eqn (1)). A high diastereoselectivity (>10:1) was also obtained in the reaction of 17-methylene steroid A-30 (eqn (2)). Thus, the above azidofluorination provides a convenient and efficient entry to β-fluorinated alkyl azides,13 which can be readily converted to the corresponding β-fluorinated amines by reduction (see also ESI‡).13 More importantly, by taking advantage of the versatility of alkyl azides in organic synthesis,14 β-fluoroalkyl azides can be elaborated into a variety of fluorinated molecules via cycloaddition, rearrangement, nitrene chemistry, etc., thus expanding the application of the above azidofluorination reactions (see also ESI‡).
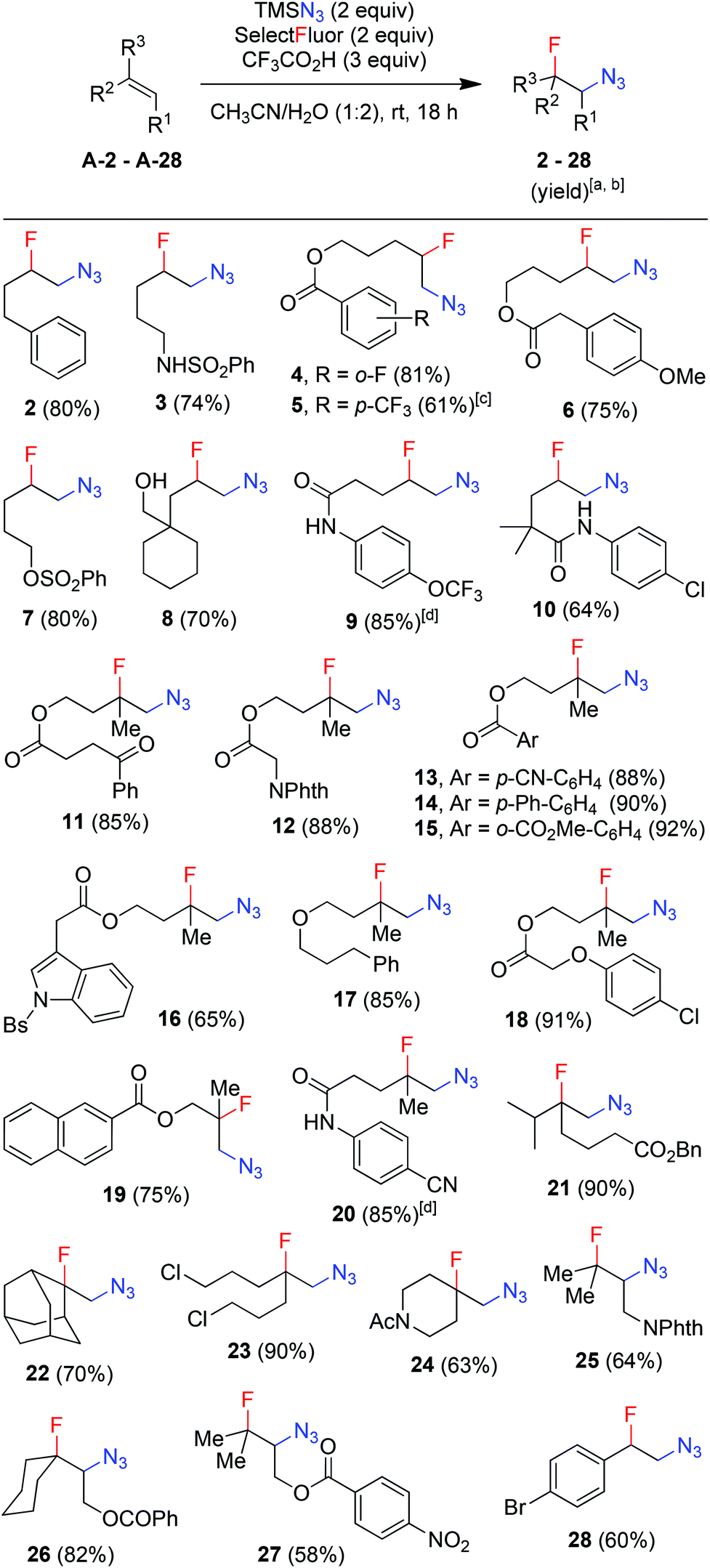 | ||
| Scheme 1 Azidofluorination of alkenes. [a] Reaction conditions: alkene (0.3 mmol), TMSN3 (0.6 mmol), Selectfluor (0.6 mmol), CF3CO2H (0.9 mmol), CH3CN (1 mL), H2O (2 mL), RT, 18 h. [b] Isolated yield based on the substrate alkene. [c] The substrate alkene was recovered in 26% yield. [d] AcOH (1 mL) and H2O (2 mL) were used as the solvent. | ||
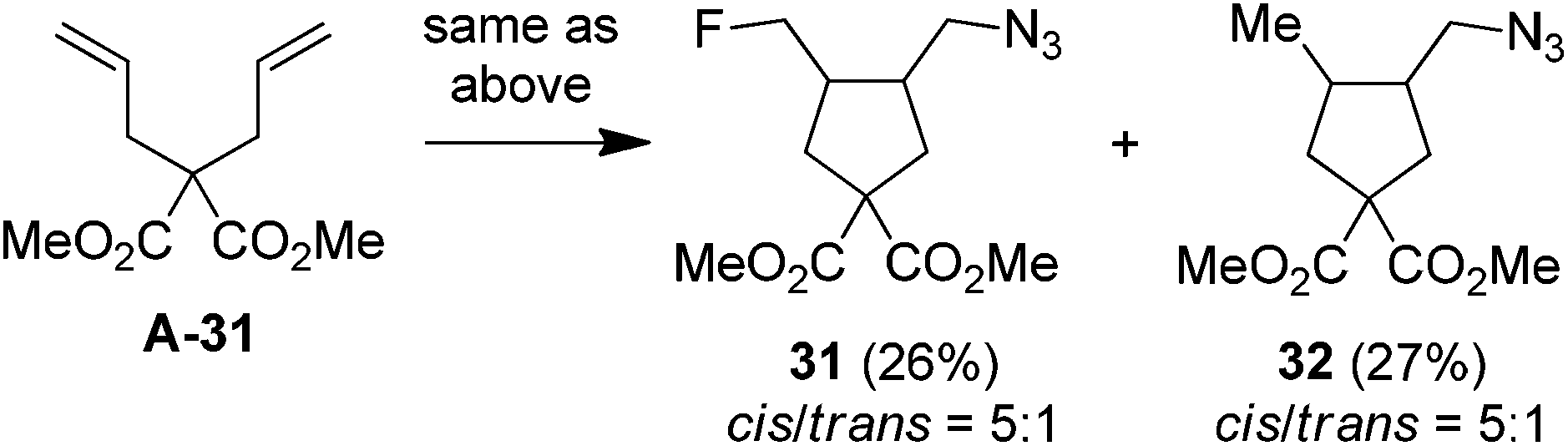
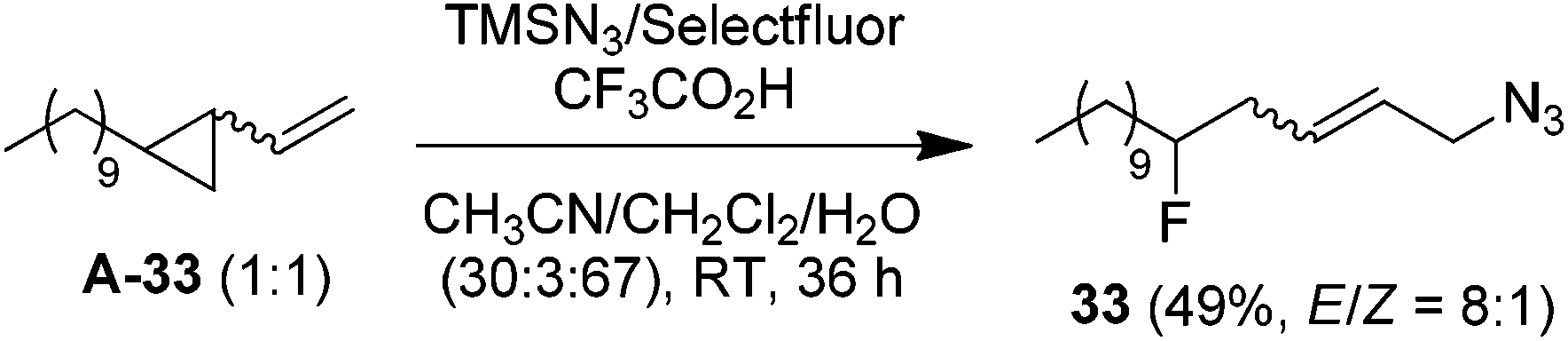
It should be noted that, while a small amount (∼5%) of bis-azidation products similar to 1D could be observed in the cases of monosubstituted alkenes, such byproducts were further suppressed using di- and tri-substituted alkenes and the yields of β-fluoroalkyl azides were generally higher in the reactions of di-substituted terminal alkenes. Presumably the second-step azidation (to give bis-azidation products) is more sensitive to steric hindrance than the fluorination (to give azido-fluorination products). Moreover, a number of substrates capable of electrophilic fluorocyclization15 with Selectfluor failed to display this competitive behaviour, yielding only the azidofluorination products (3, 8–10, 20) in high yields. These results also shed light on the mechanism, indicating that the fluorination does not proceed through a carbocationic intermediate. To gain more insight into the mechanism, the reaction of 1,6-diene A-31 was carried out. Under the above optimized conditions, the cyclized products 31 (26%) and 32 (27%) were isolated, both in preference for the cis-configuration (eqn (3)). This result strongly suggests the azidyl radical-mediated free radical mechanism for azidofluorination.16 Furthermore, vinyl cyclopropane A-33 was employed as a radical probe.16 The reaction of A-33 under the above optimized conditions was slow probably because of its poor solubility in aqueous CH3CN. When a small amount of CH2Cl2 was added to improve the solubility of A-33, the reaction cleanly afforded the ring-opening product, 33, in 49% yield along with 23% recovered A-33 (eqn (4)). This experiment provides evidence for the involvement of carbon-centered radicals in the above azidofluorination.
 | (1) |
 | (2) |
 | (3) |
 | (4) |
Although the detailed mechanism remains unclear, a tentative mechanism was proposed based on the above results, as shown in Fig. 1. In aqueous acidic solution TMSN3 is quickly decomposed to hydrazoic acid,14b,17 which then undergoes single electron transfer (SET) with Selectfluor to generate an azidyl radical18–21 and a Selectfluor radical anion.22 The electrophilic23 azidyl radical adds to an alkene to give the nucleophilic β-azidoalkyl radical. The subsequent fluorine atom transfer from the Selectfluor radical anion to the β-azidoalkyl radical affords the azidofluorination product.24 The formation of bis-azidation products18 such as 1D might result from the coupling between the β-azidoalkyl radical and another azidyl radical. The addition of TFA slows down the SET process and thus the concentration of azidyl radical is kept in a low concentration range. As a result, the bis-azidation byproduct is significantly inhibited.
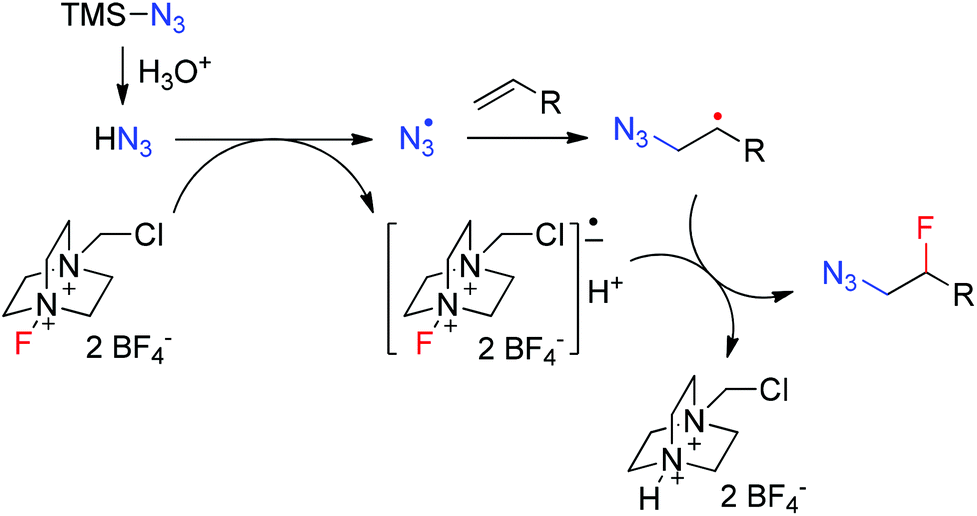 | ||
| Fig. 1 Proposed mechanism of azidofluorination. | ||
In conclusion, we have successfully developed a transition-metal-free regioselective azidofluorination reaction of unactivated alkenes with TMSN3 and Selectfluor, providing rapid and efficient access to β-fluorinated alkyl azides. The reaction proceeds in aqueous media at room temperature and exhibits broad substrate scope, wide functional group compatibility as well as good stereoselectivity. The above results further expand the scope of radical fluorination,10–12 which is emerging as a versatile and powerful tool for C(sp3)–F bond formation. In view of the importance of fluorine and the rich chemistry of alkyl azides, the azidofluorination should find broad applications in organic synthesis.
Experimental section
Typical procedure for the azidofluorination of unactivated alkenes
2-(Pent-4-en-1-yl)-isoindoline-1,3-dione (A-1, 64.5 mg, 0.3 mmol) and Selectfluor (212 mg, 0.6 mmol) were placed in a Schlenk tube under a nitrogen atmosphere. CF3CO2H (69 μL, 0.9 mmol), TMSN3 (78 μL, 0.6 mmol), CH3CN (1 mL) and water (2 mL) were then added successively at RT. The reaction mixture was stirred at RT for 18 h. The resulting mixture was extracted with CH2Cl2 (15 mL × 3). The combined organic phase was dried over anhydrous Na2SO4. After the removal of the solvent under reduced pressure, the crude product was purified by column chromatography on silica gel with hexane–ethyl acetate (7:1, v/v) as the eluent to give the pure product 2-(5-azido-4-fluoropentyl)isoindoline-1,3-dione (1) as a yellow oil. Yield: 68 mg (83%).
Acknowledgements
This project was supported by the NSFC (grant no. 21072211, 21228202, 21272259, 21290180 and 21361140377) and by the National Basic Research Program of China (973 Program) (grant no. 2011CB710805).References
- (a) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed; (b) D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC; (c) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC; (d) K. L. Kirk, Org. Process Res. Dev., 2008, 12, 305–321 CrossRef CAS.
- (a) P. W. Miller, N. J. Long, R. Vilar and A. D. Gee, Angew. Chem., 2008, 120, 9136–9172 ( Angew. Chem. Int. Ed. , 2008 , 47 , 8998–9033 ) CrossRef; (b) S. M. Ametamey, M. Horner and P. A. Schubiger, Chem. Rev., 2008, 108, 1501–1516 CrossRef CAS PubMed.
- For the latest selected reviews, see: (a) V. V. Grushin, Acc. Chem. Res., 2010, 43, 160–171 CrossRef CAS PubMed; (b) T. Furuya, J. E. M. N. Klein and T. Ritter, Synthesis, 2010, 1804–1821 CAS; (c) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470–477 CrossRef CAS PubMed; (d) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., 2013, 125, 8372–8423 ( Angew. Chem. Int. Ed. , 2013 , 52 , 8214–8264 ) CrossRef.
- (a) C. D. Cox, et al. , J. Med. Chem., 2008, 51, 4239–4252 CrossRef CAS PubMed; (b) J. T. Welch and S. Eswarakrishman, Fluorine in Bioorganic Chemistry, Wiley, New York, 1991 Search PubMed.
- D. Seebach, et al. , Helv. Chim. Acta, 2008, 91, 1736–1786 CrossRef CAS.
- For a review, see: J. M. Percy, Sci. Synth., 2005, 34, 379–416 Search PubMed . For the latest example of the synthesis of β-fluorinated amines via ring opening of aziridines, see: J. A. Kalow and A. G. Doyle, Tetrahedron, 2013, 69, 5702–5709 CrossRef CAS PubMed.
- (a) T. Wu, G. Yin and G. Liu, J. Am. Chem. Soc., 2009, 131, 16354–16355 CrossRef CAS PubMed; (b) O. Lozano, G. Blessley, T. M. del Campo, A. L. Thompson, G. T. Giuffredi, M. Bettati, M. Walker, R. Borman and V. Gouverneur, Angew. Chem., 2011, 123, 8255–8259 ( Angew. Chem. Int. Ed. , 2011 , 50 , 8105–8109 ) CrossRef; (c) T. Xu, X. Mu, H. Peng and G. Liu, Angew. Chem., 2011, 123, 8326–8329 ( Angew. Chem. Int. Ed. , 2011 , 50 , 8176–8179 ) CrossRef; (d) Q. Wang, W. Zhong, X. Wei, M. Ning, X. Meng and Z. Li, Org. Biomol. Chem., 2012, 10, 8566–8569 RSC; (e) T. Xu and G. Liu, Org. Lett., 2012, 14, 5416–5419 CrossRef CAS PubMed; (f) H.-T. Huang, T. C. Lacy, B. Blachut, G. X. Ortiz Jr. and Q. Wang, Org. Lett., 2013, 15, 1818–1821 CrossRef CAS PubMed; (g) H. P. Shunatona, N. Früh, Y.-M. Wang, V. Rauniyar and F. D. Toste, Angew. Chem., 2013, 125, 7878–7881 ( Angew. Chem. Int. Ed. , 2013 , 52 , 7724–7727 ) CrossRef.
- (a) S. P. Vincent, M. D. Burkart, C.-Y. Tsai, Z. Zhang and C.-H. Wong, J. Org. Chem., 1999, 64, 5264–5279 CrossRef CAS; (b) S. Manandhar, R. P. Singh, G. V. Eggers and J. M. Shreeve, J. Org. Chem., 2002, 67, 6415–6420 CrossRef CAS PubMed; (c) J. S. Yadav, B. V. S. Reddy, D. N. Chary and D. Chandrakanth, Tetrahedron Lett., 2009, 50, 1136–1138 CrossRef CAS PubMed; (d) S. Qiu, T. Xu, J. Zhou, Y. Guo and G. Liu, J. Am. Chem. Soc., 2010, 132, 2856–2857 CrossRef CAS PubMed; (e) C. Appayee and S. E. Brenner-Moyer, Org. Lett., 2010, 12, 3356–3359 CrossRef CAS PubMed; (f) W. Kong, P. Feige, T. de Haro and C. Nevado, Angew. Chem., 2013, 125, 2529–2533 ( Angew. Chem. Int. Ed. , 2013 , 52 , 2469–2473 ) CrossRef.
- (a) R. P. Singh and J. M. Shreeve, Acc. Chem. Res., 2004, 37, 31–44 CrossRef CAS PubMed; (b) P. T. Nyffeler, S. G. Durón, M. D. Burkart, S. P. Vincent and C.-H. Wong, Angew. Chem., 2005, 117, 196–217 ( Angew. Chem. Int. Ed. , 2005 , 44 , 192–212 ) CrossRef.
- (a) F. Yin, Z. Wang, Z. Li and C. Li, J. Am. Chem. Soc., 2012, 134, 10401–10404 CrossRef CAS PubMed; (b) Z. Li, L. Song and C. Li, J. Am. Chem. Soc., 2013, 135, 4640–4643 CrossRef CAS PubMed; (c) C. Zhang, Z. Li, L. Zhu, L. Yu, Z. Wang and C. Li, J. Am. Chem. Soc., 2013, 135, 14082–14085 CrossRef CAS PubMed.
- For a highlight on radical fluorination, see: M. P. Sibi and Y. Landais, Angew. Chem., 2013, 125, 3654–3656 ( Angew. Chem. Int. Ed. , 2013 , 52 , 3570–3572 ) CrossRef.
- (a) M. Rueda-Becerril, C. C. Sazepin, J. C. T. Leung, T. Okbinoglu, P. Kennepohl, J.-F. Paquin and G. M. Sammis, J. Am. Chem. Soc., 2012, 134, 4026–4029 CrossRef CAS PubMed; (b) J. C. T. Leung, C. Chatalova-Sazepin, J. G. West, M. Rueda-Becerril, J.-F. Paquin and G. M. Sammis, Angew. Chem., 2012, 124, 10962–10965 ( Angew. Chem. Int. Ed. , 2012 , 51 , 10804–10807 ) CrossRef; (c) S. Bloom, C. R. Pitts, D. C. Miller, N. Haselton, M. G. Holl, E. Urheim and T. Lectka, Angew. Chem., 2012, 124, 10732–10735 ( Angew. Chem. Int. Ed. , 2012 , 51 , 10580–10583 ) CrossRef; (d) W. Liu, X. Huang, M.-J. Cheng, R. J. Nielsen, W. A. Goddard III and J. T. Groves, Science, 2012, 337, 1322–1325 CrossRef CAS PubMed; (e) T. J. Barker and D. L. Boger, J. Am. Chem. Soc., 2012, 134, 13588–13591 CrossRef CAS PubMed; (f) S. Mizuta, et al. , Org. Lett., 2013, 15, 2648–2651 CrossRef CAS PubMed.
- β-Fluorinated alkyl azides were typically synthesized from classical nucleophilic substitution reactions of NaN3 or the conversion of alcohols to fluorides by DAST. For examples, see: (a) J. A. D. Good, F. Wang, O. Rath, H. Y. K. Kaan, S. K. Talapatra, D. Podgorski, S. P. MacKay and F. Kozielski, J. Med. Chem., 2013, 56, 1878–1893 CrossRef CAS PubMed; (b) V. Mascitti, et al. , Bioorg. Med. Chem. Lett., 2011, 21, 1306–1309 CrossRef CAS PubMed; (c) F. Xue, J. Fang, W. W. Lewis, P. Martasek, L. J. Roman and R. B. Silverman, Bioorg. Med. Chem. Lett., 2010, 20, 554–557 CrossRef CAS PubMed; (d) R. Sasson and S. Rozen, J. Fluorine Chem., 2006, 127, 962–965 CrossRef CAS PubMed; (e) S. C. Annedi, W. Li, S. Samson and L. P. Kotra, J. Org. Chem., 2003, 68, 1043–1049 CrossRef CAS PubMed; (f) S. M. Andersen, M. Ebner, C. W. Ekhart, G. Gradnig, G. Legler, I. Lundt, A. E. Stutz, S. G. Withers and T. Wrodnigg, Carbohydr. Res., 1997, 301, 155–166 CrossRef CAS; (g) T. Kajimoto, K. K.-C. Liu, R. L. Pederson, Z. Zhong, Y. Ichikawa, J. A. Proco Jr. and C.-H. Wong, J. Am. Chem. Soc., 1991, 113, 6187–6196 CrossRef CAS.
- For selected reviews on organic azides, see: (a) S. Bräse, C. Gil, K. Knepper and V. Zimmermann, Angew. Chem., 2005, 117, 5320–5374 ( Angew. Chem. Int. Ed. , 2005 , 44 , 5188–5240 ) CrossRef; (b) Organic Azides: Syntheses and Applications, ed. S. Bräse and K. Banert, Wiley, Chichester, UK, 2010 Search PubMed.
- For selected examples of electrophilic fluorocyclization, see: (a) V. Rauniyar, A. D. Lackner, G. L. Hamilton and F. D. Toste, Science, 2011, 334, 1681–1684 CrossRef CAS PubMed; (b) Y. A. Serguchev, L. F. Lourie, M. V. Ponomarenko, E. B. Rusanov and N. V. Ignat'ev, Tetrahedron Lett., 2011, 52, 5166–5169 CrossRef CAS PubMed; (c) S. C. Wilkinson, O. Lozano, M. Schuler, M. C. Pacheco, R. Salmon and V. Gouverneur, Angew. Chem., 2009, 121, 7217–7220 ( Angew. Chem. Int. Ed. , 2009 , 48 , 7083–7086 ) CrossRef; (d) L. F. Lourie, Y. A. Serguchev, G. V. Shevchenko, M. V. Ponomarenko, A. N. Chernega, E. B. Rusanov and J. A. K. Howard, J. Fluorine Chem., 2006, 127, 377–385 CrossRef CAS PubMed.
- M. Newcomb, in Radicals in Organic Synthesis, ed. P. Renaud and M. P. Sibi, Wiley-VCH, Weinheim, Germany, 2001, vol. 1, pp. 317–336 Search PubMed.
- M. Jafarzadeh, Synlett, 2007, 2144–2145 CrossRef CAS PubMed.
- For reviews on the oxidative generation of azidyl radicals, see: (a) S. D. Jong, D. G. Nosal and D. J. Wardrop, Tetrahedron, 2012, 68, 4067–4105 CrossRef PubMed; (b) F. Minisci, Acc. Chem. Res., 1975, 8, 165–171 CrossRef CAS.
- For selected examples of oxidative generation of azidyl radicals from TMSN3, see: (a) P. Magnus, M. B. Roe and C. Hulme, J. Chem. Soc., Chem. Commun., 1995, 263–265 RSC; (b) P. Magnus, J. Lacour, P. A. Evans, M. B. Roe and C. Hulme, J. Am. Chem. Soc., 1996, 118, 3406–3418 CrossRef CAS; (c) C. Tang and N. Jiao, J. Am. Chem. Soc., 2012, 134, 18924–18927 CrossRef CAS PubMed; (d) K. Matcha and A. P. Antonchick, Angew. Chem., 2013, 125, 2136–2140 ( Angew. Chem. Int. Ed. , 2013 , 52 , 2082–2086 ) CrossRef; (e) K. Matcha, R. Narayan and A. P. Antonchick, Angew. Chem., 2013, 125, 8143–8147 ( Angew. Chem. Int. Ed. , 2013 , 52 , 7985–7989 ) CrossRef.
- For a review on radical azidation reactions, see: C. Jimeno and P. Renaud, in Organic Azides: Syntheses and Applications, ed. S. Bräse and K. Banert, Wiley, Chichester, UK, 2010, pp. 239–267 Search PubMed.
- For the latest example of the generation of azidyl radicals via single electron transfer, see: B. Zhang and A. Studer, Org. Lett., 2013, 15, 4548–4551 CrossRef CAS PubMed.
- X. Zhang, Y. Liao, R. Qian, H. Wang and Y. Guo, Org. Lett., 2005, 7, 3877–3880 CrossRef CAS PubMed.
- M. S. Workentin, B. D. Wagner, J. Lusztyk and D. D. M. Wayner, J. Am. Chem. Soc., 1995, 117, 119–126 CrossRef CAS.
- The F-abstraction of β-azidoalkyl radicals from Selectfluor reagent is unlikely in aqueous solution based on our previous mechanistic study. For details, see ref. 10a.
Footnotes |
| † Dedicated to Professor Max Malacria on the occasion of his 65th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c3qo00037k |
| This journal is © the Partner Organisations 2014 |