Asymmetric total synthesis of (−)-cebulactam A1†
Shouliang
Yang
a,
Yumeng
Xi
a,
Jia-Hua
Chen
*a and
Zhen
Yang
*ab
aKey Laboratory of Bioorganic Chemistry and Molecular Engineering of Ministry of Education and Beijing National Laboratory for Molecular Science (BNLMS), and Peking-Tsinghua Center for Life Sciences, Peking University, Beijing 100871, China. E-mail: jhchen@pku.edu.cn; zyang@pku.edu.cn
bLaboratory of Chemical Genomics, School of Chemical Biology and Biotechnology, Peking University Shenzhen Graduate School, Shenzhen, 518055, China
First published on 8th January 2014
Abstract
The total synthesis of (−)-cebulactam A1 (3) has been achieved for the first time in 18 steps. The key steps in this synthesis included an asymmetric chelation-controlled vinylogous Mukaiyama aldol reaction for the stereoselective synthesis of the stereogenic centers at the C8 and C9 positions, an intramolecular SmI2-mediated Reformatsky reaction for the formation of a macrocyclic lactam, and an SN2′ reaction for the stereoselective formation of the (E)-double bond linked tetrahydropyran moiety of cebulactam A1 (3).
Introduction
(−)-Q-1047H-R-A (1), (−)-Q-1047H-A-A (2),1 cebulactam A1 (3)2 and tetrapetalone A (4)3 represent a unique class of natural products (Fig. 1), which display a broad range of biological activities, such as antioxidant activity1,4 of compound 1 and soybean lipoxygenase inhibitory activity5 for compound 4.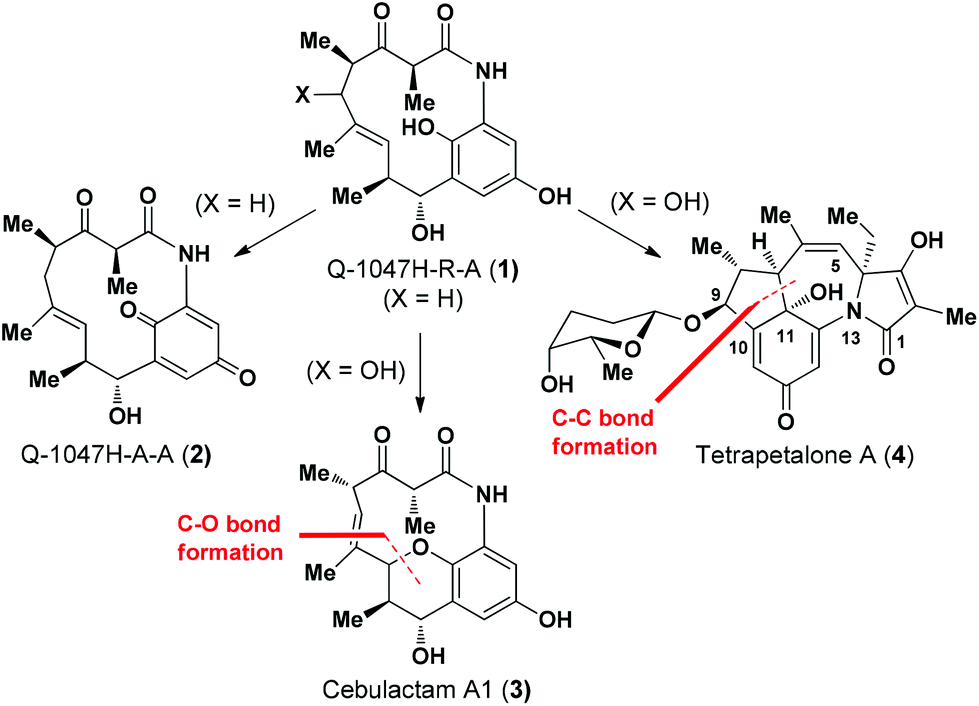 | ||
| Fig. 1 Naturally occurring biologically active products. | ||
In our previous communication,6 we achieved the total synthesis of Q-1047H-R-A (1) from Q-1047H-A-A (2) via a Na2S2O4-mediated reduction (Fig. 1). In that report, we also proposed a biogenetic strategy7 for the total synthesis of natural products 3 and 4 from 1, involving the use of C–O and C–C bond forming reactions as the key synthetic steps, respectively (Fig. 1).
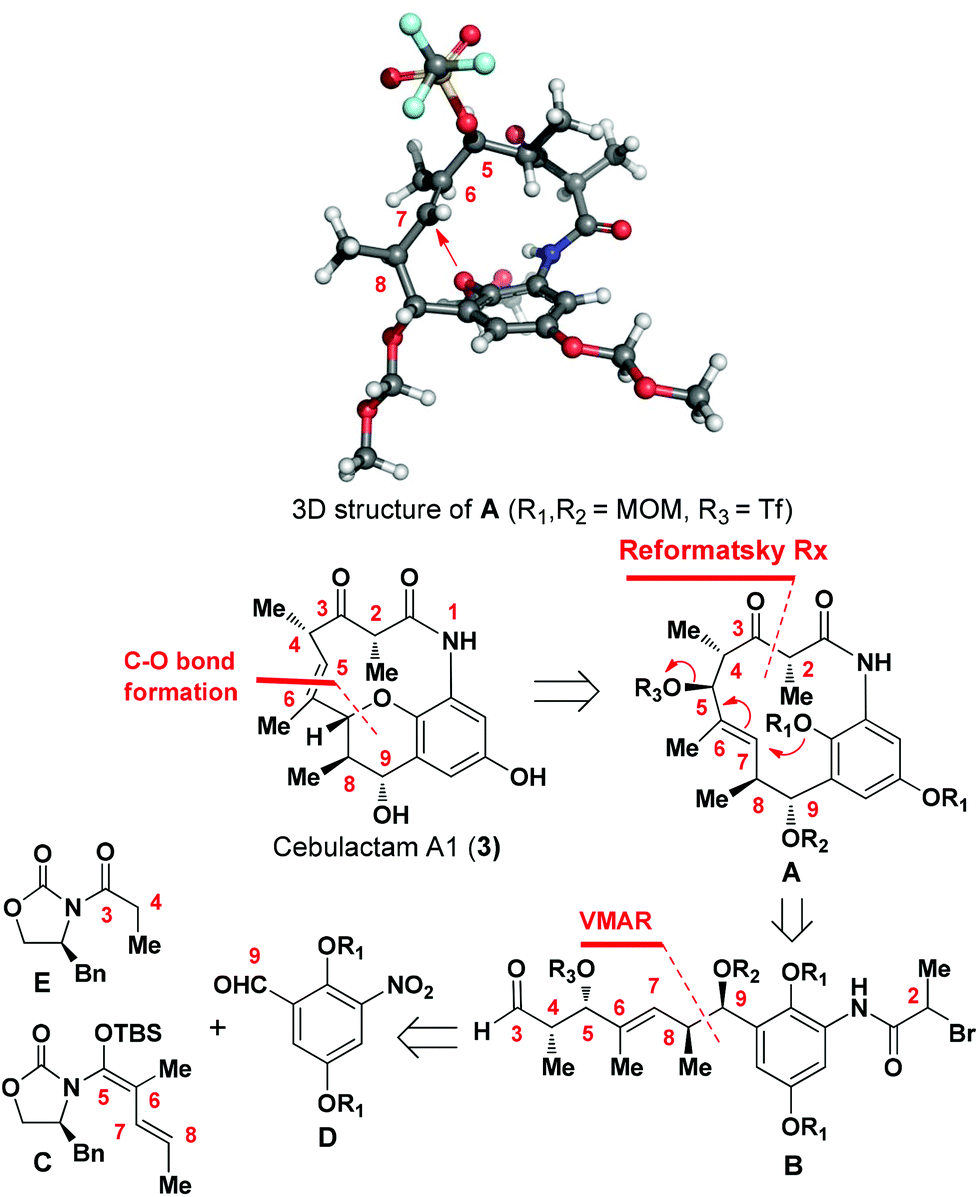
Cebulactam A1 (3) is an 11-membered macrocyclic lactam that possesses several interesting structural features, including five stereogenic centers, an isolated trisubstituted (E)-double bond, a tetrahydropyran (THP), and a hydroquinone ring system. Synthetically, it was envisaged that the THP moiety could be regio- and stereoselectively constructed from the η-hydroxy allylic alcohol in lactam A using a transition metal-mediated O-allylation8 or a formal SN2′ reaction9 (Fig. 2).
 | ||
| Fig. 2 Strategy for synthesis of cebulactam A1 (3). | ||
The most challenging aspect of this particular approach, however, would be the development of a transformation capable of discriminating the heterotopic-faces of lactam A to allow the selective installation of the C7 stereogenic center in cebulactam A1 (3). Conformational analysis of the 3D structure of lactam A (see Fig. 2) suggested that the methyl group at the C8 chiral center could be used to control the attack of the oxygen to the extent that it would occur from the backside. This facial selectivity would allow for the stereochemistry of the THP, as well as its connected double bond could be installed regio- and stereoselectively.
The realization of this strategy would allow us to adopt the chemistry that we had developed previously for our total syntheses of Q-1047H-R-A (1) and Q-1047H-A-A (2),6 because the 13-membered lactam A could be formed via the SmI2-mediated Reformatsky reaction10 from B which could in turn be derived from C, D and E with sequential chelation-controlled vinylogous Mukaiyama aldol (VMAR)11 and Evans aldol12 reactions being used as the key steps for the stereoselective construction of the four stereogenic centers at C8 and C9, C4 and C5. It was also envisaged that the C2 chiral center of cebulactam A1 (3) could be stereoselectively installed during the latter stages of the total synthesis because of its tendency to epimerize under acidic, basic or even neutral conditions.6 Herein, we report the stereocontrolled total synthesis of (−)-cebulactam A1 (3) according to a strategy involving the SmI2-mediated intramolecular Reformatsky-type reaction of bromoaldehyde B to form a macrolactam, and an SN2′ reaction for the formation of the THP moiety, as shown in Fig. 2.
Results and discussion
Synthesis of Reformatsky precursor 13
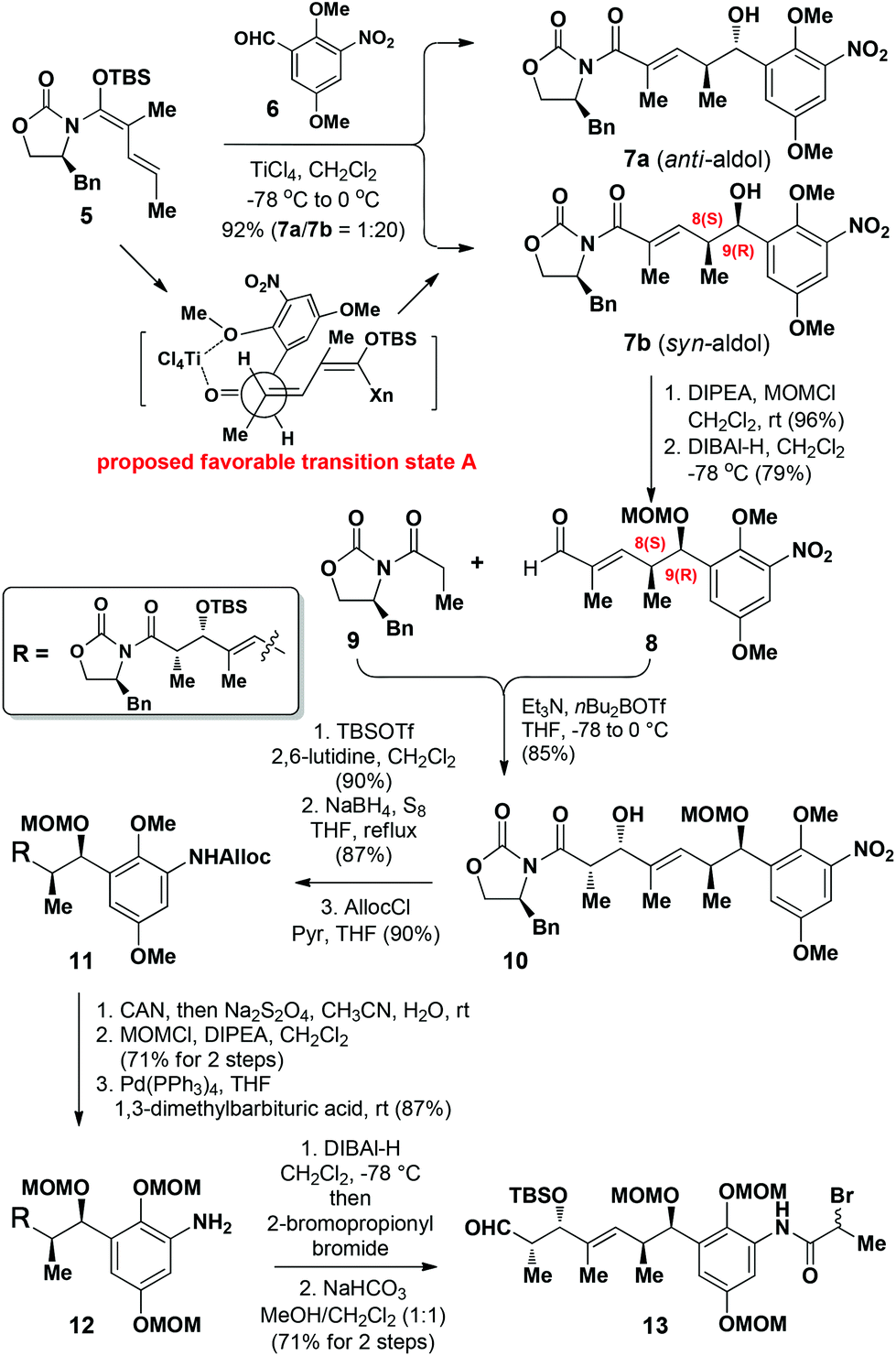
Our synthesis started with the construction of amide 13 (Scheme 1). The C8 and C9 chiral centers in compound 7b were efficiently installed using our previously developed chelation-controlled VMAR.13 The reaction of N,O-acetal 513a with aldehyde 6 under the optimized conditions13b in the presence of TiCl4 (1.1 equiv.) in CH2Cl2 afforded the anti-aldol adduct 7a and the syn-aldol adduct 7b in an isolated yield of 92% as an inseparable mixture of diastereomers (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20) at the C9-position. The favored formation of 7b was rationalized according to the formation of the chelation complex A, where the formation of a 6-membered chelation complex with TiCl4 would be preferred, leading to the formation of the syn-aldol product 7b as the major product.6
:20) at the C9-position. The favored formation of 7b was rationalized according to the formation of the chelation complex A, where the formation of a 6-membered chelation complex with TiCl4 would be preferred, leading to the formation of the syn-aldol product 7b as the major product.6
 | ||
| Scheme 1 Asymmetric synthesis of amide 13. | ||
We then proceeded with the straightforward elaboration of compound 7b to amide 13 (Scheme 2). 7b was initially protected as its methoxymethyl (MOM) ether prior to being reduced with DIBAl-H to afford aldehyde 8, and the minor diastereoisomer generated in the first step could be separated after reduction. To allow the installation of the required chirality at the C4-position, compound 8 was subsequently reacted with Evans’ oxazolidinone 912 to afford 10 as a single diastereoisomer in 85% yield. Compound 10 was then protected as the corresponding TBS ether using TBSOTf/2,6-lutidine, and its nitro group subsequently reduced with NaBH4/S814 to give the corresponding amine, which was protected with an allyloxycarbonyl (Alloc) group using AllocCl/pyridine to afford 11.
 | ||
| Scheme 2 Synthesis of intermediate 15. | ||
The methyl protecting groups of the phenol moieties in substrate 11 were converted to the corresponding MOM groups via an oxidative demethylation/reduction/protection sequence. The resulting MOM protected compound was then subjected to a Pd-catalyzed deprotection reaction in the presence of 1,3-dimethylbarbituric acid to allow the removal of the Alloc group15 to give 12 in 62% yield over three steps. The reduction of 12 with DIBAl-H followed by the acylation of the aniline with 2-bromopropionyl bromide and subsequent removal of the Evans chiral auxiliary gave 13 in 71% yield over the two steps.
Completion of the total synthesis
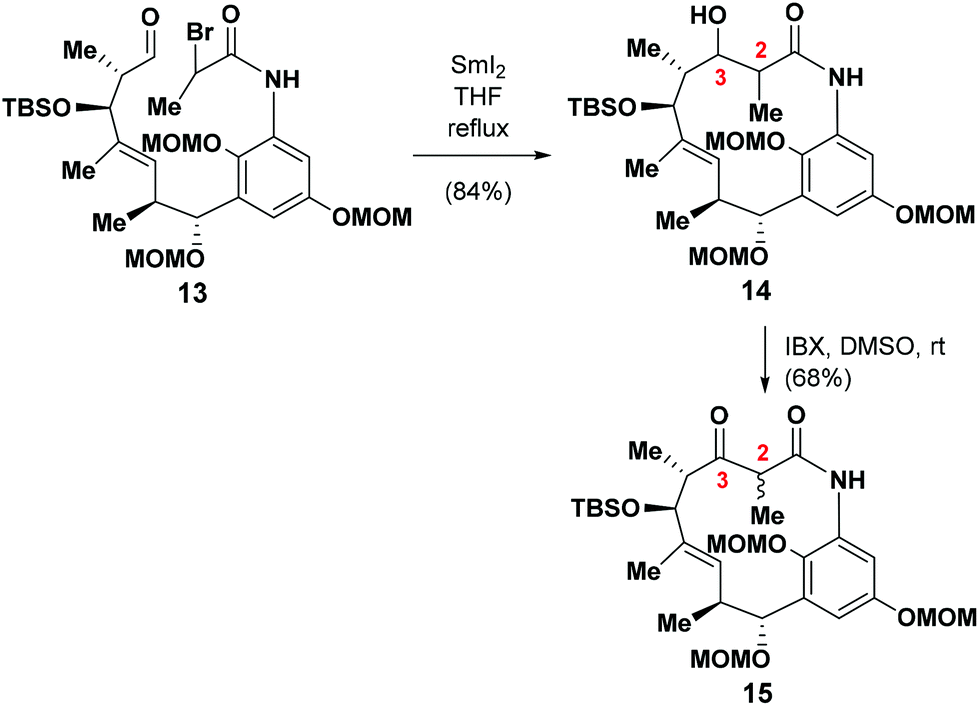
With 13 in hand, we proceeded to evaluate its performance in the SmI2-mediated intramolecular Reformatsky reaction.16The treatment of 13 with SmI2 in THF under the optimized conditions6 at a concentration of 0.005 M under refluxing conditions gave the desired product 14 in 84% yield as a couple of diastereoisomers. We then began to explore the remainder of the total synthesis. The annulated mixture of 14 was oxidized with IBX to afford 15 as a single diastereoisomer in 68% yield. The relative stereochemistry at C2 was not assigned at this stage, because it was envisaged that this chiral center would exist in its natural form as a consequence of the epimerization process outlined above.
To complete the total synthesis, we started to investigate the development of a method for the synthesis of the THP moiety in cebulactam A1 (3), with the proposed transition metal-mediated cyclization reaction8 of the η-hydroxy allylic alcohol in 17 being our preferred method (Scheme 3). A variety of different methods17 were evaluated for removal of the MOM groups in 16, but none of the desired products was obtained, and it was assumed that the resulting phenol species are unstable under the reaction conditions.
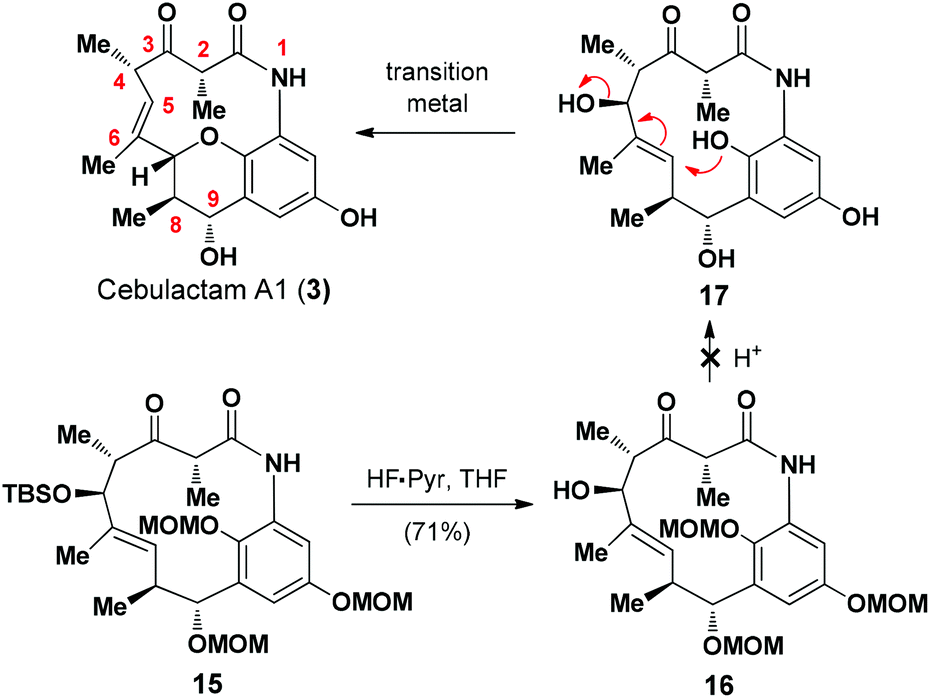 | ||
| Scheme 3 Attempt to synthesise cebulactam A1 (3) from 15. | ||
We then proceeded to develop an annulation/deprotection strategy for the formation of the THP ring using the activated η-hydroxy allylic alcohol as the substrate. The hydroxyl group in 16 was converted to the corresponding mesylate 18, which was subsequently treated with TFA in CH2Cl2 to induce the annulation/deprotection process. Unfortunately, however, the desired reaction did not proceed effectively under any of the reaction conditions tested, with only trace amounts of cebulactam A1 (3) being observed (Scheme 4).
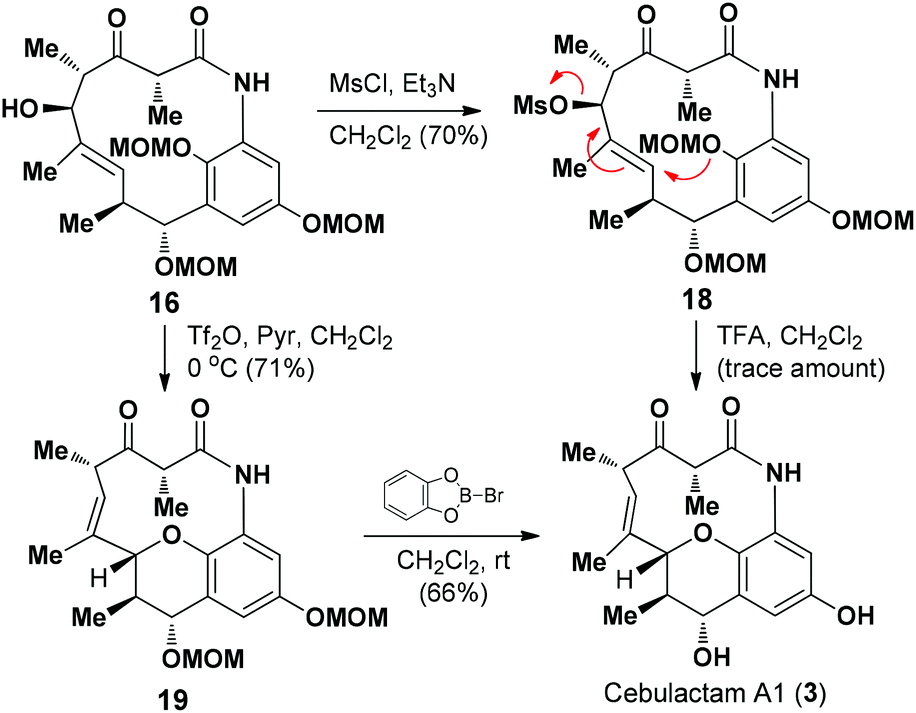 | ||
| Scheme 4 Completion of the total synthesis. | ||
We then attempted to convert the hydroxy group in 16 into the corresponding triflate. Pleasingly, when alcohol 16 was added to a solution of Tf2O in CH2Cl2 in the presence of pyridine at 0 °C, the THP product 19 was obtained in 71% yield via a triflation/annulation/deprotection process. To complete the total synthesis, compound 19 was treated with B-bromocatecholborane at room temperature to remove its two MOM groups and give cebulactam A1 (3) in 66% yield.
The 1H and 13C NMR spectral data for the synthesized cebulactam A1 (3) were in good agreement18 with those reported for the natural material.2 The optical rotation of our synthesized cebulactam A1 (3) was also determined {[α]20D = −66.1° (c 1.0 in acetone)}, although similar data were not reported in the original isolation paper.
Conclusions
The first total synthesis of (−)-cebulactam A1 (3) had been completed in 18 synthetic steps, with an overall yield of 3.2% from 2,5-dimethoxy-3-nitrobenzaldehyde 6. The key synthetic transformations involved in this synthesis include a SmI2-mediated reductive cyclization to generate the highly functionalized 13-membered lactam 14, as well as an SN2′ reaction to assemble the double bond connected to the THP moiety of (−)-cebulactam A1 (3).Experimental section
General methods
Unless otherwise mentioned, all reactions were carried out under a nitrogen atmosphere using Schlenk techniques. Solvent purification was conducted according to Purification of Laboratory Chemicals (D. D. Peerrin, W. L. Armarego, and D. R. Perrins, Pergamon Press, Oxford, 1980). Compound 5 was synthesized from L-phenylglycinol according to the literature.19 Compound 6 was synthesized from 2-hydroxy-5-methoxybenzaldehyde according to the literature.20 All other reagents were purchased from commercial sources and used as received. Yields refer to chromatographically and spectroscopically (1H NMR) homogeneous materials. Reactions were monitored by thin layer chromatography on plates (GF254) supplied by Yantai Chemicals (China) using UV light as a visualizing agent and iodine as a developing agent. If not specially mentioned, flash column chromatography uses silica gel (200–300 mesh) supplied by Tsingtao Haiyang Chemicals (China).NMR spectra were recorded on Bruker Ascend 500 and Ascend 400 instruments. TMS was used as an internal standard for 1H NMR (0 ppm), and solvent signal was used as a reference for 13C NMR (CDCl3 = δ 77.0 ppm, acetone-d6 = δ 29.84, 206.26 ppm). The following abbreviations were used to explain the multiplicities: s = singlet, d = doublet, br = broad, dd = double doublet, m = multiplet. Infrared (IR) spectra were recorded on a Thermo Nicolet Avatar 330 FT-IR spectrometer. Mass spectra were recorded on a Bruker Apex IV FTMS mass spectrometer using ESI (electrospray ionization).
:1:1) to give colorless oil product 7b (2.870 g, 92%). Rf = 0.25 (hexane–ethyl acetate = 2:1); IR vmax (neat)/cm−1: 3506 (br), 2931, 1785, 1685, 1531, 1352, 1298, 1217, 1048, 995, 773, 762, 703; 1H NMR (400 MHz, CDCl3): δ 7.42–7.25 (m, 5H), 7.25–7.13 (m, 2H), 6.02 (dd, J = 9.3, 1.4 Hz, 1H), 5.25 (dd, J = 3.9, 3.8 Hz, 1H), 4.89–4.78 (m, 1H), 4.32 (dd, J = 8.8, 8.7 Hz, 1H), 4.20 (dd, J = 9.0, 6.5 Hz, 1H), 3.87 (s, 3H), 3.82 (s, 3H), 3.34–3.19 (m, 2H), 3.13–2.98 (m, 1H), 2.90 (dd, J = 13.6, 1 Hz), 2.08 (d, J = 1.3 Hz, 3H), 0.85 (d, J = 6.8 Hz, 3H); 13C NMR (101 MHz, CDCl3: δ 171.3, 155.0, 154.2, 143.8, 143.5, 143.2, 139.3, 134.7, 130.2, 129.4, 128.9, 127.4, 119.0, 108.6, 70.9, 66.3, 62.5, 55.9, 55.0, 37.9, 37.4, 13.4, 12.4; HRMS (ESI): calcd for C25H28N2NaO8 [M + Na+] 507.1738; found 507.1734. [α]20D = +17 (1.0, CHCl3).
:1:1) to give product 8a (5.34 g, 96%) as a yellowish oil. Rf = 0.40 (hexane–ethyl acetate = 2:1); IR vmax (neat)/cm−1: 2972, 1786, 1685, 1534, 1350, 1305, 1215, 1048, 1029, 763, 703; 1H NMR (400 MHz, CDCl3): δ 7.36–7.22 (m, 5H), 7.21–7.16 (m, 2H), 5.90 (dd, J = 9.9, 1.4 Hz, 1H), 4.95 (d, J = 5.5 Hz, 1H), 4.68–4.53 (m, 3H), 4.23 (dd, J = 8.8, 8.0 Hz, 1H), 4.14 (dd, J = 9.0, 4.8 Hz, 1H), 3.87 (s, 3H), 3.84 (s, 3H), 3.35 (s, 3H), 3.34–3.28 (m, 1H), 2.94–2.76 (m, 2H), 1.85 (d, J = 1.4 Hz, 3H), 1.07 (d, J = 6.8 Hz, 3H); 13C NMR (101 MHz, CDCl3): δ 171.6, 155.1, 152.7, 145.0, 143.7, 138.5, 138.2, 135.1, 131.3, 129.4, 128.9, 127.3, 119.2, 108.9, 95.3, 74.6, 66.3, 62.8, 55.9, 55.9, 55.4, 38.8, 37.5, 14.5, 13.6; HRMS (ESI): calcd for C27H33N2O9 [M + H+] 529.2181; found 529.2184. [α]20D= +68 (1.0, CHCl3).
:1:1) to give product 8 (1.05 g, 79%) as a yellowish oil. Rf = 0.51 (hexane–ethyl acetate = 2:1); IR vmax (neat)/cm−1: 2943, 1778, 1687, 1534, 1482, 1352, 1228, 1152, 1100, 1049, 1027, 993, 830, 776; 1H NMR (400 MHz, CDCl3): δ 9.35 (s, 1H), 7.29 (d, J = 3.1 Hz, 1H), 7.21 (d, J = 3.2 Hz, 1H), 6.41 (dd, J = 10.1, 1.3 Hz, 1H), 4.98 (d, J = 5.9 Hz, 1H), 4.60 (d, J = 6.9 Hz, 1H), 4.55 (d, J = 6.9 Hz, 1H), 3.86 (s, 3H), 3.84 (s, 3H), 3.37 (s, 3H), 3.12–3.03 (m, 1H), 1.67 (d, J = 1.4 Hz, 3H), 1.14 (d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 195.1, 155.2, 154.8, 145.0, 143.6, 139.1, 137.7, 119.2, 108.9, 94.8, 74.0, 62.8, 56.0, 39.6, 14.8, 9.2; HRMS (ESI): calcd for C17H23NNaO7 [M + Na+] 376.1367; found 376.1369; [α]20D= +80.4 (1.0, CHCl3).
:2 v/v, 111 mL). The resulting slurry was then stirred vigorously at room temperature for 1 h. After removing the solvent, the reaction mixture was extracted with Et2O (3 × 100 mL), and the combined organic extracts were washed with water (2 × 50 mL), a saturated solution of NaHCO3 (2 × 20 mL), and brine (2 × 20 mL), then dried over Na2SO4. The solvent was removed under vacuum, and the residue was purified by flash column chromatography on silica gel (hexane–ethyl acetate = 2:1 to 1:1) to give product 10 (10.44 g, 85%) as a colourless oil. Rf = 0.30 (hexane–ethyl acetate = 2:1); IR vmax (neat)/cm−1: 3498, 2938, 1778, 1701, 1533, 1362, 1210, 1099, 1031, 918, 763, 751, 738, 706, 677; 1H NMR (400 MHz, CDCl3): δ 7.36–7.28 (m, 3H), 7.24–7.16 (m, 4H), 5.45 (d, J = 10.2 Hz, 1H), 4.81 (d, J = 7.9 Hz, 1H), 4.72–4.62 (m, 1H), 4.55 (dd, J = 19.6, 6.8 Hz, 2H), 4.29–4.14 (m, 3H), 3.87 (s, 3H), 3.85–3.76 (m, 4H), 3.35 (s, 3H), 3.21 (dd, J = 13.4, 2.9 Hz, 1H), 2.97 (s, 1H), 2.86–2.73 (m, 2H), 1.42 (s, 3H), 1.12 (d, J = 6.5 Hz, 3H), 0.79 (d, J = 7.0 Hz, 3H); 13C NMR (101 MHz, CDCl3): δ 176.8, 155.1, 152.8, 145.1, 143.7, 139.2, 134.9, 133.8, 129.3, 128.8, 127.3, 126.9, 118.8, 108.7, 94.7, 75.1, 74.5, 66.0, 62.8, 55.9, 55.7, 55.0, 39.6, 39.2, 37.5, 17.1, 13.2, 9.5; HRMS (ESI): calcd for C30H38N2NaO10 [M + Na+] 609.2419; found 609.2423; [α]20D = +63.7 (1.0, CHCl3).
:1 to 4:1) to give product 11a (19.23 g, 90%) as a colourless oil. Rf = 0.68 (hexane–ethyl acetate = 1:1); IR vmax (neat)/cm−1: 2956, 2927, 2359, 1781, 1700, 1534, 1362, 1211, 1103, 1032, 844, 838, 777, 722, 685; 1H NMR (400 MHz, CDCl3): δ 7.37–7.27 (m, 3H), 7.25 (d, J = 3.2 Hz, 1H), 7.20 (d, J = 7.2 Hz, 2H), 7.17 (d, J = 3.1 Hz, 1H), 5.30 (d, J = 10.0 Hz, 1H), 4.81 (d, J = 7.3 Hz, 1H), 4.52 (dd, J = 16.7, 6.9 Hz, 3H), 4.21–4.05 (m, 3H), 4.00–3.91 (m, 1H), 3.87 (s, 3H), 3.81 (s, 3H), 3.35 (s, 3H), 3.24 (dd, J = 13.3, 2.6 Hz, 1H), 2.87–2.70 (m, 2H), 0.99 (d, J = 6.3 Hz, 6H), 0.84 (s, 9H), −0.11 (s, 3H), −0.27 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 174.7, 155.1, 152.9, 145.4, 143.8, 138.9, 135.9, 135.2, 129.4, 128.9, 127.8, 127.3, 119.1, 109.0, 94.8, 77.6, 75.0, 65.8, 62.8, 55.9, 55.6, 42.1, 38.6, 37.6, 25.7, 17.9, 16.8, 12.1, 11.9, −5.0, −5.7; HRMS (ESI): calcd for C36H52N2NaO10Si [M + Na+] 723.3283; found 723.3272; [α]20D = +64.0 (1.0, CHCl3).
:1 to 2:1) to give product 11b (1.242 g, 87%) as a yellowish oil. Rf = 0.48 (hexane–ethyl acetate = 2:1); IR vmax (neat)/cm−1: 2955, 1781, 1700, 1616, 1495, 1380, 1210, 1034, 837, 776, 761, 679; 1H NMR (400 MHz, CDCl3): δ 7.37–7.25 (m, 3H), 7.20 (d, J = 7.1 Hz, 2H), 6.24 (s, 1H), 6.17 (d, J = 2.4 Hz, 1H), 5.33 (d, J = 9.8 Hz, 1H), 4.72 (d, J = 7.6 Hz, 1H), 4.59–4.44 (m, 3H), 4.21–4.04 (m, 3H), 4.00–3.88 (m, 1H), 3.70 (d, J = 4.5 Hz, 8H), 3.38 (s, 3H), 3.23 (d, J = 13.1 Hz, 1H), 2.87–2.69 (m, 2H), 1.53 (s, 3H), 1.02 (d, J = 6.5 Hz, 3H), 0.97 (d, J = 6.6 Hz, 3H), 0.86 (s, 9H), −0.10 (s, 3H), −0.21 (s, 3H); 13C NMR (101 MHz, CDCl3): δ 174.9, 156.5, 152.9, 140.5, 140.2, 135.3, 134.7, 134.4, 129.4, 129.1, 128.9, 127.3, 101.4, 101.3, 94.2, 77.9, 75.4, 65.8, 59.7, 55.7, 55.6, 55.3, 42.2, 38.6, 37.6, 25.7, 18.0, 17.1, 11.9, 11.8, −4.9, −5.7. HRMS (ESI): calcd for C36H54N2NaO8Si [M + Na+] 693.3542; found 693.3531. [α]20D = +68.3 (1.0, CHCl3).
:1 to 4:1) to give product 11 (8.357 g, 85%) as a colourless oil. Rf = 0.60 (hexane–ethyl acetate = 2:1); IR vmax (neat)/cm−1: 3428 (br), 2952, 1781, 1736, 1524, 1463, 1361, 1235, 1212, 1032, 839, 776, 703, 678; 1H NMR (400 MHz, CDCl3): δ 7.63 (s, 1H), 7.36–7.26 (m, 3H), 7.23–7.12 (m, 3H), 6.56 (d, J = 3.0 Hz, 1H), 6.06–5.93 (m, 1H), 5.38 (dd, J = 17.2, 1.3 Hz, 1H), 5.32–5.24 (m, 2H), 4.73–4.65 (m, 3H), 4.56–4.47 (m, 3H), 4.18–4.06 (m, 3H), 3.97–3.87 (m, 1H), 3.76 (s, 3H), 3.73 (s, 3H), 3.37 (s, 3H), 3.23 (dd, J = 13.3, 2.6 Hz, 1H), 2.86–2.72 (m, 2H), 1.51 (s, 3H), 1.03 (d, J = 6.6 Hz, 3H), 0.97–0.90 (m, 3H), 0.85 (s, 9H), −0.12 (s, 3H), −0.25 (s, 3H); 13C NMR (101 MHz, CDCl3): δ 174.7, 156.3, 152.9, 152.9, 140.6, 135.2, 135.0, 134.3, 132.3, 131.8, 129.3, 128.8, 128.4, 127.2, 118.1, 106.8, 103.8, 93.9, 77.6, 75.1, 65.7, 61.1, 55.6, 55.5, 55.4, 42.0, 38.8, 37.5, 25.6, 17.9, 17.2, 11.8, −5.0, −5.8; HRMS (ESI): calcd for C40H58N2NaO10Si [M + Na+] 777.3753; found 777.3777; [α]20D = +50.3 (1.0, CHCl3).
:1, 200 mL) was added a solution of CAN (19.8 g, 36.11 mmol) in a mixed solvent of CH3CN and H2O (1:1, 50 mL) at 0 °C, and the mixture was then stirred at the same temperature for 30 min. The reaction was worked up by addition of Na2S2O4 (13.67 g, 78.51 mmol) and NaHCO3 (4.40 g, 52.37 mmol), and the reaction mixture was warmed up to room temperature, and stirred for an additional 10 min. To this mixture was added H2O (100 mL), and the mixture was then extracted with Et2O (3 × 100 mL), and the combined organic extracts were washed with brine (2 × 50 mL), and finally dried over anhydrous MgSO4. The solvent was removed under vacuum, and the residue was dissolved in dry CH2Cl2 (100 mL) and the formed mixture was cooled to 0 °C. To this solution were added iPr2NEt (15.8 mL, 95.59 mmol) and MOMCl (6.54 mL, 86.93 mmol), and the formed mixture was stirred at room temperature overnight. The reaction was quenched by addition of a saturated aqueous solution of NH4Cl (60 mL), and the mixture was then extracted with CH2Cl2 (3 × 60 mL). The combined organic extracts were first washed with brine (2 × 20 mL), and then dried over anhydrous Na2SO4. The solvent was removed under vacuum, and the residue was purified by flash column chromatography on silica gel (hexane–ethyl acetate = 8:1 to 4:1) to give product 12a (5.05 g, 71%) as a colourless oil. Rf = 0.65 (hexane–ethyl acetate = 2:1); IR vmax (neat)/cm−1: 3649, 3423 (br), 2957, 1785, 1741, 1540, 1473, 1455, 1209, 1076, 1029, 966, 837, 762; 1H NMR (400 MHz, CDCl3): δ 7.95 (s, 1H), 7.74 (s, 1H), 7.37–7.27 (m, 3H), 7.20 (d, J = 7.1 Hz, 2H), 6.68 (d, J = 2.8 Hz, 1H), 6.13–5.82 (m, 1H), 5.36 (dd, J = 17.2, 1.2 Hz, 1H), 5.29–5.21 (m, 2H), 5.12 (dd, J = 14.8, 6.6 Hz, 2H), 5.02 (d, J = 6.0 Hz, 1H), 4.90 (d, J = 6.0 Hz, 1H), 4.66 (d, J = 5.4 Hz, 2H), 4.59 (d, J = 8.1 Hz, 1H), 4.57–4.49 (m, 1H), 4.48 (s, 2H), 4.18–4.06 (m, 3H), 3.93–3.85 (m, 1H), 3.63 (s, 3H), 3.45 (s, 3H), 3.33 (s, 3H), 3.24 (dd, J = 13.3, 2.5 Hz, 1H), 2.89–2.70 (m, 2H), 1.49 (s, 3H), 1.01 (d, J = 6.6 Hz, 3H), 0.89 (d, J = 6.7 Hz, 3H), 0.86 (s, 9H), −0.13 (s, 3H), −0.24 (s, 3H); 13C NMR (101 MHz, CDCl3): δ 174.8, 154.2, 153.1, 152.9, 140.4, 135.3, 135.1, 134.9, 132.6, 132.5, 129.4, 128.9, 128.3, 127.3, 117.7, 100.7, 94.7, 94.3, 65.9, 65.6, 57.4, 55.9, 55.7, 55.7, 42.1, 38.7, 37.6, 25.8, 18.1, 17.4, 11.9, 11.6, −4.9, −5.7; HRMS (ESI): calcd for C42H62N2NaO12Si [M + Na+] 837.3964; found 837.3966; [α]20D = +62.7 (1.0, CHCl3).
:1 to 2:1) to give product 12 (2.087 g, 87%) as a colourless oil. Rf = 0.53 (hexane–ethyl acetate = 2:1); IR vmax (neat)/cm−1: 2930, 1781, 1701, 1618, 1491, 1454, 1361, 1210, 1149, 1074, 1029, 968, 838, 778, 703; 1H NMR (400 MHz, CDCl3): δ 7.41–7.31 (m, 3H), 7.25 (d, J = 7.8 Hz, 2H), 6.42 (d, J = 1.4 Hz, 1H), 6.37 (d, J = 2.6 Hz, 1H), 5.38 (d, J = 10.0 Hz, 1H), 5.15–5.07 (m, 2H), 4.98 (dd, J = 22.7, 5.7 Hz, 2H), 4.73 (d, J = 7.5 Hz, 1H), 4.62–4.49 (m, 3H), 4.27–4.11 (m, 3H), 4.03 (s, 2H), 4.00–3.93 (m, 1H), 3.66 (s, 3H), 3.48 (s, 3H), 3.41 (s, 3H), 3.30 (d, J = 13.0 Hz, 1H), 2.94–2.76 (m, 2H), 1.58 (s, 3H), 1.05 (d, J = 6.6 Hz, 3H), 1.00 (d, J = 6.7 Hz, 3H), 0.92 (s, 9H), −0.04 (s, 3H), −0.14 (s, 3H); 13C NMR (101 MHz, CDCl3): δ 174.8, 154.4, 152.9, 140.8, 138.6, 135.3, 135.2, 134.6, 129.4, 128.9, 127.3, 104.5, 103.2, 99.5, 94.6, 94.2, 77.6, 75.8, 65.8, 57.2, 55.7, 55.7, 55.6, 42.1, 38.4, 37.6, 25.7, 18.0, 17.1, 11.8, 11.6, −4.9, −5.7; HRMS (ESI): calcd for C38H59N2O10Si [M + H+] 731.3934; found 731.3951. [α]20D = +61.9 (1.0, CHCl3).
:1, 60 mL). To this mixture was added NaHCO3 (1.04 g, 12.38 mmol) at room temperature, and the resulting mixture was stirred at the same temperature for 5 h. The reaction was worked up by removal of the solvent under vacuum, and the residue was purified by flash column chromatography on silica gel (hexane–ethyl acetate = 8:1 to 4:1) to give product 13 (3.055 g, 71%) as a colourless oil. Rf = 0.70, 0.72 (hexane–ethyl acetate = 1:1); IR vmax (neat)/cm−1: 3344 (br), 2931, 2855, 1702, 1604, 1534, 1453, 1398, 1152, 1101, 1030, 960, 837, 777; 1H NMR (400 MHz, CDCl3): δ 9.56 (d, J = 1.3 Hz, 1H), 9.01 (s, 1H), 7.98 (d, J = 2.8 Hz, 1H), 6.77 (d, J = 2.8 Hz, 1H), 5.28 (d, J = 10.1 Hz, 1H), 5.17–5.03 (m, 3H), 4.91 (d, J = 5.7 Hz, 1H), 4.61 (d, J = 8.4 Hz, 1H), 4.52–4.42 (m, 3H), 4.19 (d, J = 4.5 Hz, 1H), 3.62 (s, 3H), 3.43 (s, 3H), 3.32 (s, 3H), 2.88–2.75 (m, 1H), 2.34–2.23 (m, 1H), 1.92 (d, J = 7.0 Hz, 3H), 1.42 (s, 3H), 1.08 (d, J = 6.5 Hz, 3H), 0.82 (s, 9H), 0.70 (d, J = 6.8 Hz, 3H), −0.10 (s, 3H), −0.20 (s, 3H); 13C NMR (101 MHz, CDCl3): δ 204.5, 167.3, 153.9, 140.7, 134.9, 134.6, 131.8, 128.1, 110.5, 108.3, 100.9, 94.6, 94.2, 76.4, 75.8, 57.7, 55.9, 55.6, 50.4, 44.7, 39.1, 25.7, 22.4, 17.9, 17.5, 12.8, 7.7, −4.7, −5.6; HRMS (ESI): calcd for C31H52BrNNaO9Si [M + Na+] 712.2487; found 712.2493.
:1 to 1:1) to give product 14 (0.85 g, 84%) as a mixture of diastereoisomers. Rf = 0.22, 0.24 (hexane–ethyl acetate = 1:1); IR vmax (neat)/cm−1: 2953, 2901, 2363, 1659, 1473, 1401, 1249, 1150, 1073, 1028, 966, 923, 835, 774; 1H NMR (400 MHz, CDCl3): δ 7.49 (s, 1H), 7.11 (d, J = 2.9 Hz, 1H), 6.87 (d, J = 2.9 Hz, 1H), 5.44 (d, J = 10.7 Hz, 1H), 5.14 (dd, J = 15.9, 6.8 Hz, 2H), 5.05 (d, J = 6.0 Hz, 1H), 4.78 (d, J = 6.0 Hz, 1H), 4.62 (s, 2H), 4.58 (d, J = 9.9 Hz, 1H), 3.56 (s, 3H), 3.45 (s, 3H), 3.41–3.35 (m, 4H), 3.05–2.93 (m, 2H), 2.64–2.55 (m, 1H), 2.51–2.40 (m, 2H), 1.29 (s, 3H), 1.22 (d, J = 7.2 Hz, 3H), 1.19 (d, J = 6.5 Hz, 3H), 1.00 (d, J = 7.0 Hz, 3H), 0.88 (s, 9H), −0.04 (s, 3H), −0.10 (s, 3H); 13C NMR (101 MHz, CDCl3): δ 178.3, 153.8, 146.7, 138.5, 138.2, 131.1, 127.9, 115.1, 113.7, 100.5, 95.0, 94.7, 79.4, 76.4, 71.5, 57.3, 55.9, 55.8, 46.7, 42.2, 40.5, 25.9, 18.1, 17.9, 16.5, 14.1, 12.7, −4.4, −5.2; HRMS (ESI): calcd for C31H54NO9Si [M + H+] 612.3562; found 612.3571; [α]20D = +36.5 (1.0, CHCl3).
:1, 3 × 80 mL). The combined organic extracts were washed with brine (2 × 30 mL), and then dried over anhydrous Na2SO4. The solvent was removed under vacuum, and the residue was purified by flash column chromatography on silica gel (hexane–ethyl acetate = 2:1 to 1:1) to give product 15 (593 mg, 68%) as white solids. Rf = 0.70 (hexane–ethyl acetate = 1:1); IR vmax (neat)/cm−1: 3676, 2989, 2971, 2901, 2357, 1405, 1394, 1250, 1241, 1076, 1066, 1028 m, 879, 837, 742, 683; 1H NMR (400 MHz, CDCl3): δ 8.37 (s, 1H), 7.17 (d, J = 2.9 Hz, 1H), 6.83 (d, J = 2.9 Hz, 1H), 5.68 (d, J = 10.9 Hz, 1H), 5.12 (dd, J = 23.8, 6.8 Hz, 2H), 4.97 (d, J = 5.9 Hz, 1H), 4.88 (d, J = 5.8 Hz, 1H), 4.65–4.57 (m, 3H), 3.64–3.57 (m, 4H), 3.41 (s, 3H), 3.37 (s, 3H), 2.94 (dq, J = 13.6, 6.7 Hz, 1H), 2.81 (q, J = 6.8 Hz, 1H), 2.44–2.32 (m, 1H), 1.17–1.12 (m, 6H), 1.08 (d, J = 6.7 Hz, 3H), 0.89 (s, 9H), 0.88 (s, 3H), 0.00 (s, 3H), −0.10 (s, 3H); 13C NMR (101 MHz, CDCl3): δ 209.2, 174.8, 153.9, 146.9, 138.5, 136.7, 131.6, 128.7, 116.5, 114.8, 100.4, 95.1, 94.6, 76.3, 57.3, 55.9, 55.8, 53.7, 51.4, 42.2, 25.8, 18.1, 18.0, 16.3, 15.5, 12.6, −4.6, −5.2; HRMS (ESI): calcd for C31H52NO9Si [M + H+] 610.34059; found 610.34212; [α]20D= +75.3 (1.0, CHCl3).
:1 to 1:2) to give product 16 (49 mg, 71%) as a colourless oil. Rf = 0.45 (hexane–ethyl acetate = 1:2); IR vmax (neat)/cm−1: 2989, 2901, 2364, 1716, 1669, 1457, 1404, 1150, 1077, 1041, 960, 759, 753, 725, 687, 681; 1H NMR (500 MHz, CDCl3): δ 7.59 (s, 1H), 7.17 (d, J = 3.0 Hz, 1H), 6.82 (d, J = 3.0 Hz, 1H), 5.60 (d, J = 10.7 Hz, 1H), 5.18–5.08 (m, 3H), 4.82 (d, J = 6.2 Hz, 1H), 4.62 (s, 2H), 4.58 (d, J = 10.0 Hz, 1H), 3.80 (d, J = 9.1 Hz, 1H), 3.61 (s, 3H), 3.44 (s, 3H), 3.37 (s, 3H), 3.09–3.01 (m, 1H), 2.94–2.86 (m, 1H), 2.51–2.38 (m, 1H), 1.24 (d, J = 6.9 Hz, 3H), 1.21–1.16 (m, 6H), 0.97 (s, 3H); 13C NMR (126 MHz, CDCl3): δ 208.7, 173.7, 154.0, 147.1, 138.4, 137.9, 131.7, 127.5, 116.4, 114.7, 100.7, 95.0, 94.6, 76.1, 57.4, 55.9, 55.8, 50.7, 42.0, 29.7, 18.2, 15.5, 12.9; HRMS (ESI): calcd for C25H38NO9 [M + H+] 496.2541; found 496.2539; [α]20D = +108.3 (1.0, CHCl3).
:1 to 1:1) to give product 18 (108 mg, 69%) as a colourless oil. Rf = 0.68 (CH2Cl2–MeOH = 10:1); IR vmax (neat)/cm−1: 2930, 1719, 1676, 1672, 1654, 1363, 1176, 1150, 1026, 947; 1H NMR (400 MHz, CDCl3): δ 7.58 (s, 1H), 7.16 (d, J = 3.0 Hz, 1H), 6.83 (d, J = 3.0 Hz, 1H), 5.72 (d, J = 10.7 Hz, 1H), 5.19–5.08 (m, 3H), 4.81 (d, J = 6.2 Hz, 1H), 4.64–4.55 (m, 3H), 3.61 (s, 3H), 3.44 (s, 3H), 3.37 (s, 3H), 3.17–3.10 (m, 1H), 3.01 (q, J = 6.9 Hz, 1H), 2.91 (s, 3H), 2.50–2.38 (m, 1H), 1.27 (d, J = 7.0 Hz, 3H), 1.24 (d, J = 6.8 Hz, 3H), 1.18 (d, J = 6.4 Hz, 3H), 1.07 (d, J = 1.0 Hz, 3H); 13C NMR (126 MHz, CDCl3): δ 206.56, 173.34, 154.29, 146.80, 138.2, 133.06, 131.68, 131.35, 116.27, 114.56, 100.92, 95.01, 94.6, 85.6, 75.8, 57.5, 55.9, 55.9, 51.5, 51.2, 42.3, 38.9, 17.7, 15.8, 15.8, 13.4; HRMS (ESI): calcd for C26H40NO11S [M + H+] 574.2317; found 574.2313; [α]20D = +81.4 (1.0, CHCl3).
:2) to give trace amounts of product 3 as yellowish solids. Rf = 0.35 (hexane–ethyl acetate = 1:2); IR vmax (neat)/cm−1: 3675, 3263, 2989, 2901, 2355, 1714, 1652, 1602, 1394, 1250, 1066, 879, 870, 783, 694, 679; 1H NMR (400 MHz, D6-acetone) δ 8.33 (s, 1H), 7.91 (s, 1H), 7.02 (d, J = 2.8 Hz, 1H), 6.65 (d, J = 2.8 Hz, 1H), 5.00 (dd, J = 10.0, 1.0 Hz, 1H), 4.80 (d, J = 6.4 Hz, 1H), 4.38 (dd, J = 10.0, 6.4 Hz, 1H), 4.27 (d, J = 8.5 Hz, 1H), 3.42 (q, J = 6.8 Hz, 1H), 3.22–3.15 (m, 1H), 1.87 (d, J = 1.6 Hz, 3H), 1.85–1.77 (m, 1H), 1.22 (d, J = 6.8 Hz, 3H), 1.20 (d, J = 6.4 Hz, 3H), 1.01 (d, J = 7.2 Hz, 3H); HRMS (ESI): calcd for C19H24NO5 [M + H+] 346.1649; found 346.1654.
:1 to 2:1) to give product 19 (20.2 mg, 71%) as a colourless oil. Rf = 0.70 (hexane–ethyl acetate = 1:2); IR vmax (neat)/cm−1: 3694, 2968, 2931, 2360, 1718, 1683, 1482, 1378, 1147, 1079, 1028, 738, 735, 708, 683; 1H NMR (500 MHz, CDCl3): δ 7.08 (d, J = 2.5 Hz, 1H), 6.87–6.81 (m, 2H), 5.19 (d, J = 6.9 Hz, 1H), 5.12 (d, J = 6.9 Hz, 1H), 4.94 (q, J = 6.7 Hz, 2H), 4.71 (d, J = 9.4 Hz, 1H), 4.43 (d, J = 10.5 Hz, 1H), 4.30 (d, J = 8.2 Hz, 1H), 3.55 (s, 3H), 3.48 (s, 3H), 3.35 (q, J = 6.8 Hz, 1H), 3.33–3.25 (m, 1H), 1.96–1.85 (m, 4H), 1.35 (d, J = 6.8 Hz, 3H), 1.23 (d, J = 6.5 Hz, 3H), 1.02 (d, J = 7.1 Hz, 3H); 13C NMR (126 MHz, CDCl3): δ 205.7, 172.8, 153.1, 143.1, 139.7, 137.1, 129.2, 123.3, 114.3, 111.3, 97.5, 95.2, 86.3, 75.9, 56.5, 56.0, 46.5, 45.3, 40.4, 18.7, 17.1, 16.00, 15.8; HRMS (ESI): calcd for C23H32NO7 [M + H+] 434.2173; found 434.2185; [α]20D= −8.2 (1.0, CHCl3).
:2) to give product 3 (5.3 mg, 66%) as yellowish solids. Rf = 0.35 (hexane–ethyl acetate = 1:2); IR vmax (neat)/cm−1: 3675, 3263, 2989, 2901, 2355, 1714, 1652, 1602, 1394, 1250, 1066, 879, 870, 783, 694, 679; 1H NMR (500 MHz, acetone) δ 8.24 (s, 1H), 7.84 (s, 1H), 7.02 (d, J = 2.5 Hz, 1H), 6.65 (d, J = 3.0 Hz, 1H), 4.98 (d, J = 10.0 Hz, 1H), 4.72 (d, J = 6.5 Hz, 1H), 4.39 (dd, J = 10.5, 6.0 Hz, 1H), 4.27 (d, J = 8.0 Hz, 1H), 3.42 (q, J = 7.0 Hz, 1H), 3.22–3.16 (m, 1H), 1.87 (d, J = 1.0 Hz, 3H), 1.85–1.78 (m, 1H), 1.23 (d, J = 6.5 Hz, 3H), 1.20 (d, J = 6.5 Hz, 3H), 1.01 (d, J = 7.0 Hz, 3H); 13C NMR (126 MHz, D6-acetone) δ 206.5, 173.4, 153.9, 142.3, 140.4, 139.9, 130.5, 124.3, 113.6, 110.6, 86.9, 70.5, 47.8, 45.6, 41.9, 18.8, 17.3, 16.2, 16.2; 1H NMR (400 MHz, D6-acetone): δ 8.33 (s, 1H), 7.91 (s, 1H), 7.02 (d, J = 2.8 Hz, 1H), 6.65 (d, J = 2.8 Hz, 1H), 5.00 (dd, J = 10.0, 1.0 Hz, 1H), 4.80 (d, J = 6.4 Hz, 1H), 4.38 (dd, J = 10.0, 6.4 Hz, 1H), 4.27 (d, J = 8.5 Hz, 1H), 3.42 (q, J = 6.8 Hz, 1H), 3.22–3.15 (m, 1H), 1.87 (d, J = 1.6 Hz, 3H), 1.85–1.77 (m, 1H), 1.22 (d, J = 6.8 Hz, 3H), 1.20 (d, J = 6.4 Hz, 3H), 1.01 (d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, D6-acetone) δ 206.5, 173.4, 153.9, 142.3, 140.4, 139.9, 130.5, 124.3, 113.6, 110.6, 86.9, 70.5, 47.8, 45.6, 41.9, 18.8, 17.3, 16.2, 16.1; HRMS (ESI): calcd for C19H24NO5 [M + H+] 346.1649; found 346.1654. [α]20D = −66.1 (1.0, acetone).
Acknowledgements
This work has been supported by the National Science Foundation of China (21072006, 21072011 and 21272015), and National 863 Program (S2013AA090203).Notes and references
- (a) H. Yazawa, H. Imai, K. Suzuki, S. Kadota and T. Saito, U. S. Patent, 4,912,215, 1990 Search PubMed; (b) Y. Imai, S. Yazawa and T. Saito, Japanese Patent, JP01168671, 1989 Search PubMed; (c) Y. Imai, S. Yazawa, K. Suzuki, Y. S. Yamaguchi, Y. Shibazaki and M. Saito, Japanese Patent, JP01106884, 1989 Search PubMed.
- (a) S. M. Pimentel-Elardo, T. A. M. Gulder, U. Hentschel and G. Bringgman, Tetrahedron Lett., 2008, 49, 6889 CrossRef CAS PubMed; (b) J. Clardy and C. Walsh, Nature, 2004, 432, 829 CrossRef CAS PubMed.
- (a) T. Komoda, Y. Sugiyama, N. Abe, M. Imachi, H. Hirota and A. Hirota, Tetrahedron Lett., 2003, 44, 1659 CrossRef CAS; (b) T. Komoda, Y. Sugiyama, N. Abe, M. Imachi, H. Hirota, H. Koshino and A. Hirota, Tetrahedron Lett., 2003, 44, 7417 CrossRef CAS PubMed.
- (a) F. E. Koehn and G. T. Carter, Nat. Rev. Drug Discovery, 2005, 4, 206 CrossRef CAS PubMed; (b) I. Paterson and E. A. Anderson, Science, 2005, 310, 451 CrossRef PubMed; (c) W. Fenical and P. R. Jensen, Nat. Chem. Biol., 2006, 2, 666 CrossRef CAS PubMed.
- (a) T. Komoda, K. Yoshida, N. Abe, Y. Sugiyama, M. Imachi, H. Hirota, H. Koshino and A. Hirota, Biosci. Biotechnol., Biochem., 2004, 68, 104 CrossRef CAS; (b) T. Komoda, M. Kishi, N. Abe, Y. Sugiyama and A. Hirota, Biosci. Biotechnol., Biochem., 2004, 68, 903 CrossRef CAS.
- S.-L. Yang, Y.-M. Xi, R. Zhu, L. Wang, J.-H. Chen and Z. Yang, Org. Lett., 2013, 15, 812 CrossRef CAS PubMed.
- (a) S. M. Pimentel-Elardo, Novel anti-infective secondary metabolites and biosynthetic gene clusters from actinomycetes associated with marine sponges, dissertation towards Ph.D., University of Würzburg, 2008, ch. 4, p. 126 Search PubMed; (b) H. Tae, E. B. Kong and K. Park, BMC Bioinf., 2007, 8, 327 CrossRef PubMed.
- (a) T. Ohshima, Y. Miyamoto, J. Yasuhito, Y. Nakahara, M. Utsunomiya and K. Mashima, J. Am. Chem. Soc., 2009, 131, 14317 CrossRef CAS PubMed; (b) G. W. Kabalka, G. Dong and B. Venkataiah, Org. Lett., 2003, 5, 893 CrossRef CAS PubMed; (c) A. B. Zaitsev, S. Gruber, P. A. Pluss, P. S. Pregosin, L. F. Veiros and M. Wórle, J. Am. Chem. Soc., 2008, 130, 11604 CrossRef CAS PubMed; (d) H. Qin, N. Yamagiwa, S. Matsunaga and M. Shibasaki, Angew. Chem., Int. Ed., 2007, 46, 409 CrossRef CAS PubMed; (e) A. Aponick and B. Biannic, Org. Lett., 2011, 13, 1330 CrossRef CAS PubMed.
- For selected references, see: (a) Y. Hattori, S. Furuhata, M. Okajima, H. Konno, M. Abe, H. Miyoshi, T. Goto and H. Makabe, Org. Lett., 2008, 10, 717 CrossRef CAS PubMed; (b) J. M. Ketcham and A. Aponick, Top. Heterocycl. Chem., 2013, 32, 157 Search PubMed; (c) A. Guérinot, A. Serra-Muns, C. Gnamm, C. Bensoussan, S. Reymond and J. Cossy, Org. Lett., 2010, 12, 1808 CrossRef PubMed; (d) K. Miyata, H. Katsuna, S. Kawakami and M. Kitamura, Angew. Chem., Int. Ed., 2011, 50, 4649 CrossRef CAS PubMed; (e) S. Gronert and S. R. Kass, J. Org. Chem., 1997, 62, 7991 CrossRef CAS PubMed.
- A. Fürstner, In Organozinc Reagents, ed. P. Knochel and P. Jones, Oxford University Press, New York, 1999, p. 287 Search PubMed.
- For a review of the vinylogous aldol reaction, see: G. Casiraghi, F. Zanardi, G. Appendino and G. Rassu, Chem. Rev., 2000, 100, 1929 CrossRef CAS PubMed.
- D. A. Evans, M. D. Ennis and D. J. Mathre, J. Am. Chem. Soc., 1982, 104, 1737 CrossRef CAS.
- (a) Y. Liang, L. Wang, R. Zhu, L. Deng, Y. Yang, J. Quan, J. Chen and Z. Yang, Adv. Synth. Catal., 2010, 352, 2387 CrossRef CAS; (b) L. Wang, Y. Xi, S. Yang, R. Zhu, Y. Liang, J. Chen and Z. Yang, Org. Lett., 2011, 13, 74 CrossRef CAS PubMed.
- J. M. Lalancette, A. Frêche, J. R. Brindle and M. Laliberté, Synthesis, 1972, 526 CrossRef CAS.
- S. Canova, V. Bellosta, A. Bigot, P. Mailliet, S. Mignani and J. Cossy, Org. Lett., 2007, 9, 145 CrossRef CAS PubMed.
- Experimental details of the SmI2-mediated intramolecular Reformatsky reaction using the analog of 13 as a substrate is provided in the ESI.†.
- (a) M. E. Jung and T. A. Blumenkopf, Tetrahedron Lett., 1978, 19, 3657 CrossRef; (b) M. Demuynck, P. De Clercq and M. Vandewalle, J. Org. Chem., 1979, 44, 4863 CrossRef CAS; (c) S. Hanessian, D. Delorme and Y. Dufresn, Tetrahedron Lett., 1984, 25, 2515 CrossRef CAS; (d) W. Q. Yang and T. Kitahara, Tetrahedron, 2000, 56, 1451 CrossRef CAS; (e) L. A. Paquette, Z. Gao, Z. Ni and G. F. Smith, Tetrahedron, 1997, 38, 1271 CrossRef CAS; (f) R. E. Ireland and M. D. Varney, J. Org. Chem., 1986, 51, 635 CrossRef CAS; (g) J. H. Han, Y. E. Kwon, J.-H. Sohn and D. H. Ryu, Tetrahedron, 2010, 66, 1673 CrossRef CAS PubMed; (h) E. D. Moher, P. A. Grieco and J. L. Collins, J. Org. Chem., 1993, 58, 3789 CrossRef CAS; (i) T. Nakata, G. Schmid, B. Vranesic, M. Okigawa, T. Smith-Palmer and Y. Kishi, J. Am. Chem. Soc., 1978, 100, 2933 CrossRef CAS; (j) H. Monti, G. Leandri, M. Klos-Ringquet and C. Corriol, Synth. Commun., 1983, 13, 1021 CrossRef CAS; (k) A. I. Meyers, J. L. Durandetta and R. Munavu, J. Org. Chem., 1975, 40, 2025 CrossRef CAS; (l) R. B. L. Woodward, E. Logusch, K. P. Nambiar, K. Sakan, D. E. Ward, B. W. Au-Yeng, P. Balaram, L. J. Browne, P. J. Card and C. H. Chen, J. Am. Chem. Soc., 1981, 103, 3210 CrossRef; (m) J. Auerbach and S. M. Weinreb, J. Chem. Soc., Chem. Commun., 1974, 298 RSC.
- A systematic shift of the 13C NMR data between the synthesized (−)-cebulactam A1 and the reported natural product was observed, which is presumably caused by the variation of different instruments.
- K. C. Nicolaou, R. Guduru, Y. P. Sun, B. Banerji and David Y.-K. Chen, Angew. Chem., Int. Ed., 2007, 46, 5896 CrossRef CAS PubMed.
- J. S. Panek and F. Xu, J. Am. Chem. Soc., 1995, 117, 10587 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3qo00036b |
| This journal is © the Partner Organisations 2014 |