 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Formic acid electrooxidation on thallium-decorated shape-controlled platinum nanoparticles: an improvement in electrocatalytic activity
Carlos
Busó-Rogero
,
Juan V.
Perales-Rondón
,
Manuel J. S.
Farias
,
Francisco J.
Vidal-Iglesias
,
Jose
Solla-Gullon
,
Enrique
Herrero
and
Juan M.
Feliu
*
Instituto de Electroquímica, Universidad de Alicante, Ap. 99 E-03080, Alicante, Spain. E-mail: juan.feliu@ua.es
First published on 27th February 2014
Abstract
Thallium modified shape-controlled Pt nanoparticles were prepared and their electrocatalytic activity towards formic acid electrooxidation was evaluated in 0.5 M sulfuric acid. The electrochemical and in situ FTIR spectroscopic results show a remarkable improvement in the electrocatalytic activity, especially in the low potential region (around 0.1–0.2 V vs. RHE). Cubic Pt nanoparticles modified with Tl were found to be more active than the octahedral Pt ones in the entire range of Tl coverages and potential windows. In situ FTIR spectra indicate that the promotional effect produced by Tl results in the inhibition of the poisoning step leading to COads, thus improving the onset potential for the complete formic acid oxidation to CO2. Chronoamperometric experiments were also performed at 0.2 V to evaluate the stability of the electrocatalysts at constant potential. Finally, experiments with different concentrations of formic acid (0.05–1 M) were also carried out. In all cases, Tl-modified cubic Pt nanoparticles result to be the most active. All these facts reinforce the importance of controlling the surface structure of the electrocatalysts to optimize their electrocatalytic properties.
Introduction
Formic acid is among the most promising fuels for anodic reactions in electrochemical energy conversion devices such as Polymer Electrolyte Membrane (PEM) fuel cells.1,2 Assuming that the change in the Gibbs free energy for the reaction in the fuel cell (HCOOH + 1/2O2 → CO2 + H2O) is ΔGo ≈ −285 kJ mol−1,3 the standard electrode potential for the formic acid/CO2 redox couple is ca. −0.25 V. However, formic acid electro-oxidation takes place at potentials that are 0.6 V higher than the reversible thermodynamic potential on platinum electrodes.4,5 From an electrocatalytic point of view, this large overpotential is mainly due to the high resistance to the oxidation of intermediate products that diminish the performance of platinum as an anode catalyst by blocking surface sites.6 In this sense, many studies on formic acid electro-oxidation reaction aim at developing new electrocatalytic materials which present higher selectivity than platinum electrodes for the formic acid dissociation. In this respect, the mechanism of formic acid electro-oxidation occurs through two parallel routes on platinum electrodes.7–11 In one of these pathways, HCOOH is oxidized through an active intermediate, which decomposes to yield CO2. In the other reaction pathway, i.e. the indirect one, the reaction mechanism occurs through the non-faradaic dehydration reaction of formic acid yielding COads and water molecules. The latter reaction also takes place at open circuit potential on platinum12 and requires more surface sites than the active intermediate reaction path.13 Since the oxidation of COads requires a large overpotential, this species is considered to be a poisoning intermediate.It is also known that the formic acid electro-oxidation reaction is very sensitive to the local surface structure.5,14,15 Concerning this point, it was shown that the Pt(100) electrode was the most active among platinum basal planes, while the activity was observed for Pt(111) for both routes.5 To modify the electrocatalytic activity and/or selectivity of bare substrates for the different routes, the presence of other species such as foreign atoms is a powerful strategy. In fact, many studies on modified single crystal Pt electrodes with Sb,16 Bi,17 Pb,18 Pd19 and Te20 show an improvement in the catalytic performance of these materials towards formic acid oxidation. These electrocatalytic improvements by foreign atoms are possible due to: (i) a third-body effect, where adatoms block neighboring sites required for COads formation;13 (ii) electronic effects, where the foreign adatoms modify the adsorption energy between the adsorbate and the substrate facilitating the oxidation reaction;12,13,21 and (iii) a bifunctional mechanism, where, in this case, the presence of foreign atoms can reduce the overpotential for COads oxidation providing easily activated oxygen-species at lower potentials than those on the corresponding bare substrate.22
From an applied point of view, the use of single crystal surfaces is not feasible and nanomaterials with a much higher surface to volume ratio are required. Since the development of methods to synthesize shape-controlled Pt nanoparticles23 and their first use in electrocatalysis,24 many studies have been focused on the evaluation of the electrocatalytic properties of these shaped Pt particles for oxidation reactions of promising fuels, such as formic acid.25,26 For this reaction, shape-controlled Pt nanoparticles have confirmed the structural electrocatalytic dependence related to the presence of facets with (100) and (111) symmetry. Thus, Pt nanoparticles rich in (100) facets are the most electroactive applicable system towards formic acid oxidation. Unfortunately, shaped Pt nanoparticles also show a high CO poisoning rate.26
As has been done with platinum single crystals, in order to increase the activity and/or to overcome the poisoning effect, Pt nanoparticles have been decorated with adatoms such as Pd,27,28 Bi,29,30 or Sb.31 In addition, Pt/Pd alloyed32 and PtBi intermetallic nanoparticles33 have also been tested. Based on the reported results in ref. 28 and 29 it can be proposed that the improvement in electrocatalytic activity seems to be mainly related to the geometry of the surface atoms of the shaped nanoparticles and the chemical nature of foreign adatoms.
Studies in the 1980s showed that thallium (Tl) UPD (under potential deposition) on both polycrystalline platinum and gold increases the rate of formaldehyde electro-oxidation.34,35 However, even considering these promising results, almost no studies have been devoted to the impact of Tl in the electrocatalysis of formic acid.36 For that reason, we report in this paper an electrochemical and spectroscopic (in situ FTIR – Fourier Transform Infrared) study on the electrocatalytic properties of Tl-decorated shape-controlled Pt nanoparticles towards HCOOH electrooxidation.
Experimental
Platinum single crystal surfaces were prepared following the method developed by Clavilier et al.37 from single crystal beads (ca. 2 mm in diameter). Before the experiments, working electrodes were flame annealed during 30 s, cooled down in a H2/Ar atmosphere and quenched in ultrapure water in equilibrium with this atmosphere before they were transferred to the electrochemical cell.38 Cubic and octahedral shape-controlled Pt nanoparticles, denoted as (100)Pt and (111)Pt respectively, were employed. The synthesis of these shape-controlled Pt nanoparticles was performed as previously described in detail.39 Transmission Electron Microscopy (TEM) measurements were performed to estimate both particle size and morphology/shape.39For electrochemical and in situ FTIR measurements, the nanoparticles dispersed in water were deposited on a polished gold disk and dried under argon atmosphere. Thereafter, the nanoparticles were cleaned by CO (N47, Air Liquide) monolayer adsorption and stripping treatment before the experiments, avoiding high potential excursions to preserve surface order. The active area of platinum nanoparticles was calculated by measuring the charge involved in the so-called hydrogen UPD region after the subtraction of the conventional double-layer contribution in 0.5 M H2SO4 solution, using a reference value of 0.23 mC cm−2.40 A small quantity of nanoparticles (with an active area of ca. 0.05–0.1 cm2) was deposited in all the experiments in order to avoid/minimize transport problems of the reactant species to the surface of the nanoparticles.41
The deposition of Tl on Pt was performed using dilute concentrations of 10−4–10−5 M Tl2SO4 (Aldrich® 99.995%) in 0.5 M H2SO4 solution, cycling the electrode potential between 0.06 V and 0.90 V at 0.05 V s−1. The Tl coverage was monitored through the decrease of the hydrogen adsorption/desorption peaks, according to the equation:
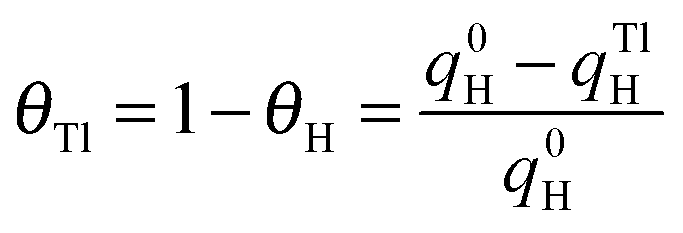
A platinum wire was used as a counter electrode, and a reversible hydrogen (N50, Air Liquide) electrode (RHE) was employed as a reference electrode. All potentials used in this paper are referenced to the RHE. Experiments were carried out in 0.5 M H2SO4 (Merck KGaA Suprapur® 96%), prepared in ultrapure water (Elga PureLab Ultra 18.2 MΩ cm), and deareated with Ar (N50, Air Liquide). Formic acid (Merck KGaG 98%) oxidation experiments were performed in 0.5 M H2SO4.
For electrochemical measurements, cyclic voltammetry and chronoamperometry were employed using a waveform generator (EG&G PARC 175) together with a potentiostat (eDAQ EA161) and a digital recorder (eDAQ ED401).
In situ FTIR spectra were acquired using a Nicolet (Model 8700) spectrometer, equipped with an MCT (Mercury–Cadmium–Telluride) detector. The spectroelectrochemical cell was fitted with a prismatic 60° CaF2 window, as described previously.42 The spectra, obtained from the average of 100 interferograms using a resolution of 8 cm−1, were collected at intervals of 50 mV between 0.05 V to 0.90 V vs. RHE. For all experiments, p-polarized light was employed, which allows detecting changes in the concentration of active species on the electrode surface and electrolyte solution.42 Spectra are presented in absorbance units 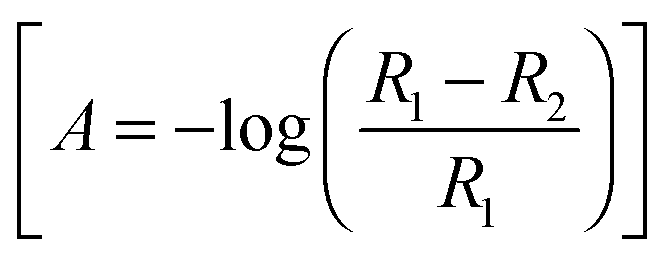 in which R2 and R1 are referred to as the reflectance values of the single beam spectra corresponding to the sample and the reference potentials, respectively. Positive bands in the spectra are related to species that have been formed at the sample potential whereas negative bands correspond to species consumed. All the experiments were performed at room temperature.
in which R2 and R1 are referred to as the reflectance values of the single beam spectra corresponding to the sample and the reference potentials, respectively. Positive bands in the spectra are related to species that have been formed at the sample potential whereas negative bands correspond to species consumed. All the experiments were performed at room temperature.
Results and discussion
Formic acid oxidation on Tl-modified Pt single crystal electrodes
Preliminary experiments with Pt(100) and Pt(111) single crystal electrodes were performed to evaluate the effects of the Tl adsorption on these Pt model surfaces both in the absence and presence of formic acid. Fig. 1 shows voltammetric results for Tl modified Pt(100) and Pt(111) single crystal electrodes in a 0.5 M H2SO4 solution. The effect of Tl deposition can be easily monitored by the changes in the cyclic voltammetric profile. In addition, in order to avoid possible Tl dissolution, the upper potential limits were set at 0.65 and 0.6 V for Pt(100) and Pt(111) electrodes, respectively. For the Pt(100) electrode (Fig. 1a), the adsorption/desorption states between 0.2 and 0.5 V (which are associated with the competitive adsorption between hydrogen and anions) remarkably decrease during Tl deposition, whereas a small peak appears at about 0.15 V at Tl coverages close to the full blockage. In the case of the Pt(111) electrode, Fig. 1b, similar effects are observed for the diminution of the charge in the hydrogen adsorption/desorption region (between 0.06 and 0.35 V) and the (bi)sulfate adsorption (between 0.35 and 0.65 V). Again, a new small peak for this electrode appears at about 0.43 V at high coverages of Tl. This peak is associated with the adsorption of sulfate on the modified electrode, because it is absent in perchloric acid solutions. Although it appears in the same potential region that the spike in sulfuric acid solutions for the Pt(111) electrode, the origin is not the same. The spike at 0.45 V diminishes upon Tl deposition and at a given Tl coverage the new peak appears. It is then probably related to structural changes in the adlayer that changes the interaction of sulfate with the electrode surface.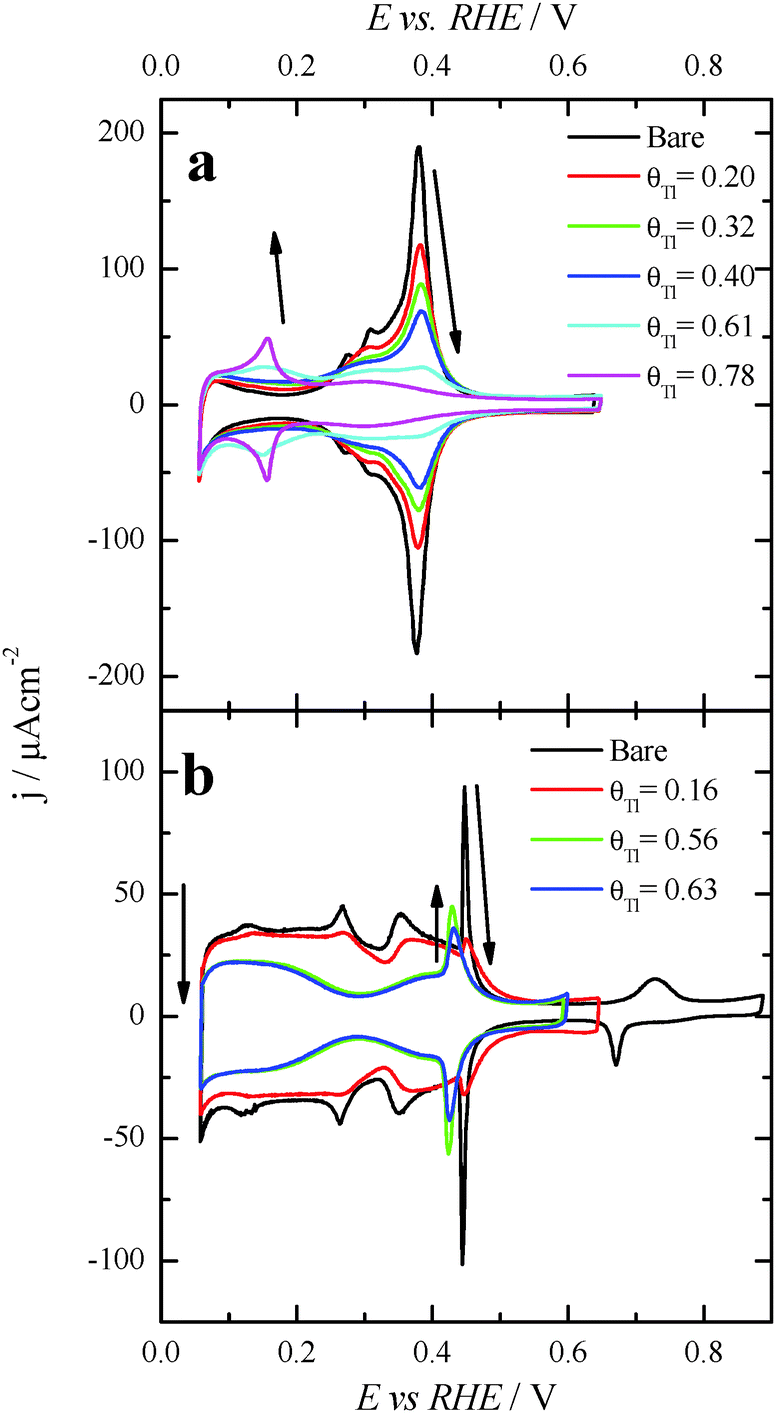 | ||
| Fig. 1 Evolution of the voltammetric profiles in 0.5 M H2SO4 with increasing amounts of Tl on (a) Pt(100) and (b) Pt(111). Scan rate: 0.05 V s−1. Arrows indicate changes associated with increasing Tl amounts at the surface. | ||
The positive going sweep voltammetric profiles of formic acid oxidation for the same Tl-modified Pt single crystal electrodes from Fig. 1 are presented in Fig. 2, using 0.1 M HCOOH in 0.5 M H2SO4 as test solution. As expected, very low currents are observed for the bare Pt(100) electrode as a consequence of the well-established CO poisoning process coming from the dehydration of formic acid, which is favored on Pt(100).11 With increasing amounts of Tl, the indirect path (through CO poisoning) is hindered. Then formic acid oxidation can take place through the direct path and oxidation currents increase very significantly. The results suggest an optimum Tl coverage at values close to 0.4. At higher coverages, the onset potential increases and the current diminishes.
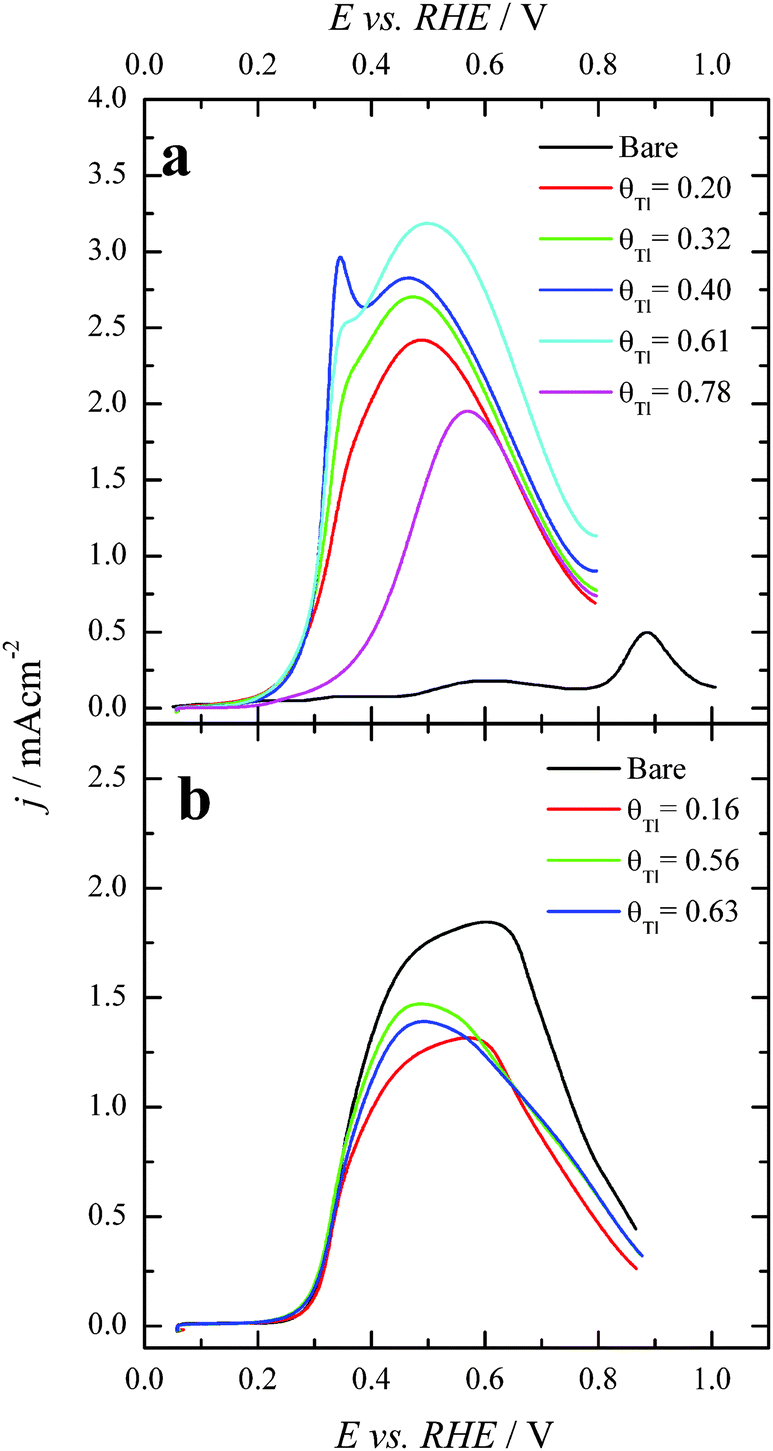 | ||
| Fig. 2 Positive sweep voltammetric profiles for 0.1 M formic acid oxidation in 0.5 M H2SO4 with and without Tl for (a) Pt(100) and (b) Pt(111) single crystals. Scan rate: 0.02 V s−1. | ||
However, in the case of the Pt(111) electrode (Fig. 2b), for which it is well-established that the CO poisoning is almost negligible,11 the addition of Tl does not provide significant changes in the electrooxidation process. These results seem to indicate that Tl acts as a third body preventing the dehydration reaction. For the Pt(111) electrode, owing to the almost negligible CO formation rate,5 the presence of Tl does not improve its catalytic activity. The current diminution for this electrode is just the result of the diminution of the Pt free sites. On the other hand, the increase of activity for the Pt(100) electrode can be related to the diminution of the COads coverage which leads to an increase of the oxidation through the active intermediate reaction path. However, electronic effects cannot be discarded for this electrode. It should be noted that the onset for the direct oxidation reaction in the absence of poison on the Pt(100) electrode is 0.30 V (see ref. 43), whereas in the presence of Tl the onset is ca. 0.20 V. This diminution of the onset potential at a Tl coverage of ca. 0.4 suggests the possible presence of an electronic effect. These results also point out the importance of controlling the surface structure of the Pt substrate, the Pt(100) square structure being much more convenient than the hexagonal Pt(111). Thus, in terms of the particle shape, it would be preferable to use cubic Pt nanoparticles in which the number of {100} surface sites is maximized.44
Electrochemical characterization of Tl modified Pt nanoparticles
Fig. 3 displays the voltammetric profiles of the (100)Pt and (111)Pt nanoparticles before and after Tl incorporation. Before the Tl decoration, the voltammetric profiles of the cubic and octahedral Pt nanoparticles are those that are expected from their shape–surface structure relationship.45 Thus, the cubic Pt nanoparticles show the main features related to a preferential {100} surface structure, that is, a sharp peak at ∼0.27 V related to {100} edge/corner sites and a signal around 0.37 V associated with wide {100} terrace sites. In contrast, the octahedral nanoparticles show the characteristics of a preferential {111} surface structure, with a broad peak at about 0.52 V related to sulfate adsorption on relatively wide two-dimensional {111} terrace sites. A detailed analysis of the characteristic voltammetric features of similar shape-controlled Pt nanoparticles was described in previous contributions.44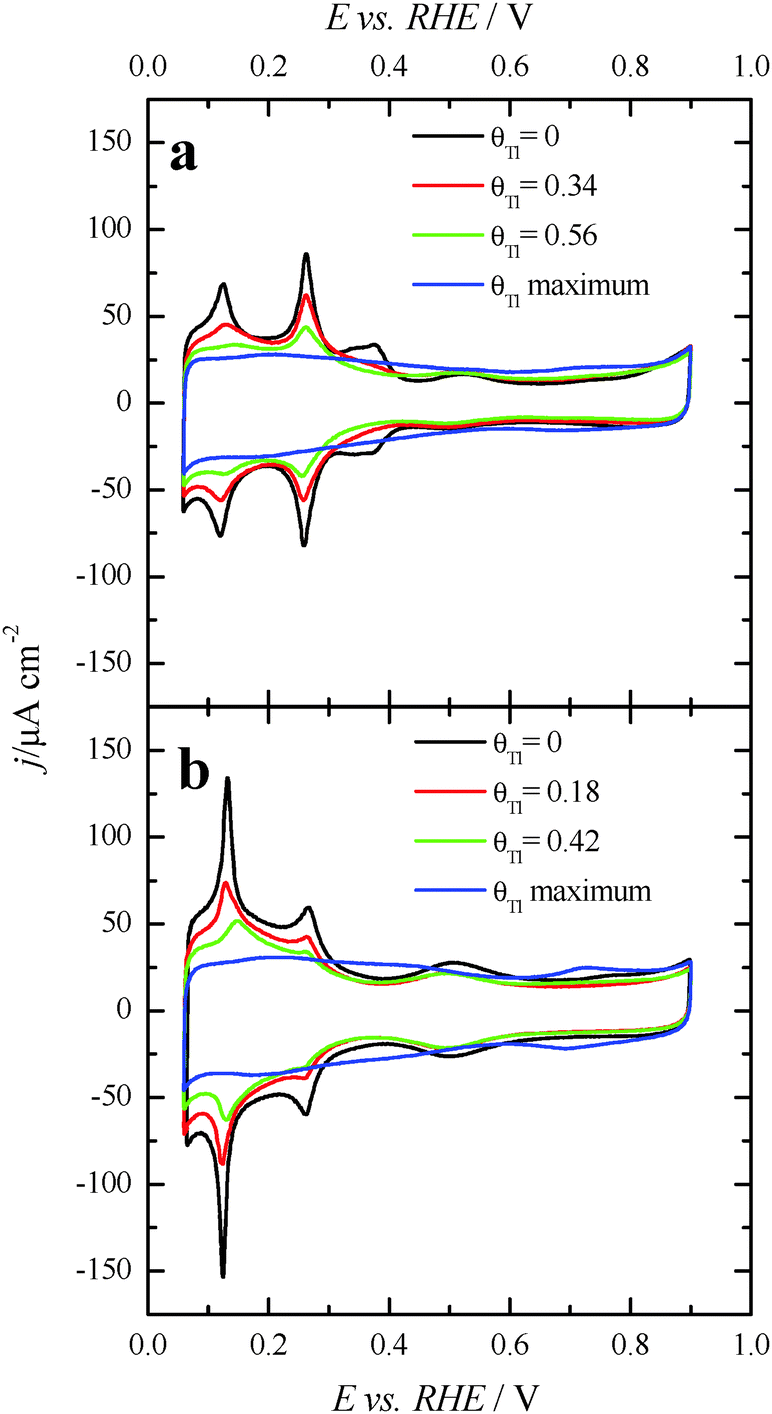 | ||
| Fig. 3 Voltammetric profiles for increasing amounts of Tl coverages on (a) (100)Pt and (b) (111)Pt nanoparticles in 0.5 M H2SO4 + 10−4–10−5 M Tl2SO4. Scan rate: 0.05 V s−1. | ||
Fig. 3 also shows the voltammetric responses of the (100)Pt and (111)Pt nanoparticles after partial and maximum deposition of Tl. The evolution of the voltammetric profiles is similar to those previously reported for Pd and Bi on shape-controlled Pt nanoparticles28,29 or after deposition of Bi(III), Te(IV) and Se(IV) on stepped Pt surfaces.46 In addition, the Tl adsorption process does not produce appreciable changes in the charge density on the so-called double layer region until high thallium coverages are reached. As described in the experimental section, the Tl coverage can be calculated considering the expression θTl = 1 − θH, where θTl and θH are the thallium and hydrogen coverages, respectively. However, at the maximum thallium coverage on both Pt nanoparticles, Fig. 3, an important increase of the double layer region is observed. This fact prevents an accurate determination of the Tl coverage for highly covered surfaces that cannot be properly calculated. For that reason, the maximum coverage for each sample will be referred to as “maximum” and no numeric value will be given. In addition, it is important to note the absence of additional voltammetric features at high coverages and remark that only a small broad peak is visible at about 0.70 V, particularly in the preferential (111)Pt nanoparticles. This feature can be linked to the thallium desorption from {111} terrace sites, as pointed out for Tl UPD in Pt(111) reports.47,48
Formic acid oxidation on Tl modified shape controlled Pt nanoparticles
After each particular Tl adsorption, the modified Pt nanoparticles were transferred to a second electrochemical cell containing 0.1 M formic acid + 0.5 M H2SO4. Fig. 4 compares the positive going sweep voltammetric responses obtained for the bare and Tl modified shaped nanoparticles. For the bare cubic (100)Pt nanoparticles (Fig. 4a, black line), the positive going scan shows almost zero current density at potentials below 0.35 V, due to CO poisoning effects. The HCOOH oxidation starts at around 0.35 V, and the current density increases slowly until ∼0.80 V. Above this potential, the current density starts to grow rapidly due to CO oxidation and therefore in the reverse scan the current density would be much higher as the surface is CO-free. As before, these negative-going results are not shown, because they are irrelevant in electrocatalytic studies.26,39 Interestingly, for Tl modified cubic (100) Pt nanoparticles with θTl = 0.34 and 0.56, Fig. 4a (red and green lines, respectively), the onset potential of HCOOH oxidation remarkably decreases to about 0.1 V. Moreover, at the maximum coverage (blue line), the onset potential is even lower (about 0.08 V). This feature clearly evidences the positive impact of thallium on the electro-oxidation of formic acid in the low potential region. | ||
| Fig. 4 Positive sweep voltammetric profiles for formic acid electro-oxidation with increasing amounts of Tl coverages for (a) (100)Pt and (b) (111)Pt nanoparticles. Scan rate: 0.02 V s−1. Test solution: 0.1 M formic acid in 0.5 M H2SO4. | ||
For the (111)Pt nanoparticles, Fig. 4b, a similar effect on the onset potential is observed after surface decoration with Tl. Taking into account that (i) the {111} domains do not suffer the same strong CO poisoning effect as that observed with the {100} domains,11 (see Fig. 2), and (ii) the {111} domains have a low catalytic activity for the direct oxidation,5 the electrocatalytic enhancement observed is lower than that reported for the (100)Pt nanoparticles. Anyhow, the onset potential diminishes when θTl increases, decreasing to around 0.1 V at the maximum coverage (blue line). Also, the onset for the formic acid oxidation observed for the Tl modified nanoparticles at high coverages is ca. 0.2–0.3 V lower than the onset for the direct oxidation route in the unmodified nanoparticles.26
Finally, with the aim of comparing the activity of both samples in the whole potential range, Fig. 5 displays the HCOOH activity ratio between the bare and Tl modified (maximum coverage) Pt nanoparticles. From Fig. 5, it is evident that the “improvement factor” due to the presence of Tl for the (100)Pt nanoparticles is larger than that obtained with the (111)Pt nanoparticles in the entire potential range. In fact, the highest ratio between both activities is observed at ca. 0.3 V, the potential at which the {100} domains have the greatest activity, in agreement with the large catalytic activity of Tl on the {100} domains (Fig. 2a). In addition, in terms of maximum currents, the electrooxidation activity of the Tl-modified (100)Pt nanoparticles is always higher (about 1.5 times) than that of the (111)Pt nanoparticles, Fig. 6.
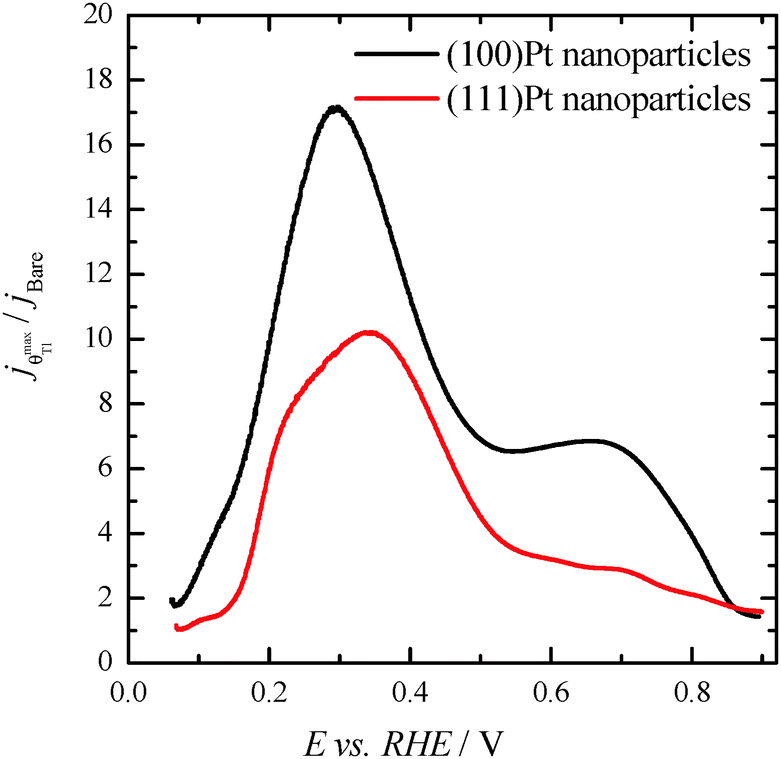 | ||
| Fig. 5 Ratio between the HCOOH oxidation current densities obtained using the bare and Tl-decorated (maximum coverage) shape-controlled Pt nanoparticles. Data taken from Fig. 4. | ||
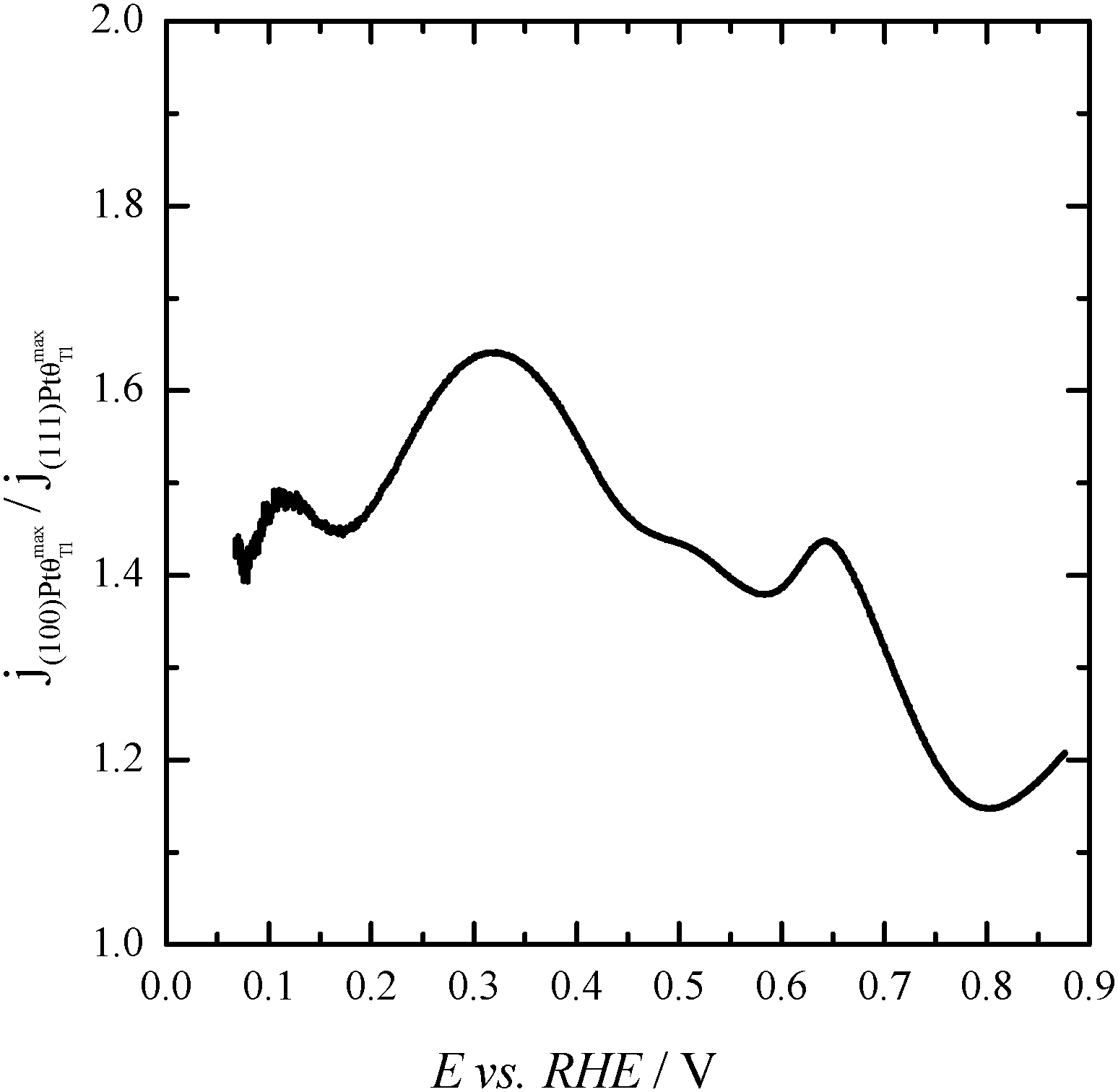 | ||
| Fig. 6 Ratio between the HCOOH oxidation current densities obtained using Tl-modified (maximum coverage) (100) and (111)Pt nanoparticles. Data taken from Fig. 4. | ||
In order to better evaluate the activity as well as the stability of the Tl-modified samples, current transient responses for each modified electrode were recorded at 0.2 V, (Fig. 7), a reasonably low potential value. In these experiments, a pretreatment consisting of a potential step at 0.85 V for 10 s prior to the potential step at 0.2 V was performed in order to oxidize all the possible CO blocking surface sites.26 Current transients for the bare nanoparticles are not reported, as current densities for both samples are almost zero. This is due to the fact that the CO formation rate is very high and the surface is immediately covered by CO after the cleaning step. For Tl modified (100)Pt nanoparticles (Fig. 7a), the current densities after 600 s are 0.002, 0.010 and 0.014 mA cm−2 at Tl coverages of 0.25, 0.48 and the maximum one, respectively. For Tl modified (111)Pt nanoparticles (Fig. 7b), reactivity is lower and current densities of 0.002, 0.004 and 0.006 mA cm−2 were obtained at coverages of 0.43, 0.57 and the maximum one, respectively. These results indicate that Tl adsorbed on the surface successfully hinders the CO adsorption through the poisoning pathway, and this effect increases with the Tl coverage.
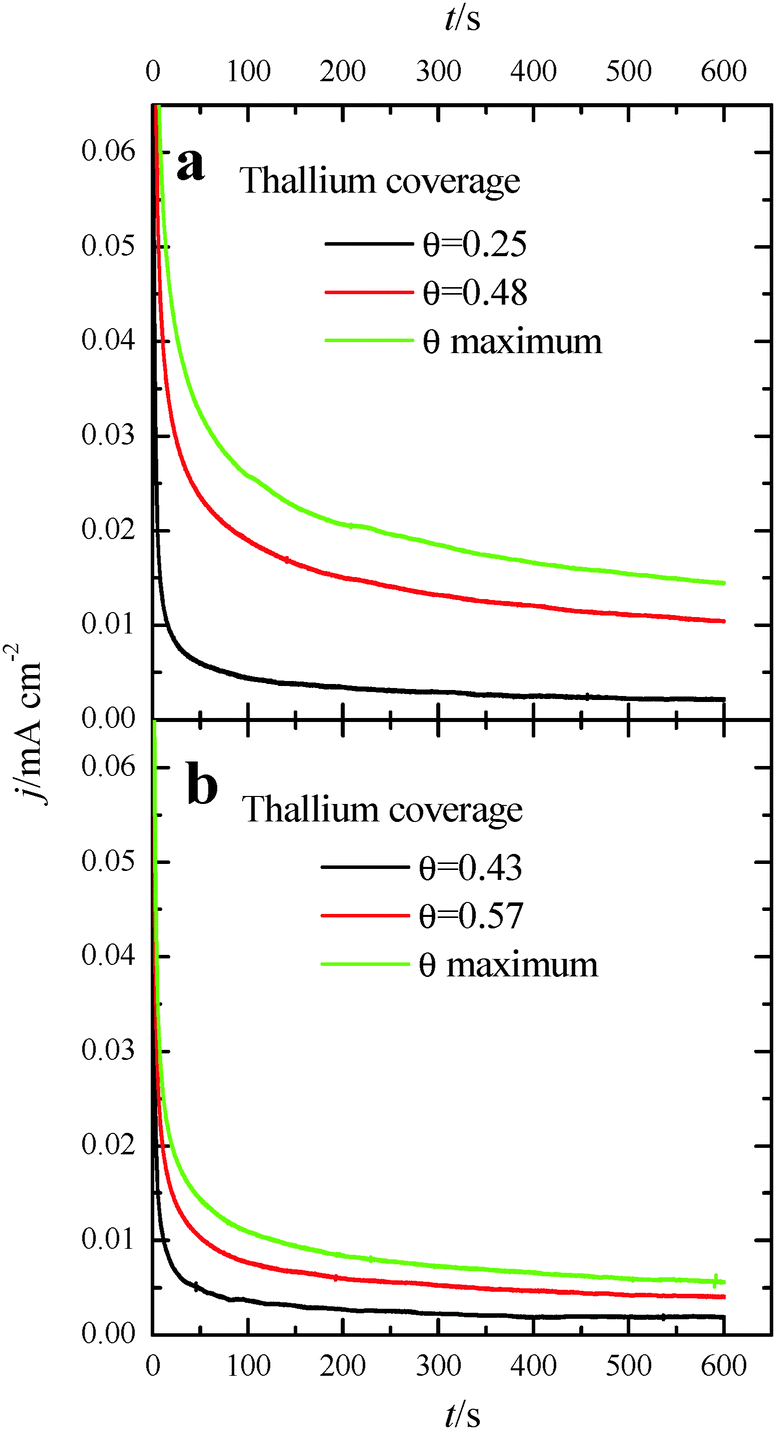 | ||
| Fig. 7 Current transients recorded at 0.2 V for formic acid electrooxidation for (a) (100)Pt and (b) (111)Pt nanoparticles with different Tl coverages. Test solution: 0.1 M formic acid in 0.5 M H2SO4. | ||
FTIR experiments of formic acid oxidation on Tl-decorated Pt nanoparticles
In situ FTIR spectra were measured in order to better understand the role of Tl in the enhancement of formic acid oxidation on the modified nanoparticles. Only the (100)Pt nanoparticles were studied because these nanoparticles present more COads than the (111)Pt nanoparticles,39,49 and then, it is easier to determine the electrocatalytic effects induced by the presence of Tl. Spectra were collected in 0.05 M formic acid in 0.1 M H2SO4 on non-modified and Tl-modified electrodes (at maximum thallium coverage). Formic acid concentration was diminished with the aim of decreasing the huge amount of CO2 formed during formic acid oxidation, which destabilizes the thin-layer configuration. Sulfuric acid concentration was also reduced in order to avoid possible damage in the CaF2 prism. For all spectra presented, the spectrum acquired at 0.05 V was used as a reference.Fig. 8a displays the spectra for formic acid oxidation on (100)Pt bare nanoparticles. From 0.2 V to 0.6 V, the spectra show the linearly adsorbed CO band between 2050 and 2060 cm−1.42 At 0.2 V, the band frequency is around 2052 cm−1, whereas at 0.6 V the frequency shifts to 2060 cm−1, likely due to the Stark effect.50 Between 0.4 and 0.5 V, the band shows a slightly bipolar character, meaning that adsorbed CO was present in the reference potential (0.05 V).42 At around 0.2 V, a new band at 2343 cm−1 appears, which is associated with the start of CO2 formation.42 The spectra do not show any signal band that could be attributed to bridge adsorbed CO, which should appear at around 1820 cm−1 as was observed for formic acid oxidation in ATR-SEIRA (Attenuated Total Reflection – Surface Enhanced Infra-Red Absorption) experiments on thin-film polycrystalline platinum electrodes at 0.35 V vs. RHE.9
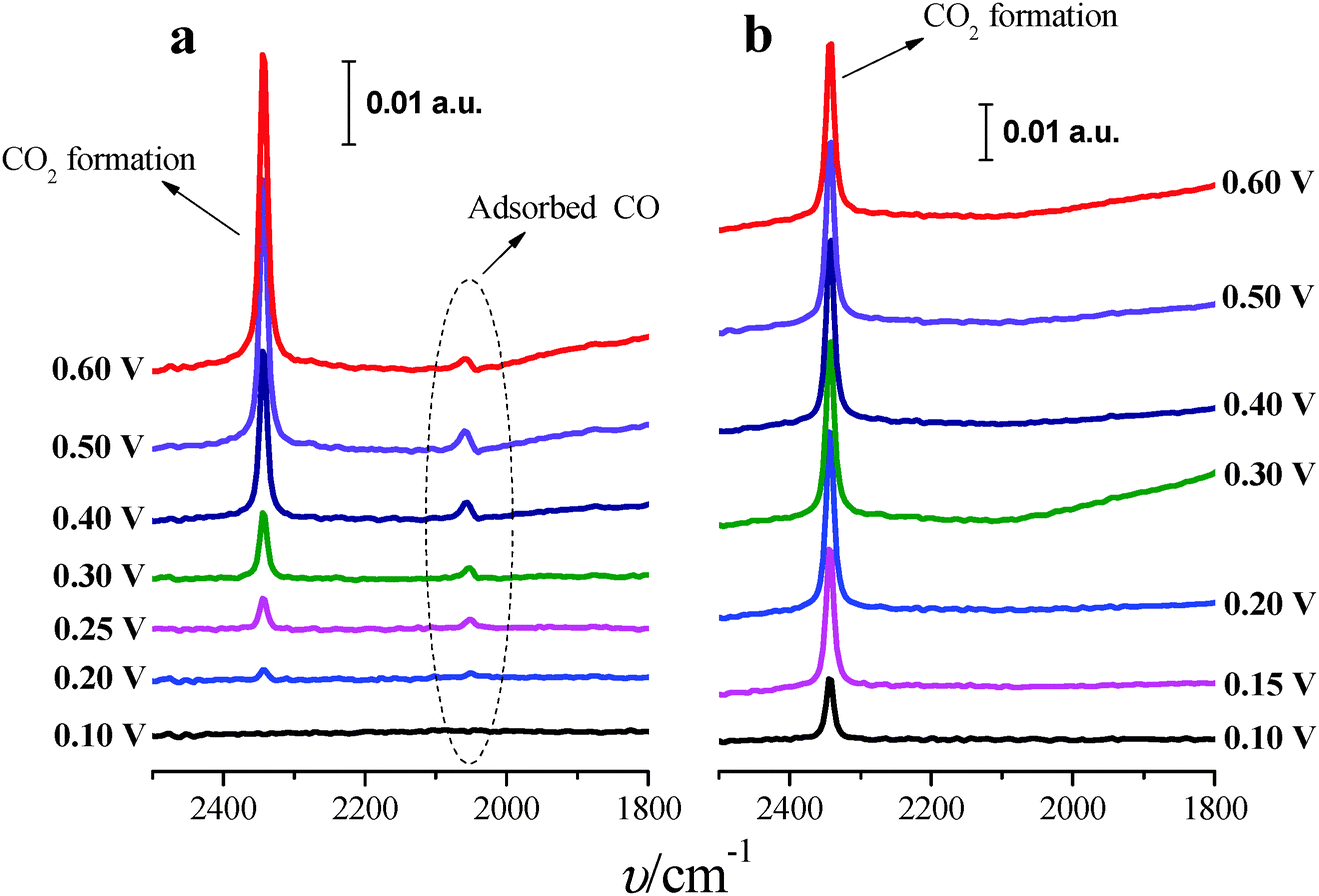 | ||
| Fig. 8 In situ FTIR spectra for formic acid electro-oxidation on (100)Pt nanoparticles: (a) bare Pt and (b) maximum Tl coverage. Reference spectra were taken at 0.05 V. Test solution: 0.05 M formic acid in 0.1 M H2SO4. | ||
Fig. 8b shows the spectra for formic acid oxidation on Tl-modified (100)Pt nanoparticles. The band for CO2 can be clearly observed at 0.1 V, and the COads band is absent for all the spectra, pointing out that Tl enhances the electrocatalytic activity (Fig. 4a) by hindering the poisoning pathway thus allowing formic acid oxidation to proceed through the direct pathway, which also explains why CO2 is formed at low potentials.
Effect of formic acid concentration
Once the catalytic effect of Tl over Pt nanoparticles for formic acid oxidation has been confirmed, experiments using different formic acid concentrations were also performed with the maximum Tl coverage to evaluate its possible effect on the electrocatalytic activity. The corresponding potentiodynamic responses are shown in Fig. 9, for both (100)Pt and (111)Pt nanoparticles. In the two cases, current densities increase with the concentration of formic acid, especially at low potentials. In principle, it would be expected that currents were directly proportional to the concentration of formic acid, that is, j ∝ Co*. Thus, the representation of log(j) at a given potential vs. log(Co*) should be linear with a slope of 1, the expected reaction order for the formic acid concentration. The plots are linear in the range between 0.2 and 0.7 V in both scan directions (inset of Fig. 9b). However, the slope is 0.75 ± 0.03 in this potential range for both electrodes, which would indicate that the reaction order is 0.75. This fact clearly suggests that the reaction mechanism is complex. It could be argued that the complexity is due to the presence of the two routes for the reaction. However, it is known that the CO formation reaction occurs in the potential window lower than 0.5 V.26 Thus, the oxidation reaction in the negative scan at E > 0.5 V would take place on a clean surface and only through the active intermediate. Moreover, the presence of Tl has suppressed the route through CO significantly. The constant value for the reaction order in the whole range, then, suggests that the complexity is not due to the CO route, but there is an intrinsic factor in the direct oxidation route. This fact would indicate that an additional species (not only formic acid) is involved in the reaction mechanism.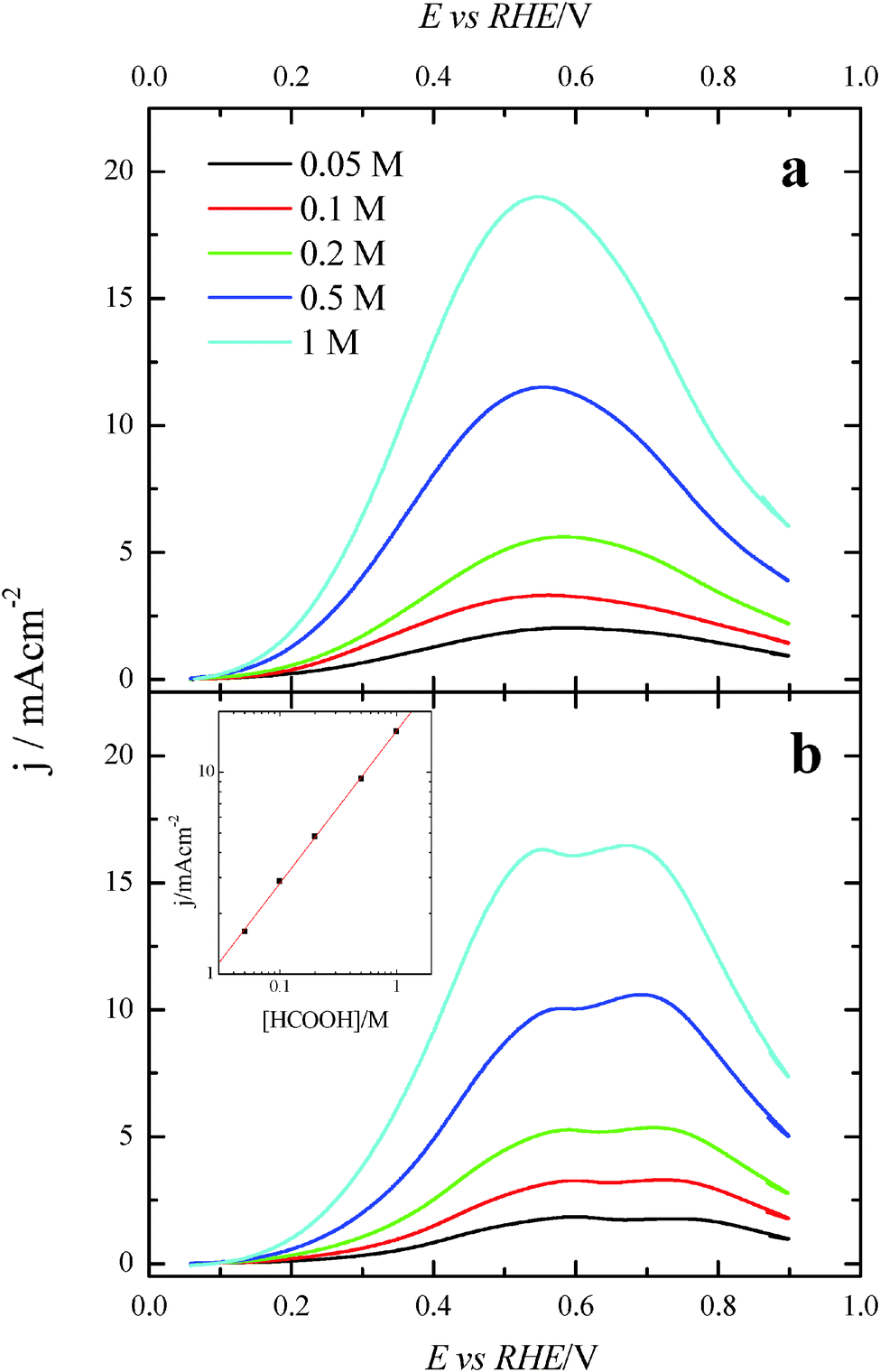 | ||
| Fig. 9 Positive sweep voltammetric profiles for (a) (100)Pt and (b) (111)Pt nanoparticles with maximum Tl coverage in different concentrations of formic acid in 0.5 M H2SO4. Scan rate: 0.02 V s−1. Inset: double logarithmic plot of the current density at 0.5 V for the (111)Pt nanoparticles vs. formic acid concentration. | ||
Additional evidence on that is found in the chronoamperometric responses after 600 s of reaction at 0.2 V for both nanoparticles at maximum Tl coverage at different formic acid concentrations (Fig. 10). In the case of (100)Pt nanoparticles (Fig. 10a), there is nearly no clear difference between the final current density for 0.05 M and 0.1 M (around 0.015–0.020 mA cm−2), but for the other concentrations, current density increases with the amount of formic acid in solution, increasing up to 0.145 mA cm−2 for 1 M formic acid concentration. A similar behavior was observed for (111)Pt nanoparticles in Fig. 10b, with a final current density of 0.049 mA cm−2 for 1 M formic acid. As for voltammetric curves, chronoamperometric currents become higher when the concentration of formic acid increases. Fig. 11 summarizes the differences observed for the final current density after 10 min at 0.2 V for both samples at maximum coverage, clearly indicating that the Tl-modified (100)Pt nanoparticles are much more active than the (111)Pt nanoparticles in the range of HCOOH concentration under study (0.05–1 M HCOOH). Currents recorded after 10 minutes reflect a nearly stationary situation. For the currents at 10 min the reaction order is similar to that measured by voltammetry, which reinforces the fact that the reaction mechanism through the active intermediate is complex.
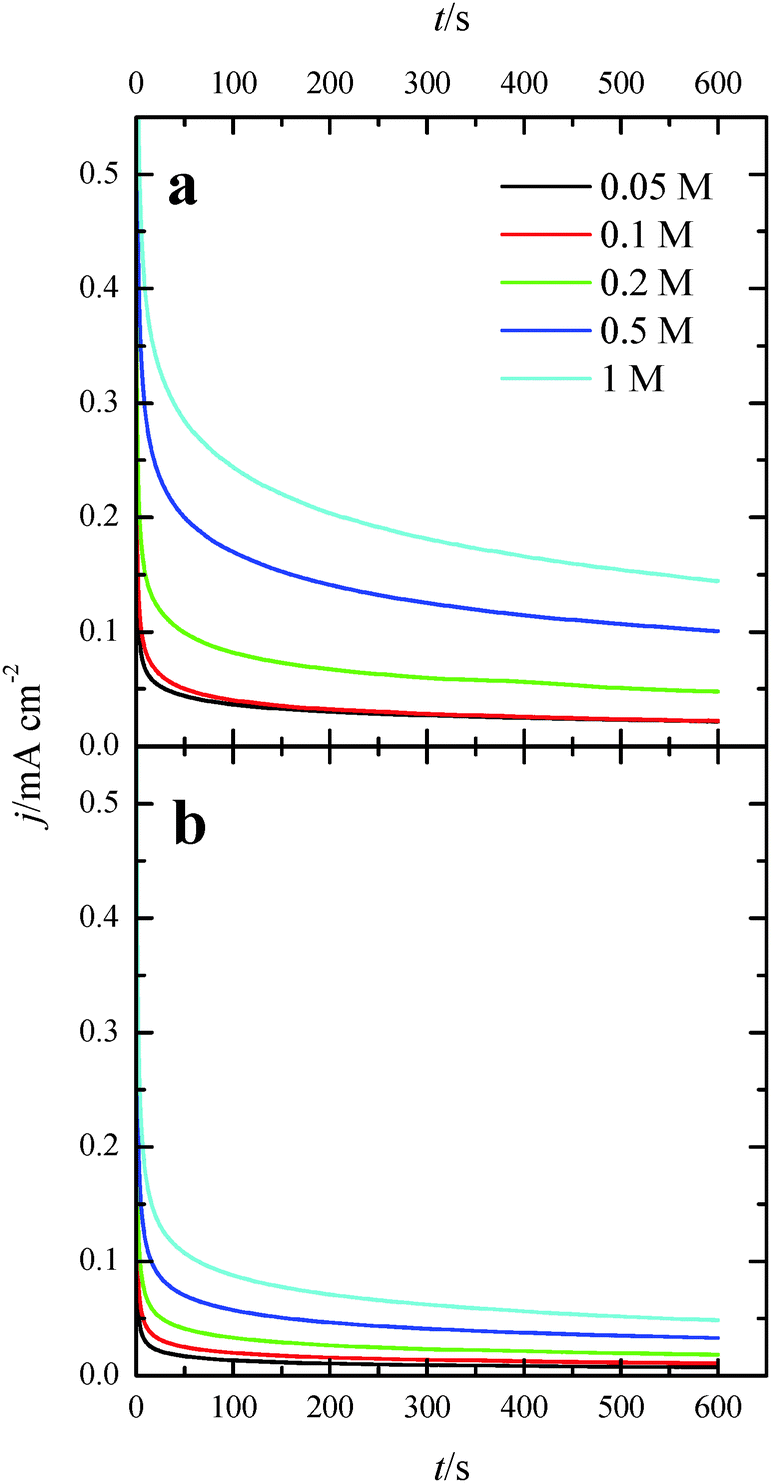 | ||
| Fig. 10 Current transients for (a) (100)Pt and (b) (111)Pt nanoparticles with maximum Tl coverage recorded at 0.20 V for different formic acid concentrations in 0.5 M H2SO4. | ||
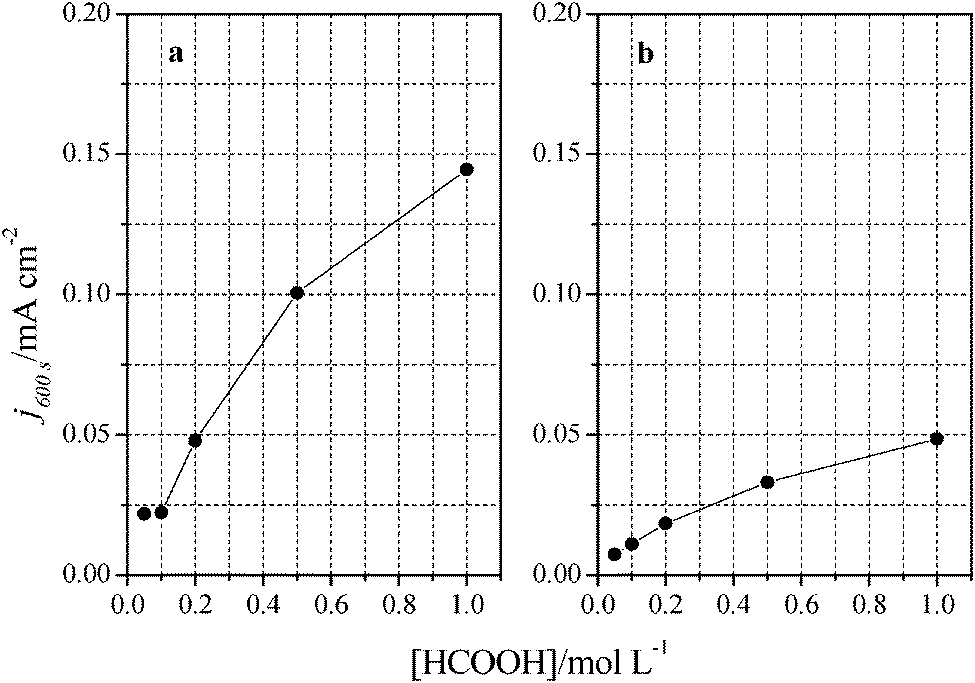 | ||
| Fig. 11 Comparison between formic acid concentrations and current densities obtained after 10 minutes at 0.2 V. Data extracted from Fig. 9 for (a) (100)Pt and (b) (111)Pt nanoparticles. | ||
Conclusions
The deposition of Tl on (100)Pt and (111)Pt nanoparticles remarkably improves their electrocatalytic activity towards formic acid oxidation, especially in the low potential region. The activity enhancement is also dependent on the surface structure of the Pt substrate. In agreement with fundamental data from Pt single crystal studies, the use of (100)Pt nanoparticles is clearly more beneficial than the use of (111)Pt nanoparticles. Voltammetric and in situ FTIR data indicate that Tl decoration prevents the formation of adsorbed COads. Consequently, it can be concluded that the enhancement of formic acid oxidation with Tl-decorated platinum nanoparticles (especially with (100)Pt nanoparticles) is due to the hindrance of the indirect pathway (third body effect). So, the reaction mainly takes place through the direct pathway (via non-COads). However, electronic effects cannot be discarded, due to the diminution of the onset potential for the formic acid oxidation reaction.Acknowledgements
This work has been financially supported by the MICINN (Spain) (project CTQ2010-16271) and Generalitat Valenciana (project PROMETEO/2009/045 – FEDER). Farias M.J.S. would also like to thank CNPq, Brazil.References
- C. Rice, R. I. Ha, R. I. Masel, P. Waszczuk, A. Wieckowski and T. Barnard, J. Power Sources, 2002, 111, 83–89 CrossRef CAS.
- N. V. Rees and R. G. Compton, J. Solid State Electrochem., 2011, 15, 2095–2100 CrossRef CAS PubMed.
- S. Uhm, H. J. Lee and J. Lee, Phys. Chem. Chem. Phys., 2009, 11, 9326–9336 RSC.
- G. Q. Lu, A. Crown and A. Wieckowski, J. Phys. Chem. B, 1999, 103, 9700–9711 CrossRef CAS.
- V. Grozovski, V. Climent, E. Herrero and J. M. Feliu, Phys. Chem. Chem. Phys., 2010, 12, 8822–8831 RSC.
- C. Rice, S. Ha, R. I. Masel and A. Wieckowski, J. Power Sources, 2003, 115, 229–235 CrossRef CAS.
- A. Capon and R. Parsons, J. Electroanal. Chem., 1973, 45, 205–231 CrossRef CAS.
- M. Osawa, K. Komatsu, G. Samjeske, T. Uchida, T. Ikeshoji, A. Cuesta and C. Gutierrez, Angew. Chem., Int. Ed., 2011, 50, 1159–1163 CrossRef CAS PubMed.
- A. Cuesta, G. Cabello, M. Osawa and C. Gutiérrez, ACS Catal., 2012, 2, 728–738 CrossRef CAS.
- Y. X. Chen, M. Heinen, Z. Jusys and R. J. Behm, Langmuir, 2006, 22, 10399–10408 CrossRef CAS PubMed.
- J. M. Feliu and E. Herrero, in Handbook of Fuel Cells – Fundamentals, Technology and Applications, ed. W. Vielstich, H. Gasteiger and A. Lamm, John Wiley & Sons, Ltd., Chichester, 2003, vol. 2 Search PubMed.
- E. Herrero, A. Fernández-Vega, J. M. Feliu and A. Aldaz, J. Electroanal. Chem., 1993, 350, 73–88 CrossRef CAS.
- E. Leiva, T. Iwasita, E. Herrero and J. M. Feliu, Langmuir, 1997, 13, 6287–6293 CrossRef CAS.
- S. G. Sun and Y. Y. Yang, J. Electroanal. Chem., 1999, 467, 121–131 CrossRef CAS.
- A. Cuesta, M. a. Escudero, B. Lanova and H. Baltruschat, Langmuir, 2009, 25, 6500–6507 CrossRef CAS PubMed.
- Y.-Y. Yang, S.-G. Sun, Y.-J. Gu, Z.-Y. Zhou and C.-H. Zhen, Electrochim. Acta, 2001, 46, 4339–4348 CrossRef CAS.
- S. P. E. Smith, K. F. Ben-Dor and H. D. Abruna, Langmuir, 1999, 15, 7325–7332 CrossRef CAS.
- H. W. Lei, H. Hattori and H. Kita, Electrochim. Acta, 1996, 41, 1619–1628 CrossRef CAS.
- F. J. Vidal-Iglesias, J. Solla-Gullón, E. Herrero, A. Aldaz and J. M. Feliu, J. Appl. Electrochem., 2006, 36, 1207–1214 CrossRef CAS.
- E. Herrero, M. J. Llorca, J. M. Feliu and A. Aldaz, J. Electroanal. Chem., 1995, 383, 145–154 CrossRef.
- M. Shibata, N. Furuya, M. Watanabe and S. Motoo, J. Electroanal. Chem., 1989, 263, 97–108 CrossRef CAS.
- M. Watanabe and S. Motoo, J. Electroanal. Chem., 1975, 60, 275–283 CrossRef CAS.
- T. S. Ahmadi, Z. L. Wang, T. C. Green, A. Henglein and M. A. El-Sayed, Science, 1996, 272, 1924–1926 CAS.
- F. J. Vidal-Iglesias, J. Solla-Gullón, P. Rodríguez, E. Herrero, V. Montiel, J. M. Feliu and A. Aldaz, Electrochem. Commun., 2004, 6, 1080–1084 CrossRef CAS PubMed.
- N. Tian, Z.-Y. Zhou, S.-G. Sun, Y. Ding and Z. L. Wang, Science, 2007, 316, 732–735 CrossRef CAS PubMed.
- V. Grozovski, J. Solla-Gullon, V. Climent, E. Herrero and J. M. Feliu, J. Phys. Chem. C, 2010, 114, 13802–13812 CAS.
- P. Waszczuk, T. M. Barnard, C. Rice, R. I. Masel and A. Wieckowski, Electrochem. Commun., 2002, 4, 599–603 CrossRef CAS.
- F. J. Vidal-Iglesias, J. Solla-Gullon, E. Herrero, A. Aldaz and J. M. Feliu, Angew. Chem., Int. Ed., 2010, 49, 6998–7001 CrossRef CAS PubMed.
- Q. S. Chen, Z. Y. Zhou, F. J. Vidal-Iglesias, J. Solla-Gullon, J. M. Feliu and S. G. Sun, J. Am. Chem. Soc., 2011, 133, 12930–12933 CrossRef CAS PubMed.
- A. Saez, E. Exposito, J. Solla-Gullon, V. Montiel and A. Aldaz, Electrochim. Acta, 2012, 63, 105–111 CrossRef CAS PubMed.
- F. J. Vidal-Iglesias, A. López-Cudero, J. Solla-Gullón and J. M. Feliu, Angew. Chem., Int. Ed., 2013, 52, 964–967 CrossRef CAS PubMed.
- H. Lee, S. E. Habas, G. A. Somorjai and P. Yang, J. Am. Chem. Soc., 2008, 130, 5406–5407 CrossRef CAS PubMed.
- E. Casado-Rivera, Z. Gal, A. C. D. Angelo, C. Lind, F. J. DiSalvo and H. D. Abruna, ChemPhysChem, 2003, 4, 193–199 CrossRef CAS PubMed.
- M. D. Spasojevic, R. R. Adzic and A. R. Despic, J. Electroanal. Chem., 1980, 109, 261–269 CrossRef CAS.
- R. R. Adzic and M. L. Avramovivic, J. Electroanal. Chem., 1982, 134, 177–180 CrossRef CAS.
- A. Kelaidopoulou, E. Abelidou and G. Kokkinidis, J. Appl. Electrochem., 1999, 29, 1255–1261 CrossRef CAS.
- J. Clavilier, D. Armand, S. G. Sun and M. Petit, J. Electroanal. Chem., 1986, 205, 267–277 CrossRef CAS.
- J. Clavilier, K. El Achi, M. Petit, A. Rodes and M. A. Zamakhchari, J. Electroanal. Chem., 1990, 295, 333 CrossRef CAS.
- J. Solla-Gullón, F. J. Vidal-Iglesias, A. López-Cudero, E. Garnier, J. M. Feliu and A. Aldaz, Phys. Chem. Chem. Phys., 2008, 10, 3689–3698 RSC.
- Q. S. Chen, J. Solla-Gullon, S. G. Sun and J. M. Feliu, Electrochim. Acta, 2010, 55, 7982–7994 CrossRef CAS PubMed.
- S. Chumillas, C. Busó-Rogero, J. Solla-Gullón, F. J. Vidal-Iglesias, E. Herrero and J. M. Feliu, Electrochem. Commun., 2011, 13, 1194–1197 CrossRef CAS PubMed.
- T. Iwasita and F. C. Nart, Prog. Surf. Sci., 1997, 55, 271–340 CrossRef CAS.
- V. Grozovski, V. Climent, E. Herrero and J. M. Feliu, ChemPhysChem, 2009, 10, 1922–1926 CrossRef CAS PubMed.
- J. Solla-Gullón, P. Rodríguez, E. Herrero, A. Aldaz and J. M. Feliu, Phys. Chem. Chem. Phys., 2008, 10, 1359–1373 RSC.
- Z. Y. Zhou, N. Tian, Z. Z. Huang, D. J. Chen and S. G. Sun, Faraday Discuss., 2008, 140, 81–92 RSC.
- E. Herrero, V. Climent and J. M. Feliu, Electrochem. Commun., 2000, 2, 636–640 CrossRef CAS.
- J. Clavilier, J. P. Ganon and M. Petit, J. Electroanal. Chem., 1989, 265, 231–245 CrossRef CAS.
- R. R. Adzic, J. X. Wang, O. M. Magnussen and B. M. Ocko, J. Phys. Chem., 1996, 100, 14721–14725 CrossRef CAS.
- C. Buso-Rogero, V. Grozovski, F. J. Vidal-Iglesias, J. Solla-Gullon, E. Herrero and J. M. Feliu, J. Mater. Chem. A, 2013, 1, 7068–7076 CAS.
- D. K. Lambert, Electrochim. Acta, 1996, 41, 623–630 CrossRef CAS.
| This journal is © the Owner Societies 2014 |