DOI:
10.1039/B917958E
(Communication)
Chem. Commun., 2010,
46, 130-132
Synthesis of 3-hydroxyoxindoles by Pd-catalysed intramolecular nucleophilic addition of aryl halides to α-ketoamides†‡
Received
(in Cambridge, UK)
2nd September 2009
, Accepted 7th October 2009
First published on
3rd November 2009
Abstract
Pd/PtBu3-catalysed intramolecular nucleophilic addition of aryl halides to α-ketoamides in the presence of nBuOH and base has been realized with high yields, providing a new, direct, and efficient synthetic strategy to obtain 3-hydroxyoxindoles.
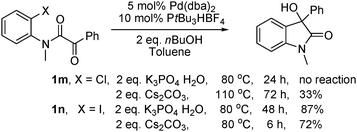
The predominantly electrophilic reactivity of aryl and vinyl organopalladium intermediates derived from the corresponding halides by an oxidative addition process with a Pd(0) catalyst is well established.1 Conversely their use as nucleophiles has received much less attention although literature precedent exists and includes reactions with aldehydes,2 ketones,3 imines,4 esters and amides,5 anhydrides6 and nitriles.7 The direct catalytic addition reaction of aryl and vinyl halides to electrophilic partners is attractive since no extra organometallic reagents are needed. In the context of our interest in the synthesis of oxindoles8 we envisioned that it might be possible to obtain 3-hydroxyoxindoles by an intramolecular addition of aryl halides to α-ketoamides using a Pd catalyst. This would provide a direct access to these compounds avoiding the use of Grignard-, organoborane-, or organolithium-reagents (Scheme 1).9 Herein, we report the preliminary results of this new synthetic strategy to oxindoles.
 |
| | Scheme 1 New route to 3-hydroxyoxindoles. | |
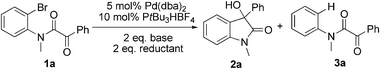
The model substrate 1a was prepared by condensation of N-methyl o-bromoaniline and 2-oxo-2-phenylacetyl chloride. Initial attempts to use Pd(PPh3)4 as a catalyst were not efficient for the transformation 1a → 2a and attempts to improve results by modifications of base, solvent, and additives were not successful. The best result was a 17% yield of 2a with 5 mol% Pd(PPh3)4 in toluene at 110 °C for 24 h in the presence of 2 eq. Na2CO3 and 10 eq. NEt3. Using nBuOH as solvent afforded 3a and only trace amounts of 2a. We then focused on the use of electron-rich ligands,10 which should favour oxidative addition of the catalyst to the aryl bromide and, more importantly, facilitate the nucleophilic attack of the Pd–aryl intermediate on the ketone. Indeed, when 10 mol% PtBu3·HBF4 was used as a ligand precursor, the reaction of 1a proceeded smoothly in the presence of 5 mol% Pd(dba)2, 2 eq. Cs2CO3, and 2 eq. nBuOH in toluene at 50 °C giving oxindole 2a in 76% isolated yield along with 19% of the corresponding hydrodehalogenation side product 3a (Table 1, entry 1). We note that these conditions are milder than those reported previously for this type of reaction—e.g. 100–150 °C for intramolecular additions to ketones.3 Solvent examination revealed toluene to be the best choice as low yields (<15%) were observed in ether solvents such as THF, DME (1,2-dimethoxy ethane), and 1,4-dioxane. In DCE (1,2-dichloroethane) product yield was 54% after 20 h, while in DMF the reaction was completely suppressed. Although the best result was obtained when nBuOH was used as an additive, MeOH and EtOH afforded slightly lower yields whereas iPrOH gave a poor product yield (entries 4–6). In the absence of an additive, the reaction fails, as has been also reported by the Yamamoto group.3b Interestingly, instead of an alcohol H2 (1 bar) could be used to promote this reaction with comparable yield (entry 7). Moderate yields of 2a were observed when nBu3SnH and Et3SiH were used and for the former no side product 3a was detected (entries 8, 9).
Table 1 Optimization of reaction conditionsa
| Entry |
Base |
Solvent |
Reductant |
Temp./°C |
Time/h |
Yb (%) (2a/3a) |
|
0.4 mmol scale in 8 mL solvent.
Isolated yield. nd = not determined.
With a hydrogen balloon.
10 mol% PCy3 was used instead of PtBu3·HBF4.
|
| 1 |
Cs2CO3 |
Toluene |
nBuOH |
50 |
7 |
76/19 |
| 2 |
Cs2CO3 |
DCE |
nBuOH |
50 |
20 |
54/nd |
| 3 |
Cs2CO3 |
tBuOMe |
nBuOH |
50 |
20 |
29/nd |
| 4 |
Cs2CO3 |
Toluene |
MeOH |
50 |
7 |
69/nd |
| 5 |
Cs2CO3 |
Toluene |
EtOH |
50 |
7 |
71/nd |
| 6 |
Cs2CO3 |
Toluene |
iPrOH |
50 |
16 |
21/nd |
| 7c |
Cs2CO3 |
Toluene |
H2 |
100 |
12 |
62/28 |
| 8 |
Cs2CO3 |
Toluene |
nBu3SnH |
100 |
24 |
43/0 |
| 9 |
Cs2CO3 |
Toluene |
Et3SiH |
50 |
12 |
58/23 |
| 10 |
K2CO3 |
Toluene |
nBuOH |
80 |
24 |
23/nd |
| 11 |
K3PO4·H2O |
Toluene |
nBuOH |
50 |
30 |
85/9 |
| 12 |
K3PO4·H2O |
Toluene |
nBuOH |
80 |
7 |
88/7 |
| 13d |
K3PO4·H2O |
Toluene |
nBuOH |
80 |
9 |
85/7 |
Variation of the base revealed this to be an important factor. No reactions took place in the presence of Cy2NMe, KF, KOAc, Li2CO3, and Na2CO3. With K2CO3 as base, oxindole 2a could be isolated in 23% yield after 24 hours (entry 10). Finally, K3PO4·H2O was found to be the best base giving the highest yield of the product albeit a longer reaction time was needed to achieve full conversion at 50 °C (entry 11). Increasing the temperature to 80 °C resulted in complete reaction in 7 hours and 2a was isolated in 88% yield along with 7% side product 3a (entry 12). Further investigation of the ligand showed that PCy3 was also suitable (entry 13), while dppe (1,2-bis(diphenylphosphino)ethane) and 1,3-bis(2,6-diisopropyl-phenyl)-4,5-dihydroimidazolium chloride (SIPr·HCl) as ligand or ligand precursor were inefficient in this reaction.
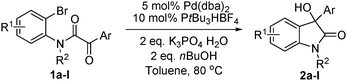
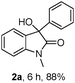
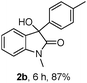
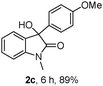
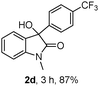
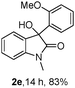
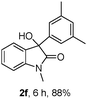
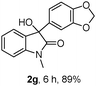
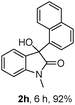
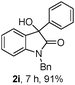
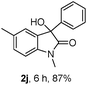
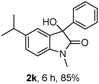
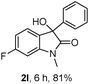
The optimum reaction conditions were then applied to the substrates shown in Table 2. Yields are generally good (81–92%). It is noteworthy that the reaction to oxindole 2d with CF3 as an electron-withdrawing group was complete in 3 h, while 14 h were required for the reaction of substrate 1e with an o-MeO group. Not only N-Me substrates but also an N-benzyl amide (2i) could be used, but no reaction took place for the free N-H substrate (R1 = R2 = H, Ar = Ph).
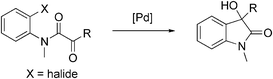
The reaction is sensitive to the aryl halide used (eqn (1)). In comparison with 1a, no reaction took place for the analogous chloride substrate 1m. Noteworthy, the reaction of the iodo substrate 1n was slower than that of the bromo substrate 1a and 48 h were needed to achieve full conversion. Since the reaction of 1a with Cs2CO3 as base was faster than with K3PO4·H2O, we introduced Cs2CO3 to the reactions of these two substrates. Product 2a was isolated in 72% yield from 1n after 6 h at 80 °C and in 33% yield from 1m after 72 h at 110 °C. Interestingly, the reaction of 1a under standard conditions was suppressed in the presence of 3.0 eq. LiI as additive and only traces of oxindole 2a were detected after 48 h.
| |  | (1) |
Vinyl bromide substrate
1o was synthesized and applied in the reaction (
eqn (2)).
Pyrrolidinone product
2m could be obtained; yields were low (16% for K
3PO
4·H
2O as base, 29% for Cs
2CO
3).
| | 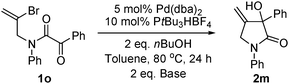 | (2) |
Next, experiments designed to shed light on mechanistic aspects of the reaction were carried out. As shown in
eqn (3),
oxindole 2a could be isolated in 92% yield with stoichiometric Pd(0)/P
tBu
3 complex in the absence of
nBuOH, thus demonstrating that Pd(0) can perform the same role as Mg in
Grignard reactions. As mentioned above,
catalytic use of Pd requires an additive and
primary alcohols performed best. An earlier study of an intramolecular aryl addition to ketones, catalysed by Pd with added
1-hexanol,
3b reported that the alcohol was recovered. Our findings are different. Using
piperonyl alcohol, as shown in
eqn (4), piperonyl aldehyde was isolated in 95% yield. This confirms the role of the alcohol as reductant in the reaction and thus closing the
catalytic cycle. Also, the deuterated
dehalogenation side product was detected when MeOH-d
4 was used (
eqn (5)).
| | 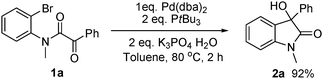 | (3) |
| | 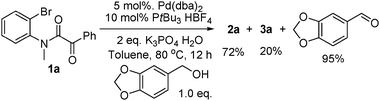 | (4) |
| | 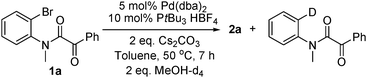 | (5) |
Based on the above, we propose the mechanism shown in
Fig. 1. The
oxindole is formed by intramolecular addition of the oxidative addition intermediate
I to the
ketone. Alkoxyligand exchange
II (cycle A) liberates product
2a and the Pd(0)
catalyst is regenerated by β-H elimination (
aldehyde formation) followed by base assisted reductive elimination. For the hydrodehalogenation side product
3a we propose
halide–alcoholate ligand exchange in
I to give
III (cycle B). β-Elimination followed by reductive elimination affords
3a (cycle B). The observation of the deuterated side product shown in
eqn (5) supports this pathway.
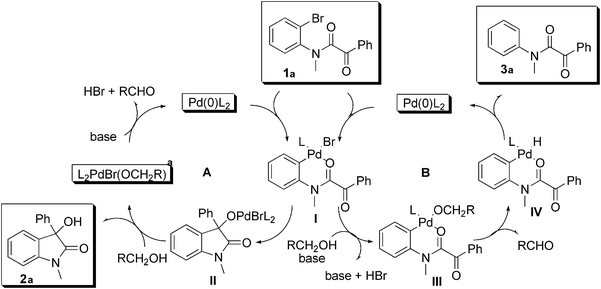 |
| | Fig. 1 Proposed mechanism. | |
In summary, we have developed a new and efficient procedure to synthesize 3-hydroxyoxindole compounds in high yields by applying palladium-catalysed direct addition of aryl halides to α-ketoamides. Both, electron-rich phosphine ligands and a reductant (alcohol, H2) are crucial. The formation of a new stereogenic center in 2 is another important aspect that is receiving attention in ongoing research.
Notes and references
-
J. Tsuji, Palladium Reagents and Catalysis—New Perspectives for the 21st Century, Wiley, Weinheim, 2004 Search PubMed.
-
(a) R. C. Larock, M. J. Doty and S. Cacchi, J. Org. Chem., 1993, 58, 4579–4583 CrossRef CAS;
(b) V. Gevorgyan, L. G. Quan and Y. Yamamoto, Tetrahedron Lett., 1999, 40, 4089–4092 CrossRef;
(c) J. Vicente, J. A. Abad, B. Lopez-Pelaez and E. Martinez-Viviente, Organometallics, 2002, 21, 58–67 CrossRef CAS;
(d) Y. B. Zhao, B. Mariampillai, D. A. Candito, B. Laleu, M. Z. Li and M. Lautens, Angew. Chem., Int. Ed., 2009, 48, 1849–1852 CrossRef CAS.
-
(a) L. G. Quan, V. Gevorgyan and Y. Yamamoto, J. Am. Chem. Soc., 1999, 121, 3545–3546 CrossRef CAS;
(b) L. G. Quan, M. Lamrani and Y. Yamamoto, J. Am. Chem. Soc., 2000, 122, 4827–4828 CrossRef CAS;
(c) D. Sole, L. Vallverdu, X. Solans, M. Font-Bardia and J. Bonjoch, J. Am. Chem. Soc., 2003, 125, 1587–1594 CrossRef CAS.
-
(a) K. R. Roesch and R. C. Larock, Org. Lett., 1999, 1, 553–556 CrossRef CAS;
(b) H. Ohno, A. Aso, Y. Kadoh, N. Fujii and T. Tanaka, Angew. Chem., Int. Ed., 2007, 46, 6325–6328 CrossRef CAS.
-
(a) D. Sole and O. Serrano, Angew. Chem., Int. Ed., 2007, 46, 7270–7272 CrossRef CAS;
(b) D. Sole and O. Serrano, J. Org. Chem., 2008, 73, 9372–9378 CrossRef CAS.
- S. Cacchi, G. Fabrizi, F. Gavazza and A. Goggiamani, Org. Lett., 2003, 5, 289–291 CrossRef CAS.
-
(a) C. C. Yang, P. J. Sun and J. M. Fang, J. Chem. Soc., Chem. Commun., 1994, 2629–2630 RSC;
(b) R. C. Larock, Q. P. Tian and A. A. Pletnev, J. Am. Chem. Soc., 1999, 121, 3238–3239 CrossRef CAS;
(c) A. A. Pletnev and R. C. Larock, J. Org. Chem., 2002, 67, 9428–9438 CrossRef CAS;
(d) A. A. Pletnev, Q. P. Tian and R. C. Larock, J. Org. Chem., 2002, 67, 9276–9287 CrossRef CAS;
(e) Q. P. Tian, A. A. Pletnev and R. C. Larock, J. Org. Chem., 2003, 68, 339–347 CrossRef CAS;
(f) C. X. Zhou and R. C. Larock, J. Org. Chem., 2006, 71, 3551–3558 CrossRef.
-
(a) E. P. Kündig, T. M. Seidel, Y.-X. Jia and G. Bernardinelli, Angew. Chem., Int. Ed., 2007, 46, 8484–8487 CrossRef;
(b) Y.-X. Jia, J. M. Hillgren, E. L. Watson, S. P. Marsden and E. P. Kündig, Chem. Commun., 2008, 4040–4042 RSC;
(c) Y.-X. Jia and E. P. Kündig, Angew. Chem., Int. Ed., 2009, 48, 1636–1639 CrossRef CAS.
- 3-Hydroxyoxindoles can be obtained by addition of aryl Grignard reagents or aryl lithium reagents (both are prepared from aryl halides) to isatin:
(a) M. A. Sukari and J. M. Vernon, J. Chem. Soc., Perkin Trans. 1, 1983, 2219–2223 RSC;
(b) S. J. Angyal, E. Bullock, W. G. Hanger, W. C. Howell and A. W. Johnson, J. Chem. Soc., 1957, 1592–1602 RSC and ref. cit. Catalytic asymmetric synthesis of 3-hydroxyoxindole: Pd catalysed intramolecular arylation of amides;
(c)
ref. 8b; M-catalysed addition of aryl boronic acids to isatin;
(d) R. Shintani, M. Inoue and T. Hayashi, Angew. Chem., Int. Ed., 2006, 45, 3353–3356 CrossRef CAS;
(e) H. S. Lai, Z. Y. Huang, Q. Wu and Y. Qin, J. Org. Chem., 2009, 74, 283–288 CrossRef CAS; Lewis acid-catalysed asymmetric hydroxylation of oxindoles;
(f) T. Ishimaru, N. Shibata, J. Nagai, S. Nakamura, T. Toru and S. Kanemasa, J. Am. Chem. Soc., 2006, 128, 16488–16489 CrossRef.
- Although PCy3 has been used as a ligand in a similar addition reaction of simple ketone substrates (ref. 3b), no reaction took place from 1a to 2a using the reported conditions (5 mol% Pd(OAc)2, 10 mol% PCy3, 2 eq. Na2CO3, 5 eq. 1-hexanol in DMF).
Footnotes |
| † This article is part of a ChemComm ‘Catalysis in Organic Synthesis’™ web-theme issue showcasing high quality research in organic chemistry. Please see our website (http://www.rsc.org/chemcomm/organicwebtheme2009) to access the other papers in this issue. |
| ‡ Electronic supplementary information (ESI) available: Experimental details, spectral characterisation and analytical data of reported compounds. See DOI: 10.1039/b917958e |
|
| This journal is © The Royal Society of Chemistry 2010 |
Click here to see how this site uses Cookies. View our privacy policy here.