Dioxygen mediated hydroacylation of vinyl sulfonates and sulfones on water†
Vijay
Chudasama
,
Richard J.
Fitzmaurice
,
Jenna M.
Ahern
and
Stephen
Caddick
*
Department of Chemistry, University College London, London, UK WC1H 0AJ. E-mail: s.caddick@ucl.ac.uk; Fax: +44 (0)20 7679 4603; Tel: +44 (0)20 7679 7482
First published on 13th November 2009
Abstract
Herein we report a mild, facile method for the preparation of 1,4-keto-sulfonates and sulfones on water. Further synthetic manipulations can result in products that are not readily accessed by hydroacylation of electron rich alkenes.
The development of new, efficient and environmentally benign synthetic methodologies for the construction of complex molecules, and in particular C–C bonds, is an important target in modern organic synthesis.1–3 In view of this, many recent methods have focused on the use of sub-stoichiometric reagents or catalysts which tend to offer enhanced atom-economy and minimisation of waste.4 Furthermore, significant advances in the development of aqueous organic transformations and organocatalysts, which take advantage of the unique properties of water, have been made in recent years.5–11Water is generally perceived as a desirable, ‘green’ solvent for organic transformations owing to cost, safety and environmental acceptability.2,3 Furthermore, on water reactions, have been demonstrated to offer several other advantages as a consequence of hydrogen bonding and/or hydrophobic effects accelerating the rate and/or yield of certain chemical reactions.8
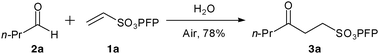
Recently, we have reported the hydroacylation of electron poor olefins such as pentafluorophenyl (PFP) vinyl sulfonate and trichlorophenyl (TCP) vinyl sulfonate with a variety of aldehydes.12 Although the conditions previously developed offer an attractive method for hydroacylation of olefins we have an aspiration to develop reagent-free conditions using only water and air for carbon–carbon bond formation.
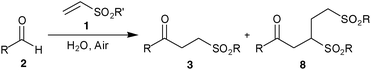
We were inspired by the recent work of Shapiro and Vigalok11 describing the aerial oxidation of aldehydes to carboxylic acids and considered that we might be able to utilise similar conditions for the generation and trapping of acyl radicals for carbon–carbon formation. Thus we rapidly identified an operationally simple protocol for the hydroacylation of vinyl sulfonate 1a with butanal2a on water at room temperature to give hydroacylation product 3a in good yield (Scheme 1). It should be noted that the reaction requires no particularly specific conditions, although we have found that it is important to have a reasonably concentrated reaction mixture, efficient stirring (stirrer bar) and to ensure that the reaction is exposed to air (further details in ESI† ). Although we have not examined scalability in detail, we have carried out the reaction of 1a with 2a on a preparatively useful 2 g scale with isolated yields of >70%.
In order to gather evidence for the general nature of this process we examined a small selection of aliphatic aldehydes (Table 1). Although structurally similar, aldehydes were specifically chosen to test the limits of our methodology with respect to aldehyde auto-oxidation rate (Table 2). The yields for the hydroacylation reactions are comparable to those utilising peroxide and superior to the analogous reactions in the 1,4-dioxane conditions that we have previously described.12 The tolerance of the reaction to α-branched and lengthy alkyl chain aliphatic aldehydes was particularly encouraging. Moreover, the good yields obtained with the application of only 2 equivalents of aldehyde with no additional catalyst are competitive with existing hydroacylation protocols for electron poor alkenes.13 However, as in our previous study, we note a disappointing yield for the hydroacylation of vinyl sulfonate 1a with isobutyraldehyde2b (Table 1, entry 2).
| Entry | R = | Time/h | Yield 3b (%) | Solubilityd /mass% | 2 : 4e |
|---|---|---|---|---|---|
a Conditions: aldehyde (2 eq.) and alkene (1 eq.) were stirred at 300 rpm in water at 21 °C.
b 100% conversion of 1a.
c 60% conversion of 1a.
d Solubility of aldehyde in water at 30 °C.14
e Ratio determined by 1H NMR integration in D2O.
f Due to poor solubility of aldehyde in D2O, a DMSO–D2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture was used. :1) mixture was used.
|
|||||
| 1 | –(CH2)2CH3, 2a | 3 | 78, 3a | 5.48 | 1:1.04 |
| 2 | –CH(CH3)2, 2b | 3 | 40, c3b | 4.57 | 1:0.86 |
| 3 | –CH2CH(CH3)2, 2c | 3 | 74, 3c | 1.78 | 1:0.66 |
| 4 | –(CH2)4CH3, 2d | 6 | 75, 3d | 0.44 | 1:0.98 |
| 5 | –c-C6H11, 2e | 3 | 74, 3e | — | 1:0.54 |
| 6 | –CH(C2H5)(CH2)3CH3, 2f | 3 | 83, 3f | 0.05 | 1:0.03f |
| 7 | –(CH2)8CH3, 2g | 6 | 62, 3g | 0.02 | 1:0.12f |
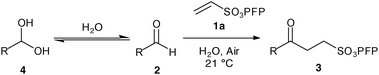
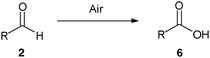
As with our previously described work we invoke a radical mechanism based on the observed retardation by addition of 2,6-di-tert-butyl-4-methylphenol (BHT).12 We envisaged that hydration of aldehyde may play a role and whilst NMR spectroscopy confirmed that the aldehydes were in equilibrium with their hydrate4 under the reaction conditions (Table 1) we could find no correlation between aldehyde solubility in water14 or aldehyde hydration on the yield of product 3. In view of this, and the low conversion observed in the absence of water for the hydroacylation of 1a with butanal2a (ca. 25% in 3 h), we believe water to influence the transformation through a hydrophobic effect and postulate that the reaction proceeds via the mechanism given in Scheme 2. The aldehyde is aerobically transformed to acyl radical 5, which can auto-oxidise to acid 6 or add to the alkene to form 7. Adduct radical 7 then abstracts an aldehydic hydrogen to form product 3 and regenerate acyl radical 5. No deuterium incorporation was observed when reaction of vinyl sulfonate 1a and butanal2a was performed in deuterium oxide; this indicates that the hydrogen atom in 3 is likely to come from abstraction of an aldehydic C–H.
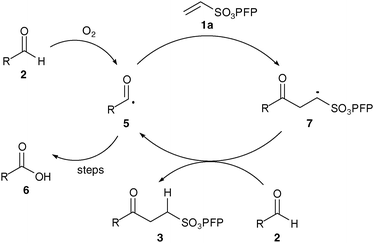 | ||
| Scheme 2 Postulated mechanism for hydroacylation. | ||
To gain further insight into the rate of the auto-oxidation process for different aldehydes a brief study was carried out (Table 2). The results from this study correlate with the reaction times observed on reaction of the aldehydes with 1a in that the aldehydes that auto-oxidised at the slowest rate required prolonged reaction times (Table 1, entries 4 and 7, and Table 2, entries 6 and 7). Moreover, the low conversion observed for isobutyraldehyde could be attributed to its propensity to rapidly auto-oxidise (Table 2, entry 1).
| Entry | R = | 2 : 6a |
|---|---|---|
| a Ratio of aldehyde2 to acid 6 after 2 h stirring at 300 rpm determined through comparison of the 1H NMR integration. It should also be noted that not stirring significantly slowed the rate of auto-oxidation. | ||
| 1 | –CH(CH3)2, 2b | 1:5.11 |
| 2 | –CH(C2H5)(CH2)3CH3, 2f | 1:2.16 |
| 3 | –CH2CH(CH3)2, 2c | 1:1.20 |
| 4 | –(CH2)2CH3, 2a | 1:1.08 |
| 5 | –c-C6H11, 2e | 1:0.66 |
| 6 | –(CH2)4CH3, 2d | 1:0.37 |
| 7 | –(CH2)8CH3, 2g | 1:0.04 |
We have extended the olefin tolerance to both ethyl and phenyl vinyl sulfonates and, most pleasingly, vinyl sulfones (Table 3). Although this required the use of 5 equivalents of aldehyde and needs further optimisation to make it synthetically efficient, it indicates that the trapping of acyl radical intermediates in the aerial oxidation of aldehydes is not entirely predicated on a very electron poor vinyl sulfonate.12 Nonetheless, the application of a large excess of one of the reagents is not unknown in hydroacylation reactions,13 even those that employ polarity reversal catalysts.15,16
| Entry | R = | R′ = | Yield 3b,c (%) |
|---|---|---|---|
| a Conditions: aldehyde (5 eq.) and alkene (1 eq.) were stirred at 300 rpm in water at 21 °C unless stated otherwise. b All reactions proceeded with 100% conversion of 1. c Yields given in parentheses refer to 1H NMR yields based on pentachlorobenzene as an internal standard. d Heating to 60 °C was required. | |||
| 1 | –(CH2)2CH3, 2a | OEt, 1b | 55 (64), 3ab |
| 2 | –c-C6H11, 2e | 52 (62), 3eb | |
| 3 | –(CH2)2CH3, 2a | OPh, 1c | 52, 3ac |
| 4 | –c-C6H11, 2e | 57, 3ec | |
| 5 | –(CH2)2CH3, 2a | Et, 1d | 64, d3ad |
| 6 | –c-C6H11, 2e | 57, d3ed | |
| 7 | –(CH2)2CH3, 2a | Ph, 1e | 56, d3ae |
| 8 | –c-C6H11, 2e | 61, d3ee | |
Although an excess of aldehyde was required to permit vinyl sulfonate 1 to compete effectively with molecular oxygen for acyl radical 5, it ensured high conversion and minimisation of the formation of higher order products such as 8. Indeed, in the case of reaction of 2a with 1b the use of 2 equivalents of 2a resulted in a 2:1 ratio of products 3 and 8. Whereas at a 5:1 reaction stoichiometry a 5:1 ratio was observed (Table 3, entry 1). It appears that in the case of the alkenes1b–1e the acyl radical trapping and radical chain propagation are less efficient when compared to 1a. A consequence of the high reaction stoichiometry is formation of significant amounts of acid which could be recovered if required in appreciable amounts (65% based on starting aldehyde for 2e with 1b).
Prior work from this laboratory17–20 has shown that sulfonates are useful alternatives to sulfonyl chlorides in the synthesis of sulfonamides. However β-keto-sulfonates offer specific opportunities for additional transformations. Thus using a modification of our usual protocol,19,20 we were able to convert β-keto-sulfonate 3a into sulfonamide9 in good yield. Moreover, due to the presence of the β-keto functionality, we were also able to synthesise sultam10 (Scheme 3) in excellent yield through a reductive cyclisation of 9 under acidic conditions.
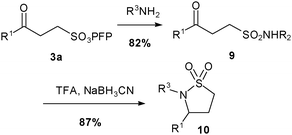 | ||
| Scheme 3 Synthesis of sultam10, where R1 = nBu and R2 = nhexyl. | ||
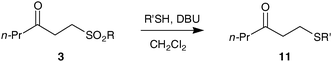
Furthermore, treatment of 3a with DBU led to clean elimination to the enone (not shown) demonstrating an overall conversion of an aldehyde to an enonevia hydroacylation and elimination. We envisage that this hydroacylation–elimination approach is complementary to current methods for the generation of enones from aldehydes and avoids the use of potent nucleophiles and oxidising conditions commonly employed.21 The opportunities afforded by the vinyl sulfonate and hence β-keto-sulfonate derived from hydroacylation are enhanced by this elimination protocol, because the enone can undergo a wide range of conjugate addition reactions.22 For example, in the alkene hydroacylation arena, there is a significant limitation associated with electron rich alkenes.16 Thus, using a simple sequence involving hydroacylation, elimination and addition, it should be possible to access a variety of unsymmetrical functionalised ketones from aldehydes which would offer a significant challenge to current alkene hydroacylation methodologies.16 In order to demonstrate this concept we have made β-keto-sulfides, the synthesis of which, to the best of our knowledge, has not been reported by direct hydroacylation of vinyl sulfides.23 Thus, treatment of sulfonates 3a, 3ab and 3ac with DBU and a thiol in dichloromethane led to a smooth and high yielding transformation to 11, presumably via an elimination–addition sequence (Table 4, entries 1–6). As the elimination of sulfones to give alkenes is well documented,24 the elimination conditions were applied to sulfones3ad and 3ae (Table 4, entries 7–10) and were successful, albeit in poorer yields. We believe the inferior yields observed for β-keto-sulfones3ad and 3ae highlight a specific advantage of β-keto-sulfonates 3a, 3ab and 3ac.
| Entry | R = | R′ = | Yield 9 (%) |
|---|---|---|---|
| 1 | –OPFP, 3a | –(CH2)5CH3 | 98, 11a |
| 2 | –CH2C6H4CH3 | 97, 11b | |
| 3 | –OEt, 3ab | –(CH2)5CH3 | 89, 11a |
| 4 | –CH2C6H4CH3 | 90, 11b | |
| 5 | –OPh, 3ac | –(CH2)5CH3 | 92, 11a |
| 6 | –CH2C6H4CH3 | 90, 11b | |
| 7 | –Et, 3ad | –(CH2)5CH3 | 46, 11a |
| 8 | –CH2C6H4CH3 | 34, 11b | |
| 9 | –Ph, 3ae | –(CH2)5CH3 | 78, 11a |
| 10 | –CH2C6H4CH3 | 75, 11b |
In summary, we have developed a novel method for the effective hydroacylation of vinyl sulfonates and sulfones on water using only air to promote C–H activation. The previously unexplored β-keto-PFP-sulfonates, which have been prepared via a relatively efficient hydroacylation pathway, show promise as reagents for β-keto-sulfonamide and sultam formation. Their ability to also undergo efficient elimination, and hence for the in situ generation of enones, provides opportunities for the synthesis of molecules that may not be readily accessible via the hydroacylation of electron rich alkenes.
We gratefully acknowledge the EPSRC and UCL for funding and the EPSRC Mass Spectrometry Service for provision of spectra.
Notes and references
- P. T. Anastas and M. M. Kirchhoff, Acc. Chem. Res., 2002, 35, 686 CrossRef CAS.
- C. J. Li, Chem. Rev., 1993, 93, 2023 CrossRef CAS.
- U. M. Lindstrom, Chem. Rev., 2002, 102, 2751 CrossRef.
- R. A. Sheldon, Green Chem., 2008, 10, 359 RSC.
- A. El-Batta, C. C. Jiang, W. Zhao, R. Anness, A. L. Cooksy and M. Bergdahl, J. Org. Chem., 2007, 72, 5244 CrossRef CAS.
- D. Gonzalez-Cruz, D. Tejedor, P. de Armas and F. Garcia-Tellado, Chem.–Eur. J., 2007, 13, 4823 CrossRef.
- S. Y. Jung and R. A. Marcus, J. Am. Chem. Soc., 2007, 129, 5492 CrossRef CAS.
- S. Narayan, J. Muldoon, M. G. Finn, V. V. Fokin, H. C. Kolb and K. B. Sharpless, Angew. Chem., Int. Ed., 2005, 44, 3275 CrossRef CAS.
- M. C. Pirrung and K. Das Sharma, J. Am. Chem. Soc., 2004, 126, 444 CrossRef CAS.
- M. C. Pirrung, Chem.–Eur. J., 2006, 12, 1312 CrossRef CAS.
- N. Shapiro and A. Vigalok, Angew. Chem., Int. Ed., 2008, 47, 2849 CrossRef CAS.
- R. J. Fitzmaurice, J. M. Ahern and S. Caddick, Org. Biomol. Chem., 2009, 7, 235 RSC.
- For lead references to hydroacylation strategies see: M. Christmann, Angew. Chem., Int. Ed., 2005, 44, 2632 Search PubMed; D. Seebach, Angew. Chem., Int. Ed. Engl., 1979, 18, 239 CrossRef CAS; C. Chatgilialoglu, D. Crich, M. Komatsu and I. Ryu, Chem. Rev., 1999, 99, 1991 CrossRef.
- R. M. Stephenson, J. Chem. Eng. Data, 1993, 38, 630 CrossRef CAS.
- H. S. Dang and B. P. Roberts, J. Chem. Soc., Perkin Trans. 1, 1998, 67 RSC.
- S. Tsujimoto, T. Iwahama, S. Sakaguchi and Y. Ishii, Chem. Commun., 2001, 2352 RSC; S. Tsujimoto, S. Sakaguchi and Y. Ishii, Tetrahedron Lett., 2003, 44, 5601 CrossRef CAS.
- S. Caddick, J. D. Wilden and D. B. Judd, J. Am. Chem. Soc., 2004, 126, 1024 CrossRef CAS.
- S. Caddick, J. D. Wilden, H. D. Bush, S. N. Wadman and B. Judd, Org. Lett., 2002, 4, 2549 CrossRef CAS.
- J. D. Wilden, D. B. Judd and S. Caddick, Tetrahedron Lett., 2005, 46, 7637 CrossRef CAS.
- S. Caddick, J. D. Wilden and D. B. Judd, Chem. Commun., 2005, 2727 RSC.
- For examples see: R. T. Lewis, W. B. Motherwell and M. Shipman, J. Chem. Soc., Chem. Commun., 1988, 948 Search PubMed; E. Negishi, S. Ma, T. Sugihara and Y. Noda, J. Org. Chem., 1997, 62, 1922 RSC.
- For example see: P. Perlmutter, Conjugate Addition Reactions in Organic Synthesis, Tetrahedron Organic Chemistry Series, Pergamon, Oxford, 1992, vol. 9 Search PubMed.
- We note that Willis and co-workers have reported the synthesis of β-keto-sulfidesvia metal catalysed alkene and alkyne hydroacylation incorporating the sulfide into the aldehyde component, see for example: G. L. Moxham, H. Randell-Sly, S. K. Brayshaw, A. S. Weller and M. C. Willis, Chem.–Eur. J., 2008, 14, 8383 Search PubMed.
- For example see: B. R. Langlois, T. Billard, J.-C. Mulatier and C. Yezeguelian, J. Fluorine Chem., 2007, 128, 851 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of experimental procedures and data for novel compounds. See DOI: 10.1039/b914563j |
| This journal is © The Royal Society of Chemistry 2010 |