
Improving the expression of taxadiene synthase to enhance the titer of taxadiene in Saccharomyces cerevisiae†
Chenglong
Zhang
ab,
Jia
Wang
ab,
Yi
Shi
ab,
Nan
Wu
ab,
Xia
Li
ab,
Ying
Wang
 ab,
Bingzhi
Li
ab,
Wenhai
Xiao
*abcd,
Mingdong
Yao
*ab and
Yingjin
Yuan
ab
ab,
Bingzhi
Li
ab,
Wenhai
Xiao
*abcd,
Mingdong
Yao
*ab and
Yingjin
Yuan
ab
aFrontier Science Center for Synthetic Biology and Key Laboratory of Systems Bioengineering (Ministry of Education), School of Chemical Engineering and Technology, Tianjin University, Tianjin 300072, China. E-mail: mingdong.yao@tju.edu.cn
bFrontier Research Institute for Synthetic Biology, Tianjin University, China
cSchool of Life Sciences, Faculty of Medicine, Tianjin University, China
dGeorgia Tech Shenzhen Institute, Tianjin University, Shenzhen 518071, China
First published on 13th September 2024
Abstract
Taxadiene is an important precursor of paclitaxel. However, its low yield in eukaryotic systems limits its biosynthesis. This study found that the low yield of taxadiene is likely due to the degradation of expressed taxadiene synthase (TS) in Saccharomyces cerevisiae. The TS expression degradation problem was improved by the knockout of protease PRB1, and the yield of taxadiene was increased by 97% from 19.8 mg L−1 to 39.2 mg L−1. Furthermore, multi-copy integration of the TS gene and enhancement of the geranylgeranyl diphosphate (GGPP) precursor pathway increased the taxadiene titer to 282.4 mg L−1 in a shake flask. Interestingly, enhancing TS expression also decreased the competitive synthesis of geranylgeraniol (GGOH). Finally, the taxadiene titer reached 878.5 mg L−1 after optimising the fed-batch fermentation, which is the highest taxadiene titer reported for eukaryotic microbes. This study alleviated the problems associated with the expression and degradation of heterologous proteins and provided an efficient and green strategy to produce complex natural compounds.
1. Introduction
Paclitaxel is a natural product biosynthesized from the bark extract of Taxus brevifolia (evergreen needle-leaf trees of Taxaceae).1 Owing to its unique chemical structure and pharmacological characteristics, this compound exhibits potent anti-proliferative activity. Hence, clinically approved chemotherapeutic agents have proved to be effective in treating refractory ovarian carcinoma, metastatic breast malignancy, advanced non-small cell lung cancer, and other solid tumours. Paclitaxel was originally isolated from the bark of T. brevifolia; however, this species has a prolonged growth period, short cycling, poor regeneration capacity, and an extremely low paclitaxel content in the bark.2 Chemical and semi-chemical synthesis methods were used to produce paclitaxel.3 Concurrently, the chemical synthesis of paclitaxel faces obstacles such as more than 20 steps of the synthetic route, low yields, stringent reaction conditions, and significant generation of toxic waste streams, rendering it economically unfeasible for commercial production.4 Currently, the industrial production of paclitaxel primarily adopts a semi-synthetic approach.5 This involves isolating the key intermediate (10-deacetylbaccatin III) first from the needles and leaves of the yew tree, followed by a four-step chemical transformation to synthesize paclitaxel.6 However, owing to the structural complexity of paclitaxel, the precursors used for its semi-chemical synthesis are still derived from the extraction of artificially planted trees and, thus, are still unable to alleviate the shortage of raw materials for paclitaxel synthesis.In recent years, the utilisation of microbes to produce high-value plant compounds has become a promising alternative approach owing to rapid advances in synthetic biology and metabolic engineering.7,8 Furthermore, compared to the production processes of natural product chemical synthesis, microbial synthesis utilizes renewable biomass to produce the target compounds via green and sustainable means.9 Taxadiene is the key intermediate in the paclitaxel biosynthetic pathway and high-yield taxadiene production is an essential prerequisite for efficient paclitaxel biosynthesis.10 Taxadiene is formed by the cyclisation of geranylgeranyl diphosphate (GGPP) by taxadiene synthase (TS). Ajikumar et al. used a multivariate modular pathway engineering approach for dividing taxadiene synthesis into an upstream native IPP synthesis module and downstream GGPP and taxadiene synthesis modules. By combining these two modules with different promoters and gene copy numbers, they identified an optimal combination that increased taxadiene production in a 2 L bioreactor by 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold to 1 g L−1.11 In the paclitaxel biosynthetic pathway, the intermediate taxadiene undergoes a series of complex reactions, including hydroxylation, acetylation, and epoxidation.12 Compared to prokaryotic systems, the eukaryotic system Saccharomyces cerevisiae (S. cerevisiae) possesses complete membrane systems and post-translational protein modification machinery, giving it an advantage for expressing P450 enzymes.13 Therefore, S. cerevisiae have tremendous potential as a platform for the green bio-synthesis of paclitaxel.
000-fold to 1 g L−1.11 In the paclitaxel biosynthetic pathway, the intermediate taxadiene undergoes a series of complex reactions, including hydroxylation, acetylation, and epoxidation.12 Compared to prokaryotic systems, the eukaryotic system Saccharomyces cerevisiae (S. cerevisiae) possesses complete membrane systems and post-translational protein modification machinery, giving it an advantage for expressing P450 enzymes.13 Therefore, S. cerevisiae have tremendous potential as a platform for the green bio-synthesis of paclitaxel.
Although taxadiene production in Escherichia coli (E. coli) has made significant progress, its yield in S. cerevisiae remains very low. Compared to other terpene synthases, TS not only has lower expression levels but also relatively lower activity, with a turnover rate 70-fold lower than that of plant diterpene synthases with higher sequence homology.14 Engels et al. introduced GGPP synthase (SaGGPPS) from Sulfolobus acidocaldarius and TS from Taxus into S. cerevisiae and overexpressed tHMG1. Through TS codon optimisation, the final taxadiene titer reached 8.7 mg L−1.15 According to previous studies, the expression of soluble TS in S. cerevisiae is poor. Nowrouzi et al. increased the expression of soluble TS by utilising solubility enhancement tags, thereby increasing the taxadiene titer in S. cerevisiae to 129 mg L−1.16 TS protein expression was enhanced by the fusion of GGPPS and TS in our previous study.17 Although previous studies have proposed various engineering strategies to improve TS protein expression and activity, including N-terminal truncation, solubility-enhancing tags, subcellular relocalization, and copy number amplification, taxadiene production has not yet reached the expected level in S. cerevisiae. Progress has been made regarding the downstream modification pathways of taxadiene to paclitaxel and the latest studies have revealed the key missing enzymes in the biological pathway of paclitaxel, clarified a new mechanism of oxidative rearrangement in paclitaxel oxetane formation, and discovered multiple related genes in the heterogeneous synthesis pathway of paclitaxel.18 The exploration of taxadiene and its downstream pathways in eukaryotic systems has garnered considerable attention.19 Therefore, finding new strategies and tools to improve the expression in S. cerevisiae and increase the taxadiene yield is worthy of further investigation.
Many studies have addressed the problem of low heterologous protein expression in S. cerevisiae. To increase the yield of haemoglobin in S. cerevisiae, Ishchuk et al. knocked out the VPS1 gene encoding the vacuolar protein sorting receptor and the PEP4 gene encoding vacuolar protease A to reduce the degradation of haemoglobin.20 Moreover, disrupting the PAH1 encoding phospholipid phosphatase by CRISPR/Cas9 caused endoplasmic reticulum stress (ERS), thereby stimulating the production of recombinant triterpene biosynthetic enzymes and ultimately promoting the accumulation of triterpene compounds and triterpene saponins.21 Yap et al. knocked out PRB1 and PEP4 genes in S. cerevisiae BJ5464, improved the expression of sesquiterpene synthetase derived from tiger milk mushroom Lignosus rhinocerotis, and increased the yield of α-cadinol.22 Tang et al. enhanced the expression of monacolin-j acid (MJA) biosynthesis genes and increased the MJA titer to 75 mg L−1 by deleting PYC2, PRB1, and PEP4 proteases.23 The productivity of heterologous proteins in S. cerevisiae can be effectively improved by inhibiting the activity of related proteases to prevent their degradation.24
In previous studies, we had successfully constructed a high-producing GGPP strain by screening metabolic pathway genes and controlling key gene promoters, thereby providing an excellent basis for taxadiene biosynthesis.25 In this study, we aimed to verify the abnormal protein expression of TS and the phenomenon of TS protein cleavage in the expression system of S. cerevisiae. Subsequently, we knocked out the endogenous protease PRB1 of S. cerevisiae and improved the expression level of the intact TS protein, which resulted in a significant increase in the production of taxadiene. Then, we further increased the titer of taxadiene to 282.4 mg L−1 in a shaking bottle by enhancing the GGPP precursor pathway and integrating the multi-copy TS gene into the TY1 site. Finally, the titer of taxadiene was increased to 878.5 mg L−1 in a 5 L bioreactor under optimised fed-batch fermentation conditions, which is the highest taxadiene yield reported in eukaryotes (Fig. 1).
 | ||
| Fig. 1 Biosynthesis pathway of taxadiene and optimization of key enzyme expression. HMG-CoA, 3-hydroxy-3-methylglutaryl coenzyme A; tHMG1, a truncated HMG-CoA reductase 1; IPP, isopentenyl pyrophosphate; DMAPP, dimethylallyl pyrophosphate; GGPP, geranylgeranyl diphosphate; TS, taxadiene synthase; GGOH, geranylgeraniol. | ||
2. Materials and methods
2.1 Strains and culture media
The yeast strains used in this study are listed in Table 1. Plasmid construction and amplification were performed using E. coli Top10 (TransGen Biotech, Beijing, China). S. cerevisiae CEN.PK2-1C (MAT a, ura3-52, trp1-289, leu2-3,112, his3Δ1, MAL2-8C, SUC2) (Table 1), obtained from EUROSCARF (Frankfurt, Germany), was employed for the development of taxadiene producing strains as listed in Table 1.26| Strains | Host strain | Description | Source |
|---|---|---|---|
| CEN.PK2-1C | MAT a; ura3-52, trp1-289, leu2-3,112, his3Δ1, MAL2-8C, SUC2 | This lab | |
| yZCL078 | CEN.PK2-1C | MAT a; ura3-52, trp1-289, leu2-3,112, his3Δ1, MAL2-8C, SUC2; Delta22::CYC1t-TDH1p-BST1-ERG20-TEF1t-PYC1p-tHMG1-HXT7t; ΔYL1062W; Δgal80 | This study |
| yZCL080 | yZCL078 | pZCL106 | This study |
| yZCL081 | yZCL078 | ΔCYM1::LEU2 | This study |
| yZCL082 | yZCL078 | ΔPEP4::LEU2 | This study |
| yZCL083 | yZCL078 | ΔPRB1::LEU2 | This study |
| yZCL084 | yZCL078 | ΔYAP3::LEU2 | This study |
| yZCL089 | yZCL078 | pZCL096 | This study |
| yZCL090 | yZCL081 | pZCL096 | This study |
| yZCL091 | yZCL082 | pZCL096 | This study |
| yZCL092 | yZCL083 | pZCL096 | This study |
| yZCL093 | yZCL084 | pZCL096 | This study |
| yZCL099 | yZCL081 | pZCL106 | This study |
| yZCL100 | yZCL082 | pZCL106 | This study |
| yZCL101 | yZCL083 | pZCL106 | This study |
| yZCL102 | yZCL084 | pZCL106 | This study |
| yZCL107 | yZCL078 | pZCL116 | This study |
| yZCL108 | yZCL081 | pZCL116 | This study |
| yZCL109 | yZCL082 | pZCL116 | This study |
| yZCL110 | yZCL083 | pZCL116 | This study |
| yZCL111 | yZCL084 | pZCL116 | This study |
| yZCL115 | yZCL083 | ΔPRB1::LEU2; G418-100 mg; TY1::PKK1t-GAL1p-2× His-TS-GPM1t-PYC1p-KanMX-TEF1p | This study |
| yZCL117 | yZCL083 | ΔPRB1::LEU2; G418-200 mg; TY1::PKK1t-GAL1p-2× His-TS-GPM1t-PYC1p-KanMX-TEF1p | This study |
| yZCL118 | yZCL083 | ΔPRB1::LEU2; G418-400 mg; TY1::PKK1t-GAL1p-2× His-TS-GPM1t-PYC1p-KanMX-TEF1p | This study |
| yZCL119 | yZCL083 | ΔPRB1::LEU2; G418-400 mg; TY1::PKK1t-GAL1p-2× His-TS-GPM1t-PYC1p-KanMX-TEF1p | This study |
| yZCL124 | yZCL083 | ΔPRB1::LEU2; G418-800 mg; TY1::PKK1t-GAL1p-2× His-TS-GPM1t-PYC1p-KanMX-TEF1p | This study |
| yZCL126 | yZCL083 | ΔPRB1::LEU2; G418-1600 mg; TY1::PKK1t-GAL1p-2× His-TS-GPM1t-PYC1p-KanMX-TEF1p | This study |
| yZCL136 | yZCL124 | ΔPRB1::LEU2; G418-800 mg; Delta22::CYC1t-TDH1p-BST1-ERG20-TEF1t-PYC1p-tHMG1-HXT7t; HO::CYC1t-TDH1p-BST1-ERG20-TEF1t-PYC1p-tHMG1-HXT7t; | This study |
| yZCL137 | yZCL136 | ΔPRB1::LEU2; G418-800 mg; Delta22::CYC1t-TDH1p-BST1-ERG20-TEF1t-PYC1p-tHMG1-HXT7t; HO::CYC1t-TDH1p-BST1-ERG20-TEF1t-PYC1p-tHMG1-HXT7t; TKL2::CYC1t-TDH1p-BST1-ERG20-TEF1t-PYC1p-tHMG1-HXT7t | This study |
The E. coli top 10 strain was cultured in LB medium (0.5% yeast extract, 1% tryptone, and 1% NaCl) supplemented with either 50 μg mL−1 kanamycin or 100 μg mL−1 ampicillin at a temperature of 37 °C for the propagation of recombinant plasmids. S. cerevisiae strains devoid of plasmids were cultured in yeast extract-peptone-dextrose (YPD) medium. The selected plasmids carrying nutritional screening tags were introduced into S. cerevisiae cells and cultured in a synthetic complete (SC) medium lacking the corresponding amino acids. All medium formulations were the same as those used in a previous study.17
2.2 Plasmid construction
All plasmids and primers used in this study are listed in Tables S1 and S2,† respectively. The yeast expression plasmids pRS416 and pRS426, obtained from ADDGENE (USA), were used for the construction of all the cassettes used in this study. The TS gene (Accession Q41594) was codon optimised for expression in S. cerevisiae and synthesised using GenScriptInc (China) (Table S3†). The yeast strains were transformed with plasmids using the LiAc/ssDNA carrier DNA/PEG3350 method.27 The S. cerevisiae strains used in the present study are listed in Table 1.2.3 Knockout of yeast endogenous protease
According to the gene sequences of the proteases PEP4, YAP3, PRB1, and CYM1 in S. cerevisiae, 500 bp were taken from both ends of the genes as homologous arms, a Leu2 nutritional tag was used to replace the related protease genes in the genome, and monoclonal strains were screened in SC-Leu nutritive deficiency medium.2.4 Expression and purification of TS protein in S. cerevisiae
The TS expression plasmid (PRS416-t60TS or PRS426-t60TS) was transformed into the PRB1 single-knock chassis and the control chassis. Colonies were selected on synthetic dextrose minus leucine medium (SD-Ura, 2% dextrose, 0.67% yeast nitrogen base, 1× amino acids minus Ura). Seed activation and culture of the proteome detection strain were consistent with the culture conditions described in section 2.6. Yeast cells were harvested by centrifugation at 5000g for 10 min at 40 h when OD600 reached about 10 (ethanol consumption phase). The centrifuged yeast cells were washed three times with PBS buffer. The harvested cells were suspended in buffer A (50 mM Tris-HCl pH 7.5, 200 mM NaCl, 5% glycerol) and were lysed by sonication on ice. The supernatant was obtained after centrifugation at 12000g for 30 min and was then applied onto a Ni2+-nitrilotriacetate affinity resin (Ni-NTA, GENvien). Impurities were washed away with buffer B (50 mM Tris-HCl pH 7.5, 200 mM NaCl, 5% glycerol, 20 mM imidazole) and buffer C (50 mM Tris-HCl pH 7.5, 200 mM NaCl, 5% glycerol, 40 mM imidazole). The TS protein was eluted using buffer D (20 mM Tris-HCl pH 7.5, 200 mM NaCl, 5% glycerol, 300 mM imidazole). The elution protein was estimated by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE).28
2.5 Western blot
The procedure for protein extraction and western blotting analysis was based on our previous research scheme.29 1 mL of fermentation broth (cultured for 40 hours) was centrifuged to remove the supernatant, and then washed with pre-cooled 0.01 M PBS, centrifuged at 4 °C 3000g for 5 min, and the supernatant was removed. An appropriate amount of lysis buffer was added to the cell precipitation, blown and mixed, and placed on ice for lysis for 30 min. After centrifugation to remove the supernatant, the lysed cells were washed with 1 mL of sterile water. Finally, the collected cells were resuspended in 100 μL of SDS sample buffer, heated at 100 °C for 10 minutes, and after centrifugation, the supernatant was the prepared sample of the target protein. The protein concentration of the prepared sample can be quantified using a Coomassie (Solarbio) protein assay kit compatible with detergents.After diluting the total protein concentrations to the same level across different experimental groups, 40 μL of protein sample was taken and mixed thoroughly with 10 μL of 5× SDS-PAGE protein sample buffer. The protein sample was heated in a boiling water bath for 10 minutes to denature the protein completely, then the sample was placed on ice to cool to room temperature and centrifuged for 5 minutes. Equal volumes of each sample containing the same amount of total protein were loaded into 15% SurePAGE gel. Protein electrophoresis was divided into two stages. In the first step, a constant voltage of 80 V was applied for 20 min and in the second step, a constant voltage of 120 V was applied for 60–70 minutes. For transfer (blotting), the conditions were: a constant voltage of 110 V for approximately 70 minutes. After sealing the PVDF membrane with 5% BSA solution at room temperature for 4 h, a mixture of 20 μL of the primary antibody (anti-His mouse monoclonal antibody for the target protein and anti-GAPDH mouse monoclonal antibody for the reference gene GAPDH) at a ratio of 1:2000 was added into the same 5% BSA solution and incubated overnight at 4 °C. The PVDF membrane was washed three times with 40 mL of pre-cooled TBST buffer. Subsequently, the PVDF membrane was incubated with goat anti-mouse IgG(H + L) and HRP conjugate (diluted at 1:3000) in TBST buffer at room temperature for 3 hours. After incubation with the secondary antibody, the membrane was washed three times with buffer C. The PVDF membranes were washed three times with pre-cooled TBST buffer (Solarbio, China) and incubated for 50 s using the EasySee western-blotting kit (TransGen, China). Then protein expression was detected using an Azure Biosystems C280 chemiluminescent blot imaging system (USA). The gray values of protein bands were quantitatively analyzed using ImageJ.
2.6 Preparation of samples for protein group analysis
The seed culture of the proteome detection strain was consistent with the culture conditions described in section 2.8. The seed culture solution was transferred to a shaker and cultured for 16 hours at 220 rpm until the logarithmic growth phase was reached. The temperature was then reduced to 20 °C while maintaining 220 rpm agitation speed. Taxadiene production was induced by the addition of dodecane 20% (v/v) and galactose (20 g L−1). The samples were collected at time points when taxadiene production differed substantially between the control and experimental groups, as determined by the product accumulation curves. The cells were harvested by centrifugation (with consistent cell pellet volumes between the control and experimental groups), washed rapidly three times with pre-chilled phosphate-buffered saline (PBS), and then centrifuged at 4 °C and 5000 rpm for 5 min after each wash. The supernatant was discarded completely, and the cell pellets were collected in 2 mL cryovials, snap-frozen in liquid nitrogen for 30 min, and stored at −80 °C until transport on dry ice.All samples were sent to Novozymes (Tianjin, China) for data-independent acquisition (DIA) quantitative proteomics analysis. Significantly differentially expressed proteins were screened according to the criteria of an expression fold change of more than 1.2 times (up-regulated more than 1.2 times or down-regulated less than 0.83 times) and P-value <0.05. Proteomic DIA sequencing data were obtained from three biological replicates. Raw data files were searched against the Baker's yeast (S. cerevisiae ATCC 204508/S288c) reference proteome database downloaded from UniProt (UP000002311, 6060 sequences, 20211018) and supplemented with QJX58297.1(NCBI), which is a frequently observed contaminant. At least two peptides were required for protein identification, of which at least one peptide was required to be unique in the database. Identified proteins were quantified with MaxQuant's LFQ algorithm. Proteomic samples were tested using Novo, and the experimental results were processed and analysed using Novogene (Novogene, Tianjin, China).
2.7 Multiple-copy site integration
The TY1 multiple-copy sites in the S. cerevisiae genome were selected and the TS gene expression module was coupled with the G418 (Geneticin) expression module induced by the weak promoter pyc1. The high-copy strains of the TS gene were screened on the plate with different concentrations of G418.30 The concentration of taxadiene was determined.2.8 Fermentation conditions
For shake-flask fermentation, a single colony of the engineered strain was selected from YPD or SC agar plates and inoculated into tubes containing 4 mL of YPD or SC medium and cultured for 24 hours at 30 °C and 250 rpm. Subsequently, the overnight cultures from 0.05 mL of YPD medium or 0.1 mL of SC medium were inoculated into tubes containing 4 mL of fresh YPD or SC medium and cultured again for 14–16 hours at 30 °C and 250 rpm. Then, the initial YPD or SC seed suspension (OD600 = 0.2) was transferred to 250 mL flasks containing 25 mL of fresh YPD or SC medium for growth and fermentation. These flasks were cultured at 30 °C and 220 rpm. After 10 hours of fermentation, a two-phase culture strategy was implemented by adding 20% (v/v, 10 mL) sterile n-dodecane to the fermentation broth to promote the extraction of products into the organic phase and reduce the toxic effects on cells.25The feeding batch fermentation conditions were the same as those used for shaking bed fermentation, with a seed inoculation amount of 10% (v/v) in the fermentation tank. Subsequently, 200 mL of the seed culture was transferred to 1.8 L of YPD medium containing 2% (w/v) glucose, 1% (w/v) yeast extract, and 2% (w/v) peptone and inoculated at an OD600 of 1. The initial culture conditions were set at 300 rpm with a stirring-dissolved oxygen linkage mode to maintain the dissolved oxygen at 30%. The temperature was maintained at 30 °C. Oxygen was supplied to the fermentation tank by supplying 2 vvm of gas, and the pH was adjusted to 6.0 using 5 M NaOH. In addition, adding nitrogen sources to batches could increase the fermentation biomass and late accumulation of taxadiene. Glucose was used as the carbon source, and forty-four hours before fermentation, 500 g L−1 glucose was added to the fermentation tank to maintain its concentration below 5.0 g L−1. A nitrogen source containing 1% (w/v) yeast extract was added every 8 h. After 44 hours of fermentation, the temperature was lowered to 20 °C and 20% (v/v) n-dodecane was added for two-phase extraction fermentation, switching the carbon source from glucose to ethanol. Cell growth and glucose and ethanol concentrations were continuously monitored throughout fermentation. The organic layer was selected for taxadiene analysis. After centrifugation at 12000 rpm for 10 min, an appropriate amount of anhydrous sodium sulphate was added to remove the water. After filtering through an organic 0.22 μm filter to remove impurities, the sample could be directly used for GC-MS analysis.
2.9 Analytical and preparation methods
Taxadiene was extracted as previously described. Before gas chromatography-mass spectrometry (GC-MS) analysis, the processed organic layer (40 μL) was diluted with n-hexane (360 μL). Subsequently, the sample was subjected to analysis using a GC/MS system comprising of a SHIMADZU GC-2010 PLUS gas chromatography instrument equipped with a fused-silica capillary column (30 m × 0.25 mm, 0.25 mm DB-5MS, J&W Scientific, Folsom) and a SHIMADZU GCMS-QP2020 mass spectrometer. The relevant parameters for the sample detection method were designed and set. The injector temperature was set to 260 °C, the ion source temperature was set to 250 °C, and the mass scanning range was set between 50 and 800 m/z.The split ratio for the GC-TOF/MS analysis of taxadiene and GGOH was set to 50:1. The column temperature was initially held constant at 70 °C for 1 min, followed by a ramping rate of 30 °C min−1 to reach 180 °C, which was maintained for an additional minute. Subsequently, the temperature was increased to 265 °C at a rate of 10 °C min−1 and held steady for a duration of 4 minutes. The total run time encompassed a period of precisely 19.95 minutes. Taxadiene identification relied on mass fragments with m/z values of 109 and 122, with peak elution occurring at approximately 12.24 min. In contrast, GGOH detection involved mass fragments with m/z values of 69, 93, and 119 (Fig. S1†). The peak retention time corresponded to approximately 13.46 min.25 The standard curve of GGOH (Solarbio) was constructed as shown in Fig. S2.†
After fermentation, the organic phase containing taxadiene products was collected by centrifugation. The taxadiene sample in the organic phase was purified using modified silica gel flash chromatography, followed by concentration under reduced pressure at room temperature to obtain taxadiene. After 1H NMR and 13C NMR testing, comparison with literature carbon spectra confirmed the isolated compound as taxadiene (Fig. S3, S4 and Table S4†). And the purity of the prepared taxadiene was up to 98.45%, which was confirmed by gas chromatography according to the peak area normalization (Fig. S5†). Finally, the purified taxadiene was regarded as the standard to construct a standard curve to determine the taxadiene yield (Fig. S6†).
3. Results and discussion
3.1 Abnormal TS expression in S. cerevisiae
TS gene fragments from sequence-optimised Taxus brevifolia (t60TS) were inserted into the expression vector PRS416 to construct the PRS416-t60TS plasmid. The plasmid was introduced into the chassis of the high-yielding GGPP (yZCL078), resulting in the construction of the primary taxadiene-producing strain yZCL080 (Fig. 2A). Strain yZCL080 was then cultured in SC-Ura medium for 5 days. And taxadiene and geranylgeraniol (GGOH) were detected in the production strain yZCL080 by GC-MS. The GC-MS results confirmed that taxadiene was successfully obtained in the strain yZCL080 with a distinct peak at 12.24 min, the same retention time as the taxadiene standard (Fig. 2B). The MS results for the taxadiene standard and the production of the strain yZCL080 were also shown (Fig. 2C). | ||
| Fig. 2 Abnormal TS expression in taxadiene biosynthesis pathway. (A) Schematic of the expression plasmids for taxadiene production. (B) Gas chromatogram results for the taxadiene standard, GGOH standard, and starting strain yZCL080. (C) The MS results for the production of the strain yZCL080 and taxadiene standard are also shown. (D) TS protein expression in Saccharomyces cerevisiae (S. C) and Escherichia coli (E. coli) chassis. The black and red arrows represent intact and cleaved TS protein bands, respectively. (E) Western blotting analysis of TS protein expression in strain yZCL080. GAPDH represents the internal reference protein band. The black and red arrows represent intact and cleaved TS protein bands, respectively. | ||
Meanwhile, the collected yeast cells of strain yZCL080 were washed, cleaved, purified by nickel affinity, and identified by SDS-PAGE. The control group included the expression and purification of the TS gene in E. coli BL21(DE3), and the SDS-PAGE results showed an obvious intact band of TS, which was consistent with the theoretical molecular weight (MW) of TS (MW: 93KD) (Fig. 2D). While in S. cerevisiae, SDS-PAGE results for TS protein showed three distinct cleaved bands (MW: 53KD, 35KD and 15KD) besides the intact bands of TS (Fig. 2D). Therefore, it is speculated that the TS protein expressed in S. cerevisiae (TS-S.C) may be degraded. The four protein bands of the TS-S.C sample were analyzed by protein mass spectrometry, and the residue sequence of each protein band sample matched the continuous specific sequence of the target protein TS within a certain range, which suggested that these protein samples should contain fragments of TS sequences. However, the low coverage scores of the residue sequence of the four protein band samples of TS-S.C were obtained by matching the full-length sequence of the TS protein, and the coverage scores were only 39.4%, 15.5%, 7.5% and 8.7% respectively (Fig. S7†). This result may be due to the unsatisfactory purity of SDS-PAGE protein bands for the TS-S.C sample. Therefore, further validation by western blotting (WB) is required, and the results of WB confirmed the phenomenon of TS protein cleavage in the expression system of S. cerevisiae. In Fig. 2E, four distinct protein bands are observed, including an intact TS band and three cleaved bands, and the proportion of the intact TS band is only 13.7% (intact TS/TS-S.C). In our opinion, the abnormal expression of TS will limit the synthesis of taxadiene in S. cerevisiae which may be the key to the low production of taxadiene in S. cerevisiae.
Compared to prokaryotic expression systems (e.g. E. coli), S. cerevisiae has more abundant post-protein modification and degradation systems.31 Moreover, TS is a heterologous complex protein with fused αβγ multi-domain structures and a class I terpene cyclase catalytic motif (DXDD), which is even more unfavourable for its correct folding and expression in S. cerevisiae.32,33 For the above reasons, the heterozyme TS introduced in S. cerevisiae may be easily degraded by proteases.
3.2 Effects of knockout of proteases on TS expression and taxadiene synthesis in S. cerevisiae
In this study, we successfully obtained a strain capable of taxadiene synthesis. However, the TS protein was disrupted and abnormally expressed, resulting in limited production of taxadiene. We speculated that the proteases in S. cerevisiae may have degraded the heterologous TS protein (Fig. 3A). Studies using proteases to improve heterologous protein expression have yielded promising results. Guan et al. showed a 2.58-fold increase in bFGF levels after knocking out four yeast proteases: PEP4, CYM1, YAP3, and PRB1. Knocking out endogenous proteases in yeast helps reduce cytokine degradation in S. cerevisiae cells and promotes cytokine expression and accumulation.24 Moreover, taxadiene synthase is a plant-sourced protein and its intrinsically disordered sequence at the N-terminus may trigger degradation by yeast proteases when expressed in S. cerevisiae.34 | ||
| Fig. 3 Knock-out of endogenous proteases to enhance TS expression and taxadiene synthesis in S. cerevisiae. (A) Schematic of the TS protein degradation in S. cerevisiae. (B) Diagram of endogenous protease knockout fragments. The protease gene fragment was replaced with the LEU2 tag constructed via homologous recombination. (C) Effect of endogenous protease knockout on taxadiene production. In the control strains and single knockout strains of endogenous proteases CYM1, PEP4, PRB1 and YAP3, by introducing the single-copy pZCL106 plasmid related to taxadiene synthesis, protease single knockout strains yZCL080, yZCL099, yZCL100, yZCL101, and yZCL102 for taxadiene synthesis were successfully constructed. **P < 0.01, *P < 0.05, Student's t-test. (D) Screening for an endogenous protease knockout chassis to enhance taxadiene synthesis using a TS multi-copy expression plasmid. In the control strains and single knockout strains of endogenous proteases CYM1, PEP4, PRB1 and YAP3, by introducing the multi-copy pZCL116 plasmid related to taxadiene synthesis, protease single knockout strains yZCL107, yZCL108, yZCL109, yZCL110, and yZCL111 for taxadiene synthesis were successfully constructed. **P < 0.01, *P < 0.05, Student's t-test. (E) Western blotting analysis of the TS protein expression difference between control strains yZCL107 and PRB1 protease single knockout strains yZCL110. GAPDH represents the internal reference protein band. The black and red arrows represent intact and cleaved TS protein bands, respectively. (F) The gray values of the intact TS protein in control strains yZCL107 (intact TS/action, 10.3%) and PRB1 protease single knockout strains yZCL110 (intact TS/action, 48.6%). | ||
To investigate the impact of the four proteases (CYM1, PEP4, PRB1, and YAP3) on the expression of taxadiene synthase and taxadiene synthesis, single-deletion strains were generated by disrupting the endogenous proteases CYM1, PEP4, PRB1, and YAP3 individually using homologous fragments containing a LEU2 nutritional marker (Fig. 3B). As shown in Fig. 3C, the taxadiene yield was increased to 39.2 mg L−1 in the PRB1 protease knockout (ΔPRB1) strain yZCL101, with approximately 97% higher than that of the control strain (yZCL080). Subsequently, we also inserted the t60TS gene into the multi-copy plasmid PRS426, then introduced it into the PRB1 protease knockout chassis and constructed the ΔPRB1 strain yZCL110. As shown in Fig. 3D, the taxadiene yield was further increased to 78.4 mg L−1 in the ΔPRB1 strain yZCL110, which was approximately 74% higher than that of the control strain (yZCL107) with the multi-copy plasmid of the t60TS gene. In addition, we also evaluated the expression levels of the TS protein in the ΔPRB1 strain yZCL110 and the control strain yZCL107 by western blotting assay (Fig. 3E). The expression level of the intact TS protein was significantly improved by knocking out the endogenous protease PRB1 of S. cerevisiae, even though the cleavage of TS was not completely eliminated (Fig. 3E). And the relative intensity analysis showed that the expression of the intact TS protein increased by 371% in PRB1 protease single knockout chassis, compared with that in the control strains (Fig. 3F), which resulted in a significant increase in the production of taxadiene. Moreover, the quantification of the expression of TS by RFP is consistent with the above results (Fig. S8†). Therefore, PRB1 protease knockout will promote the stable expression of taxadiene synthase and increase the yield of taxadiene in S. cerevisiae.
3.3 Proteomic analysis of the effect of protease PRB1 knockout on the taxadiene synthesis pathway
Recent studies have shown that knocking out relevant proteases can effectively improve the stable and efficient expression of target proteins. Deletion of the vacuolar protease PEP4p has been demonstrated to significantly increase the production of several proteins, whereas similar deletions of other proteases have also led to improved accumulation of the desired proteins.35 Yoshinori Ohsumi et al. found that vacuolar hydrolases PEP4 and PRB1 are involved in the activation of other hydrolytic enzymes and are essential for the degradation of the autophagic material.36 Proteomic analysis is of great importance to elucidate the mechanisms underlying protein synthesis and stabilised expression after PRB1 knockout.To further characterise the effects of PRB1 protease knockout on taxadiene biosynthesis, we performed proteomic analysis of the expression of key enzymes in the MVA pathway and taxadiene synthase in the ΔPRB1 strain yZCL110 and the control strain yZCL107. We first conducted differential analysis of proteins closely related to product synthesis pathways in S. cerevisiae (Fig. S9†). The introduction of a heterologous terpenoid synthesis pathway may lead to acetyl-CoA deficiency. Moreover, acetyl-CoA may be involved in cofactor balance and redox balance in S. cerevisiae.37 GO enrichment analysis (Fig. 4A) revealed significant changes in important biological processes. Enhanced carbohydrate metabolism generates energy and provides intermediates for biosynthesis. Additionally, enhanced redox processes are likely related to an increased acetyl-CoA supply, achieving a balance between anabolism and catabolism (Fig. S10†). The heatmap (Fig. 4B) shows that enzymes related to glycolysis were upregulated in the PRB1 protease-knockout strain, consistent with acetyl-CoA being mainly derived from glycolysis and taxadiene biosynthesis, potentially draining metabolic flux from glycolysis. Compared to the control, tHMG1, ERG20 and BST1 were endogenous proteins of S. cerevisiae, and their expression was relatively stable without significant improvement. The abnormal expression of the TS protein may be more closely related to the PRB1 protease. TS expression was enhanced in the PRB1 knockout strain, alleviating the key problem of low TS expression in S. cerevisiae (Fig. 4C).
 | ||
| Fig. 4 Comparative proteomic analysis of taxadiene-producing strains. (A) The GO enrichment analysis results of the upregulated proteins in the control strain yZCL107 and the PRB1 knockout multi-copy strain yZCL110 are presented using a histogram. (B) Heatmap of differential proteome analysis related to the central metabolic and MVA pathways in the engineered strains yZCL107 and yZCL110. (C) Absolute quantification of key metabolites, including N-terminally truncated HMG-CoA reductase 1 (tHMG1), isopentenyl diphosphate delta-isomerase 1 (IDI1), bifunctional (2E,6E)-farnesyl diphosphate (ERG20), farnesyl transferase (BST1), and taxadiene synthase (TS). The identified proteins were quantified using the MaxQuant LQF algorithm. **P < 0.01, *P < 0.05, Student's t-test. | ||
Although PRB1 protease knockout can enhance the expression of genes related to the taxadiene synthesis pathway and taxadiene synthase, the excess taxadiene precursor GGPP still accumulates, producing large amounts of the by-product GGOH. Therefore, increasing the TS protein expression in the PRB1 knockout strain to reduce the accumulation of GGOH may have a positive effect on the taxadiene yield. Endogenous proteases are essential for the maintenance of normal cell growth and viability. Environmental and intracellular factors can cause DNA damage. Proteases play a crucial role in DNA repair.38 The ubiquitin–proteasome system plays an important role in the DNA repair pathway in S. cerevisiae.39 Abnormal folding of proteins occurs because of folding failure and mutations in coding genes during protein translation. These abnormally folded proteins are ubiquitinated and eventually degraded by the proteasome, thereby ensuring the stability of S. cerevisiae.40 Therefore, several protease-knockout modifications could not be performed. Subsequently, the expression of the TS protein should be further improved.
3.4 Multi-copy integration enhanced TS expression and the supply of GGPP precursors, improving the yield of taxadiene
According to previous studies, the expression level of TS protein in S. cerevisiae is low and increasing the copy number of TS can significantly improve the taxadiene yield. In our previous study, by enhancing the expression of the GGPPS-t60TS fusion protein module with a multi-copy plasmid, the taxadiene yield increased by 38.9%.17 Xu et al. increased the taxadiene yield by 173% after three rounds of iterative integration to increase the copy number of taxadiene synthase.41 Although the multi-copy plasmid improved the taxadiene yield, the GGPP precursor in the chassis was not fully converted, resulting in the accumulation of GGOH. To increase the copy number of taxadiene more effectively, we designed a high-copy screening system to integrate the expression module of taxadiene synthase TS into the yeast genome for stable expression.42 The G418 resistance gene screening module driven by the weak promoter pyc1 was combined with the TS expression module and integrated at the multi-copy site Ty1 in the yeast genome (Fig. 5A). The high-copy TS integrated strains were screened for resistance to different concentrations of G418. Strains with high copy numbers were screened using the weak promoter Pyc1 carrying a G418 resistance tag and selected from plates containing different concentrations of G418. The taxadiene yield of each strain was determined using a fermentation culture. The results showed the screening concentration of G418 to be 800 mg L−1, and the taxadiene yield reached the highest value of 131.97 mg L−1 (Fig. 5B). Compared to the multi-copy PRB1 strain (yZCL110), the yield increased by 93%. Moreover, the yield of taxadiene in strain yZCL124 exceeded that of the precursor by-product GGOH. A decrease in GGPP, the precursor of taxadiene synthesis in S. cerevisiae, may limit taxadiene synthesis in the high-copy TS strain.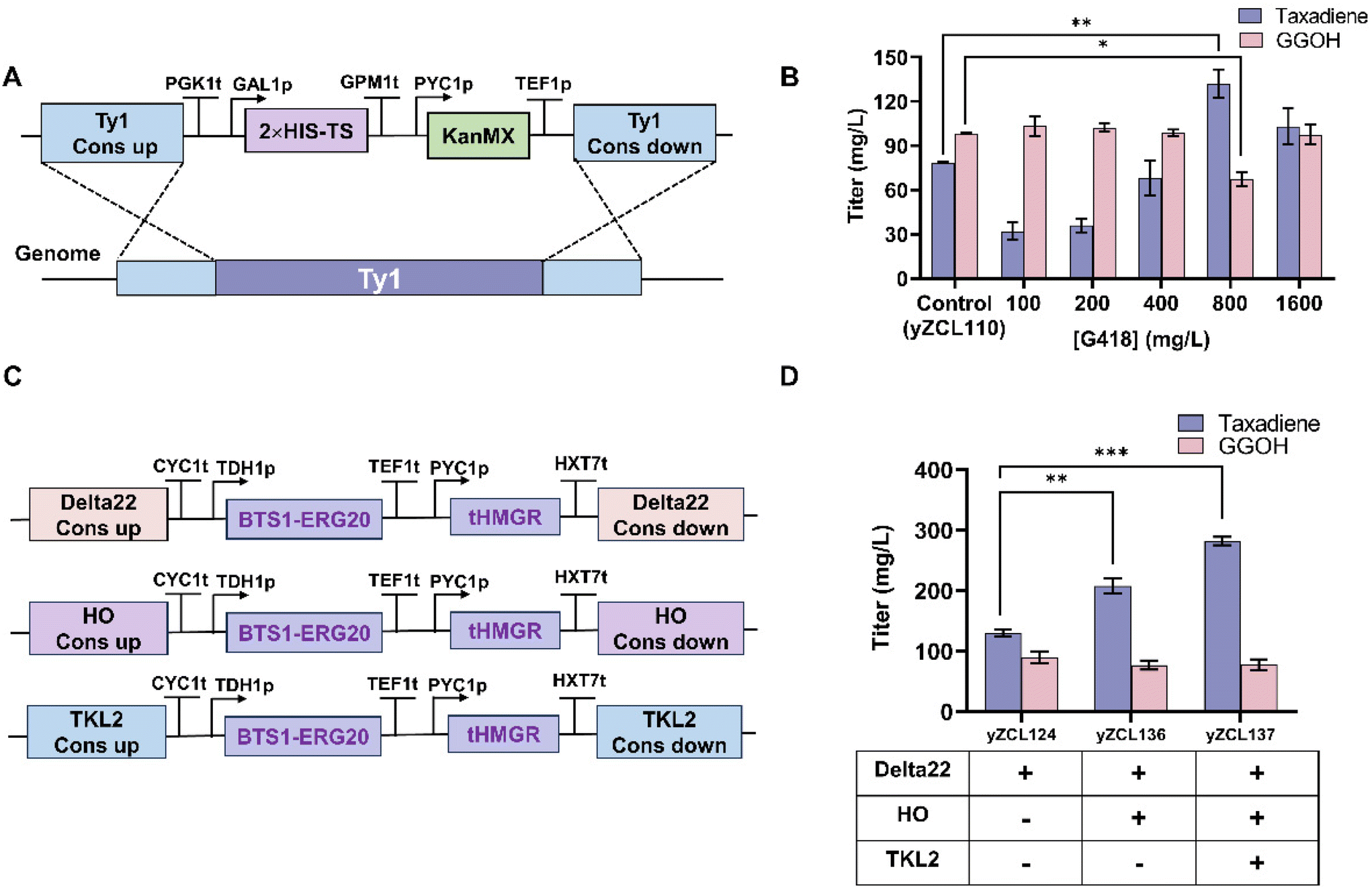 | ||
| Fig. 5 Multi-copy integration enhanced TS expression and GGPP synthesis precursor supply to improve the yield of taxadiene. (A) Schematic of the TS and G418 co-expression cassette at the Ty1 multi-copy site of the yeast genome. (B) Screening of high-yielding taxadiene using the G418 concentration gradient. (C) Schematic of the yeast genome Delta22, HO and TKL2 site integrating the GGPP precursor pathway. (D) Enhancements of GGPP synthesis-critical pathway genes and increments of taxadiene production. **P < 0.01, *P < 0.05, Student's t-test. | ||
N-terminally truncated HMG1 (tHMG1) effectively relieves feedback inhibition and increases the production of monoterpenes, sesquiterpenes, and triterpenes in yeast.43 Ohto et al. promoted GGPP synthesis by fusing the FPP synthase ERG20 with BTS1 while reducing the loss of the ERG20 catalytic product FPP to the squalene synthesis pathway.44 After TS multi-copy integration, we constructed a precursor enhancement expression module, GGPP (Fig. 5C), and gradually integrated the GGPP precursor synthesis module into the HO and TKL2 sites of the high-producing PRB1 strain yZCL124, successfully constructing the GGPP module-enhanced strains yZCL136 and yZCL137. The results showed that with an increase in the copy number of the GGPP precursor module, the taxadiene yield increased significantly. The taxadiene yield was increased by 113.9% from 131.97 mg L−1 to 282.4 mg L−1 gradually (Fig. 5D). The highest taxadiene titer in S. cerevisiae was obtained. Simultaneously, with the enhanced GGPP synthesis pathway, the yield of GGOH increased from 76.9 mg L−1 to 110.4 mg L−1.
3.5 Highest reported titer of taxadiene in yeast by fed-batch fermentation
To evaluate the production performance of the optimal taxadiene-producing strain (yZCL137) in fed-batch fermentation, a 5 L bioreactor was used with YPD as the batch medium. As shown in Fig. 6, the total titer reached 878.5 mg L−1. The maximum taxadiene titer was obtained at an OD600 of 133, which is the highest titer reported in eukaryotic cells. The temperature was controlled at 30 °C with an initial glucose concentration of 20 g L−1. Glucose feeding was started after 8 h of cultivation. The taxadiene strain entered the rapid growth phase and reached the stationary phase after 44 h. Since glucose represses the GAL promoter expression of the TS protein, the carbon source was switched from glucose to ethanol to favour taxadiene synthesis and accumulation.45 Additionally, 20% dodecane extractant was added to the fermenter, and the temperature was reduced to 20 °C to induce TS protein expression at low temperatures and increase TS stability. After 180 hours of cultivation, the taxadiene titer reached 878.5 mg L−1 (Fig. 6), representing a 5.7-fold increase over the initial shake-flask titer. In the product preparation system, the recovery rate of taxadiene was 57.3%. However, under nutrient-rich fermentation conditions, the taxadiene strain biomass was slightly lower than expected, potentially due to the growth effects of the PRB1 protease knockout on the long-pathway product-synthesis strain. This study successfully achieved the highest taxadiene titer reported for S. cerevisiae. | ||
| Fig. 6 Fed-batch fermentation of the strain yZCL137 in a 5 L bioreactor. Fed-batch fermentations were performed at a 2.0 L scale using YPD medium and the engineered S. cerevisiae strain yZCL137. OD600 and glucose, ethanol, and taxadiene curves during fed-batch fermentation. Black arrows indicate the addition of n-dodecane and galactose to the medium. Error bars represent the means ± SD from three biological replicates. | ||
4. Conclusion
In this study, we improved the chassis environment for TS expression in the taxadiene biosynthesis pathway by knocking out the endogenous protease PRB1 of S. cerevisiae. Sequentially, by multi-copy integration of the TS gene and enhancement of the precursor supply pathway, a high-yield taxadiene strain, yZCL137, was obtained with a titer of 282.4 mg L−1. Interestingly, enhancing the expression of TS not only improved the target product yield of taxadiene but also decreased the competitive synthesis of the by-product GGOH. Ultimately, in 5 L fed-batch fermentation, the titer of taxadiene reached 878.5 mg L−1.Author contributions
Chenglong Zhang: conceptualization, investigation, methodology, formal analysis, visualization, and writing – original draft; Jia Wang: investigation and validation; Yi Shi: methodology and validation; Nan Wu: methodology; Xia Li: funding acquisition; Ying Wang: supervision; Bingzhi Li: supervision; Wenhai Xiao: conceptualization, funding acquisition, project administration, and supervision. Mingdong Yao: conceptualization, funding acquisition, writing – review & editing, and supervision. Yingjin Yuan: supervision.Data availability
The data are available from the corresponding author on reasonable request.Conflicts of interest
The authors declare no competing interests.Acknowledgements
We are grateful for the financial support from the National Key Research and Development Program of China (2021YFC2101500). The authors would like to thank Nan Wu for the original chassis sharing.References
- E. L. van Rozendaal, G. P. Lelyveld and T. A. van Beek, Phytochemistry, 2000, 53, 383–389 CrossRef CAS.
- M. Nadeem, H. C. Rikhari, A. Kumar, L. M. S. Palni and S. K. Nandi, Phytochemistry, 2002, 60, 627–631 Search PubMed.
- D. J. Pollard and J. M. Woodley, Trends Biotechnol., 2007, 25, 66–73 CrossRef CAS PubMed.
- Y.-J. Hu, C.-C. Gu, X.-F. Wang, L. Min and C.-C. Li, J. Am. Chem. Soc., 2021, 143, 17862–17870 CrossRef CAS PubMed.
- S. Zhang, T. Ye, Y. Liu, G. Hou, Q. Wang, F. Zhao, F. Li and Q. Meng, Molecules, 2023, 28, 7517 CrossRef CAS.
- J. Liu, X. Liu, J. Wu and C.-C. Li, Chem, 2020, 6, 579–615 CAS.
- A. Madhavan, K. B. Arun, R. Sindhu, P. Binod, S. H. Kim and A. Pandey, Biochim. Biophys. Acta, Proteins Proteomics, 2019, 1867, 140262 CrossRef CAS.
- R. N. Patel, Curr. Opin. Biotechnol, 2001, 12, 587–604 CrossRef CAS PubMed.
- J. S. Cho, G. B. Kim, H. Eun, C. W. Moon and S. Y. Lee, JACS Au, 2022, 2, 1781–1799 CrossRef CAS PubMed.
- M. Sabzehzari, M. Zeinali and M. R. Naghavi, Biotechnol. Adv., 2020, 43, 107569 CrossRef CAS PubMed.
- P. K. Ajikumar, W.-H. Xiao, K. E. J. Tyo, Y. Wang, F. Simeon, E. Leonard, O. Mucha, T. H. Phon, B. Pfeifer and G. Stephanopoulos, Science, 2010, 330, 70–74 CrossRef CAS.
- M. E. Ondari and K. D. Walker, J. Am. Chem. Soc., 2008, 130, 17187–17194 CrossRef CAS PubMed.
- X. Zhang, J. Guo, F. Cheng and S. Li, Nat. Prod. Rep., 2021, 38, 1072–1099 RSC.
- Y. Jin, D. C. Williams, R. Croteau and R. M. Coates, J. Am. Chem. Soc., 2005, 127, 7834–7842 CrossRef CAS.
- B. Engels, P. Dahm and S. Jennewein, Metab. Eng., 2008, 10, 201–206 CrossRef CAS.
- B. Nowrouzi, R. A. Li, L. E. Walls, L. d'Espaux, K. Malcı, L. Liang, N. Jonguitud-Borrego, A. I. Lerma-Escalera, J. R. Morones-Ramirez, J. D. Keasling and L. Rios-Solis, Microb. Cell Fact., 2020, 19, 200 CrossRef CAS PubMed.
- C. Zhang, W. Chen, T. Dong, Y. Wang, M. Yao, W. Xiao and B. Li, Front. Bioeng. Biotechnol., 2023, 11, 1141272 CrossRef.
- Y. Zhao, F. Liang, Y. Xie, Y.-T. Duan, A. Andeadelli, I. Pateraki, A. M. Makris, T. G. Pomorski, D. Staerk and S. C. Kampranis, J. Am. Chem. Soc., 2024, 146, 801–810 CrossRef CAS PubMed.
- M. C. Y. Chang and J. D. Keasling, Nat. Chem. Biol., 2006, 2, 674–681 CrossRef CAS.
- O. P. Ishchuk, A. T. Frost, F. Muñiz-Paredes, S. Matsumoto, N. Laforge, N. L. Eriksson, J. L. Martínez and D. Petranovic, Metab. Eng., 2021, 66, 259–267 CrossRef CAS PubMed.
- P. Arendt, K. Miettinen, J. Pollier, R. De Rycke, N. Callewaert and A. Goossens, Metab. Eng., 2017, 40, 165–175 CrossRef CAS PubMed.
- H.-Y. Y. Yap, M. J. Muria-Gonzalez, B.-H. Kong, K. A. Stubbs, C.-S. Tan, S.-T. Ng, N.-H. Tan, P. S. Solomon, S.-Y. Fung and Y.-H. Chooi, Microb. Cell Fact., 2017, 16, 103 CrossRef PubMed.
- C. M. Bond and Y. Tang, Metab. Eng., 2019, 51, 1–8 CrossRef CAS.
- Q. Lei, J. Ma, G. Du, J. Zhou and X. Guan, Food Res. Int., 2023, 170, 113017 CrossRef CAS.
- T.-Q. Song, M.-Z. Ding, F. Zhai, D. Liu, H. Liu, W.-H. Xiao and Y.-J. Yuan, Sci. Rep., 2017, 7, 14991 CrossRef PubMed.
- K.-D. Entian and P. Kötter, in Methods in Microbiology, ed. I. Stansfield and M. J. Stark, Academic Press, 2007, vol. 36, pp. 629–666 Search PubMed.
- R. D. Gietz, Yeast Protocols, 2014, pp. 33–44 Search PubMed.
- S. V. Chittur, Y. Chen and V. J. Davisson, Protein Expression Purif., 2000, 18, 366–377 CrossRef CAS PubMed.
- G. Jiang, M. Yao, Y. Wang, W. Xiao and Y. Yuan, Metab. Eng., 2021, 66, 51–59 CrossRef CAS PubMed.
- W. Czaja, D. Bensasson, H. W. Ahn, D. J. Garfinkel and C. M. Bergman, PLoS Genet., 2020, 16, e1008632 CrossRef CAS PubMed.
- J. K. K. Low and M. R. Wilkins, FEBS J., 2012, 279, 4423–4443 CrossRef CAS.
- M. Chen, G. G. Harris, T. A. Pemberton and D. W. Christianson, Curr. Opin. Struct. Biol., 2016, 41, 27–37 CrossRef CAS PubMed.
- R. L. Wiseman, J. S. Mesgarzadeh and L. M. Hendershot, Mol. Cell, 2022, 82, 1477–1491 CrossRef CAS.
- I. Kats, A. Khmelinskii, M. Kschonsak, F. Huber, R. A. Knieß, A. Bartosik and M. Knop, Mol. Cell, 2018, 70, 488–501 CrossRef CAS.
- V. Gast, A. Sandegren, F. Dunås, S. Ekblad, R. Güler, S. Thorén, M. T. Mohedano, M. Molin, M. K. M. Engqvist and V. Siewers, Microb. Cell Fact., 2022, 21, 36 CrossRef CAS.
- Y. Kagohashi, M. Sasaki, A. I. May, T. Kawamata and Y. Ohsumi, J. Cell Biol., 2023, 222, e202306120 CrossRef CAS.
- Q. Zhang, W. Zeng, S. Xu and J. Zhou, Bioresour. Technol., 2021, 342, 125978 CrossRef CAS.
- R. J. Dohmen, I. Willers and A. J. Marques, Biochim. Biophys. Acta, 2007, 1773, 1599–1604 CrossRef CAS.
- W. Liu, X. Tang, X. Qi, X. Fu, S. Ghimire, R. Ma, S. Li, N. Zhang and H. Si, Int. J. Mol. Sci., 2020, 21, 2894 CrossRef CAS.
- A. Folger and Y. Wang, Cells, 2021, 10, 2835 CrossRef CAS PubMed.
- M. Xu, W. Xie, Z. Luo, C.-X. Li, Q. Hua and J. Xu, Synth. Syst. Biotechnol., 2023, 8, 331–338 CrossRef CAS PubMed.
- J. Lian, R. Jin and H. Zhao, Biotechnol. Bioeng., 2016, 113, 2462–2473 CrossRef CAS PubMed.
- N. Gohil, G. Bhattacharjee, K. Khambhati, D. Braddick and V. Singh, Front. Bioeng. Biotechnol., 2019, 7, 50 CrossRef.
- C. Ohto, M. Muramatsu, S. Obata, E. Sakuradani and S. Shimizu, Appl. Microbiol. Biotechnol., 2010, 87, 1327–1334 CrossRef CAS.
- G.-Z. Jiang, M.-D. Yao, Y. Wang, L. Zhou, T.-Q. Song, H. Liu, W.-H. Xiao and Y.-J. Yuan, Metab. Eng., 2017, 41, 57–66 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc03079f |
| This journal is © The Royal Society of Chemistry 2024 |