
Donor–acceptor organogels and xerogels from C3-symmetric pyrene and naphthalene-diimide components†
Fanny
Peigneguy‡
,
Cristina
Oliveras-González‡
,
Marie
Voltz
,
Nagham
Ibrahim
 ,
Marc
Sallé
*,
Narcis
Avarvari
* and
David
Canevet
*
,
Marc
Sallé
*,
Narcis
Avarvari
* and
David
Canevet
*
Univ Angers, CNRS, MOLTECH-Anjou, SFR MATRIX, F-49000, Angers, France. E-mail: marc.salle@univ-angers.fr; narcis.avarvari@univ-angers.fr; david.canevet@univ-angers.fr
First published on 22nd July 2022
Abstract
Construction of gel- or xerogel-based materials by associating several components has increasingly attracted the attention of chemists over the last decade. It constitutes a valuable strategy to access gels with tailored properties, notably in the field of photo- and/or electroactive materials. In this context, we recently demonstrated that a C3-symmetric gelator (C3Pyr) endowed with three electron-rich pyrene units forms gels in the presence of electron poor additives. More than affecting the gelling behaviour, these derivatives strongly influenced the spectroscopic properties (absorption and emission) of the corresponding materials. This assessment prompted us to design a new C3-symmetric derivative (C3NDI) grafted with electron poor naphthalene diimide (NDI) units, to explore the well-established electronic complementarity with pyrene functional groups and promote the supramolecular co-polymerisation of C3Pyr and C3NDI into alternate stacks. Thereby, the corresponding bi-component organogels were characterized through a wide range of techniques, including NMR, UV-visible absorption and fluorescence spectroscopies, variable-concentration and variable-temperature measurements, and scanning electron and confocal microscopies. According to the solvent of preparation, remarkably different behaviours were observed, both gelators acting in either an antagonist or a synergetic manner. Since charge transfer interactions between pyrene and NDI units were set up along the gel formation, the formation and the dissociation of the aggregates could be probed through variable temperature measurements. The latter notably highlighted the extent to which the presence of a C3-symmetric platform organising the NDI units contributes to the enhancement of the melting temperature. Finally, this study highlights how relevant modern confocal microscopy can be used to study such co-assembly.
Introduction
Low molecular weight gelators (LMWGs)1–7 constitute a class of precursors for self-assembled nanomaterials, which have attracted tremendous current interest.8–13 These derivatives are able to generate a three-dimensional network of entangled fibres, trapping the solvent and thus, ensuring gel phase stability.This particular behaviour results from the creation of non-covalent bonds between gelling molecules and leads to well-organized and defined materials at the mesoscopic scale. This proves to be true for gels and their corresponding xerogels, once the gelled solvent is evaporated.
Over the last few decades, much effort has been dedicated to the rationalisation of gelling properties.5,14–16 Though scientists can still discover exotic supramolecular behaviours, non-covalent design principles have been proposed and accepted to build supramolecular polymers17,18 eventually behaving as gelators.19,20 The role of the solvent also has to be emphasized, since this component appears critical with regard to the gelling ability, the structure of the corresponding xerogel materials and their physicochemical properties.7,21–33 To control the latter, introducing relevant functional units also appears of utmost importance. This explains why a myriad of functionalities have been introduced in gel-based materials and have largely broadened the scope of possibilities. For instance, supramolecular gelators have demonstrated their relevance in very diverse fields, including biomaterials,34 self-healable materials,35–37 crystallization of pharmaceutics,38 sensing,39 organic electronics and photonics.3,40–44
According to the literature, these important achievements were mainly reported thanks to single component gelators. More recently, scientists have increasingly considered the possibility of tuning the physicochemical properties of gels and xerogels: (i) by adding non-gelling additives, (ii) by forming supramolecular polymers from complementary monomers unable to assemble individually, or (iii) by combining gelators in the same materials.45–48
Whereas single-component gelators essentially assemble by means of self-complementary hydrogen bonds, aromatic and solvophobic interactions, or van der Waals forces, two-component organogels are supported by a significantly different conceptual approach. In this case, the self-assembly largely relies on the interaction between two distinct and complementary subunits that subsequently self-assemble into a supramolecular polymer.
Several advantages are expected from this approach: (i) the directional interaction occurring between both components in the initial step provides an additional level of control over the hierarchical self-assembly process,48 a key parameter in order to tune the properties at the bulk material scale; (ii) structural modifications can be conducted on either one or both components, which readily enables the introduction of specific functions (e.g. photo- or electroactive) within the materials; and (iii) the ratio of both components can be varied,49 which offers additional possibilities of tuning the properties and the morphology of the material.49 For instance, two-component organogels often present a wide solvent tolerance, allowing to reach higher mechanical strengths, and lead to lower critical gelation concentration (CGC) in comparison to their individual subcomponents.48
Since nature is a constant source of inspiration for chemists, it is not surprising that two-component organogels based on hydrogen bonding have been extensively studied for three decades.45,50–53 In this context, most research efforts were led by taking advantage of homo- or heterodimerization processes of base pairs,53 ureidopyrimidinone,54 melamine derivatives and imides,55 or Hamilton type complexes,56 for instance. Alternatively, charge-transfer (CT) complexes present similarities to H-bonded complexes, since they are directional by nature and are supported by two complementary counter-parts, namely the Donor (D) and Acceptor (A) moieties.57 Accordingly, the CT interaction can be used to construct electron–donor–acceptor-mediated gels,58,59 as reported by Maitra et al. more than twenty years ago.60
In parallel, outstanding supramolecular nanostructures were obtained by Meijer and coworkers with a singular family of C3-symmetric compounds.61 These systems are composed of a central benzene ring functionalized at the 1,3,5 positions by three N-monoacylated 3,3′-diamino-2,2′-bipyridine units (Scheme 1).62–64 This platform induces strong intermolecular and intramolecular interactions leading to an enhanced stability of the mesophases. Indeed, the 3,3′-diamino-2,2′-bipyridine fragments adopt a transoid conformation via intramolecular hydrogen bonding and are slightly tilted vs. the central benzene ring due to steric hindrance, thus giving the molecule a propeller-like conformation. Intermolecular hydrogen bonds and π–π stacking lead to columnar stacks of molecules with a helical architecture, an original supramolecular organization which has been widely explored since.
 | ||
| Scheme 1 Chemical structures of C3Pyr and C3NDI. | ||
These columnar aggregates can also be seen as C3-symmetric scaffolds to organize functional units in helical stacks. This prompted some of us to graft tetrathiafulvalene redox-active units21,65–67 in order to design supramolecular conducting nanofibers or porphyrin macrocycles to reach luminescent chiral supramolecular polymers.68 In the same line, the gelling and spectroscopic properties of C3Pyr (Scheme 1), which is endowed with three electron-rich and luminescent pyrene units, were explored.69,70 The corresponding studies highlighted that adding electron poor and non-gelling additives to C3Pyr solutions leads to increased gelling ability and drastically different spectroscopic properties, which result from donor–acceptor interactions. Herein, the concept that we explore goes one step further by mixing C3-symmetric organogelators, which display electronic complementarity. Beyond the synthesis and characterizations of a new C3-symmetric organogelator, this work focuses on (i) the impact of D–A interactions, (ii) the differences between the D–A gels and the corresponding individual components, (iii) solvent effects and eventually, (iv) the relative distribution of both subcomponents in the xerogel state.
Results and discussion
Design and synthesis
Among the large variety of possible combinations regarding the acceptor and donor structures, we have designed C3NDI as the acceptor. The latter will be combined with the previous C3-symmetric C3Pyr to provide an original mixture of C3-symmetry derivatives (Scheme 1 and Fig. 1). Indeed, according to the literature, the association constant between the respective parent derivatives (pyrene and NDI) is among the highest for donor–acceptor couples.71,72 It is also worth noting that the crystallographic analysis of the corresponding charge-transfer complex shows columnar stacking of the DADA type.71 In addition, according to our previous work,70 the two-component association of C3Pyr and N,N-dimethylnaphthalene diimide affords a robust thermosensitive/thermoreversible gel with an intense red colour, highlighting the formation of a charge-transfer complex at room temperature. | ||
| Fig. 1 Schematic representation of a columnar aggregate obtained through alternate D–A organisation of electroactive C3-symmetric building blocks. | ||
The synthesis of C3NDI was carried out in three steps starting from naphthalene tetracarboxylic dianhydride (NDA) and 3,3′-diamino-2,2′-bipyridine 270,73 (Scheme 2). Under microwave activation, NDA was first converted into its monoimide analogue grafted with an ethylpropyl chain, and subsequently treated with 4-aminobutanoic acid to afford NDI derivative 1 with a 28% yield. The corresponding acyl chloride was prepared upon reaction with excess thionyl chloride in dichloromethane. A subsequent addition–elimination reaction involving 3,3′-diamino-2,2′-bipyridine 2 in the presence of triethylamine led to the formation of amine 3 with a 40% yield. Finally, the reaction between trimesic acid chloride (1 equiv.), 3 (5 equiv.), and triethylamine (5 equiv.) in dry dichloromethane allowed for isolating the target C3NDI (58%) after purification by flash chromatography (CHCl3/EtOH, 96/4 v/v) and recycling preparative HPLC. Concomitantly, C3Pyr was prepared as described in our earlier report.69
 | ||
| Scheme 2 Synthesis of C3NDI. | ||
Gelation studies
C3Pyr vs. C3NDI. The characterization of C3NDI in CDCl3 by 1H NMR spectroscopy showed clear variations of chemical shifts for most Ar-H protons, confirming the contribution of aromatic interactions to the aggregation process (Fig. S1, ESI†). However, the signals of NDI protons (δ = 8.60 and 8.63 ppm) did not experience any variation, which suggests a rather moderate contribution of NDI units to supramolecular polymerisation in these solvents and concentration ranges.To be able to draw comparisons between C3Pyr,§69,70C3NDI and their mixtures, the gelation ability of C3NDI was assessed according to the ‘inverted-vial method’.30,74 Among the twelve tested solvents, C3NDI showed gelling properties in chloroform, 1,1,2,2-tetrachloroethane (TCE), chlorobenzene (CB), tetraline, o-dichlorobenzene (oDCB) and dimethylformamide (DMF) (Fig. S2 and Table S1, ESI†). Therefore, the solvents gelled either by C3NDI or C3Pyr belong to the same region of Hansen space with the exception of DMF, which could not be gelled by C3Pyr despite several attempts at varying the concentration and the cooling rate. It is also interesting to note that the grafted π-functional units sometimes have a major influence on the critical gelation concentration (CGC, i.e. the minimal concentration allowing for gelation). For instance, when switching from pyrene to naphthalene diimide, the CGC is divided by 30 in tetraline, and ca. 6 in oDCB.
Mixing C3Pyr and C3NDI
Three organic solvents with different properties (see Table 1 for their respective physicochemical parameters) were selected to study the gelling properties of the two-component organogels. o-Dichlorobenzene is a chlorinated, aromatic, aprotic, and polar solvent. N,N-Dimethylformamide is an aprotic and polar solvent. Lastly, 1,1,2,2-tetrachloroethane is a chlorinated, aliphatic, aprotic, and moderately polar solvent, which (i) affords solutions of C3Pyr and C3NDI at moderate concentrations and (ii) previously showed that the critical gelation concentration of C3Pyr was dramatically lowered in the presence of π-functional acceptors.69,70 To run the corresponding experiments, equimolar amounts of both gelators were placed in a vial, sonicated for five minutes, heated until complete solubilisation and allowed to cool down to room temperature. In both chlorinated solvents, both gelators get dissolved and afford transparent and slightly yellow solutions at high temperature. Upon cooling, the medium progressively turns to red showing the formation of charge-transfer complexes and affording a gel (Fig. 2 and Fig. S3, Movie S1, ESI†). The critical gelation concentrations (CGCs, i.e. minimal concentrations allowing for gelation) were determined for individual gelling components and their equimolar mixtures. In TCE, C3Pyr and C3NDI displayed CGCs of 65 (39.3 mM) and 60 mg mL−1 (33.7 mM) and act synergistically upon mixing, affording gels above a total gelator concentration of CT = [C3Pyr] + [C3NDI] = 10.5 mM ([C3Pyr] = [C3NDI] = 5.25 mM). Similar experiments were led in oDCB and DMF (Fig. S3, ESI†). In oDCB, heating the samples systematically led to the solubilization of the gelators, before forming red gels at room temperature. Unlike TCE- and oDCB-containing samples, red aggregates were observed when mixing C3Pyr and C3NDI in DMF at high temperatures. This particular behaviour is likely to result from weaker solubilities in DMF and/or stronger interactions between the donor and acceptor moieties in a polar solvent.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture
:1 mixture
| CGC in TCE | CGC in oDCB | CGC in DMF | |
|---|---|---|---|
| Hansen solubility parameters according to ref. 71. | |||
| C3Pyr | 39.3 mM (65 mg mL−1) | 5.2 mM (8 mg mL−1) | Suspension |
| C3NDI | 33.7 mM (60 mg mL−1) | 0.6 mM (1.2 mg mL−1) | 6 mM (12 mg mL−1) |
| C3Pyr/C3NDI | [C3Pyr] = [C3NDI] = 5.25 mM (8/10 mg mL−1) | [C3Pyr] = [C3NDI] = 2.3 mM (3.5/4.4 mg mL−1) | [C3Pyr] = [C3NDI] = 4.3 mM (6.5/8.2 mg mL−1) |
| bp (°C) | 146 | 179 | 153 |
| E | 8.5 | 10.1 | 38.2 |
| μ (D) | 1.3 | 2.5 | 3.8 |
| δ H | 5.3 | 3.3 | 11.3 |
| δ D | 18.8 | 19.2 | 17.4 |
| δ P | 5.1 | 6.3 | 13.7 |
 | ||
| Fig. 2 Top. Donor–acceptor organogel obtained from C3NDI and C3Pyr in TCE-d2 ([C3Pyr] = [C3NDI] = 5.2 mM). Middle. Solution of C3NDI in TCE (5.2 mM). Bottom. Solution of C3Pyr in TCE (5.2 mM). | ||
Contrary to the case of TCE, C3Pyr contributed to the disruption of the C3NDI-based network in oDCB and DMF and led to increased critical gelation concentrations.
1H NMR spectroscopy
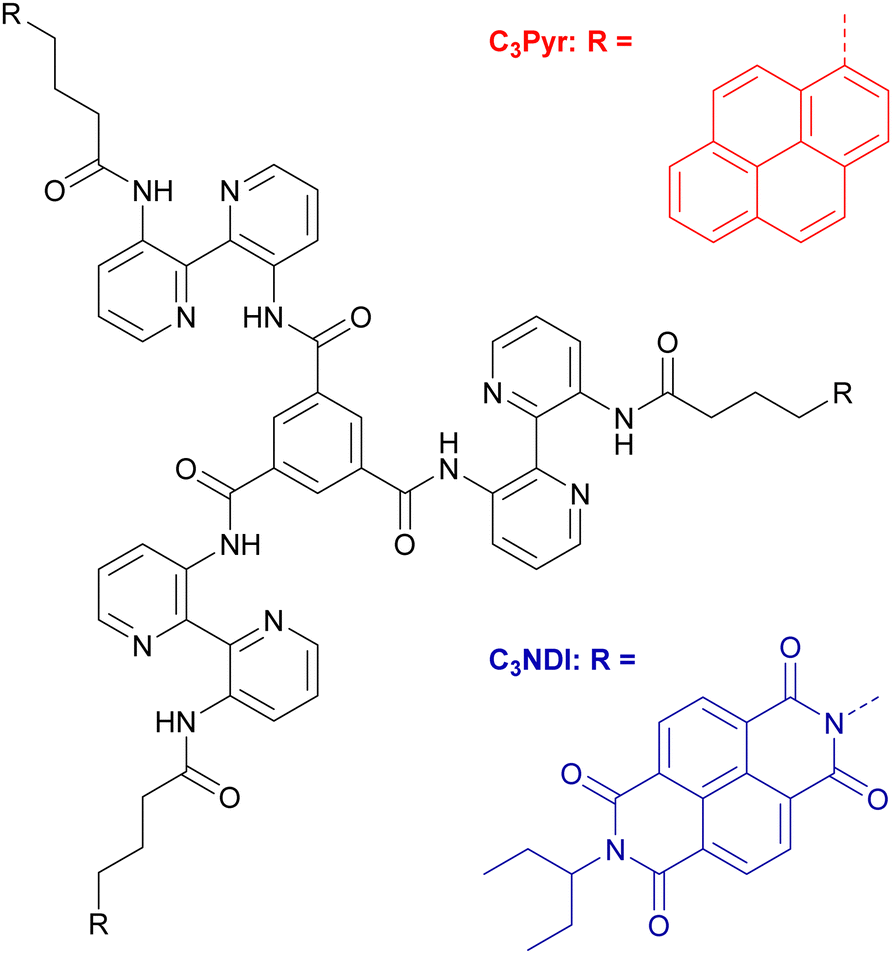
To get insight into the intermolecular interactions involved along the supramolecular polymerization process, variable-temperature NMR experiments were performed for both individual gelling components C3Pyr and C3NDI as well as for their 1:1 mixture in TCE, a solvent which ensures sufficient solubilities of all species at room temperature (Fig. 3, Fig. S4 and S5, ESI†). In all cases, lowering the temperature led to a clear deshielding of the NH chemical shifts (Δδ ∼ 0.3 ppm), which is associated with the strengthening of intramolecular hydrogen bonds. For individual gelators and the corresponding mixture ([C3Pyr] = [C3NDI] = 5.2 mM), the temperature had little effect on the resolution and the chemical shifts (Fig. S4 and S5, ESI†), whereas a major difference was observed regarding resolution and integrals in the case of mixtures. Indeed, upon cooling a warm solution of C3Pyr/C3NDI in TCE-d2 (5.2 mM each), the resolution of the spectra and the signal-to-noise ratio progressively decreased (Fig. 3 and Fig. S6, ESI†), which logically results from the co-aggregation of both gelators. Noteworthily, the intensities of C3Pyr signals decreased faster than those of C3NDI (Fig. S6, ESI†), suggesting that the equilibrium between solvated C3 derivatives and aggregated ones is more shifted towards the solvated state in the case of C3NDI.
 | ||
| Fig. 3 Variable temperature 1H NMR spectroscopy of a C3Pyr/C3NDI (1/1) mixture in TCE-d2 ([C3Pyr] = [C3NDI] = 5.2 mM). | ||
UV-Visible absorption spectroscopy
UV-Visible absorption spectra of individual gelators and their equimolar mixture C3Pyr/C3NDI were recorded in different solvents at room temperature (Fig. 4). Given the strong molar extinction coefficients of C3Pyr70 and C3NDI (Fig. S7, ESI†) below 400 nm (π–π* transitions) and the high concentrations required to observe aggregation, visible absorption spectra were recorded above 400 nm. As evidenced by Fig. 4 (top), a broad charge-transfer absorption band appeared in the visible range upon preparing an equimolar sample of both gelators (5.2 mM) in TCE. This observation, which confirms the occurrence of aromatic interactions between pyrene and naphthalene diimide units,75 was also made in oDCB and DMF (CT = CGC – Fig. S8, ESI†). These measurements notably allowed highlighting the stronger propensity of DMF to promote charge transfer interactions, which is likely to explain why C3Pyr and C3NDI aggregate even at high temperatures in this polar solvent (δP = 13.7 MPa1/2). In both chlorinated solvents, the charge transfer absorption bands have similar molar extinction coefficients, which shows that similar concentrations of pyrene–NDI stacks are formed in the medium and hence, that the nature of the solvent mostly influences the interactions between the C3 symmetric cores. | ||
| Fig. 4 Top. Visible absorption spectra of the individual compounds C3Pyr (5.2 mM), C3NDI (5.2 mM) and their equimolar mixture C3Pyr/C3NDI (CT = 10.5 mM) in TCE. Bottom. Visible absorption spectra of C3Pyr/C3NDI (1:1) organogels in TCE, oDCB and DMF (CT = CGC). | ||
Variable-temperature visible absorption spectroscopy constitutes a relevant technique to probe the strength of charge transfer interactions between donor and acceptor units in supramolecular systems. This prompted us to evaluate the impact of temperature over the absorption spectra of individual components and C3Pyr–C3NDI gels. Regarding individual components (Fig. S9, ESI†), these experiments were led at the concentrations allowing for gelation upon addition of the complementary C3 derivative. In these conditions, the spectrum of C3Pyr does not evolve between 100 and 20 °C in TCE (5.2 mM) but does in oDCB (2.3 mM) or DMF (4.3 mM), with the appearance of a shoulder above 400 nm that results from intermolecular interactions. Unlike C3Pyr, C3NDI proved to aggregate upon lowering the temperature in TCE (5.2 mM) and DMF (4.3 mM) and not in oDCB (2.3 mM). Setting up NDI–NDI interactions was again associated with the appearance of a shoulder above 400 nm.76
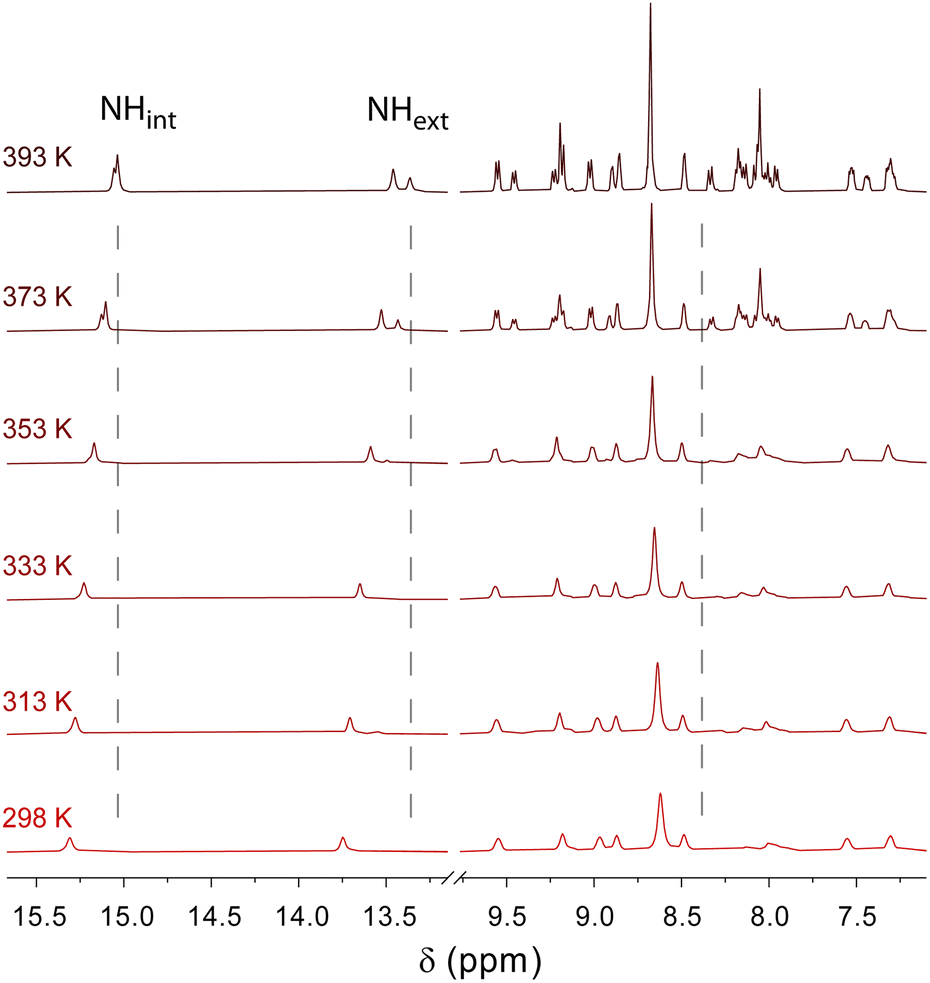
When performing these experiments with C3NDI–C3Pyr mixtures (Fig. 5 and Fig. S10, S11, ESI†), a first important observation lies on the persistence of the CT band at temperatures as high as 100 °C (373 K), which highlights the robustness of C3NDI/C3Pyr-based supramolecular aggregates. This was further confirmed by plotting the evolution of the absorbance at λ = 525 nm as a function of temperature and fitting the corresponding data to assess melting temperature values (Tm, temperatures at which half of the charge transfer complexes are dissociated). The latter proved to be as high as 152 °C (425 K) for TCE-containing gels and 121 °C (394 K) for oDCB ones. As for the DMF-containing gel at the critical gelation, the melting temperature was too high to be determined.77 This again evidences the occurrence of stronger aromatic interactions between pyrene and naphthalene diimide in this solvent. Consequently, similar experiments were led in more diluted conditions (CT = CGC/10 – Fig. S12, ESI†) and allowed determining a melting temperature of 128 °C (401 K) (CT = 0.86 mM, DMF).
 | ||
| Fig. 5 Evolution of the visible absorption spectrum of a C3Pyr/C3NDI (1:1) mixture in TCE ([C3Pyr] = [C3NDI] = 5.2 mM) upon decreasing the temperature from 100 °C (red curve) to 20 °C (blue curve) (interval of 10 °C between each curve). Inset. Corresponding evolution of the absorbance at λ = 525 nm. | ||
More than offering a way to assess the contribution of pyrene and NDI fragments to the supramolecular polymerization process, these variable temperature experiments also allow a comparison of the association of both C3-symmetric compounds C3Pyr and C3NDI, and the previously reported C3Pyr(NDI)3 system, which was obtained by mixing C3Pyr and three equivalents of the parent N,N-dimethylnaphthalene diimide NDI (Scheme S1, ESI†). A first important difference was observed regarding the spectrum shape since C3Pyr(NDI)3 displayed a significantly more intense charge-transfer band at the same concentrations of π-functional units (either pyrene or NDI). This could result from a weaker concentration of pyrene–NDI associations when mixing C3Pyr and C3NDI, which is possibly associated with the formation of non-fully alternated stacks. Another explanation could lie on the relative conformations of pyrene and NDI units: in the case of C3Pyr(NDI)3 it appears reasonable to consider that NDI additives will adopt the optimal arrangement with regard to pyrene to promote the formation of charge transfer complexes; on the contrary, linking NDI units to the C3-symmetric core in C3NDI produces a geometric constraint, which may impede the adoption of the ideal conformation and hence, leads to weaker absorption.70 It is worth noting that both systems also display a major difference regarding their temperature-dependent behaviour: heating the C3Pyr(NDI)3 composite ([C3Pyr] = [NDI]/3 = 5.2 mM) to above 80 °C (353 K) in TCE leads to the complete disappearance of the CT band, while this absorption band was hardly affected when heating a mixture of C3Pyr (5.2 mM) and C3NDI (5.2 mM) in TCE at 100 °C (373 K). This highlights the extent to which the C3-symmetric core acts as a preorganising scaffold to stabilise pyrene–NDI interactions. This conclusion was further confirmed by comparing the estimated melting temperatures, which are about 50 °C for the C3Pyr(NDI)3 system and 150 °C for the C3Pyr–C3NDI system. To establish the thermosensitive nature of the CT interaction and since the heating limits of the spectrometer were reached, we additionally led a complementary experiment consisting in heating and cooling an equimolar solution of C3Pyr and C3NDI ([C3Pyr] = [NDI]/3 = 5.2 mM, TCE) at a temperature close to the boiling point of the solvent (146 °C). In this manner, Movie S1 (ESI†) definitely shows that approaching the boiling point of TCE allows for breaking pyrene–NDI interactions.
Scanning electron microscopy of xerogels
The micro and nanostructures formed by assembling C3Pyr and/or C3NDI were studied by scanning electron microscopy to get insight into the effects of (i) mixing C3-symmetric gelators, (ii) solvents, and (iii) deposition methods on the corresponding solid-state materials. To do so, films were prepared by depositing pieces of gels (CT = CGC, Fig. 6) or drop casting warm solutions on glass slides (Fig. S13, ESI†), evaporation of the solvent and metallization with platinum. Though the deposition method may have a dramatic impact on the corresponding structures, similar trends were observed by gel deposition or drop casting. | ||
| Fig. 6 SEM micrographs of samples prepared by gel deposition of the individual compounds C3Pyr (8 mg mL−1, a), C3NDI (10 mg mL−1, b) and their equimolar mixture C3Pyr/C3NDI (identical concentrations, c) in TCE. | ||
In TCE, the structures formed from individual C3Pyr or C3NDI were poorly defined in comparison to the nanofibers formed upon mixing both gelators. Hence, in the latter case, the presence of well-defined monodimensional aggregates, which display diameters comprised between 40 and 120 nm and lengths up to 1.5 μm, confirm the occurrence of interactions between both gelators. In DMF, C3Pyr-based materials did not display a fibrous aspect whatever the casting technique, which is in line with its non-gelling ability (Table S1, ESI†). On the other hand, C3NDI formed particularly compact materials by gel deposition and networks of nanofibers by drop casting. Upon mixing both C3-symmetric precursors in DMF, the network formed by drop casting resembled those formed by individual C3NDI, while the structures formed by C3Pyr alone were not observed. On the other hand, the deposition of a C3Pyr–C3NDI gel and subsequent evaporation of the solvent afforded a particularly well-defined network of nanofibers. This observation, which is in sharp contrast to the observations made for individual components, confirms a social behaviour and hence, the formation of alternate stacks of C3Pyr and C3NDI. Eventually, we found that samples prepared from oDCB had a significantly different behaviour. Indeed, all samples showed networks of nanofibers with similar dimensions and aspects (Fig. 7), which did not provide any significant hint regarding the effect of mixing C3Pyr and C3NDI.
 | ||
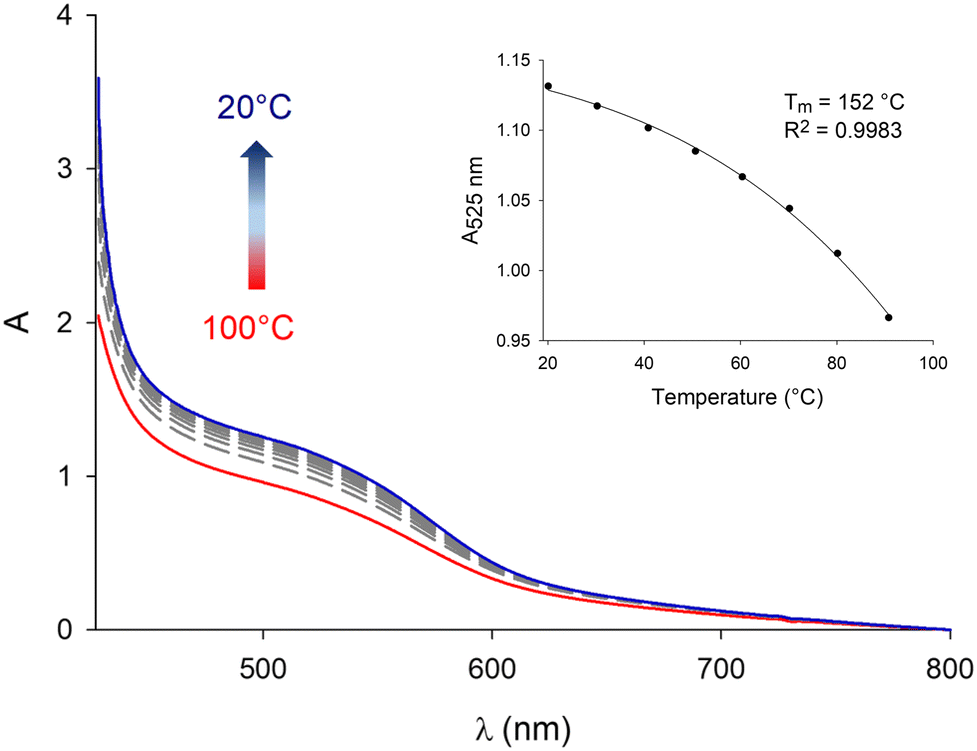
| Fig. 7 Top. Emission spectra of C3Pyr (5.2 mM), C3NDI (5.2 mM) and an equimolar mixture of both components ([C3Pyr] = [C3NDI] = 5.2 mM – gel state) in TCE upon excitation at 405 nm (the exact same conditions were used to perform these measurements). Bottom. Corresponding normalized spectra. | ||
Fluorescence spectroscopy
In a former report,70 we have demonstrated that pyrene–NDI exciplexes could be detected at a specific wavelength by luminescence spectroscopy upon mixing C3Pyr with a NDI additive. Considering the particular case of samples originating from DMF and the absence of the ‘mixing effect’ on the structures obtained from oDCB, studying the luminescent properties of the different gels and xerogels and imaging the latter by confocal microscopy appeared relevant. This is the reason why individual compounds C3Pyr, C3NDI and their equimolar mixture C3Pyr/C3NDI were first studied by fluorescence spectroscopy. Fig. 7 shows the emission spectra of C3Pyr (5.2 mM), C3NDI (5.2 mM) and their equimolar mixture ([C3Pyr] = [C3NDI] = 5.2 mM) in TCE. For pure C3Pyr and C3NDI, intense emission bands (λ = 517 and 537 nm), which correspond to radiative relaxations of excited bipyridyl motifs69 and NDI excimers,78,79 were respectively observed. More importantly, this comparison shows to which extent the luminescent properties of C3Pyr and C3NDI are quenched upon mixing both components (Fig. 7) and demonstrates that non-radiative relaxation processes are promoted upon mixing both derivatives. Given the presence of a CT absorption band in the spectrum of the C3Pyr–C3NDI mixture, a rational explanation lies in the generation of a charge-separated state, which notably returns to the ground-state through vibrational and rotational processes. Comparing the corresponding normalized emission spectra allows for evidencing an emission band centred at λmax = 639 nm when mixing both derivatives, which corresponds to the formation of pyrene–NDI exciplexes.Whatever the solvent under consideration (Fig. 8), emission spectra obtained for λexc = 405 nm and λexc = 525, 512, and 523 nm (CT band) show a single emission band centred at 624–640 nm, which matches the emission wavelength of {C3Pyr–C3NDI}* exciplexes. Noteworthily, this typical emission band was detected in all three solvents (Fig. 8 and Fig. S14, ESI†), including TCE and DMF which do not form exciplexes with NDI derivatives, unlike aromatic solvents such as toluene.79
 | ||
| Fig. 8 Normalized emission spectra of C3Pyr/C3NDI (1:1) organogels (C = CGC) upon excitation at different wavelengths (bottom right corner: irradiations at λexc = 525 (TCE), 512 (oDCB) and 523 nm (DMF)). | ||
Along these fluorescence measurements, samples containing both C3-symmetric precursors showed important variations of emission spectra upon varying the excitation wavelength. This shows that different radiative relaxation pathways are followed after excitation. Furthermore, the maximum emission wavelength associated with bipyridyl moieties does not significantly vary with the solvent, while variations of up to 30 nm were observed for the exciplex-related emission band. Though the maximum emission wavelengths were difficult to evaluate in DMF for λexc = 344 and 360 nm, the same trend was most likely followed whatever the excitation wavelength: the emission band in oDCB was systematically the most blue-shifted and TCE afforded the samples emitting at the lowest energies. Since no clear relationship between this trend and the physicochemical parameters of the solvents (Table 1) could be established, the formation of different supramolecular arrangements affording different relative orientations of chromophores appears as a rational explanation to account for this behaviour.
Confocal microscopy
Confocal microscopy constitutes a particularly interesting technique to study materials including luminescent derivatives.80,81 It may provide valuable information related to the distribution of chromophores in films and, provided the use of an appropriate setup, may furnish point-by-point emission spectra. Taking advantage of such a device, films of C3Pyr, C3NDI and their equimolar mixture (CT = CGC) were prepared from TCE (Fig. 9 and Fig. S15, ESI†), oDCB and DMF (Fig. 10) by gel deposition and studied. Though making reliable quantitative comparisons may appear difficult by confocal microscopy, the samples obtained from C3Pyr alone were clearly more luminescent than the others under the same conditions (Fig. S15–S17, ESI†), which is in line with the analyses performed in the liquid or the gel state. Whatever the solvent under consideration, films prepared from individual gelators respectively showed emission bands centred at 519 (C3Pyr) and 552 nm (C3NDI) upon excitation at 405 nm. This appears consistent with the observations made in solution and in the gel state, with a moderate shift in the case of C3NDI, which could result from a slightly different stacking of NDI units. Irradiation at the same wavelength of mixed xerogels prepared from TCE and DMF produced a single emission band centred at 628 nm, characterizing the {C3Pyr–C3NDI}* exciplex. In oDCB, the situation proved slightly different with a second emission band at higher energies. This suggests that C3Pyr and C3NDI are partially segregated in this case and do not present a fully social behaviour in this solvent (Fig. 10).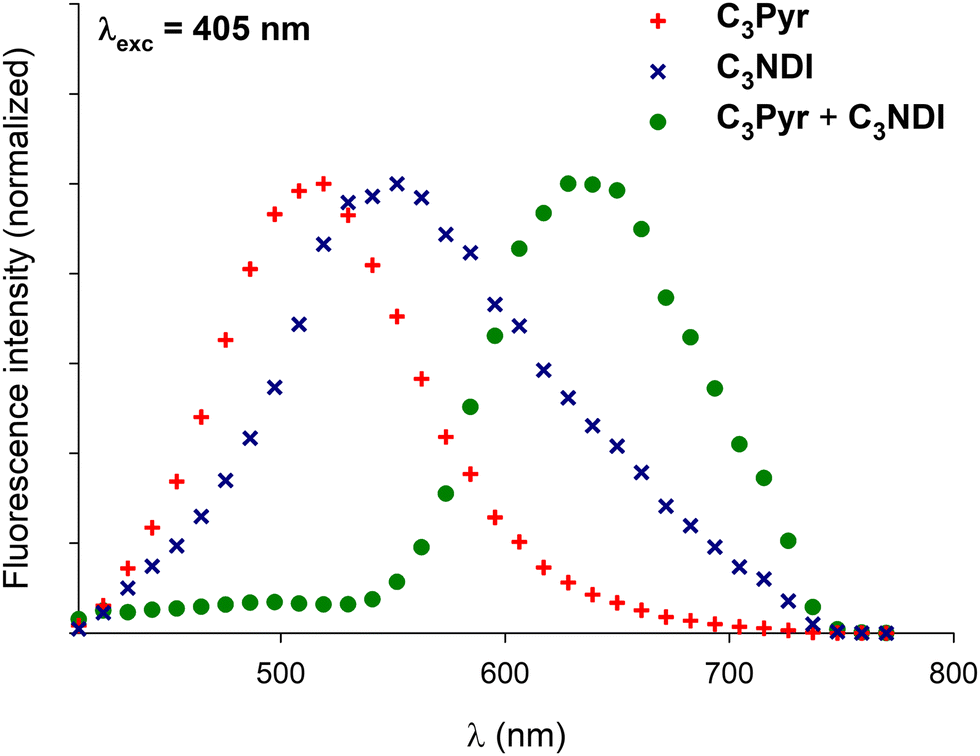 | ||
| Fig. 9 Normalized emission spectra of xerogels prepared by gel deposition of individual compounds C3Pyr (8 mg mL−1), C3NDI (10 mg mL−1) and their equimolar mixture C3Pyr/C3NDI (identical concentrations) in TCE for λexc = 405 nm. | ||
 | ||
| Fig. 10 Normalized emission spectra of xerogels prepared by gel deposition of C3Pyr/C3NDI (1:1) mixtures in TCE, oDCB, and DMF for λexc = 405 nm. | ||
The preparation method of the samples also proved to have a strong impact on the morphology of the xerogels. The films resulting from the deposition of a piece of gel showed the systematic presence of heterogeneities (Fig. 11 left), while the xerogels obtained by drop casting warm solutions of C3Pyr and C3NDI appeared more homogeneous (Fig. 11 right).
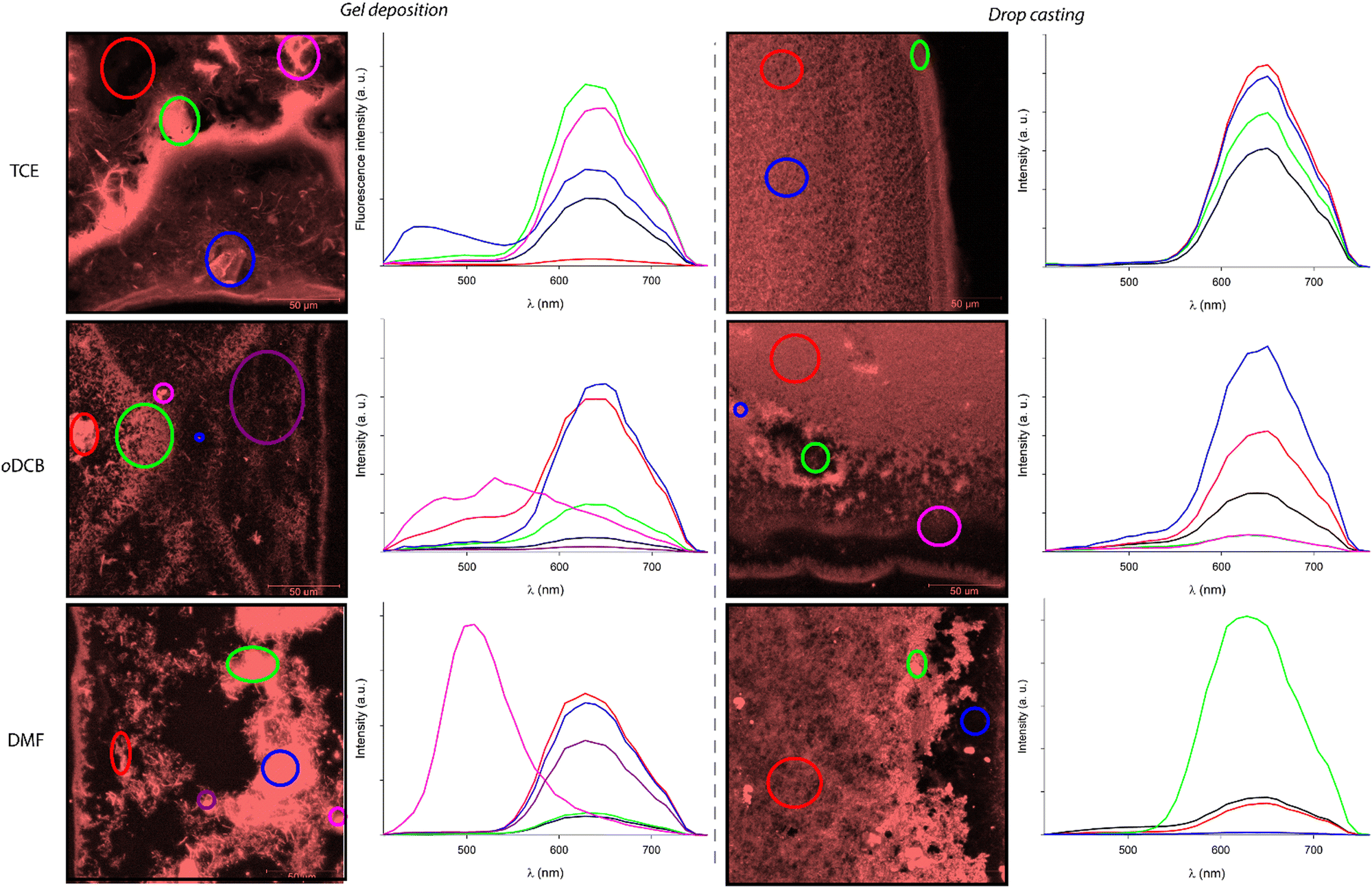 | ||
| Fig. 11 Emission spectra of the whole images (black line) and specific areas (highlighted with different colours in the corresponding images) obtained by confocal microscopy of xerogels prepared by gel deposition (left) or by drop casting a hot solution (right) of the equimolar mixture C3Pyr/C3NDI ([C3Pyr] = 8 mg mL−1, [C3NDI] = 10 mg mL−1) in TCE for λexc = 405 nm. | ||
In this context, the possibility to measure the emission spectra in a point-by-point manner was particularly valuable. It notably allowed the plotting of the average emission spectra of selected areas. Thereby, Fig. 11 gathers the emission spectra of various regions of the presented micrographs (λexc = 405 nm), the black curves corresponding to the average emission spectra of the whole studied areas. These spectra confirm that all samples display similar emission profiles that are typical for the “pyrene–NDI exciplex” emission. When comparing the confocal micrographs, one can also clearly observe that the samples obtained by gel deposition present heterogeneities with drastically different emission spectra. According to the emission profiles of films prepared from pure C3Pyr or C3NDI, the additional shoulder observed in the case of oDCB (λ = 520 nm, red curve) corresponds to the radiative relaxation of individual C3Pyr. When plotting the emission spectra of specific regions of the samples (circled in various colours), one can also observe different emission profiles that probably correspond to an impurity (TCE, blue circle) and to a particle of undissolved C3Pyr (DMF, purple circle). In both cases, these microstructures remained scarce throughout the sample and their contribution to the average emission spectrum (black curves) was not significant.
On the contrary, all the studied regions of drop-cast samples afforded the typical emission spectra of pyrene–NDI exciplexes, confirming the efficient mixing of both C3-symmetric gelators. This observation is in line with an efficient supramolecular copolymerization of C3Pyr and C3NDI. It also shows that preparing such supramolecular polymers by drop casting warm solutions of gelators constitutes a more relevant method than gel deposition to obtain homogeneous samples.
Conclusions
As previously demonstrated, C3Pyr displays improved gelling ability in the presence of electron poor derivatives. This prompted us to design and synthesize the electron deficient C3NDI to form co-assemblies with C3Pyr through aromatic donor–acceptor interactions. The gelling abilities of individual gelators were determined in solvents of different natures, which allowed for highlighting the detrimental or synergistic effect of mixing gelators. These experiments also showed that charge-transfer complexes are formed when mixing gelators, as emphasized by the intense red colour of the samples. UV-Visible absorption spectroscopy showed that the charge transfer band was particularly intense in DMF, which suggests a particularly strong interaction and is in line with the poor solubility observed, when C3Pyr and C3NDI are dissolved in this solvent. After evaporation of the solvents, the films of individual gelators and the corresponding C3Pyr–C3NDI xerogels were studied through scanning electron microscopy, which clearly evidenced the formation of new structures, be it through gel deposition or drop casting warm solutions. Besides, the deposition method once again proved to have a strong influence over the morphology of the samples. To get insight into the distribution of chromophores within the films, studying the xerogels through confocal microscopy was particularly valuable for probing the presence of heterogeneity in the materials. In particular, the possibility of measuring the emission spectra in a point-by-point manner constituted a particularly relevant feature.Altogether, the results presented herein emphasize the role of the solvent in the establishment of D–A interactions and the importance of the deposition method to reach homogeneous materials. This study also highlights the interest in associating complementary C3-symmetric units to tune the emissive properties of the corresponding self-assemblies. As precursors of helical fibres, chiral C3-symmetric derivatives could constitute interesting precursors in the field of chiroptics.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The French National Research Agency (ANR-16-CE09-0013 FOGEL project) is acknowledged for funding. The authors are also thankful to the SCIAM platform and the SFR Matrix (University of Angers).Notes and references
- P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133–3159 CrossRef CAS PubMed.
- D. J. Abdallah and R. G. Weiss, Adv. Mater., 2000, 12, 1237–1247 CrossRef CAS.
- N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821–836 RSC.
- R. G. Weiss and P. Terech, ed., Molecular gels: materials with self-assembled fibrillar networks, Springer: Dordrecht, 2006 Search PubMed.
- P. Dastidar, Chem. Soc. Rev., 2008, 37, 2699–2715 RSC.
- G. Yu, X. Yan, C. Han and F. Huang, Chem. Soc. Rev., 2013, 42, 6697–6722 RSC.
- J.-M. Guenet, Organogels: Thermodynamics, Structure, Solvent Role, and Properties, Springer, 2016 Search PubMed.
- E. R. Draper and D. J. Adams, Chemistry, 2017, 3, 390–410 CrossRef CAS.
- P. R. A. Chivers and D. K. Smith, Nat. Rev. Mater., 2019, 4, 463–478 CrossRef CAS.
- V. Nele, J. P. Wojciechowski, J. P. K. Armstrong and M. M. Stevens, Adv. Funct. Mater., 2020, 30, 2002759 CrossRef CAS.
- Y. Gu, J. Zhao and J. A. Johnson, Angew. Chem., Int. Ed., 2020, 59, 5022–5049 CrossRef CAS PubMed.
- Y. Zhuo, J. Chen, S. Xiao, T. Li, F. Wang, J. He and Z. Zhang, Mater. Horiz., 2021, 8, 3266–3280 RSC.
- S. Panja and D. J. Adams, Chem. Soc. Rev., 2021, 50, 5165–5200 RSC.
- J. H. van Esch, Langmuir, 2009, 25, 8392–8394 CrossRef CAS PubMed.
- C. Kulkarni, S. Balasubramanian and S. J. George, Chem. Phys. Chem., 2013, 14, 661–673 CrossRef CAS PubMed.
- R. van Lommel, W. M. de Borggraeve, F. de Proft and M. Alonso, Gels, 2021, 7, 87 CrossRef CAS PubMed.
- L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071–4098 CrossRef CAS PubMed.
- E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Chem. Rev., 2016, 116, 13752–13990 CrossRef CAS PubMed.
- N. Zweep and J. H. van Esch, in Soft Matter Series, ed. B. Escuder and J. F. Miravet, Royal Society of Chemistry: Cambridge, 2013, pp. 1–29 Search PubMed.
- P. Dastidar, Gels, 2019, 5, 15 CrossRef CAS PubMed.
- I. Danila, F. Riobé, F. Piron, J. Puigmartí-Luis, J. D. Wallis, M. Linares, H. Ågren, D. Beljonne, D. B. Amabilino and N. Avarvari, J. Am. Chem. Soc., 2011, 133, 8344–8353 CrossRef CAS PubMed.
- Y. Lan, M. G. Corradini, R. G. Weiss, S. R. Raghavan and M. A. Rogers, Chem. Soc. Rev., 2015, 44, 6035–6058 RSC.
- J. Van Esch, S. De Feyter, R. M. Kellogg, F. De Schryver and B. L. Feringa, Chem. – Eur. J., 1997, 3, 1238–1243 CrossRef CAS.
- S. S. Babu, S. Mahesh, K. K. Kartha and A. Ajayaghosh, Chem. – Asian J., 2009, 4, 824–829 CrossRef CAS PubMed.
- F. Aparicio, F. Garcia and L. Sanchez, Chem. – Eur. J., 2013, 19, 3239–3248 CrossRef CAS PubMed.
- V. Palermo, S. Morelli, C. Simpson, K. Müllen and P. Samorì, J. Mater. Chem., 2006, 16, 266–271 RSC.
- J. Puigmarti-Luis, A. Pérez Del Pino, V. Laukhin, L. N. Feldborg, C. Rovira, E. Laukhina and D. B. Amabilino, J. Mater. Chem., 2010, 20, 466–474 RSC.
- D. Dasgupta, S. Srinivasan, C. Rochas, A. Ajayaghosh and J.-M. Guenet, Soft Matter, 2011, 7, 9311–9315 RSC.
- D. Canevet, A. Pérez Del Pino, D. B. Amabilino and M. Sallé, J. Mater. Chem., 2011, 21, 1428–1437 RSC.
- T.-L. Lai, D. Canevet, Y. Almohamed, J.-Y. Mévellec, R. Barillé, N. Avarvari and M. Sallé, New J. Chem., 2014, 38, 4448–4457 RSC.
- F. Aparicio, L. Faour, D. Gindre, D. Canevet and M. Sallé, Soft Matter, 2016, 12, 8480–8484 RSC.
- A. B. Marco, F. Aparicio, L. Faour, K. Iliopoulos, Y. Morille, M. Allain, S. Franco, R. Andreu, B. Sahraoui, D. Gindre, D. Canevet and M. Sallé, J. Am. Chem. Soc., 2016, 138, 9025–9028 CrossRef CAS PubMed.
- A. B. Marco, D. Gindre, K. Iliopoulos, S. Franco, R. Andreu, D. Canevet and M. Sallé, Org. Biomol. Chem., 2018, 16, 2470–2478 RSC.
- S. Varela-Aramburu, G. Morgese, L. Su, S. M. C. Schoenmakers, M. Perrone, L. Leanza, C. Perego, G. M. Pavan, A. R. A. Palmans and E. W. Meijer, Biomacromolecules, 2020, 21, 4105–4115 CrossRef CAS PubMed.
- X. Yu, L. Chen, M. Zhang and T. Yi, Chem. Soc. Rev., 2014, 43, 5346 RSC.
- M. Zhang, D. Xu, X. Yan, J. Chen, S. Dong, B. Zheng and F. Huang, Angew. Chem., Int. Ed., 2012, 51, 7011–7015 CrossRef CAS PubMed.
- D. L. Taylor and M. in het Panhuis, Adv. Mater., 2016, 28, 9060–9093 CrossRef CAS PubMed.
- D. K. Kumar and J. W. Steed, Chem. Soc. Rev., 2014, 43, 2080–2088 RSC.
- A. Sandeep, V. K. Praveen, K. K. Kartha, V. Karunakaran and A. Ajayaghosh, Chem. Sci., 2016, 7, 4460–4467 RSC.
- S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973–2129 CrossRef CAS PubMed.
- J. Puigmarti-Luis and D. B. Amabilino, in Functional Molecular Gels, B. Escuder, J. F. Miravet, ed., The Royal Society of Chemistry, 2014, vol. 1, pp. 195–254 Search PubMed.
- K. G. Cho, J. I. Lee, S. Lee, K. Hong, M. S. Kang and K. H. Lee, Adv. Funct. Mater., 2020, 30, 1907936 CrossRef CAS.
- Z. Li, X. Ji, H. Xie and B. Z. Tang, Adv. Mater., 2021, 33, 2100021 CrossRef CAS PubMed.
- S. Prasanthkumar, S. Ghosh, V. C. Nair, A. Saeki, S. Seki and A. Ajayaghosh, Angew. Chem., Int. Ed., 2015, 54, 946–950 CrossRef CAS PubMed.
- T. Haino, Polym. J., 2013, 45, 363–383 CrossRef CAS.
- L. E. Buerkle and S. J. Rowan, Chem. Soc. Rev., 2012, 41, 6089–6102 RSC.
- S. Bhattacharya and S. K. Samanta, Chem. Rev., 2016, 116, 11967–12028 CrossRef CAS PubMed.
- A. R. Hirst and D. K. Smith, Chem. – Eur. J., 2005, 11, 5496–5508 CrossRef CAS PubMed.
- A. R. Hirst, J. F. Miravet, B. Escuder, L. Noirez, V. Castelletto, I. W. Hamley and D. K. Smith, Chem. – Eur. J., 2009, 15, 372–379 CrossRef CAS PubMed.
- S. Yagai, J. Photochem. Photobiol., C, 2006, 7, 164–182 CrossRef CAS.
- W. Binder, ed., Hydrogen Bonded Polymers, Springer Berlin Heidelberg, Berlin, Heidelberg, 2007, vol. 207 Search PubMed.
- A. J. Wilson, Soft Matter, 2007, 3, 409–425 RSC.
- J. L. Sessler, C. M. Lawrence and J. Jayawickramarajah, Chem. Soc. Rev., 2007, 36, 314–325 RSC.
- J. H. K. K. Hirschberg, R. A. Koevoets, R. P. Sijbesma and E. W. Meijer, Chem. – Eur. J., 2003, 9, 4222–4231 CrossRef CAS PubMed.
- C. Fouquey, J.-M. Lehn and A.-M. Levelut, Adv. Mater., 1990, 2, 254–257 CrossRef CAS.
- V. Berl, M. Schmutz, M. J. Krische, R. G. Khoury and J.-M. Lehn, Chem. – Eur. J., 2002, 8, 1227 CrossRef CAS PubMed.
- W. Hayes and B. W. Greenland, in Supramolecular Polymer Networks and Gels, ed. S. Seiffert, Springer International Publishing; Cham, 2015, vol. 268, pp. 143–166 Search PubMed.
- A. Das and S. Ghosh, Angew. Chem., Int. Ed., 2014, 53, 2038–2054 CrossRef CAS PubMed.
- X. Peng, L. Wang and S. Chen, J. Inclusion Phenom. Macrocyclic Chem., 2021, 99, 131–154 CrossRef CAS.
- U. Maitra, P. V. Kumar, N. Chandra, L. J. D’Souza, M. D. Prasanna and A. R. Raju, Chem. Commun., 1999, 595–596 RSC.
- S. Cantekin, T. F. A. de Greef and A. R. A. Palmans, Chem. Soc. Rev., 2012, 41, 6125 RSC.
- A. R. A. Palmans, J. A. J. M. Vekemans, E. E. Havinga and E. W. Meijer, Angew. Chem., Int. Ed. Engl., 1997, 36, 2648–2651 CrossRef CAS.
- A. R. A. Palmans, J. A. J. M. Vekemans, H. Fischer, R. A. Hikmet and E. W. Meijer, Chem. – Eur. J., 1997, 3, 300–307 CrossRef CAS PubMed.
- J. J. van Gorp, J. A. J. M. Vekemans and E. W. Meijer, J. Am. Chem. Soc., 2002, 124, 14759–14769 CrossRef CAS PubMed.
- I. Danila, F. Riobé, J. Puigmartí-Luis, Á. Pérez Del Pino, J. D. Wallis, D. B. Amabilino and N. Avarvari, J. Mater. Chem., 2009, 19, 4495–4504 RSC.
- I. Danila, F. Pop, C. Escudero, L. N. Feldborg, J. Puigmarti-Luis, F. Riobé, N. Avarvari and D. B. Amabilino, Chem. Commun., 2012, 48, 4552–4554 RSC.
- F. Pop, C. Melan, I. Danila, M. Linares, D. Beljonne, D. B. Amabilino and N. Avarvari, Chem. – Eur. J., 2014, 20, 17443–17453 CrossRef CAS PubMed.
- C. Oliveras-González, M. Linares, D. B. Amabilino and N. Avarvari, ACS Omega, 2019, 4, 10108–10120 CrossRef PubMed.
- T.-L. Lai, F. Pop, C. Melan, D. Canevet, M. Sallé and N. Avarvari, Chem. – Eur. J., 2016, 22, 5839–5843 CrossRef CAS PubMed.
- A. Gainar, T.-L. Lai, C. Oliveras-González, F. Pop, M. Raynal, B. Isare, L. Bouteiller, M. Linares, D. Canevet, N. Avarvari and M. Sallé, Chem. – Eur. J., 2021, 27, 2410–2420 CrossRef CAS PubMed.
- M. D. Gujrati, N. S. S. Kumar, A. S. Brown, B. Captain and J. N. Wilson, Langmuir, 2011, 27, 6554–6558 CrossRef CAS PubMed.
- M.-Y. Yeh and H.-C. Lin, Phys. Chem. Chem. Phys., 2014, 16, 24216–24222 RSC.
- L. S. Kaczmarek and P. Nantka-Namirski, Acta Pol. Pharm., 1979, 36, 629–634 CAS.
- J. Brinksma, B. L. Feringa, R. M. Kellogg, R. Vreeker and J. van Esch, Langmuir, 2000, 16, 9249–9255 CrossRef CAS.
- C. R. Martinez and B. L. Iverson, Chem. Sci., 2012, 3, 2191 RSC.
- T. Choisnet, D. Canevet, M. Sallé, E. Nicol, F. Niepceron, J. Jestin and O. Colombani, Chem. Commun., 2019, 55, 9519–9522 RSC.
- Differential scanning calorimetry experiments were led up to T = bp-20 K. No significant thermal event was observed within this temperature range.
- S. Basak, N. Nandi, S. Paul and A. Banerjee, ACS Omega, 2018, 3, 2174–2182 CrossRef CAS PubMed.
- P. Lasitha, ChemistrySelect, 2021, 6, 7936–7943 CrossRef CAS.
- A. Aliprandi, M. Mauro and L. De Cola, Nat. Chem., 2016, 8, 10–15 CrossRef CAS PubMed.
- S. Onogi, H. Shigemitsu, T. Yoshii, T. Tanida, M. Ikeda, R. Kubota and I. Hamachi, Nat. Chem., 2016, 8, 743–752 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tc01799g |
| ‡ Contributed equally to this work. |
| § The gelation properties of C3Pyr were depicted in ref. 69 and 70. |
| This journal is © The Royal Society of Chemistry 2022 |