
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLinking interfacial work of deformation from deconvoluted macro-rheological spectrum to early stage healing in selected polyurethanes†
Vincenzo
Montano
 *,
Michele
Senardi
,
Sybrand
van der Zwaag
and
Santiago J.
Garcia
*,
Michele
Senardi
,
Sybrand
van der Zwaag
and
Santiago J.
Garcia
Novel Aerospace Materials group, Faculty of Aerospace Engineering, Delft University of Technology, Kluyverweg 1, 2629 HS, Delft, The Netherlands. E-mail: v.montano@tudelft.nl
First published on 17th September 2020
Abstract
The use of rheology and terminal flow relaxation times to predict healing behavior at long healing times is by now quite well accepted. In this work we go one step further and explore the use of macro-rheology (in particular the stored work of deformation) to predict the early stage interfacial healing properties (fracture resistance) of a set of self-healing polyurethanes. The interfacial healing is measured by single edge notch fracture experiments, using short healing times and a low healing temperature to exclude the effect of long range molecular motion on mechanical properties restoration. The systems based on aromatic diisocyanates show high fracture resistance after healing, while very limited restoration of the mechanical properties is observed for aliphatic and cycloaliphatic based polyurethanes. Linear sweep rheology and time-temperature-superposition allow obtaining the macro-rheological master curve and the mechanical relaxation spectra (H(t)). The application of a recently established deconvolution protocol to the H(t) gives the characteristic relaxation times and stored works of deformation associated to individual dynamic processes such as segmental motion, reversible bonds, and terminal flow. It is found that the calculated stored works of deformation related to the reversible bond relaxation reproduce the trend observed by fracture resistance at healed interfaces and reveal a qualitative correspondence between reversible bonds work of deformation and interfacial healing fracture resistance. Moreover, the method seems to point to the existence of a threshold interfacial work of deformation below which no efficient load transfer can be observed.
1. Introduction
The field of self-healing polymers has rapidly grown in recent times driven by increasing environmental concerns related to plastic overconsumption that boost the scientific research towards the development of alternatives to current commodities polymers, including functional and self-healing systems.Many proofs of concepts have been presented exploiting the intrinsic healing approach. Intrinsic healing is addressed by the molecular design of the network in opposition to extrinsic systems which rely on the present of discretely dispersed carriers containing the healing agent.1,2 In order to exploit the intrinsic self-healing ability two requirements have to be fulfilled: (i) high density of reversible bonds to deconstruct and reconstruct the broken interface and (ii) sufficient chain mobility to facilitate local flow at the damage site. A number of reviews summarize how the incorporation of different reversible chemical and physical bonds can successfully lead to the production of self-healing polymers.3–6 A careful analysis of these works evidences that the use of van der Waals,7,8 π–π stacking9 and/or microphase separation10 could be used to turn commodity-like systems into self-healing variants without the introduction of severe chemical modifications.
The healing action in intrinsic systems occurs in two successive stages: initial interface restoration by reversible bonds swapping and subsequent interphase healing by macromolecular diffusion.11 In this work we aim to clarify the contribution of non-covalent reversible bonds to the initial recovery of interfacial strength. In a previous study we showed evidences that the incorporation of multiple reversible moieties (namely hydrogen bonding and disulphide exchange) with different association/dissociation kinetics strongly influence the early steps of healing, in which fast hydrogen bonding relaxation dynamics determines initial tack, and affect long term healing, when the opening of sulphur–suplhur bonds allows long range motion.12 This issue is critical because it defines the design boundaries between systems with a very high density of reversible bonds but a sluggish terminal relaxation and polymers with fast terminal relaxation but a sparse physical network. Macro-rheology has been used to estimate the timescale of interphase diffusion and macroscopic crack closure by comparing the kinetics of these processes to the supramolecular bond lifetime (τb = 2π/ωcr) of embedded reversible interactions.7,13 However, this method does not offer insights on the processes that govern the initial interface strength restoration. In this work we try to bridge this gap and explore how to extract more information from the rheological tests that can be used to predict early stage macroscopic healing strength. To do this we extend our deconvolution protocol of the mechanical relaxation spectrum constructed by rheology that allow to isolate the dynamics of individual molecular features,14 including the calculation of the stored work of deformation.
We explore the use of this approach to predict early stage interfacial healing properties of a set of polyurethanes, for which the recombination of reversible bonds at the deconstructed interface defines the initial tack. To this purpose, we use four diisocyanates (diphenylmethane 4,4′ diisocynate MDI, 1,4-phenylene diisocyanate PPDI, hexamethylene diisocyanate HDI, 4,4′-methylenebiscyclohexyl isocyanate HMDI) of increasing aromaticity to selectively incorporate multiple reversible interactions in the network (hydrogen bonding from the urethane linkage and aromatic interactions from the isocyanates) and to study the effect of the monomer symmetry on the fracture resistance restoration using the single edge notch tensile (SENT) protocol.15–17
2. Experimental section
2.1. Materials
2-Ethyl-1,3-hexanediol (EHD, 99%), diphenylmethane 4,4′ diisocynate (MDI, 98%), 1,4-phenylene diisocyanate (PPDI), hexamethylene diisocyanate (HDI, >98%), 4,4′-methylenebis(cyclohexyl isocyanate) (HMDI, mixture of isomers 90%), dibutyltin dilaurate (DBTDL, 95%) were purchased from Sigma-Aldrich. CroHeal 2000 (equivalent molecular weight 2000 g mol−1) was kindly provided from Croda Nederland B.V.2.2. Synthesis of segmented polyurethanes
Four polyurethanes were synthesized by single shot technique reacting CroHeal 2000, EHD and different diiscoyanates (MDI, PPDI, HDI, HMDI). The diisocyanates were selected considering increasing level of aromaticity HMDI = HDI < PPDI < MDI, and symmetry HMDI ≅ MDI < PPDI ≅ HDI. For all the polymers the molar ratio of the hydroxyl chemical functionalities OH (CroHeal2000)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :OH (EHD) was kept fixed at 1:0.6. 10% molar excess of NCO was used to carry the reaction to full completion. Samples are coded by the name of the isocyanate used followed by the addition ‘-p’ to distinguish the polymer from the monomer (e.g. MDI based polymer is coded as MDI-p). Below we report the synthesis of polymer MDI-p. Analogous procedures were followed for the other polymers in this study. Details on their syntheses are reported in the ESI.† The molecular structure of the segmented polyurethanes is schematically represented in Fig. 1.
:OH (EHD) was kept fixed at 1:0.6. 10% molar excess of NCO was used to carry the reaction to full completion. Samples are coded by the name of the isocyanate used followed by the addition ‘-p’ to distinguish the polymer from the monomer (e.g. MDI based polymer is coded as MDI-p). Below we report the synthesis of polymer MDI-p. Analogous procedures were followed for the other polymers in this study. Details on their syntheses are reported in the ESI.† The molecular structure of the segmented polyurethanes is schematically represented in Fig. 1.
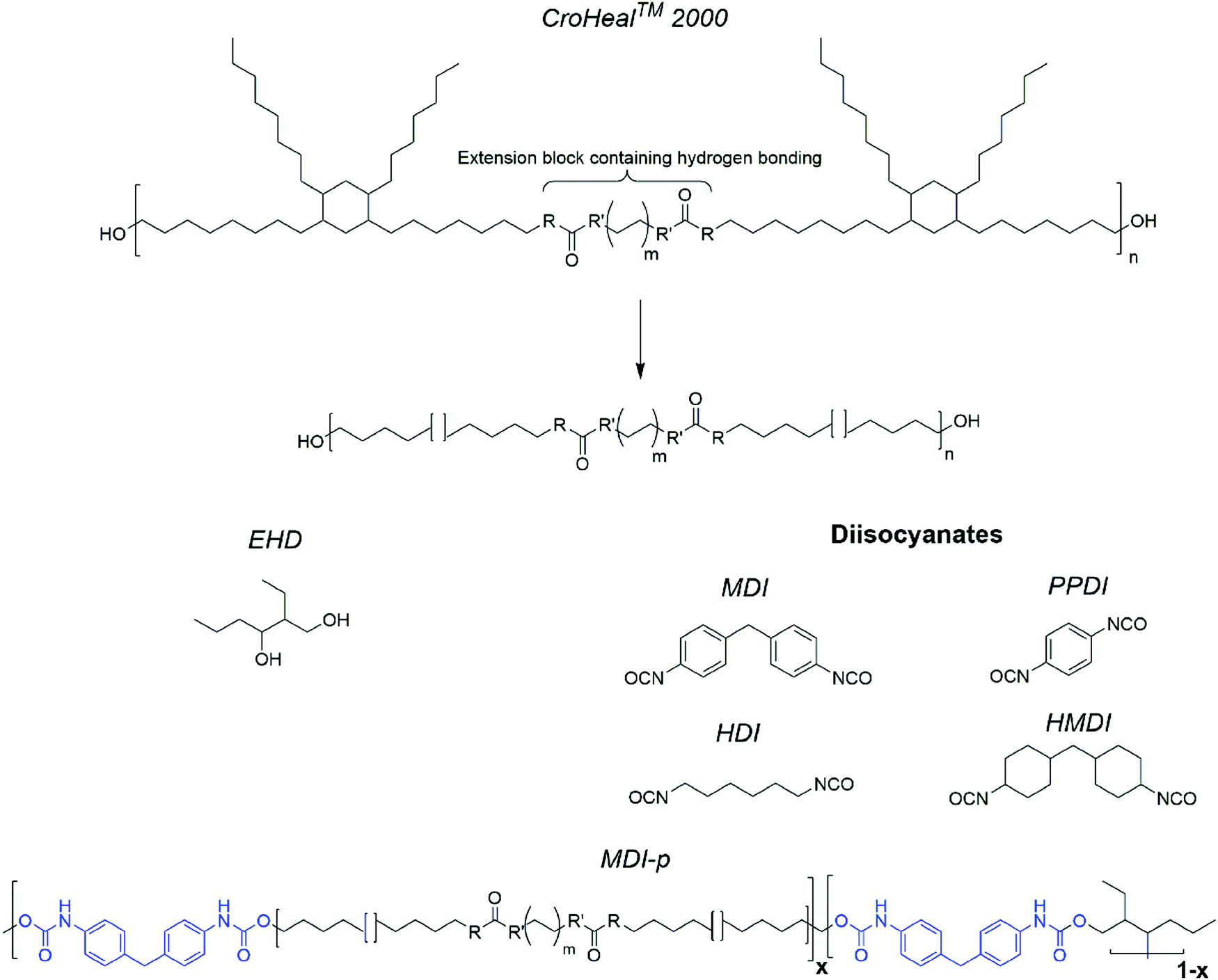 | ||
| Fig. 1 Monomers used and molecular structure of synthesized segmented polyurethanes (MDI-p is showed as mode of example). | ||
CroHeal 2000 was heated for 1 hour at 90 °C to reduce the intrinsic viscosity of the monomer (melting the crystalline domains). Subsequently 90.0 g (45.0 mmol) were transferred to a 300 mL polypropylene cup. 3.95 g of EHD (27.0 mmol) were then added to the cup. 31.64 g (79.20 mmol) of MDI were weighted in a separate 25 mL polypropylene cup, purged under nitrogen inert environment and heated 1 hour at 60 °C to reduce its intrinsic viscosity (melting crystalline domains). The MDI was rapidly poured into the main reaction cup (containing CroHeal 2000 and EHD). The mixture was sheared under vacuum at 2300 rpm for 135 seconds using a vacuum assisted high speed mixer (SpeedMixerTM DAC 400.2 VAC-P). The application of high vacuum (pressure below 100 mbar) turned out to be critical for the formation of bubble-free polymers. The mixture was then transferred to a 20 × 20 × 0.2 cm PTFE mold, equilibrated for 30 minutes at ambient conditions and subsequently cured overnight at 60 °C. The obtained bulk polymers were then equilibrated for 1 week at ambient conditions before testing.
The degree of completion of the polyurethane synthesis was monitored by Attenuated Total Reflectance Fourier transform Infrared (ATR-FTIR) analysis by monitoring the appearance of the characteristic carbonyl stretching absorption peak at ≈1700 cm−1 and the disappearance of isocyanate absorption peak at about ≈2270 cm−1. ATR-FTIR spectra of all synthesized polymers are shown in Fig. S1 (ESI†).
2.3. Characterization methods
The mechanical relaxation spectrum H(t) was constructed applying the non-linear regression method developed by Honerkamp and Weese.18,19 The source code of the algorithm can be found in the CPC Program library.20 In this protocol, the minimum and maximum relaxation times are pre-selected by the user. In our case we simply set  and
and  where ωmax and ωmin are the highest and the lowest angular frequency in the modulus data, as suggested in the library user manual. The deconvolution protocol was performed following the procedure reported in ref. 14. For all the deconvoluted spectra the coefficients of determination of the reconstructed mastercurve (|rG′2| and |rG′′2|) exceed 0.9 confirming the high quality of the fit. Details of the protocol, including the calculation of stored work of deformation (W) are reported in the ESI.†
where ωmax and ωmin are the highest and the lowest angular frequency in the modulus data, as suggested in the library user manual. The deconvolution protocol was performed following the procedure reported in ref. 14. For all the deconvoluted spectra the coefficients of determination of the reconstructed mastercurve (|rG′2| and |rG′′2|) exceed 0.9 confirming the high quality of the fit. Details of the protocol, including the calculation of stored work of deformation (W) are reported in the ESI.†
The mechanical tests were performed at a cross-head speed of 0.5 mm s−1, allowing detailed observation of crack tip opening. The testing temperature was controlled using an environmental chamber (Instron) with a glass window using evaporating liquid nitrogen and was set at 10 °C. Optical snapshots at a frequency of 10 Hz were taken during testing using an Optomotive Velociraptor camera in order to observe crack opening and propagation. The camera was placed on a vibration damping tripod at ≈50 cm of distance from the sample.
The fracture properties were determined using the J-integral analysis method, as described elsewhere.12,15,21 Critical fracture resistance values, JC were calculated as:

The healed samples were mechanically tested following the SENT procedure described in Section 2.3.3 to obtain a value for the critical fracture resistance after healing JinterfacialC.
Table 1 reports the results of TGA analyses and the DSC-Tg (glass transition). All polymers reported a high thermal stability (5% weight loss at T > 350 °C).
| Polymer | TGA-5% weight loss (°C) | DSC-Tg (°C) |
|---|---|---|
| MDI-p | 353 | 5.2 |
| PPDI-p | 361 | 10.0 |
| HMDI-p | 353 | −5.5 |
| HDI-p | 344 | 0.4 |
All systems showed a single endothermic peak in the DSC curve located between −5 and 10 °C which is attributed to the main chain segmental mobility. The relatively low content of EHD monomer feed is responsible for the absence of a high temperature first order thermal transition, generally attributed to the melting of the hard segment (chain extender + isocyanate).22,23
Principal component analysis (PCA) was applied to identify spectral heterogeneities. The PCA is based on the generation of 8 principal spectral scores. The analysis was performed using the built-in function of the ImageSpectra software (PerkinElmer).
3. Results and discussion
3.1 Deconvolution protocol to unveil individual relaxations and related work of deformation
The first step to establish the relationship between the dynamics and energy storage of individual relaxation processes and the interfacial healing fracture resistance is the application of the deconvolution protocol to the mechanical relaxation spectrum following previous reports.14The resulting deconvoluted H(t) with identified individual time constants are shown in Fig. 2 for the aromatic-based polyurethanes (MDI-p and PPDI-P) and in Fig. 3 for the aliphatic-based polyurethanes (HDMI-p, HDI-p). Starting from a known polymer architecture the method allows to assign physical meaning to the relaxation mechanism identified through the deconvolution. Fig. 2A shows that five dominant relaxation mechanisms are obtained for MDI-p. Polyurethanes synthesized from a mixture of a high molecular weight diol (polyol), a low molecular weight diol (chain extender) and a diisocyanate may present a segmented structure. The segmented structure is physically described as hard phase regions, enriched with chain extender + diisocyanate segments, intercalated in a soft matrix, enriched of polyol + diisocyanate segments.24,25 The phase separated architecture is responsible for the elastomeric properties of this class of material, where the hard phase works as stable physical crosslinks of the polymer network. These preliminary considerations are essential to infer the physical meaning of the five relaxation mechanisms deconvoluted for MDI-p. The first dynamic event (R1) is associated to main chain segmental motion occurring in the soft matrix of the segmented polyurethane microstructure. As inferred from the molecular structure of this system, reversible bonds (hydrogen bonding and aromatic π–π stacking) are present in the soft phase. They are responsible of the second relaxation event (R2). The transition part of the spectrum (103 s < τ < 105 s) is dominated by relaxation of the hard blocks dispersed in the segmented microstructure. Hard blocks segmental motion (R3) is followed by hydrogen bonds and aromatic π–π stacking relaxations (R4). Thermoplastic elastomers present a strong but transient crosslinked network, therefore terminal relaxation can be observed but is very sluggish (R5). The elastomeric nature of MDI-p is confirmed by its mechanical behaviour, tested by SENT in the pristine state and shown as auxiliary data in Fig. 2A. Analogous arguments follow the deconvolution of PPDI-p (Fig. 2B) which shows five dominant relaxation mechanisms and mechanical features typical of a thermoplastic elastomer.
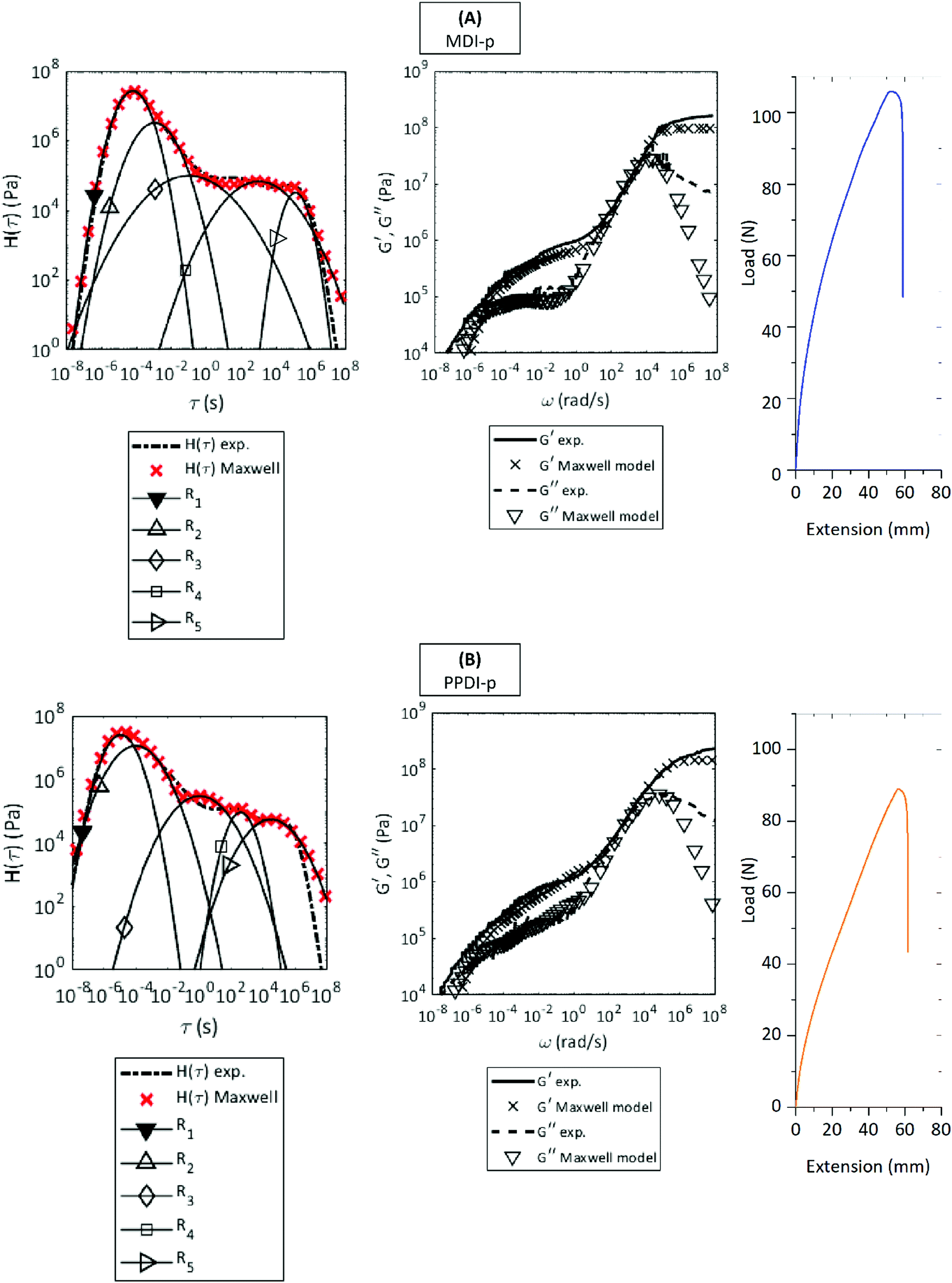 | ||
| Fig. 2 Deconvoluted relaxation spectra, mastercurve reconstruction and SENT load–displacement data of MDI-p and PPDI-p. (A) MDI-p (B) PPDI-p. For both polymers the method identifies five dominant relaxation mechanisms. R1 = soft phase segmental mobility. R2 = soft phase reversible bonds disjunction. R3 = hard phase segmental mobility. R4 = hard phase reversible bonds disjunction. R5 = terminal relaxation. Mechanical properties are typical of thermoplastic elastomers. The mastercurves are shifted at the reference temperature TR = 30 °C. | ||
 | ||
| Fig. 3 Deconvoluted relaxation spectra, mastercurve reconstruction and SENT results of HMDI-p and HDI-p. (A) HMDI-p. The deconvolution method identifies three dominant relaxation mechanisms. R1 = main chain segmental mobility. R2 = reversible bonds disjunction. R3 = terminal relaxation. (B) HDI-p. The deconvolution method identifies three dominant relaxation mechanisms. R1 = main chain segmental mobility. R2 and R3 = reversible bonds disjunction and quasi-crystalline melting. R4 = terminal relaxation. The mechanical properties are typical of thermoplastic polymers. | ||
The deconvolution procedure applied to HMDI-p results in a radically different outcome (Fig. 3A). In this case only three main relaxation mechanisms can be identified with a complete disappearance of the wide transient region previously observed for MDI-p and PPDI-p. The deconvolution spectrum resembles the one of lightly crosslinked systems reported elsewhere.14 The first dynamic process is attributed to main chain segmental motion (R1), followed by a temporary restriction of mobility associated to entanglements and/or physical interactions (R2) (e.g. hydrogen bonds) and rapid terminal relaxation (R3). The absence of segmented structure and rubbery elasticity for HMDI-p is confirmed from static mechanical properties (auxiliary data in Fig. 3A). Moreover, extensional DMA data for HMDI-p (Fig. S4, ESI†) show a complete absence of the rubbery plateau and terminal flow occurring at ≈60 °C, a typical feature of linear unentangled/lightly entangled networks.9,26 A similar description applies to the deconvolution of HDI-p, for which an optimal fit is found when assuming four relaxation mechanisms (Fig. 3B). The additional relaxation mechanism located in the transient regime is related to the stacking of highly linear and symmetric HDI segments that determines crystalline-like ordered structures.27 Nevertheless in analogy with HMDI-p, because of no aromatic entities being present there is no segmented microstructure. In parallel with HMDI-p, the mechanical SENT data of the pristine HDI-p sample shows the typical behaviour of linear networks and appearance of terminal flow in DMA analysis at about ≈100 °C (Fig. S5, ESI†).
Table 2 shows the quantitative data extracted from the deconvolution protocol in terms of kinetics of the relaxation transition (τ) and stored work of deformation (W). The information regarding the physical nature of the individual relaxation mechanisms is conveyed through the parameters subscript and superscripts: Wsegsp (soft phase segmental mobility) Wrevsp (soft phase reversible bonds) Wsephp (hard phase segmental mobility) Wrevhp (hard phase reversible bonds) Wt (terminal relaxation). Winterfacial corresponds to the sum of all the works of deformation associated to reversible ligands relaxation (Winterfacial = Wrevsp + Wrevhp). This choice is based on the assumption that segmental motion/short range conformational re-arrangements do not contribute to the reconstruction of the broken interface. MDI-p and PPDI-p present comparable soft and hard segmental relaxation timescales. The slightly lower values for the kinetic parameters of PPDI-p are attributed to the higher rigidity and symmetry of the PPDI diisocyanate monomer when compared to MDI. The work of deformation related to reversible bonds in the soft phase (Wrevsp) is higher for PPDI-p. Such an increase can be explained considering the rigid segment design and in particular to the reduced periodic distance among reversible bonding moieties (aromatic units and urethane linkages).
| τ segsp (s) | τ revsp(s) | τ seghp (s) | τ revhp (s) | τ t (s) | W segsp (kJ m−3) × 10−1 | W revsp (kJ m−3) × 10−1 | W seghp (kJ m−3) × 10−1 | W revhp (kJ m−3) × 10−1 | W t (kJ m−3) × 10−1 | W interfacial (kJ m−3) × 10−1 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| MDI-p | 6.2 × 10−5 | 1.1 × 10 −5 | 0.1 | 1.0 × 10 3 | 1.3 × 105 | 579 | 120 | 9.6 | 12.9 | 3.4 | 132.9 |
| PPDI-p | 1.1 × 10−5 | 1.0 × 10 −4 | 1.0 | 0.3 × 10 3 | 3.1 × 104 | 481 | 407 | 25.1 | 6.5 | 11.8 | 413.5 |
| HMDI-p | 2.3 × 10−5 | 1.0 × 10 −3 | — | — | 0.92 | 512 | 73.1 | — | — | 19.5 | 73.1 |
| HDI-p | 1.0 × 10−6 | 1.0 × 10 −4 | — | 0.10 | 1.0 × 103 | 356 | 23.6 | — | 12.2 | 1.5 | 35.8 |
Beyond the reduced number of relaxation mechanisms obtained by the deconvolution process, HMDI-p deviates from MDI-p and PPDI-p in its rapid terminal relaxation kinetics (τt ≅ 1 s) and the reduced interfacial work of deformation (Winterfacial), an expected result when considering the absence of π–π stacking interactions among aromatics and therefore the formation of a weaker transient network. For the second aliphatic polyurethane (HDI-p), as qualitatively discussed in the previous paragraph, the slower terminal relaxation (τt ≅ 103 s) when compared to HMDI-p is attributed to HDI monomer stacking and the strong chemical compatibility between HDI monomer and the CroHeal 2000 segment, which yields a denser transient network (higher chain packing and friction). Nevertheless, the two aliphatic systems maintain comparable levels of Winterfacial indicating that the physically crosslinked networks constituted by fully aliphatic chains are able to store similar amount of energy related to reversible interactions.
The effect of the diisocyanate chemistry on the macromolecular segmentation in turn responsible for microphase separation in polyurethanes was further investigated by dedicated μATR-FTIR analysis. Fig. 4 reports the infrared spectra maps obtained upon application of principal component analysis of the four polyurethanes. The aromatic-based polyurethanes (MDI-p and PPDI-p) show a continuous phase (reported in grey color) intercalated by a micron-sized dispersed phase (yellow color). The aliphatic and linear polyurethane (HDI-p) presents a homogeneous microstructure (no dispersed phase is probed), while HMDI-p shows some phase separation, yet to a lower extent and smaller dimension than in the case of MDI-p and PPDI-p. The analysis of the infrared spectra traces corresponding to the continuous (points numbered as ‘1’ in Fig. 4) and the dispersed phases (points numbered as ‘2’ in Fig. 4) gives insights into the molecular differences between the two probed regions. All shifts and intensity variations point towards a microstructure composed of a soft continuous phase (enriched in CroHeal + MDI segments) and hard and urea-rich heterogeneities (enriched in EHD + MDI segments). For MDI-p the carbonyl band (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) probed for the continuous phase at 1695 cm−1 shifts to 1643 cm−1 due to increase in bidentate urea ligands and aromatic conjugation.28 The N–H band located at 3324 cm−1 broadens and shifts to 3274 cm−1 indicating a stronger hydrogen bonding state. The aliphatic C–H bands at 2852 and 2919 cm−1 undergo a strong decrease in intensity due to the scarcity of highly aliphatic branch in the hard heterogeneities. The C–O–C stretch peak loses intensity and shifts from 1220 cm−1 to 1234 cm−1 as result of loss of urethane linkages in favor of formation of urea units. The C–H stretch in the aromatic rings clearly visible for the soft phase at 1600 cm−1, only appear as a shoulder of the intense and broader main carbonyl peak in the hard phase spectrum. For HDI-p no substantial shift is observed when scanning through the probed infrared map. Only a slight shoulder of the carbonyl CO peak located at 1680 cm−1 appears at about 1630 cm−1 as a sign of a possible increase in the hydrogen bonding strength in the small (size < 2 μm) yellow-grey regions. HMDI-p presents similar spectral feature when compared to MDI-p and PPDI-p, yet the band associated to the C–O–C stretch at 1253 cm−1 does not undergo any shift nor decrease in intensity suggesting a minimal difference in the urethane linkages content. The higher content in bidentate urea and the stronger hydrogen bonded state of the segregated phase might be responsible for a slower molecular reorganization which, in turn, determines a local higher Tg.29
O) probed for the continuous phase at 1695 cm−1 shifts to 1643 cm−1 due to increase in bidentate urea ligands and aromatic conjugation.28 The N–H band located at 3324 cm−1 broadens and shifts to 3274 cm−1 indicating a stronger hydrogen bonding state. The aliphatic C–H bands at 2852 and 2919 cm−1 undergo a strong decrease in intensity due to the scarcity of highly aliphatic branch in the hard heterogeneities. The C–O–C stretch peak loses intensity and shifts from 1220 cm−1 to 1234 cm−1 as result of loss of urethane linkages in favor of formation of urea units. The C–H stretch in the aromatic rings clearly visible for the soft phase at 1600 cm−1, only appear as a shoulder of the intense and broader main carbonyl peak in the hard phase spectrum. For HDI-p no substantial shift is observed when scanning through the probed infrared map. Only a slight shoulder of the carbonyl CO peak located at 1680 cm−1 appears at about 1630 cm−1 as a sign of a possible increase in the hydrogen bonding strength in the small (size < 2 μm) yellow-grey regions. HMDI-p presents similar spectral feature when compared to MDI-p and PPDI-p, yet the band associated to the C–O–C stretch at 1253 cm−1 does not undergo any shift nor decrease in intensity suggesting a minimal difference in the urethane linkages content. The higher content in bidentate urea and the stronger hydrogen bonded state of the segregated phase might be responsible for a slower molecular reorganization which, in turn, determines a local higher Tg.29
 | ||
| Fig. 4 μATR-FTIR mapping of self-healing polyurethane elastomers. Principal component analysis reveals a phase separated microstructure consisting in a continuous matrix (grey color) intercalated by heterogeneities (yellow color). Right figures show the local IR spectra of locations 1 (homogeneous) and 2 (dispersoids). | ||
3.2. Interfacial work of deformation relation to early-stage healing fracture resistance
The deconvolution results were not only used to obtain information on the physical state of the network. Starting from the hypothesis that interfacial healing is governed by the reconstruction of reversible bonds at the broken interface and using SENT data collected for all the polymers in the early stage of healing, we explore the existence of a relationship between the interfacial work of deformation (obtained by melt rheology) and fracture resistance (a mechanical measure of interfacial healing obtained by SENT analysis). All the mechanical raw data and optical snapshots used for fracture resistance calculation are reported in the ESI† (Fig. S6 MDI-p, Fig. S7 PPDI-p, Fig. S8 HDMI-p and Fig. S9 HDI-p). Fig. 5 displays the relation between the calculated interfacial work of deformation (Winterfacial) from rheology and JinterfacialC from the SENT tests of the polymers healed for 3.6 × 103 s (1 h) at 30 °C. A closer look to the data (Table 2) reveals that higher Winterfacial of the PPDI-p and MDI-p samples is mainly related to the higher work of deformation assigned to the relaxation of the reversible ligands (concurrent relaxation of hydrogen bonds and aromatics) in the soft phase (Wrevsp). Since the soft phase undergoes fast conformational re-arrangements, its reversible bonds reshuffling are expected to be the main contributors to the reconstruction of the broken interface at short healing times.10,11,30 In this framework the use of the deconvolution protocol offers a critical advance to the understanding and optimization of interfacial healing, since its results are successfully sensitive to the incorporation of multiple reversible ligands, their symmetry and their energetic contribution to the reconstruction of the broken interface. | ||
| Fig. 5 Comparison between interfacial healing fracture resistance and interfacial work of deformation as obtained by macro-rheology. While qualitatively an increase in Winterfacial corresponds to higher JinterfacialC, from a quantitative point of view the work of deformation does not capture the dramatic drop of fracture resistance for the aliphatic polyurethanes (HDI-p, HMDI-p). HDI-p point data does not include error bars due to immediate breakage of the samples tested upon application of the minimal sample alignment load of 3 N. | ||
Quantitatively, for short healing times (3600 seconds at 30 °C), both the aliphatic systems (HMDI-p and HDI-p) show a virtual absence of tensile fracture resistance (JinterfacialC HMDI-p = 0.18 kJ m−2, JinterfacialC HDI-p = 0.03 kJ m−2), as can be easily inferred from optical snapshots acquired during SENT testing (Fig. S8 and S9, ESI†). In this sense the data provided by macro-rheology seems to point to the existence of a limiting Winterfacial that must be surpassed to get sufficient recovery of fracture resistance upon interfacial healing. However, more systems, ideally incorporating different types and numbers of reversible ligands, should be tested in a wide range of Winterfacial to confirm and locate the presence of such threshold.
Another possible explanation to absence of fracture resistance for HMDI-p and HDI-p at the early stage of healing can be found in the characteristics of the different polyurethanes microstructures. The aromatic polymers present a segmented microstructure and, as probed by μFTIR-ATR, show some high Tg segregated heterogeneities at the microscale (Fig. 4 and Table S1, ESI†). The high Tg of these segregates implies, locally, a much slower reorganization and an improved directionality of the reversible ligands. This effect, when occurring at a damage site, leads to a more efficient interfacial re-bonding/healing. The lack of such structures in the aliphatic polymers causes a dramatic drop of JinterfacialC. In a recent work Yang et al.31 show similar beneficial effects of micro-segregated structures on scratch-healing behavior of PU coatings, attributing the healing enhancement to a more efficient entropic energy storage during local deformation. Furthermore, Chen et al.10 have shown that a multiphase microstructure in self-healing thermoplastic elastomers is crucial to the rapid re-arrangement of the polymer soft phase leading to high healing.
Long range dynamics is expected to determine interphase randomisation and to lead the system to higher degree of property restoration at longer healing times. The deconvolution procedure provides kinetics of terminal relaxation in the range τt = 100 to 103 s for HDI-p and HMDI-p and very sluggish terminal flow for MDI-p and PPDI-p (τt > 105 s). Fig. 6 shows a quantitative impression of the effect of long term healing on the polyurethane systems used for this study. The aliphatic polymers show a significant recovery of fracture resistance going from being virtually absent for short healing times (3.6 × 103 seconds) to a moderate mechanical stability at longer healing times (after 106 seconds JC HMDI-p = 11.4 kJ m−2, JC HDI-p = 1.8 kJ m−2). An immediate feedback regarding the increase in fracture properties when going from short to longer healing times can be obtained when looking at the optical snapshots acquired during SENT testing and used for JC calculation (Fig. S8 and S9, ESI†), in which a higher deformation can be seen before fracture at the healed interface. Moreover, in accordance with kinetics predictions obtained by rheology deconvolution, MDI-p and PPDI-p do not show any significant improvement in the fracture resistance which remains virtually constant over long holding times at the healing temperature of 30 °C.
 | ||
| Fig. 6 Long range dynamics effect on fracture resistance recovery. In accordance with time constants obtained from deconvolution, the beneficial effect of long range dynamics is only observed for aliphatic polyurethanes (HDI-p, HMDI-p). Healing temperature is 30 °C. | ||
4. Conclusions
In this work we demonstrate that the intelligent use of macro-rheology data can provide semi-quantitative information on the degree of interfacial healing in polymers. In this framework, we apply a deconvolution protocol of the mechanical relaxation spectrum derived from macro-rheology to a set of self-healing polyurethanes and explore the relation between the derived stored work of deformation and the degree of mechanical restoration in the early stage of polymer healing.The deconvolution protocol is found to be sensitive to the variations in the polymer architecture and in particular to the addition of multiple reversible ligands, to the symmetry of the rigid backbone segments and to their individual energetic contribution to the relaxation process.
The work of deformation associated to the disjunction of reversible moieties reflects in a semi-quantitative way the recovery of fracture resistance in the early stage of healing. It results higher for polymers endowed of multiple reversible ligands that coherently show a higher degree of mechanical restoration. The results points at the existence of a so far unidentified threshold of interfacial work of deformation marking the transition between healing (i.e. sufficient reversible bonds at interface to transfer load along the fracture plane) and no healing. Furthermore, the time constants capture the kinetics of the entire healing process, predicting the access to long range motion for less physically crosslinked systems.
These insights on the relation between polymer structure, rheology and interfacial healing benefit the design of novel systems showing high mechanical restoration in the early stage of healing.
Conflicts of interest
The authors report no conflicts of interest.Acknowledgements
The authors acknowledge the financial support of Croda Nederland BV and the Dutch National Organization for Scientific Research, Domain Applied and Engineering Sciences (NWO-TTW) under the grant number 15010. The authors acknowledge Dr Angela Smits and Dr Wouter Vogel (Croda) for helpful discussions and valuable feedbacks.References
- D. Y. Zhu, M. Z. Rong and M. Q. Zhang, Prog. Polym. Sci., 2015, 49–50, 175–220 Search PubMed.
- S. J. Garcia, Effect of polymer architecture on the intrinsic self-healing character of polymers, Eur. Polym. J., 2014, 53, 118–125 Search PubMed.
- M. D. Hager, P. Greil, C. Leyens, S. van der Zwaag and U. S. Schubert, Self-healing materials, Adv Mater., 2010, 22, 5424–5430 Search PubMed.
- S. J. García, H. R. Fischer and S. Van Der Zwaag, A critical appraisal of the potential of self healing polymeric coatings, Prog. Org. Coat., 2011, 72, 211–221 Search PubMed.
- Y. Yang, X. Ding and M. W. Urban, Prog. Polym. Sci., 2015, 49–50, 34–59 Search PubMed.
- A. Campanella, D. Döhler and W. H. Binder, Macromol. Rapid Commun., 2018, 39, 1700739 Search PubMed.
- A. Susa, R. K. Bose, A. M. Grande, S. van der Zwaag and S. J. Garcia, Effect of the Dianhydride/Branched Diamine Ratio on the Architecture and Room Temperature Healing Behavior of Polyetherimides, ACS Appl. Mater. Interfaces, 2016, 8, 34068–34079 Search PubMed.
- V. Montano, M. M. B. Wempe, S. M. H. Does, J. C. Bijleveld, S. van der Zwaag and S. J. Garcia, Controlling Healing and Toughness in Polyurethanes by Branch-Mediated Tube Dilation, Macromolecules, 2019, 52, 8067–8078 Search PubMed.
- A. Susa, J. Bijleveld, M. Hernandez Santana and S. J. Garcia, Understanding the Effect of the Dianhydride Structure on the Properties of Semiaromatic Polyimides Containing a Biobased Fatty Diamine, ACS Sustainable Chem. Eng., 2018, 6, 668–678 Search PubMed.
- Y. Chen, A. M. Kushner, G. A. Williams and Z. Guan, Multiphase design of autonomic self-healing thermoplastic elastomers, Nat. Chem., 2012, 4, 467–472 Search PubMed.
- Y. H. Kim and R. P. Wool, A Theory of Healing at a Polymer Polymer Interface, Macromolecules, 1983, 16, 1115–1120 Search PubMed.
- A. M. Grande, R. Martin, I. Odriozola, S. van der Zwaag and S. J. Garcia, Effect of the polymer structure on the viscoelastic and interfacial healing behaviour of poly(urea-urethane) networks containing aromatic disulphides, Eur. Polym. J., 2017, 97, 120–128 Search PubMed.
- R. K. Bose, N. Hohlbein, S. J. Garcia, A. M. Schmidt and S. van der Zwaag, Connecting supramolecular bond lifetime and network mobility for scratch healing in poly(butyl acrylate) ionomers containing sodium, zinc and cobalt, Phys. Chem. Chem. Phys., 2015, 17, 1697–1704 Search PubMed.
- V. Montano, S. J. Picken, S. van der Zwaag and S. J. Garcia, A deconvolution protocol of the mechanical relaxation spectrum to identify and quantify individual polymer feature contributions to self-healing, Phys. Chem. Chem. Phys., 2019, 21, 10171–10184 Search PubMed.
- A. M. Grande, S. J. Garcia and S. van der Zwaag, On the interfacial healing of a supramolecular elastomer, Polymer, 2015, 56, 435–442 Search PubMed.
- F. Maes, D. Montarnal, S. Cantournet, F. Tournilhac, L. Corté and L. Leibler, Activation and deactivation of self-healing in supramolecular rubbers, Soft Matter, 2012, 8, 1681–1687 Search PubMed.
- A. M. Grande, J. C. Bijleveld, S. J. Garcia and S. Van Der Zwaag, A combined fracture mechanical - rheological study to separate the contributions of hydrogen bonds and disulphide linkages to the healing of poly(urea-urethane) networks, Polymer, 2016, 96, 26–34 Search PubMed.
- J. Honerkamp and J. Weese, A nonlinear regularization method for the calculation of relaxation spectra, Rheol. Acta, 1993, 32, 65–73 Search PubMed.
- I. McDougall, N. Orbey and J. M. Dealy, Inferring meaningful relaxation spectra from experimental data, J. Rheol., 2014, 58, 779–797 Search PubMed.
- T. Roths, M. Marth, J. Weese and J. Honerkamp, A generalized regularization method for nonlinear ill-posed problems enhanced for nonlinear regularization terms, Comput. Phys. Commun., 2001, 279–296 Search PubMed.
- G. Ramorino, S. Agnelli, R. De Santis and T. Riccò, Investigation of fracture resistance of natural rubber/clay nanocomposites by J-testing, Eng. Fract. Mech., 2010, 77, 1527–1536 Search PubMed.
- A. Eceiza, M. D. Martin, K. De La Caba, G. Kortaberria, N. Gabilondo, M. A. Corcuera and I. Mondragon, Thermoplastic polyurethane elastomers based on polycarbonate diols with different soft segment molecular weight and chemical structure: Mechanical and thermal properties, Polym. Eng. Sci., 2008, 48, 297–306 Search PubMed.
- Z. Yang, H. Peng, W. Wang and T. Liu, Crystallization behavior of poly(ε-caprolactone)/layered double hydroxide nanocomposites, J. Appl. Polym. Sci., 2010, 116, 2658–2667 Search PubMed.
- I. Yilgör, E. Yilgör and G. L. Wilkes, Critical parameters in designing segmented polyurethanes and their effect on morphology and properties: A comprehensive review, Polymer, 2015, 58, A1–A36 Search PubMed.
- J. P. Sheth, D. B. Klinedinst, G. L. Wilkes, I. Yilgor and E. Yilgor, Role of chain symmetry and hydrogen bonding in segmented copolymers with monodisperse hard segments, Polymer, 2005, 46, 7317–7322 Search PubMed.
- T. Aida, E. W. Meijer and S. I. Stupp, Functional supramolecular polymers, Science, 2012, 335, 813–817 Search PubMed.
- R. L. McKiernan, S. P. Gido and J. Penelle, Synthesis and characterization of polyethylene-like polyurethanes derived from long-chain, aliphatic α,ω-diols, Polymer, 2002, 43, 3007–3017 Search PubMed.
- P. J. Woodward, D. H. Merino, B. W. Greenland, I. W. Hamley, Z. Light, A. T. Slark and W. Hayes, Hydrogen bonded supramolecular elastomers: Correlating hydrogen bonding strength with morphology and rheology, Macromolecules, 2010, 43, 2512–2517 Search PubMed.
- N. Luo, D. N. Wang and S. K. Ying, Hydrogen-bonding properties of segmented polyether poly(urethane urea) copolymer, Macromolecules, 1997, 30, 4405–4409 Search PubMed.
- R. P. Wool and K. M. O’Connor, A theory of crack healing in polymers, J. Appl. Phys., 1981, 52, 5953–5963 Search PubMed.
- Y. Yang, D. Davydovich, C. C. Hornat, X. Liu and M. W. Urban, Leaf-Inspired Self-Healing Polymers, Chem, 2018, 4, 1928–1936 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cp03776a |
| This journal is © the Owner Societies 2020 |