
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Novel green fabrication of stable hydrogel beads from industrial waste lignin for efficient Pb(II) ion removal†
Xiwen
Hu
ab,
Muhammad Amirul
Islam
*ab,
Aria
Khalili
ab,
Amir
Aghaei
ab,
Jae-Young
Cho
b and
Mohtada
Sadrzadeh
 *a
*a
aDepartment of Mechanical Engineering, 10-367 Donadeo Innovation Center for Engineering, Advanced Water Research Lab (AWRL), University of Alberta, Edmonton, AB T6G 1H9, Canada. E-mail: sadrzade@ualberta.ca; maislam@ualberta.ca
bQuantum and Nanotechnologies Research Centre, National Research Council Canada, 11421 Saskatchewan Drive, Edmonton, AB T6G 2M9, Canada. E-mail: Muhammad.Islam@nrc-cnrc.gc.ca
First published on 8th January 2025
Abstract
The adsorption of water contaminants using renewable biopolymer hydrogel beads represents an environmentally friendly and economically viable solution for pollution control. Traditional methods for preparing these beads are often not green, are slow, and lack versatility in producing beads with specific biopolymer compositions. We utilized a novel, green and efficient approach using liquid nitrogen for the instantaneous cooling of solution droplets, facilitating the rapid formation of gelatin–lignin biopolymer beads. This process enables the production of beads from low-concentration solutions with various lignin-to-gelatin ratios, which is challenging with other techniques. The rapid cooling results in beads with a thin shell and a highly porous gel network, enhancing their stability under harsh chemical conditions. These beads exhibit superior lead (Pb(II)) ion adsorption performance, maintaining stability across 10 adsorption–regeneration cycles in both acidic and basic environments. The optimal bead composition of 0.45 lignin/0.55 gelatin achieved approximately 155 mg g−1 Pb(II) ion adsorption. The optimal beads exhibited a 3.5-fold higher adsorption capacity than gelatin-only beads, which disintegrated after three cycles, highlighting lignin's crucial role in enhancing bead stability and adsorption performance. The results further demonstrated that a dosage of 0.1 g was sufficient to achieve 96% removal of Pb(II) from 50 mL of solution with an initial concentration of 100 mg L−1. The long-term stability and mechanical integrity of the beads were further enhanced through amide and ester crosslinking of lignin and gelatin using EDC as a zero-length crosslinker. A concentration of 1 mM EDC improved the adsorption capacity by 32% compared to the non-crosslinked beads over 10 adsorption–regeneration cycles. The influence of fabrication methods and swelling–deswelling cycles on bead morphology was analyzed using advanced cryo-SEM imaging. This innovative approach offers a more efficient, sustainable, and versatile method for producing high-value hydrogel beads from industrial waste byproducts.
Sustainability spotlightIn a breakthrough for sustainable water treatment, we present a novel method for fabricating hydrogel beads from industrial lignin waste, showcasing a green, rapid, and scalable solution to heavy metal pollution. Utilizing liquid nitrogen for instantaneous cooling, the study pioneers a zero-waste process that transforms sulfonated kraft lignin—a pulp industry byproduct—into highly porous, stable adsorbents capable of removing toxic Pb(II) ions from water. These gelatin–lignin beads achieve a remarkable adsorption capacity of 155 mg g−1 while maintaining performance across 10 regeneration cycles in challenging acidic and basic environments. The method aligns with circular economy principles by upcycling lignin, minimizing environmental impact, and advancing efficient pollutant mitigation strategies. This work highlights the potential of integrating renewable resources and green chemistry for sustainable water treatment applications, offering transformative solutions for industrial and municipal wastewater management. |
1. Introduction
The increasing levels of toxic heavy metal lead (Pb) in water bodies – stemming from improper battery disposal, paint scraps, mining and refining waste, and aging pipes – pose significant risks to ecosystems, organisms, and human health.1,2 Pb ions pose significant health risks, including neurological and digestive damage in adults and developmental, cognitive, and physical impairments in children.3 Consequently, the maximum acceptable lead concentration in drinking water is set at 5 μg L−1 by Health Canada and 10 μg L−1 by both the European Union directives and World Health Organization (WHO).4Common treatment methods for Pb(II) ion removal include chemical precipitation and coagulation, membrane filtration, electrochemical methods, and adsorption.5,6 Among these methods, adsorption stands out due to its cost-effectiveness, low energy consumption, ability to operate at lower concentrations, and lack of toxic byproducts, and is often biocompatible.7,8
In adsorption-based separation processes, one commonly used method involves polymers with functional groups capable of ionic or coordination interaction. Sulfonated kraft lignin (SKL), an abundant byproduct from the pulp industry, is particularly effective for Pb(II) ion adsorption due to its diverse functional groups, such as phenolic hydroxyl (–OH), carboxylic (–COOH), sulfonic (–SO3H), and methoxy (–OCH3) groups.9,10 These groups actively participate in the adsorption process via ionic and/or coordination interaction with Pb(II) ions, thereby increasing the efficiency of Pb(II) ion removal from contaminated water. Moreover, SKL's functional groups allow further modification and integration with other polymers or nanomaterials, creating composite adsorbents with expanded applications.11 It is also highly chemically and thermally stable since it is recovered as a stable byproduct after harsh chemical (alkali, sodium sulfide, and sulfuric acid) and thermal treatment (steam cooking) in pulp and paper processing.12,13 The renewable and cost-effective nature of SKL, along with its waste-reducing potential and high stability, make it ideal for large-scale, sustainable adsorption applications. However, to ensure the successful deployment of SKL-based adsorption, other key factors, such as adsorption efficiency, recyclability, and process feasibility, must be enhanced. This can be achieved by optimizing physical properties (e.g., surface area), facilitating adsorbent regeneration, and designing suitable adsorption setups (e.g., batch vs. column adsorption).
Hydrogels, known for their high surface area, moldability, and functional groups, are particularly suited for enhancing adsorption efficiency.14 Bead-shaped hydrogels offer additional advantages, such as improved surface area, ease of separation, and flexibility in batch and column adsorption processes.15 Gelatin (GE) is a natural, biocompatible polymer that provides a stable matrix for forming SKLGE hydrogel beads. This protein-based natural polymer is water soluble, can mold into spherical beads by simply changing temperature, and provides a larger surface area for adsorption. GE is self-adhesive, offering good stability, and its abundant functional groups (e.g., amine, carboxylic acid, amide) interact strongly with the functional groups on SKL, stabilizing the SKLGE hydrogel's porous structure.16 The branched structure of SKL provides enhanced physical crosslinking between SKL and GE, improving the mechanical and chemical stability of the SKLGE beads. The abundance of polar functional groups on GE also enhances the SKLGE bead's capacity for Pb(II) ion adsorption.
The method for gel bead formation should be cost-effective, versatile, reproducible, relatively straightforward, and eco-friendly.17 The most common technique for preparing hydrogel beads is extrusion, where the hydrogel solution is extruded dropwise through a syringe or pipette into the gelation medium. Beads form via physical or chemical crosslinking in the gelation medium between the polymers and fillers.18 Physical crosslinking is preferred over chemical crosslinking, as it is simpler and avoids using costly or toxic chemicals. However, physically crosslinked gel beads may lack stability during adsorption and regeneration. Therefore, selecting suitable hydrogel components and fabrication methods is essential to create stable beads without chemical crosslinking.
Physical crosslinking often involves dripping the hydrogel solution droplets into a medium that contains metal ions (e.g., Ca2+), with different pH or temperature, or is made of anti-solvent.19,20 For GE beads, the most straightforward method is cooling hot droplets in cold water or oil.21 However, lower SKL and GE concentrations (ca. 10 wt%) cause the polymer film to spread across the water surface due to rapid mixing, and hydrogel droplets coalesce if injected into the oil. Higher-concentration solutions, however, become too viscous for easy extrusion. In an alternative method, using DMSO as the solvent and methanol as the gelation medium for lignin/sericin bead formation, low concentrations lead to SKLGE film precipitation, while higher concentrations (20 wt%) create bulk gel in DMSO.22
We hypothesized that hydrogel droplets could be effectively formed by injecting them into a non-polar, low-density, low-freezing-point oil (e.g., kerosene). This would prevent water in the hydrogel from mixing with the cooling medium, allowing the droplets to sink and solidify. However, cooling and gelation was too slow, limiting scalability. To address this, we utilized liquid nitrogen (LN2) as a rapid cooling medium, which instantaneously solidifies droplets as they sink, enabling a rapid fabrication process. LN2 also offers an environmentally friendly option as it vaporizes into harmless N2 gas, maintaining green fabrication from the gelator material choice to fabrication method. One of the prominent aspects of LN2-induced cooling is the formation of highly porous gel beads with less porous, thin, and compact skin.23,24 The rapid and steep temperature gradient creates numerous ice crystals, and the temperature-driven Marangoni effect drives fluid flow from warmer (bead interior) to colder regions (bead exterior), evenly redistributing hydrogel materials onwards and creating a dense skin layer.25 The high porosity of the interior gel enhances the accessibility and surface area of the adsorbent for Pb(II) ion capture, while the dense outer skin layer improves bead stability due to increased SKL and GE concentration and stronger interactions in this region.
This study reports on LN2-induced hydrogel bead formation, emphasizing its advantages in achieving rapid and stable bead fabrication with a protective skin layer. The SKLGE hydrogel beads were characterized using cryo-SEM, FTIR, XPS and TGA. Their Pb(II) adsorption performance was evaluated through batch adsorption experiments, including adsorption equilibrium, adsorbent dosage, Pb(II) ion concentration studies, adsorption kinetics, and isotherm models. Stability and recyclability were assessed over ten adsorption–regeneration cycles. Finally, bead stability was further enhanced through post-fabrication zero-length crosslinking using EDC/NHS coupling, a biocompatible method widely used in biomedical applications.
2. Experimental methods
2.1 Materials
Sulfonated Kraft Lignin (SKL), with a molecular weight of 5–8 kDa, was provided by West Fraser Mills Ltd. (British Columbia, Canada). To purify the material, 100 g of SKL at a concentration of 50 g L−1 was mixed for 48 hours and sonicated for 15 minutes before centrifuging at 5000 rpm for 60 minutes. The samples were then dried in an 80 °C oven and ground into a powder. Type A gelatin (GE) was purchased from Sigma Aldrich with a molecular weight of 50–100 kDa. Lead nitrate (Pb(NO3)2) was purchased from Anachemia Canada Inc. and dissolved in Milli-Q water to make the stock metal solution, with the pH adjusted to 6 using 0.1 M sodium hydroxide (NaOH), provided by Fisher Scientific. Other chemicals, including 4-(2-pyridylazo)resorcinol (PAR), disodium tetraborate (Na2B4O7·10H2O), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC·HCl, C8H17N3·HCl), N-hydroxysuccinimide (NHS, C4H5NO3) and N,N-diisopropylethylamine (DIPEA, [(CH3)2CH]2NC2H5) were purchased from Sigma Aldrich. All the reagents were of reagent grade and were used as received. Deionized (DI) water from a Milli-Q water purification system was used to prepare solutions throughout the experiments.2.2 Fabrication of SKLGE beads
SKLGE beads with variable ratios of SKL to GE were prepared by rapid cooling of solution droplets injected from a heated syringe into LN2. To fabricate SKL45 beads with 45% SKL and 55% GE, 0.9 g of SKL and 1.1 g of GE were separately dissolved in two 9 mL portions of water by stirring for 30 minutes at 60 °C, which were sealed with parafilm. Next, the SKL solution was added to the GE solution and stirred for an additional 30 minutes at the same temperature. Meanwhile, a 50 mL glass syringe was wrapped with a temperature-controlled heating pad, covered with cotton wool for insulation, and clamped vertically on a stand with the needle facing downward. The injection syringe, equipped with a 20G needle, was preheated to 80 °C for 30 minutes before transferring the SKLGE mixed solution into it. The solution was injected dropwise into the LN2, which was placed in a 2 L dewar. The remaining LN2 was left to evaporate from the beads in the fume hood, and they were allowed to dry overnight at room temperature. Finally, the beads were transferred to a vial and subjected to high vacuum drying for 24 h to remove any residual water, then stored in a desiccator to prevent humidity until use. The SKLGE beads with other SKL to GE ratios were prepared using the same process, as detailed in Table S1.† A schematic of the fabrication process of SKLGE beads is provided in Fig. S1.†To create EDC/NHS cross-coupled beads, additional steps were integrated into the original fabrication process. The beads were stirred in the EDC/NHS solution for 24 hours at room temperature and 50 rpm to enable cross-coupling, as specified in Table S2.† Afterward, the beads were rinsed five times with Milli-Q water, followed by high vacuum drying for 24 hours. The cross-coupled beads were then stored in a desiccator to prevent them from absorbing moisture until further use.
2.3 Characterization of SKLGE beads
2.4 Adsorption experiments
The adsorption of Pb(II) ions using SKLGE beads was tested by preparing stock solutions of Pb(NO3)2 in DI water at various concentrations. Each stock solution was adjusted to pHs of 2, 3, 4, 5, 6, and 7 using 0.1 N HNO3 and 0.1 M NaOH to study the effect of pH on Pb(II) ion adsorption. To avoid interference from Pb(II) hydroxide precipitation, a pH of 6 was chosen as optimal for the adsorption experiments.26 The dry beads (e.g., 0.05 g) were weighed and placed in a 100 mL round glass jar. After adding 50 mL of Milli-Q water, the beads were allowed to swell in the solution for 24 hours to remove any unbound or excess chemicals. Once the swelling was complete, the beads were gently rinsed three times with Milli-Q water. They were then treated with 20 mL of 0.1 M NaOH for 20 minutes, followed by thorough rinsing with Milli-Q water. Fresh Milli-Q water (50 mL) was added, and the beads were left to sit overnight. The next day, the water was discarded, and the beads were rinsed again with Milli-Q water prior to the adsorption test.For the batch adsorption test, 50 mL of Pb(NO3)2 solution was added to the beads, and the mixture was stirred at 75 rpm for 4 hours. At the end of the experiment, 10 mL of the solution was collected in a glass vial for UV-Vis analysis, and the remaining solution was disposed of in the appropriate waste container. All adsorption experiments were conducted at room temperature, except for the thermal stability test, where experiments were carried out at 40 °C, 50 °C, and 60 °C.
The adsorption capacity (Qe, mg g−1) and removal efficiency (E, %) were calculated using eqn (1) and (2):
 | (1) |
 | (2) |
2.5 Analyzing Pb(II) ion concentration
The concentration of Pb(II) ions was analyzed using a UV-Vis method, which involved the following steps:The calibration curve solution was prepared across a concentration range of 0 to 5 mg L−1, as shown in Fig. S2.† The same preparation method was applied for testing solutions, with only the Pb(II) concentration adjusted as needed. To ensure accurate measurements, the absorbance value of the blank (0 mg L−1) was subtracted to correct the background interference.
| A = ε × c × l | (3) |
2.6 Desorption and regeneration experiments
After each adsorption test, excess Pb(II) solution was poured into the appropriate waste container, and the Pb(II)-loaded beads were thoroughly rinsed three times with Milli-Q water. Desorption was performed by adding 20 mL of 0.1 M HNO3 and stirring at 75 rpm for 5 hours. The beads were then treated with 20 mL of 0.1 M NaOH for 20 minutes. Thorough rinsing with Milli-Q water was conducted after each step. After rinsing, 50 mL of DI water was added, and the beads were left to soak overnight to allow rearrangement. The next adsorption cycle began the following day. This adsorption–regeneration procedure was repeated for 10 consecutive cycles. A schematic of the adsorption–regeneration cycles is provided in Fig. S3.†3. Results and discussion
3.1 Bead fabrication
To fabricate SKLGE beads with an optimal spherical morphology, a comprehensive evaluation of various methodologies was undertaken, including the injection of heated hydrogelator solution droplets into water and oil,20,29 as well as the introduction of heated gelator solution droplets in DMSO into methanol,22 as outlined in Table S4.† However, none of these methodologies worked for our SKLGE bead compositions. For example, while gelatin beads could be readily formed by injecting hot gelatin solution droplets into the water at a 20 wt% concentration, a lower wt% resulted in mixing gelatin with water. Attempting to inject SKLGE solutions with concentrations exceeding 10 wt% proved challenging, as the high viscosity caused the solution to extrude from the needle as fibers rather than discrete droplets. At lower wt% concentrations, the water in the SKLGE solution was mixed with the water used as a cooling medium, resulting in a film of SKLGE floating on the surface of the gelation medium. While lignin and sericin beads were successfully prepared by injecting a 20 wt% solution in DMSO into a methanol gelation medium, SKLGE at 20 wt% formed a gel in DMSO that could not be transferred into the injection syringe, and lower wt% solutions again resulted in film formation over methanol.23 The most widely employed method, involving injection into oil, led to droplet coalescence.In our final hypothesis, we sought to engineer precise temperature, density, and miscibility gradients between the injected solution droplet and the gelation medium. This strategic methodology aimed to preserve the spherical shape of the droplets through controlled immiscibility between the two liquid phases while simultaneously accelerating gelation as the denser droplets sank into the colder gelation medium. The approaches explored included injecting hot solution droplets into room-temperature kerosene, freezer-cold kerosene, and LN2, as detailed in Table S4.† Among these, LN2 emerged as the most effective medium for forming spherical droplets from the 60 °C SKLGE polymer solution, owing to its ability to induce a rapid and uniform temperature drop—essential for achieving the desired bead morphology (Fig. 1). Other methods were ineffective due to insufficient cooling rates and challenges, such as premature fusion of the beads before solidification, which compromised the consistency, reproducibility, and scalability of the process. Additionally, LN2-induced gelation offered advantages beyond rapid fabrication, including the development of an enhanced porous interior and dense protective skin on the beads, as noted by Dey et al. in their efforts to enhance hydrogel porosity via LN2 cooling and lyophilization.24 To assess the impact of cooling rate on porosity and skin layer thickness, cryo-SEM images were acquired of beads formed under varying cooling conditions in LN2, freezer-cold kerosene, and room-temperature kerosene, as shown in Fig. 2a.
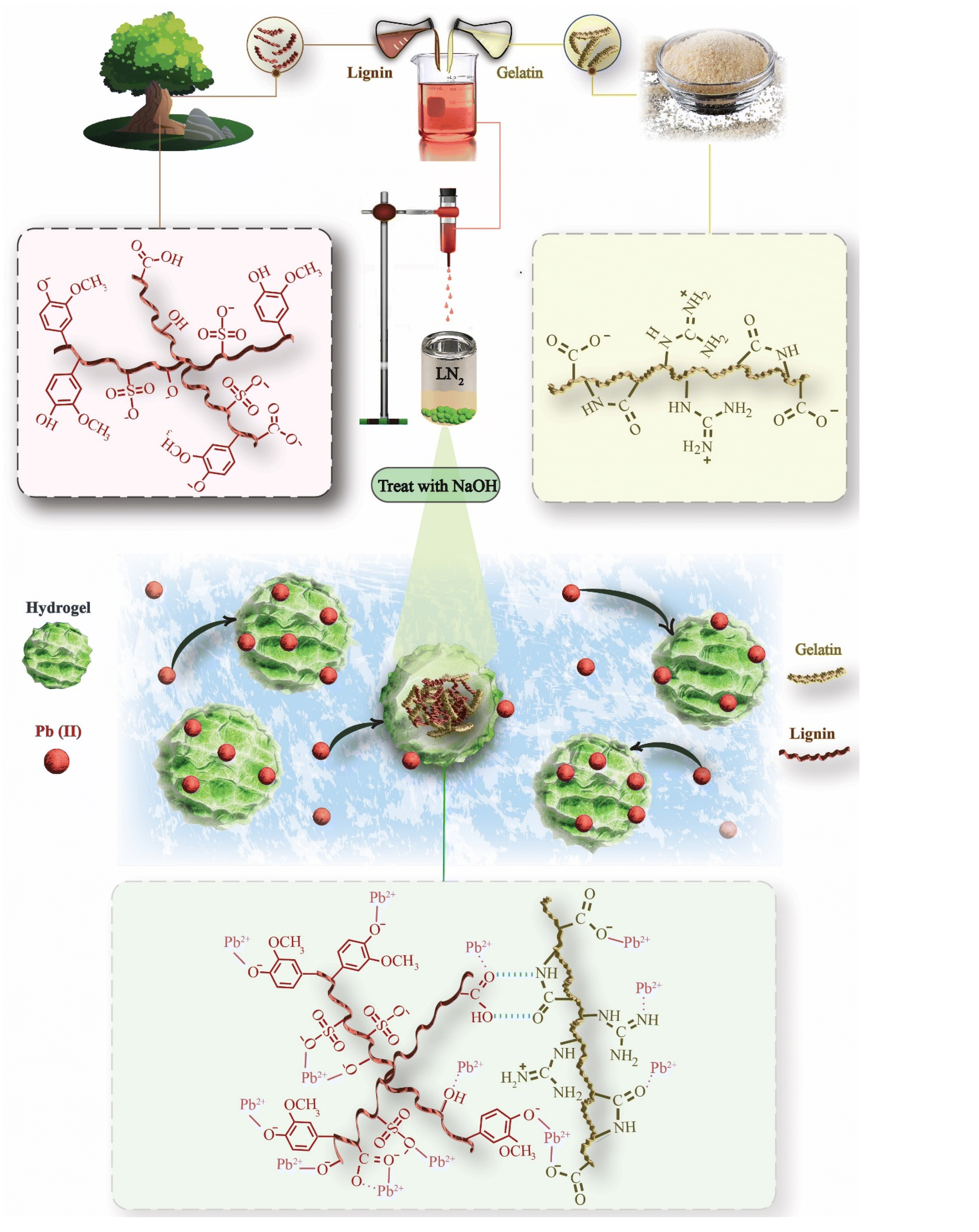 | ||
| Fig. 1 Conceptual illustration of the SKLGE bead fabrication process and the mechanism of Pb(II) adsorption. | ||
 | ||
| Fig. 2 (a) Effect of gelling medium and temperature on the porosity and skin layer formation of the SKLGE beads and (b) impact of SKL ratio on swelling of SKLGE beads, showing SKL hydrophilicity before and after Pb(II) adsorption. | ||
When comparing polymer distribution in SKLGE beads subjected to LN2versus freezer-cooled kerosene, a significant difference in shell thickness is evident. Beads cooled in LN2 show a shell thickness of 2.6 μm, while those cooled in freezer-cold kerosene exhibit a 2.1 μm shell, marking a 23.8% increase in the LN2-treated beads. Additionally, LN2-cooled beads display significantly reduced porosity both at the core and surface. Rapid injection of a 60 °C hydrogel solution into liquid nitrogen (−196 °C) creates a steep temperature gradient between the warmer droplet interior and colder exterior. This difference induces a Marangoni effect, where fluid flows from the warmer, lower surface tension interior to the colder, higher surface tension exterior, redistributing hydrogel materials like water and polymer chains towards the bead's surface.25 As these components rapidly freeze, a dense, solid outer shell forms, while the bead's interior becomes porous due to ice crystal formation and material depletion.
At −20 °C, freezer-cooled kerosene has a slower cooling rate, causing the hydrogel's viscosity to increase gradually and limit fluid flow from the bead's interior to its surface, leading to a less dense outer shell. Although −20 °C is sufficient to freeze the water within the beads, preserving the spherical integrity of the injected droplet and forming water crystal-induced porous structures, the slower fluid redistribution rate compared to the LN2 case results in larger and less uniform pores. Conversely, at room temperature, gelation proceeds at a sluggish pace due to the insufficient temperature gradient. Although the hydrophobic nature of kerosene temporarily confines the water within the injected droplet and the density of the hydrogel causes the semi-liquid bead to settle at the bottom of the kerosene, leaching of water from the semi-liquid gel bead leaves behind a densely polymerized structure. This room-temperature cooling method fails to produce the desired porous interior or a distinct skin layer on the bead. As previously noted, the skin layer serves as a crucial protective barrier, enhancing the stability of hydrogel beads during recycling processes.
In summary, the methodologies explored for SKLGE bead fabrication faced significant challenges when using conventional gelation media, such as water, oil, and methanol, primarily due to insufficient cooling rates and miscibility issues. Solutions dispersed in water or methanol resulted in film formation rather than bead formation, while oil-based methods led to droplet coalescence before solidification. Even with kerosene cooled to −20 °C, the cooling rate was inadequate to produce beads with scalability, uniform morphology and protective skin layers, and the process generated significant kerosene waste. Considering factors such as fabrication speed, bead shape uniformity, reproducibility, interior porosity, and dense protective skin layer formation, the LN2-based fabrication method was deemed optimal. After gelation, the beads were dried at room temperature within a fume hood, where the high laminar airflow facilitated the removal of water as mist along with LN2, promoting enhanced interaction between SKL and GE polymer chains within the dried and compact structure (Fig. 1), imparting additional stability. It is important to note that high vacuum drying induces lyophilization, preventing gel bead shrinkage and mitigating the interaction between SKL and GE polymer chains.
3.2 Optimization of the SKL to GE ratio
The ratios of lignin to gelatin were systematically varied to optimize the SKLGE bead formulation. This optimization aimed to maximize the number of functional groups (e.g., phenolic OH, carboxyl, sulfonate, and ethers) for Pb(II) ion adsorption and to enhance swelling in water, thereby increasing access to adsorption sites by utilizing the maximum amount of SKL (Fig. 1) while ensuring bead stability, primarily governed by the gelatin component. The SKL ratios were incrementally adjusted from 2.5% to 7.5%, resulting in samples designated SKL25 through SKL75, each maintaining a total weight percentage of 10%. Regardless of the SKL to GE ratio, all formulations successfully formed beads through the LN2 cooling method. Post-fabrication swelling tests revealed that beads with higher SKL content exhibited significantly greater swelling, as shown in Fig. 2b. The high hydrophilicity of SKL, due to its abundant ionic and polar functional groups, enhances water absorption. This enhanced swelling is advantageous as it facilitates increased accessibility to adsorption sites for Pb(II) ions. This effect is qualitatively evidenced by the compaction of the beads following Pb(II) ion adsorption. Initially, the beads with high SKL content were significantly larger; however, they compacted to a size comparable to or smaller than that of beads with lower SKL45 content. This compaction results from the Pb(II) ions occupying more ionic and polar sites (Fig. 1), reducing the beads' hydrophilicity and, consequently, their swelling.Challenges emerged with samples having higher SKL content. Starting from SKL50, increased viscosity caused flow issues, leading to solidification at the needle tip and blockages during the injection process. The increased viscosity of the SKLGE solution with increasing SKL content reflects stronger interactions between SKL and GE, driven by ionic and hydrogen bonding interactions between the polar functional groups on both components (Fig. 1). However, SKL60, and SKL75 beads demonstrated deformation and partial fusion during drying and dissolved in water during the swelling phase before Pb(II) ion adsorption experiments. This instability resulted from excessive hydrophilicity and insufficient gelatin to maintain the gel matrix. Hence, five bead formulations (SKL25, SKL30, SKL35, SKL40, and SKL45) were selected for further analysis to assess their performance and stability regarding Pb(II) ion adsorption and regeneration cycles.
3.3 FTIR characterization of GE10, SKL powder, and SKL45 bead
The FTIR spectrum of the representative SKL45 bead was recorded and compared with those of the GE10 bead (gelatin-only bead) and SKL powder, as presented in Fig. 3. This analysis aimed to elucidate the presence of SKL and GE components in the SKLGE beads and to investigate the interactions among the functional groups, including hydrogen bonding and ionic interactions. The FTIR spectra of SKL powder reveal characteristic absorption bands: 3650–3150 cm−1 corresponding to the highly hydrogen-bonded O–H stretching of phenols and aliphatic alcohols; 1580 cm−1 for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of carboxylate groups and CC stretching of aromatic rings; 1263 cm−1 for guaiacyl ring breathing coupled with C–O stretching; 1210 cm−1 for C–C stretching alongside C–O and CO stretching; 1127 cm−1 for SO stretching of sulfonate groups; and 1030 cm−1 for C–O stretching related to ether linkages.11,30 The FTIR spectra of the GE10 bead reveal characteristic gelatin amide bands: amide A (N–H stretching) at 3285 cm−1, amide I (CO stretching) at 1629 cm−1, amide II (N–H bending) at 1518 cm−1, and amide III (C–N stretching) at 1232 cm−1.31 Notably, the frequencies of the amide I and amide II bands at 1629 cm−1 and 1518 cm−1, respectively, indicate the presence of a β-sheet secondary structure stabilized by intermolecular hydrogen bonding.32 A striking difference is observed for the FTIR spectra of SKL45 compared to those for SKL powder and GE10. The characteristic SKL bands at 1263 cm−1, 1127 cm−1, and 1030 cm−1 observed in the FTIR spectra of SKL45 beads indicate the inclusion of SKL into the GE matrix. The CO band at 1580 cm−1, associated with carboxylate groups on SKL, merged with the amide I CO band of GE, significantly enhancing its intensity relative to the N–H band. Since both CO and N–H bands originate from the same amide structure (–HN–CO), the increased intensity of the CO band for SKL45 beads suggests a contribution from the CO group of SKL. The FTIR bands for amide A, amide I, amide II, and amide III were observed to shift to longer wavenumbers at 3302 cm−1, 1647 cm−1, 1534 cm−1, and 1236 cm−1, respectively. The shifts of the amide I and amide II bands to 1647 cm−1 and 1534 cm−1 suggest the presence of a random coil structure.32,33 The observed shifts in all amide bands and the transition from a β-sheet to a random coil structure indicate interactions between SKL and GE, weakening intermolecular hydrogen bonding among GE chains.33
O stretching of carboxylate groups and CC stretching of aromatic rings; 1263 cm−1 for guaiacyl ring breathing coupled with C–O stretching; 1210 cm−1 for C–C stretching alongside C–O and CO stretching; 1127 cm−1 for SO stretching of sulfonate groups; and 1030 cm−1 for C–O stretching related to ether linkages.11,30 The FTIR spectra of the GE10 bead reveal characteristic gelatin amide bands: amide A (N–H stretching) at 3285 cm−1, amide I (CO stretching) at 1629 cm−1, amide II (N–H bending) at 1518 cm−1, and amide III (C–N stretching) at 1232 cm−1.31 Notably, the frequencies of the amide I and amide II bands at 1629 cm−1 and 1518 cm−1, respectively, indicate the presence of a β-sheet secondary structure stabilized by intermolecular hydrogen bonding.32 A striking difference is observed for the FTIR spectra of SKL45 compared to those for SKL powder and GE10. The characteristic SKL bands at 1263 cm−1, 1127 cm−1, and 1030 cm−1 observed in the FTIR spectra of SKL45 beads indicate the inclusion of SKL into the GE matrix. The CO band at 1580 cm−1, associated with carboxylate groups on SKL, merged with the amide I CO band of GE, significantly enhancing its intensity relative to the N–H band. Since both CO and N–H bands originate from the same amide structure (–HN–CO), the increased intensity of the CO band for SKL45 beads suggests a contribution from the CO group of SKL. The FTIR bands for amide A, amide I, amide II, and amide III were observed to shift to longer wavenumbers at 3302 cm−1, 1647 cm−1, 1534 cm−1, and 1236 cm−1, respectively. The shifts of the amide I and amide II bands to 1647 cm−1 and 1534 cm−1 suggest the presence of a random coil structure.32,33 The observed shifts in all amide bands and the transition from a β-sheet to a random coil structure indicate interactions between SKL and GE, weakening intermolecular hydrogen bonding among GE chains.33
 | ||
| Fig. 3 FTIR analysis of SKL powder, GE10, and SKL45 as the representative bead. | ||
3.4 Effect of pH on adsorption
To evaluate the effect of pH on the Pb(II) ion adsorption capacity of the beads, Pb(II) adsorption was assessed using 100 mg L−1 Pb(II) and 0.05 g of SKL45 beads across a pH range of 2–7, as pH values above 7 lead to the precipitation of Pb(II) as Pb(OH)2, potentially affecting the adsorption results.26 The effect of pH on Pb(II) adsorption and zeta potential measurements reveals a consistent relationship between surface charge dynamics and adsorption efficiency. As shown in Fig. S4,† lower adsorption capacity was observed under acidic conditions, which can be attributed to the reduced negative surface charge of the beads. At lower pH levels, protonation of functional groups limited adsorption by reducing the availability of –COO− groups and introducing competition between H+ ions and Pb(II) for binding sites. This behavior was further supported by the zeta potential measurements (Fig. S5†), which showed a lower negative surface charge at lower pH levels and positive surface charge below pH 3.97 (i.e., pHzpc). As the pH increased, fewer H+ ions were present, allowing the carboxyl groups to deprotonate to –COO−, which attracted and bound Pb(II) more effectively. A higher negative zeta potential at pH 6 (−21.2 mV) supports improved adsorption efficiency, highlighting the hydrogel's suitability for Pb(II) removal in near neutral pH environments.26 At pH 7, the adsorption capacity suddenly increased due to the interference caused by the precipitation of Pb(OH)2, rather than actual adsorption by the beads.26 Therefore, to accurately evaluate the adsorption capacity without this interference, further experiments were conducted at pH 6.3.5 Examining the bead pretreatment and regeneration process
This study aimed to maximize Pb(II) adsorption by optimizing the use of SKL while ensuring the mechanical stability of the beads during operation and facilitating ease of fabrication. Among the materials tested, SKL45 best aligned with these goals, making it the ideal candidate for evaluating NaOH pretreatment and regeneration with HNO3 and NaOH. The NaOH pretreatment is crucial for negatively ionizing the functional groups, enhancing the binding of positively charged Pb(II) ions. Additionally, post-regeneration NaOH treatment re-ionizes these groups following HNO3 exposure. However, prolonged acid or base exposure can degrade the beads, which rely on physical interactions like ionic and hydrogen bonding. Thus, the volumes of 0.1 M NaOH and 0.1 M HNO3 and treatment durations were optimized for stability and effectiveness.Initially, 0.05 g of dried beads was soaked in 50 mL of Milli-Q water for 24 hours, followed by treatment with 20, 30, or 40 mL of 0.1 M NaOH. The beads were then subjected to Pb(II) ion adsorption from 50 mL of a 100 mg L−1 solution in a 4 hour batch adsorption study at 75 rpm. The regeneration step for the second and third cycle adsorption studies involved treating the Pb(II)-adsorbed beads with 20 mL of 0.1 M HNO3 for 3 hours at 75 rpm, followed by ionization with 20, 30, or 40 mL of 0.1 M NaOH.
Adsorption capacity was determined by measuring initial and 4 hour concentrations via UV-Vis spectroscopy. Results from three consecutive cycles (Fig. S6a†) showed that beads treated with 0.1 M NaOH consistently achieved higher adsorption capacity than untreated beads, with 20 mL NaOH yielding the highest performance across all cycles. Consequently, this volume was selected to study the effect of treatment time, revealing that a treatment duration of 20 minutes consistently resulted in high adsorption capacity across all cycles. Therefore, a 20 mL solution of 0.1 M NaOH and a treatment time of 20 minutes were deemed optimal for negatively ionizing the functional groups while minimizing the impact on bead stability.
For Pb(II) ion desorption and regeneration of beads for the subsequent adsorption cycles, 0.1 M HNO3 was used. While higher HNO3 volumes and longer exposure enhance Pb(II) removal, acidic conditions can degrade the gelatin matrix, making optimization essential. In this study, the amount of adsorbent was kept constant at 0.05 g. The volumes of 0.1 M HNO3 were varied at 20 mL, 30 mL, and 40 mL for desorption, with stirring conducted for 3 hours at 75 rpm. Overall, the Qe was the highest and most consistent with the 20 mL volume, which performed similarly to the 30 mL volume while using less acid (Fig. S6b†). Therefore, 20 mL was considered optimal, as it minimized damage to the SKLGE bead structure without significantly compromising adsorption capacity. For contact time, three batches were tested with desorption durations of 3 hours, 5 hours, and 8 hours. Both the 3 hour and 8 hour contact times resulted in a decrease in the Qe value. The drop observed at 3 hours was due to insufficient removal of absorbed Pb(II) ions from the first adsorption cycle, leading to a 31% reduction in Qe. In the case of 8 hours, the decrease, though smaller, was attributed to prolonged exposure to the acidic environment, which damaged the active sites and network structure. Thus, a 5 hour desorption time was determined to be the optimal choice for maintaining a consistent Qe value. To summarize, the optimal regeneration conditions were determined to be 20 mL each of 0.1 M NaOH and 0.1 M HNO3, with 20 minute NaOH and 5 hour HNO3 treatment durations for effective ionization and regeneration of the beads.
3.6 Identifying the adsorption kinetics
An equilibrium experiment was conducted to elucidate the adsorption rate and investigate the impact of adsorption time on the results. SKL45 was utilized to establish consistent contact time for the adsorption tests, as illustrated in Fig. S7.† The increase in Qt plateaued after 3 hours, ultimately reaching 48.6 mg g−1 in 4 hours. Data was collected over 6 hours to confirm the attainment of a stable phase. Consequently, an adsorption time of 4 hours was selected as optimum for all subsequent adsorption studies.Pseudo-first-order and pseudo-second-order kinetic models were applied to experimental data to further investigate the adsorption mechanism.34
Pseudo-first-order:
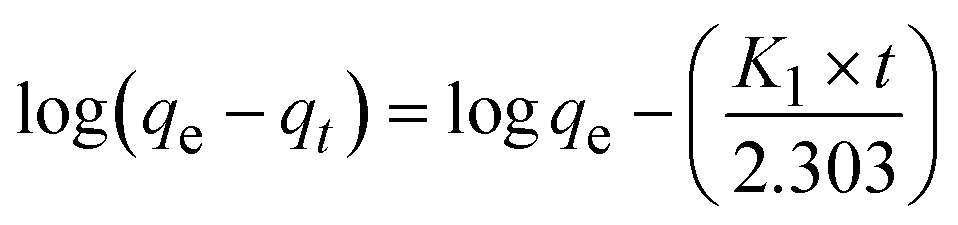 | (4) |
Pseudo-second-order:
 | (5) |
Pseudo-first-order:
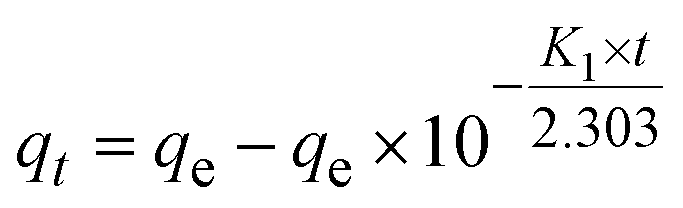 | (6) |
Pseudo-second-order:
 | (7) |
The pseudo-first-order model implies that the adsorption rate is directly proportional to the number of unoccupied sites. In contrast, the pseudo-second-order model suggests that the rate of adsorption site occupation is proportional to the square of the number of unoccupied sites. The equilibrium data were analyzed for both models in Origin, and the results are presented in Table 1.
| Pseudo first order | Pseudo second order | Experimental | ||||
|---|---|---|---|---|---|---|
| q e (mg g−1) | K 1 (min−1) | R 2 | q e (mg g−1) | K 2 (g mg−1 min−1) | R 2 | q e (mg g−1) |
| 47.35 | 0.034 | 0.984 | 51.48 | 0.001 | 0.996 | 48.55 |
Based on the results from Table 1, the Pb(II) ion adsorption by SKLGE beads aligns more closely with the pseudo-second-order model, as evidenced by an R2 value exceeding 0.996 compared to 0.984 for the pseudo-first-order model. This indicates that the adsorption process is primarily governed by chemisorption, involving strong chemical interactions, particularly the complexation between phenolate, sulfonate, and carboxylate groups on the SKLGE beads and Pb(II) ions. The preference for the pseudo-second-order model suggests that the adsorption capacity of the beads and the availability of adsorption sites play a crucial role, emphasizing chemisorption, where strong chemical bonds form between the adsorbate and the adsorbent surface.35 This is further supported by the presence of ionic and polar functional groups in gelatin and lignin, including phenoxide (Ph–O−), sulfonate (R-SO3−), carboxylate (R-COO−), amines (R-NH2), amides (–HN–CO), ethers (C–O–C), and aromatic π electrons. These functional groups can participate in Lewis acid–base coordination bond formation with Pb(II) ions. The rapid initial increase in adsorption over time, followed by a slower rate of rise, underscores the heterogeneity of the adsorption sites. Ionized functional groups, such as phenoxide (Ph–O−), sulfonate (R–SO3−), and carboxylate (R–COO−), exhibit stronger Lewis basicity compared to non-ionic polar functional groups like amines (R–NH2), amides (–HN–CO), and ethers (C–O–C). Consequently, coordination interactions with the ionized functional groups occur more rapidly, facilitating swift adsorption, while adsorption involving the non-ionic polar functional groups and aromatic π electrons progresses at a slower rate.
3.7 Optimal bead composition for adsorption capacity and recyclability
The objective of preparing SKLGE beads with varying ratios of GE and SKL was to identify a composition that exhibits high Pb(II) ion adsorption while maintaining stability under harsh adsorption and regeneration conditions involving acids and bases. Regeneration ability is crucial for cost-effective adsorption-based Pb(II) removal. To this end, SKLGE beads with five different SKL-to-GE ratios were examined over 10 adsorption and regeneration cycles, using an initial Pb(II) concentration of 100 mg L−1. As presented in Fig. 4a and b, a clear trend emerges: as the SKL content increases, so does the adsorption capacity (Qe). The enhanced adsorption of Pb(II) ions with increased SKL content is further evidenced by the greater compaction of the pre-swelled beads after Pb(II) ion adsorption, as illustrated in Fig. 2b. SKL45 was identified as the optimal composition for further study, demonstrating the highest and most stable adsorption performance with minimal standard deviation. | ||
| Fig. 4 (a) Box chart showing the average adsorption capacity with standard deviation for SKL25–SKL45 beads, (b) adsorption performance over ten continuous adsorption and regeneration cycles, (c) effect of SKLGE adsorbent dosage on removal efficiency (C0: 100 mg L−1 and 300 mg L−1, volume: 50 mL, pH: 6.0, stirring rate: 75 rpm, duration: 4 hours), and (d) effect of initial Pb(II) ion concentration on adsorption using 0.05 g SKL45 adsorbent (C0: 30, 100, 300, 600, and 1000 mg L−1, volume: 50 mL, pH: 6.0, stirring rate: 75 rpm, duration: 4 hours), (e) Langmuir and (f) Freundlich adsorption isotherm linear models for the SKL45 bead's Pb(II) ion adsorption. | ||
After finding the optimal SKL-to-GE blend ratio (45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :55), three additional adsorption and regeneration cycles were conducted to optimize the total SKLGE weight percentage relative to solvent weight. SKL45 beads were fabricated with total SKLGE weight percentages of 5%, 7.5%, and 12.5%, in addition to the 10% used in the 10-cycle test. The 12.5 wt% concentration presented slight challenges with needle ejection during fabrication, while the 5 wt% formulation failed to maintain individual spherical shapes during drying, leading to bead coalescence. Therefore, adsorption and regeneration cycles were continued with the 7.5 wt% and 12.5 wt% beads. As shown in Fig. S8,† the 12.5 wt% beads exhibited a similar Qe value to the 10 wt% beads, reaching 42.5 mg g−1, while the 7.5 wt% beads demonstrated a lower capacity of 34 mg g−1, with stable adsorption over three cycles. Considering visual observations and adsorption results, the 10 wt% formulation was confirmed as the optimal weight percentage for SKL45. Since SKL45 was found to be optimum, a total of 3 repetitions of the adsorption experiments were conducted under the same conditions: C0 = 100 mg L−1, volume = 50 mL, pH = 6.0, stirring rate = 75 rpm, and duration = 4 hours, to evaluate the repeatability of the adsorption results. The mean adsorption capacity is 44.03 mg g−1, with a low standard deviation of 1.59.
:55), three additional adsorption and regeneration cycles were conducted to optimize the total SKLGE weight percentage relative to solvent weight. SKL45 beads were fabricated with total SKLGE weight percentages of 5%, 7.5%, and 12.5%, in addition to the 10% used in the 10-cycle test. The 12.5 wt% concentration presented slight challenges with needle ejection during fabrication, while the 5 wt% formulation failed to maintain individual spherical shapes during drying, leading to bead coalescence. Therefore, adsorption and regeneration cycles were continued with the 7.5 wt% and 12.5 wt% beads. As shown in Fig. S8,† the 12.5 wt% beads exhibited a similar Qe value to the 10 wt% beads, reaching 42.5 mg g−1, while the 7.5 wt% beads demonstrated a lower capacity of 34 mg g−1, with stable adsorption over three cycles. Considering visual observations and adsorption results, the 10 wt% formulation was confirmed as the optimal weight percentage for SKL45. Since SKL45 was found to be optimum, a total of 3 repetitions of the adsorption experiments were conducted under the same conditions: C0 = 100 mg L−1, volume = 50 mL, pH = 6.0, stirring rate = 75 rpm, and duration = 4 hours, to evaluate the repeatability of the adsorption results. The mean adsorption capacity is 44.03 mg g−1, with a low standard deviation of 1.59.
To evaluate the effectiveness of lignin in enhancing adsorption efficiency and bead stability, a batch of beads was fabricated using 10 wt% gelatin (GE10) only and subjected to the same adsorption procedure as a control experiment. These beads exhibited significantly less swelling compared to SKL45, resulting in a notably lower adsorption capacity of 12 mg g−1. Furthermore, they showed increased fragility and instability during regeneration, causing a gradual decline in Qe values and ultimately dissolving completely by the third cycle. This underscores the critical role of SKL in enhancing both the stability and adsorption performance of the gel beads.
3.8 Effect of adsorbent dosage on the adsorption capacity
To assess the effect of adsorbent dosage on adsorption capacity (Qe) and removal efficiency at a constant initial Pb(II) ion concentration (C0) of 100 mg L−1, SKLGE dry bead dosages were incrementally increased to 0.025, 0.05, 0.10, 0.15, and 0.20 g per 50 mL of the initial Pb(II) solution. The batch adsorption process was conducted at 75 rpm for 4 hours. A dosage of 0.05 g achieved the highest Qe of 60.2 mg g−1 (Fig. S9a†), indicating a positive correlation between Qe and dosages up to 0.05 g, followed by a decline at higher dosages. Thus, 0.05 g per 50 mL yielded the optimal Qe. The removal efficiency revealed that a dosage of 0.1 g was sufficient to remove 97% of Pb(II) from a 50 mL solution of 100 mg L−1, plateauing beyond this dosage (Fig. 4c and S9a, b†). Since 0.1 g was deemed effective in removing the maximum percentage of lead, a total of 3 repetitions were conducted under this condition to ensure repeatability, resulting in a mean value of 95.77% removal efficiency with a standard deviation of 0.66.When the initial Pb(II) concentration was increased to 300 mg L−1, the removal efficiency did not plateau; instead, it increased consistently with higher dosages, achieving a maximum of 87% removal with 0.2 g of adsorbent (Fig. 4c). Notably, this same 0.2 g dosage resulted in a 10% decrease in removal efficiency when the Pb(II) ion concentration increased from 100 mg L−1 to 300 mg L−1, indicating the need for a higher dosage to maintain efficient removal at elevated Pb(II) levels. These trends can be attributed to the increased availability of active sites for Pb(II) ions as the adsorbent dosage rises, establishing a positive correlation between dosage and removal efficiency. However, as adsorption proceeds, the slightly negatively charged SKLGE beads become neutralized by the positively charged Pb(II) ions, reducing electrostatic attraction. This neutralization can create a partially positively charged shell or lead to surface saturation, limiting further adsorption and causing removal efficiency to plateau beyond a certain dosage threshold.
3.9 Effect of Pb(II) ion initial concentration on adsorption
To determine the maximum adsorption capacity of the 0.05 g SKL45 adsorbent, the initial Pb(II) ion concentration was incrementally increased to 30, 100, 300, 600, and 1000 mg L−1. The initial Pb(II) concentration range was selected arbitrarily to identify the level that maximizes adsorptive capacity while avoiding errors caused by Pb2+ precipitation. At concentrations exceeding 1000 mg L−1, the Pb(NO3)2 solution became slightly opaque, indicating inhomogeneity that could compromise the accuracy of adsorption results. Consequently, the study was limited to a maximum concentration of 1000 mg L−1. As shown in Fig. 4d, there is a clear correlation between the initial Pb(II) concentration and the adsorption capacity (Qe). Notably, Qe rises sharply as the concentration increases from 30 mg L−1 to 300 mg L−1, with a substantial increase of 82 mg g−1 within this range. However, the rate of increase in Qe diminishes between 300 mg L−1 and 1000 mg L−1, where a 700 mg L−1 increment in Pb(II) ion concentration results in only a 45 mg g−1 increase in Qe. The maximum Qe observed was 155 mg g−1 at an initial Pb(II) concentration of 1000 mg L−1. At higher Pb(II) concentrations, more ions are adsorbed onto the surface, occupying active sites and neutralizing negatively charged functional groups. However, as concentration increases further, repulsive forces between the adsorbed Pb(II) ions on the surface and those in the bulk solution make it increasingly difficult to access the remaining vacant sites. To ensure repeatability, the adsorption experiments were conducted a total of 3 times for the Pb(II) concentration of 1000 mg L−1, at which the adsorption capacity was maximum. The mean adsorption capacity was 154.5 mg g−1, with a standard deviation of 1.92 mg g−1.Finally, the Pb(II) ion adsorption capacity of the SKL45 beads was compared with that of other spherical lignin-based adsorbents, including beads, microspheres, and nanospheres (Table S5†).26,36–43 Three types of adsorbents, namely lignin–sodium alginate beads (LSAB; 191.0 mg g−1),36 dual-modified lignin–sodium alginate microspheres (DMLSAM; 187.5 mg g−1),37 and carboxymethyl lignin nanospheres (CLNPs; 333.26 mg g−1),26 exhibited higher adsorption capacity than SKL45 beads, while porous polyamine lignin microspheres (PPALM; 156.82 mg g−1)38 showed similar adsorption capacity under comparable experimental conditions. The adsorption experiments were conducted at 30 °C for CLNPs, DMLSAM, and PPALM, higher than the 23 °C used in this study. Therefore, absolute comparison is not reliable, as higher temperatures typically result in increased capacity.36,39 Additionally, microspheres and nanospheres are generally more difficult to recover after adsorption and regeneration than beads. LSAB beads are characterized by costly lignin extraction, slow bead fabrication, and the use of sodium alginate, a popular Pb(II) ion adsorbent, leading to the assumption that sodium alginate, rather than lignin, is the main adsorbent. DMLSAM entails extensive chemical modification of lignin and uses sodium alginate (which dominates Pb adsorption), with very low stability, as evidenced by a decline in removal efficiency from 98.82% to 69.34% after three cycles. CLNPs involve high commercial lignin cost, chemically and instrumentally demanding synthesis and purification, with high adsorptivity due to carboxymethyl modification, but exhibit lower stability, showing a 27% loss in adsorption capacity after 10 cycles. PPALM also involves a highly chemically demanding synthesis and demonstrates lower stability, as evidenced by a decrease in adsorption capacity from 156.82 to 120 mg g−1 after five cycles. In contrast, the SKL45 beads investigated in this study were prepared through a simple cooling-induced self-assembly process, utilizing the industrial byproduct SKL. This approach promotes lignin waste recycling for value-added applications while enabling rapid fabrication, stability over 10 cycles, and superior adsorptivity compared to many lignin-based spherical adsorbents, as shown in Table S5.†
3.10 Langmuir and Freundlich isotherm models
The Langmuir and Freundlich isotherm models were employed to elucidate the Pb(II) adsorption mechanism, specifically examining the interaction between the adsorbent and Pb(II) ions, the homogeneity or heterogeneity of the adsorption sites, monolayer versus multilayer adsorption, and the influence of Pb(II) concentration on the adsorption rate. The Langmuir isotherm equation (eqn (8) and (9)) is:Linear:
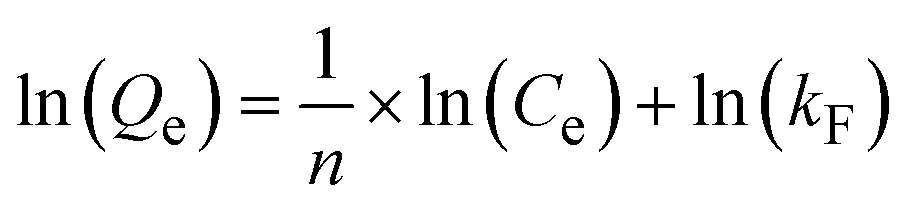 | (8) |
Nonlinear:
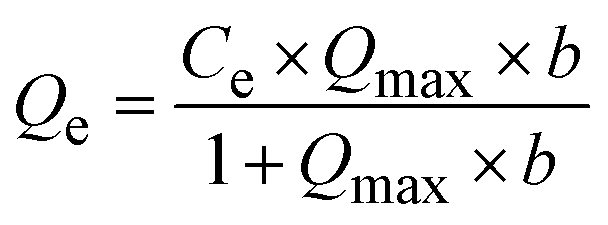 | (9) |
Linear:
 | (10) |
Nonlinear:
 | (11) |
 ) suggests favorable adsorption conditions, with increasing adsorbate concentration enhancing adsorbent capacity, indicative of a heterogeneous surface with sites of varying affinities.45 In contrast, n < 1 implies a decrease in adsorption capacity as concentration rises due to uniformly occupied sites. When n = 1, this indicates a linear adsorption relationship, where the adsorption capacity is directly proportional to the adsorbate concentration.
) suggests favorable adsorption conditions, with increasing adsorbate concentration enhancing adsorbent capacity, indicative of a heterogeneous surface with sites of varying affinities.45 In contrast, n < 1 implies a decrease in adsorption capacity as concentration rises due to uniformly occupied sites. When n = 1, this indicates a linear adsorption relationship, where the adsorption capacity is directly proportional to the adsorbate concentration.
The correlation coefficient in Fig. 4e and f in the linear form and Fig. S10† in nonlinear form indicates that the Freundlich model provides a better fit for the adsorption process of the SKL45 sample compared to the Langmuir model. With an n value of 3.536 (greater than 1), the adsorption process is confirmed to be favorable, suggesting the presence of heterogeneous surface sites. This observation supports a pseudo-second-order adsorption rate, characterized by a rapid initial uptake followed by a slower increase. This aligns with the adsorption mechanism, where ionic functional groups coordinate rapidly, while non-ionic polar groups and aromatic π-electrons coordinate more slowly.
3.11 XPS evaluation of the Pb(II) adsorption process on SKL45
XPS analysis indicates chemisorption of Pb(II) ions on SKLGE beads, as shown by shifts in the binding energy of peaks corresponding to elements involved in Pb(II) adsorption. Fig. 5a shows the elemental composition of SKL45 before Pb(II) adsorption: C (64.76%), O (22.60%), N (8.99%), S (2.20%), and Na (1.46%). After adsorption, Na was fully replaced by Pb (1.79%), confirming that Pb(II) was chemisorbed onto –COOH, –SO3H, and Ph–OH groups, forming coordination complexes. Shifts in Pb 4f7/2 and Pb 4f5/2 binding energies from 139.5 eV and 144.4 eV (for Pb(NO3)2) to 139.1 eV and 143.9 eV further validate chemisorption (Fig. 5b).46 The C 1s peak analysis (Fig. 5c, d and Table 2) reveals a reduction in C1 peak area for C–C and CC bonds after Pb(II) adsorption and an increase in C2 peak area for C–O and C–N bonds. This suggests that complexation with Pb(II) ions disrupts resonance stabilization in phenoxide groups, withdrawing electron density from aromatic rings and shifting the CC bond peak to higher binding energies, which merges with the C–O peak. An alternative interpretation is that Pb(II) complexation removes negative charges from carboxylate and phenoxide C–O− ions, shifting their peaks back to positions for non-ionized forms and increasing the C2 peak area.
 | ||
| Fig. 5 XPS evaluation of the Pb(II) ion adsorption process on SKL45 including (a) survey spectra presenting the elemental composition; (b) wide scan region of the Pb4f for SKL45Pb; (c) wide scan region of the C 1s for SKL45; (d) wide scan region of the C 1s for SKL45Pb; (e) wide scan region of the O 1s for SKL45; and (f) wide scan region of the O 1s for SKL45 Pb. | ||
| Sample | % C in different environments | % O in different environments | ||||||
|---|---|---|---|---|---|---|---|---|
| C1 | C2 | C3 | C2/C1 | C2/C3 | O1 | O2 | O2/O1 | |
| SKL45 | 52.5 | 32.0 | 15.5 | 0.6 | 2.1 | 63.1 | 36.9 | 0.6 |
| SKL45 Pb | 38.0 | 40.5 | 21.5 | 1.1 | 1.9 | 41.8 | 58.2 | 1.4 |
The O 1s region (Fig. 5e and f) also shows notable changes. Deconvolution of the O 1s spectrum into O1 (531.4 eV) and O2 (532.7 eV) regions, representing ionized and non-ionized C–O and S–O bonds,47–49 shows a doubled O2/O1 ratio in SKL45 after Pb(II) adsorption, supporting Pb(II) complexation with ionic groups. These findings indicate chemisorption of Pb(II) ions onto SKLGE beads, predominantly involving SKL functional groups, which are crucial for effective adsorption. A comprehensive interpretation of the XPS results is provided in the ESI.†
3.11 Effect of Pb(II) adsorption on the thermal decomposition of SKL45
Adsorption of Pb(II) ions onto the beads resulted in a size reduction, as shown in Fig. 2b. This decrease could be associated with Pb-induced crosslinking of the SKL and GE components, which is expected to stabilize the beads and increase their thermal decomposition temperature. TGA was conducted to evaluate the effect of Pb(II) ion adsorption on the thermal decomposition of SKL45 (Fig. 6). As the temperature increases, all samples exhibit weight loss, typically through three phases of change: water evaporation, rapid decomposition, and residue formation.50 | ||
| Fig. 6 Effect of Pb(II) adsorption on the thermal decomposition of SKL45 beads on (a) TGA curves and (b) DTG curves. | ||
A striking contrast is evident in the rapid decomposition phase of SKL45 before and after Pb(II) ion adsorption. Prior to adsorption, SKL45 exhibited single-step decomposition with a peak at 320 °C. After adsorption, SKL45 displayed two distinct decomposition peaks at 267 °C and 350 °C, corresponding to the catalytic thermal decomposition of less stable bonds (e.g., ether, carboxylic, sulfonic) and more stable bonds (e.g., aromatic rings), respectively.51,52 Overall, Pb(II) ion adsorption increased thermal stability, as evidenced by the higher decomposition temperature of 350 °C, despite the catalytic effect of the metal ion. This increased thermal stability indicates crosslinking between SKL and GE via Pb(II) ion chemisorption (i.e., coordination).
3.12 Zero-length EDC cross-coupling to enhance bead stability
Although the beads demonstrated good performance in their current form, enhancing their stability was essential for long-term diverse applications. To achieve this, EDC cross-coupling was employed. EDC acts as a cross-coupling agent that facilitates the formation of stable covalent bonds between the carboxyl and phenolic groups of lignin and the amine and carboxyl groups of gelatins. Specifically, EDC enables the formation of amide bonds between the carboxyl groups (–COOH) of lignin and the amino groups (–NH2) of gelatin, as well as ester bonds between the phenolic (Ph–OH) groups on lignin and the carboxyl (–COOH) groups on gelatin. This dual covalent bonding mechanism enhances the stability of the final SKLGE beads. Additionally, EDC, recognized as a zero-length crosslinker, differs from traditional crosslinkers such as glutaraldehyde and radical crosslinkers, as it is not incorporated into the final product. Nevertheless, EDC plays a vital role in stabilizing the hydrogel network and enhancing its mechanical properties without introducing synthetic chemical loads, thereby preserving the integrity of the biopolymer's chemical composition.As shown in Table S2,† various amounts of crosslinker were utilized to identify the optimal balance between adsorption capacity and stability, as EDC coupling replaces numerous adsorption sites (R-COOH, R-NH2, Ph–OH) through the formation of chemical bonds. Fig. S7† illustrates that under similar conditions, the equilibrium curve for SKL45 exhibited a maximum adsorption capacity of 48.5 mg g−1 at 4 hours. SKL45 with EDC cross-coupling generally achieved stable adsorption after 3 hours of contact time, with capacities of 52.0, 49.6, 39.7, and 30.0 mg g−1 corresponding to increasing EDC concentrations of 0.5 mM, 1 mM, 2 mM, and 4 mM, respectively (Fig. S7†).
As EDC concentration increased, the adsorption rate decelerated before reaching a plateau. This behavior can be attributed to the increasing crosslinking, which led to a more compact structure of the SKLGE beads (Fig. 7a), consequently reducing the available surface area for Pb(II) ions and diminishing the number of active negative sites for cation binding. As a result, the adsorption process took longer to achieve equilibrium. This is further evidenced by the absence of significant compaction in bead size after Pb(II) ion adsorption, a phenomenon typically observed in non-crosslinked SKL45 beads (Fig. 2b). Although the EDC cross-coupled beads reached a stable phase more quickly, the adsorption time was maintained at 4 hours for comparison with SKL45 without EDC in further experiments. The equilibrium curve confirmed that 4 hours was sufficient for the adsorption process to reach a plateau (Fig. S7†).
 | ||
| Fig. 7 (a) Influence of EDC concentration on bead swelling and compaction: (A) fresh beads before adsorption and (B) beads after adsorption. (b) Box plot illustrating Pb(II) ion adsorption capacity over 10 adsorption–regeneration cycles for EDC-crosslinked beads, and (c) bar chart comparing adsorption capacity between SKL45 beads crosslinked with 1 mM EDC and non-crosslinked SKL45 beads. | ||
A comparative cycling test was conducted using SKL45 beads cross-coupled with EDC at four concentrations to identify the optimal level for maximum adsorptivity and stability. Increasing EDC concentrations enhanced cross-coupling but reduced available active sites, leading to lower Qe values, especially at 4 mM (Fig. 7b and S11†). In contrast, 0.5 mM and 1 mM concentrations yielded similar Qe values around 50 mg g−1, significantly higher than those at greater concentrations. These results indicate that while higher EDC concentrations improve bead stability and handling, 1 mM EDC offers the best balance between covalent crosslinking and active site availability for Pb(II) ions. Additionally, EDC reduces fluctuations in adsorption capacities and enhances regeneration stability (Fig. 7c), making EDC-treated SKL45 a superior choice for scale-up applications due to its consistent output.
3.13 Stability of the gel in EDC-crosslinked and non-crosslinked SKL45 beads
The cryo-SEM imaging technique was employed to evaluate the stability of gel morphology in non-crosslinked SKL45 beads compared to SKL45 beads crosslinked with 1 mM EDC (Fig. 8). Focusing on the internal structure, C1_A and C2_A reveal a well-defined polymer network, with the C2 sample showing notably more compact overall porosity, likely due to EDC cross-coupling. The crosslinking also accounts for the further reduction in pore size at stage B, as Pb(II) ions are adsorbed and bonded within the structure. The subsequent treatment with HNO3 effectively dissociates the adsorbed Pb(II), restoring the pore structure in stage C and preparing the beads for a new cycle. By stage E, after 10 adsorption–regeneration cycles, more torn fibers are observed, and the previously well-defined porous morphology of SKL45 without EDC crosslinking has altered. However, the network remains intact and functional, as indicated by the stable Pb(II) ion adsorption capacity in the 10th cycle. Crosslinking with EDC helped maintain the porous structure, although the pores increased in size to some extent. This indicates that EDC crosslinking effectively enhanced the stability of the gel beads for long-term adsorption regeneration. | ||
| Fig. 8 Cryo-SEM images of cross-sections at the interior and surface of SKL45 beads: (C1 and S1) for non-crosslinked SKL45 beads and (C2 and S2) for SKL45 beads crosslinked with 1 mM EDC during the following stages: fresh (A), cycle 1 adsorption (B), cycle 1 regeneration (C), cycle 10 adsorption (D), and cycle 10 regeneration (E). | ||
Cryo-SEM images at the surfaces (S1 and S2) clearly illustrate the formation of a surface shell influenced by the LN2-induced Marangoni effect. In images S1_A and S2_A, which depict fresh beads after NaOH pretreatment but before Pb(II) ion adsorption, the pores near the shell appear smaller than those deeper within the bead. This difference is less pronounced in stage B, where Pb(II) ion adsorption compacted the gel due to increased Pb(II)-induced crosslinking and reduced swelling in water, as the negatively charged hydrophilic groups were occupied by Pb(II) ions. By stage C, following one regeneration cycle, a thicker shell is observed compared to stage A. This thickening results from the slight removal of gelatin and SKL due to acid and base treatments during regeneration, leading to a looser network of interactions and an overall expansion of shell thickness. After 10 cycles, no significant physical damage is noted in stages D and E; however, S2_E demonstrates a superior porous and shell structure compared to S1_E, indicating that EDC crosslinking enhanced the stability of the protective skin layer.
The regeneration performance of the SKL45 adsorbent was also evaluated at higher temperatures (40 °C, 50 °C, and 60 °C) over three regeneration cycles, as shown in Fig. S12a.† The adsorption capacity decreases slightly with each cycle but remains relatively high, particularly at 40 °C, where the decline was less than that at the higher temperatures. Additionally, the SKL45 beads maintained their structural integrity during the first cycle, even at 60 °C, without liquefying. However, in the second and third cycles, the beads gradually lost their structural integrity, becoming increasingly fragile under physical forces. The slow decline in the stability of SKL 45 beads is not surprising, since the solubility of GE increases significantly at higher temperatures. Eventually, all the hydrogel solutions were prepared by mixing SKL and GE at 60 °C for only 30 minutes before injecting them into LN2 for rapid cooling, which induced bead formation. It is rather interesting to observe that gel bead formation increased thermal stability, indicating enhanced interactions between SKL and GE in the beads, as well as the stabilizing effect of the skin layer created by the LN2-induced fabrication method. In contrast to SKL45 beads without EDC cross-coupling, SKL45 beads cross-coupled with EDC demonstrated improved stability, retaining their shape and experiencing less surface and structural damage compared to SKL45 beads without EDC treatment, as shown in Fig. S12b–e.†
4. Conclusion
In this study, we present the fabrication of SKLGE hydrogel beads using a green and innovative method, along with an investigation into their adsorption capabilities for toxic Pb(II) ions. Using SKL is pivotal in recycling waste from the pulp and paper industry, enabling value-added applications that contribute to a circular economy. Additionally, the abundant availability of SKL from various natural, agricultural, and industrial processes positions it as a cost-effective adsorbent for large-scale applications. Our approach not only addresses environmental sustainability but also leverages SKL's properties to enhance the efficiency of Pb(II) ion adsorption, showcasing its potential for practical applications in water treatment and pollution mitigation. This LN2-facilitated fabrication method enables rapid production, generates no waste, and is entirely green. It results in porous gel bead interiors and the formation of a protective high-density skin layer.These beads are fabricated from various blend ratios of SKL and GE, with SKL45 at a 45:55 ratio of SKL to GE at 10 wt% identified as the optimal composition for achieving a balance between adsorptivity, hydrophilicity and stability. The maximum adsorption capacity reached an impressive 155 mg g−1, as determined through batch adsorption tests. The adsorption process follows a pseudo-second-order rate and is best described by the Freundlich model, indicating the presence of heterogeneous adsorption sites capable of chemisorption at variable rates. The beads demonstrated remarkable stability over 10 cycles of adsorption and regeneration under harsh acidic and basic conditions. Notably, EDC cross-coupling significantly enhances both the adsorption capacity and stability of the beads.
Future studies could explore the adsorption of other positively charged heavy metal ions using SKLGE beads and investigate modifications to enhance selectivity. Potential applications are extensive, including integration into municipal water treatment systems and household filters for Pb(II) removal, as well as use in industrial wastewater treatment, agricultural irrigation processes, soil and groundwater remediation, and valuable metal recovery systems. These advancements could yield significant environmental benefits and broaden the utility of SKLGE beads in addressing water pollution challenges.
Data availability
The datasets supporting the findings of this study are available from the corresponding author upon reasonable request. The specific data include:(1) Raw and processed data:
◦ Raw adsorption data for Pb(II) ion concentrations under different experimental conditions.
◦ Processed adsorption isotherm and kinetics data.
◦ Data used for generating plots and tables presented in the manuscript.
(2) Characterization data:
◦ Cryo-SEM, FTIR, TGA, and XPS data files, including raw spectra and processed datasets used for analysis.
◦ Detailed parameters and conditions for all analytical techniques applied.
(3) Supplementary data:
◦ ESI† containing additional experimental details and derived parameters for adsorption models.
◦ Requests for data access will be considered within the framework of institutional policies and ethical considerations to protect intellectual property and sensitive proprietary information.
For inquiries or data requests, please contact the corresponding author at E-mail: sadrzade@ualberta.ca
Conflicts of interest
The authors declare no competing interests.Acknowledgements
The authors would like to acknowledge the financial and technical support from the National Research Council (NRC) of Canada, the Advanced Water Research Lab (AWRL) at the University of Alberta, the Natural Science and Engineering Research Council of Canada (NSERC) and Canada's Oil Sands Innovation Alliance (COSIA).References
- S. R. Dhokpande, S. M. Deshmukh, A. Khandekar and A. Sankhe, Sep. Purif. Technol., 2024, 350, 127868 CrossRef CAS.
- S. Tong, Y. E. Von Schirnding and T. Prapamontol, Bull. W. H. O., 2000, 78(9), 1068–1077 CAS.
- C. Yu, J. C. Shao, W. J. Sun and X. N. Yu, Arabian J. Chem., 2020, 13(1), 3474–3483 CrossRef CAS.
- I. R. Chowdhury, S. Chowdhury, M. A. J. Mazumder and A. Al-Ahmed, Appl. Water Sci., 2022, 12(8), 185 CrossRef CAS PubMed.
- N. A. A. Qasem, R. H. Mohammed and D. U. Lawal, npj Clean Water, 2021, 4(1), 1–15 CrossRef.
- A. Aghaei, K. Suresh, M. D. Firouzjaei, M. Elliott, A. Rahimpour and M. Sadrzadeh, in Advanced Technologies in Wastewater Treatment: Oily Wastewaters, 2023 Search PubMed.
- A. M. Badran, U. Utra, N. S. Yussof and M. J. K. Bashir, Separations, 2023, 10(11), 565 CrossRef CAS.
- Y. Ge and Z. Li, ACS Sustainable Chem. Eng., 2018, 6(5), 7181–7192 CrossRef CAS.
- M. Sajjadi, F. Ahmadpoor, M. Nasrollahzadeh and H. Ghafuri, Int. J. Biol. Macromol., 2021, 178, 394–423 CrossRef CAS PubMed.
- K. Suresh, M. A. Islam, M. Rastgar, A. Mohammadnezhad, B. A. Fleck and M. Sadrzadeh, Cellulose, 2022, 29, 3351–3374 CrossRef CAS.
- L. Shamaei, B. Khorshidi, M. A. Islam and M. Sadrzadeh, J. Cleaner Prod., 2020, 256, 120304 CrossRef CAS.
- L. Kouisni, P. Holt-Hindle, K. Maki and M. Paleologou, J. Sci. Technol. For. Prod. Processes, 2012, 2, 6–10 Search PubMed.
- T. Aro and P. Fatehi, ChemSusChem, 2017, 10, 1861–1877 CrossRef CAS PubMed.
- E. M. Ahmed, J. Adv. Res., 2015, 6(2), 105–121 CrossRef CAS PubMed.
- V. Van Tran, D. Park and Y. C. Lee, Environ. Sci. Pollut. Res., 2018, 25, 24569–24599 CrossRef CAS PubMed.
- Y. H. Teow, L. M. Kam and A. W. Mohammad, J. Environ. Chem. Eng., 2018, 6(4), 4588–4597 CrossRef CAS.
- M. N. Saqib, B. M. Khaled, F. Liu and F. Zhong, Food Hydrocolloids Health, 2022, 2, 100073 CrossRef.
- P. Sánchez-Cid, M. Jiménez-Rosado, A. Romero and V. Pérez-Puyana, Polymers, 2022, 14, 3023 CrossRef PubMed.
- I. Yosha, A. Shani and S. Magdassi, J. Agric. Food Chem., 2008, 56, 8045–8049 CrossRef CAS PubMed.
- X. Song, H. Wu, J. Shi, X. Wang, W. Zhang, Q. Ai and Z. Jiang, J. Mol. Catal. B: Enzym., 2014, 100, 49–58 CrossRef CAS.
- G. Viscusi and G. Gorrasi, J. Polym. Environ., 2021, 29(11), 3756–3769 CrossRef CAS.
- H. W. Kwak, M. Shin, H. Yun and K. H. Lee, Int. J. Mol. Sci., 2016, 17(9), 1466 CrossRef PubMed.
- S. Van Vlierberghe, V. Cnudde, P. Dubruel, B. Masschaele, A. Cosijns, I. De Paepe, P. J. S. Jacobs, L. Van Hoorebeke, J. P. Remon and E. Schacht, Biomacromolecules, 2007, 8(2), 331–337 CrossRef CAS PubMed.
- K. Dey, S. Agnelli and L. Sartore, Micro, 2023, 3(2), 434–457 CrossRef.
- R. Chebbi, ACS Omega, 2023, 8, 37196–37201 CrossRef CAS PubMed.
- C. Liu, Y. Li and Y. Hou, Molecules, 2019, 24(15), 2704 CrossRef CAS PubMed.
- I. M. M. Rahman, Y. Furusho, Z. A. Begum, R. Sato, H. Okumura, H. Honda and H. Hasegawa, Cent. Eur. J. Chem., 2013, 11, 672–678 CAS.
- S. Haider and S. Y. Park, J. Membr. Sci., 2009, 328(1–2), 90–96 CrossRef CAS.
- H. Tokuyama and N. Yazaki, React. Funct. Polym., 2010, 70(12), 967–971 CrossRef CAS.
- V. Hemmilä, R. Hosseinpourpia, S. Adamopoulos and A. Eceiza, Waste Biomass Valorization, 2020, 11, 5835–5845 CrossRef.
- S. R. Derkach, N. G. Voron’ko, N. I. Sokolan, D. S. Kolotova and Y. A. Kuchina, J. Dispersion Sci. Technol., 2019, 41(5), 690–698 CrossRef.
- D. M. Hashim, Y. B. C. Man, R. Norakasha, M. Shuhaimi, Y. Salmah and Z. A. Syahariza, Food Chem., 2010, 118(3), 856–860 CrossRef CAS.
- S. Wang, K. Li and Q. Zhou, Sci. Rep., 2020, 10(1), 17842 CrossRef CAS PubMed.
- J. Wang and X. Guo, J. Hazard. Mater., 2020, 390, 122156 CrossRef CAS PubMed.
- Y. Ge, L. Qin and Z. Li, Mater. Des., 2016, 95, 141–147 CrossRef CAS.
- S. I. Mohammadabadi and V. Javanbakht, Int. J. Biol. Macromol., 2020, 164, 1133–1148 CrossRef CAS PubMed.
- Z. Zhang, Y. Chen and C. Wu, Polymers, 2022, 14(14), 2824 CrossRef CAS PubMed.
- Y. Pang, P. Lin, Z. Chen, M. Zhou, D. Yang, H. Lou and X. Qiu, Int. J. Biol. Macromol., 2023, 253, 127026 CrossRef CAS PubMed.
- Y. Ge, L. Qin and Z. Li, Mater. Des., 2016, 95, 141–147 CrossRef CAS.
- W. Zheng, Q. Zhao, M. J. Malkmes, G. Gao, J. He, L. Zheng and L. Jiang, J. Appl. Microbiol., 2022, 132, 2080–2092 CrossRef CAS PubMed.
- Y. Meng, C. Li, X. Liu, J. Lu, Y. Cheng, L. P. Xiao and H. Wang, Sci. Total Environ., 2019, 685, 847–855 CrossRef CAS PubMed.
- X. Li and X. Luo, Adv. Mater. Res., 2012, 549, 423–427 CAS.
- X. Shen, Y. Xie, Q. Wang, X. Yi, J. L. Shamshina and R. D. Rogers, Cellulose, 2019, 26, 4005–4019 CrossRef CAS.
- J. Xu, L. Wang and Y. Zhu, Langmuir, 2012, 28(22), 8418–8425 CrossRef CAS PubMed.
- B. H. Hameed, J. Hazard. Mater., 2008, 154(1–3), 204–212 CrossRef CAS PubMed.
- B. Fu and F. Xie, Environ. Sci. Pollut. Res., 2020, 27, 5108–5121 CrossRef CAS PubMed.
- K. H. Song, S. K. Jeong, B. H. Jeong, K. Y. Lee and H. J. Kim, Catalysts, 2020, 10(10), 1149 CrossRef CAS.
- K. Radhakrishnan, P. Panneerselvam and M. Marieeswaran, Anal. Methods, 2019, 11(4), 490–506 RSC.
- Z. Tian, L. Zong, R. Niu, X. Wang, Y. Li and S. Ai, J. Appl. Polym. Sci., 2015, 132(25), 42057 CrossRef.
- Q. Yan, C. R. Boardman and Z. Cai, Thermochim. Acta, 2020, 690, 178659 CrossRef CAS.
- Q. Yan, J. Li, X. Zhang, E. B. Hassan, C. Wang, J. Zhang and Z. Cai, J. Nanopart. Res., 2018, 20, 1–20 CrossRef CAS.
- F. H. Isikgor and C. R. Becer, Polym. Chem., 2015, 6(25), 4497–4559 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Comprehensive details of the fabrication, optimization, and performance evaluation of SKLGE beads for Pb(II) adsorption, including experimental methodologies, adsorption kinetics, isotherm models, bead stability, regeneration processes, and thermal stability analyses. See DOI: https://doi.org/10.1039/d4su00732h |
| This journal is © The Royal Society of Chemistry 2025 |