
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElucidating the mechanism of solid-state energy release from dianthracenes via auto-catalyzed cycloreversion†
Cijil
Raju‡
 a,
Zhenhuan
Sun‡
a,
Ryo
Koibuchi‡
b,
Ji Yong
Choi
c,
Subhayan
Chakraborty
a,
Jihye
Park
c,
Hirohiko
Houjou
*b,
Klaus
Schmidt-Rohr
*a and
Grace G. D.
Han
*a
a,
Zhenhuan
Sun‡
a,
Ryo
Koibuchi‡
b,
Ji Yong
Choi
c,
Subhayan
Chakraborty
a,
Jihye
Park
c,
Hirohiko
Houjou
*b,
Klaus
Schmidt-Rohr
*a and
Grace G. D.
Han
*a
aDepartment of Chemistry, Brandeis University, 415 South Street, Waltham, MA 02453, USA. E-mail: gracehan@brandeis.edu; srohr@brandeis.edu
bInstitute of Industrial Science, University of Tokyo, 4-6-1 Komaba, Meguro-ku, Tokyo 153-8505, Japan. E-mail: houjou@iis.u-tokyo.ac.jp
cDepartment of Chemistry, University of Colorado Boulder, 215 UCB, Boulder, CO 80309, USA
First published on 4th September 2024
Abstract
The unique and complex heat release processes of dianthracene-based molecular solar thermal energy storage compounds were elucidated by comprehensive solid-state analyses of their chemical and physical changes using isothermal time-dependent DSC, PXRD, and solid-state NMR. Our study reveals that dianthracenes undergo chemical dissociation, formation of a mixed intermediate phase, and phase transition to the final anthracene crystal, during the triggered energy release process. The solid-state kinetic analysis shows auto-catalyzed cycloreversion and heat release from the energy storage compounds, which is facilitated by the partially cooperative transformation of molecules in the crystalline state. The chemical and physical transformations contribute to the overall energy release to different extents, unveiled by the calculation of lattice energy and dissociation energy changes during the cycloreversion. The fundamental insights into the solid-state transformations will aid in designing advanced energy storage materials and controlling energy release processes.
Introduction
Molecular solar thermal (MOST) energy storage in light-responsive molecules has emerged as an effective method of storing solar photon energy in chemical bonds and releasing the energy on demand in the form of heat.1–3 Common MOST compounds including norbornadienes,4,5 azo(hetero)arenes,6–8 hydrazones,9 dihydroazulenes,10,11 and fulvalene diruthenium derivatives12,13 typically isomerize in the solution state, forming metastable isomers in which the photon energy is stored. The energy-releasing reverse isomerization from the metastable to the thermodynamically stable isomer can be triggered by light irradiation, heating, chemical catalysis, and electrocatalysis.14,15 Recently, MOST systems that operate in condensed phases, particularly in the solid-state, have been developed to increase the gravimetric and volumetric energy storage densities of the systems and to prevent the leakage of liquid phase during energy storage.16 However, the low conformational freedom of common molecular switches in crystalline phases limits their photoisomerization, thus constraining their potential for MOST energy storage applications in the solid form.17 Exceptional systems that are engineered to enable solid-state photoswitching have been reported,18–23 but their ability to store energy has been either uninvestigated or insufficient. This necessitates the development of specific molecular designs that can perform energy storage and release reversibly in the solid state. Reversible topochemical cycloaddition reactions,24–33 which occur completely in crystalline states, can be promising candidates, because they produce metastable cycloadducts that can store energy. Compared to molecular switches that undergo large structural changes during isomerizations, topochemical reactions are accompanied by smaller structural changes during the bond formation and dissociation among prearranged molecules in the crystals.34–36Recent investigations of reversible solid-state photochemical reactions, e.g., [2 + 2] or [4 + 4] intermolecular cycloadditions,37,38 have opened up opportunities to achieve MOST energy storage in crystalline states. The topochemical [2 + 2] photocycloaddition of styrylpyryliums and [4 + 4] photocycloaddition of anthracenes occur via crystal-to-crystal transformations, storing photon energy in strained cycloadducts.37,38 The cycloreversion of the cycloadducts is triggered by thermal activation or UV irradiation, which releases the stored energy. Notably, a recently reported anthracene system with a donor and an acceptor substituent at the 9- and 10-position, respectively, exhibits remarkable energy storage and release (∼0.2 MJ kg−1),38 compared to the styrylpyrylium system with energy storage densities lower than 0.1 MJ kg−1.37 This advance is attributed to the dearomatization of the highly conjugated anthracenes upon photo-induced dimerization, which enlarges the energy difference between the anthracene and dianthracene states. In addition, the energy storage values of dianthracenes are comparable to the activation energies of their thermal cycloreversion, which enables the novel observation of self-activated energy release from the dianthracenes.
The reported dianthracene derivatives (1-D, 2-D, and 3-D) have been revealed to undergo topochemical cycloreversion to anthracenes (1-A, 2-A, and 3-A) upon heating (Fig. 1a), supported by their crystal structure analysis. Interestingly, the thermally induced cycloreversion of the dianthracenes was discovered to show unusual non-Gaussian exothermic features in differential scanning calorimetry (DSC) (Fig. 1b–d). Conventionally, exotherms from thermal isomerizations (e.g., Z-to-E back conversion of azo(hetero)arenes) or solid-state topochemical reactions (e.g., cycloreversion of cyclobutanes to styrylpyryliums and azide–alkyne cycloaddition to triazole rings) display Gaussian-shaped DSC profiles, owing to the simultaneous chemical transformations and molecular rearrangements in condensed phases.37,39,40 Thus, the prominent multiple exothermic features observed during the dianthracene cycloreversion may be attributed to the formation of a transient intermediate mixed phase that is stable enough to be detected (Fig. 1a). However, so far, any mechanistic or kinetic understanding of the remarkable energy release processes in the solid state has not been achieved due to the challenging analysis of convoluted chemical and physical transformations that occur during the cycloreversion. Herein, we examine the solid-state cycloreversion mechanism of dianthracenes, employing a suite of comprehensive solid-state analyses, including isothermal DSC, time-dependent powder X-ray diffraction (PXRD), and time-dependent solid-state NMR spectroscopy, which enables elucidating the intricate energy release processes of the solid-state MOST system.
 | ||
| Fig. 1 (a) A proposed mechanism of thermally induced cycloreversion of dianthracene (D) to anthracene (A) via an intermediate mixed crystalline phase. (b) A broad and unsymmetrical exothermic peak of 1-D, (c) a sharp and distorted exothermic peak of 2-D, and (d) multiple exothermic peaks of 3-D measured by DSC, during their cycloreversion. Complete DSC profiles showing exothermic cycloreversion and subsequent melting of anthracene are available in Fig. S1.† | ||
Results and discussion
In order to delineate the solid-state mechanism that contributes to the large energy release during the cycloreversion of dianthracenes, we first monitored the overall process under a mild thermal condition that allows for the slow and gradual solid-state conversion over hours. We selected a thermal activation temperature slightly below 50 °C, which can be consistently applied in multiple solid-state analysis methods with a small margin of error. The isothermal DSC traces of all dianthracenes measured at 48 °C show non-Gaussian exothermic curves (Fig. 2a–c), similar to the variable-temperature DSC results (Fig. 1b–d), indicating the presence of multiple exothermic events over the course of their cycloreversion to anthracenes at a constant temperature. To correlate the exothermic events and the structural changes of compounds during the cycloreversion, we also monitored the time-dependent PXRD of the dianthracenes heated at 49 ± 1 °C (Fig. 2d–f). For the first 4 hours of heating for 1-D and 2-D, and 2 hours for 3-D, only minor PXRD peak shifts were observed, which are marked by orange arrows, indicating small changes to the overall crystal structures during the initial exothermic process. New PXRD signals corresponding to the final anthracene crystalline phase emerge only after 4 hours of heating for 1-D and 2-D, and 2 hours for 3-D, which are marked by the blue arrows. These results suggest that the initial exothermic chemical transformation (i.e., bond dissociation of dianthracene) leads to the formation of a transient intermediate phase of anthracene resembling the dianthracene crystal, which then undergoes a gradual phase transition to the final anthracene crystalline state, as proposed in Fig. 1a. Importantly, through comparison of DSC and PXRD data both the chemical transformation and phase transition are revealed to contribute to the overall energy release during the cycloreversion. | ||
| Fig. 2 Isothermal DSC plots of dianthracene (a) 1-D, (b) 2-D and (c) 3-D obtained at 48 °C, and the isothermal PXRD patterns of dianthracene (d) 1-D, (e) 2-D and (f) 3-D obtained at 49 ± 1 °C. | ||
To understand the chemical composition of such a transient intermediate phase, we employed isothermal solid-state NMR spectroscopy. We note that the initial dianthracenes do not contain any anthracenes and are stable at room temperature in the solid state; however, they completely convert to anthracenes upon heating over 3–10 hours, as confirmed by solid-state NMR (Fig. S2–S4†). The produced anthracenes are clean, in the absence of any thermal decomposition byproducts, and can be irradiated in solid again to generate dianthracenes via [4 + 4] cycloaddition. Solution-state NMR, on the other hand, does not accurately assess the composition of samples (Fig. S5–S10†), due to the solvation-induced D-to-A reversion, which directed us to using solid-state NMR spectroscopy. Time-dependent solid-state NMR spectroscopy of dianthracenes subject to in situ heating at 49 ± 1 °C (Fig. 3) also enabled the analysis of the reaction kinetics of dianthracene cycloreversion to anthracene (vide infra). First, the gradual decrease in the 13C NMR signal intensity of dianthracene with the concurrent formation of new signals assigned to anthracene confirms the successful cycloreversion process. In particular, the disappearance of dianthracene quaternary carbon signals (marked as b’ and c’ in Fig. 3) at <90 ppm along with the emergence of new 13C NMR signals, for instance of the C9 carbon of anthracene near 100 ppm (marked as c in Fig. 3), validates the aromatization of the central ring of the product. Inspection further shows that the signal positions of the R substituents on the C10 carbon (marked as b in Fig. 3) are often slightly different between dianthracene and anthracene. The sharpness of the major 13C NMR signals throughout the process indicates the preserved crystallinity of solid compounds during the reaction and supports the notion of a crystal-to-crystal phase transition as observed in PXRD analysis.
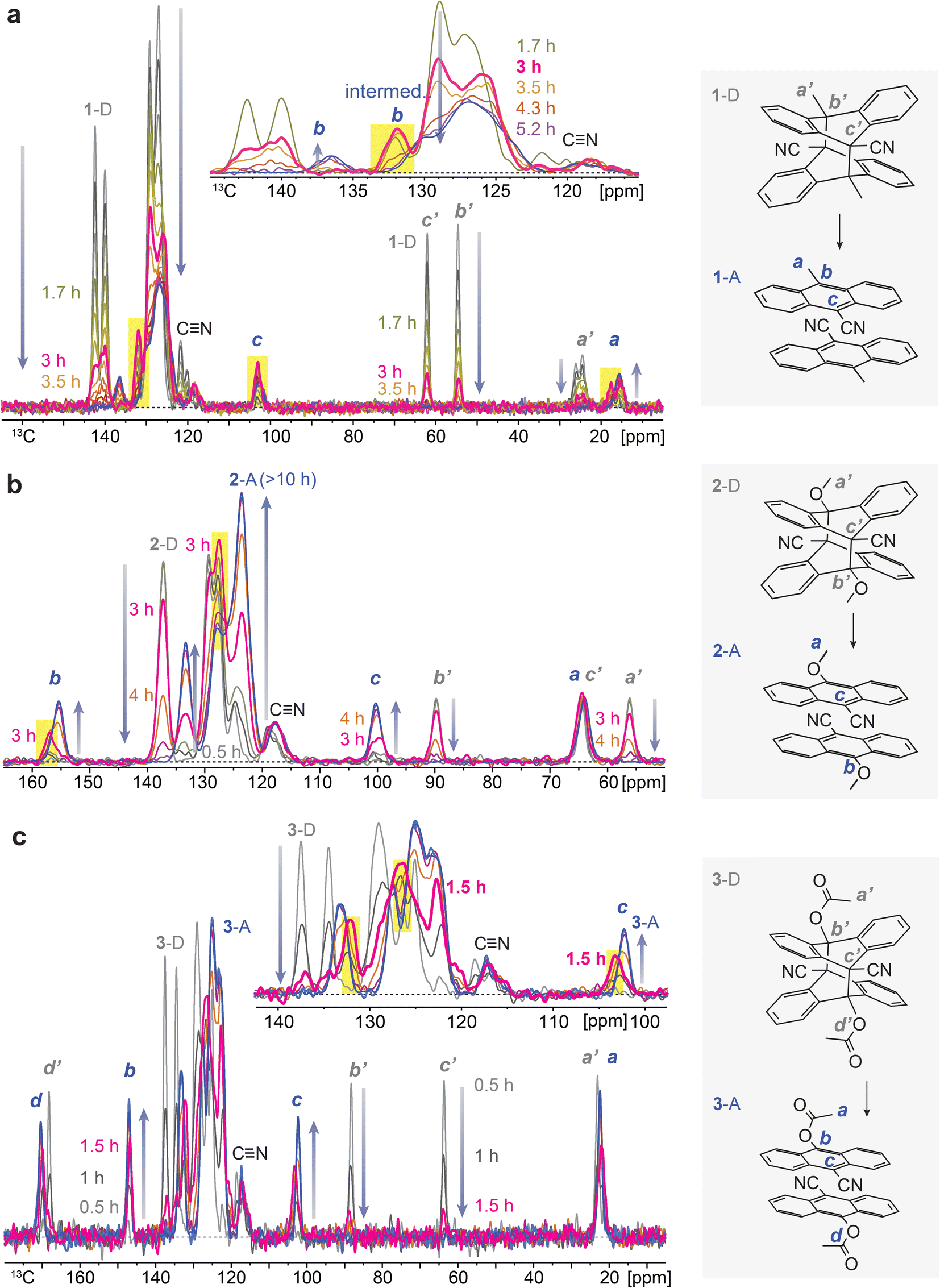 | ||
| Fig. 3 Series of isothermal solid-state 13C NMR spectra showing the conversion of (a) 1-D to 1-A, (b) 2-D to 2-A and (c) 3-D to 3-A at 49 ± 1 °C. The expanded view of the aromatic-carbon signals in (a) and (c) shows the appearance of an intermediate form of the anthracene with a maximum at 3 h for compound 1 and 1.5 h for compound 3, i.e., before the final anthracene crystal formation. The peaks of the intermediate phase that initially grow and then disappear are highlighted in the yellow boxes. | ||
Closer inspection of the NMR spectral series, in particular in the inset of Fig. 3a and c, reveals that some of the anthracene peaks have two components that are more or less resolved, which offers insight into the transient intermediate phase. For 1-A two anthracene signals with different time courses are seen most clearly at 137 and 132 ppm (C10, marked as b in Fig. 3a), for 2-A at 158 and 156 ppm (C10, marked as b in Fig. 3b), and for 3-A at 132 and 133 ppm (bridgehead carbons) as well as 103 and 102 ppm (C9, marked as c in Fig. 3c). The first anthracene signal appears early, passes through an intensity maximum and then disappears, while the appearance of the other component is delayed; its chemical shifts are those of the final anthracene crystal. The first component matches the definition of an intermediate in chemical kinetics, confirming the hypothesis from isothermal DSC and PXRD. Based on the chemical shifts, the transient species are identified as anthracenes. Therefore, they can be convincingly attributed to pairs of anthracenes formed by dianthracene decomposition. By necessity, these anthracene pairs are initially mostly surrounded by dianthracene molecules in a dianthracene crystal. This packing environment is different than in a neat anthracene crystal, resulting in slightly different chemical shifts of the newly formed anthracene molecules. In other words, the intermediate phase consists of a dianthracene crystal filled with a mix of dianthracenes and anthracene pairs, with the latter becoming more abundant with time.
This variable composition is reflected in the gradual shift of the Bragg peaks in PXRD at early to intermediate times (Fig. 2). In compounds 1 and 3, the intermediate component becomes the dominant species at intermediate times, which is confirmed by the quantitative analysis of the NMR data (vide infra). At that point, the dianthracene peaks broaden (e.g., near 140 ppm, see Fig. 3a), which can be attributed to the various packing environments of the remaining dianthracene molecules surrounded by a mix of new anthracene pairs and other residual dianthracenes. The signals of neat anthracene crystal increase significantly only when the intermediate peaks pass their maximum concentration, which is at different levels for compounds 1, 2 and 3.
The time dependence of the fractions of the three components (dianthracene, intermediate anthracene, and final anthracene) detected by NMR in the three samples is shown in Fig. 4. 1-D and 2-D undergo about 80% cycloreversion within 4 hours of heating at 49 ± 1 °C, and the anthracene in the transient intermediate phase, which first grows and then disappears, is detected before the formation of the stable anthracene crystalline phase (Fig. 4a and b). This intermediate anthracene, which is marked in orange, shows different maximum fractions for each dianthracene. The degree of accumulated intermediate phase reflects the activation energy barrier for the transition from the intermediate to the final anthracene phase. The continued heating beyond 4 hours induced the conversion of the intermediate phase to the final crystalline anthracene phase, marked in blue. A similar mechanism of cycloreversion was detected for 3-D, involving the growth of anthracene in an intermediate phase, followed by the conversion of the intermediate phase to the stable anthracene crystals (Fig. 4c). For 3-D, which reverts more rapidly than the other two dianthracenes, solid-state NMR measurements were also conducted at 25 °C to follow the process in more detail (Fig. S11†).
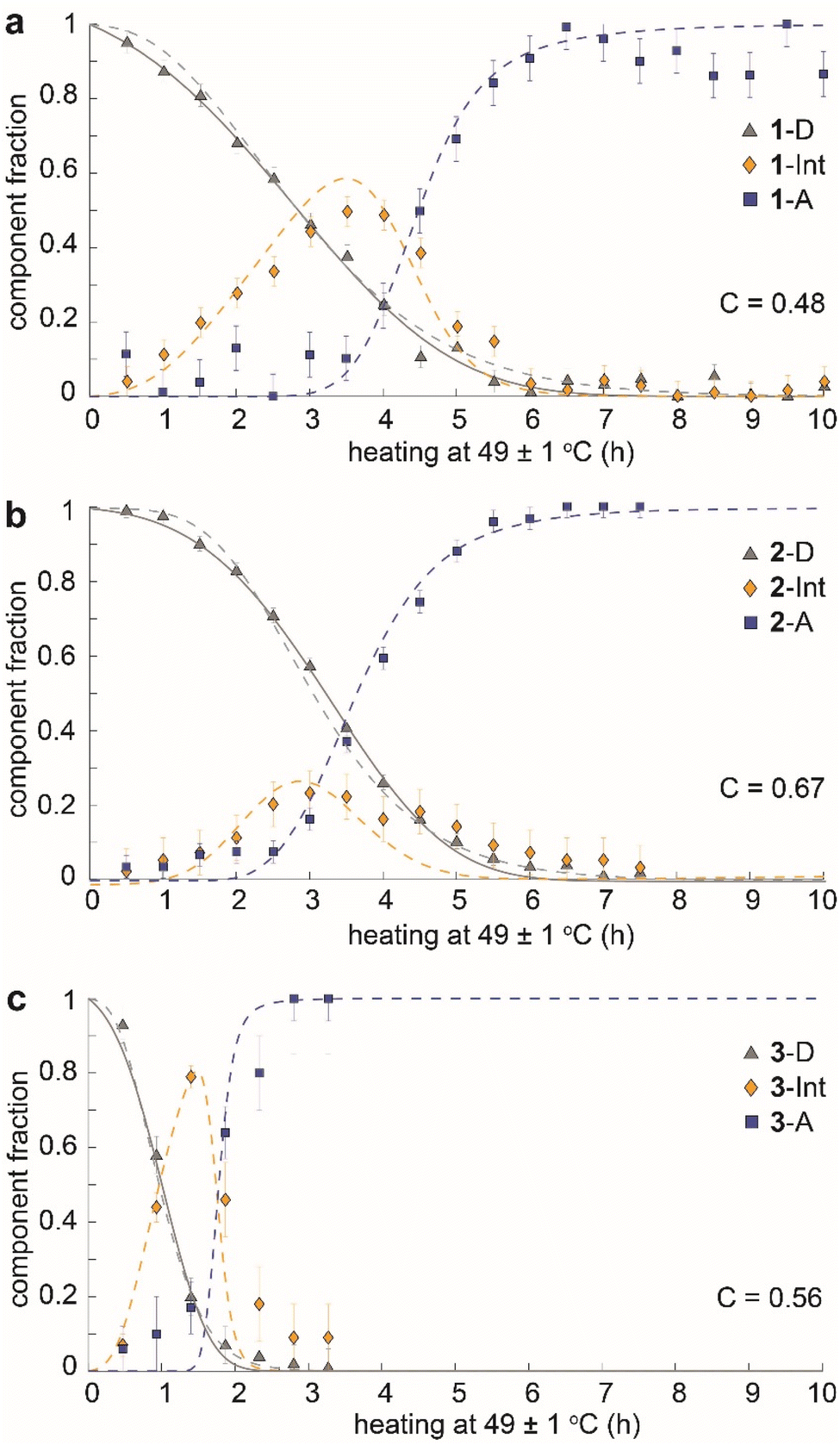 | ||
| Fig. 4 Time dependence of the fractions of dianthracene (grey triangles), intermediate anthracene in dianthracene crystals (orange diamonds), and final anthracene crystals (blue squares) during the cycloreversion of (a) 1-D to 1-A, (b) 2-D to 2-A, and (c) 3-D to 3-A, as derived from solid-state NMR. The dashed fit curves were obtained by the microscopically detailed models described below. The solid curve has been obtained from a modification of Avrami theory with partial cooperativity, and a cooperativity parameter (C) has been derived from it for each cycloreversion process. | ||
We also note that the sigmoidal shape of dianthracene conversion over time (grey line) reflects its auto-catalyzed cycloreversion in the solid state, and the kinetic analysis reveals different levels of molecular cooperativity for the cycloreversion of 1-D, 2-D, and 3-D. The decay curves of dianthracenes were fitted with the analytical equation of a partially cooperative 3D Avrami theory,41VD = exp(Kt04) × exp(−K(t + t0)4), where K is the Avrami constant and t0 is the pre-nucleation time42 that controls the ratio between random conversion and cooperative growth. The cooperativity parameter can be calculated as C = exp(−√2 × Kt01/4),41 ranging from 0 where the system approaches noncooperative first-order kinetics, to 1 where growth is fully cooperative. Compound 1 (R = CH3) showed the lowest cooperativity (C = 0.48), while compounds 2 (R = OCH3) and 3 (R = OAc) exhibited higher cooperativity (C = 0.67 and 0.56, respectively). In highly cooperative solid-state reactions, the local formation of intermediates and/or products facilitates further reactions, as a result of structural changes in the solid that lower the activation energy of reaction for the neighboring molecules.43,44
The observed behavior was then visualized by modeling the microscopically detailed, partially cooperative conversion in the dianthracene crystal, followed by conversion of anthracene-rich regions to neat anthracene crystals. Snapshots at five different time points are shown in Fig. 5. The spatial distribution and growth of the intermediate phases (yellow regions) are more uniform for compounds 1 and 3 (note that 3 at 1 h resembles 1 at 3 h) than compound 2, corroborating their relative cooperativity. Compound 2 displays a clustered formation of the intermediate phase, reflecting its high molecular cooperativity for the solid-state reaction, which then rapidly generates the final anthracene crystalline phase (Fig. 5b, blue region at 2 h). Such locally formed anthracene crystals propagate until the entire area is filled with the final anthracene phase.
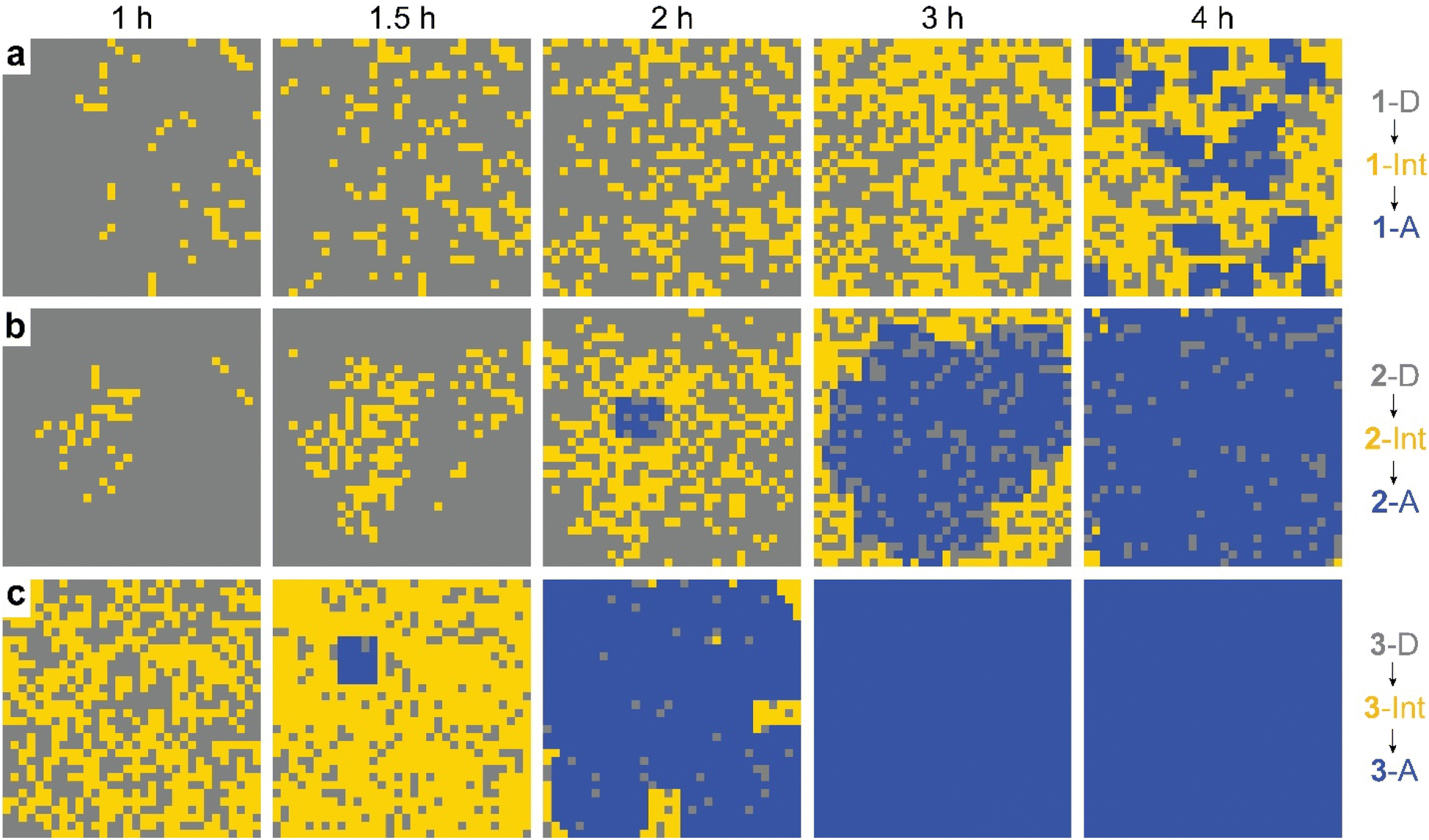 | ||
| Fig. 5 Snapshots of simulated conversion of dianthracene (grey) to anthracene pairs (yellow) in dianthracene crystals (i.e., an intermediate phase) followed by nucleation and growth of anthracene crystals (blue) for (a) 1-D, (b) 2-D, and (c) 3-D, producing the fit curves to the NMR data shown in Fig. 4. | ||
Finally, we investigated the relative contributions of chemical and physical transformations to the total energy release process, using density functional theory (DFT) calculations carried out using Quantum ESPRESSO (QE) code.45 The energetics of the overall cycloreversion can be described by the following three energy terms: ΔĒcry, ΔĒunit and ΔĒlatt. Based on Hess's law, only the initial dianthracene crystals and the final anthracene crystals were analyzed, without consideration of the intermediate phase structure.46 The following equations were developed to define each energy term (see ESI Section S6 for details†):
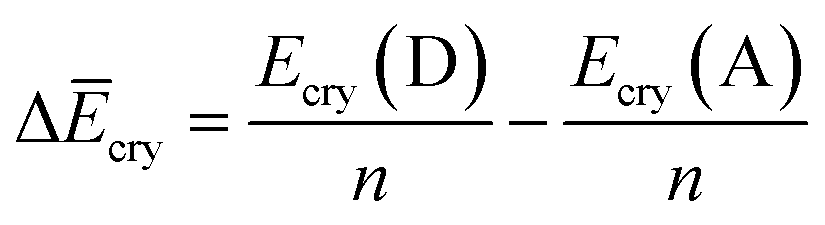 | (1) |
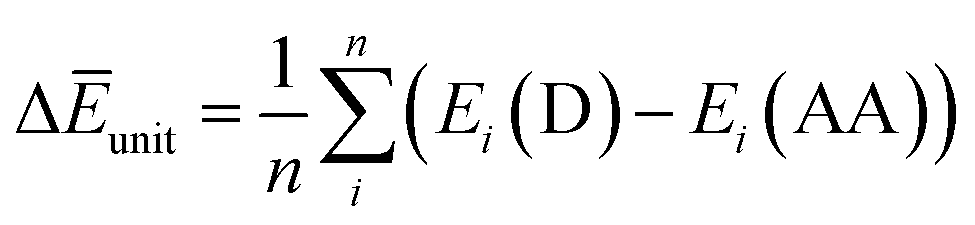 | (2) |
| ΔĒlatt = ΔĒcry − ΔĒunit | (3) |
E cry: the energy of the crystallographic unit cell containing multiple dianthracenes (Ecry(D)) or anthracenes (Ecry(A)).
ΔĒcry: the difference between the averaged energy of anthracene and dianthracene unit in crystals (Ecry divided by the number of cycloaddition units (n) in the unit cell) (Fig. S12–S14†).
ΔĒunit: the averaged dissociation energy of an individual dianthracene that is isolated by the cubic unit cell with the edge of 20 Å after the optimization using QE (Fig. S12–S14†).
ΔĒlatt: the difference in the lattice energy, which represents the environmental effect in the crystalline state during the cycloreversion process.
As per theoretical calculations, ΔĒcry represents the total energy released during the dianthracene cycloreversion, which is described as the sum of chemical dissociation energy, ΔĒunit, and crystal lattice stabilization energy, ΔĒlatt. Table 1 shows that for all compounds, the chemical dissociation serves as the main contributor to the overall energy release, whereas the lattice energy change during the cycloreversion offers a minor extra energy release. Even though the calculated values of total energy release are larger than the experimental ones, the relative degrees of ΔĒunit and ΔĒlatt provide important insights on the solid-state energy storage in the anthracene system, which can guide the further design of solid-state MOST energy storage systems.
| ΔĒcry | ΔĒunit | ΔĒlatt | |
|---|---|---|---|
| 1 | 191.7 | 157.6 | 34.1 |
| 2 | 185.9 | 169.2 | 16.7 |
| 3 | 187.4 | 154.1 | 33.3 |
Conclusions
The initial formation of a transient intermediate phase, featuring anthracene pairs in dianthracene crystals, was discovered and followed by the subsequent phase transition to the final anthracene crystals through isothermal time-dependent DSC, PXRD, and solid-state NMR. The fundamental understanding of the convoluted process allowed for the qualitative and calculation-based quantitative distinction between the chemical transformation and phase transition, both of which contribute to the overall energy storage and release in the anthracene-based MOST compounds. The solid-state kinetic analysis of cycloreversion revealed the molecular cooperativity in the auto-catalytic reaction for the anthracene derivatives. Solid-state NMR and PXRD probed the formation of an intermediate phase containing progressively more anthracene pairs within the dianthracene crystal, which at high anthracene loading exothermically converts to the stable anthracene crystal. The fundamental insights gained and the solid-state analytical methods discussed in this work will shed light on designing and studying novel solid-state MOST energy storage compounds that store solar photon energy and release heat through auto-catalysis.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
C. R. performed DSC and PXRD analyses. Z. S. and K. S.-R. conducted solid-state NMR analyses and Avrami theory modeling. R. K. and H. H. performed energy calculations. J. Y. C. and J. P. contributed crystal structure simulations based on PXRD. S. C. synthesized all compounds. G. G. D. H. conceived the project and refined the manuscript. All authors discussed the results and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based upon work supported by the Air Force Office of Scientific Research under Award Number FA9550-22-1-0254. G.G.D.H. acknowledges the NSF CAREER award (DMR-2142887), Alfred P. Sloan Foundation (FG-2022-18328), and the Camille and Henry Dreyfus Foundation (TC-23-028). S.C. was partly supported by Brandeis MRSEC (DMR-2011846). PXRD patterns were obtained on a powder X-ray diffractometer funded by the AFOSR DURIP award (FA9550-23-1-0072). The solid-state NMR spectrometer used in this project was funded by the National Science Foundation Major Research Instrumentation Program (Award 1726346). Computation time was provided by the SuperComputer System, Institute for Chemical Research, Kyoto University.References
- C. L. Sun, C. Wang and R. Boulatov, Applications of Photoswitches in the Storage of Solar Energy, ChemPhotoChem, 2019, 3(6), 268–283 CrossRef.
- Z. Wang, P. Erhart, T. Li, Z.-Y. Zhang, D. Sampedro, Z. Hu, H. A. Wegner, O. Brummel, J. Libuda, M. B. Nielsen and K. Moth-Poulsen, Storing energy with molecular photoisomers, Joule, 2021, 5(12), 3116–3136 CrossRef.
- A. Giménez-Gómez, L. Magson, C. Merino-Robledillo, S. Hernáez-Troya, N. Sanosa, D. Sampedro and I. Funes-Ardoiz, State-of-the-art and challenges towards a Molecular Solar Thermal (MOST) energy storage device, React. Chem. Eng., 2024, 9(7), 1629–1640 RSC.
- J. Orrego-Hernández, A. Dreos and K. Moth-Poulsen, Engineering of Norbornadiene/Quadricyclane Photoswitches for Molecular Solar Thermal Energy Storage Applications, Acc. Chem. Res., 2020, 53(8), 1478–1487 CrossRef PubMed.
- R. Schulte, S. Afflerbach, T. Paululat and H. Ihmels, Bis- and Tris-norbornadienes with High Energy Densities for Efficient Molecular Solar Thermal Energy Storage, Angew. Chem., Int. Ed., 2023, 62(38), e202309544 CrossRef.
- L. Dong, Y. Feng, L. Wang and W. Feng, Azobenzene-based solar thermal fuels: design, properties, and applications, Chem. Soc. Rev., 2018, 47(19), 7339–7368 RSC.
- A. Kunz, A. H. Heindl, A. Dreos, Z. Wang, K. Moth-Poulsen, J. Becker and H. A. Wegner, Intermolecular London Dispersion Interactions of Azobenzene Switches for Tuning Molecular Solar Thermal Energy Storage Systems, ChemPlusChem, 2019, 84(8), 1145–1148 CrossRef PubMed.
- M. Le and G. G. D. Han, Stimuli-Responsive Organic Phase Change Materials: Molecular Designs and Applications in Energy Storage, Acc. Mater. Res., 2022, 3(6), 634–643 CrossRef.
- Q. Qiu, S. Yang, M. A. Gerkman, H. Fu, I. Aprahamian and G. G. D. Han, Photon Energy Storage in Strained Cyclic Hydrazones: Emerging Molecular Solar Thermal Energy Storage Compounds, J. Am. Chem. Soc., 2022, 144(28), 12627–12631 CrossRef PubMed.
- Z. Wang, J. Udmark, K. Börjesson, R. Rodrigues, A. Roffey, M. Abrahamsson, M. B. Nielsen and K. Moth-Poulsen, Evaluating Dihydroazulene/Vinylheptafulvene Photoswitches for Solar Energy Storage Applications, ChemSusChem, 2017, 10(15), 3049–3055 CrossRef PubMed.
- M. Brøndsted Nielsen, N. Ree, K. V. Mikkelsen and M. Cacciarini, Tuning the dihydroazulene – vinylheptafulvene couple for storage of solar energy, Russ. Chem. Rev., 2020, 89(5), 573–586 CrossRef.
- Y. Kanai, V. Srinivasan, S. K. Meier, K. P. C. Vollhardt and J. C. Grossman, Mechanism of Thermal Reversal of the (Fulvalene)tetracarbonyldiruthenium Photoisomerization: Toward Molecular Solar–Thermal Energy Storage, Angew. Chem., Int. Ed., 2010, 49(47), 8926–8929 CrossRef PubMed.
- A. Lennartson, A. Lundin, K. Börjesson, V. Gray and K. Moth-Poulsen, Tuning the photochemical properties of the fulvalene-tetracarbonyl-diruthenium system, Dalton Trans., 2016, 45(21), 8740–8744 RSC.
- J. Usuba and G. G. D. Han, Photoswitch designs for molecular solar thermal energy storage, Trends Chem., 2023, 5(8), 577–580 CrossRef.
- Z. Wang, H. Hölzel and K. Moth-Poulsen, Status and challenges for molecular solar thermal energy storage system based devices, Chem. Soc. Rev., 2022, 51(17), 7313–7326 RSC.
- A. Gonzalez, E. S. Kengmana, M. V. Fonseca and G. G. D. Han, Solid-state photoswitching molecules: structural design for isomerization in condensed phase, Mater. Today Adv., 2020, 6, 100058 CrossRef.
- X. Li, S. Cho, J. Wan and G. G. D. Han, Photoswitches and photochemical reactions for optically controlled phase transition and energy storage, Chem, 2023, 9(9), 2378–2389 CAS.
- S. Kobatake, S. Takami, H. Muto, T. Ishikawa and M. Irie, Rapid and reversible shape changes of molecular crystals on photoirradiation, Nature, 2007, 446(7137), 778–781 CrossRef CAS.
- R. Klajn, Spiropyran-based dynamic materials, Chem. Soc. Rev., 2014, 43(1), 148–184 RSC.
- D. Kitagawa, K. Kawasaki, R. Tanaka and S. Kobatake, Mechanical Behavior of Molecular Crystals Induced by Combination of Photochromic Reaction and Reversible Single-Crystal-to-Single-Crystal Phase Transition, Chem. Mater., 2017, 29(17), 7524–7532 CrossRef.
- M. Samperi, B. Bdiri, C. D. Sleet, R. Markus, A. R. Mallia, L. Pérez-García and D. B. Amabilino, Light-controlled micron-scale molecular motion, Nat. Chem., 2021, 13(12), 1200–1206 CrossRef.
- G. R. Wilson, K. C. Park, G. C. Thaggard, C. R. Martin, A. R. Hill, J. Haimerl, J. Lim, B. K. P. Maldeni Kankanamalage, B. J. Yarbrough and K. L. Forrester, et al., Cooperative and Orthogonal Switching in the Solid State Enabled by Metal-Organic Framework Confinement Leading to a Thermo-Photochromic Platform, Angew. Chem., Int. Ed., 2023, 62(37), e202308715 CrossRef.
- M. Clerc, S. Sandlass, O. Rifaie-Graham, J. A. Peterson, N. Bruns, J. Read de Alaniz and L. F. Boesel, Visible light-responsive materials: the (photo)chemistry and applications of donor-acceptor Stenhouse adducts in polymer science, Chem. Soc. Rev., 2023, 52(23), 8245–8294 RSC.
- J. W. Chung, Y. You, H. S. Huh, B.-K. An, S.-J. Yoon, S. H. Kim, S. W. Lee and S. Y. Park, Shear- and UV-Induced Fluorescence Switching in Stilbenic π-Dimer Crystals Powered by Reversible [2 + 2] Cycloaddition, J. Am. Chem. Soc., 2009, 131(23), 8163–8172 CrossRef PubMed.
- L. Zhu, R. O. Al-Kaysi and C. J. Bardeen, Reversible photoinduced twisting of molecular crystal microribbons, J. Am. Chem. Soc., 2011, 133(32), 12569–12575 CrossRef CAS PubMed.
- G. K. Kole, T. Kojima, M. Kawano and J. J. Vittal, Reversible Single-Crystal-to-Single-Crystal Photochemical Formation and Thermal Cleavage of a Cyclobutane Ring, Angew. Chem., Int. Ed., 2014, 53(8), 2143–2146 CrossRef CAS.
- P. Kissel, D. J. Murray, W. J. Wulftange, V. J. Catalano and B. T. King, A nanoporous two-dimensional polymer by single-crystal-to-single-crystal photopolymerization, Nat. Chem., 2014, 6(9), 774–778 CrossRef CAS PubMed.
- M. J. Kory, M. Wörle, T. Weber, P. Payamyar, S. W. van de Poll, J. Dshemuchadse, N. Trapp and A. D. Schlüter, Gram-scale synthesis of two-dimensional polymer crystals and their structure analysis by X-ray diffraction, Nat. Chem., 2014, 6(9), 779–784 CrossRef CAS.
- F.-L. Hu, H.-F. Wang, D. Guo, H. Zhang, J.-P. Lang and J. E. Beves, Controlled formation of chiral networks and their reversible chiroptical switching behaviour by UV/microwave irradiation, Chem. Commun., 2016, 52(51), 7990–7993 RSC.
- T. Nishiuchi, K. Kisaka and T. Kubo, Synthesis of Anthracene-Based Cyclic π-Clusters and Elucidation of their Properties Originating from Congested Aromatic Planes, Angew. Chem., Int. Ed., 2021, 60(10), 5400–5406 CrossRef CAS.
- T. J. Gately, W. Sontising, C. J. Easley, I. Islam, R. O. Al-Kaysi, G. J. O. Beran and C. J. Bardeen, Effect of halogen substitution on energies and dynamics of reversible photomechanical crystals based on 9-anthracenecarboxylic acid, CrystEngComm, 2021, 23(34), 5931–5943 RSC.
- C. Sun, J. J. Oppenheim, G. Skorupskii, L. Yang and M. Dincă, Reversible topochemical polymerization and depolymerization of a crystalline 3D porous organic polymer with C–C bond linkages, Chem, 2022, 8(12), 3215–3224 Search PubMed.
- G. C. George and K. M. Hutchins, Solid-State [4+4] Cycloaddition and Cycloreversion with Use of Unpaired Hydrogen-Bond Donors to Achieve Solvatomorphism and Stabilization, Chem.–Eur. J., 2023, 29(65), e202302482 CrossRef.
- K. Hema, A. Ravi, C. Raju, J. R. Pathan, R. Rai and K. M. Sureshan, Topochemical polymerizations for the solid-state synthesis of organic polymers, Chem. Soc. Rev., 2021, 50(6), 4062–4099 RSC.
- K. Biradha and R. Santra, Crystal engineering of topochemical solid state reactions, Chem. Soc. Rev., 2013, 42(3), 950–967 RSC.
- G. Campillo-Alvarado, C. Li and L. R. MacGillivray, Topochemistry Meets Supramolecular Chemistry: Opportunities for Targeted Organic Synthesis in Molecular Crystals. in Reactivity in Confined Spaces, ed. Lloyd, G. and Forgan, R. S., The Royal Society of Chemistry, 2021 Search PubMed.
- S. Cho, J. Usuba, S. Chakraborty, X. Li and G. G. D. Han, Solid-state photon energy storage via reversible [2 + 2] cycloaddition of donor-acceptor styrylpyrylium system, Chem, 2023, 9(11), 3159–3171 CAS.
- S. Chakraborty, H. P. Q. Nguyen, J. Usuba, J. Y. Choi, Z. Sun, C. Raju, G. Sigelmann, Q. Qiu, S. Cho, S. M. Tenney, K. E. Shulenberger, K. Schmidt-Rohr, J. Park and G. G. D. Han, Self-activated energy release cascade from anthracene-based solid-state molecular solar thermal energy storage systems, Chem, 2024, 10, 1–14 Search PubMed.
- A. Gonzalez, M. Odaybat, M. Le, J. L. Greenfield, A. J. P. White, X. Li, M. J. Fuchter and G. G. D. Han, Photocontrolled Energy Storage in Azobispyrazoles with Exceptionally Large Light Penetration Depths, J. Am. Chem. Soc., 2022, 144(42), 19430–19436 CrossRef CAS.
- C. Raju, S. Kunnikuruvan and K. M. Sureshan, Topochemical Cycloaddition Reaction between an Azide and an Internal Alkyne, Angew. Chem., Int. Ed., 2022, 61(37), e202210453 CrossRef CAS PubMed.
- Z. Sun, G. G. D. Han and K. Schmidt-Rohr, Partially Cooperative Phase Conversions and Noninteger Avrami Exponents, under review.
- Y. Ishibashi and Y. Takagi, Note on Ferroelectric Domain Switching, J. Phys. Soc. Japan, 1971, 31(2), 506–510 CrossRef CAS.
- T. Salzillo, A. Brillante, T. Weber and A. D. Schlüter, What Changes in Topochemistry when Going from Small Molecule Dimerizations to Polymerizations in Single Crystals?, Helv. Chim. Acta, 2023, 106(3), e202200168 CrossRef CAS.
- A. E. Keating and M. A. Garcia-Garibay, Photochemical Solid-to-Solid Reactions, in Molecular and Supramolecular Photochemistry, ed. Ramamurthy, V. and Schanze, K. S., Marcel Dekker, 1998, vol. 2, pp. 195–248 Search PubMed.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni and I. Dabo, et al., QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials, J. Phys.: Condens.Matter, 2009, 21(39), 395502 CrossRef.
- J. Davik, Teaching Hess's Law, J. Chem. Educ., 1980, 57(12), 895 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta05282j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |