
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA mixed proton–electron-conducting cathode with a Ru nanoparticle catalyst for electrochemical ammonia synthesis based on a proton-conducting BZCYYb electrolyte†
Jiaqi
Chen
ab,
Wenbo
Gao
 *bc,
Liangzhu
Zhu
d,
Haoliang
Tao
d,
Sheng
Feng
b,
Hujun
Cao
bc,
Jianping
Guo
bc,
Yanxia
Chen
*a and
Ping
Chen
bc
*bc,
Liangzhu
Zhu
d,
Haoliang
Tao
d,
Sheng
Feng
b,
Hujun
Cao
bc,
Jianping
Guo
bc,
Yanxia
Chen
*a and
Ping
Chen
bc
aHefei National Research Center for Physical Sciences at the Microscale, Department of Chemical Physics, University of Science and Technology of China, Hefei 230026, P. R. China. E-mail: yachen@ustc.edu.cn
bDalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, P. R. China. E-mail: gaowenbo@dicp.ac.cn
cCenter of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing 100049, P. R. China
dNingbo Institute of Material Technology and Engineering, Chinese Academy of Science, Ningbo 315200, P. R. China
First published on 3rd September 2024
Abstract
Electrochemical ammonia synthesis (EAS) has emerged as a promising alternative to the traditional Haber–Bosch process. Indeed, N2 activation in room-temperature EAS systems remains a formidable challenge due to the strong N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond. Solid oxide proton conductor EAS (PCEAS) electrolysis cells operating at intermediate temperatures offer a promising solution by utilizing both temperature and potential. In this process, the design of the cathode is crucial, requiring abundant proton and electron conduction channels, along with highly active catalysts. Herein, we design a cathode composed of ruthenium–La0.6Sr0.4Co0.2Fe0.8O3−δ–BaZr0.1Ce0.7Y0.1Yb0.1O3−δ (Ru–LSCF–BZCYYb) to meet the aforementioned requirements for PCEAS. LSCF and BZCYYb form a porous skeleton at the cathode, with Ru nanoparticles dispersed on the surface of this structure. This configuration features numerous triple-phase boundaries (TPBs), facilitating the contact between activated N2, H+, and e−, thereby promoting electrochemical ammonia synthesis. The impregnated Ru–LSCF–BZCYYb|BZCYYb|Ni–BZCYYb PCEAS electrolysis cell exhibited a maximum NH3 formation rate of 5.14 × 10−11 mol s−1 cm−2 and a maximum Faraday efficiency (FE) of 0.128% at 400 °C and −0.2 V with H2 and N2 as feedstock gases. Its yield surpassed those of the mixed Ru–LSCF–BZCYYb|BZCYYb|Ni–BZCYYb and the impregnated Ru–BZCYYb|BZCYYb|Ni–BZCYYb by a factor of 3.9 and 11.5, respectively. The authenticity of ammonia synthesis is confirmed using the 15N2 isotope combined with NMR detection. This study also achieved EAS using water as the hydrogen source. This approach would better meet the future demand for EAS by directly using N2 and H2O.
N bond. Solid oxide proton conductor EAS (PCEAS) electrolysis cells operating at intermediate temperatures offer a promising solution by utilizing both temperature and potential. In this process, the design of the cathode is crucial, requiring abundant proton and electron conduction channels, along with highly active catalysts. Herein, we design a cathode composed of ruthenium–La0.6Sr0.4Co0.2Fe0.8O3−δ–BaZr0.1Ce0.7Y0.1Yb0.1O3−δ (Ru–LSCF–BZCYYb) to meet the aforementioned requirements for PCEAS. LSCF and BZCYYb form a porous skeleton at the cathode, with Ru nanoparticles dispersed on the surface of this structure. This configuration features numerous triple-phase boundaries (TPBs), facilitating the contact between activated N2, H+, and e−, thereby promoting electrochemical ammonia synthesis. The impregnated Ru–LSCF–BZCYYb|BZCYYb|Ni–BZCYYb PCEAS electrolysis cell exhibited a maximum NH3 formation rate of 5.14 × 10−11 mol s−1 cm−2 and a maximum Faraday efficiency (FE) of 0.128% at 400 °C and −0.2 V with H2 and N2 as feedstock gases. Its yield surpassed those of the mixed Ru–LSCF–BZCYYb|BZCYYb|Ni–BZCYYb and the impregnated Ru–BZCYYb|BZCYYb|Ni–BZCYYb by a factor of 3.9 and 11.5, respectively. The authenticity of ammonia synthesis is confirmed using the 15N2 isotope combined with NMR detection. This study also achieved EAS using water as the hydrogen source. This approach would better meet the future demand for EAS by directly using N2 and H2O.
 Wenbo Gao | Wenbo Gao received his PhD in January 2020 from Dalian Institute of Chemical Physics (DICP), Chinese Academy of Sciences, under the supervision of Prof. Ping Chen. Shortly thereafter, he received the Outstanding Young Doctoral Talent award and was appointed as an Associate Professor at DICP in October 2020. He has pioneered a novel metal hydride/imide/amide-mediated chemical looping process for ammonia synthesis and decomposition and has developed a series of highly efficient alkali (and alkaline earth) metal hydride catalysts for low-temperature thermo-catalytic ammonia synthesis. His current research is dedicated to advancing green ammonia synthesis, including thermo-catalysis, chemical looping, and electrochemistry. |
1. Introduction
Ammonia is an essential feedstock for industry and agriculture.1,2 Moreover, due to its high energy density and carbon-free nature, ammonia has garnered significant interest as a promising energy carrier.3–6 Currently, ammonia production relies predominantly on the Haber–Bosch (H–B) process, characterized by its harsh reaction conditions of high temperature and pressure. This process is not only energy-intensive, consuming 1–2% of the global energy production, but also responsible for the emission of about 500 Mt of CO2 annually.7,8 Consequently, there is a critical need to explore sustainable methods for ammonia synthesis. Electrochemical ammonia synthesis (EAS) offers a promising alternative, as it can be operated under milder conditions and powered by renewable electricity.Recently, significant advancements have been made in the field of EAS based on various electrolytes. Substantial research has been conducted in aqueous solutions, leading to the development of a wide range of catalysts.9,10 However, aqueous-electrolyte EAS has been questioned due to the presence of contaminants and its low ammonia yield.11,12 In addition, lithium-mediated EAS, which uses ionic liquids or organic solvents with dissolved lithium salts as electrolytes and can proceed at room temperature, has been proven to be reliable.13,14 However, this system often struggles to form a stable solid electrolyte interphase (SEI) layer, leading to poor long-term stability.15,16 Only a few systems can be operated for several hundred hours.17,18 Moreover, molten-salt-electrolyte EAS operates at elevated temperatures, facilitating N2 activation. Nevertheless, it is similarly constrained by system stability issues, primarily due to the strong corrosiveness of molten salt at high temperatures.19–21 It should be noted that the low solubility of N2 in solutions for the aforementioned liquid electrolyte systems represents an issue that needs to be addressed. Employing all-solid-state electrolytes, such as solid oxide oxygen-ion conductors or proton conductors, which operate at elevated temperatures, can address the issues of N2 activation and supply.22,23 Among these, solid-state proton conductors can work at around 400–500 °C,24 which is close to the operating temperature of thermal catalytic ammonia synthesis, thereby presenting greater potential.
A solid oxide proton-conductor EAS (PCEAS) electrolysis cell encompasses three principal components: the proton-conducting electrolyte, the anode layer, and the cathode layer. As shown in Fig. 1, a PCEAS electrolysis cell can utilize H2 or H2O as the hydrogen source.25 Protons are produced at the anode, and are then conducted through the electrolyte to the cathode. At the cathode, a nitrogen reduction reaction (NRR) occurs, where electrons, protons, and N2 react to form NH3 with the help of electrocatalysts. The reactions involved are illustrated in eqn (1)–(3).
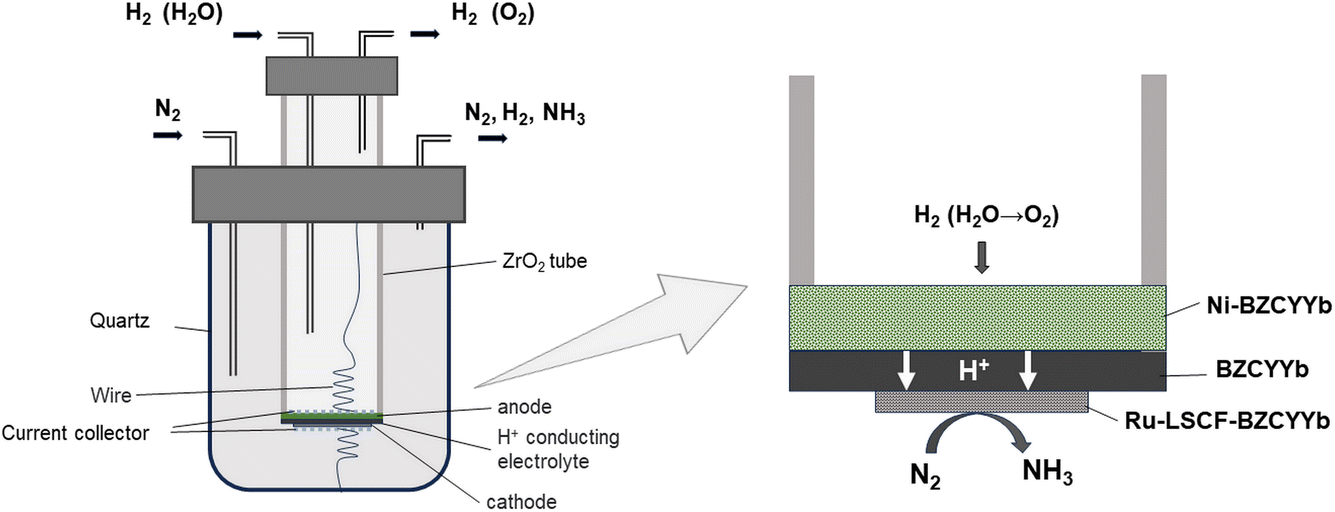 | ||
| Fig. 1 Schematic diagram of a device for a PCEAS electrolysis cell. | ||
Anode:
| 6H2 (6H2O) → 12H+ + 12e− (+ 3O2) | (1) |
Cathode:
| 2N2 + 12H+ + 12e− → 4NH3 | (2) |
Overall:
| 2N2 + 6H2 (6H2O) → 4NH3 (+ 3O2) | (3) |
A series of advances have been made in the electrolyte and anode, laying the foundation for designing efficient PCEAS electrolysis cells.26–30 However, there is a lack of a suitable cathode for efficiently reducing N2 and producing ammonia in the PCEAS process, which is crucial for the selectivity and activity of EAS. Since the pioneering work by Stoukides, a variety of cathodes have been explored in the PCEAS process.25,31–33 Compared to the earlier use of precious metals (Ag, Pd, etc.) as catalysts that operated at temperatures of about 700 °C, Fe-based and Ru-based catalysts, which are efficient in thermochemical ammonia synthesis, could also effectively activate N2 under electrochemical conditions, thereby lowering the temperature required for the PCEAS process.34–38 Furthermore, the design of composite cathodes enhances the ammonia production activity of PCEAS electrolysis cells.38,39 This may be attributed to the extension of the three-phase boundary (TPB). The significance of the TPB has also been confirmed in other electrochemical processes.40–42 The TPB, where the proton-conducting electrolyte, electron conductor, gas reactant, and catalyst nanoparticles simultaneously come into contact, offers the active site for electrochemical reactions and ultimately determines the performance. Hence, optimizing the design of the three-phase interface and incorporating efficient NRR catalysts may create a highly efficient cathode, facilitating an effective PCEAS process.
In this study, BaZr0.1Ce0.7Y0.1Yb0.1O3−δ (BZCYYb) was selected as the electrolyte due to its high ionic conductivity (∼0.005 S cm−1 at 400 °C) and wide application in fuel cells and water electrolysis.43,44 Ni is widely used as an anode material in high-temperature solid oxide cells, due to its high hydrogen electrocatalytic oxidation activity, excellent compatibility with electrolytes, and its inherent high conductivity as a metal.45 Based on the BZCYYb|Ni–BZCYYb half-cells, we investigated the impact of cathode design on the performance of PCEAS electrolysis cells. In the cathode, we employed Ru as the catalyst, BZCYYb as the proton conductor, and La0.6Sr0.4Co0.2Fe0.8O3−δ (LSCF) as the electron conductor. This choice is based on the compatibility between LSCF and BZCYYb.46,47 A cathode consisting of an LSCF–BZCYYb composite skeleton loaded with dispersed Ru nanoparticles via an impregnation method exhibits enhanced activity attributable to its expanded TPB. Furthermore, the feasibility of steam electrolysis at the anode to provide protons for the PCEAS is also demonstrated. This suggests potential for further application.
2. Experimental section
2.1 Materials synthesis and characterization
BZCYYb powders were synthesized by the solid-state reaction. BaCO3 (SCR Co., Ltd, China), ZrO2 (Aladdin), CeO2 (Aladdin), Y2O3 (Innochem Co., Ltd, China), and Yb2O3 (Aladdin) were mixed in a stoichiometric ratio and ball-milled in ethanol for 24 h. The dried samples were calcined at 1100 °C for 12 h. The process of ball-milling and sintering was repeated once to obtain pure phase BZCYYb.LSCF powders were synthesized by the sol–gel method. La(NO3)3·6H2O (SCR Co., Ltd, China), Sr(NO3)2 (Kermel Co., Ltd, China), Co(NO3)2·6H2O (SCR Co., Ltd, China), and Fe(NO3)3·9H2O (SCR Co., Ltd, China) were dissolved in a stoichiometric ratio in deionized water, along with ammonium citrate (SCR Co., Ltd, China) as a chelating reagent (molar ratio of ammonium citrate![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :metal cations = 1.5:1). This solution was continuously stirred at 80 °C to evaporate the water until a transparent gel was obtained. The gel was then heated to 300 °C, and then the resulting ashes were calcined at 800 °C for 2 h.
:metal cations = 1.5:1). This solution was continuously stirred at 80 °C to evaporate the water until a transparent gel was obtained. The gel was then heated to 300 °C, and then the resulting ashes were calcined at 800 °C for 2 h.
2.2 Cell fabrication
Anode-supported electrolysis cells with the configurations of impregnated Ru–LSCF–BZCYYb (I-Ru–LSCF–BZCYYb)|BZCYYb|Ni–BZCYYb, impregnated Ru–BZCYYb (I-Ru–BZCYYb)|BZCYYb|Ni–BZCYYb, and mixed Ru–LSCF–BZCYYb (M-Ru–LSCF–BZCYYb)|BZCYYb|Ni–BZCYYb were used in the electrochemical experiment.First, a mixture of BZCYYb, NiO (Macklin), starch (Macklin) (weight ratio of 65:35:20), and binder was ball-milled in ethanol. After drying, the powder was pressed into a 16 mm diameter pellet. Then, the green pellet was sintered at 1200 °C for 2 h to form an anode support. The suspension of BZCYYb was dropped on one side of the pellets and the organic matter was removed by heat treatment. Finally, the anode–electrolyte assemblies were sintered at 1450 °C for 18 h to form the BZCYYb|NiO–BZCYYb half-cell. For more details refer to the study by Zhu et al.48 LSCF, BZCYYb, and starch (mass ratio of 6:4:2) were then mixed thoroughly with a 6 wt% ethylcellulose–terpineol (Aladdin) binder to prepare the cathode slurry which was painted on the BZCYYb electrolyte. This pellet was calcined at 1100 °C and it took 2 h to form a porous skeleton. The skeleton was impregnated using 1 M Ru(NO)(NO3)3 (Alfa) solution (a mixture of water and ethanol, volume ratio of 1:1), and the amount of impregnated Ru accounts for 6.5 wt% of the cathode layer which was confirmed by inductively coupled plasma optical emission spectrometry (ICP-OES, Agilent 5110). Subsequently, this cell was dried under vacuum and sintered under an air atmosphere at 500 °C for 1 h to decompose all organics and Ru(NO)(NO3)3. Before electrochemical experiments and characterization, the prepared cathode consisting of impregnated RuO2–LSCF–BZCYYb was reduced to I-Ru–LSCF–BZCYYb by pure H2 for 2 h. The area of the cathode was about 0.28 cm2. As a comparison sample, the I-Ru–BZCYYb|BZCYYb|Ni–BZCYYb electrolysis cell was prepared in a similar way to the above process, except that LSCF was not added. It should be noted that due to the poor sintering properties of BZCYYb, the sintering temperature was increased to 1300 °C.
Ru powder, another comparison sample, was directly mixed with LSCF, BZCYYb, starch, and binder. This slurry was painted on one BZCYYb electrolyte and then sintered at 1100 °C for 2 h. The prepared cell was also reduced by pure H2 for 2 h to produce the M-Ru–LSCF–BZCYYb|BZCYYb|Ni–BZCYYb electrolysis cell.
2.3 Characterization
The crystalline phases of materials were verified through X-ray diffraction (XRD, X'Pert, PANalytical B.V., Netherlands). The microstructures of the Ru–LSCF–BZCYYb catalyst were detected by transmission electron microscopy (TEM, JEM-2100X, JEOL, Japan). The structures and morphologies of the different cathodes were observed by scanning electron microscopy (SEM, JMS-7800F, JEOL, Japan).2.4 Electrochemical measurements
The setup for the electrochemical measurements is shown in Fig. 1. The cell was sealed on the end of the ZrO2 tube using a dielectric ceramic adhesive (552-VFG, Aremco). A silver electric adhesive (DAD-87, Shanghai, China) was used as the current collector, and Ag wires were used to maintain the electrical connection. Before measurement, both the inner and outer tubes were filled with H2 at 600 °C for 2 h, reducing the metal oxide of Ni and Ru to the pure metallic state. Then, the gaseous mixture of H2 and Ar (5% H2–95% Ar) was fed into the inner tube to work as an anodic chamber (counter electrode), with a flow rate of 30 mL min−1. The N2 gas was fed into the outer tube to work as a cathodic chamber (working electrode), with a flow rate of 60 mL min−1.49 Pure N2 gas should be fed at least 2 h before the experiment to remove residual H2. The AC impedance was measured at open circuit voltage (OCV) with a frequency range of 0.1 Hz to 1 MHz. The value at high frequency was used for iR compensation (90%). The polarization curves were recorded in the potential range (OCV to−0.8 V) with a sweep rate of 10 mV s−1. The effects of temperature (300–500 °C) and potential (0 to −0.6 V) on the activity of ammonia formation were measured through chronoamperometry. The chronoamperometry lasted more than 10 minutes for each potential, and the NH3 in the outlet gas was collected by 1 mM H2SO4 solution (6 mL). To confirm the ammonia concentration, 2 mL of this solution was tested using the indophenol blue colorimetry method with a UV-visible spectrometer.50–52 The standard curve of absorbance at 655 nm and concentration is shown in Fig. S1.† After each potentiostatic measurement, the tail gas without electricity was also collected as blank. To avoid the interference of potential trace leakage of hydrogen with the electrocatalytic ammonia formation, the value of the blank was subtracted from the results of UV-vis measurements, although ammonia production was not observed without electricity below 500 °C. During the coupled electrolysis of steam, the anode gas was substituted with wet argon gas, and the potential range of the experiment was adjusted (OCV to −1.6 V). The remaining parameters remained consistent.The yield rate of NH3 generation and its faradaic efficiency (FE) during chronoamperometry are calculated using eqn (4) and (5):
 | (4) |
 | (5) |
2.5 15N2 isotopic labeling experiment
To identify the source of NH3, N2 isotope labeling experiments were conducted using 15N2 (Linde Industrial Gases) as the feed gas. Electrolysis was performed for 2 h under optimal conditions. To conserve the expensive 15N2 gas, its flow rate was adjusted to 6 mL min−1, which is lower than the usual test conditions, while the flow rate of H2 was kept constant. 1 mL H2SO4 solution was used to collect the NH3 in the outlet gas. For the NMR studies, the pH of the solutions to be tested was adjusted to 2 using concentrated hydrochloric acid aqueous solution. A solution of 5 vol% dimethyl sulfoxide d6 (99.9 atom% D, 0.03% (v/v) tetramethylsilane) was added for deuterium locking and referencing. The solution was then taken out for 1H NMR measurement (JEOL, JNM-ECZ400S, 400 MHz). The quantification of 15NH3 or 14NH3 was performed using the method established in our previous research.533. Results and discussion
3.1 XRD and microscopic analysis of cathodes
The as-synthesized materials of LSCF and BZCYYb and three different cathodes of I-Ru–LSCF–BZCYYb, I-Ru–BZCYYb, and M-Ru–LSCF–BZCYYb were characterized by XRD. The successful synthesis of pure LSCF and BZCYYb is confirmed by the peaks observed in the XRD patterns, consistent with those reported in previous literature (Fig. S2†).43,54 The LSCF–BZCYYb composite, as the support of the I-Ru–LSCF–BZCYYb cathode, displays diffraction peaks that matched well with those of both LSCF and BZCYYb (Fig. S2†), indicating that the interaction between these two materials is negligible. Fig. 2 shows the XRD patterns of the three cathodes. After loading the ruthenium catalyst, the XRD pattern of I-Ru–LSCF–BZCYYb shows additional diffraction peaks at 38.3° and 44.0° compared to the LSCF–BZCYYb composite, corresponding to the crystal planes of Ru (100) and Ru (101). The original diffraction peaks of LSCF and BZCYYb are unchanged, indicating the structure of the LSCF–BZCYYb is preserved through the impregnation process. The XRD pattern of the I-Ru–BZCYYb cathode reveals a discernible diffraction peak attributable to Ru at 44.0°, while the diffraction peaks of BZCYYb also remain essentially unchanged. However, the M-Ru–LSCF–BZCYYb displays diffraction peaks that are significantly different from those of Ru, LSCF, and BZCYYb. It should be noted that in the preparation of the impregnated cathode, the Ru precursor and LSCF–BZCYYb were calcined at 500 °C and then reduced in H2 gas at 600 °C, while in the preparation of the mixed cathode, metallic Ru, LSCF, and BZCYYb were co-calcined at 1100 °C and then reduced in H2 gas at 600 °C. Hence, Ru, LSCF, and BZCYYb might react to produce new substances upon calcination at higher temperatures. However, the phase structure has not yet been identified. | ||
| Fig. 2 XRD patterns of the I-Ru–LSCF–BZCYYb, I-Ru–BZCYYb, and M-Ru–LSCF–BZCYYb cathodes and the standard XRD pattern of Ru. | ||
The morphology and microstructure of the cathodes were then characterized using SEM and TEM. The SEM image of the LSCF–BZCYYb composite reveals a porous structure, which could potentially serve as a skeleton for loading the Ru catalyst (Fig. S3†). Fig. 3a–c show the SEM images of the I-Ru–LSCF–BZCYYb, M-Ru–LSCF–BZCYYb, and I-Ru–BZCYYb cathodes, respectively. Both the I-Ru–LSCF–BZCYYb and M-Ru–LSCF–BZCYYb cathodes maintain a porosity similar to that of LSCF–BZCYYb (Fig. 3a and b). In contrast, the I-Ru–BZCYYb cathode exhibits lower porosity, possibly due to the sintering of BZCYYb in the cathode at high temperatures (Fig. 3c). Fig. 3d displays a higher magnification SEM image of the I-Ru–LSCF–BZCYYb cathode, showing that small particles ranging from a few to tens of nanometers in size exist on the surface of the LSCF–BZCYYb skeleton, which may be nano-sized Ru particles. However, nearly no Ru particles are observed on the M-Ru–LSCF–BZCYYb cathode, indicating that Ru is almost entirely integrated into the LSCF–BZCYYb skeleton (Fig. 3e). This is consistent with the XRD results, which show no diffraction peaks of Ru (Fig. 2). For the I-Ru–BZCYYb cathode, there are numerous regions where Ru aggregates (Fig. 3f). Among the three cathodes discussed, the I-Ru–LSCF–BZCYYb cathode may be more suitable for use in the PCEAS electrolysis cell due to its pore structure being conducive to gas diffusion and providing a large number of active sites. Additionally, the uniformly dispersed Ru particles could be beneficial for N2 activation. So further TEM studies were conducted on the I-Ru–LSCF–BZCYYb cathode. Fig. 3g shows that particles of around 10 nm are present on the surface of the I-Ru–LSCF–BZCYYb cathode. The lattice spacing of these nanoparticles is 0.209 nm, corresponding to the (101) lattice plane of Ru, consistent with the XRD results. EDX analysis further confirms that these particles located at the edge of the material are metallic Ru (Fig. S4†). Additionally, lattice spacings at around 0.281 nm and 0.316 nm could also be observed, which likely correspond to the (110) lattice plane of LSCF and the (002) lattice plane of BZCYYb, respectively (Fig. 3h).54–57 A SEM image of the cross-section of the whole cell with the I-Ru–LSCF–BZCYYb cathode is shown in Fig. 3i. This suggests good contact between the electrolyte and both electrodes, alongside the presence of a porous structure in the cathode. In summary, a porous cathode was successfully prepared with mixed proton and electron conduction based on BZCYYb and LSCF, and nano-sized Ru particles were uniformly dispersed into the skeleton through impregnation.
 | ||
| Fig. 3 SEM images of (a and d) the I-Ru–LSCF–BZCYYb cathode, (b and e) the M-Ru–LSCF–BZCYYb cathode, and (c and f) the I-Ru–BZCYYb cathode. (g and h) HR-TEM images of the I-Ru–LSCF–BZCYYb cathode. (i) SEM image of the cross-section of the I-Ru–LSCF–BZCYYb|BZCYYb|Ni–BZCYYb electrolysis cell. | ||
3.2 Electrochemical analysis of the PCEAS electrolysis cells
We investigated the electrochemical properties of the I-Ru–LSCF–BZCYYb|BZCYYb|Ni–BZCYYb, M-Ru–LSCF–BZCYYb|BZCYYb|Ni–BZCYYb, and I-Ru–BZCYYb|BZCYYb|Ni–BZCYYb electrolysis cells. The investigation was carried out in the reactor displayed in Fig. 1, using N2 and 5% H2–95% Ar as reaction gases for the cathode and anode, respectively. Good electrochemical properties may be a prerequisite for high performance in electrochemical ammonia synthesis, so we first explored the electrochemical properties of these cells. However, it should be noted that this system involves both the NRR and hydrogen evolution reaction (HER), and the performance of electrochemical ammonia synthesis is influenced not only by the electrochemical properties of the cell but also by the electrocatalysts in the electrode. The electrochemical ammonia synthesis performance will be discussed in the next section.Fig. 4a–c display the voltammograms of these cells at 300, 400, and 500 °C. Among them, the I-Ru–LSCF–BZCYYb cathode exhibits superior performance, achieving a remarkable current density of 150 mA cm−2 at a potential of −0.8 V at 500 °C. This current density is slightly higher than that of the M-Ru–LSCF–BZCYYb cathode and nearly 4–5 times higher than that of the I-Ru–BZCYYb cathode. Even at a lower temperature of 300 °C, the cell with the I-Ru–LSCF–BZCYYb cathode exhibits a current density of 10 mA cm−2. Furthermore, the voltammetric characteristic of a PCEAS electrolysis cell with the LSCF–BZCYYb skeleton as a cathode was tested (Fig. S5†). At 400 °C and −0.8 V, the current density for the cell utilizing the I-Ru–LSCF–BZCYYb cathode is nearly 5.5 times higher than that of the LSCF–BZCYYb cathode. In summary, the cathode components of LSCF, BZCYYb, and Ru, and the preparation method collectively influence the electrochemical performance of the PCEAS electrolysis cell.
 | ||
| Fig. 4 (a–c) j–E curves with a scan rate of 10 mV s−1, and (d–f) EIS at OCV of the I-Ru–LSCF–BZCYYb|BZCYYb|Ni–BZCYYb, M-Ru–LSCF–BZCYYb|BZCYYb|Ni–BZCYYb, and I-Ru–BZCYYb|BZCYYb|Ni–BZCYYb electrolysis cells at different temperatures. Reaction gas: pure N2 at the working electrode; 5% H2–95% Ar at the counter electrode (reference electrode). | ||
The AC impedance spectra of the three aforementioned PCEAS electrolysis cells were measured under open circuit voltage at 300, 400, and 500 °C, to further analyze the effect of different electrode designs on electrochemical properties, and the results are displayed in Fig. 4d–f. We fitted the impedance spectrum with the equivalent circuit as shown in Fig. S6.† The fitted values of ohmic resistance (RΩ) and polarization resistance (RP, total of RP1 and RP2) are shown in Table 1. The RΩ is primarily associated with the ion and electron transport properties of the electrolyte material, electrode layer, and the interface between the electrode and electrolyte. Given that the electrolyte and anode layers are identical across all three cells, variations in RΩ can be attributed to differences in the cathode layer and its interface with the electrolyte. The RΩ of electrolysis cells with I-Ru–LSCF–BZCYYb and M-Ru–LSCF–BZCYYb cathodes is significantly lower than that of the I-Ru–BZCYYb cathode at equivalent temperatures. Specifically, the RΩ of the electrolysis cell using the I-Ru–LSCF–BZCYYb cathode is only 29.9, 8.1, and 5.6 Ω cm2 at 300, 400, and 500 °C, respectively. The decrease of RΩ may be attributed to the incorporation of LSCF, which introduces more conductive pathways for electrons within the porous skeletal structure of the cathode. Conversely, for the I-Ru–BZCYYb cathode, ruthenium, serving as the sole electron conductor, tends to aggregate into large and isolated particles on the surface of the dense BZCYYb electrolyte (Fig. 3c). These particles may impair physical contact with the substrate, potentially hindering the formation of an effective conductive network within the electrode.58
| Temperature | I-Ru–LSCF–BZCYYb | M-Ru–LSCF–BZCYYb | I-Ru–BZCYYb | |||
|---|---|---|---|---|---|---|
| R Ω | R P | R Ω | R P | R Ω | R P | |
| 300 °C | 29.9 | 407.4 | 18.6 | 326.1 | 52.3 | 758.2 |
| 400 °C | 8.1 | 185.9 | 8.3 | 77.6 | 12.5 | 322.4 |
| 500 °C | 5.6 | 90.3 | 5.7 | 40.8 | 9.3 | 197.2 |
The RP is primarily related to the electrochemical activity of the electrode materials and the mass transport properties of the reactants. The RP values of electrolysis cells using I-Ru–LSCF–BZCYYb and M-Ru–LSCF–BZCYYb cathodes are only 90.3 and 40.8 Ω cm2 at 500 °C, respectively, which are 1.2 and 3.8 times lower than that of the I-Ru–BZCYYb cathode at the same temperature. This suggests that the electrochemical processes occur with greater ease on the cathodes composed of I-Ru–LSCF–BZCYYb and M-Ru–LSCF–BZCYYb configurations. For the I-Ru–LSCF–BZCYYb cathode, the highly dispersed Ru nanoparticles and the porous skeleton structure of the cathode (Fig. 3a and d) provide abundant active sites for nitrogen or proton activation, resulting in low polarization resistance. Additionally, the M-Ru–LSCF–BZCYYb cathode also presents a similarly porous skeleton structure (Fig. 3b), and the integration of Ru into the LSCF–BZCYYb framework (Fig. 2 and 3e) may form highly efficient active sites for these electrochemical reactions, resulting in lower polarization resistance. Subsequent studies indicate that these active sites are more inclined towards the hydrogen evolution reaction.
3.3 Ammonia synthesis performance of the PCEAS electrolysis cells
Due to the fine electrochemical behavior and definite composition of the I-Ru–LSCF–BZCYYb cathode, we first studied the performance of the I-Ru–LSCF–BZCYYb|BZCYYb|Ni–BZCYYb electrolysis cell in EAS under various temperature and potential conditions. The electrochemical reaction was carried out in the reactor as shown in Fig. 1, where the exhaust gas passes through a dilute sulfuric acid solution, and the ammonia content in the solution is detected using the UV-visible colorimetric method. Fig. 5a illustrates the ammonia synthesis rate and FE of this cell under −0.2 V at 300, 400, and 500 °C. Both the ammonia production rate and FE exhibit volcano-shaped curve characteristics, reaching their maximum values at 400 °C with a rate of 5.14 × 10−11 mol s−1 cm−2 and FE of 0.128%. More data about the j–t curves and colorimetric results can be found in Fig. S7 and S8 and Tables S1–S4.† | ||
| Fig. 5 NH3 formation rate and Faraday efficiency of the I-Ru–LSCF–BZCYYb|BZCYYb|Ni–BZCYYb electrolysis cell (a) at different temperatures and an applied potential of −0.2 V, and (b) at 400 °C and different potentials. Reaction gas: pure N2 at the working electrode; 5% H2–95% Ar at the counter electrode (reference electrode). | ||
Electrochemical testing in the prior section shows that significant ohmic and polarization resistances are present at a low temperature of 300 °C. Hence, the low rate can be attributed to the low ion conductivity of the electrolyte and the reduced efficiency of Ru in activating N2 at a low temperature. At an elevated temperature of 500 °C, the decline in the rate of ammonia synthesis could be attributed to ammonia decomposition, a phenomenon that has been previously reported in the literature.59 The volcano-shaped curve relationship of FE may be attributed to the differing temperature sensitivities of the HER and the NRR, as well as the influence of the ammonia decomposition side reaction at a high temperature. Fig. 5b demonstrates the relationship between the ammonia synthesis rate and the applied potential at 400 °C. It can be seen that as the applied potential becomes more negative, both the rate of ammonia synthesis and the FE initially increase and then decrease, reaching their maximum values at −0.2 V. At higher potentials, the decrease in the rate of ammonia synthesis and FE can be attributed to the enhanced HER side reaction. Additionally, more hydrogen may occupy active sites on the surface of the cathode at higher potentials, impeding the NRR.
The electrochemical ammonia synthesis activity of the M-Ru–LSCF–BZCYYb|BZCYYb|Ni–BZCYYb and I-Ru–BZCYYb|BZCYYb|Ni–BZCYYb electrolysis cells was also evaluated, with the results shown in Fig. S9–S13 and Tables S1–S4.†Fig. 6a compares the ammonia synthesis rate and FE of cells configured with different cathodes under conditions of 400 °C and −0.2 V, while Fig. 6b presents the performance of these cells under their optimal working conditions. Although the M-Ru–LSCF–BZCYYb cathode exhibits excellent electrochemical performance, its ammonia synthesis activity is notably poor, with only weak activity observed at 400 °C and −0.1 V. In contrast, the I-Ru–BZCYYb electrode, despite having lower electrochemical performance across all potential and temperature ranges, demonstrates higher electrochemical ammonia synthesis activity. Meanwhile, the I-Ru–LSCF–BZCYYb cathode exhibits the highest activity, with its ammonia synthesis rate being 3.9 times higher than that of the I-Ru–BZCYYb cathode. As a reference, the LSCF–BZCYYb cathode was also tested, showing almost no electrochemical ammonia synthesis activity, which indicates that metallic Ru catalysts in the cathode could play a significant role. Combining the characterization results from the prior section, the low ammonia synthesis activity of the M-Ru–LSCF–BZCYYb may be due to the reaction between Ru and LSCF, BZCYYb in the cathode to generate new substances, resulting in almost no presence of metallic Ru. In addition, the new substances may possess excellent HER activity, leading to low polarization resistance. This explains the excellent electrochemical performance but poor ammonia synthesis activity of the M-Ru–LSCF–BZCYYb cathode. Considering the characterization results of the cathodes (Fig. 3), we speculate that there may be three possible reasons for the best performance of the I-Ru–LSCF–BZCYYb cathode: (1) the pore structure of the LSCF–BZCYYb in both the I-Ru–LSCF–BZCYYb cathodes likely enhances the transport of reactants, a feature not present in the I-Ru–BZCYYb cathode. (2) The LSCF–BZCYYb composite cathode provides ample conductive pathways for both protons and electrons, while Ru nanoparticles offer active sites for N2 activation. These factors contribute to an increased three-phase interface. (3) For the I-Ru–BZCYYb cathode, the aggregation of Ru could result in fewer reduced active sites.
 | ||
| Fig. 6 Comparison of NH3 formation rate and Faraday efficiency of the I-Ru–LSCF–BZCYYb|BZCYYb|Ni–BZCYYb, the M-Ru–LSCF–BZCYYb|BZCYYb|Ni–BZCYYb, and the I-Ru–BZCYYb|BZCYYb|Ni–BZCYYb electrolysis cells (a) at 400 °C and −0.2 V, and (b) under the optimum conditions. Reaction gas: pure N2 at the working electrode; 5% H2–95% Ar at the counter electrode (reference electrode). | ||
To confirm the source of ammonia from N2 activation rather than N-containing contaminants, 15N2 isotope labeling experiments were conducted. When 15N2 gas was used, the doublet splitting peaks in the 1H NMR spectra revealed that15 NH3 was produced (Fig. S14†). In contrast, when 14N2 gas was used, the 1H NMR spectra showed triplet splitting peaks, indicating the formation of 14NH3 (Fig. S14†). Additionally, when argon was introduced into the cathode instead of nitrogen, no ammonia production was detected (Fig. S15†). These results confirm that the ammonia was indeed produced from the electrochemical reduction of nitrogen. To quantify the 15NH3 yield using 1H NMR, the production rate was determined to be 2.06 × 10−11 mol s−1 cm−2, which is lower than the results (5.14 × 10−11 mol s−1 cm−2) from the previous 14N2 experiments. This reduction can mainly be attributed to the lower flow rate used in the 15N2 experiments, which limits gas mass transfer in the cathode layer, affects gas distribution near the interface, and shifts the local chemical equilibrium. Additionally, prolonged electrolysis time may result in a slight loss of activity.
3.4 Discussion
Ru is one of the best catalysts in thermochemical ammonia synthesis and has also shown commendable performance in certain electrochemical ammonia synthesis systems.53,60,61 In thermo-catalysis, it is suggested that Ru catalysts show the size effect, with 2.5 nm Ru particles presenting an increased number of B5 active sites, leading to enhanced performance.62 Our work found that the I-Ru–LSCF–BZCYYb cathode with the small, uniformly dispersed Ru particles yields superior results. Nevertheless, achieving precise control over the size of Ru particles through the impregnation method remains challenging. Existing literature indicates that the in situ exsolution method can generate nanoparticles of uniform size on the electrode, showcasing effective performance SOFCs.63,64 This technique holds potential for future application in our system.Moreover, our findings indicate that the cathode design is pivotal in the performance of PCEAS electrolysis cells. By combining LSCF and BZCYYb, we established a mixed proton and electron conduction pathway, which could generate more active sites. The porous structure of the cathode may enhance gas transport. In the future, directly integrating mixed proton and electron ionic conductors with a multi-level porous cathode design could substantially enhance performance.24,65,66
Furthermore, our results indicate that improvements in electrochemical performance do not necessarily lead to increased electrochemical ammonia synthesis activity. For instance, the M-Ru–LSCF–BZCYYb cathode demonstrates good electrochemical performance but poor ammonia synthesis activity. This discrepancy is primarily due to the presence of the HER side reaction in this system. Nitrogen activation is difficult, and Ru (most metal catalysts) is inherently more prone to adsorbing H atoms than N2. Moreover, the NRR is complex, involving 6 electrons and 6 protons, while the HER only necessitates 2 electrons and 2 protons. Consequently, the HER demonstrates a clear kinetic advantage. The existence of the scaling relation restricts the improvement of FE in ammonia synthesis.67 Earlier studies have shown that hydride-based catalysts show significant nitrogen activation in the medium temperature range (>150 °C) and may separate the active sites of H and N, partially circumventing the scaling relation, thereby enhancing the activity of ammonia synthesis.68 Additionally, theoretical calculation predicts that early transition metals such as Sc, Y, Ti, and Zr may be in favor of the conversion of N instead of H, possessing a stronger capability for the NRR compared to the HER.69 Nitride catalysts such as VN, ZrN, and NbN are generally believed to operate via the Mars–Van Krevelen (MVK) mechanism, in which the nitrogen vacancies are considered to be the active site of nitrogen activation, and prefer to bind N rather than H. Single-atom catalysts may show a selectivity advantage due to their unique structure, which allows only the top site to adsorb hydrogen. This ensemble effect results in the atomic surface displaying positive relative energies for hydrogen adsorption on the top site.70 This offers some choices for future development of electrocatalytic materials.
Significantly, PCEAS can employ both hydrogen gas and water as hydrogen sources.71,72 Opting for water as the hydrogen source brings about environmental sustainability, process simplification, and increased safety. The performance of an electrolysis cell featuring an I-Ru–LSCF–BZCYYb cathode and using water as the hydrogen source is shown in Fig. 7 (more details are shown in Fig. S16 and S17 and Tables S5 and S6†). It is observed that the reaction of N2 and H2O requires a higher potential due to the more challenging thermodynamics compared to the reaction of N2 and H2. A volcanic curve relationship is also apparent between the ammonia synthesis rate and FE. At 400 °C, the ammonia production rate and FE reach 2.7 × 10−11 mol s−1 cm−2 and 0.116%, respectively, at −1.4 V. Furthermore, at 500 °C, the optimal potential for peak rate (4.6 × 10−11 mol s−1 cm−2) and FE (0.313%) drops to −1.0 V, likely due to diminished polarization and ohmic resistances at higher temperatures.
 | ||
| Fig. 7 NH3 formation rate and Faraday efficiency of the I-Ru–LSCF–BZCYYb|BZCYYb|Ni–BZCYYb electrolysis cell at (a) 400 °C and (b) 500 °C. Reaction gas: pure N2 at the working electrode; wet Ar at the counter electrode (reference electrode). | ||
4. Conclusion
We constructed a protonic ceramic electrolyte electrochemical ammonia synthesis cell with a configuration of Ru–LSCF–BZCYYb|BZCYYb|Ni–BZCYYb. The cathode comprises an LSCF–BZCYYb porous skeleton with uniformly dispersed Ru nanoparticles as catalysts. This cathode design enables effective activation of N2 gas by the Ru particles, facilitates electron and proton conduction via LSCF and BZCYYb, respectively, and benefits from a porous structure that enhances gas transport, thereby improving the performance of electrochemical ammonia synthesis. The Ru–LSCF–BZCYYb cathode prepared by an impregnation method achieves a maximum NH3 formation rate of 5.14 × 10−11 mol s−1 cm−2 and a maximum FE of 0.128% at 400 °C and −0.2 V using H2 gas as a hydrogen source. Additionally, we have successfully achieved ammonia synthesis using water as the hydrogen source based on this cell.Data availability
The data supporting this article have been included as part of the ESI.† Any additional data or materials required for replication of the results can also be provided by the corresponding author.Conflicts of interest
There is no conflict to declare.Acknowledgements
The authors are grateful for the financial support from the National Key R&D Program of China (2022YFB4002400), National Natural Science Foundation of China (Grant No. 22109158, 22279131, 21988101, 22172151), and Youth Innovation Promotion Association of CAS (No. Y2022060 and 2022180).Notes and references
- J. W. Erisman, M. A. Sutton, J. Galloway, Z. Klimont and W. Winiwarter, Nat. Geosci., 2008, 1, 636–639 CrossRef CAS.
- J. Lim, C. A. Fernández, S. W. Lee and M. C. Hatzell, ACS Energy Lett., 2021, 6, 3676–3685 CrossRef CAS.
- J. Guo and P. Chen, Chem, 2017, 3, 709–712 CAS.
- A. Valera-Medina, H. Xiao, M. Owen-Jones, W. I. David and P. Bowen, Prog. Energy Combust. Sci., 2018, 69, 63–102 CrossRef.
- D. R. MacFarlane, P. V. Cherepanov, J. Choi, B. H. Suryanto, R. Y. Hodgetts, J. M. Bakker, F. M. F. Vallana and A. N. Simonov, Joule, 2020, 4, 1186–1205 CrossRef CAS.
- F. Chang, W. Gao, J. Guo and P. Chen, Adv. Mater., 2021, 33, 2005721 CrossRef CAS PubMed.
- J. G. Chen, R. M. Crooks, L. C. Seefeldt, K. L. Bren, R. M. Bullock, M. Y. Darensbourg, P. L. Holland, B. Hoffman, M. J. Janik and A. K. Jones, Science, 2018, 360, eaar6611 CrossRef PubMed.
- W. David, Ammonia: Zero-Carbon Fertiliser, Fuel and Energy Store, The Royal Society, 2020 Search PubMed.
- G. Qing, R. Ghazfar, S. T. Jackowski, F. Habibzadeh, M. M. Ashtiani, C.-P. Chen, M. R. Smith III and T. W. Hamann, Chem. Rev., 2020, 120, 5437–5516 CrossRef CAS PubMed.
- W. Guo, K. Zhang, Z. Liang, R. Zou and Q. Xu, Chem. Soc. Rev., 2019, 48, 5658–5716 RSC.
- S. Z. Andersen, V. Čolić, S. Yang, J. A. Schwalbe, A. C. Nielander, J. M. McEnaney, K. Enemark-Rasmussen, J. G. Baker, A. R. Singh, B. A. Rohr, M. J. Statt, S. J. Blair, S. Mezzavilla, J. Kibsgaard, P. C. K. Vesborg, M. Cargnello, S. F. Bent, T. F. Jaramillo, I. E. L. Stephens, J. K. Nørskov and I. Chorkendorff, Nature, 2019, 570, 504–508 CrossRef CAS PubMed.
- J. Choi, B. H. Suryanto, D. Wang, H.-L. Du, R. Y. Hodgetts, F. M. Ferrero Vallana, D. R. MacFarlane and A. N. Simonov, Nat. Commun., 2020, 11, 5546 CrossRef CAS PubMed.
- B. H. Suryanto, K. Matuszek, J. Choi, R. Y. Hodgetts, H.-L. Du, J. M. Bakker, C. S. Kang, P. V. Cherepanov, A. N. Simonov and D. R. MacFarlane, Science, 2021, 372, 1187–1191 CrossRef CAS PubMed.
- N. Lazouski, M. Chung, K. Williams, M. L. Gala and K. Manthiram, Nat. Catal., 2020, 3, 463–469 CrossRef CAS.
- J. A. Schwalbe, M. J. Statt, C. Chosy, A. R. Singh, B. A. Rohr, A. C. Nielander, S. Z. Andersen, J. M. McEnaney, J. G. Baker and T. F. Jaramillo, ChemElectroChem, 2020, 7, 1542–1549 CrossRef CAS.
- S. Z. Andersen, M. J. Statt, V. J. Bukas, S. G. Shapel, J. B. Pedersen, K. Krempl, M. Saccoccio, D. Chakraborty, J. Kibsgaard and P. C. Vesborg, Energy Environ. Sci., 2020, 13, 4291–4300 RSC.
- S. Li, Y. Zhou, X. Fu, J. B. Pedersen, M. Saccoccio, S. Z. Andersen, K. Enemark-Rasmussen, P. J. Kempen, C. D. Damsgaard, A. Xu, R. Sažinas, J. B. V. Mygind, N. H. Deissler, J. Kibsgaard, P. C. K. Vesborg, J. K. Nørskov and I. Chorkendorff, Nature, 2024, 629, 92–97 CrossRef CAS PubMed.
- X. Fu, J. B. Pedersen, Y. Zhou, M. Saccoccio, S. Li, R. Sažinas, K. Li, S. Z. Andersen, A. Xu and N. H. Deissler, Science, 2023, 379, 707–712 CrossRef CAS PubMed.
- T. Murakami, T. Nishikiori, T. Nohira and Y. Ito, J. Am. Chem. Soc., 2003, 125, 334–335 CrossRef CAS PubMed.
- I. J. McPherson, T. Sudmeier, J. P. Fellowes, I. Wilkinson, T. Hughes and S. E. Tsang, Angew. Chem., 2019, 131, 17594–17602 CrossRef.
- S. Feng, W. Gao, J. Guo, H. Cao and P. Chen, ACS Energy Lett., 2023, 8, 1567–1574 CrossRef CAS.
- G. Marnellos and M. Stoukides, Science, 1998, 282, 98–100 CrossRef CAS PubMed.
- S. Gunduz, D. J. Deka and U. S. Ozkan, J. Catal., 2020, 387, 207–216 CrossRef CAS.
- C. Duan, J. Tong, M. Shang, S. Nikodemski, M. Sanders, S. Ricote, A. Almansoori and R. O'Hayre, Science, 2015, 349, 1321–1326 CrossRef CAS PubMed.
- V. Kyriakou, I. Garagounis, E. Vasileiou, A. Vourros and M. Stoukides, Catal. Today, 2017, 286, 2–13 CrossRef CAS.
- A. Atkinson, S. Barnett, R. J. Gorte, J. T. S. Irvine, A. J. McEvoy, M. Mogensen, S. C. Singhal and J. Vohs, Nat. Mater., 2004, 3, 17–27 CrossRef CAS PubMed.
- S. Dwivedi, Int. J. Hydrogen Energy, 2020, 45, 23988–24013 CrossRef CAS.
- K.-D. Kreuer, Annu. Rev. Mater. Res., 2003, 33, 333–359 CrossRef CAS.
- Y. Zhang, R. Knibbe, J. Sunarso, Y. Zhong, W. Zhou, Z. Shao and Z. Zhu, Adv. Mater., 2017, 29, 1700132 CrossRef PubMed.
- D. Vignesh and E. Rout, Energy Fuels, 2023, 37, 3428–3469 CrossRef CAS.
- B. Wang, T. Li, F. Gong, M. H. D. Othman and R. Xiao, Fuel Process. Technol., 2022, 235, 107380 CrossRef CAS.
- R. Li, Q. Zhen, S. Bashir and J. Liu, in Nanostructured Materials for Sustainable Energy: Design, Evaluation, and Applications, ACS Publications, 2022, pp. 155–184 Search PubMed.
- I. Garagounis, V. Kyriakou, A. Skodra, E. Vasileiou and M. Stoukides, Front. Energy Res., 2014, 2, 1 Search PubMed.
- G. Marnellos, S. Zisekas and M. Stoukides, J. Catal., 2000, 193, 80–87 CrossRef CAS.
- Z. Lei, J. Jing, J. Pang, R. Hu, X. Shi, Z. Yang and S. Peng, Int. J. Hydrogen Energy, 2020, 45, 8041–8051 CrossRef CAS.
- M. Li, B. Hua, W. Wu, L.-C. Wang, Y. Ding, M. M. Welander, R. A. Walker and D. Ding, Mater. Today, 2022, 60, 31–40 CrossRef CAS.
- S. Klinsrisuk and J. Irvine, Catal. Today, 2017, 286, 41–50 CrossRef CAS.
- F. Kosaka, T. Nakamura and J. Otomo, J. Electrochem. Soc., 2017, 164, F1323 CrossRef CAS.
- F. Kosaka, N. Noda, T. Nakamura and J. Otomo, J. Mater. Sci., 2017, 52, 2825–2835 CrossRef CAS.
- L. Yang, Z. Liu, S. Wang, Y. Choi, C. Zuo and M. Liu, J. Power Sources, 2010, 195, 471–474 CrossRef CAS.
- S. B. Adler, Chem. Rev., 2004, 104, 4791–4844 CrossRef CAS PubMed.
- M. Papac, V. Stevanović, A. Zakutayev and R. O'Hayre, Nat. Mater., 2021, 20, 301–313 CrossRef CAS PubMed.
- L. Yang, S. Wang, K. Blinn, M. Liu, Z. Liu, Z. Cheng and M. Liu, Science, 2009, 326, 126–129 CrossRef CAS PubMed.
- C. Duan, J. Huang, N. Sullivan and R. O'Hayre, Appl. Phys. Rev., 2020, 7, 011314 CAS.
- J. Kim, S. Sengodan, S. Kim, O. Kwon, Y. Bu and G. Kim, Renewable Sustainable Energy Rev., 2019, 109, 606–618 CrossRef CAS.
- H. Shimada, Y. Yamaguchi, H. Sumi and Y. Mizutani, J. Electrochem. Soc., 2020, 167, 124506 CrossRef CAS.
- S. Sun and Z. Cheng, J. Electrochem. Soc., 2017, 164, F3104 CrossRef CAS.
- L. Zhu, C. Cadigan, C. Duan, J. Huang, L. Bian, L. Le, C. H. Hernandez, V. Avance, R. O'Hayre and N. P. Sullivan, Commun. Chem., 2021, 4, 121 CrossRef CAS PubMed.
- K. Wang, H. Chen, S.-D. Li and Z. Shao, J. Mater. Chem. A, 2022, 10, 24813–24823 RSC.
- J. J. Giner-Sanz, G. Leverick, V. Pérez-Herranz and Y. Shao-Horn, J. Electroanal. Chem., 2021, 896, 115250 CrossRef CAS.
- L. Li, C. Tang, B. Xia, H. Jin, Y. Zheng and S.-Z. Qiao, ACS Catal., 2019, 9, 2902–2908 CrossRef CAS.
- M. Weatherburn, Anal. Chem., 1967, 39, 971–974 CrossRef CAS.
- Q. Wang, J. Pan, J. Guo, H. A. Hansen, H. Xie, L. Jiang, L. Hua, H. Li, Y. Guan and P. Wang, Nat. Catal., 2021, 4, 959–967 CrossRef CAS.
- S. Wang, M. Katsuki, M. Dokiya and T. Hashimoto, Solid State Ionics, 2003, 159, 71–78 CrossRef CAS.
- L. da Conceicao, A. M. Silva, N. F. Ribeiro and M. M. Souza, Mater. Res. Bull., 2011, 46, 308–314 CrossRef CAS.
- A. VahidMohammadi and Z. Cheng, J. Electrochem. Soc., 2015, 162, F803 CrossRef CAS.
- Z. Liu, Y. Song, X. Xiong, Y. Zhang, J. Cui, J. Zhu, L. Li, J. Zhou, C. Zhou, Z. Hu, G. Kim, F. Ciucci, Z. Shao, J.-Q. Wang and L. Zhang, Nat. Commun., 2023, 14, 7984 CrossRef CAS PubMed.
- Z. Teng, Z. Xiao, G. Yang, L. Guo, X. Yang, R. Ran, W. Wang, W. Zhou and Z. Shao, Mater. Today Energy, 2020, 17, 100458 CrossRef.
- S. Yin, B. Xu, X. Zhou and C. Au, Appl. Catal., A, 2004, 277, 1–9 CrossRef CAS.
- M. Kitano, Y. Inoue, Y. Yamazaki, F. Hayashi, S. Kanbara, S. Matsuishi, T. Yokoyama, S.-W. Kim, M. Hara and H. Hosono, Nat. Chem., 2012, 4, 934–940 CrossRef CAS PubMed.
- G. Weng, S. Lei, R. Wang, K. Ouyang, J. Dong, X. Lin, J. Xue, L.-X. Ding and H. Wang, Joule, 2023, 7, 1333–1346 CrossRef CAS.
- K. Honkala, A. Hellman, I. Remediakis, A. Logadottir, A. Carlsson, S. Dahl, C. H. Christensen and J. K. Nørskov, science, 2005, 307, 555–558 CrossRef CAS PubMed.
- J.-h. Myung, D. Neagu, D. N. Miller and J. T. Irvine, Nature, 2016, 537, 528–531 CrossRef CAS PubMed.
- H. Han, J. Park, S. Y. Nam, K. J. Kim, G. M. Choi, S. S. Parkin, H. M. Jang and J. T. Irvine, Nat. Commun., 2019, 10, 1471 CrossRef PubMed.
- E. Fabbri, I. Markus, L. Bi, D. Pergolesi and E. Traversa, Solid State Ionics, 2011, 202, 30–35 CrossRef CAS.
- R. Merkle, M. F. Hoedl, G. Raimondi, R. Zohourian and J. Maier, Annu. Rev. Mater. Res., 2021, 51, 461–493 CrossRef CAS.
- J. H. Montoya, C. Tsai, A. Vojvodic and J. K. Nørskov, ChemSusChem, 2015, 8, 2180–2186 CrossRef CAS PubMed.
- P. Wang, F. Chang, W. Gao, J. Guo, G. Wu, T. He and P. Chen, Nat. Chem., 2017, 9, 64–70 CrossRef CAS PubMed.
- E. Skulason, T. Bligaard, S. Gudmundsdóttir, F. Studt, J. Rossmeisl, F. Abild-Pedersen, T. Vegge, H. Jónsson and J. K. Nørskov, Phys. Chem. Chem. Phys., 2012, 14, 1235–1245 RSC.
- G. F. Chen, S. Ren, L. Zhang, H. Cheng, Y. Luo, K. Zhu, L. X. Ding and H. Wang, Small Methods, 2019, 3, 1800337 CrossRef.
- A. Skodra and M. Stoukides, Solid State Ionics, 2009, 180, 1332–1336 CrossRef CAS.
- R. Li, T. Li, X. Liu, C. Xie, Q. Zhen, S. Bashir and J. L. Liu, Energy Sci. Eng., 2023, 11, 2293–2301 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta04520c |
| This journal is © The Royal Society of Chemistry 2024 |