
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pathway control in metallosupramolecular polymerization of a monoalkynylplatinum(II) terpyridine complex through competitive complex formation†
Minhye
Kim
a,
Heekyoung
Choi
a,
Minjoo
Kim
a,
Seonghan
Kim
c,
Seohyeon
Yun
d,
Eunji
Lee
 d,
Jaeheung
Cho
c,
Sung Ho
Jung
*ab and
Jong Hwa
Jung
*ab
d,
Jaeheung
Cho
c,
Sung Ho
Jung
*ab and
Jong Hwa
Jung
*ab
aDepartment of Chemistry, Gyeongsang National University, Jinju 52828, Korea. E-mail: jonghwa@gnu.ac.kr
bResearch Institute of Advanced Materials Chemistry, Gyeongsang National University, Jinju 52828, Korea
cDepartment of Chemistry, Ulsan National Institute of Science and Technology, Ulsan 44919, Korea
dDepartment of Chemistry and Advanced Materials, Gangneung-Wonju National University, Gangneung 25457, Korea
First published on 6th November 2024
Abstract
Understanding the pathway complexity of supramolecular polymerization in biomimetic systems has been a challenging issue due to its importance in the development of rationally controlled materials and insight into self-assembly in nature. We herein report a kinetic trapping strategy as a new methodology on how to control the pathway of metallosupramolecular polymerization by employing secondary metal ions and/or ligands which form competitive complex species. For this, we proposed monoalkynylplatinum(II) metalloligand (Pt-L1) derived from a bis(amideterpyridine) receptor with one unoccupied terpyridyl terminal as a coordination site for the secondary metal ion (Ag+ or Fe2+). The inherent pathway complexity intrinsic to the Pt-L1-anchored supramolecular polymerization has been modulated through the incorporation of Ag+ or Fe2+. During the supramolecular polymerization of Pt-L1 in the presence of Ag+ and Fe2+, the added secondary ligand bpy (4,4′-dimethyl-2,2′-bipyridine) or DA18C6 (1,14-diaza-18-crown-6) form complexes as kinetic species, thereby inhibiting spontaneous polymerizations. The supramolecular polymer (SP-I), with a spherical structure composed of Pt-L1 in the absence of metal ions as a kinetic product, did not transform into the thermodynamic product, namely supramolecular polymer (SP-III) with a left-handed fiber structure, due to a high energy barrier. However, the supramolecular polymer (SP-II) with a left-handed fiber structure, which was formed by Pt-L1 in the presence of AgNO3, converted to SP-III upon the addition of NaCl. Additionally, SP-II transformed into supramolecular polymer (SP-IV) upon the addition of Fe(BF4)2, through an on-pathway process. Both the morphological and emissive characteristics of the resulting supramolecular polymers can be fine-tuned via the Pt⋯Pt or Ag⋯Ag interactions as well as through the changes of the coordination geometry depending on the existing Ag+ or Fe2+ ions. The present results have important implications in expanding the scope of pathway complexity to produce a variety of products via kinetically controlled processes involving secondary metal ions and ligands.
Introduction
In both natural and artificial supramolecular systems, the formation of complicated multi-functional structures is often derived from concerted non-covalent interactions.1–13 The performance of specific functions in these systems requires particular molecular and supramolecular organization. Such specificity can be especially challenging due to the coexistence of multiple competitive pathways, known as pathway complexity.14–16 In the realm of biomolecules, such pathways frequently give rise to multifaceted systems characterized by an array of structures and functionalities.17–19 Therefore, the concept of pathway complexity in supramolecular polymers (hereafter ‘SPs’) has emerged as an invaluable paradigm for mimicking inherent natural processes. In other words, the strategic modulation of pathway complexity holds the potential to herald the development of functionally advanced SPs with enhanced architectural sophistication.20,21Recently, several researchers have utilized the nature of pathway complexity to regulate the morphology and length of SPs.22–27 For example, some innovative attempts have been made in kinetically controlled supramolecular polymerization, emerging as pioneering strategies to yield self-assembled structures with properties surpassing those derived from equilibrium states.28–34 A promising alternative to attain kinetic control is the strategic design of monomers that are kinetically trapped with inactive conformations. Würthner and coworkers reported a perylene bisimide derivative where the monomeric state is stabilized by hydrogen bonding.35 They further demonstrated a novel strategy to control supramolecular polymerization via a chaperone-like two components system as a new kinetic trapping method, utilizing a squaraine dye.36 Similarly, Sánchez and colleagues reported the kinetically governed self-assembly behaviors of a suite of N-annulated perylenebisimides.37 They employed the trialkoxybenzamide group, linked to the N-annulated perylene fragment comprising a variable count of methylene units to optimize the formation of an H-bonded pseudocycle, thereby moderating the supramolecular polymerization. In contrast, Sugiyasu, Takeuchi and coworkers elucidated that porphyrin entities spatially align in a J-aggregate, typified by face-to-face stacking but with substantial offset, as a kinetic manifestation.38,39 Following an initial latency period, this J-aggregate form transitioned to the H-aggregate, characterized by face-to-face stacking but with augmented overlap. These kinetic controls have opened new pathways, catalyzing programmable self-assembly that generates specific aggregated species marked by regulated polydispersity, particularly evident in block copolymerization scenarios.40 To date, efforts to navigate the pathway complexity in purely organic π-conjugated systems have predominantly revolved around the integration of the amide functionality within the molecular blueprint, as well as the judicious alteration of temperature and solvent geometries to control aggregation modes.
Metallo-SPs incorporating d8 metal ions – namely, Pt(II), Pd(II) and Au(III) – and interfaced with ligands like terpyridine, pyridine-triazole, and pyridine-benzimidazole, have attracted substantial interest.41,42 This intrigue arises from their inherent photophysical properties, notably luminescence, coupled with their propensity for metal–metal and π–π interactions.43–46 With specific regard to the Pt(II) complex, Manners and colleagues pioneered their first attempt in 2015 to regulate the length of metallo-SPs based on tridentate pyridine-bis-tetrazole ligand.47 The Yam, Eisenberg, Tung, and Chen groups have probed alkynyl terpyridine-centric tridentate ligands in homo-metal complex systems, motivated by their captivating photophysical characteristics and their ensuing diverse applicative potential spanning molecular recognition, pH sensing, biomolecular labeling and beyond.48–59 Yam and coworkers, in particular, have offered pivotal insights, elucidating the nexus between self-assembly dynamics and structure–property relation in the supramolecular polymerization of alkynylplatinum(II) terpyridine complexes.60–63
In particular, bis-type ligands not only dictate the selective formation of mononuclear complexes, so-called metalloligands, depending on the binding affinity for metal ions but also produce a variety of complexes with different stoichiometric ratios. Among these, some complexes may act as intermediates in supramolecular polymerization. Moreover, introducing a secondary ligand during supramolecular polymerization of the metalloligands might induce the formation of kinetic species due to competitive conditions which may suppress the spontaneous nucleation of monomers.
Thus, we envisioned that the modification of metalloligands based on bis-type amideterpyridine may not only induce the formation of SPs but also might introduce a kinetic trapping strategy in competitive conditions. To probe this hypothesis, in this work, we employed monoalkynylplatinum(II) diterpyridine complex (hereafter ‘Pt-L1’) with one terpyridine terminal remaining unoccupied (Fig. 1). In addition to the self-assembly of Pt-L1 alone, as depicted, the strategy involves the secondary metal ions (Ag+ and Fe2+) and ligands (bispyridine (hereafter ‘bpy’) and diaza-18-crown-6 (hereafter ‘DA18C6’)) to modulate the inherent pathway complexity associated with the Pt-L1 based supramolecular polymerization. In particular, we have emphasized the influence of secondary metal ions and ligands on the new kinetically controlled process because this is the first example of employing this strategy systemically in metallosupramolecular polymerization to control the pathway. The details of our approaches and the related investigations are discussed below.
 | ||
Fig. 1 (A) Chemical structures of Pt-L1 and its reference compounds Pt-L2 and Pt-L3. (B) Chemical structures of the secondary ligands (bpy and DA18C6). (C) Proposed formation mechanism of SPs from Pt-L1 in the (a) absence and (b and c) presence of secondary metal ions (Ag+ and Fe2+); (a) SP-I formed by an isodesmic model and (b) SP-II (Pt-L1/Ag+ = 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) and (c) SP-IV (Pt-L1/Fe2+ = 2:1) with a left-handed helical structure formed by a cooperative model. (d and e) Partial chemical structures of the Pt-L1 complexes with (d) Ag+ and (e) Fe2+ ions obtained by DFT calculation. :2) and (c) SP-IV (Pt-L1/Fe2+ = 2:1) with a left-handed helical structure formed by a cooperative model. (d and e) Partial chemical structures of the Pt-L1 complexes with (d) Ag+ and (e) Fe2+ ions obtained by DFT calculation. | ||
Results and discussion
Synthesis of a monomeric building block (Pt-L1) and its reference compounds (Pt-L2 and Pt-L3)
As a versatile metalloligand, Pt-L1 was designed for supramolecular polymerization to be driven synergistically via a concerted combination of π–π stacking, intermolecular H-bond, and Pt⋯Pt interaction (Fig. 1). Incorporating an extended alkyl chain into the phenylacetylene framework is expected to promote fiber elongation, enhancing the flexibility of SPs. The unoccupied terpyridine site can be occupied by the secondary metal ion (e.g. Ag+), facilitating a metal–metal interaction, which may yield a pronounced helical structure.The synthetic routes for the bis-type ligand and target metalloligands are shown in Scheme S1.† The chiral precursor R-L2 incorporating an R-alanine moiety was prepared using a known procedure.64 The bis-type ligand R-L1 was synthesized by the reaction of R-L2 with decanedioyl dichloride in the presence of triethylamine in dichloromethane. The precursor Pt-L3 (also as a reference compound) was prepared via the reaction of R-L1 with dichloro(1,5-cyclooctadiene)platinum(II) to yield monochloroplatium(II) complex PtCl-L2, followed by the anion exchange with NH4PF6. The main target metalloligand Pt-L1 was synthesized through the coupling of Pt-L3 with N-(4-ethylphenyl)stearamide (R-1, Scheme S2†) in the presence of CuI and diisopropylamine in dichloromethane. Another reference compound Pt-L2 was obtained by reacting phenylacetylene in an identical condition.
Complexation behaviors of Pt-L1 with the secondary metal ions (Ag+ and Fe2+)
As a preliminary study, the complexation stoichiometries between Pt-L1 and secondary metal ions were investigated by high-resolution electrospray ionization mass spectrometry (HR-ESI-MS) in DMSO/H2O (9:1 v/v). In the presence of AgNO3 (1.5–3.0 equiv.), the intense peak corresponding to a 1:1 species ([(Pt-L1)Ag]2+, m/z 731.2876) was observed (Fig. S1–S5†). Some minor peaks corresponding to a 2:1 and a 3:2 species have vanished after 24 h ageing, reflecting the binding of one silver(I) to the free terpyridine moiety, regardless of the AgNO3 concentration. When Fe(BF4)2 (1.5 equiv.) was used as a secondary metal ion source, initially [(Pt-L1)2Fe]4+ (major, m/z 691.8191) and [(Pt-L1)Fe(H2O)]3+ (minor) coexists (Fig. S6†). After 5 h, the major peak intensity for the 2:1 species enhanced, indicating that both terpyridine sites are occupied by Fe2+, forming a distorted octahedral coordination geometry as the thermodynamic product.
The complexation behaviors were also monitored by UV-vis measurements. The spectrum of Pt-L1 displays prominent high-energy absorption bands spanning 290–370 nm and lower-energy bands in the 430–480 nm range (Fig. S7a†). These can be ascribed to the intraligand π–π* transition and the metal-to-ligand charge transfer (MLCT), respectively, with a superimposed ligand-to-ligand charge transfer (LLCT). In the presence of AgNO3 (1.5 equiv.), an alteration in the absorption tail in the 525–570 nm range is evident (Fig. S7a†), indicative of a metal-metal-to-ligand charge transfer (MMLCT) stemming from Pt⋯Pt interactions.65 Interestingly, in the presence of Fe(BF4)2 (1.5 equiv.) (Fig. S7b†), a pronounced shoulder band in the 520–580 nm range appears unlike that of AgNO3 probably due to the stronger MLCT interactions66 with Fe2+ over Ag+, similar to the results obtained from the HR-ESI-MS.
Supramolecular polymerization of Pt-L1 without and with AgNO3
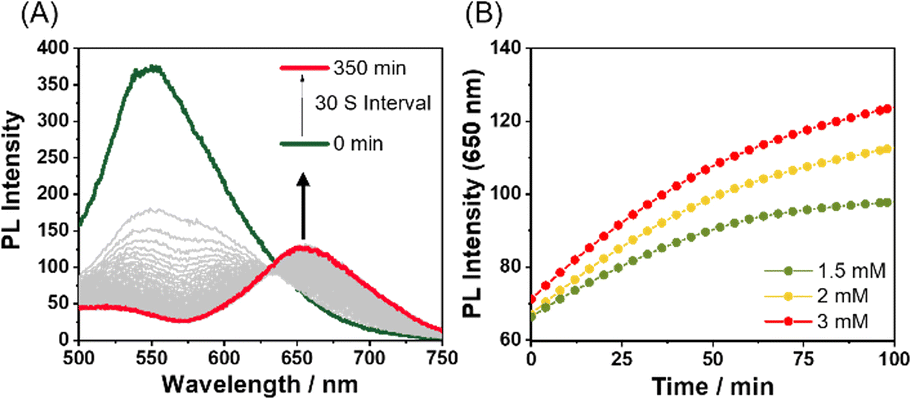
In monitoring the supramolecular polymerization process including the mechanisms, time- and/or concentration-dependent measurements to follow the transition from dissolved monomeric species to the self-assembled aggregated state have been the mainstream. As shown in Fig. 2, the photoluminescence (PL) spectrum of Pt-L1 exhibits a strong emission band with a maximum at ∼565 nm dominated by the MLCT transition, indicating that the typical aggregation of the monomeric species occurs to form a supramolecular polymer (hereafter ‘SP-I’, see Fig. 1). In addition, the time-dependent spectra show no significant changes with time at 293 K, reflecting that SP-I is formed without Pt⋯Pt interactions, which we will discuss in the later part.67 We also observed temperature-dependent PL spectral changes of SP-I in DMSO and H2O (9:1 v/v). The PL band of SP-I observed in DMSO and H2O (9:1 v/v) at 298 K increased as the temperature rose (Fig. S8A†), which can be attributed to the π–π stacking interactions between the terpyridine moieties of Pt-L1 being disrupted as SP-I dissociates into monomeric species. This observation indicates that the PL band obtained at approximately 565 nm in the DMSO and H2O (9:1 v/v) mixture is associated with the MLCT of SP-I. Additionally, an intense PL band of Pt-L1 was observed at 560 nm in pure DMSO (Fig. S8B†). These observations suggest that the PL band detected at approximately 565 nm in the DMSO and H2O (9:1 v/v) mixture is associated with the MLCT of SP-I. The absorbance of SP-I in DMSO and H2O (9:1 v/v) around 280 nm is lower than that of monomeric Pt-L1 in pure DMSO (Fig. S8C†), due to the formation of SP-I with nanoparticles, driven by π–π stacking interactions.
 | ||
| Fig. 2 (A) Time-dependent PL spectra of Pt-L1 (3.0 mM, λex = 420 nm) in DMSO/H2O (9:1 v/v) at 293 K. (B) Plots for times vs. PL intensity (565 nm) of different concentrations of Pt-L1 (1.5–3.0 mM). | ||
The influences of the secondary metal ions on the formation of SPs have been investigated. After the addition of AgNO3 (1.5 equiv.) to Pt-L1 (1.0–3.0 mM), interestingly, a new emission band around 620 nm appears and gradually increases with time (Fig. 3). The red-shift (from ∼560 to ∼620 nm, Δλ = ∼60 nm) with the intensity increase is mainly attributed to the Pt⋯Pt interactions characterized by MMLCT.68 The PL profile with a plot of the intensity vs. time shows typical sigmoidal curves with a well-recognized lag phase. This result seems to be associated with the transient existence of [(Pt-L1)3Ag2]5+ and [(Pt-L1)2Ag]3+, as evidenced by HR-ESI-MS data (Fig. S1†). Taken collectively, the above UV-vis, mass, and PL results for the complexations and supramolecular polymerizations suggest that SP-II generated in the presence of Ag+ seems to be derived from the aggregation of [(Pt-L1)2Ag2]4+ featuring the Pt⋯Pt interactions. Moreover, as the concentrations of Pt-L1 increase in the 1.5–3.0 mM range, a decrease in the duration of the lag phase is noted from 8 to 2 min (Fig. 3A and B), which could be related to the increased ratio of monomeric [(Pt-L1)2Ag2]4+ (Fig. S5†). These observations suggest a buffered release and self-nucleation of the monomer from SP-II, followed by autocatalytic growth of the remaining monomers via an on-pathway mechanism. When AgBF4 and AgPF6 were also employed in the same procedures, the lag times increased in the order of NO3− < PF6− < BF4− (Fig. S9†), implying a kinetic tunability of the supramolecular polymerization depending on the anionic environment. We recorded the CD spectra of SP-I and SP-II in a DMSO/H2O mixture (9:1 v/v) (Fig. S10†). A very weak positive CD signal for SP-I was detected around 325 nm, while a relatively stronger positive CD signal for SP-II was observed at approximately 335 nm in the presence of AgNO3 (1.5 equiv.). These results suggest that SP-II forms a helical molecular arrangement, leading to the induction of chiral supramolecular polymers.
 | ||
| Fig. 3 (A) Time-dependent PL spectra of Pt-L1 (2.0 mM, λex = 420 nm) in the presence of AgNO3 (1.5 equiv.) in DMSO/H2O (9:1 v/v) at 293 K. (B) Plots for PL intensity at 620 nm vs. time at different concentrations of Pt-L1 (1.5–3.0 mM) in the presence of AgNO3 (1.5 equiv.). | ||
Distinct morphologies of the SPs were elucidated using atomic force microscopy (AFM). The SP-I assembled from Pt-L1 alone features nanoparticles with a height of 3 nm (Fig. 4A). The SP-I with nanoparticles is formed from the monomeric Pt-L1 in the DMSO and H2O (9:1 v/v) mixture, where one portion of the terpyridine is coordinated with Pt(II) ion while another remains a free ligand. This asymmetric structure may contribute to the micellar property due to the higher polarity of the Pt(II)-coordinated portion compared to the free terpyridine portion, which shows a hydrophobic effect in the aqueous environment. Whereas SP-II assembled from Pt-L1 with AgNO3 (1.5 equiv.) shows a left-handed fiber structure with a height of 4 nm on the HOPG surface (Fig. 4B and S11†), probably due to the Ag⋯Ag interactions within a tetrahedral structure, as demonstrated by DFT calculations (Fig. 1C(d)).
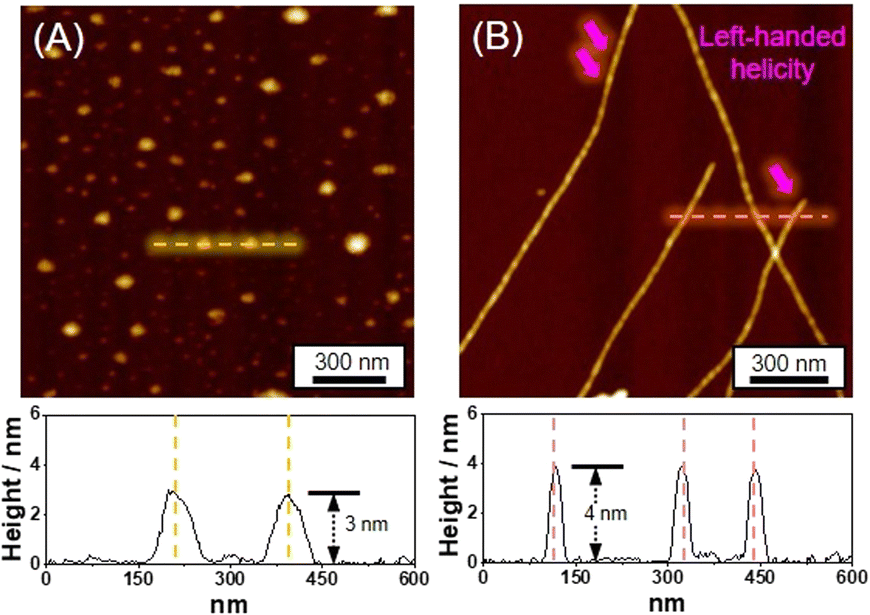 | ||
| Fig. 4 AFM images and height profiles of (A) SP-I (assembled from Pt-L1 (3.0 mM) only), and (B) SP-II (assembled from Pt-L1 (2.0 mM) + AgNO3 (1.5 equiv.)) in DMSO and H2O (9:1 v/v). | ||
Removal of Ag+ from SP-II: two different SPs with the same component
Having obtained SP-I and SP-II of types [Pt-L1]n and [(Pt-L1)2Ag2]n4+ respectively, by direct methods, we propose an indirect formation of a new SP of type [Pt-L1]n by removal of Ag+ from SP-II. Indeed, it is challenging to compare the new SP (hereafter ‘SP-III’, see Scheme 1) with SP-I because both products originate from different routes but contain the same component. For this, an excess amount of Cl− (as NaCl) was added to SP-II solution to remove the bounded Ag+ (Scheme 1A). Upon addition, interestingly, the PL intensity at 620 nm increased by approximately 2-fold (Fig. S12†). This result is associated with the enhanced Pt⋯Pt interactions upon removal of Ag+. The distinct changes in the PL properties represent the appearance of a novel pathway. Moreover, SP-III exhibited a loosely packed left-handed fiber structure with a height of 6 nm (Fig. S13†) compared to SP-II. Furthermore, to demonstrate the presence of Ag+ ions in SP-II and SP-III before and after treatment with NaCl, the helical fibers were analyzed using scanning electron microscopy (SEM) combined with energy-dispersive spectrometry (EDS) microanalysis (Fig. S14†). The fibers of SP-II, prior to NaCl treatment, prominently displayed a characteristic peak associated with silver (Fig. S14B†). In contrast, the silver peak disappeared in the fiber of SP-III obtained after treatment with NaCl (6.0 equiv.) (Fig. S14D†). These findings suggest that SP-III consists solely of Pt-L1 as a building block, without the presence of an Ag+ complex. Considering the combined results, the supramolecular polymerization of Pt-L1 initially afforded SP-I as a kinetically controlled product. Meanwhile, SP-III obtained from SP-IIvia the removal of bounded Ag+ is thermodynamically stable (Scheme 1A). Consequently, the proposed give-and-take-away approach of the secondary metal ion is versatile for the controlled preparation of the kinetic and thermodynamic products.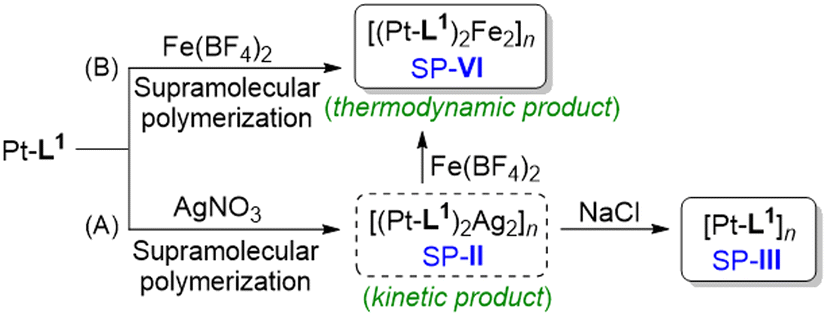 | ||
| Scheme 1 Proposed formation mechanism of SP-II, SP-III and SP-VI in the presence of secondary metal ions: (A) AgNO3 and (B) Fe(BF4)2, upon the addition of NaCl or Fe(BF4)2 to SP-II. | ||
Supramolecular polymerization of Pt-L1 in the presence of Fe(BF4)2
When Fe2+ was used as a secondary metal ion by adding Fe(BF4)2 (1.5 equiv., Scheme 1B) to Pt-L1, the time-dependent PL spectra showed a larger red-shift (from ∼560 nm to ∼650 nm, Δλ = ∼90 nm) than that of Ag+ and stabilized within approximately 100 min, with the intensity decrease (Fig. 5A, hereafter ‘SP-IV’, see Fig. 1C). This pronounced red shift to 650 nm is ascribed to the formation of excimers within the [(Pt-L1)2Fe]4+ complex during the polymerization process.69 Notably, the PL profiles displayed no lag phase (Fig. 5B), despite the coexistence of [(Pt-L1)Fe]3+ and [(Pt-L1)2Fe]4+ with 1:1 and 2:1 stoichiometric ratios, respectively. According to our UV-vis titrations to determine the stability constants between Pt-L1 and Fe2+, the above result could be explained by the higher stability of the 2:1 complex (logK = 11.87) than the 1:1 complex (logK = 5.57) (Fig. S15 and S16†). The stability constant of [(Pt-L1)2Fe]4+ is approximately 106 times higher than that of the 1:1 complex, [(Pt-L1)Fe]3+. Consequently, the 1:1 complex [(Pt-L1)Fe]3+ rapidly converts to the 2:1 complex. Thus, no lag phase was observed in the presence of Fe2+ ions during the supramolecular polymerization because the 1:1 complex did not act as a kinetically trapped species in this process. SP-IV exhibits a left-handed fiber structure with a height of 5 nm (Fig. S17†). Consequently, the excimer [(Pt-L1)2Fe]4+ is an active precursor in forming SP-IV. The positive CD signal of SP-IV was also observed (Fig. S10c†), similar to that of SP-II, although its intensity was slightly lower compared to SP-II.
 | ||
| Fig. 5 (A) Time-dependent PL spectra of Pt-L1 (2.0 mM, λex = 420 nm) in the presence of Fe(BF4)2 (1.5 equiv.) in DMSO/H2O (9:1 v/v) at 293 K. (B) Plots for PL intensity at 650 nm vs. time at different concentrations of Pt-L1 (1.5–3.0 mM) in the presence of Fe(BF4)2 (1.5 equiv.). | ||
Supramolecular polymerization of Pt-L1 in the presence of Ag+ and Fe2+via on-pathway process
Having obtained the results with Ag+ and Fe2+ separately, a competitive metal system was performed. In this approach, SP-II of type [(Pt-L1)2Ag2]n4+ was prepared and then exposed to Fe2+ to induce the consecutive reaction in the competition condition (Scheme 1B). Practically, the time-dependent PL spectral evolution of SP-II was recorded in the presence of Fe(BF4)2 in DMSO/H2O (9:1 v/v) (Fig. 6A and B). The added Fe2+ induced a notable red-shift from 620 nm to 630 nm and subsequently to 650 nm with an isosbestic point at 638 nm. The appearance of a new PL band near 631 nm is attributed to the metastable [(Pt-L1)Fe]3+ complex via the replacement of Ag+ with Fe2+. The higher stability of the complex of Pt-L1 with Fe2+ (logK = 11.87 for a 2:1 complex) than with Ag+ (logK = 7.70 for a 1:1 complex) supports the above spectroscopic results (Fig. S16†). In other words, the proposed competition condition allows the conversion of SP-II to SP-IVvia the metastable intermediate [(Pt-L1)Fe]3+. Also, the duration of the lag phase from the sigmoidal PL profiles reduces with the increase of Fe2+ concentrations (Fig. 6B), indicating that the formation of SP-IV proceeds via an on-pathway mechanism retaining the left-handed helical shape (Fig. S18†). As a result, SP-IV can be generated in two routes; one is a direct method (via Pt-L1 + Fe2+) and another is a two-step method including competition condition (via SP-II + Fe2+) (Scheme 1). Consequently, in a scenario where both Ag+ and Fe2+ coexisted, the resulting SP-IV was identified to be a thermodynamic product (Scheme 1), whereas SP-II existed as the kinetic product in the presence of Fe2+. The left-handed helical fiber structure of SP-IV (Fig. S18†) is attributed to the formation of an octahedral coordination complex in the presence of Fe2+. The related thermodynamic studies are discussed in the later part. These results clearly show the decisive roles of the secondary metal ions in the pathway complexity of supramolecular polymerization depending on their relative reactivity.
 | ||
| Fig. 6 (A) Time-dependent PL spectra of SP-II (assembled from Pt-L1 + 1.5 equiv. of AgNO3, λex = 420 nm) upon addition of Fe(BF4)2 (3.0 equiv.) in DMSO/H2O (9:1 v/v) at 293 K, λex = 420 nm. (B) Plots of PL intensity (at 650 nm) vs. times at different concentrations of Fe(BF4)2. | ||
Comparative studies with the reference compounds (Pt-L2 and Pt-L3)
To support the supramolecular polymerization behaviors of Pt-L1 observed in the absence and presence of secondary metal ions, similar experiments were repeated using reference compounds Pt-L2 and Pt-L3, in which phenylacetylene and PF6 (as an R group) are bound to the Pt(II) center, respectively (Fig. 1). In the time-dependent PL spectra of Pt-L2 and Pt-L3, each prominent MLCT band centered at ∼560 nm shows no significant changes with time (Fig. S19†). In the presence of AgNO3 or Fe(BF4)2, it is also noted that no appreciable spectral changes were observed (Fig. S20†), despite the ESI-MS results showing the formation of complexes of type [(Pt-L2)Ag]2+, [(Pt-L3)Ag]2+, [(Pt-L2)2Fe]4+ or [(Pt-L3)2Fe]4+ (Fig. S21–S24†), similar to the case of Pt-L1. These findings demonstrate that the flexibility of the extended alkyl chain in Pt-L1 plays a pivotal role in promoting the formation of Pt⋯Pt interaction during supramolecular polymerization. The AFM images for the supramolecular aggregates of Pt-L2 in both the absence and the presence of AgNO3 or Fe(BF4)2 show a sheet structure with a thickness of 3 nm (Fig. S25†). Similarly, SPs of Pt-L3 without a metal ion showed a sheet with a height of approximately 3 nm (Fig. S26†). These results suggest that the Pt⋯Pt and Ag⋯Ag interactions in Pt-L1 play an essential role in forming the helical structure during supramolecular polymerization.IR, Raman, and WXRD studies
To understand the reasons underlying the substantial enhancement of PL signal of the self-assembled Pt-L1 with metal ions, we employed various rcharacterization techniques such as FT-IR spectroscopy, Raman spectroscopy, and wide-angle X-ray diffraction (WXRD). In the FT-IR study, SP-I showed typical vibration bands of amide I and II at 1681 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch) and 1578 cm−1 (Fig. S27b†), respectively. For SP-II, the amide I and II bands shifted to lower wavenumbers of 1645 (CO stretch) and 1554 cm−1, respectively (Fig. S27c†), probably due to the enhanced intermolecular H-bonds.70,71 In addition, the vibration bands of SP-IV appeared at 1651 and 1550 cm−1 for amide I and II, respectively (Fig. S27d†). Additionally, the two NH stretching bands observed in the FT-IR spectra could be ascribed to the presence of two different types of amide bonds within the structures (Fig. S27a†). These results indicate that the intermolecular H-bonds observed in SP-II or SP-IV with a helical fiber structure are stronger than those observed in SP-I. The wide-angle X-ray diffraction (WXRD) patterns of SP-I show one small peak at d = 3.20, corresponding to π–π stacking (Fig. S28†). While, SP-II shows three peaks at d = 3.20, 2.82 and 2.79, which corresponds to π–π stacking, Pt–Pt, and Ag–Ag interactions, respectively. The Ag–Ag interaction in SP-II was observed at 120 cm−1 by the Raman spectroscopy (Fig. S29†).
O stretch) and 1578 cm−1 (Fig. S27b†), respectively. For SP-II, the amide I and II bands shifted to lower wavenumbers of 1645 (CO stretch) and 1554 cm−1, respectively (Fig. S27c†), probably due to the enhanced intermolecular H-bonds.70,71 In addition, the vibration bands of SP-IV appeared at 1651 and 1550 cm−1 for amide I and II, respectively (Fig. S27d†). Additionally, the two NH stretching bands observed in the FT-IR spectra could be ascribed to the presence of two different types of amide bonds within the structures (Fig. S27a†). These results indicate that the intermolecular H-bonds observed in SP-II or SP-IV with a helical fiber structure are stronger than those observed in SP-I. The wide-angle X-ray diffraction (WXRD) patterns of SP-I show one small peak at d = 3.20, corresponding to π–π stacking (Fig. S28†). While, SP-II shows three peaks at d = 3.20, 2.82 and 2.79, which corresponds to π–π stacking, Pt–Pt, and Ag–Ag interactions, respectively. The Ag–Ag interaction in SP-II was observed at 120 cm−1 by the Raman spectroscopy (Fig. S29†).
Thermodynamic studies for the formation of SPs
In the presence of AgNO3 and Fe(BF4)2, as can be seen in the above time-dependent PL and other studies, Pt-L1 exhibited different supramolecular polymerization behaviors compared to the conditions without metal ions. To support or reconfirm these behaviors, temperature-dependent PL spectral changes were also studied. The heating profile of SP-I (1.0–3.0 mM) in the absence of metal ions displayed a sigmoidal trend (Fig. S30A and S31†), reflecting an immediate disassembly into monomers and the supramolecular polymerization proceeds via an isodesmic model. Conversely, in the presence of AgNO3 and Fe(BF4)2, a marked reduction in the intensity at 620 or 650 nm was observed at 310 K and 320 K, respectively (Fig. S30B and C†). At the same time, there was an elevation in the intensity of the MLCT band centered around 560 nm (Fig. S30B and C†). Upon heating the solutions containing SP-II and SP-IV, the melting curves manifested a non-sigmoidal pattern (Fig. 7A and S32†). Taken collectively, these results suggest that both SP-II and SP-IV are formed via a cooperative model governed by a nucleation–elongation mechanism.Upon determining the elongation binding constants (Ke) and/or elongation temperature (Te) via curve fitting with cooperative model,72 the thermodynamic parameters (ΔGe, ΔHe and ΔS) were calculated using the van't Hoff plot (Fig. 7 and S31C†). For example, the negative value of ΔGe for SP-III (−19.1 kJ mol−1) is larger than that of SP-I (−17.8 kJ mol−1) (Table 1), indicating that the SP-III with a left-handed helical structure is the thermodynamically favored product, while SP-I with a spherical structure is a kinetically trapped product. On the other hand, the elongation temperatures for SP-IV, when formed in the presence of Fe2+, were roughly 9–12 K higher than those recorded for the Ag+ system (Fig. 7B). In particular, the ΔGe value (−21.5 kJ mol−1) surpasses that obtained in the presence of Ag+ (−19.6 kJ mol−1), suggesting that SP-IV is thermodynamically more stable than SP-II. Consequently, it is possible that the SP-II formed with Ag+ can be spontaneously converted to SP-IV upon the introduction of Fe2+, following an on-pathway mechanism as shown in Scheme 1.
:1 v/v)
| ΔHe (kJ mol−1) | ΔS (J K−1 mol−1) | ΔGe (kJ mol−1) | |
|---|---|---|---|
| SP-I | −165.0 | −494.2 | −17.8 |
| SP-II | −88.7 | −231.6 | −19.6 |
| SP-IV | −75.8 | −182.0 | −21.5 |
 | ||
| Fig. 7 (A) Degree of aggregation (α) in different concentrations of Pt-L1 (2.0 mM) with 1.5 equiv. of (a) AgNO3 and (b) Fe(BF4)2 as a function of temperature at 620 and 650 nm in DMSO/H2O (9:1 v/v). (B) Natural logarithm of the reciprocal CT as a function of the reciprocal Te showing the linear relationship. | ||
Pathway complexity of SPs
Based on the above observations, we proposed an energy landscape representing the complexity of pathways and mechanism for the supramolecular polymerizations (Fig. 8). In the absence of metal ions, both the heating and cooling curves of SP-I exhibited a well-defined sigmoidal shape without hysteresis (Fig. S33†), indicating the supramolecular polymerization of Pt-L1 to SP-Ivia an isodesmic model. In the transformation to SP-II or SP-IV, with increasing concentrations of Pt-L1 in the presence of AgNO3 or Fe(BF4)2, sigmoidal kinetic profiles were observed, and the duration of the lag phase decreased (Fig. 3B). Furthermore, an increase in the concentration of Pt-L1 in the presence of Fe(BF4)2 rapidly led to reaching an equilibrium state (Fig. 5B). These observations indicate a buffered release and self-nucleation of the monomeric [(Pt-L1)2Ag2]4+ and [(Pt-L1)2Fe]4+ under pre-equilibrium between kinetic products, followed by autocatalytic growth of the remaining monomers via an on-pathway mechanism (Fig. 8). In particular, an increase in the concentration of Fe(BF4)2 in the transformation of SP-II to SP-IV led to a decrease in the lag time (Fig. 6B), indicating that increasing concentration of Fe(BF4)2 accelerates the conversion of SP-II to SP-IV. Therefore, SP-II acts as a kinetically trapped state in the presence of Fe(BF4)2. | ||
| Fig. 8 Proposed energy landscapes for the formation of SP-I, SP-II, SP-III and SP-IV: Pt-L1 (blue) only, with AgNO3 (pink), with Fe(BF4)2 (green). | ||
Interestingly, in contrast to the SP-I, the hidden pathway of SP-III in Pt-L1 was obtained by adding NaCl to SP-II under kinetically controlled supramolecular polymerization. This is evidenced by the observation that heating and then cooling SP-III resulted in the formation of SP-I, and the obtained SP-III showed a non-sigmodal heating curve with Te (315 K), indicating cooperative growth (Fig. S34†). Moreover, the SP-I did not transform into SP-III within the experimental timescale. This outcome suggests that the formation of SP-I and SP-III from Pt-L1 occurred through distinct pathways and mechanisms. Therefore, these results indicate that the pathway of Pt-L1 into SP-III was prevented due to a higher activation barrier than the pathway for SP-I. A rational strategy employing the complexation behavior of Pt-L1 with secondary metal ions revealed a hidden pathway to SP-III (Fig. 8).
Kinetic control of supramolecular polymerization by secondary ligands
In the complexations of the mixed ligand system, there is the possibility of forming a variety of complexes including kinetically controlled products. Motivated by this trend, we guess that metallosupramolecular polymerizations can also be kinetically regulated in the presence of a competing coligand. To elucidate the concept of kinetic control in such systems, we monitored the time-dependent PL spectra of Pt-L1 in the presence of secondary ligands (bpy or DA18C6) upon coordination with AgNO3 or Fe(BF4)2 (Fig. 9). When bpy was used, the time-dependent PL intensity at 620 nm showed sigmoidal progressions (Fig. 9B), reflecting the cooperative pathway formation. The PL peak intensity at 620 nm diminished in comparison to that observed in the absence of bpy. The lag time increases by 1.5 to 3.0-fold with increasing bpy concentrations attributed to the [(bpy)2Ag]+ complex formation (Scheme 2) as evidenced by HR-ESI-MS (Fig. S35†). Consequently, some complexed species including [(bpy)2Ag]+ (logK = 7.10, Fig. S36†) and [(Pt-L1)(bpy)Ag]2+ serve as kinetic species during the supramolecular polymerization to build up SP-II of type [(Pt-L1)2Ag2]n4+ as a thermodynamic product (Fig. S37†). For comparison, pyridine was also employed as a secondary ligand. However, no significant changes were observed in the lag time for the supramolecular polymerization of a mixture containing Pt-L1 and pyridine in the presence of AgNO3 or Fe(BF4)2 (Fig. S38†), compared to pure Pt-L1 with metal ions (Fig. 3 and 5). The result suggests that the pyridine complex has not served as an intermediate in controlling the kinetics, due to its weaker coordinating ability than bpy. In the parallel experiments with DA18C6 (Fig. S39†), a similar trend with bpy was observed and the elongated lag phases exceeded those observed with bpy, probably due to the formation of a 1:1 complex, [(DA18C6)Ag]+ (Scheme 2), as a kinetic complex with higher stability (logK = 7.22) than that of bpy (Fig. S40 and S41†). As a result, no secondary ligands were incorporated into the formation of SPs due to the absence of driving forces for the self-assembly. However, the secondary ligands competitively influence the kinetic control of supramolecular polymerization.
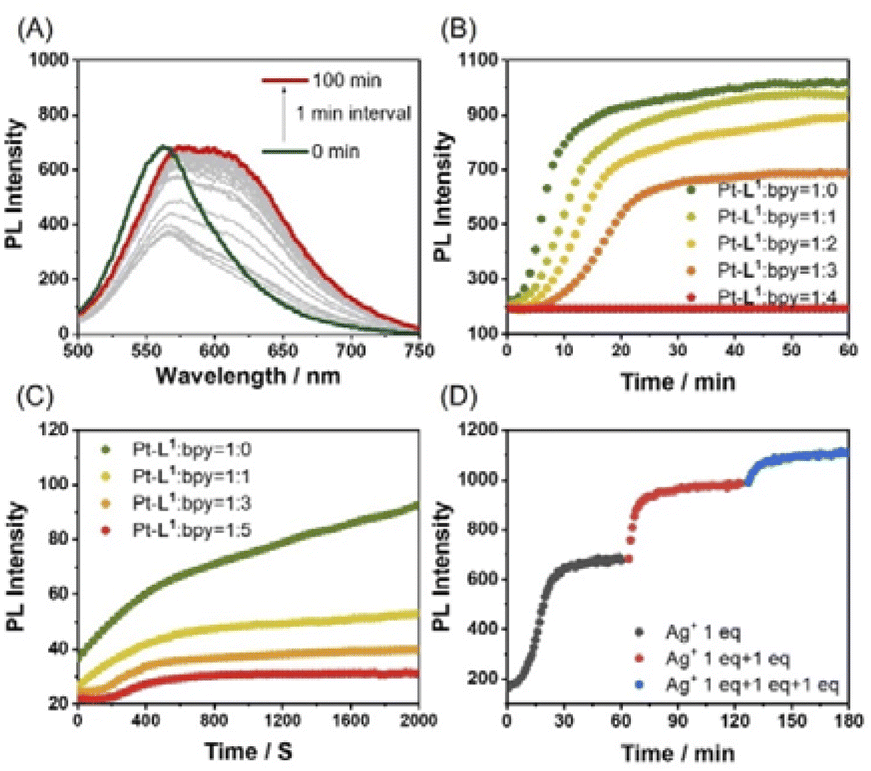 | ||
| Fig. 9 (A) Time-dependent PL spectra of a mixture of Pt-L1 (2.0 mM), bpy (6 mM), and AgNO3 (1.5 equiv.) in DMSO/H2O (9:1 v/v) at 293 K, λex = 420 nm. (B and C) Plots of the PL intensity at 620 or 650 nm vs. times for supramolecular polymerization in the mixed (B) Pt-L1 and bpy with AgNO3 (1.5 equiv.), and (C) the mixed Pt-L1 and bpy with Fe(BF4)2 (1.5 equiv.) in DMSO/H2O (9:1 v/v). (D) Plot showing the PL intensity (at 620 nm) vs. time of Pt-L1 (2 mM) with stepwise addition of AgNO3 (1.0–3.0 equiv.). | ||
 | ||
| Scheme 2 Proposed formation mechanism of SP-II in the presence of either (A) bpy and Ag+ or (B) DA18C6 and Ag+ as secondary ligands. | ||
More interestingly, in the presence of bpy or DA18C6 and Fe2+, distinct lag phases (t50 = 2.5 min with bpy and 3.5 min with DA18C6) were observed in the formation of SP-IV of [(Pt-L1)2Fe]4+ (Fig. 9C). The duration of the lag phase was kinetically modulated due to the formation of [(bpy)3Fe]2+ or [(DA18C6)Fe]2+ as a kinetic complex (Fig. S42–S45†), influenced by the concentration of the second ligands. The PL spectral changes exhibited a sigmoidal progression, reflecting a cooperative polymerization pathway governed by a nucleation–elongation mechanism. The observed lag phase is attributed to the stable formation of [(bpy)3Fe]2+ (logK = 11.56) (Fig. S46†), which are kinetically trapped species. Additionally, upon the addition of bpy to the equilibrium states of SP-II and SP-IV, no significant changes in PL intensity were observed (Fig. S47†). This result implied that bpy contributed to the kinetic pathway in supramolecular polymerization. Again, it is concluded that secondary ligand complexes can significantly influence kinetics as effective intermediates in metallosupramolecular polymerization.
To realize the kinetic growth pathway of the cooperative supramolecular polymerization, the addition of AgNO3 was modified. For example, 3.0 equiv. of AgNO3 or Fe(BF4)2 was added in three divided steps with an equal amount (1.0 equiv. in each step) to the mixture of Pt-L1 and bpy. As shown in Fig. 9D and S48,† the first addition resulted in a sigmoidal pattern, while the second and third were non-sigmoidal, suggesting that the SP formed during the first batch served as a nucleation seed. As shown in Fig. 9D, notably the magnitude of aggregation is reduced during the second and third batches (red and blue curved lines) compared to the first one (black curved line). The observed difference in the aggregation degree (α) during subsequent additions may be attributed to the in situ formation of kinetic products with the secondary ligand across the batches.
Conclusions
In summary, we have demonstrated a novel strategy for controlling the pathway for the supramolecular polymerization of a monoalkynylplatinum(II) terpyridine complex (Pt-L1) by introducing secondary metal ions and ligands. By employing a kinetic trapping strategy through the formation of complexes with various coordination numbers in the presence of secondary metal ions, we were able to regulate spontaneous metallosupramolecular polymerization. The introduction of the secondary metal ion to the Pt-L1 solution produced kinetic complexes, which act as a key factor in the kinetic control. In particular, the addition of Ag+ induced a large red-shift due to the Pt⋯Pt and Ag⋯Ag interactions during the formation of supramolecular polymer. These Pt⋯Pt and/or Ag⋯Ag interactions during supramolecular polymerization also act as additional driving forces. Furthermore, the self-assembly process was precisely and kinetically controlled by introducing secondary ligands to create competition complexes. In the presence of secondary ligands, some kinetic complexes that form complexes with the secondary ligand act as efficient kinetic species. Therefore, it can be concluded that SP-I, as a kinetical product, is formed by an isodesmic model, whereas SP-II, SP-III and SP-IV are generated by a cooperative model via a nucleation–elongation mechanism. In particular, SP-II obtained in the presence of Ag+ ion transforms to SP-III or SP-IV through an on-pathway mechanism upon the introduction of NaCl or Fe(BF4)2. As a result, we have shown a novel methodology on how to control the pathway of metallosupramolecular polymerization precisely by employing secondary metal ions and/or ligands which form competitive complex species in the self-assembly process. Consequently, this study provides new insights into the molecular design that rationally governs the aggregation pathways of multi-component systems, with implications for the development of optical materials.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
M. K., H. C. and M. K. contributed to the design and performed the preparation and characterization of supramolecular polymers. S. K. and J. C. contributed ESI-MS observation. S. Y. and E. L. determined stability constants. J. H. J. and S. H. J. directed the project. All authors contributed to the discussion and the preparation of the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (2022R1A4A1022252, 2021R1A2C2007664, and RS-2023-00241926). The results for DFT calculation were obtained by the high-performance computing resources of the UNIST Supercomputing Center.Notes and references
- T. F. A. D. Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef PubMed.
- C. Rest, R. Kandanelli and G. Fernandez, Chem. Soc. Rev., 2015, 44, 2543–2572 RSC.
- S. I. Stupp, Nano Lett., 2010, 10, 4783–4786 CrossRef CAS PubMed.
- E. W. M. T. Aida and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef PubMed.
- S. Sarkar, R. Laishram, D. Deb and S. J. George, J. Am. Chem. Soc., 2023, 145, 22009–22018 CrossRef CAS PubMed.
- Q. Wan, X. S. Xiao, W. P. To, W. Lu, Y. Chen, K. H. Low and C. M. Che, Angew. Chem., Int. Ed., 2018, 57, 17189–17193 CrossRef CAS PubMed.
- F. Wang and C. L. Feng, Angew. Chem., Int. Ed., 2018, 57, 5655–5659 CrossRef CAS PubMed.
- S. Yagai, J. Photochem. Photobiol., C, 2006, 7, 164–182 CrossRef CAS.
- S. Yagai, Y. Kitamoto, S. Datta and B. Adhikari, Acc. Chem. Res., 2019, 52, 1325–1335 CrossRef CAS PubMed.
- S. L. Higashi and M. Ikeda, JACS Au, 2021, 1, 1639–1646 CrossRef CAS PubMed.
- Y. J. Wang, X. Y. Shi, P. Xing and S. Q. Zang, JACS Au, 2023, 3, 565–574 CrossRef CAS PubMed.
- B. S. Morozov, A. S. Oshchepkov, I. Klemt, A. M. Agafontsev, S. Krishna, F. Hampel, H. G. Xu, A. Mokhir, D. Guldi and E. Kataev, JACS Au, 2023, 3, 964–977 CrossRef CAS PubMed.
- C. Li, J. Chen, T. Man, B. Chen, J. Li, Q. Li, X. Yang, Y. Wan, C. Fan and J. Shen, JACS Au, 2024, 4, 1125–1133 CrossRef CAS PubMed.
- P. A. Korevaar, S. J. George, A. J. Markvoort, M. M. Smulders, P. A. Hilbers, A. P. Schenning, T. F. De Greef and E. W. Meijer, Nature, 2012, 481, 492–496 CrossRef CAS PubMed.
- J. Matern, Y. Dorca, L. Sánchez and G. Fernandez, Angew. Chem., Int. Ed., 2019, 58, 16730–16740 CrossRef CAS PubMed.
- A. Sorrenti, J. Leira-Iglesias, A. J. Markvoort, T. F. A. de Greef and T. M. Hermans, Chem. Soc. Rev., 2017, 46, 5476–5490 RSC.
- E. Mattia and S. Otto, Nat. Nanotechnol., 2015, 10, 111–119 CrossRef CAS PubMed.
- J. F. Stoddart, Nat. Chem., 2009, 1, 14–15 CrossRef CAS.
- G. M. Whitesides and R. F. lsmagilov, Science, 1999, 284, 89–92 CrossRef CAS PubMed.
- A. Langenstroer, K. K. Kartha, Y. Dorca, J. Droste, V. Stepanenko, R. Q. Albuquerque, M. R. Hansen, L. Sánchez and G. Fernandez, J. Am. Chem. Soc., 2019, 141, 5192–5200 CrossRef CAS.
- N. Sasaki, M. F. J. Mabesoone, J. Kikkawa, T. Fukui, N. Shioya, T. Shimoaka, T. Hasegawa, H. Takagi, R. Haruki, N. Shimizu, S. I. Adachi, E. W. Meijer, M. Takeuchi and K. Sugiyasu, Nat. Commun., 2020, 11, 3578 CrossRef PubMed.
- A. Jain, S. Dhiman, A. Dhayani, P. K. Vemula and S. J. George, Nat. Commun., 2019, 10, 450 CrossRef CAS PubMed.
- S. Ogi, C. Grzeszkiewicz and F. Würthner, Chem. Sci., 2018, 9, 2768–2773 RSC.
- I. Helmers, G. Ghosh, R. Q. Albuquerque and G. Fernandez, Angew. Chem., Int. Ed., 2021, 60, 4368–4376 CrossRef CAS PubMed.
- S. Ogi, T. Fukui, M. L. Jue, M. Takeuchi and K. Sugiyasu, Angew. Chem., Int. Ed., 2014, 53, 14363–14367 CrossRef CAS PubMed.
- J. Matern, K. K. Kartha, L. Sánchez and G. Fernandez, Chem. Sci., 2020, 11, 6780–6788 RSC.
- E. Weyandt, L. Leanza, R. Capelli, G. M. Pavan, G. Vantomme and E. W. Meijer, Nat. Commun., 2022, 13, 248 CrossRef CAS PubMed.
- L. Borsdorf, L. Herkert, N. Baumer, L. Rubert, B. Soberats, P. A. Korevaar, C. Bourque, C. Gatsogiannis and G. Fernandez, J. Am. Chem. Soc., 2023, 145, 8882–8895 CrossRef CAS.
- P. Khanra, A. K. Singh, L. Roy and A. Das, J. Am. Chem. Soc., 2023, 145, 5270–5284 CrossRef CAS PubMed.
- G. Liu, M. G. Humphrey, C. Zhang and Y. Zhao, Chem. Soc. Rev., 2023, 52, 4443–4487 RSC.
- J. J. B. van der Tol, G. Vantomme and E. W. Meijer, J. Am. Chem. Soc., 2023, 145, 17987–17994 CrossRef CAS PubMed.
- F. Wang, R. Liao and F. Wang, Angew. Chem., Int. Ed., 2023, 62, e202305827 CrossRef CAS PubMed.
- F. Xu, S. Crespi, G. Pacella, Y. Fu, M. C. A. Stuart, Q. Zhang, G. Portale and B. L. Feringa, J. Am. Chem. Soc., 2022, 144, 6019–6027 CrossRef CAS PubMed.
- F. Xu, S. Crespi, L. Pfeifer, M. C. A. Stuart and B. L. Feringa, CCS Chem., 2022, 4, 2212–2220 CrossRef CAS.
- S. Ogi, V. Stepanenko, K. Sugiyasu, M. Takeuchi and F. Würthner, J. Am. Chem. Soc., 2015, 137, 3300–3307 CrossRef CAS.
- L. Kleine-Kleffmann, V. Stepanenko, K. Shoyama, M. Wehner and F. Würthner, J. Am. Chem. Soc., 2023, 145, 9144–9151 CrossRef CAS PubMed.
- C. Naranjo, S. Adalid, R. Gomez and L. Sánchez, Angew. Chem., Int. Ed., 2023, 62, e202218572 CrossRef CAS.
- S. Ogi, K. Sugiyasu, S. Manna, S. Samitsu and M. Takeuchi, Nat. Chem., 2014, 6, 188–195 CrossRef CAS PubMed.
- T. Fukui, S. Kawai, S. Fujinuma, Y. Matsushita, T. Yasuda, T. Sakurai, S. Seki, M. Takeuchi and K. Sugiyasu, Nat. Chem., 2017, 9, 493–499 CrossRef CAS.
- N. Sasaki, J. Kikkawa, Y. Ishii, T. Uchihashi, H. Imamura, M. Takeuchi and K. Sugiyasu, Nat. Chem., 2023, 15, 922–929 CrossRef CAS.
- A. Aliprandi, M. Mauro and L. De Cola, Nat. Chem., 2016, 8, 10–15 CrossRef CAS.
- Q. Wan, W.-P. To, X. Chang and C.-M. Che, Phys. Chem., 2020, 6, 945–967 CAS.
- S. Carrara, A. Aliprandi, C. F. Hogan and L. De Cola, J. Am. Chem. Soc., 2017, 139, 14605–14610 CrossRef CAS.
- A. Galstyan, A. R. Naziruddin, C. Cebrián, A. Iordache, C. G. Daniliuc, L. De Cola and C. A. Strassert, Eur. J. Inorg. Chem., 2015, 2015, 5822–5831 CrossRef CAS.
- A. Haque, L. Xu, R. A. Al-Balushi, M. K. Al-Suti, R. Ilmi, Z. Guo, M. S. Khan, W.-Y. Wong and P. R. Raithby, Chem. Soc. Rev., 2019, 48, 5547–5563 RSC.
- J. K. Poon, Z. Chen, S. Y. Leung, M. Y. Leung and V. W. Yam, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2022829118 CrossRef PubMed.
- M. E. Robinson, D. J. Lunn, A. Nazemi, G. R. Whittell, L. De Cola and I. Manners, Chem. Commun., 2015, 51, 15921–15924 RSC.
- Y. Chen, C. M. Che and W. Lu, Chem. Commun., 2015, 51, 5371–5374 RSC.
- P. Du, K. Knowles and R. Eisenberg, J. Am. Chem. Soc., 2008, 130, 2576–12577 Search PubMed.
- P. Du, J. Schneider, P. Jarosz and R. Eisenberg, J. Am. Chem. Soc., 2006, 128, 7726–7727 CrossRef CAS PubMed.
- X. Han, L. Z. Wu, G. Si, J. Pan, Q. Z. Yang, L. P. Zhang and C. H. Tung, Chemistry, 2007, 13, 1231–1239 CrossRef CAS PubMed.
- E. C. H. Kwok, M. Y. Chan, K. M. C. Wong, W. H. Lam and V. W. W. Yam, Chem.–Eur. J., 2010, 16, 12244–12254 CrossRef CAS PubMed.
- M. Mauro, A. Aliprandi, D. Septiadi, N. S. Kehr and L. De Cola, Chem. Soc. Rev., 2014, 43, 4144–4166 RSC.
- W.-S. Tang, X.-X. Lu, K. M.-C. Wong and V. W.-W. Yam, J. Mater. Chem., 2005, 15, 2714–2720 RSC.
- K. M.-C. Wong and V. W.-W. YAM, Acc. Chem. Res., 2011, 44, 424–434 CrossRef CAS PubMed.
- V. W. Yam, V. K. Au and S. Y. Leung, Chem. Rev., 2015, 115, 7589–7728 CrossRef CAS PubMed.
- V. W. Yam, K. H. Chan, K. M. Wong and N. Zhu, Chem.–Eur. J., 2005, 11, 4535–4543 CrossRef PubMed.
- D. Zhang, L.-Z. Wu, L. Zhou, X. Han, Q.-Z. Yang, L.-P. Zhang and C.-H. Tung, J. Am. Chem. Soc., 2004, 126, 3440–3441 CrossRef CAS PubMed.
- K. Zhang, M. C. Yeung, S. Y. Leung and V. W. Yam, ACS Appl. Mater. Interfaces, 2020, 12, 8503–8512 CrossRef CAS PubMed.
- H.-Z. Wang, M. H.-Y. Chan, Z. Chen, Z.-Y. Chen, M.-Y. Leung and V. W.-W. Yam, Chem.–Eur. J., 2023, 9, 3573–3587 CAS.
- S. K. Leung, A. K. Chan, S. Y. Leung, M. Y. Leung and V. W. Yam, Chem. Sci., 2020, 11, 499–507 RSC.
- S. Y. Leung, K. M. Wong and V. W. Yam, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 2845–2850 CrossRef CAS PubMed.
- V. W.-W. Yam, K. M.-C. Wong and N. Zhu, J. Am. Chem. Soc., 2002, 124, 6506–6507 CrossRef CAS PubMed.
- M. Ok, K. Y. Kim, H. Choi, S. Kim, S. S. Lee, J. Cho, S. H. Jung and J. H. Jung, Chem. Sci., 2022, 13, 3109–3117 RSC.
- S. G. Kang, K. Y. Kim, Y. Cho, D. Y. Jeong, J. H. Lee, T. Nishimura, S. S. Lee, S. K. Kwak, Y. You and J. H. Jung, Angew. Chem., Int. Ed., 2022, 61, e202207310 CrossRef CAS PubMed.
- S. M. Fatur, S. G. Shepard, R. F. Higgins, M. P. Shores and N. H. Damrauer, J. Am. Chem. Soc., 2017, 139, 4493–4505 CrossRef CAS PubMed.
- K. M.-C. Wong, W.-S. Tang, X.-X. Lu, N. Zhu and V. W.-W. Yam, Inorg. Chem., 2005, 44, 1492–1498 CrossRef CAS PubMed.
- M. Kim, M. Ok, C. Li, K. Go, S. Kim, J. Kim, J. H. Jung and S. H. Jung, Inorg. Chem. Front., 2023, 10, 768–775 RSC.
- P. Pander, A. Sil, R. J. Salthouse, C. W. Harris, M. T. Walden, D. S. Yufit, J. A. G. Williams and F. B. Dias, J. Mater. Chem. C, 2022, 10, 15084–15095 RSC.
- W. Ji, C. Yuan, P. Chakraborty, S. Gilead, X. Yan and E. Gazit, Commun. Chem., 2019, 2, 1–11 CrossRef CAS.
- H. Jiang, M. O. Guler and S. I. Stupp, Soft Matter, 2007, 3, 454–462 RSC.
- M. M. Smulders, M. M. Nieuwenhuizen, T. F. de Greef, P. van der Schoot, A. P. Schenning and E. W. Meijer, Chem.–Eur. J., 2010, 16, 362–367 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc06083k |
| This journal is © The Royal Society of Chemistry 2024 |