
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Energy exchange between Nd3+ and Er3+ centers within molecular complexes†
Diamantoula
Maniaki
 ab,
Annika
Sickinger
d,
Leoní A.
Barrios
ab,
David
Aguilà
ab,
Olivier
Roubeau
e,
Yannick
Guyot
f,
François
Riobé
dg,
Olivier
Maury
d,
Laura
Abad Galán
*c and
Guillem
Aromí
*ab
ab,
Annika
Sickinger
d,
Leoní A.
Barrios
ab,
David
Aguilà
ab,
Olivier
Roubeau
e,
Yannick
Guyot
f,
François
Riobé
dg,
Olivier
Maury
d,
Laura
Abad Galán
*c and
Guillem
Aromí
*ab
aDepartament de Química Inorgànica i Orgànica, Universitat de Barcelona, Diagonal 645, 08028 Barcelona, Spain. E-mail: aromi@ub.edu
bInstitute of Nanoscience and Nanotechnology of the University of Barcelona (IN2UB), Barcelona, Spain
cDepartamento de Química Inorgánica, Fac. CC. Químicas, Universidad Complutense de Madrid, Avda. Complutense s/n, 28040 Madrid, Spain. E-mail: laabad03@ucm.es
dUniv Lyon, ENS Lyon, CNRS, UMR 5182, Laboratoire de Chimie, F69342 Lyon, France
eInstituto de Nanociencia y Materiales de Aragón (INMA), CSIC and Universidad de Zaragoza, Plaza San Francisco s/n, 50009, Zaragoza, Spain
fUniv. Lyon, Institut Lumière Matière, UMR 5306 CNRS-Université Claude Bernard, Lyon 1, 10 rue Ada Byron, F-69622 Villeurbanne Cedex, France
gUniv. Bordeaux, CNRS, Bordeaux INP, ICMCB UMR 5026, F-33600 Pessac, France
First published on 18th October 2024
Abstract
Developing controlled and reproducible molecular assemblies incorporating lanthanide centers is a crucial step for driving forward up- and down-conversion processes. This challenge calls for the development of strategies to facilitate the efficient in situ segregation of different Ln metal ions into distinct positions within the molecule. The unique family of pure [LnLn′Ln] heterometallic coordination compounds previously developed by us represents an ideal platform for studying the desired Ln-to-Ln′ energy transfer (ET). In this context, we report here the new pure one-step synthetically produced [ErNdEr] (3) complex, which allows for the first time at the molecular level to study the mechanisms behind Nd-to-Er energy transfer. To further assess the photophysical properties of this complex, the analogous [LuNdLu] (1) and [ErLaEr] (2) complexes have also been prepared and photophysically studied. Efficient sensitization via the two β-diketones employed as main ligands was probed for both Nd3+ and Er3+ ions, resulting in highly resolved emission spectra and sufficiently long excited state lifetimes, which allowed further assessment of the Ln-to-Ln′ ET. This intermetallic transfer was first detected by comparing the emission spectra of iso-absorbant solutions and demonstrated by comparing the lifetime values with or without the lanthanide quencher (Er3+), as well as with a deep analysis of the excitation spectrum of the three complexes. Thus, a very unique phenomenon was discovered, consisting of a mutual Nd-to-Er and Er-to-Nd ET with no net increase of brightness by any metal; while Nd3+ transfers the energy received from the antenna to Er3+, the sensitization of the latter results in back-transfer to Nd3+ into a non-emissive, thus silent, state.
Introduction
Lanthanides in their trivalent form (Ln3+) are well known for their characteristic optical (and also magnetic) properties. The specific electronic configuration 4fn5s25p6 (n = 0–14) when zero-valent, in which the 4f inner shell is well shielded by the 5s and 5p orbitals, gives rise to long-lived excited states with sharp 4f emission bands that span from the near-infrared (NIR) to the visible and UV regions. These optimal photoluminescence properties account for their application in medical imaging,1–3 telecommunications,4–6 light emitting devices7 and solar energy harvesting8–10 among other areas.11 Ln ions have the major drawback that their intraconfigurational f–f electronic transitions are mostly forbidden by the selection rules, resulting in very low absorption coefficients. This can be overcome by the presence of a light-harvesting antenna which efficiently transfers the absorbed energy to the lanthanide ions, bringing them into excited states. Conjugated organic groups12,13 and charge transfer states of d-metal complexes14–16 are often presented as the most efficient vectors of lanthanide sensitization. In addition, the electronic configuration of the Ln atoms leads to the presence of electronic levels and ladder-like energy diagrams, thus, setting up a perfect platform of possible lanthanide to lanthanide (Ln-to-Ln′, Ln ≠ Ln′) energy transfer events resulting in up- and down-conversion processes.17 While these processes have been extensively studied in solids18–20 and more recently in nanoparticles,21–24 there is a growing interest in exploring their potential at the molecular scale, using stable, well defined and reproducible molecular assemblies.25 Regarding up-conversion processes, improvements have been recently obtained with complexes presenting mainly the following pairs: Tb/Yb,26,27 Eu/Yb28 and Yb/Er.29–31 This anti-Stokes luminescence process (lower-to-higher energy transfer) bears significant potential for various applications that require or utilize NIR radiation, including deep-tissue imaging, cancer therapy, nano-thermometry, biosensing, display technologies, and solar cells.21,32–34 By contrast, down-conversion (higher-to-lower energy transfer) remains less investigated with the majority of the processes studied in solution.35 Probing Ln-to-Ln′ energy transfer (ET) within molecules is challenging because of the necessity to engineer well-defined multi-metallic compounds mixing lanthanide ions with seemingly identical coordination chemistry.36,37 Therefore, one-step self-assembly reactions involving different lanthanides often result in mixtures of metal distributions within the molecule38–41 and thus, very tedious sequential methodologies, such as covalent linkage of preformed coordination complexes,36,42–45 are typically employed to obtain site-selective heterometallic Ln molecules. The synthetic procedures that are thermodynamically controlled stem from the ability to discriminate between the ionic radii of different metals (rLn). Along these lines, we discovered a system capable to coordinate, with remarkable selectivity, two different lanthanide metals by generating two distinct coordination sites, one able to bind the larger ion and the other the smaller one.46–48 These structures were observed with heteroleptic complexes using multitopic ligands combining ONO chelates (dipicolinate type) with diketonate moieties. This principle has been successfully replicated with a different architecture thus underlining its great potential.49 The new molecular structure was revealed by mixing two ligands (Fig. 1), both with dipicolinate (O, N, O) and diketonate (O, O) units (H2LA, 2,6-bis[(3-oxo-3-naphthalene-2-yl)-propionyl]pyridine; H2LB, 6-(3-(naphthalene-2-yl)-3-oxopropanoyl)-picolinic acid), with certain combinations of two different Ln(NO3)3 salts. As a result, both metals are selectively positioned in a [LnLn′Ln] topology. The selectivity presented by these systems, as well as a distance between centers of ∼3.8 Å, allowed us to use this platform to study the intramolecular ET processes. Indeed, we succeeded in studying the Nd-to-Yb ET in the Nd/Yb pair by isolating the [NdYb]50 and [YbNdYb]51 complexes. We noted that in the case of the trinuclear analogue, the existence of two acceptors per donor dramatically enhances the efficiency of this photophysical process. These findings provide noteworthy examples of down conversion processes in heteronuclear complexes, very little studied so far. The down conversion intramolecular energy transfer has only been presented in the visible region with the Tb/Eu,52–54 and Dy/Tb43 pairs and, more interestingly, in the NIR region with the Eu/Nd,55 Nd/Yb,50,51,56,57 Tb/Yb58 and Yb/Er pairs.52,59–61
electronic levels and ladder-like energy diagrams, thus, setting up a perfect platform of possible lanthanide to lanthanide (Ln-to-Ln′, Ln ≠ Ln′) energy transfer events resulting in up- and down-conversion processes.17 While these processes have been extensively studied in solids18–20 and more recently in nanoparticles,21–24 there is a growing interest in exploring their potential at the molecular scale, using stable, well defined and reproducible molecular assemblies.25 Regarding up-conversion processes, improvements have been recently obtained with complexes presenting mainly the following pairs: Tb/Yb,26,27 Eu/Yb28 and Yb/Er.29–31 This anti-Stokes luminescence process (lower-to-higher energy transfer) bears significant potential for various applications that require or utilize NIR radiation, including deep-tissue imaging, cancer therapy, nano-thermometry, biosensing, display technologies, and solar cells.21,32–34 By contrast, down-conversion (higher-to-lower energy transfer) remains less investigated with the majority of the processes studied in solution.35 Probing Ln-to-Ln′ energy transfer (ET) within molecules is challenging because of the necessity to engineer well-defined multi-metallic compounds mixing lanthanide ions with seemingly identical coordination chemistry.36,37 Therefore, one-step self-assembly reactions involving different lanthanides often result in mixtures of metal distributions within the molecule38–41 and thus, very tedious sequential methodologies, such as covalent linkage of preformed coordination complexes,36,42–45 are typically employed to obtain site-selective heterometallic Ln molecules. The synthetic procedures that are thermodynamically controlled stem from the ability to discriminate between the ionic radii of different metals (rLn). Along these lines, we discovered a system capable to coordinate, with remarkable selectivity, two different lanthanide metals by generating two distinct coordination sites, one able to bind the larger ion and the other the smaller one.46–48 These structures were observed with heteroleptic complexes using multitopic ligands combining ONO chelates (dipicolinate type) with diketonate moieties. This principle has been successfully replicated with a different architecture thus underlining its great potential.49 The new molecular structure was revealed by mixing two ligands (Fig. 1), both with dipicolinate (O, N, O) and diketonate (O, O) units (H2LA, 2,6-bis[(3-oxo-3-naphthalene-2-yl)-propionyl]pyridine; H2LB, 6-(3-(naphthalene-2-yl)-3-oxopropanoyl)-picolinic acid), with certain combinations of two different Ln(NO3)3 salts. As a result, both metals are selectively positioned in a [LnLn′Ln] topology. The selectivity presented by these systems, as well as a distance between centers of ∼3.8 Å, allowed us to use this platform to study the intramolecular ET processes. Indeed, we succeeded in studying the Nd-to-Yb ET in the Nd/Yb pair by isolating the [NdYb]50 and [YbNdYb]51 complexes. We noted that in the case of the trinuclear analogue, the existence of two acceptors per donor dramatically enhances the efficiency of this photophysical process. These findings provide noteworthy examples of down conversion processes in heteronuclear complexes, very little studied so far. The down conversion intramolecular energy transfer has only been presented in the visible region with the Tb/Eu,52–54 and Dy/Tb43 pairs and, more interestingly, in the NIR region with the Eu/Nd,55 Nd/Yb,50,51,56,57 Tb/Yb58 and Yb/Er pairs.52,59–61
 | ||
| Fig. 1 Ligands 2,6-bis-((3-oxo-3-naphthalene-2-yl)propionyl)-pyridine (H2LA) and 6-(3-(naphthalene-2-yl)-3-oxopropanoyl)-picolinic acid (H2LB). | ||
This work presents, to our knowledge, the first case of direct energy transfer between Nd3+ and Er3+ centers in molecular complexes, specifically, in pure heterometallic molecules. This transfer was first observed in glasses in the 1990s,62,63 but had not been studied further in molecular systems. Hereby, we synthesized and determined the structure of a new compound, [Er2Nd(LA)2(LB)2(H2O)2(py)](NO3), here termed also [ErNdEr] (3), which effectively promotes intramolecular ET from Er to Nd. The photophysical properties of this complex, as well as those of the previously reported [Lu2Nd(LA)2(LB)2(H2O)2(py)](NO3)51 or [LuNdLu] (1) and [Er2La(LA)2(LB)2(H2O)2(py)](NO3)64 or [ErLaEr] (2) were thoroughly investigated to best characterize the desired Nd-to-Er ET.
Results and discussion
Synthesis
The new complex [Er2Nd(LA)2(LB)2(H2O)2(py)](NO3) (3), which allowed unveiling the phenomenon reported here, was obtained as single crystals from a one-step reaction between the stoichiometric amounts of Er(NO3)3, Nd(NO3)3, H2LA and H2LB in pyridine, following the diffusion of heptane. The chemical process is amenable to a description with a balanced chemical equation that invokes the presence of adventitious water and the association of the released protons with pyridine molecules from the solvent, as the sole base available (eqn (1)):| 2Er(NO3)3 + Nd(NO3)3 + 2H2LA + 2H2LB +2H2O + 9py → [Er2Nd(LA)2(LB)2(H2O)2(py)](NO3) + 8HpyNO3 | (1) |
Formation of suitable single crystals of 3 required the addition of one equivalent of CuCl2, which does not participate in the reaction but presumably plays a role of a modulator of the crystallization (see ESI† for details).65 The formulation of 3 was established by single-crystal X-ray diffraction (SCXRD, see below). The uniqueness of this compound is its strict and well defined heterometallic nature, exhibiting two Er3+ atoms per atom of Nd3+. Indeed, multinuclear Ln complexes from one step reactions frequently feature quasi-statistical distributions of the different metal types within the molecule, since they have very similar chemical behavior.38 In the present case, the scaffold generated by LA2− and LB2− generates coordination sites of two distinct types (the central and the peripheral ones) that favor longer metal-to-donor bond distances in the central location, driving the binding of the larger metal to this position (see structural details below). The purity of the bulk material is supported by C, H, and N microanalysis and inductively coupled plasma (ICP) metal content determination, the latter providing a molar Nd/Er ratio of 0.53. The formulation was also consistent with the response from variable temperature molar paramagnetic susceptibility and variable field magnetization measurements (details in ESI and Fig. S1†), which are ascribed to the presence of one Nd3+ and two Er3+ ions within a molecule with the molecular mass of 3. The persistence of the architecture of complex 3 in solution was established by mass spectrometry (MS), which unveiled three dominant signals from the trinuclear complex containing its four bridging ligands (Fig. S2–S5†). The absence of any trinuclear moiety with a metal composition other than [ErNdEr] reveals that no metal scrambling occurs.
Structure
Detailed information on the molecular structure of 3 was obtained from SCXRD data collected at 100 K. Its crystal lattice belongs to the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The asymmetric unit is made of one formula unit of 3 and 10 molecules of pyridine. The main [ErNdEr] complex (Fig. 2 and S6†) features a quasi-linear trimetallic Er⋯Nd⋯Er array (angle of 174.17°) with the metals bridged together by two μ3-LA2− and two μ-LB2− ligands that chelate them with two types of pockets, bis-β-diketonate units (O, O) and dipicolinate-like sites (O, N, O). The intramolecular Er⋯Nd separations are 3.944 and 3.947 Å, while the Er⋯Er distance within the complex is 7.881 Å. Interestingly, a shorter intermolecular Er⋯Er distance (6.059 Å) was observed between the symmetry equivalent complexes, associated by complementary H-bonding interactions via their water ligands (Fig. S7†). In addition to this intermolecular interaction, each complex interacts with a second one through a π⋯π interaction involving the central pyridine groups of two LB2− ligands (Fig. S8†). Each molecule is related to a third complex through another set of π⋯π contacts involving a total of seven short distances between sp2 C-atoms from LA2− ligands (Fig. S9†).
. The asymmetric unit is made of one formula unit of 3 and 10 molecules of pyridine. The main [ErNdEr] complex (Fig. 2 and S6†) features a quasi-linear trimetallic Er⋯Nd⋯Er array (angle of 174.17°) with the metals bridged together by two μ3-LA2− and two μ-LB2− ligands that chelate them with two types of pockets, bis-β-diketonate units (O, O) and dipicolinate-like sites (O, N, O). The intramolecular Er⋯Nd separations are 3.944 and 3.947 Å, while the Er⋯Er distance within the complex is 7.881 Å. Interestingly, a shorter intermolecular Er⋯Er distance (6.059 Å) was observed between the symmetry equivalent complexes, associated by complementary H-bonding interactions via their water ligands (Fig. S7†). In addition to this intermolecular interaction, each complex interacts with a second one through a π⋯π interaction involving the central pyridine groups of two LB2− ligands (Fig. S8†). Each molecule is related to a third complex through another set of π⋯π contacts involving a total of seven short distances between sp2 C-atoms from LA2− ligands (Fig. S9†).
 | ||
| Fig. 2 Representation of the cation [Er2Nd(LA)2(LB)2(H2O)2(py)]+ of 3 (also representing those of 1 and 2). The carbon atoms of ligands LA2− and LB2− are darker and lighter gray, respectively. Er is orange, Nd is green, O is red, N is purple and C is gray. H atoms are not shown. The inset shows the coordination polyhedrons of Nd3+ and Er3+, using the same color codes (both Er3+centers have very similar polyhedrons, despite not being crystallographically equivalent, thus, only one is shown). | ||
The coordination environment of each Er3+ ion is made up of one (O, N, O) and two (O, O) coordination pockets in addition to one molecule of H2O, thus featuring a coordination number (CN) of 8. The Nd3+ centers exhibit two (O, O) and two (O, N, O) chelates and one pyridine ligand, yielding CN 11. The program SHAPE66 was used to determine the ideal polyhedron that represents best the coordination geometry around each metal (Table S4†). For Er3+ it is a biaugmented trigonal prism with normalized distances (in a 0 to 100 scale) of 1.639 (Er1) and 1.487 (Er2). Nd is best represented by the figure of a capped pentagonal antiprism, with a calculated distance from it of 6.267. The bond distances to the metals were compared using the Ln–O average distances at each center. These average values are 2.32/2.30 and 2.60 Å for Er1/Er2 and Nd, respectively. Thus, the bond distances to the central metal are about 15% longer than to the peripheral metals.
Photophysical properties
To assist the investigation of the intermolecular Nd-to-Er energy transfer within the [ErNdEr] (3) molecule, the analogous [LuNdLu] (1) and [ErLaEr] (2) complexes were also studied under the same conditions. The experiments were performed in dilute solutions (10−4 M) of MeOH-d4 and DMSO-d6 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to avoid intermolecular transfers both at room temperature and at 77 K. Studies in the solid state were also performed to ascertain that the complexes in solution coincide with those described by SCXRD (see below). The main photophysical data extracted from this study are compiled in Table 1.
-d4:DMSO-d6 (1:1) at room temperature and 77 K (between brackets)
:1) to avoid intermolecular transfers both at room temperature and at 77 K. Studies in the solid state were also performed to ascertain that the complexes in solution coincide with those described by SCXRD (see below). The main photophysical data extracted from this study are compiled in Table 1.
-d4:DMSO-d6 (1:1) at room temperature and 77 K (between brackets)
In previous studies, we established the energy of the triplet excited state of ligands H2LA and H2LB to be about 18950 cm−1 and 19050 cm−1, respectively.51 Both energies are sufficiently high to sensitize the 4F3/2 state of Nd3+ centered at, approximately, 11260 cm−1 and the 4I11/2 or the 4I13/2 states of Er3+ at 10150 cm−1 and ∼6500 cm−1, respectively (see below). Therefore, emissions at 1058 nm and 1530 nm can be expected from compounds 1 and 2, respectively. These complexes were thus first investigated to confirm the mentioned antenna effect and to study their luminescence properties.
The photophysics of complex 1 was previously studied in non-deuterated solutions.51 We thus now repeated the measurements under deuterated conditions to compare its properties with those of complex 3 (Fig. 3 and S10†). As expected, upon excitation in the ligand transition, 1 presents the characteristic Nd3+ transitions at ∼880 nm, ∼1058 nm and ∼1530 nm assigned to 4F3/2 → 4IJ (J = 9/2, 11/2, 13/2), respectively. An increase of the excited state lifetime at room temperature was observed (3.99 μs vs. 1.8 μs) due to a decrease of the non-radiative relaxation, especially C–H and O–H vibrational quenching, by the use of deuterated solvents.67,68 The excitation spectrum (λem = 1056 nm) at 77 K shows two types of sensitization of the excited states of Nd3+, the mentioned antenna effect by energy transfer from the triplet states of the ligands (∼375 nm) in addition to that caused by direct excitations as the f–f transitions (properly assigned in Fig. 3).
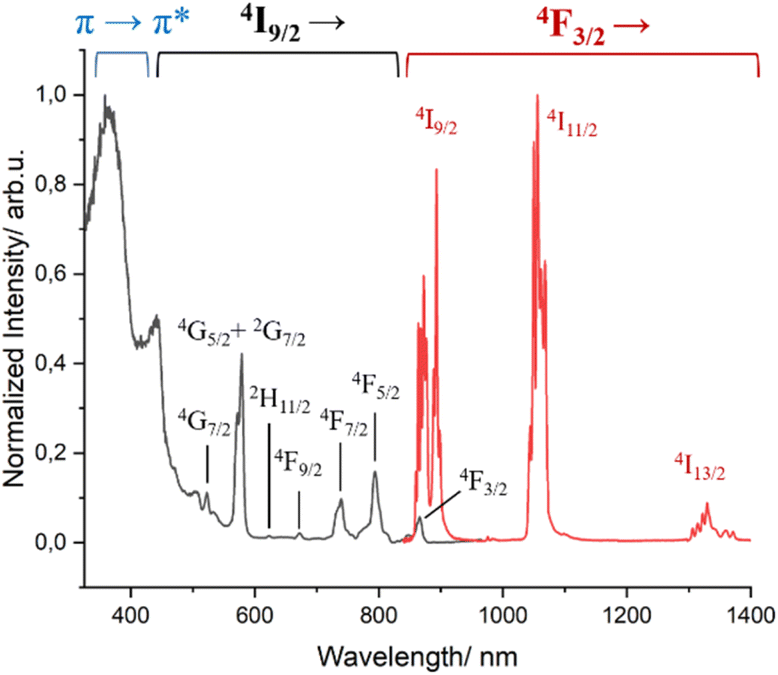 | ||
| Fig. 3 Excitation (λem = 1058 nm, black trace) and emission (λexc = 400 nm, red trace) spectra of complex 1 in MeOH-d4:DMSO-d6 (1:1) at 77 K. | ||
Complex 2 presents the characteristic broad emission band of the 4I13/2 → 4I15/2 transition of Er3+ centered at 1530 nm (Fig. S10†), often solely observed in coordination complexes.69,70 When this spectrum was measured at 77 K, the intricate fine splitting of the 4I15/2 state could be observed (Fig. S11†) in addition to the disappearance of the “hot bands” at higher energy of the emissive 4I13/2 state.71 The same fine splitting was observed in frozen solution, indicating that the first coordination environment of Er3+ observed in the solid state is kept in solution (Fig. S10 and S11†). The excited-state lifetime decay measured at 1530 nm was fitted with a mono exponential function both at room temperature and 77 K, giving values of 2.85 μs and 3.50 μs, respectively. As seen, lowering the temperature has only a subtle positive effect on the lifetime value by slightly minimizing the non-radiative deactivation. These values are in the typical range of other β-diketonate-based Er3+ complexes.69,72,73 The respective excitation spectra in solution at low temperature show, as for complex 1, the antenna effect and the effect of the direct excitation of the 4I15/2 state of Er3+, as depicted in Fig. 4.
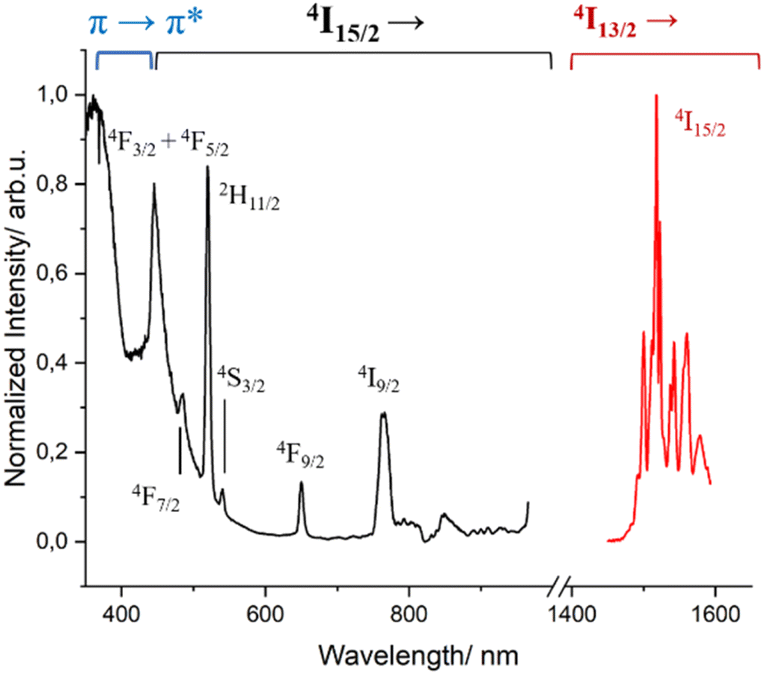 | ||
| Fig. 4 Excitation (λem = 1530 nm, black trace) and emission (λexc = 400 nm, red trace) spectra of complex 2 in MeOH-d4:DMSO-d6 (1:1) at 77 K. | ||
Complex 3 [ErNdEr] was then analyzed to investigate the possible photophysical intercommunication between the two different centers present within the same molecule. The emission spectra upon excitation of the singlet state of the ligands (λexc = 400 nm), both in solution and the solid state, show luminescence coming from both the 4F3/2 state of Nd3+ and the 4I13/2 one of Er3+ (Fig. 5 and S12†). However, this does not allow confirming the ET between both centers. It only demonstrates an efficient antenna effect occurring when the ligands of the mixed-metal complex are brought to their excited states.
 | ||
| Fig. 5 Emission (λexc = 400 nm, red trace) spectrum of complex 3 in MeOH-d4:DMSO-d6 (1:1) at 77 K. | ||
To investigate a possible direct ET between both lanthanides, dilute solutions of complexes 1, 2 and 3 with identical optical density (OD = 0.4 at 400 nm, Fig. S13†) were compared at room temperature, which allowed evaluating changes in intensity of the emission bands. As illustrated in Fig. 6, the emission intensity of Nd3+ decreases significantly (∼80%) in going from complex 1 to 3. This pronounced decline cannot be solely attributed to the increase in the number of lanthanide acceptors (one Nd3+ in complex 1vs. two Er3+ and one Nd3+ in complex 3). The data therefore suggest that another deactivation pathway of Nd3+ may be occurring, which we anticipate could be a direct ET to Er3+. This, however, is not substantiated by a concomitant increase of the Er3+ emission in complex 3. In contrast, a clear decrease of this emission is observed in comparison to complex 2. The dilution effect is again evident when changing from complex 2 (two Er3+) to 3 (two Er3+ and one Nd3+), resulting in an emission decrease of 1/3. However, the reduction of the Er3+ emission (∼50%) is higher than expected, suggesting that a possible back ET to Nd3+ could be also occurring. The same effect was measured at 77 K. However, the decrease of the Nd3+ and Er3+ centered bands of complex 3 was less evident at low temperature (Fig. S14†). Therefore, further verification was required.
 | ||
| Fig. 6 Emission spectra (λexc = 400 nm) of iso-absorbant solutions of complexes (1) (black trace), (2) (blue trace), and (3) (red trace) in MeOD-d4:DMSO-d6 (1:1) solution at room temperature (a) with the corresponding Er3+ emission zoom (b). | ||
Since the hypothesis of a back ET is unprecedented at the molecular level, a quantitative analysis was carried out. Thus, the lifetimes of complex 3 were determined in solution at 1058 nm and 1530 nm (Fig. S15†). The excited state decay at 1058 nm (4F3/2 state of Nd3+) was satisfactorily fitted to a monoexponential function with a lifetime of 1.00 μs at room temperature. A clear shortening of the lifetime values is observed in comparison with those obtained for the [LuNdLu] (1) complex (3.99 μs). Indeed, by using the following equation:
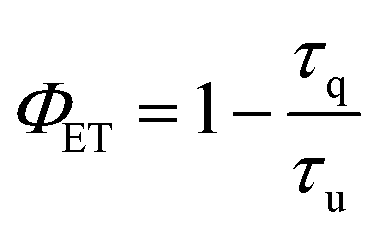 | (2) |
the reduction of the lifetime can be translated directly into the efficiency of the energy transfer, amounting to 75% at room temperature. This energy transfer (ET1) is expected to occur from the 4F3/2 state of Nd3+ to the approximately isoenergetic state/s of Er3+, i.e.4I11/2 and/or 4I9/2 (Fig. 7).74 A similar lifetime variation is observed at 77 K (Table 1) but the effect is less pronounced, leading to a ET1 efficiency of 51%. The fact that the ET1 was found to be lower at 77 K may reflect the fact that at low temperature the transfer 4F3/2 (Nd3+) → 4I9/2 (Er3+) is not efficient, since the latter (4I9/2 of Er3+) lies slightly higher in energy (13089 cm−1) than the 4F3/2 state of Nd3+ (11338 cm−1), as determined from the excitation spectra of “pure” complexes 1 and 2, respectively (see above). The possibility of the reverse effect, i.e. the energy transfer from Er3+ to Nd3+, was also investigated by lifetime determination. This was conducted by assessing the excited state decay at 1530 nm (that occurs from the state 4I13/2 of Er3+) in complex 3. The mono-exponential fitting of the decay gave lifetime values of 1.90 μs at room temperature, compared to 2.85 μs for 2 in the absence of Nd3+, suggesting an Er-to-Nd energy transfer. In this case, this remarkable transfer (ET2, Fig. 7) occurring from the 4I13/2 state of Er3+ may be taking place to the 4I15/2 or a lower lying state of Nd3+.74,75 In contrast to the first type of transfer (ET1), ET2 does not result in emission from Nd3+ but into non-radiative decay processes as the ultimate outcome of the partial deactivation of the Er3+ emission. Furthermore, this transfer was also unveiled at 77 K, where a lifetime of 2.3 μs was determined following a mono-exponential fitting, compared to 3.5 μs for the pure Er3+ complex.
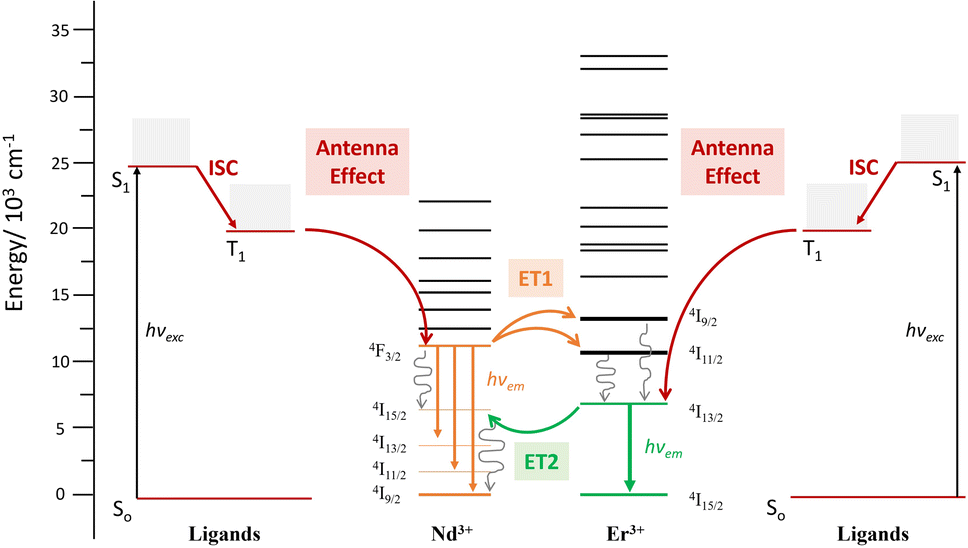 | ||
| Fig. 7 Energy diagram levels and terms involved in the ET phenomena occurring within complex 3 [ErNdEr]. | ||
Excitation spectra allowed further characterization of this rare intramolecular double ET phenomenon. These were obtained by generating f–f emission transitions within complex 3 in frozen solution. When fixing the detected emission wavelength at 1056 (Nd3+ emission), the main excitations lines could be assigned, as expected, to transitions from the 4I9/2 state of Nd3+ as for complex 1 (Fig. 8). On the other hand, no excitations of Er3+ were detected to generate any emission from Nd3+. This observation is not in contradiction with ET2 (Er-to-Nd) but rather corroborates it, as this type of transfer only results in non-radiative deactivation of Er3+ instead of any radiative emission (Fig. 8). On the other hand, when the emission was fixed at 1550 nm (Er3+ emission), two sets of lines could be identified; those from transitions arising from the 4I15/2 state of Er3+ (also seen in complex 2) but also, some from exciting the 4I9/2 state of Nd3+ (Fig. 8). This further confirms the direct ET1 (Nd-to-Er) discussed above.
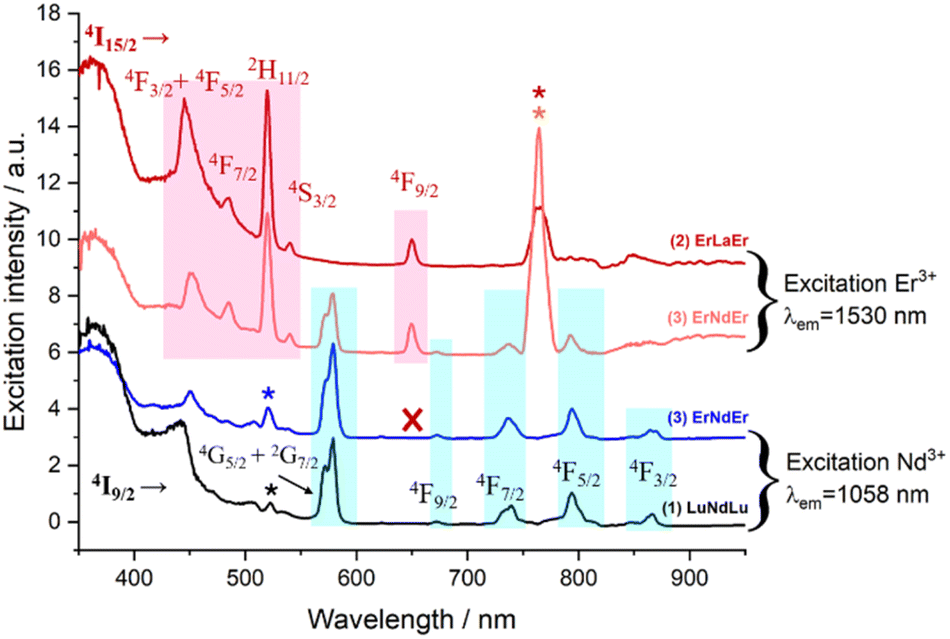 | ||
| Fig. 8 Excitation spectra of complexes (1) at λem = 1058 nm (black trace), (2) at λem = 1530 nm (red trace) and (3) at λem = 1058 nm (blue trace) and at λem = 1530 nm (orange trace) in MeOH-d4/DMSO-d6 (1:1) at 77 K. | ||
Conclusions
The pure heterometallic [ErNdEr] complex synthesized here allows unveiling the first instance of intramolecular ET between the Nd and Er centers being studied at the molecular level. The ideal distance between both ions (∼4 Å) and the specific molecular scaffold have been identified as key parameters in the analysis of this transfer. Indeed, the photophysical analysis reveals a rare intramolecular double ET phenomenon. A comparison of the excited state lifetime values in the presence and absence of the lanthanide quencher (Er3+ in the case of the Nd-to-Er or Nd3+ in the case of Er-to-Nd ET) indicated the presence of two distinct types of transfer. The first one (ET1) occurs from the 4F3/2 state of Nd3+ to the approximately isoenergetic states of Er3+, i.e.4I11/2 and/or 4I9/2, which results in the emission of the Er3+ 4I11/2 state at 1530 nm. In contrast, ET2 occurs from the 4I13/2 state of Er3+ to the 4I15/2 or lower-lying states of Nd3+ and does not result in Nd3+ emission but in non-radiative decay processes, as the ultimate outcome of the partial deactivation of the Er3+ emission. This unprecedented observation was first qualitatively detected by comparing the emission of iso-absorbant solutions and quantitatively confirmed by studying the excited state decays as well as by a critical excitation spectra analysis. The availability of a rich collection of pure heterometallic [LnLn′Ln] complexes offers a great opportunity to discover new intermetallic ET phenomena at the molecular level and their very precise analysis.Data availability
The data supporting this article, including crystallographic details, information on synthetic procedures, ESI-MS, absorption, emission, lifetime measurements and fits and NMR experiments, magnetic data treatment, and DFT calculations, have been included as part of the ESI.†Author contributions
DM, coordination chemistry synthesis and collection of photophysical data; AS, collection of photophysical data and interpretation; LAB, conceptualization, ligand synthesis and coordination chemistry synthesis; DA, conceptualization and coordination chemistry synthesis; OR, crystal structure determination and magnetic properties; YG, collection of photophysical data and interpretation; FR, conceptualization and photophysical data interpretation; OM, conceptualization and photophysical data interpretation; LAG, conceptualization, photophysical data interpretation and paper writing; GA, research coordination, conceptualization and paper writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
GA thanks the NextGenerationEU/PRTR-C17.I1 (from “Plan Complementario en Comunicación Cuántica” funded by Generalitat de Catalunya and the European Union) and Generalitat de Catalunya for the ICREA Academia 2023 prize. GA, DM, LAB, DA and OR thank the Spanish Ministry of Innovation for grants PID2020-118329RB-I00, PID2022-137764OB-I00, PDC2022-133184-I00 and TED2021-129214B-I00 (Spanish MICIN/AEI/10.13039/501100011033 and ERDF “A way of making Europe”). DM thanks the Generalitat de Catalunya for an FI-SDUR Grant (2020-FISDU-00492).Notes and references
- T. Xian, Q. Meng, F. Gao, M. Hu and X. Wang, Coord. Chem. Rev., 2023, 474, 214866 CrossRef CAS.
- J.-C. G. Bünzli, Chem. Rev., 2010, 110, 2729–2755 CrossRef PubMed.
- M. C. Heffern, L. M. Matosziuk and T. J. Meade, Chem. Rev., 2014, 114, 4496–4539 CrossRef CAS PubMed.
- J.-C. G. Bünzli and S. V. Eliseeva, J. Rare Earths, 2010, 28, 824–842 CrossRef.
- G. Tessitore, G. A. Mandl, S. L. Maurizio, M. Kaur and J. A. Capobianco, RSC Adv., 2023, 13, 17787–17811 RSC.
- J. H. S. K. Monteiro, Molecules, 2020, 25, 2089 CrossRef CAS PubMed.
- S. V. Eliseeva and J.-C. G. Bünzli, Chem. Soc. Rev., 2010, 39, 189–227 RSC.
- J.-C. G. Bünzli and A.-S. Chauvin, in Handbook on the Physics and Chemistry of Rare Earths, ed. J.-C. G. Bünzli and V. K. Pecharsky, Elsevier, 2014, vol. 44, pp. 169–281 Search PubMed.
- B. S. Richards, D. Hudry, D. Busko, A. Turshatov and I. A. Howard, Chem. Rev., 2021, 121, 9165–9195 CrossRef CAS PubMed.
- B. M. Van Der Ende, L. Aarts and A. Meijerink, Phys. Chem. Chem. Phys., 2009, 11, 11081 RSC.
- L. E. Mackenzie and R. Pal, Nat. Rev. Chem, 2020, 5, 109–124 CrossRef PubMed.
- J.-C. G. Bünzli and S. V. Eliseeva, Basics of Lanthanide Photophysics, Springer, Berlin Heidelberg, 2010, pp. 1–45, DOI:10.1007/4243_2010_3.
- A. D’Aléo, F. Pointillart, L. Ouahab, C. Andraud and O. Maury, Coord. Chem. Rev., 2012, 256, 1604–1620 CrossRef.
- S. J. A. Pope, B. J. Coe, S. Faulkner, E. V. Bichenkova, X. Yu and K. T. Douglas, J. Am. Chem. Soc., 2004, 126, 9490–9491 CrossRef CAS PubMed.
- M. D. Ward, Coord. Chem. Rev., 2010, 254, 2634–2642 CrossRef CAS.
- L.-J. Xu, G.-T. Xu and Z.-N. Chen, Coord. Chem. Rev., 2014, 273–274, 47–62 CrossRef CAS.
- A. Nadort, J. Zhao and E. M. Goldys, Nanoscale, 2016, 8, 13099–13130 RSC.
- S. Zhao, D. Yu, B. Li, S. Kanwal, T. Shen, J. Wu, S. Zhuang and D. Zhang, Adv. Opt. Mater., 2024, 12, 2302408 CrossRef CAS.
- M. Safdar, A. Ghazy, M. Lastusaari and M. Karppinen, J. Mater. Chem. C, 2020, 8, 6946–6965 RSC.
- P. S. David, P. Panigrahi, S. Raman and G. S. Nagarajan, Mater. Sci. Semicond. Process., 2021, 122, 105486 CrossRef CAS.
- C. Duan, L. Liang, L. Li, R. Zhang and Z. P. Xu, J. Mater. Chem. B, 2018, 6, 192–209 RSC.
- H. Zhao, Y. Li, X. Zhang, K. Wu, J. Lv, C. Chen, H. Liu, Z. Shi, H. Ju and Y. Liu, Biomaterials, 2022, 291, 121873 CrossRef CAS PubMed.
- E. M. Mettenbrink, W. Yang and S. Wilhelm, Adv. Photonics Res., 2022, 3, 2200098 CrossRef CAS PubMed.
- K. Malhotra, D. Hrovat, B. Kumar, G. Qu, J. V. Houten, R. Ahmed, P. A. E. Piunno, P. T. Gunning and U. J. Krull, ACS Appl. Mater. Interfaces, 2023, 15, 2499–2528 CrossRef CAS PubMed.
- L. J. Charbonnière, A. M. Nonat, R. C. Knighton and L. Godec, Chem. Sci., 2024, 15, 3048–3059 RSC.
- R. C. Knighton, L. K. Soro, L. Francés-Soriano, A. Rodríguez-Rodríguez, G. Pilet, M. Lenertz, C. Platas-Iglesias, N. Hildebrandt and L. J. Charbonnière, Angew. Chem., Int. Ed., 2022, 61, e202113114 CrossRef CAS PubMed.
- A. Nonat, S. Bahamyirou, A. Lecointre, F. Przybilla, Y. Mély, C. Platas-Iglesias, F. Camerel, O. Jeannin and L. J. Charbonnière, J. Am. Chem. Soc., 2019, 141, 1568–1576 CrossRef CAS PubMed.
- I. Hernández, N. Pathumakanthar, P. B. Wyatt and W. P. Gillin, Adv. Mater., 2010, 22, 5356–5360 CrossRef PubMed.
- T. V. Balashova, A. P. Pushkarev, A. N. Yablonskiy, B. A. Andreev, I. D. Grishin, R. V. Rumyantcev, G. K. Fukin and M. N. Bochkarev, J. Lumin., 2017, 192, 208–211 CrossRef CAS.
- J. Wang, Y. Jiang, J.-Y. Liu, H.-B. Xu, Y.-X. Zhang, X. Peng, M. Kurmoo, S. W. Ng and M.-H. Zeng, Angew. Chem., Int. Ed., 2021, 60, 22368–22375 CrossRef CAS PubMed.
- D. A. Gálico, J. S. Ovens, F. A. Sigoli and M. Murugesu, ACS Nano, 2021, 15, 5580–5585 CrossRef PubMed.
- S. Goderski, M. Runowski, P. Woźny, V. Lavín and S. Lis, ACS Appl. Mater. Interfaces, 2020, 12, 40475–40485 CrossRef CAS PubMed.
- A. Gupta, S. Ghosh, M. K. Thakur, J. Zhou, K. Ostrikov, D. Jin and S. Chattopadhyay, Prog. Mater. Sci., 2021, 121, 100838 CrossRef CAS.
- X. Qin, J. Xu, Y. Wu and X. Liu, ACS Cent. Sci., 2019, 5, 29–42 CrossRef CAS PubMed.
- S. Lis, M. Elbanowski, B. Mąkowska and Z. Hnatejko, J. Photochem. Photobiol., A, 2002, 150, 233–247 CrossRef CAS.
- D. J. Lewis, P. B. Glover, M. C. Solomons and Z. Pikramenou, J. Am. Chem. Soc., 2011, 133, 1033–1043 CrossRef CAS PubMed.
- C. Egger, L. Guénée, N. Deorukhkar and C. Piguet, Dalton Trans., 2024, 53, 6050–6062 RSC.
- F. Artizzu, F. Quochi, L. Marchiò, R. F. Correia, M. Saba, A. Serpe, A. Mura, M. L. Mercuri, G. Bongiovanni and P. Deplano, Chem.–Eur. J., 2015, 21, 3833 CrossRef.
- N. André, T. B. Jensen, R. Scopelliti, D. Imbert, M. Elhabiri, G. Hopfgartner, C. Piguet and J.-C. G. Bünzli, Inorg. Chem., 2004, 43, 515–529 CrossRef PubMed.
- S. Floquet, M. Borkovec, G. Bernardinelli, A. Pinto, L.-A. Leuthold, G. Hopfgartner, D. Imbert, J.-C. G. Bünzli and C. Piguet, Chem.–Eur. J., 2004, 10, 1091–1105 CrossRef CAS PubMed.
- F. Artizzu, F. Quochi, L. Marchiò, M. Saba, A. Serpe, A. Mura, M. L. Mercuri, G. Bongiovanni and P. Deplano, MRS Adv., 2016, 1, 2683–2688 CrossRef CAS.
- M. P. Placidi, A. J. L. Villaraza, L. S. Natrajan, D. Sykes, A. M. Kenwright and S. Faulkner, J. Am. Chem. Soc., 2009, 131, 9916–9917 CrossRef CAS PubMed.
- T. J. Sørensen, M. Tropiano, O. A. Blackburn, J. A. Tilney, A. M. Kenwright and S. Faulkner, Chem. Commun., 2013, 49, 783–785 RSC.
- J. J. Le Roy, J. Cremers, I. A. Thomlinson, M. Slota, W. K. Myers, P. H. Horton, S. J. Coles, H. L. Anderson and L. Bogani, Chem. Sci., 2018, 9, 8474–8481 RSC.
- C. D. Buch, S. H. Hansen, D. Mitcov, C. M. Tram, G. S. Nichol, E. K. Brechin and S. Piligkos, Chem. Sci., 2021, 12, 6983–6991 RSC.
- D. Aguilà, L. A. Barrios, V. Velasco, O. Roubeau, A. Repollés, P. J. Alonso, J. Sesé, S. J. Teat, F. Luis and G. Aromí, J. Am. Chem. Soc., 2014, 136, 14215–14222 CrossRef PubMed.
- J. González-Fabra, N. A. G. Bandeira, V. Velasco, L. A. Barrios, D. Aguila, S. J. Teat, O. Roubeau, C. Bo and G. Aromí, Chem.–Eur. J., 2017, 23, 5117–5125 CrossRef PubMed.
- D. Aguilà, V. Velasco, L. A. Barrios, J. González-Fabra, C. Bo, S. J. Teat, O. Roubeau and G. Aromí, Inorg. Chem., 2018, 57, 8429–8439 CrossRef PubMed.
- V. Velasco, L. A. Barrios, M. Schütze, O. Roubeau, F. Luis, S. J. Teat, D. Aguilà and G. Aromí, Chem.–Eur. J., 2019, 25, 15228–15232 CrossRef CAS PubMed.
- L. Abad Galán, D. Aguilà, Y. Guyot, V. Velasco, O. Roubeau, S. J. Teat, M. Massi and G. Aromí, Chem.–Eur. J., 2021, 27, 7288–7299 CrossRef PubMed.
- D. Maniaki, A. Sickinger, L. A. Barrios, D. Aguilà, O. Roubeau, N. S. Settineri, Y. Guyot, F. Riobé, O. Maury, L. A. Galán and G. Aromí, Inorg. Chem., 2023, 62, 3106–3115 CrossRef CAS PubMed.
- H.-B. Xu, J.-G. Deng, L.-Y. Zhang and Z.-N. Chen, Cryst. Growth Des., 2013, 13, 849–857 CrossRef CAS.
- H. Yao, G. Calvez, C. Daiguebonne, K. Bernot, Y. Suffren, M. Puget, C. Lescop and O. Guillou, Inorg. Chem., 2017, 56, 14632–14642 CrossRef CAS PubMed.
- T. Chuasaard, P. Jittipiboonwat, A. Ngamjarurojana, B. Yotnoi and A. Rujiwatra, J. Solid State Chem., 2023, 324, 124075 CrossRef CAS.
- L. Abad Galán, A. N. Sobolev, B. W. Skelton, E. Zysman-Colman, M. I. Ogden and M. Massi, Dalton Trans., 2018, 47, 12345–12352 RSC.
- F. Artizzu, A. Serpe, L. Marchiò, M. Saba, A. Mura, M. L. Mercuri, G. Bongiovanni, P. Deplano and F. Quochi, J. Mater. Chem. C, 2015, 3, 11524–11530 RSC.
- M. Oggianu, V. Mameli, M. A. Hernández-Rodríguez, N. Monni, M. Souto, C. D. S. Brites, C. Cannas, F. Manna, F. Quochi, E. Cadoni, N. Masciocchi, A. N. Carneiro Neto, L. D. Carlos and M. L. Mercuri, Chem. Mater., 2024, 36, 3452–3463 CrossRef CAS PubMed.
- S. Faulkner and S. J. A. Pope, J. Am. Chem. Soc., 2003, 125, 10526–10527 CrossRef CAS PubMed.
- F. Artizzu, F. Quochi, L. Marchiò, E. Sessini, M. Saba, A. Serpe, A. Mura, M. L. Mercuri, G. Bongiovanni and P. Deplano, J. Phys. Chem. Lett., 2013, 4, 3062–3066 CrossRef CAS.
- F. Artizzu, F. Quochi, L. Marchiò, C. Figus, D. Loche, M. Atzori, V. Sarritzu, A. M. Kaczmarek, R. Van Deun, M. Saba, A. Serpe, A. Mura, M. L. Mercuri, G. Bongiovanni and P. Deplano, Chem. Mater., 2015, 27, 4082–4092 CrossRef CAS.
- L. Song, Q. Wang, D. Tang, X. Liu and Z. Zhen, New J. Chem., 2007, 31, 506–511 RSC.
- W. Q. Shi, M. Bass and M. Birnbaum, J. Opt. Soc. Am. B, 1989, 6, 23–29 CrossRef CAS.
- O. Barbosa-García, R. A. McFarlane, M. Birnbaum and L. A. Díaz-Torres, J. Opt. Soc. Am. B, 1997, 14, 2731–2734 CrossRef.
- E. Macaluso, M. Rubín, D. Aguilà, A. Chiesa, L. A. Barrios, J. I. Martínez, P. J. Alonso, O. Roubeau, F. Luis, G. Aromí and S. Carretta, Chem. Sci., 2020, 11, 10337–10343 RSC.
- R. S. Forgan, Chem. Sci., 2020, 11, 4546–4562 RSC.
- P. Alemany, D. Casanova, S. Alvarez, C. Dryzun and D. Avnir, Rev. Comput. Chem., 2017, 289–352, DOI:10.1002/9781119356059.ch7.
- A. d. Bettencourt-Dias, Luminescence of lanthanide ions in coordination compounds and nanomaterials, Wiley, Chichester, U.K., 2014 Search PubMed.
- R. H. C. Tan, M. Motevalli, I. Abrahams, P. B. Wyatt and W. P. Gillin, J. Phys. Chem. B, 2006, 110, 24476–24479 CrossRef CAS PubMed.
- L. A. Galán, B. L. Reid, S. Stagni, A. N. Sobolev, B. W. Skelton, M. Cocchi, J. M. Malicka, E. Zysman-Colman, E. G. Moore, M. I. Ogden and M. Massi, Inorg. Chem., 2017, 56, 8975–8985 CrossRef PubMed.
- P. Martín-Ramos, M. Ramos Silva, F. Lahoz, I. R. Martín, P. Chamorro-Posada, M. E. S. Eusebio, V. Lavín and J. Martín-Gil, J. Photochem. Photobiol., A, 2014, 292, 16–25 CrossRef.
- A. Sickinger, B. Baguenard, A. Bensalah-Ledoux, Y. Guyot, L. Guy, F. Pointillart, O. Cador, M. Grasser, B. Le Guennic, F. Riobé, O. Maury and S. Guy, J. Mater. Chem. C, 2024, 12, 4253–4260 RSC.
- P. Martín-Ramos, I. R. Martín, F. Lahoz, S. Hernández-Navarro, P. S. Pereira da Silva, I. Hernández, V. Lavín and M. Ramos Silva, J. Alloys Compd., 2015, 619, 553–559 CrossRef.
- Z. Ahmed, R. E. Aderne, J. Kai, J. A. L. C. Resende, H. I. Padilla-Chavarría and M. Cremona, RSC Adv., 2017, 7, 18239–18251 RSC.
- Y. Liu, Y. Sun, Y. Wang, Z. You, Z. Zhu, J. Li and C. Tu, J. Lumin., 2018, 198, 40–45 CrossRef CAS.
- Y. Wang, J. Li, Z. Zhu, Z. You, J. Xu and C. Tu, Opt. Express, 2015, 23, 18554 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, magnetism, mass spectrometry, crystallographic details and photophysical properties. CCDC 2351760. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc03994g |
| This journal is © The Royal Society of Chemistry 2024 |