
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of low-molecular weight and branched polyethylenes via ethylene polymerization using 9-(arylimino)-5,6,7,8-tetrahydrocyclohepta-pyridylnickel precatalysts†
Zhao
Ning
ab,
Yanping
Ma
 *a,
Yanning
Zeng
*b,
Yizhou
Wang
a,
Aoqian
Xi
b and
Wen-Hua
Sun
*a
*a,
Yanning
Zeng
*b,
Yizhou
Wang
a,
Aoqian
Xi
b and
Wen-Hua
Sun
*a
aKey Laboratory of Engineering Plastics and Beijing National Laboratory for Molecular Science, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: myanping@iccas.ac.cn; whsun@iccas.ac.cn; Tel: +86-1062557955
bCollege of Materials Science and Engineering, Guilin University of Technology, Guilin 541004, China. E-mail: ynzeng@ glut.edu.cn
First published on 3rd September 2024
Abstract
Targeting pour point depressants of low-molecular weight and branched polyethylenes, a series of 9-[2,4-bis(benzhydryl)-6-R-phenylimino]-5,6,7,8-tetrahydro-cycloheptapyridine-nickel complexes (Ni1–Ni10) were developed as efficient precatalysts. Upon activation with either EASC or MAO, all nickel complex precatalysts exhibited high activity [up to 8.12 × 106 g PE (mol of Ni)−1 h−1] with single-site behavior toward ethylene polymerization, producing low-molecular weight and unimodal polyethylenes. The resultant polyethylenes possessed high branching with predominant methyl groups and longer chains, along with either internal vinylene or vinyl end groups. The activities of these complex precatalysts were heavily rationalized on the basis of the electronic and steric influences of their 6-R-substituents, with bromides following the order of Ni5 (F) > Ni4 (Cl) > Ni1 (Me) > Ni2 (Et) > Ni3 (iPr) and chlorides following the order of Ni10 (F) > Ni9 (Cl) > Ni6 (Me) > Ni7 (Et) > Ni8 (iPr). DFT calculations revealed the crucial role of agostic interactions (–Ni⋯H–C(Ph2)) between the nickel metal and the hydrogen atom of the ortho bulky group in achieving high catalytic activity and intramolecular hydrogen bonding with the fluoride atom in producing low Mw PE wax. Moreover, the organic compounds and nickel complexes were well characterized, including representative complexes Ni3 and Ni4, via single-crystal X-ray diffraction.
Introduction
The discovery of late-transition metal precatalysts for ethylene polymerization was initiated with the Brookhart α-diimino metal (palladium and nickel) complexes1 and evolved into Brookhart-Gibson bis(imino)pyridyl metal (iron and cobalt) complexes.2 Subsequently, derivatives of both α-diimino-3 and bis(imino)pyridyl-metal complexes4 were widely developed for exploring highly active and practical systems to polymerize ethylene and produce polyethylenes with either highly branched3 or highly linear features,4 which have considerable importance in academic and industrial settings. Branched polyethylene materials are particularly attractive owing to their intriguing chain-walking mechanism5 and new cost-effective processes for producing low-density polyolefins without expensive comonomers of α-olefins.6Beyond the α-diimino nickel model (A, Chart 1),1,3,6 2-iminopyridylnickel precatalysts (B, Chart 1) exhibited high activities and produced polyethylenes7 with more branches but relatively lower molecular weights. Extensively, using various pyridine-fused carbocyclic ketones to form rigid iminopyridine derivatives,8–12 their corresponding nickel complexes (C–F, Chart 1) have not only provided efficient catalytic systems but also tailored the resultant polyethylenes with significant differences in branches and molecular weights ranging from oligomers to waxes. For example, 8-arylamine-5,6,7-dihydroquinolyl nickel(II) complexes (D, Chart 1) exhibited high activity in ethylene polymerization, achieving narrow dispersive and low molecular-weight polyethylenes (Mw = 3.3–9.2 kg mol−1);9 however, their analogues with 2-substituted ligated nickel complexes only showed activity for ethylene oligomerization.10 With a further increase in their ring size, E complexes11 achieved an activity of up to 7.80 × 106 (g PE) (mol of Ni)−1 h−1; simultaneously, resultant polyethylenes showed low molecular weights in the range of thousands together with narrower distribution (PDI: 1.92–2.16). Further increasing the ring size of fused cycles, F complexes12 showed slightly lower activity, with the highest activity of 3.44 × 106 (g PE) (mol of Ni)−1 h−1 to produce branched polyethylenes with a lower molecular weight of 1.4 kg mol−1.
 | ||
| Chart 1 Representative models of N,N-bidentate nickel precatalysts. | ||
Meanwhile, bulky substituents have been introduced into ligand compounds, thus significantly enhancing the catalytic performances of their nickel precatalysts and specifically improving their catalytic activity and thermal stability.3a–c,6d–l Demanding polyethylene with low molecular weights, the E model11 was worthily revisited using various substituents to finely tune the electronic and steric influences around the nickel core and achieve higher activities together with controlling the microstructures of resultant polyethylenes. Therefore, herein, a series of anilines with 2,4-bis(benzhydryl)- and various 6-substituents was synthesized as ligand compounds, 9-[2,4-bis(benzhydryl)-6-R-phenylimino]-5,6,7,8-tetrahydrocycloheptapyridine, and their nickel complexes (G, Chart 1) were newly explored in polymerizing ethylene. It was demonstrated that these compounds are not only good catalytic systems but there was also a nice correlation between the activities and the 6-R-substituents regarding their electronic and steric influence. Moreover, DFT calculation methods were used to elucidate their influence on the properties of the catalysts.
Results and discussion
Synthesis and characterization
A series of 9-[2,4-bis(benzhydryl)-6-R-phenylimino]-5,6,7,8-tetrahydrocyclo-heptapyridines was obtained in two steps using 5,6,7,8-tetrahydrocycloheptapyridine-9-one. Firstly, a template reaction of this ketone with the corresponding aniline and zinc(II) chloride was conducted to obtain an intermediate zinc complex. Subsequently, zinc metal chloride was removed to obtain L1–L5 in high yields in the presence of a saturated aqueous solution of potassium carbonate. This demetalation approach was used to improve the yield in the condensation reactions.6l,9i All the new ligands were characterized by FT-IR, 1H NMR, 13C NMR spectroscopy and elemental analysis.Next, the metal complexes (Ni1–Ni5) and (Ni6–Ni10) were obtained in good to high yields (72.4–92.3%) by the reaction of 1 equiv. of L1–L5 with either (DME)NiBr2 or NiCl2·6H2O, respectively (Scheme 1). All ten complexes were characterized by FT-IR spectroscopy and elemental analysis. In the FT-IR spectra of L1–L5, the νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N absorption bands were visible between 1635 and 1642 cm−1. However, that of Ni1–Ni10 appeared in the range of 1599–1605 cm−1, respectively. This shift to a lower wavenumber for the complex indicates the effective coordination between the imine nitrogen atoms of the N,N-ligand and the nickel metal center. Similar observations regarding pyridylimine-nickel complexes have been reported.7–12
N absorption bands were visible between 1635 and 1642 cm−1. However, that of Ni1–Ni10 appeared in the range of 1599–1605 cm−1, respectively. This shift to a lower wavenumber for the complex indicates the effective coordination between the imine nitrogen atoms of the N,N-ligand and the nickel metal center. Similar observations regarding pyridylimine-nickel complexes have been reported.7–12
 | ||
| Scheme 1 Synthesis route for L1–L5 and corresponding nickel halide complexes Ni1–Ni10. | ||
In addition, the molecular structures of both Ni3 and Ni4 suitable for X-ray diffraction analysis are shown in Fig. 1 and 2, respectively, with selected bond lengths and angles listed in Table 1. The complexes of Ni3 and Ni4 were chloro-bridged dimers with a distorted square-pyramidal geometry around the nickel cation; this binuclear nickel model has been frequently reported in bidentate nickel halide complexes.7b–e,9a–c,h–i,11a
 | ||
| Fig. 1 ORTEP diagram of the molecular structure of complex Ni3 with the thermal ellipsoids at the 30% probability level. All hydrogen atoms are omitted for clarity. The atoms labeled with i have been generated by symmetry. | ||
 | ||
| Fig. 2 ORTEP diagram of the molecular structure of complex Ni4 with the thermal ellipsoids at the 30% probability level. All hydrogen atoms are omitted for clarity. The atoms labeled with i have been generated by symmetry. | ||
| Ni3 | Ni4 | |
|---|---|---|
| Bond lengths (Å) | ||
| Ni(1)–N(1) | 2.052(3) | 2.043(2) |
| Ni(1)–N(2) | 2.043(3) | 2.072(2) |
| Br(1)–Ni(1) | 2.4144(7) | 2.4626(5) |
| Br(2)–Ni(1) | 2.4752(7) | 2.4084(5) |
| N(2)–C(11) | 1.445(5) | 1.436(3) |
| Bond angles (°) | ||
| N(1)–Ni(1)–N(2) | 78.51(13) | 78.66(9) |
| N(1)–Ni(1)–Br(1) | 90.27(10) | 161.71(7) |
| N(1)–Ni(1)–Br(2) | 94.40(10) | 94.73(7) |
| N(2)–Ni(1)–Br(1) | 101.19(9) | 94.78(6) |
| N(2)–Ni(1)–Br(2) | 146.45(9) | 98.63(6) |
| Br(1)–Ni(1)–Br(2) | 111.69(3) | 103.169(19) |
| C(1)–N(1)–Ni(1) | 123.7(3) | 124.3(2) |
| C(5)–N(1)–Ni(1) | 114.0(2) | 114.35(17) |
| C(6)–N(2)–Ni(1) | 117.0(3) | 115.81(17) |
| C(11)–N(2)–Ni(1) | 122.7(2) | 124.12(16) |
In the case of complex Ni3, its nickel center is symmetrically bridged by two bromine atoms (Br2 and Br2i) and is further coordinated by two nitrogen atoms (N1 and N2) from the corresponding ligand. The distorted square-pyramidal geometry (Fig. 1) features a bromine atom (Br1) at the apex, with the basal plane containing two nitrogen atoms (N1 and N2) and two bromine atoms (Br2 and Br2i). The nickel atom (Ni1) is located inside the pyramidal geometry. The bond length of Ni1–N1 at 2.052(3) Å is similar to that of Ni1–N2 at 2.043(3) Å; a similar observation was reported in bidentate nickel halide complexes.7b
Complex Ni4 (Fig. 2) was found to be a centrosymmetric binuclear structure, with the nickel symmetrically bridged by two bromine atoms (Br2 and Br2i). The bond length of Ni(1)–N(1)pyridine (2.043(2) Å) is shorter than the bond length of Ni(1)–N(2)imine (2.072(2) Å), indicating the more effective coordination of the pyridine nitrogen atom compared to the imine nitrogen atom, which is similar with the reported results in the literature.7c–e
Evaluation of ethylene polymerization
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
| Entry | Co-cat. | Al/Ni | Yield (g) | Act.b |
M
wc |
M
w/Mnc |
T
md (°C) |
|---|---|---|---|---|---|---|---|
| a Conditions: 2.0 μmol of Ni5, 100 mL of toluene, 10 atm of ethylene, 30 °C, and 30 min. b 106 g PE (mol of Ni)−1 h−1. c M w: kg mol−1, Mw and Mw/Mn determined via GPC. d Determined by DSC. | |||||||
| 1 | Et2AlCl | 400 | 2.27 | 2.27 | 1.28 | 1.83 | 78.2 |
| 2 | EtAlCl2 | 400 | 1.85 | 1.85 | 1.32 | 1.82 | 69.9 |
| 3 | EASC | 400 | 2.44 | 2.44 | 1.48 | 2.34 | 83.6 |
| 4 | MAO | 2000 | 3.25 | 3.25 | 1.35 | 2.26 | 86.4 |
| 5 | MMAO | 2000 | 1.14 | 1.14 | 1.17 | 2.56 | 80.6 |
The results revealed significant variations in yield, activity, molecular weight (Mw), molecular weight distribution (Mw/Mn), and melting temperature (Tm) among the co-catalysts. MAO demonstrated the highest activity (3.25) (entry 4, Table 2), with a high melting temperature (86.4 °C) and moderate molecular weight (1.35 kg mol−1) with a molecular weight distribution (Mw/Mn) of 2.26. EASC also showed high activity (2.44) (entry 3, Table 2) with the highest molecular weight (1.48 kg mol−1) and Tm of 83.6 °C, though it had a broader molecular weight distribution (2.34). Et2AlCl and EtAlCl2 exhibited a moderate performance, with yields of 2.27 g and 1.85 g and relatively lower molecular weights (1.28 and 1.32 kg mol−1) and melting temperatures (78.2 °C and 69.9 °C), respectively. MMAO had the lowest yield and activity (1.14) but produced polyethylene with the narrowest molecular weight distribution (2.56). These results suggest that MAO and EASC are the most effective co-catalysts for ethylene polymerization with Ni5 under the tested conditions, and thus selected for the subsequent optimization of the polymerization conditions.
:Ni molar ratio, the run temperature and the ethylene pressure. The screening results are presented in Table 3.
| Entry | Precat. | Al/Ni | T (°C) | t (min) | Yield (g) | Act.b |
M
wc |
M
w/Mnc |
T
md (°C) |
|---|---|---|---|---|---|---|---|---|---|
| a Conditions: 2.0 μmol of nickel precatalyst, 100 mL of toluene, and 10 atm of ethylene. b 106 g PE (mol of Ni)−1 h−1. c M w: kg mol−1, Mw and Mw/Mn determined via GPC. d Determined via DSC. e 5 atm of ethylene. f 1 atm of ethylene. | |||||||||
| 1 | Ni5 | 200 | 30 | 30 | 1.71 | 1.71 | 1.66 | 2.18 | 68.6 |
| 2 | Ni5 | 300 | 30 | 30 | 3.15 | 3.15 | 1.62 | 2.32 | 68.9 |
| 3 | Ni5 | 400 | 30 | 30 | 2.44 | 2.44 | 1.48 | 2.34 | 83.6 |
| 4 | Ni5 | 500 | 30 | 30 | 1.94 | 1.94 | 1.14 | 2.09 | 70.9 |
| 5 | Ni5 | 600 | 30 | 30 | 1.07 | 1.07 | 1.04 | 2.04 | 69.2 |
| 6 | Ni5 | 300 | 20 | 30 | 3.82 | 3.82 | 1.75 | 2.50 | 86.2 |
| 7 | Ni5 | 300 | 40 | 30 | 1.33 | 1.33 | 1.19 | 2.10 | 86.3 |
| 8 | Ni5 | 300 | 50 | 30 | 0.46 | 0.46 | 0.98 | 2.21 | 69.2 |
| 9 | Ni5 | 300 | 20 | 5 | 1.17 | 7.02 | 1.07 | 1.72 | 74.3 |
| 10 | Ni5 | 300 | 20 | 15 | 2.23 | 4.46 | 1.30 | 1.86 | 74.1 |
| 11 | Ni5 | 300 | 20 | 45 | 4.41 | 2.94 | 1.77 | 2.59 | 80.9 |
| 12 | Ni5 | 300 | 20 | 60 | 4.64 | 2.32 | 1.89 | 2.65 | 77.6 |
| 13e | Ni5 | 300 | 20 | 30 | 1.23 | 1.23 | 1.25 | 2.15 | 69.1 |
| 14f | Ni5 | 300 | 20 | 30 | Trace | Trace | — | — | — |
Firstly, the effect of the Al/Ni molar ratio on the catalytic performance of the Ni5/EASC system was studied, maintaining the reaction temperature of 30 °C, ethylene pressure of 10 atm and reaction time of 30 min (entries 1–5, Table 3). As the Al/Ni molar ratio increased from 200 to 600, the activity of Ni5/EASC initially increased, reaching the highest activity of 3.15 at 300 (entry 2, Table 3), before declining at a higher Al/Ni ratio. Specifically, the catalytic activity decreased to 1.07 when the ratio was 600 (entry 5, Table 3). This trend was further reflected by their GPC curves (Fig. 3a). The molecular weight of the obtained polyethylene gradually decreased from 1.66 kg mol−1 at the Al/Ni ratio of 200 to 1.04 kg mol−1 at a ratio of 600 (Fig. 3b), indicating the formation of shorter polymer chains at a high Al/Ni molar ratio. This can be ascribed to the fact that the chain transfer rate competes with the chain propagation rate, resulting in the production of shorter chain polymers.11a,17a
 | ||
| Fig. 3 (a) GPC curves of polyethylene produced using Ni5/EASC at different Al:Ni molar ratios and (b) effects of Al/Ni molar ratios on catalytic activity and molecular weight of polyethylene (entries 1–5, Table 3). | ||
For a constant Al/Ni ratio of 300 and a reaction time of 30 min, varying the temperature from 20 °C to 50 °C had a substantial impact on the performance of Ni5/EASC (entries 2 and 6–8, Table 3). At 20 °C, the Ni5/EASC catalyst achieved the highest activity of 3.82 and the obtained polyethylene exhibited a low molecular weight of 1.75 kg mol−1 (entry 6, Table 3). When the reaction temperature increased to 40 °C and 50 °C (entries 7 and 8, Table 3), the activity significantly decreased to 1.33 and 0.46, respectively. This sharp decrease in catalytic performance can be attributed to the partial deactivation of the active species and/or the decrease in the solubility of the ethylene monomer in the solvent at a higher temperature.8b,9f,11,12 According to the GPC curves (Fig. 4a), the molecular weight of the polymer decreased from 1.75 to 0.98 kg mol−1 when the reaction temperature increased from 20 °C to 50 °C, which is consistent with an increase in the chain transfer rate at elevated temperatures. The trends of activity and molecular weight of the obtained polyethylene with an increase in the reaction temperature are clearly shown in Fig. 4b.
 | ||
| Fig. 4 (a) GPC curves of the polyethylene produced using Ni5/EASC at different reaction temperatures; (b) effects of reaction temperature on the catalytic activity and molecular weight of polyethylene (entries 2 and 6–8, Table 3). | ||
To investigate the effect of reaction time on the polymerization behavior for this catalytic system, the polymerization runs using Ni5/EASC were conducted for a reaction time of 5, 15, 30, 45, and 60 min with the Al:Ni molar ratio maintained at 300 and the run temperature fixed at 20 °C (entries 6 and 9–12, Table 3). The catalytic activity reached the maximum value of 7.02 when the polymerization reaction was halted after 5 min (entry 9, Table 3). Evidently, the nickel complex was rapidly activated upon the addition of the co-catalyst, initiating the ethylene polymerization reaction efficiently.9i,17 As the reaction time was extended to 60 min, the activity decreased to 2.32 (entry 12, Table 3), indicating that partial active species gradually deactivated during the ethylene polymerization.
It is worth noting that the molecular weight of the obtained polymer increased from 1.07 to 1.89 kg mol−1 and the polyethylene yield increased from 1.17 g to 4.64 g with the extension of the reaction time, indicating that the active catalyst could maintain an appreciable catalytic effect within 60 min.9i Meanwhile, the dispersity remained reasonably narrow (Mw/Mn range of 1.72–2.65), highlighting the good control exhibited by this catalyst. The GPC curves of the corresponding polymers are provided in Fig. 5a. The trend of the catalytic activity and polymer molecular weight using Ni5/EASC with a prolonged polymerization time is shown in Fig. 5b.
 | ||
| Fig. 5 (a) GPC curves of the polyethylene produced using Ni5/EASC over different reaction times; (b) effects of reaction time on the catalytic activity and molecular weight of polyethylene (entries 6 and 9–12, Table 3). | ||
With regard to the impact of the ethylene pressure on the polymerization reaction, both the activity and molecular weight of the polymer significantly decreased as the ethylene pressure decreased. With the Al:Ni molar ratio fixed at 300 and the run temperature set at 20 °C for a reaction time of 30 min, the activity decreased from 3.82 to 1.23 when the pressure was reduced from 10 to 5 atm. Similarly, the molecular weight of the resulting polymer also decreased from 1.75 to 1.25 kg mol−1 (entries 6 and 13, Table 3) and no polymer could be obtained at 1 atm (entry 14, Table 3). This indicates that a critical ethylene pressure is required to sustain the catalytic activity in these catalysts, which can impact the rate of chain growth.
Ethylene polymerization studies using Ni1–Ni10/EASC
To investigate the influence of the complex structure (specifically the variations in the 6-R groups and the halides) on the catalytic performance, all the nickel bromide complexes (Ni1–Ni5) and nickel chloride complexes (Ni6–Ni10) were evaluated for ethylene polymerization. The reaction conditions were standardized to a 30 min duration, using an Al/Ni ratio of 300 and a reaction temperature of 20 °C based on the optimal reaction conditions established for Ni5/EASC. The complete set of data including catalytic activity and corresponding polymer properties is presented in Table 4.| Entry | Precat. | Al/Ni | T (°C) | t (min) | Yield (g) | Act.b |
M
wc |
M
w/Mnc |
T
md (°C) |
|---|---|---|---|---|---|---|---|---|---|
| a Conditions: 2.0 μmol of the nickel precatalyst, 100 mL of toluene, and 10 atm of ethylene. b 106 g PE (mol of Ni)−1 h−1. c M w: kg mol−1, Mw and Mw/Mn determined via GPC. d Determined via DSC. | |||||||||
| 1 | Ni1 | 300 | 20 | 30 | 3.11 | 3.11 | 5.42 | 2.76 | 97.2 |
| 2 | Ni2 | 300 | 20 | 30 | 2.65 | 2.65 | 6.17 | 3.03 | 72.8 |
| 3 | Ni3 | 300 | 20 | 30 | 2.23 | 2.23 | 6.75 | 3.32 | 72.0 |
| 4 | Ni4 | 300 | 20 | 30 | 3.44 | 3.44 | 3.40 | 2.58 | 81.1 |
| 5 | Ni5 | 300 | 20 | 30 | 3.82 | 3.82 | 1.75 | 2.50 | 86.2 |
| 6 | Ni6 | 300 | 20 | 30 | 4.82 | 4.82 | 5.92 | 3.32 | 99.1 |
| 7 | Ni7 | 300 | 20 | 30 | 3.31 | 3.31 | 6.73 | 3.17 | 70.5 |
| 8 | Ni8 | 300 | 20 | 30 | 2.45 | 2.45 | 6.84 | 2.96 | 77.6 |
| 9 | Ni9 | 300 | 20 | 30 | 4.97 | 4.97 | 2.94 | 2.84 | 81.7 |
| 10 | Ni10 | 300 | 20 | 30 | 5.74 | 5.74 | 1.80 | 2.61 | 85.7 |
For the bromide series, the catalytic activity ranged from 2.23 to 3.82 (entries 1–5, Table 4), with the activity decreasing in the order of Ni5 (R = F) > Ni4 (R = Cl) > Ni1 (R = Me) > Ni2 (R = Et) > Ni3 (R = iPr). A similar trend was observed for the nickel chloride complex series, as follows: Ni10 (R = F) > Ni9 (R = Cl) > Ni6 (R = Me) > Ni7 (R = Et) > Ni8 (R = iPr).
Notably, the Ni10/EASC catalyst exhibited the highest activity of 5.74 (entry 10, Table 4). It is apparent that the catalytic activity of the complexes with halogen atoms at the ortho-position was higher than that of the complexes containing an alkyl substituent at the same position. Specifically, Ni4 and Ni5 demonstrated higher activity than Ni1–Ni3, and Ni9 and Ni10 were more active than Ni6–Ni8.
This implies that the catalysts bearing electron-withdrawing 6-R groups displayed higher activity than that with electron-donating 6-R groups. The electron-withdrawing nature of the halogen substituents at the ortho-position of the phenyl ring enhances the electrophilicity of the nickel center, thereby increasing the rate of ethylene insertion and affording higher activities.9i,18 Previous calculations revealed that the F or Cl substituents could influence the net charge of the active species to increase the activity.19 Additionally, the steric hindrance of the 6-R ortho substituent had a negative impact on the catalytic activities when the 2-orthor position was already occupied by a steric group (CHPh2). The order of activity for Ni1–Ni3 or Ni6–Ni8 suggests that increased steric hindrance led to a decrease in catalytic activity. Specifically, 6-isopropyl-containing Ni3 and Ni8 (entries 3 and 8, Table 4) displayed the lowest activity in their series, which is likely due to the steric hindrance exerted by the ortho-isopropyl substituents, impeding ethylene coordination and insertion into the active site, respectively.7c,d,9e,11a,12 It is worth noting that the overall activities of the nickel chloride complexes were generally higher than that of the corresponding nickel bromide complexes, indicating that the halogen atoms around the metal active center significantly impacted the catalytic activity. Similar observations were noted in our previous work.9b,h,i
Fig. 6 and 7 illustrate the GPC curves of polyethylene and comparison trends of catalytic activity and molecular weight of polyethylene produced using Ni1–Ni5 and Ni6–Ni10 with EASC as the co-catalyst, respectively. The Mw values of polyethylene generated using Ni1–Ni5 were in the range of 1.75–6.75 kg mol−1, with a relatively narrow polydispersity (Mw/Mn range of 2.50–3.32), indicating a single active center, as also confirmed by the GPC curves in Fig. 6a. The polymer molecular weight followed the order of Ni3 (R = iPr) > Ni2 (R = Et) > Ni1 (R = Me) > Ni4 (R = Cl) > Ni5 (R = F) (entries 1–5, Table 4), indicating that the presence of the bulky isopropyl in complex Ni3 is beneficial for promoting chain growth, resulting in the formation of relatively higher molecular weight polyethylene (Mw value of 6.75 kg mol−1).7d,11a,20 In contrast, the molecular weight of polyethylene obtained using Ni4 and Ni5 was only 3.40 and 1.75 kg mol−1, respectively. Similar resulted were observed for Ni6–Ni10 (Fig. 7a). The presence of the o-halide substituent near the metal center in the active catalyst promoted β-H elimination,9i which is possibly due to the weak hydrogen bonding between the o-halide and the coordinated ethylene during the insertion transition state.7d,18
 | ||
| Fig. 6 (a) GPC curves of polyethylene and (b) comparison trend of catalytic activity and molecular weight of polyethylene produced using Ni1–Ni5 with EASC as the co-catalyst (entries 1–5, Table 4). | ||
 | ||
| Fig. 7 (a) GPC curves of polyethylene and (b) comparison trend of catalytic activity and molecular weight of polyethylene produced using Ni6–Ni10 with EASC as the co-catalyst (entries 6–10, Table 4). | ||
| Entry | Precat. | Al/Ni | T (°C) | t (min) | Yield (g) | Act.b |
M
wc |
M
w/Mnc |
T
md (°C) |
|---|---|---|---|---|---|---|---|---|---|
| a Conditions: 2.0 μmol of the nickel precatalyst, 100 mL of toluene, and 10 atm of ethylene. b 106 g PE (mol of Ni)−1 h−1. c M w: kg mol−1, Mw and Mw/Mn determined via GPC. d Determined via DSC. e 5 atm of ethylene. f 1 atm of ethylene. | |||||||||
| 1 | Ni5 | 500 | 30 | 30 | 1.22 | 1.22 | 1.75 | 2.44 | 80.7 |
| 2 | Ni5 | 1000 | 30 | 30 | 2.18 | 2.18 | 1.62 | 2.53 | 92.0 |
| 3 | Ni5 | 1500 | 30 | 30 | 2.79 | 2.79 | 1.49 | 2.27 | 90.8 |
| 4 | Ni5 | 2000 | 30 | 30 | 3.25 | 3.25 | 1.35 | 2.26 | 86.4 |
| 5 | Ni5 | 2500 | 30 | 30 | 4.73 | 4.73 | 1.29 | 2.01 | 85.3 |
| 6 | Ni5 | 3000 | 30 | 30 | 3.67 | 3.67 | 1.26 | 2.17 | 85.7 |
| 7 | Ni5 | 2500 | 20 | 30 | 3.28 | 3.28 | 2.33 | 2.59 | 91.6 |
| 8 | Ni5 | 2500 | 40 | 30 | 2.81 | 2.81 | 1.25 | 1.79 | 88.6 |
| 9 | Ni5 | 2500 | 50 | 30 | 0.74 | 0.74 | 1.04 | 1.69 | 88.5 |
| 10 | Ni5 | 2500 | 30 | 5 | 1.37 | 8.22 | 0.96 | 1.68 | 77.9 |
| 11 | Ni5 | 2500 | 30 | 15 | 2.84 | 5.68 | 1.25 | 1.94 | 88.2 |
| 12 | Ni5 | 2500 | 30 | 45 | 5.28 | 3.52 | 1.39 | 2.30 | 91.2 |
| 13 | Ni5 | 2500 | 30 | 60 | 5.66 | 2.83 | 1.52 | 2.08 | 86.7 |
| 14e | Ni5 | 2500 | 30 | 30 | 1.91 | 1.91 | 1.19 | 2.00 | 75.3 |
| 15f | Ni5 | 2500 | 30 | 30 | Trace | Trace | — | — | — |
Firstly, the Al/Ni molar ratio using Ni5/MAO for ethylene polymerization sequentially increased from 500 to 3000 with the run temperature set at 30 °C and the duration at 30 min (entries 1–6, Table 5). With the ratio of 2500, the activity using Ni5/MAO reached its peak value of 4.73 (entry 5, Table 5). As the molar ratio of Al/Ni increased from 500 to 3000, the molecular weight of polyethylene gradually decreased from 1.75 to 1.26 kg mol−1, while the corresponding molecular weight distribution was maintained in the range of 2.01–2.53, indicating single-site catalysis and effective control of the polymerization. The GPC curves of polyethylene with varying molar ratios together with the trends in activity and molecular weight of the obtained polyethylene are depicted in Fig. S11a and S11b,† respectively.
Secondly, to explore the effect of temperature on the catalytic performance of Ni5/MAO, a series of polymerization reactions was conducted at temperatures in the range of 20 °C to 50 °C with the Al/Ni molar ratio maintained at 2500 (entries 5 and 7–9, Table 5). At 30 °C, the Ni5/MAO catalyst combination reached the peak activity of 4.73 (entry 5, Table 5), resulting in a molecular weight of 1.29 kg mol−1. The GPC curves of the polymers generated at different temperatures are shown in Fig. S12a.† A significant decrease in activity to 0.74 was observed as the temperature increased to 50 °C (entry 9, Table 5), with the molecular weight of the polymer decreasing from 2.33 to 1.04 kg mol−1, which is due to the higher rates of chain transfer and termination at elevated temperature.9h,10c Although the optimal temperature for Ni5/MAO was 10 °C higher than that for Ni5/EASC, similar trends can be observed in Fig. S12b.† In comparison, the melting temperature, Tm, of the polymers varied in the temperature range of 85.3 °C to 91.6 °C.
Further, to explore the catalytic efficiency with time, polymerization runs using Ni5/MAO were conducted with different reaction times of 5, 15, 30, 45 and 60 min under the optimal conditions of 30 °C and Al:Ni molar ratio of 2500 (entries 5 and 10–13, Table 5). The highest activity of 8.22 was noted after 5 min (entry 10, Table 5). Also, the activity gradually decreased with the extension of reaction time, reaching the minimum of 2.83 after 60 min (entry 13, Table 5). Similar to EASC, the active species also formed rapidly after adding the MAO co-catalyst, followed by partial deactivation as the reaction proceeded.9i,17 The molecular weight of the polymer exhibited an upward trend from 0.96 to 1.52 kg mol−1 with time, indicating the capability of the catalyst to maintain an appreciable catalytic lifetime. This catalytic behavior is consistent with that observed in nickel analogues.9a,d–f,i,11,12 All the GPC curves of the generated polymers show narrow dispersity (Mw/Mn range of 1.68–2.30), reflecting the characteristic of a well-controlled polymerization, as shown in Fig. S13a.† The changes in activity and Mw data at different reaction times are shown in Fig. S13b.†
In addition, both the molecular weight and catalytic activity showed a declining trend with a decrease in ethylene pressure and no polymer could be isolated at 1 atm (entry 15, Table 5), indicating that a critical ethylene pressure is required for these catalysts to exhibit activity. These observations are consistent with the EASC system.
| Entry | Precat. | Al/Ni | T (°C) | t (min) | Yield (g) | Act.b |
M
wc |
M
w/Mnc |
T
md (°C) |
|---|---|---|---|---|---|---|---|---|---|
| a Conditions: 2.0 μmol of the nickel precatalyst, 100 mL of toluene, and 10 atm of ethylene. b 106 g PE (mol of Ni)−1 h−1. c M w: kg mol−1, Mw and Mw/Mn determined via GPC. d Determined via DSC. | |||||||||
| 1 | Ni1 | 2500 | 30 | 30 | 3.45 | 3.45 | 2.70 | 2.63 | 80.6 |
| 2 | Ni2 | 2500 | 30 | 30 | 2.87 | 2.87 | 3.63 | 2.19 | 70.6 |
| 3 | Ni3 | 2500 | 30 | 30 | 1.22 | 1.22 | 3.88 | 2.06 | 71.2 |
| 4 | Ni4 | 2500 | 30 | 30 | 3.29 | 3.29 | 1.48 | 1.83 | 78.1 |
| 5 | Ni5 | 2500 | 30 | 30 | 4.73 | 4.73 | 1.29 | 2.01 | 85.3 |
| 6 | Ni6 | 2500 | 30 | 30 | 4.55 | 4.55 | 3.60 | 2.38 | 85.0 |
| 7 | Ni7 | 2500 | 30 | 30 | 4.06 | 4.06 | 3.66 | 2.46 | 72.0 |
| 8 | Ni8 | 2500 | 30 | 30 | 2.34 | 2.34 | 4.62 | 2.53 | 70.2 |
| 9 | Ni9 | 2500 | 30 | 30 | 7.37 | 7.37 | 2.01 | 2.29 | 81.5 |
| 10 | Ni10 | 2500 | 30 | 30 | 8.12 | 8.12 | 1.23 | 2.03 | 77.3 |
For the bromide series, the best activity of 4.73 was achieved by Ni5/MAO (entry 5, Table 6). The catalytic activities for the five nickel precatalysts decreased in the order of Ni5 (R = F) > Ni4 (R = Cl) > Ni1 (R = Me) > Ni2 (R = Et) > Ni3 (R = iPr). Also, a similar trend was observed regarding the chloride series, following the order of Ni10 (R = F) > Ni9 (R = Cl) > Ni6 (R = Me) > Ni7 (R = Et) > Ni8 (R = iPr). The Ni10/MAO catalyst achieved the best activity of 8.12 (entry 10, Table 6), indicating the positive effect of fluorine atoms on improving the catalytic performance.6a
Similar to the EASC system, this order further proved that the precatalyst bearing electron-withdrawing 6-R groups displayed higher activity than that with electron-donating 6-R groups for this series of nickel complexes. In addition, it is apparent that the steric hindrance by the ortho substituents has a significant impact on the catalytic activity according to the comparison of the order of activity of Ni1–Ni3 or Ni6–Ni8. For example, 6-isopropyl-containing Ni3 and Ni8 (entries 3 and 8, Table 6) displayed the lowest activity in their respective series, which is ascribed to the increased obstacle for ethylene coordination and insertion into the active site. Interestingly, the catalytic activity using the chloride complexes (Ni6–Ni10) was generally higher than that of the corresponding bromide series (Ni1–Ni5) containing the same ligand, indicating that the halogen atoms around the metal center have a significant impact on the catalytic activity, respectively. These phenomena are similar with the above-mentioned observations in the EASC system.
The ligand structure greatly affects the polymer properties, as shown by the GPC traces in Fig. S14a and S15a.† The corresponding polyethylene exhibited a narrow molecular weight distribution in the range of 1.83–2.63, indicating the single-site catalytic behavior of the catalysts. According to the comparison of the polymer molecular weight, it followed the order of Ni3 (6-R = iPr) > Ni2 (6-R = Et) > Ni1 (6-R = Me) > Ni4 (6-R = Cl) > Ni5 (6-R = F) (entries 1–5, Table 5), Ni8 (6-R = iPr) > Ni7 (6-R = Et) > Ni6 (6-R = Me) > Ni9 (6-R = Cl) > Ni10 (6-R = F) (entries 6–10, Tables 5). Among them, the molecular weight of the polymers generated using Ni3 and Ni8 (6-R = iPr) reached 3.88 and 4.62 kg mol−1 (entry 3 and 8, Table 5), which are the maximum values in their series, respectively. A similar finding was noted for their EASC system, which suggests again that the particular spatial properties provided by Ni3 and Ni8 (6-R = iPr) are most beneficial for promoting polymer chain growth. It is thought that the introduction of the sterically bulky substituent at the ortho-positions of the aryl ring blocked the axial sites at the metal center, suppressing chain transfer processes and increasing the chain propagation.3e,4a,d Moreover, the polyethylene produced by Ni3 and Ni8 possessed a higher molecular weight but lower Tm, indicating the formation of more highly branched polyethylene.11b A comparison of the trends of catalytic activity and molecular weight of the generated polyethylene using Ni1–Ni5 and Ni6–Ni10 is shown in Fig. S14b and S15b,† respectively.
 | ||
| Fig. 8 DFT-optimized structures of cationic nickel-alkyl species (a) Ni3-R and (b) Ni5-R. For clarity, most of the hydrogen atoms are omitted and only the main moieties are focused. | ||
The optimized structures of Ni3-R and Ni5-R in their ground states (triplet and singlet, respectively) are illustrated in Fig. 8. The zero-point energy, Ezpe, of Ni3-R and Ni5-R in both the singlet and triplet states was calculated to determine their stable ground state structures. The results revealed that the Ezpe of Ni3-R in the triplet state is 1.52 kcal mol−1 lower than that in its singlet state, whereas for Ni5-R, it is 2.37 kcal mol−1 higher in the triplet state compared to the singlet state. The great stability of Ni5-R in the singlet state indicates a strong binding nature. Notably, significant agostic interactions (–Ni⋯H–CPh2) were observed between the ortho diphenylmethyl groups and the nickel metal center. The strength of the agostic interactions was reflected by the distance between the nickel metal atom and hydrogen atom bonded to carbon atom C1 (Fig. 8) in the bulky diphenylmethyl (–CHPh2) group. In the structure of Ni5-R, the Ni⋯H–C1 distance is 1.64 Å, whereas in Ni3-R, it is 2.12 Å, which again indicates stronger intramolecular interactions in the former. This strong interaction in Ni5-R can likely interpret the higher catalytic activity observed in the Ni5 (Ni10) catalyst system compared to Ni3 (Ni8), as illustrated in Fig. 6 and 7. The closer proximity of the agostic interaction in Ni5-R suggests a more stabilized active site, facilitating the ethylene insertion and chain propagation steps of the polymerization process.
Further structural analysis indicated significant differences between Ni3-R and Ni5-R due to the substituents at the ortho 6-position. In Ni3-R, the hydrogen atoms on the two carbon atoms of the 6-position isopropyl (iPr) substituent form intramolecular agostic (iPr(C–H)⋯Ni: 2.71 Å) and hydrogen bonds (iPr(C–H)⋯N: 2.98 Å) with the metal center and one nitrogen atom in the framework, respectively. This, combined with the steric hindrance of the ortho (2-position) bulky diphenylmethyl group, causes substantial deviation of the aniline ring plane from the bidentate pyridine basal plane, with a dihedral angle of 51.8°. This deviation likely impacts the spatial arrangement and accessibility of the catalytic site, influencing the polymerization kinetics and product characteristics. In contrast, the structure of Ni5-R features an ortho (6-position) fluorine atom, which forms strong intramolecular hydrogen bonds (F⋯(H)–C0, 2.19/2.85 Å) with the hydrogen atoms on the seven fused rings of the framework. This interaction promotes a more coplanar alignment between the aniline ring plane and the bidentate pyridine basal plane, with a dihedral angle of 24.6°. The increased planarity in Ni5-R facilitates better spatial orientation and interaction with ethylene monomers, enhancing the activity of the catalyst. Furthermore, the intramolecular hydrogen bonding involving the fluorine atoms creates a more open space around the nickel atom, promoting the formation of polymers with lower molecular weights.
Thus, the presence of ortho bulky diphenylmethyl groups in the catalyst structure and their strong agostic interactions with the nickel center significantly enhance the catalytic activity. The structural differences between Ni3-R and Ni5-R, particularly in terms of their dihedral angles and intra-molecular interactions caused by the ortho 2-position, play a crucial role in determining the efficiency of the catalysts and the molecular weight of the produced polymers. These findings provide valuable insights into the design of more efficient nickel-based catalysts for ethylene polymerization, emphasizing the importance of precise structural tuning to optimize the catalytic performance.
Microstructural features of the polyethylene
According to melting temperature data for the polymers collected in Table 4 (Tm range of 70.5–99.1 °C) and Table 6 (Tm range of 70.2–85.3 °C), the polymer materials likely possessed a moderate degree of branching with a relatively high content of short chain branches. Thus, to confirm this assertion and evaluate the microstructural properties of the polyethylene products, high-temperature 1H and 13C NMR spectroscopic measurements were carried out on two representative samples, i.e., one sample prepared using Ni5/EASC at 30 °C (entry 5, Table 4) and the other using Ni5/MAO at 20 °C (entry 5, Table 6). Based on the analysis of the spectra, information about the type of branch and branching density as well as chain ends was obtained.15c,d,9iThe 1H NMR spectra of both samples (Fig. 9 and Fig. S16†) revealed the presence of two types of unsaturated groups in the obtained polyethylene, respectively. One is the vinyl (–CHCH2) end group, which is identified downfield peaks at δ 5.0 (Ha) and δ 5.9 (Hb) (integral ratio of 1:2). The other is the vinylene group (–CHCH–) with a characteristic peak at δ 5.5 (Hc/Hc′), which can be ascribed to the internal vinylene protons. These observations highlighted the role of β-H elimination. As a result, the polymer chains can be classified into three types, i.e., saturated (methyl)-end chains, vinyl-end chains, and chains containing internal vinylene groups. The relative integration of the corresponding 1H NMR resonances (Fig. 9) indicates a molar ratio of saturated-end to unsaturated chains of 0.379, and a molar ratio of vinyl-end chains to chains with internal vinylene groups of 0.348.15e The presence of characteristic downfield peaks (a, b, c, and c′) for the corresponding carbon atoms in their 13C NMR spectra (Fig. 10) supports these findings. The protons for the main –(CH2)n– repeating unit and various CH and CH2 protons are observed in the upfield region of the 1H NMR spectra.
 | ||
| Fig. 9 1H NMR spectrum of the polyethylene sample produced using Ni5/EASC at 20 °C (entry 5, Table 4), recorded at 110 °C in 1,1,2,2-tetrachloroethane-d2. | ||
 | ||
| Fig. 10 13C NMR spectrum of the polyethylene sample produced using Ni5/EASC at 20 °C (entry 5, Table 4) together with an inset showing the δ 114–140 region and a segment of the assigned polymer backbone recorded at 110 °C in 1,1,2,2-tetrachloroethane-d2. | ||
In the 13C NMR spectrum of the polyethylene prepared using Ni5/EASC at 20 °C (Fig. 10), carbon resonances corresponding to methyl, ethyl, propyl, butyl, amyl, sec-butyl, and longer chain branches are visible. By using 13C NMR spectroscopic techniques,15 the branching density was determined to be 35 branches per 1000 Cs based on the composition of methyl (43.94%), ethyl (1.21%), propyl (0.85%), butyl (4.96%), amyl (9.45%), and longer chain branches (39.59%). In comparison, the polyethylene prepared using Ni5/MAO at 30 °C contained 30 branches per 1000 Cs, including methyl (49.69%), ethyl (1.79%), propyl (0.75%), butyl (3.36%), amyl (10.66%), and longer chain branches (39.75%) (Fig. S17†).
Furthermore, it is apparent that the compositions of branch chains are similar and dominated by methyl groups and longer chain branches for both samples generated using different types of co-catalysts. Considering the similarity of the branching compositions, it seems that the relatively higher melting temperatures for the samples obtained using EASC are mainly caused by the higher molecular weight of the corresponding polymers.
Comparison with previously reported precatalysts
For the purpose of comparison, the activity and molecular weight data for the polymer generated using Ni10 were compared with that obtained for structurally related examples Ea and Eb, as shown in Fig. 11 (all polymerizations were performed under the optimal conditions at 10 atm C2H4 over 30 min). Significantly, Ni10/MAO displayed the highest activity (8.12) (entry 10, Table 6), surpassing the activities of the previously reported E.11 This enhancement is likely attributed to both the strong agostic interactions caused by the ortho bulky diphenylmethyl groups and the formation of intramolecular hydrogen bonding due to the ortho-fluorine substituents. In addition, the molecular weight of polyethylene obtained using Ni10/MAO was only 1.23 kg mol−1 lower than that of the two analogue catalysts (4.54 and 1.91 kg mol−1). This observation is also likely due to the presence of the fluorine atom substituent, which promotes β-H elimination by forming intramolecular hydrogen bonds.22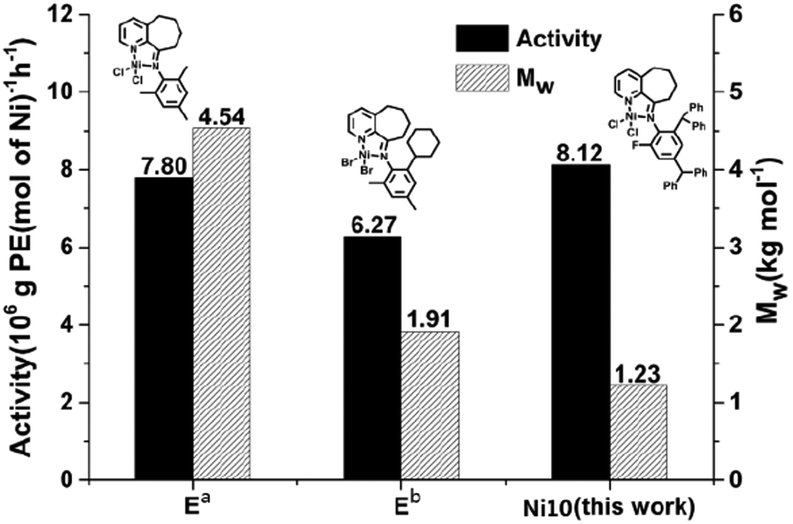 | ||
| Fig. 11 Comparative catalytic performance of Ni10 (entry 10, Table 6) with nickel-containing E (Chart 1); all polymerizations were performed under the optimal conditions at 10 atm C2H4 over 30 min. | ||
Experimental
General considerations
Manipulation of all air- and/or moisture-sensitive compounds was performed under an inert nitrogen atmosphere using standard Schlenk techniques and/or a glovebox. Toluene was heated to reflux and distilled under a nitrogen atmosphere prior to use. Methylaluminoxane (MAO, 1.46 M solution in toluene) and modified methylaluminoxane (MMAO, 1.93 M solution in n-heptane) were purchased from Anhui Botai Electronic Materials Co. Ethylaluminum dichloride (EtAlCl2), ethylaluminum sesquichloride (EASC), and diethylaluminum chloride (Et2AlCl) were purchased from Lianli Chemical. High-purity ethylene was acquired from Beijing Yanshan Petrochemical Co. All other reagents were purchased from Aladdin, Aldrich or local distributors. Five types of aniline derivatives were synthesized based on the procedure reported in the literature.13,14 The 1H and 13C nuclear magnetic resonance (NMR) spectra of all the ligand compounds were recorded on a Bruker DMX 400 MHz instrument at ambient temperature using TMS as an internal standard. A PerkinElmer System 2000 FT-IR spectrometer was used to record the FT-IR spectra. Elemental analysis was performed on a Flash EA 1112 microanalyzer. The melt temperature of the polyethylenes was measured by differential scanning calorimetry using a PerkinElmer TA-Q2000 DSC analyzer under a nitrogen atmosphere. Typically, a sample of polyethylene (4.0–6.0 mg) was heated to 150 °C at a rate of 20 °C min−1, held for 5 min at 150 °C to eliminate the thermal history, and then cooled to −20 °C at a rate of 20 °C min−1. The molecular weight (Mw) and dispersity (Mw/Mn) of the polyethylene were determined by gel permeation chromatography (GPC) using an Agilent PL-GPC 220 instrument equipped with a refractive index (RI) detector operating at 150 °C using 1,2,4-trichlorobenzene as the eluent. For the 1H NMR and 13C NMR spectra of the polyethylenes, a weighed amount of polyethylene (40–60 mg) was first dissolved in 1,1,2,2-tetrachloroethane-d2 (2 mL) at high temperature with TMS as an internal standard and the spectra recorded on a Bruker AVANCE III 500 MHz instrument at 110 °C. The branching density was calculated by integrating the corresponding peaks in the 13C NMR spectrum using the methods described in the literature.15Synthesis of 9-[2,4-bis(benzhydryl)-6-R-phenylimino]-5,6,7,8-tetrahydrocyclo-heptapyridine (L1–L5)
:1). Then, the solvent was removed under vacuum to obtain L1 as a yellow powder (0.43 g, 35.8%). 1H NMR (400 MHz, CDCl3, TMS): δ 8.66 (d, J = 4.8 Hz, 1H, Py–H), 7.43 (d, J = 7.6 Hz, 1H, Py–H), 7.25–7.21 (m, 3H, Ar–H, 1H, Py–H), 7.19–7.03 (m, 15H, Ar–H), 6.93 (d, J = 7.2 Hz, 2H, Ar–H), 6.80 (s, 1H, Ar–H), 6.62 (d, J = 1.2 Hz, 1H, Ar–H), 5.70 (s, 1H, CH(Ph)2), 5.40 (s, 1H, CH(Ph)2), 2.61–2.54 (m, 1H, CH2), 2.45–2.38 (m, 1H, CH2), 2.07 (s, 3H, CH3), 1.99–1.92 (m, 1H, CH2), 1.58–1.50 (m, 1H, CH2), 1.44–1.34 (m, 2H, CH2), 1.22–1.16 (m, 1H, CH2), 1.01–0.94 (m, 1H, CH2). 13C NMR (100 MHz, CDCl3, TMS): δ 174.2, 156.4, 148.4, 146.6, 144.9, 144.7, 144.6, 143.0, 137.8, 136.9, 134.6, 132.8, 130.3, 129.7, 129.5, 129.5, 129.5, 128.4, 128.3, 128.2, 128.1, 127.9, 126.1, 125.8, 124.5, 124.2, 56.4, 51.4, 31.4, 30.7, 25.5, 21.6, 18.2. FT-IR (cm−1): 3024 (w), 2933 (w), 2850 (w), 1638 (vCN, s), 1599 (s), 1566 (m), 1493 (vs), 1445 (vs), 1295 (w), 1285 (w), 1202 (w), 1131 (m), 1078 (m), 1029 (m), 966 (w), 911 (w), 850 (w), 799 (m), 740 (s), 696 (vs). Anal. calcd for C43H38N2 (582.8): C, 88.62; H, 6.57; N, 4.81. Found: C, 88.36; H, 6.79; N, 4.84%.
N, s), 1598 (s), 1566 (m), 1491 (vs), 1447 (vs), 1260 (w), 1200 (w), 1132 (w), 1078 (w), 1029 (m), 966 (w), 899 (w), 852 (w), 800 (m), 740 (s), 696 (vs). Anal. calcd for C44H40N2 (596.8): C, 88.55; H, 6.76; N, 4.69. Found: C, 88.64; H, 6.53; N, 4.82%.
N, s), 1598 (s), 1564 (m), 1492 (vs), 1445 (vs), 1296 (w), 1160 (w), 1118 (m), 1077 (w), 1029 (m), 962 (w), 910 (w), 851 (m), 799 (w), 739 (s), 697 (vs). Anal. calcd for C45H42N2 (610.8): C, 88.48; H, 6.93; N, 4.59. Found: C, 88.71; H, 6.59; N, 4.68%.
N, s), 1598 (s), 1562 (m), 1493 (vs), 1447 (vs), 1296 (w), 1158 (w), 1212 (w), 1104 (m), 1079 (w), 1028 (m), 965 (w), 916 (w), 887 (w), 850 (w), 799 (m), 738 (s), 697 (vs). Anal. calcd for C42H35ClN2 (603.2): C, 83.63; H, 5.85; N, 4.64. Found: C, 83.32; H, 6.03; N, 4.69%.
N, s), 1599 (s), 1566 (m), 1493 (vs), 1448 (vs), 1427 (m), 1291 (m), 1200 (w), 1120 (w), 1028 (m), 995 (m), 911 (w), 850 (w), 778 (w), 742 (s), 696 (vs). Anal. calcd for C42H35FN2 (586.8): C, 85.97; H, 6.01; N, 4.77. Found: C, 86.24; H, 6.06; N, 4.62%.
Synthesis of 9-[2,4-bis(benzhydryl)-6-R-phenylimino]-5,6,7,8-tetrahydrocyclo-heptapyridine nickel bromide complexes (Ni1–Ni5)
N, s), 1571 (s), 1493 (vs), 1447 (s), 1339 (m), 1285 (w), 1202 (w), 1179 (w), 1154 (w), 1113 (w), 1076 (m), 1029 (m), 975 (w), 915 (w), 802 (w), 744 (s), 699 (vs). Anal. calcd for C43H38Br2N2Ni (801.3): C, 64.45; H, 4.78; N, 3.50. Found: C, 64.21; H, 4.93; N, 3.69%.
N, s), 1571 (s), 1493 (vs), 1448 (s), 1338 (m), 1288 (w), 1200 (w), 1155 (w), 1116 (w), 1029 (m), 974 (w), 916 (w), 809 (m), 743 (s), 699 (vs). Anal. calcd for C44H40Br2N2Ni (815.3): C, 64.82; H, 4.95; N, 3.44. Found: C, 64.71; H, 5.13; N, 3.68%.
N, s), 1569 (s), 1494 (vs), 1447 (s), 1337 (w), 1314 (w), 1288 (w), 1248 (w), 1199 (w), 1156 (w), 1117 (m), 1077 (w), 1029 (m), 975 (w), 915 (w), 811 (m), 742 (s), 699 (vs). Anal. calcd for C45H42Br2N2Ni (829.4): C, 65.17; H, 5.10; N, 3.38. Found: C, 65.46; H, 5.25; N, 3.55%.
N, s), 1574 (s), 1493 (vs), 1449 (s), 1414 (m), 1340 (m), 1266 (w), 1181 (w), 1116 (m), 1030 (m), 980 (m), 920 (w), 885 (w), 805 (m), 742 (s), 701 (vs). Anal. calcd for C42H35Br2ClN2Ni (821.7): C, 61.39; H, 4.29; N, 3.41. Found: C, 61.17; H, 4.45; N, 3.49%.
N, s), 1574 (s), 1494 (vs), 1448 (s), 1339 (m), 1294 (w), 1183 (w), 1121 (w), 1076 (m), 1029 (m), 999 (w), 916 (w), 811 (m), 745 (s), 699 (vs). Anal. calcd for C42H35Br2FN2Ni (805.3): C, 62.65; H, 4.38; N, 3.48. Found: C, 62.37; H, 4.51; N, 3.65%.
Synthesis of 9-[2,4-bis(benzhydryl)-6-R-phenylimino]-5,6,7,8-tetrahydrocyclo-heptapyridine nickel chloride complexes (Ni6–Ni10)
N, s), 1575 (s), 1494 (vs), 1449 (s), 1264 (w), 1200 (w), 1146 (w), 1114 (w), 1082 (m), 1050 (m), 914 (w), 886 (w), 809 (m), 755 (s), 720 (vs), 702 (vs). Anal. calcd for C43H38Cl2N2Ni (712.4): C, 72.50; H, 5.38; N, 3.93. Found: C, 72.64; H, 5.21; N, 3.93%.
N, s), 1574 (s), 1494 (vs), 1450 (s), 1339 (w), 1290 (w), 1203 (w), 1116 (w), 1078 (m), 1031 (m), 916 (w), 811 (m), 745 (s), 698 (vs). Anal. calcd for C44H40Cl2N2Ni (726.4): C, 72.75; H, 5.55; N, 3.86. Found: C, 72.47; H, 5.63; N, 3.99%.
N, s), 1575 (s), 1493 (s), 1449 (s), 1264 (w), 1197 (w), 1156 (w), 1120 (m), 1080 (m), 1047 (w), 918 (w), 892 (w), 812 (m), 781 (s), 747 (s), 701 (vs). Anal. calcd for C45H42Cl2N2Ni (740.4): C, 73.00; H, 5.72; N, 3.78. Found: C, 73.23; H, 5.66; N, 3.89%.
N, s), 1576 (s), 1493 (m), 1451 (s), 1407 (w), 1336 (w), 1268 (w), 1152 (w), 1114 (m), 1082 (m), 1053 (w), 1033 (w), 916 (w), 888 (w), 809 (m), 780 (s), 753 (s), 717 (s), 711 (s), 701 (vs). Anal. calcd for C42H35Cl3N2Ni (732.8): C, 68.84; H, 4.81; N, 3.82. Found: C, 68.67; H, 4.99; N, 3.74%.
N, s), 1576 (s), 1493 (s), 1451 (s), 1427 (w), 1300 (w), 1198 (w), 1121 (w), 1301 (m), 997 (m), 915 (w), 875 (m), 812 (m), 749 (s), 702 (vs). Anal. calcd for C42H35Cl2FN2Ni (716.4): C, 70.42; H, 4.92; N, 3.91. Found: C, 70.76; H, 5.08; N, 3.73%.
X-ray diffraction studies
Single crystals of Ni3 and Ni4 suitable for X-ray diffraction studies were grown at room temperature by slow diffusion of diethyl ether into a dichloromethane solution of the corresponding complex. Each crystal was mounted on an XtaLAB Synergy-R single-crystal diffractometer equipped with a graphite monochromated Cu-Kα radiation (λ = 1.54184 Å) source and a nitrogen cold stream. The crystal was kept at 169.99(10) K during data collection. By using Olex2,16a the structures were determined by employing the ShelXT16b structure solution program using Intrinsic Phasing and refined with the ShelXL16c refinement package using least squares minimization. Details of the X-ray structure determination and refinement details are provided in Table S1.†Ethylene polymerization
The ethylene polymerization runs were conducted in a stainless-steel high-pressure reactor, with a capacity of 250 mL, equipped with a mechanical stirrer and a temperature controller. Firstly, freshly distilled toluene (25 mL) was injected into the autoclave under an ethylene atmosphere. Once the temperature stabilized, a solution of the precatalyst (2 μmol) in toluene (50 mL) was added, followed by the predetermined amount of co-catalyst, and then finally more toluene (25 mL) was introduced. The polymerization run commenced by stirring the reaction mixture at 400 rpm for the required time. Upon completion of the run, the pressure was vented and acidified ethanol added to quench the polymerization. The polymer was collected and washed with ethanol, dried under reduced pressure at 60 °C, and weighed.Conclusions
A series of 9-[2,4-bis(benzhydryl)-6-R-phenylimino]-5,6,7,8-tetrahydrocycloheptapyridine nickel complexes (Ni1–Ni10), varying in the steric and electronic properties displayed by the 6-R group, was successfully synthesized and characterized. The single crystal X-ray diffraction analysis of the Ni3 and Ni4 complexes revealed a distorted square-pyramidal geometry around the nickel center. Their catalytic performance were simultaneously investigated by both experimental and DFT calculations, reaching consistent observations. Upon activation by either EASC or MAO, all the nickel complexes exhibited active species with single-site characteristics and high activities (up to 8.12). The resultant polyethylenes possessed a moderate branching density with low molecular weights and narrow molecular weight distributions. The microstructural analysis using 13C and 1H NMR spectroscopy indicated that these polymers contained mainly methyl groups and longer chain branches with the chain end composed of vinyl and internal vinylene groups. Notably, the presence of electron-withdrawing halogen groups (R = Cl or F) resulted in higher catalytic activities but generated lower-molecular weight polymers compared to that observed for the catalysts containing o-alkyl groups (R = Me, Et, or iPr). This highlights the impact of the electronic and steric properties of the ligand frame on the catalytic performance and molecular weight of the resulting polymer. The DFT analysis further elucidated that that ortho-substituted bulky diphenylmethyl groups form strong agostic interactions with the nickel center, particularly in Ni5-R (1.64 Å) compared to Ni3-R (2.12 Å), enhancing the catalytic activity. The structural variations due to different 6-R substituents influence the spatial arrangement of the catalytic sites. In Ni5-R, intramolecular hydrogen bonding involving fluorine atoms promoted a more coplanar alignment with a 24.6° dihedral angle, facilitating better spatial orientation and interaction with the ethylene monomers, and thereby promoting the formation of polymers with a lower molecular weight. Conversely, Ni3-R showed significant deviation with a dihedral angle of 51.8°, affecting the polymerization kinetics.In conclusion, the presence of ortho bulky diphenylmethyl groups and their strong agostic interactions with the nickel center significantly enhanced the catalytic activity. The differences in the steric and electronic properties of the 6-R substituents played a crucial role in determining the efficiency of the catalysts and properties of the resultant branched polyethylene. These insights provide useful information for designing more efficient nickel-based catalysts for ethylene polymerization, emphasizing the importance of precise structural tuning to optimize the catalytic performance.
Data availability
The synthetic procedures for organic compounds (ligands) and their nickel complexes are present in the text together with analytical data, and the NMR spectra of the organic compounds are presented in the ESI.†The molecular structures and selected bond lengths and angles of nickel complexes Ni3 and Ni4 are shown in the manuscript together with their CCDC numbers; their crystal data and structure refinements are present in the ESI.† The CIF file and checkCIF are present in the ESI† for reviewers.
The detailed data regarding the catalytic performances of all nickel complex precatalysts as well as the GPC and DSC curves of the representative polyethylenes are presented in the manuscript. In addition, more detailed GPC and DSC curves are available in the ESI.†
Conflicts of interest
There are no conflicts to declareAcknowledgements
This work is supported by the National Natural Science Foundation of China (No. 52363013).References
- (a) L. K. Johnson, C. M. Killian and M. Brookhart, J. Am. Chem. Soc., 1995, 117, 6414–6415 CrossRef; (b) S. D. Ittel, L. K. Johnson and M. Brookhart, Chem. Rev., 2000, 100, 1169–1203 CrossRef PubMed.
- (a) B. L. Small, M. Brookhart and A. M. A. Bennett, J. Am. Chem. Soc., 1998, 120, 4049–4050 CrossRef; (b) G. J. P. Britovsek, V. C. Gibson, B. S. Kimberley, P. J. Maddox, S. J. McTavish, G. A. Solan, A. J. P. White and D. J. Williams, Chem. Commun., 1998, 849–850 RSC.
- (a) H. Liu, W. Zhao, J. Yu, W. Yang, X. Hao, C. Redshaw, L. Chen and W.-H. Sun, Catal. Sci. Technol., 2012, 2, 415–422 RSC; (b) L. Fan, E. Yue, S. Du, C.-Y. Guo, X. Hao and W.-H. Sun, RSC Adv., 2015, 5, 93274–93282 RSC; (c) Z. Wang, Q. B. Liu, G. A. Solan and W.-H. Sun, Coord. Chem. Rev., 2017, 350, 68–83 CrossRef; (d) I. E. Soshnikov, N. V. Semikolenova, K. P. Bryliakov, V. A. Zakharov and W.-H. Sun, Organometallics, 2015, 34, 3222–3227 CrossRef; (e) T. Wiedemann, G. Voit, A. Tchernook, P. Roesle, I. Goottker-Schnetmann and S. Mecking, J. Am. Chem. Soc., 2014, 136, 2078–2085 CrossRef PubMed; (f) D. Zhang, E. T. Nadres, M. Brookhart and O. Daugulis, Organometallics, 2013, 32, 5136–5143 CrossRef; (g) F. Wang, R. Tanaka, Z. Cai, Y. Nakayama and T. Shiono, Polymers, 2016, 8, 160 CrossRef PubMed; (h) I. D'Auria, S. Milion, T. Caruso, G. Balducci and C. Pellecchia, Polym. Chem., 2017, 8, 6443–6454 RSC.
- (a) J. Ma, C. Feng, S. Wang, K.-Q. Zhao, W.-H. Sun, C. Redshaw and G. A. Solan, Inorg. Chem. Front., 2014, 1, 14–34 RSC; (b) V. C. Gibson and G. A. Solan, Top. Organomet. Chem., 2009, 26, 107–158 CrossRef CAS PubMed; (c) Z. Wang, G. A. Solan, W. Zhang and W.-H. Sun, Coord. Chem. Rev., 2018, 363, 92–108 CrossRef CAS; (d) Z. Flisak and W.-H. Sun, ACS Catal., 2015, 5, 4713–4724 CrossRef CAS; (e) C. Bianchini, G. Giambastiani, L. Luconi and A. Meli, Coord. Chem. Rev., 2010, 254, 431–455 CrossRef CAS.
- (a) Z. Guan, P. M. Cotts, E. F. McCord and S. J. McLain, Science, 1999, 283, 2059–2062 CrossRef CAS PubMed; (b) M. D. Leatherman, S. A. Svejda, L. K. Johnson and M. Brookhart, J. Am. Chem. Soc., 2003, 125(10), 3068–3081 CrossRef CAS PubMed.
- (a) C. S. Popeney, A. L. Rheingold and Z. Guan, Organometallics, 2009, 28, 4452–4463 CrossRef CAS; (b) D. H. Camacho, E. V. Salo, Z. Guan and J. W. Ziller, Organometallics, 2005, 24, 4933–4939 CrossRef CAS; (c) D. H. Leung, J. W. Ziller and Z. Guan, J. Am. Chem. Soc., 2008, 130, 7538–7539 CrossRef CAS PubMed; (d) D. Jia, W. Zhang, W. Liu, L. Wang, C. Redshaw and W.-H. Sun, Catal. Sci. Technol., 2013, 3, 2737–2745 RSC; (e) M. Gao, S. Du, Q. Ban, Q. Xing and W.-H. Sun, J. Organomet. Chem., 2015, 798, 401–407 CrossRef CAS; (f) S. Jiang, Y. Zheng, I. V. Oleynik, Z. Yu, G. A. Solan, I. I. Oleynik, M. Liu, Y. Ma, T. Liang and W.-H. Sun, Molecules, 2023, 28, 4852 CrossRef PubMed; (g) H. Liu, W. Zhao, X. Hao, C. Redshaw, W. Huang and W.-H. Sun, Organometallics, 2011, 30, 2418–2424 CrossRef; (h) Q. Mahmood, Y. Zeng, X. Wang, Y. Sun and W.-H. Sun, Dalton Trans., 2017, 46, 6934–6947 RSC; (i) Q. Mahmood, Y. Zeng, E. Yue, G. A. Solan, T. Liang and W.-H. Sun, Polym. Chem., 2017, 8, 6416–6430 RSC; (j) R. Wu, Y. Wang, R. Zhang, C.-Y. Guo, Z. Flisak, Y. Sun and W.-H. Sun, Polymer, 2018, 153, 574–586 CrossRef; (k) M. Liu, R. Zhang, Y. Ma, M. Han, G. A. Solan, W. Yang, T. Liang and W.-H. Sun, Polym. Chem., 2022, 13, 1040–1058 RSC; (l) H. Saeed, Q. Mahmood, R. Yuan, Y. Wang, S. Zou, K. F. Tahir, Y. Ma, T. Liang and W.-H. Sun, Polym. Chem., 2024, 15, 1437–1452 RSC.
- (a) A. A. Antonov, N. V. Semikolenova, E. P. Talsi, M. A. Matsko, V. A. Zakharov and K. P. Bryliakov, J. Organomet. Chem., 2016, 822, 241–249 CrossRef; (b) W.-H. Sun, S. Song, B. Li, C. Redshaw, X. Hao, Y.-S. Li and F. Wang, Dalton Trans., 2012, 41, 11999–12010 RSC; (c) E. Yue, L. Zhang, Q. Xing, X.-P. Cao, X. Hao, C. Redshaw and W.-H. Sun, Dalton Trans., 2014, 43, 423–431 RSC; (d) M. Zada, A. Vignesh, L. Guo, R. Zhang, W. Zhang, Y. Ma, Y. Sun and W.-H. Sun, ACS Omega, 2020, 5, 10610–10625 CrossRef PubMed; (e) D. D. Sage, Q. Zhang, M. Liu, G. A. Solan, Y. Sun and W.-H. Sun, Catalysts, 2022, 12, 961 CrossRef CAS.
- (a) Z. Wang, Y. Zhang, Y. Ma, X. Hu, G. A. Solan, Y. Sun and W.-H. Sun, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3494–3505 CrossRef CAS; (b) Y. Zhang, C. Huang, X. Wang, Q. Mahmood, X. Hao, X. Hu, C.-Y. Guo, G. A. Solan and W.-H. Sun, Polym. Chem., 2017, 8, 995–1005 RSC; (c) H. Suo, Y. Zhang, Z. Ma, W. Yang, Z. Flisak, X. Hao, X. Hu and W.-H. Sun, Catal. Commun., 2017, 102, 26–30 CrossRef CAS.
- (a) J. Yu, Y. Zeng, W. Huang, X. Hao and W.-H. Sun, Dalton Trans., 2011, 40, 8436–8443 RSC; (b) X. Hou, Z. Cai, X. Chen, L. Wang, C. Redshaw and W.-H. Sun, Dalton Trans., 2012, 41, 1617–1623 RSC; (c) L. Zhang, X. Hao, W.-H. Sun and C. Redshaw, ACS Catal., 2011, 1, 1213–1220 CrossRef CAS; (d) Z. Sun, E. Yue, M. Qu, I. V. Oleynik, I. I. Oleynik, K. Li, T. Liang, W. Zhang and W.-H. Sun, Inorg. Chem. Front., 2015, 2, 223–227 RSC; (e) Y. Zeng, Q. Mahmood, X. Hao and W.-H. Sun, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 1910–1919 CrossRef; (f) J. Guo, W. Zhang, Q. Mahmood, R. Zhang, Y. Sun and W.-H. Sun, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 1269–1281 CrossRef; (g) C. Huang, Y. Zhang, T. Liang, Z. Zhao, X. Hu and W.-H. Sun, New J. Chem., 2016, 40, 9329–9336 RSC; (h) C. Huang, Y. Zeng, Z. Flisak, Z. Zhao, T. Liang and W.-H. Sun, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2071–2083 CrossRef; (i) S. Jiang, Y. Zheng, M. Liu, Z. Yu, Y. Ma, G. A. Solan, W. Zhang, T. Liang and W.-H. Sun, Organometallics, 2022, 41, 3197–3211 CrossRef.
- (a) J. Yu, X. Hu, Y. Zeng, L. Zhang, C. Ni, X. Hao and W.-H. Sun, New J. Chem., 2011, 35, 178–183 RSC; (b) X. Hou, T. Liang, W.-H. Sun, C. Redshaw and X. Chen, J. Organomet. Chem., 2012, 708, 98–105 CrossRef; (c) W. Chai, J. Yu, L. Wang, X. Hu, C. Redshaw and W.-H. Sun, Inorg. Chim. Acta, 2012, 385, 21–26 CrossRef; (d) J. Li, Q. Zhang, X. Hu, Y. Ma, G. A. Solan, Y. Sun and W.-H. Sun, Appl. Organomet. Chem., 2020, 34, e5254 CrossRef; (e) W. Lin, M. Liu, L. Xu, Y. Ma, L. Zhang, Z. Flisak, X. Hu, T. Liang and W.-H. Sun, Appl. Organomet. Chem., 2022, 36, e6596 CrossRef.
- (a) F. Huang, Z. Sun, S. Du, E. Yue, J. Ba, X. Hu, T. Liang, G. B. Galland and W.-H. Sun, Dalton Trans., 2015, 44, 14281–14292 RSC; (b) Z. Sun, F. Huang, M. Qu, E. Yue, I. V. Oleynik, I. I. Oleynik, Y. Zeng, T. Liang, K. Li, W. Zhang and W.-H. Sun, RSC Adv., 2015, 5, 77913–77921 RSC.
- R. Zhang, Z. Wang, Z. Flisak, X. Hao, Q. Liu and W.-H. Sun, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2601–2610 CrossRef.
- J. Yu, H. Liu, W. Zhang, X. Hao and W.-H. Sun, Chem. Commun., 2011, 47, 3257–3259 RSC.
- S. Meiries, K. Speck, D. B. Cordes, A. M. Z. Slawin and S. P. Nolan, Organometallics, 2013, 32, 330–339 CrossRef.
- (a) P. M. Cotts, Z. Guan, E. McCord and S. McLain, Macromolecules, 2000, 33, 6945–6952 CrossRef; (b) F. A. Kunrath, F. F. Mota, O. L. Casagrande Jr., R. S. Mauler and R. F. de Souza, Chem. Phys., 2002, 203, 2407–2411 CAS; (c) C. A. Figueira, P. S. Lopes, C. S. B. Gomes, F. Lemos and P. T. Gomes, Dalton Trans., 2018, 47, 15857–15872 RSC; (d) C. J. Stephenson, J. P. McInnis, C. Chen, M. P. Weberski Jr., A. Motta, M. Delferro and T. J. Marks, ACS Catal., 2014, 4, 999–1003 CrossRef CAS; (e) Q. Zhang, W. Lin, T. Liu, Z. Ye, T. Liang and W.-H. Sun, ACS Omega, 2021, 6, 30157–30172 CrossRef PubMed.
- (a) O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS; (b) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, A71, 3–8 CrossRef PubMed; (c) G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- (a) R. Zhang, Z. Wang, Y. Ma, G. A. Solan, Y. Sun and W.-H. Sun, Dalton Trans., 2019, 48, 1878–1891 RSC; (b) X. Wang, L. Fan, Y. Ma, C. Guo, G. A. Solan, Y. Sun and W.-H. Sun, Polym. Chem., 2017, 8, 2785–2795 RSC.
- L. Guo, D. Zhu, W. Zhang, M. Zada, G. A. Solan, X. Hao and W.-H. Sun, Eur. Polym. J., 2018, 107, 315–328 CrossRef.
- (a) W. Yang, J. Yi and W.-H. Sun, Macromol. Chem. Phys., 2015, 216, 1125–1133 CrossRef; (b) T. Zhang, D. Guo, S. Jie, W.-H. Sun, T. Li and X. Yang, Polym. Chem., 2004, 42, 4765–4774 CrossRef.
- S. Zai, H. Gao, Z. Huang, H. Hu, H. Wu and Q. Wu, ACS Catal., 2012, 2, 433–440 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford, CT, 2013.
- Q. Zhang, Z. Li, M. Han, J. Xiang, G. A. Solan, Y. Ma, T. Liang and W.-H. Sun, Catal. Sci. Technol., 2021, 11, 656–670 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: 1H/13C NMR spectra of L1–L5; GPC curves of the polyethylene produced using Ni5/MAO at different reaction conditions; GPC curves of the polyethylene produced using Ni1–Ni10/MAO; 1H/13C NMR spectra of the polyethylene sample produced using Ni5/MAO at 30 °C (entry 5, Table 6); crystal data and structure refinement for Ni3 and Ni4; coordinates of the optimized structures of Ni3 and Ni5. CCDC 2302657 (Ni3) and 2302658 (Ni4). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt02159b |
| This journal is © The Royal Society of Chemistry 2024 |