
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent advances in photocatalyst sheet development and challenges for cost-effective solar hydrogen production
Swarnava
Nandy
 a,
Takashi
Hisatomi
ab,
Tsuyoshi
Takata
a,
Tohru
Setoyama
c and
Kazunari
Domen
*ade
a,
Takashi
Hisatomi
ab,
Tsuyoshi
Takata
a,
Tohru
Setoyama
c and
Kazunari
Domen
*ade
aResearch Initiative for Supra-Materials (RISM), Interdisciplinary Cluster for Cutting Edge Research, Shinshu University, 4-17-1 Wakasato, Nagano 380-8553, Japan. E-mail: domen@shinshu-u.ac.jp
bPrecursory Research for Embryonic Science and Technology, Japan Science and Technology Agency, 4-17-1 Wakasato, Nagano-shi, Nagano 380-8553, Japan
cMitsubishi Chemical Group Science and Technology Research Center, 1000 Kamoshida, Aoba, Yokohama 227-8502, Japan
dOffice of University Professors, The University of Tokyo, 2-11-16 Yayoi, Tokyo 113-8656, Japan
eDepartment of Chemistry, Kyung Hee University, Seoul 130-701, Republic of Korea
First published on 14th September 2023
Abstract
Solar hydrogen production by photocatalytic water splitting is studied to solve energy and environmental problems. It is estimated that practical implementation of this technology will require a photocatalytic water splitting system that is not only highly efficient in solar-to-hydrogen energy conversion but also cheap and scalable. Particulate photocatalyst sheets based on immobilized particulate semiconductors are a promising approach to meet these requirements owing to their scalability without sacrificing their intrinsic water splitting activity. Nevertheless, existing sheet systems have not reached the targeted solar-to-hydrogen energy conversion efficiency yet. This is because the quality of narrow-bandgap photocatalyst materials has not been sufficiently improved, and also because methods for activating such photocatalysts in warm water under ambient pressure, which is assumed to be a practical outdoor operating condition, while suppressing reverse reactions have not been sufficiently developed. This perspective describes recent advancements in photocatalyst sheets and panel reactor systems intended for practical implementation of solar hydrogen production via the overall water splitting reaction, and discusses issues regarding the development of photocatalytic systems that can produce solar hydrogen with satisfactory performance.
 Swarnava Nandy | Swarnava Nandy received masters (2014) in Chemistry from Indian Institute of Technology Hyderabad and his PhD (2017) in Chemical System Engineering from The University of Tokyo under the supervision of Prof. Kazunari Domen. Before returning to Japan in 2020 he worked at Swiss Federal Institute of Technology Lausanne (2017–2019) as postdoctoral researcher. Since November 2022, he was promoted to project assistant professor in Shinshu University. His research interests include visible-light-driven photocatalytic materials development to produce renewable hydrogen by water splitting reaction under sunlight. |
 Takashi Hisatomi | Takashi Hisatomi received his PhD in engineering from The University of Tokyo in March 2010. He worked as a postdoctoral fellow in École Polytechnique Fédérale de Lausanne from April 2010 to March 2012. He moved to the University of Tokyo in April 2012 as a postdoctoral fellow and acquired an assistant professor position in August 2012. He moved to Shinshu University as an associate professor in April 2018 and was promoted to professor in April 2023. His major research interests include semiconductor photocatalysts for overall water splitting, kinetics of photoexcited carriers in semiconductors, and reaction systems for renewable solar hydrogen production. |
 Tsuyoshi Takata | Dr. Tsuyoshi Takata received his PhD from Tokyo Institute of Technology in 2000 under Prof. Kazunari Domen's supervision. Dr Takata had worked as an Assistant Professor at Tokyo Institute of Technology until 2004, and as an Assistant Professor and Lecturer at the University of Tokyo until 2011. Then he had worked in National Institute for Materials Science (NIMS) as a Special Researcher until 2016. Then he had worked as a senior researcher at the University of Tokyo until 2018. He has currently been an appointed professor in Shinshu University. His research area is photocatalytic water splitting with a focus on new material design and synthesis. |
 Tohru Setoyama | Tohru Setoyama is a Fellow and an Executive Officer at Mitsubishi Chemical Corporation. He received his PhD in Chemistry from The University of Tokyo. He has significantly contributed to several commercialized catalytic processes and to the launch of several businesses related to functional inorganic materials. He is regarded as one of the key persons to bridge between academia and industry in Japan. Currently, he is a Project Leader of ‘Artificial Photosynthesis Chemical Research Process (ARPChem)’ Project as a part of Green Innovation Program supported by New Energy Development Organization (NEDO). |
 Kazunari Domen | Kazunari Domen is a special contract professor at Shinshu University and university professor at the University of Tokyo, Japan. He received his PhD in science from The University of Tokyo in 1982. He joined the Tokyo Institute of Technology in 1982 as an assistant professor and was subsequently promoted to associate professor in 1990 and professor in 1996. He moved to the University of Tokyo as a professor in 2004 and was appointed by Shinshu University as a special contract professor in 2017. His research interests include heterogeneous catalysis and materials chemistry, with a particular focus on photocatalytic water splitting for solar hydrogen production. |
1. Introduction
Photocatalytic overall water splitting (OWS) has gained much attention because it has the potential to deliver renewable hydrogen from water under sunlight.1–3 Many particulate semiconductor materials have been examined as photocatalysts to drive the OWS reaction using the energy of light as the only energy input. SrTiO3 is one of the earliest examples of splitting water into hydrogen and oxygen under ultraviolet light.4 However, ultraviolet light accounts for only 5% of solar energy, and the maximum achievable solar-to-hydrogen energy conversion efficiency (STH efficiency) using wide-bandgap photocatalysts that are active only under UV light is too low to aim for practical implementation of renewable hydrogen production by photocatalytic OWS, where a STH efficiency of at least 5% is probably required.5 It is also important to develop scalable reaction systems. Photocatalytic OWS has generally been studied using suspension of photocatalysts. However, such suspensions are not practical on a large scale due to handling difficulties, such as maintaining a uniform dispersion, recovery of spent photocatalysts, and construction of large photoreactors. As an alternative approach, particulate photocatalyst sheets have emerged. Photocatalyst sheets are made of photocatalyst particles fixed on substrates and can efficiently drive OWS by both one-step and two-step excitation schemes without the need for stirring, and can be easily scaled up.6 In the case of two-step photo-excitation, the sheet structure can allow more efficient charge transfer between hydrogen and oxygen evolution photocatalysts (HEP and OEP, respectively) than the suspension system, regardless of the pH of the reaction solution. Furthermore, in this approach, the applicability of the materials is not limited by the compatibility of the photocatalysts with redox mediators (in terms of activity and stability) or by the sign of the surface charges. Nevertheless, existing sheet systems have not reached the target STH efficiency due to the inability of wide-bandgap photocatalysts to utilize visible light, or the low reactivity of narrow-bandgap photocatalysts in the photoexcited state. Effective strategies for designing highly active sheet systems and associated challenges need to be addressed so that the STH efficiency can reach commercially viable levels.In this article, we discuss advancements in the development of photocatalytic materials and their processing into particulate photocatalyst sheets for the OWS reaction. The problem of reverse reactions that would occur in the photocatalytic OWS reaction under atmospheric pressure, which is assumed in practical large-scale applications, is discussed. In relation to practicability, an estimate of the allowable cost for a system that produces solar hydrogen at a certain price by photocatalytic OWS is also given. Directions for design and development of particulate photocatalyst sheets that may lead to the achievement the target STH efficiency are then suggested.
2. Working mechanism for water-splitting photocatalysts and their sheets
The kinetic and thermodynamic aspects of the water splitting reaction on particulate photocatalysts have been extensively studied.3,7,8 Briefly, when a photocatalyst absorbs a photon that has an energy greater than the bandgap of the photocatalyst, an electron is excited from the valence band to the conduction band, and a positive hole is left behind in the valence band (Fig. 1A). These excited charge carriers generally relax to the conduction band minimum (CBM) and the valence band maximum (VBM), respectively, and migrate to surface reaction sites, where electrons and holes reduce and oxidize water into H2 and O2, respectively. The potential of the valence band maximum and the conduction band minimum influences the driving force available for the surface chemical reactions. Thermodynamically, a semiconductor can split water into H2 and O2 by one-step excitation when the potentials of the CBM and VBM are more negative and more positive than the water reduction (H+/H2: −0.41 V vs. NHE at pH 7) and oxidation (O2/H2O: +0.82 V vs. NHE at pH 7) potentials, respectively. Also, two kinds of photocatalysts, each for hydrogen and oxygen evolution, can be combined. In such two-step excitation (also known as Z-scheme) OWS systems, electrons from the CBM for an oxygen evolution photocatalyst (OEP) and holes from the VBM for a hydrogen evolution photocatalyst (HEP) recombine through a soluble or solid electron mediator (Fig. 1B). Notably, photocatalysts are applicable to Z-scheme OWS (ZOWS) as far as they are active in the hydrogen or oxygen evolution reactions. Therefore, various narrow-bandgap particulate photocatalysts can be paired to achieve ZOWS, depending on their half-reaction activity.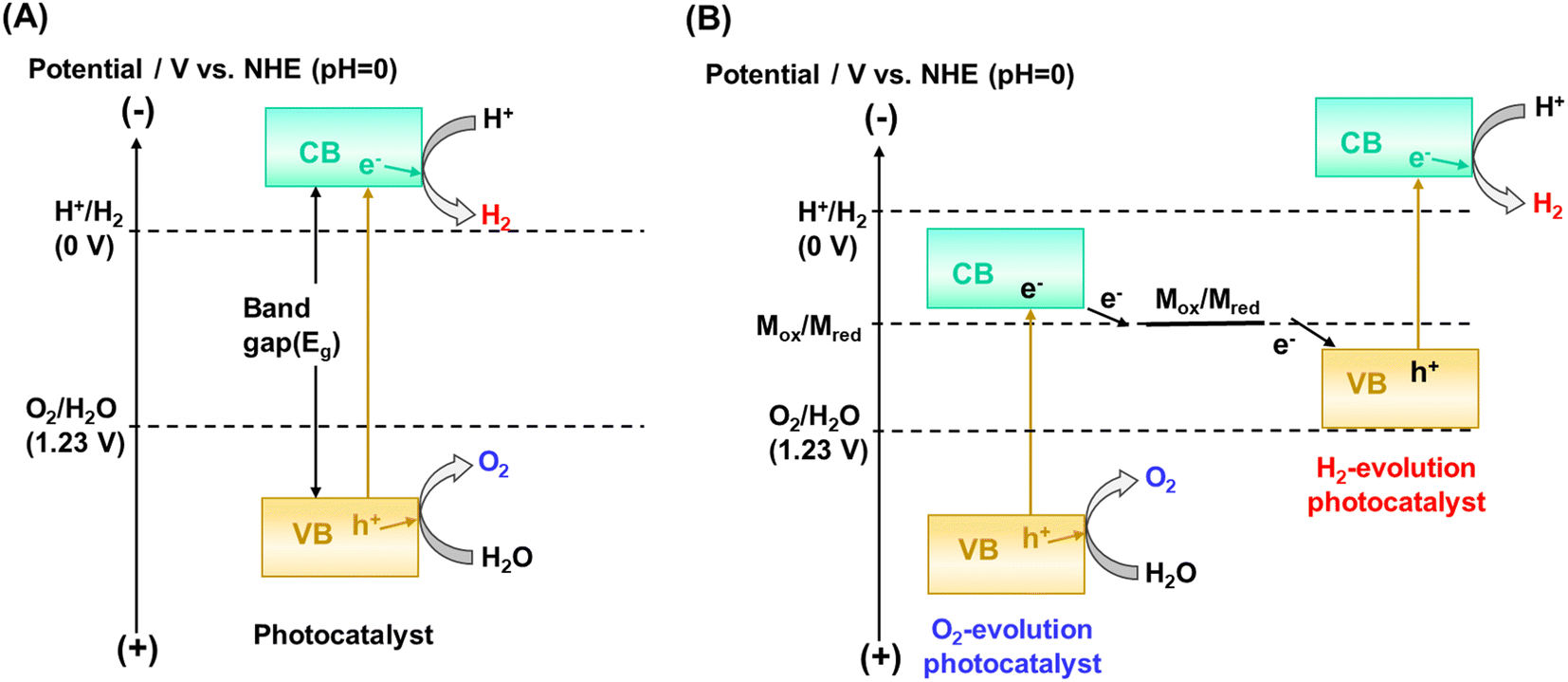 | ||
| Fig. 1 Schematic of charge transfer mechanism during OWS involving (A) one-step and (B) two-step photoexcitation. NHE, CB and VB represent the normal hydrogen electrode, conduction band and valence band, respectively. In (B) Mox and Mred represent the oxidized and reduced species of a reversible redox couple. | ||
The one-step excitation system is simple and can be driven by a photocatalyst sheet with photocatalytic particles immobilized on a substrate (Fig. 2A).6 To drive the reaction, however, it is important to facilitate the penetration of water into the particle layer and the detachment of products from it. When a photocatalyst sheet is used in ZOWS, HEPs and OEPs are well mixed and immobilized with conductive materials in close proximity to each other. Such a structure can allow efficient charge transfer between HEPs and OEPs while suppressing the solution resistance and H+/OH− concentration overpotentials, which are often issues in (photo)electrochemical systems having spatially separated hydrogen- and oxygen-evolving electrodes.9 Therefore, in principle, photocatalyst sheets can be scaled up without losing their inherent activity. In photocatalyst sheets, p-type and n-type semiconductors are commonly employed as the HEP and OEP, respectively, to achieve ZOWS efficiently, because photoexcited charge carrier migration in the photocatalyst sheet resembles that in photoelectrochemical cells based on a short-circuited photocathode and photoanode, with the HEP and OEP corresponding to the photocathode and photoanode, respectively. The working potential for the sheet is estimated from the intersection of the current–potential (i–E) curves for the photoanode and photocathode. As such, the performance of particulate photocatalysts as the photoelectrode is crucial in determining their utility in a photocatalyst sheet system.
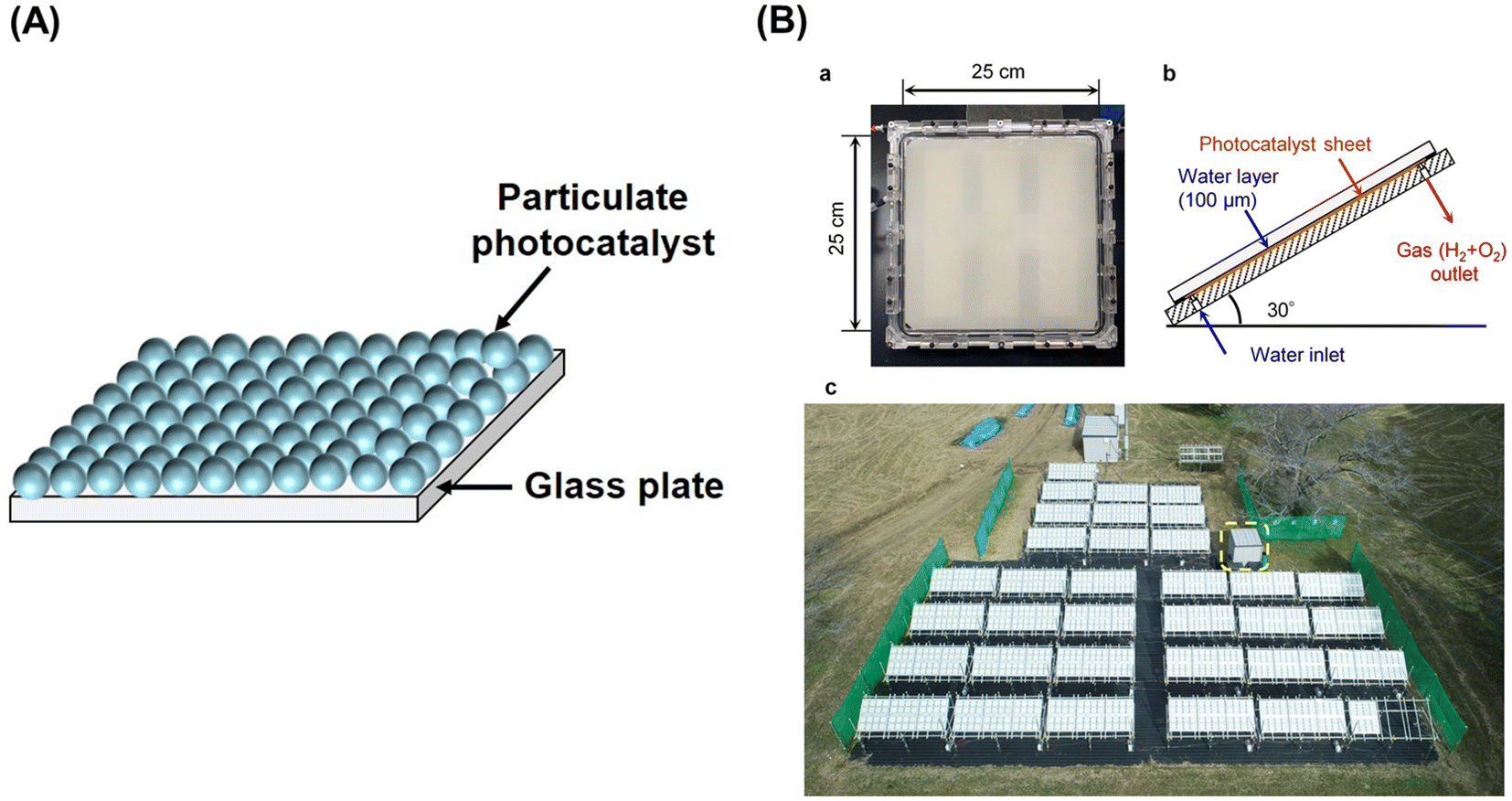 | ||
| Fig. 2 (A) Schematic diagram of one-step photoexcitation based photocatalyst sheet, (B) 100 m2-scale photocatalyst panel reactor: (a) photograph of 625 cm2 panel reactor unit, (b) schematic side view of panel reactor unit, and (c) top view of 100 m2 solar hydrogen production system consisting of 1600 panel reactor units. Reproduced from ref. 16 with permission from Springer Nature, copyright 2021. | ||
3. Cost target and necessary performance
In 2017, the Ministry of Economy, Trade and Industry (METI) of Japan set a cost target for hydrogen of 30 JPY Nm−3 (equivalent to 2.9 USD kg−1 if 1 USD equals 115 JPY) to be reached by 2030.1 In 2021, the Hydrogen and Fuel Cell Technologies Office (HFTO) of the United States Department of Energy (DOE) declared more radical targets: producing hydrogen at 2 and 1 USD kg−1 by 2026 and 2031, respectively, via renewable resources, so that hydrogen would become cost-competitive with fossil fuels.10 The cost of hydrogen produced by renewable energy-power OWS is dependent on the STH efficiency, service lifetime, and capital and running costs for the systems. These are subject to many uncertain factors that determine the market cost of H2. Therefore, it is difficult to estimate levelized costs of H2 produced by photocatalytic water splitting. However, calculations can be made for how low the capital and running costs of a system should be if certain values of STH efficiency, hydrogen and oxygen prices, and service lifetime are assumed.The allowable system and process cost for photocatalytic water splitting has been estimated to be approximately 100 USD m−2 given a STH efficiency of 10%, a hydrogen price of 3.5 USD kg−1, an oxygen price of 0.1 USD kg−1, a lifetime of 10 years, and an annual depreciation rate of 4%. The STH efficiency depends on both the wavelength of the light used by photocatalysts in the OWS reaction and the apparent quantum yield (AQY).1 Considering a constant AQY of unity, the absorption edge wavelength for the photocatalyst material must be longer than 526 nm, because below this wavelength there are too few photons to achieve the STH efficiency target of 10%. This STH efficiency requires an OWS system with an AQY of 60% in response to visible light up to 600 nm. Note that the hydrogen price assumed above is higher than the target cost. Of course, further reductions in costs, improvements to STH efficiency, and extension of the system service life are desirable. When comparing suspension and sheet systems, it appears that the sheet system is technically less difficult and costly to build and operate than the suspension system. Considering the lack of large-scale and long-term experimental demonstrations for the latter. In terms of lifespan, stirring causes long-term mechanical damage to the powder photocatalysts in the suspension system, as demonstrated with a Rh2−yCryO3/GaN:ZnO system.11 The sheet system also has an advantage that it does not require stirring because the photocatalysts are immobilised, but the influence of mass transport is still negligible.
As an alternative technology for solar hydrogen production from water, photovoltaic-assisted electrolysis (PV-E) systems may be considered. PV-E systems can reach STH efficiencies of 30% on a laboratory scale. However, extending these systems to larger scales is expensive due to high capital and balance-of-system expenses.12 It has been estimated that the balance-of-system expense alone would reach US$100 m−2 in PV-E systems.13 which highlights the need for radical innovations in system construction and operation.
4. Progress and challenges for one-step excitation systems
Al-doped SrTiO3 (SrTiO3:Al) with high crystallinity and suppressed particle growth has been synthesized by the flux method, and exhibited an AQY of 56% at 365 nm in the OWS when loaded with a RhCrOx cocatalyst.14 Subsequently, facet-selective photodeposition of Rh/Cr2O3 and CoOOH cocatalysts onto the SrTiO3:Al photocatalyst was shown to promote charge separation and led to a benchmark AQY of 91.6% at 365 nm.15 Fabrication and operation of photocatalyst sheets and panel reactors have been studied using the SrTiO3:Al photocatalyst. The photocatalyst sheet exhibited an OWS activity comparable to that of the powder suspension when silica nanoparticles were used as an inorganic binder.9 Recovery of hydrogen produced from water under natural sunlight was demonstrated using a system consisting of a 100 m2-scale panel reactor containing photocatalyst sheets based on tailored SrTiO3:Al, which exhibited a moderate AQY of approximately 50% (at 365 nm) but higher durability, and a gas-separation membrane unit.16 The modified SrTiO3:Al photocatalyst was processed into photocatalyst sheets by spraying a slurry of the photocatalyst and hydrophilic silica nanopowder onto frosted glass sheets with an active area of 625 cm2 (Fig. 2). The sheets were housed in panel reactors and connected to cover an area of 100 m2. Notably, in the panel reactor, sheets were placed with a gap of 0.1 mm between them and the window, because the OWS performance remained unaffected upon changing the water depth from the order of centimetres to 0.1 mm.The 100 m2-scale arrayed photocatalyst panel reactor split water for a period of eight months under outdoor conditions (Fig. 2B). In other trials using a small panel reactor, 80% of the peak activity was maintained after 1600 h of constant irradiation by simulated sunlight. However, the STH efficiency of the outdoor panel reactor decreased irreversibly during the winter months, because the layer of SrTiO3:Al particles peeled away from the support (due to repeated freezing and melting of water) and particles that settled at the bottom failed to absorb sunlight effectively. Thus, it is necessary to improve the chemical and mechanical stability of the panel reactors to extend their usage timescale.
The STH efficiency achieved with the above panel reactor based on SrTiO3:Al sheets was at most 0.65% because the visible part of the spectrum was not utilized. Even if UV light at wavelengths up to 400 nm was utilized for OWS at an AQY of unity, the STH efficiency would be only 1.3%. Therefore, it is essential to develop photocatalyst sheets with visible–light activity. A solid solution of GaN and ZnO (denoted as GaN:ZnO) loaded with a RhCrOx cocatalyst exhibited a maximum AQY of 5.1% at 410 nm in the OWS reaction and maintained this activity for three months.17 The RhCrOx-loaded GaN:ZnO has been processed into photocatalyst sheets,18 which exhibited an OWS activity comparable to that for a suspension when hydrophilic silica particles with appropriate sizes were incorporated into the particle layer, whereas a sheet made solely from cocatalyst-loaded GaN:ZnO exhibited lower activity. This is probably because the inclusion of such silica particles promoted the permeation of water into the photocatalyst particle layer and the release of product gases from the sheet. However, there is little scientific understanding of this, and further research is needed to clarify the extent to which the hydrophilicity of the sheet affects the performance. Recently, there have been some reports on methods for improving the photocatalytic properties of GaN:ZnO. For instance, deposition of Al2O3 on Rh-loaded GaN:ZnO by atomic layer deposition greatly suppressed the water-forming backward reaction without blocking the active sites, thus improving the AQY from 0.3% to 7.1% at 420 nm in OWS.19 Moreover, highly crystalline GaN:ZnO with a narrow bandgap energy of 2.3 eV was recently synthesized using an ammonium salt as a nitriding reagent in a sealed evacuated tube.20 This method prevented loss of ZnO during nitridation of the starting materials to form GaN:ZnO. Some non-oxide photocatalysts with bandgap energy smaller than 3.0 eV, such as Y2Ti2S2O7, SrTaO2N and TaON having a bandgap energy of 1.9, 2.1 and 2.5 eV, respectively, are active and stable in the OWS reaction via one-step photoexcitation.21–23 Such photocatalysts are expected to be applied to photocatalyst sheets for OWS given that the activity is significantly improved.
5. Progress and challenges for two-step excitation systems
In 2004, a mixed suspension of Rh-doped SrTiO3 and BiVO4 particles was reported to be active in ZOWS under visible light, where the former acted as a HEP, and the latter as an OEP.24 The photocatalytic properties of the individual photocatalysts and the mechanism for charge transfer between them have been extensively investigated to improve the activity of the ZOWS system.25 Photocatalytic sheets for ZOWS were derived during the course of the investigation into the electron transfer process from the HEP to OEP and the refinement in the photocatalyst preparation.26 A photocatalyst sheet composed of Rh, La co-doped SrTiO3 (SrTiO3:Rh,La), Mo-doped BiVO4 (BiVO4:Mo) and Au as the HEP, OEP and conductor layer, respectively, demonstrated a STH efficiency of 1.1% at a background pressure of 10 kPa and a temperature of 331 K upon loading with Cr2O3/Ru cocatalysts.27 Brief annealing of the photocatalyst sheet improved the ZOWS activity because the contact resistance between the photocatalysts and Au was reduced. This STH efficiency is one-order of magnitude higher than that achieved with the corresponding suspension system, thus highlighting the advantages of the sheet system. However, the STH efficiency decreased at elevated pressure due to oxygen reduction reaction (ORR) at the exposed Au surface. To suppress the reverse reaction, Au was replaced with C, which has a similar work function but is less active toward ORR.28 The resulting Cr2O3/Ru-modified SrTiO3:Rh,La/C/BiVO4:Mo sheet yielded a STH efficiency of 1.0% at 91 kPa and a temperature of 333 K. The working state of this sheet was simulated using a semiconductor device simulator. Under photoexcitation, electrons at the H2O/SrTiO3:Rh,La interface are transferred to the water to evolve hydrogen under the influence of an electric field generated in the depletion layer, while holes migrate to the C layer across the Schottky barrier via concentration diffusion (Fig. 3). For BiVO4:Mo, the band profile remains virtually flat due to the low majority-carrier concentration, allowing electrons to diffuse to the C layer.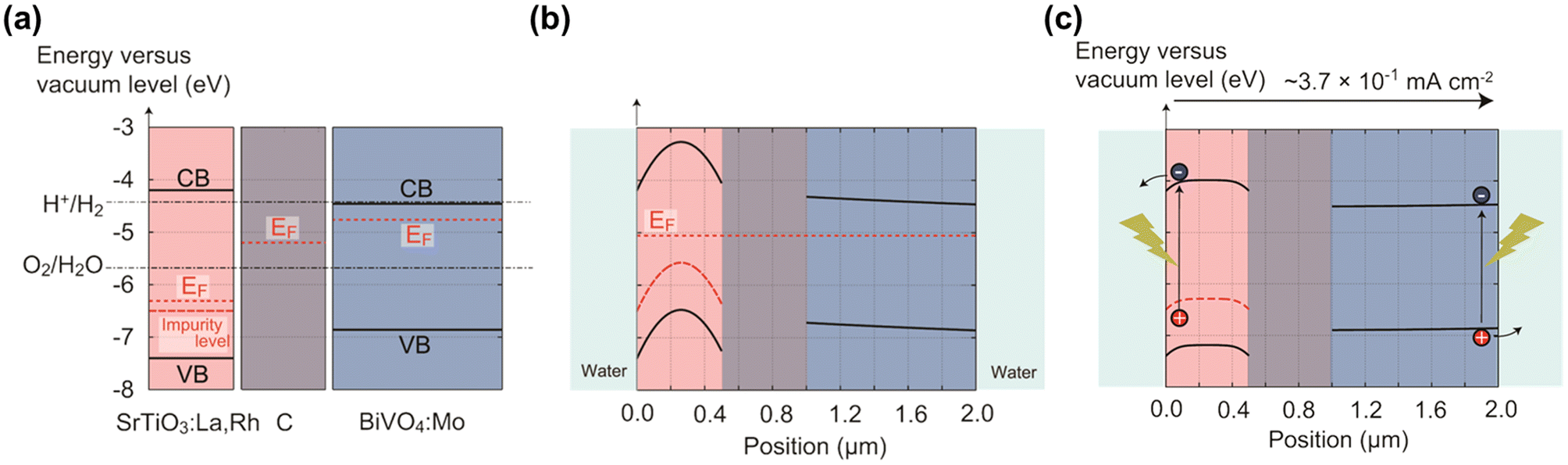 | ||
| Fig. 3 Band diagrams for SrTiO3:La,Rh/C/BiVO4:Mo sheet: (a) before contact, (b) under darkness after contact, and (c) under illumination. Reproduced from ref. 28 with permission from American Chemical Society, copyright 2017. | ||
For practical large-scale application, it is essential to avoid the use of costly particle transfer methods that require vacuum processes and to simplify the overall manufacturing process. The Z-schematic photocatalyst sheet was also fabricated by screen printing, which does not require vacuum processes as in particle transfer, using transparent indium-doped tin oxide nanoparticles (np-ITO) as a mediator.29 The STH achieved with np-ITO was 0.4% at near ambient pressure. However, further development of cheaper conductive materials and simpler manufacturing processes is desirable.
Because the absorption edge wavelengths of SrTiO3:Rh,La and BiVO4:Mo are relatively short, attempts have been made to replace these materials with narrow bandgap photocatalysts in the sheet system. Table 1 summarizes the OWS performance of particulate photocatalyst sheets employing various oxide and non-oxide semiconductors along with their detailed measurement conditions. Non-oxide photocatalysts with a longer absorption edge than SrTiO3:Rh,La have been applied as the HEP for sheet systems.30,31 In particular, La5Ti2Cu0.9Ag0.1O7S5 (LTCA) is a unique oxysulfide photocatalyst exhibiting a positive onset potential (+0.9 V vs. RHE) when used as a photocathode for photoelectrochemical hydrogen evolution.32 Cationic doping with lower-valent species at Ti4+ sites in LTCA can induce p-type semiconducting characteristics due to a positive shift of the Fermi level, thus enhancing the photocathodic current. These features make LTCA very useful as a HEP for photocatalytic sheet systems. Other narrow bandgap materials such as LaMg1/3Ta2/3O2N, and (ZnSe)0.5(CuGa2.5Se4.25)0.5 have also been investigated in sheet form as HEP in combination with BiVO4:Mo as OEP.30,33 However, the performance of LaMg1/3Ta2/3O2N in sheet form was limited due to the n-type conductivity, while most chalcogenides were not suitable for sheet application due to the negative photocurrent onset potential when evaluated as a photocathode.
| Entry | Cocatalyst/HEP (absorption edge) | Cocatalyst/OEP (absorption edge) | Mediator | AQY (wavelength/nm) | STH (pressure, temperature) | Ref. |
|---|---|---|---|---|---|---|
| a n.r.: not reported; BiVO4:Mo synthesis route: (s–l): solid–liquid reaction; (h): hydrothermal; reaction solution pH: without adjustment as that of pure water except for entry 8 where pH was adjusted to 11. | ||||||
| 1 | Cr2O3/Ru/SrTiO3:Rh,La (520 nm) | BiVO4:Mo (s–l) (520 nm) | Au | 33% (419 nm) | 1.1% (10 kPa, 331 K) | 27 |
| 2 | Cr2O3/Ru/SrTiO3:Rh,La (520 nm) | BiVO4:Mo (s–l) (520 nm) | C | 26% (419 nm) | 1.2% (10 kPa, 331 K) | 28 |
| 3 | Cr2O3/Ru/SrTiO3:Rh,La (520 nm) | BiVO4:Mo (s–l) (520 nm) | C | 19% (419 nm) | 1.0% (91 kPa, 331 K) | 28 |
| 4 | Cr2O3/Ru/SrTiO3:Rh,La (520 nm) | BiVO4:Mo (s–l) (520 nm) | np-ITO | 10.2% (420 nm) | 0.4% (91 kPa, 333 K) | 29 |
| 5 | Cr2O3/Ru/SrTiO3:Rh,La (520 nm) | BiVO4:Mo (s–l) (520 nm) | Sputter-ITO | n.r. | 0.8% (91 kPa, 333 K) | 29 |
| 6 | Pt/TiO2-CdS-(ZnSe)0.5(CuGa2.5Se4.25)0.5 (725 nm) | BiVO4:Mo (s–l) (520 nm) | Au | 1.5% (420 nm) | n.r. | 30 |
| 7 | RhCrOx/LaMg1/3Ta2/3O2N (600 nm) | BiVO4:Mo (s–l) (520 nm) | Au and RGO | 0.25% (418 nm) | 0.0035% | 33 |
| 8 | Cr2O3/Rh/LTCA:Ga (700 nm) | CoOx/LaTiO2N (600 nm) | Au | 0.04% (420 nm) | n.r. | 38 |
| 9 | Cr2O3/Rh/LTCA:Ga (700 nm) | CoOx/BiVO4:Mo (s–l) (520 nm) | Au | 11.8% (420 nm) | 0.4% (4 kPa, 301 K) | 31 |
| 10 | Cr2O3/Rh/LTCA:Mg,Al (700 nm) | CoOx/BiVO4:Mo(h) (520 nm) | Au | 16.3% (420 nm) | 0.67% (4 kPa, 301 K) | 34 |
| 11 | SiO2/Cr2O3/Rh/LTCA:Mg,Al (700 nm) | SiO2/CoOx/BiVO4:Mo(h) (520 nm) | Au | 9.7% (420 nm) | 0.41% (90 kPa, 333 K) | 34 |
Photocatalyst sheets composed of Mg,Al-doped LTCA (LTCA:Mg,Al) as the HEP and BiVO4:Mo as the OEP achieved an AQY of 16.3% (420 nm) and a STH efficiency of 0.67% under reduced pressure upon depositing a CoOx/Cr2O3/Rh cocatalyst.34 A schematic of the LTCA:Mg,Al/Au/BiVO4:Mo sheet fabrication procedure is presented in Fig. 4. Photocatalyst sheets made of short-circuited LTCA:Mg,Al/Au photocathode and BiVO4:Mo/Ti photoanode placed side by side, which serve as the HEP and OEP, respectively, exhibits lower ZOWS activity due to the solution resistance and the pH gradient. Similar phenomena were also observed in the SrTiO3:Rh,La and BiVO4:Mo systems earlier.6,28 The ZOWS activity dropped steeply with increasing Ar background pressure for both mixed and parallel types of photocatalyst sheets. This is attributed to backward ORR occurring on the bare surface of the LTCA particles and the Au layer, which was verified by investigating the (photo)electrochemical ORR activity of the relevant samples with various surface modifications (Fig. 5A). Importantly, the oxysulfide surface was not covered with a photodeposited Cr2O3 layer, which acts to suppress the ORR, while the Rh cocatalyst surface was covered. Therefore, it is essential to coat the oxysulfide surface with a material that can suppress the ORR while preserving the hydrogen evolution reaction.
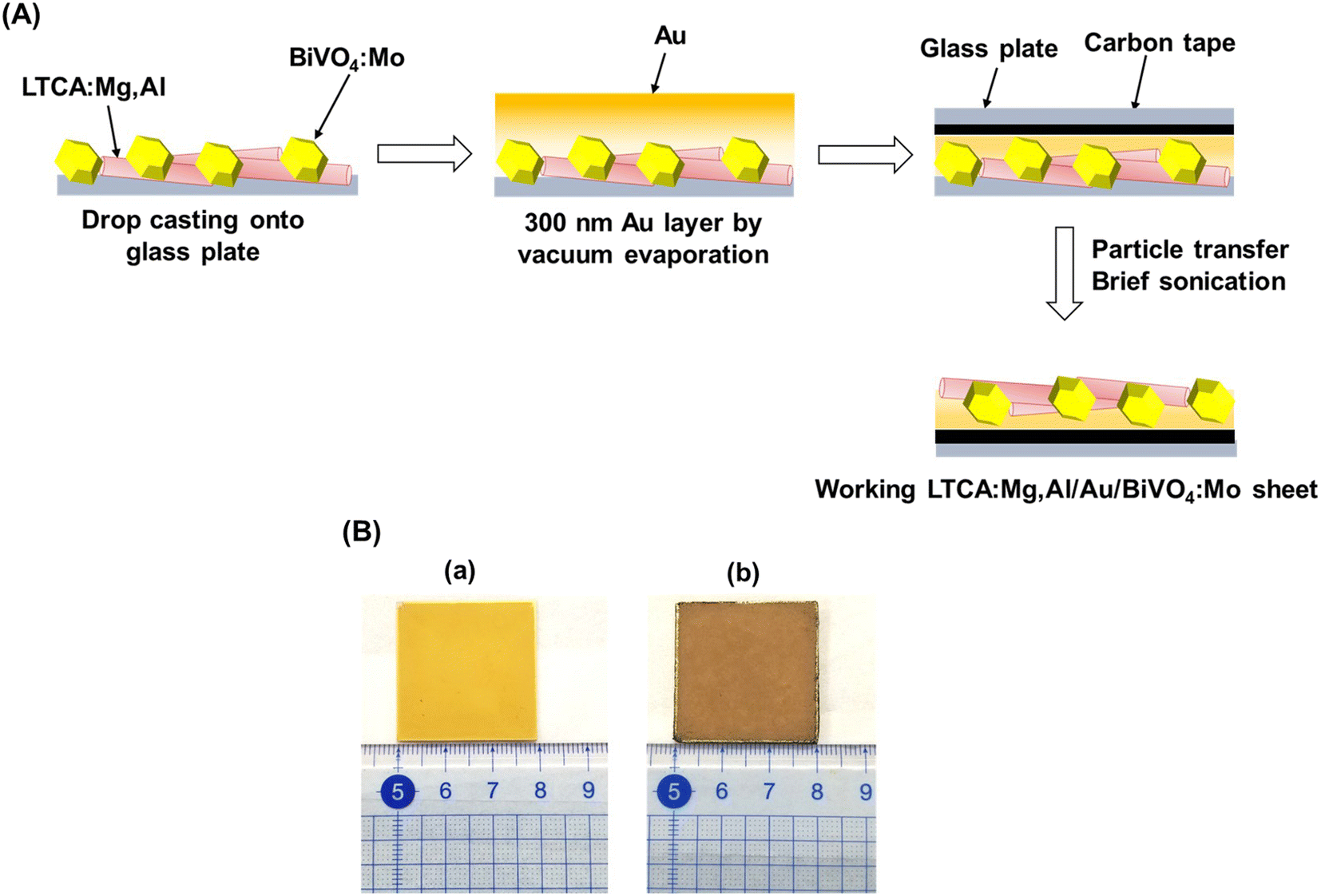 | ||
| Fig. 4 (A) Schematic illustration of the preparation procedure for photocatalyst sheet composed of LTCA:Mg,Al and BiVO4:Mo. (B) Photograph of 3 × 3 cm2 LTCA:Mg,Al/Au/BiVO4:Mo photocatalyst sheet (a) before and (b) after particle transfer. Panel (A) was reproduced from ref. 34 with permission from Elsevier Inc., copyright 2023. | ||
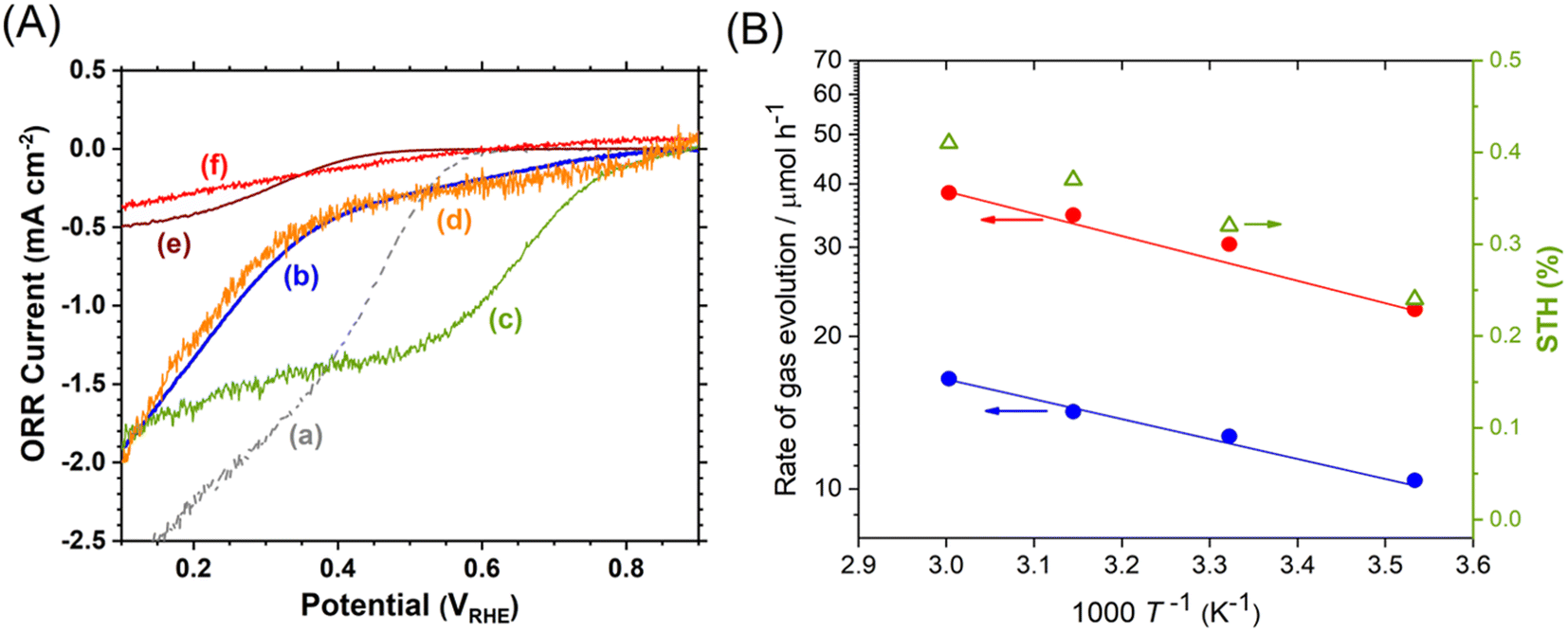 | ||
| Fig. 5 (A) Oxygen reduction current density for (a) Au/Ti, (b) LTCA:Mg,Al/Au, (c) Rh/LTCA:Mg,Al/Au, (d) Cr2O3/Rh/LTCA:Mg,Al/Au, (e) TiO2/Cr2O3/Rh/LTCA:Mg,Al/Au and (f) SiO2/Cr2O3/Rh/LTCA:Mg,Al/Au photoelectrodes. (B) Temperature (T) dependence of ZOWS activity and STH efficiency for SiO2-modified LTCA:Mg,Al/Au/BiVO4:Mo photocatalyst sheet at atmospheric pressure under simulated sunlight. Reproduced from ref. 34 with permission from Elsevier Inc., copyright 2023. | ||
A CoOx/Cr2O3/Rh-modified LTCA:Mg,Al/Au/BiVO4:Mo photocatalyst sheet coated with an amorphous SiO2 layer 5–8 nm in thickness was found to suppress the ORR and to largely maintain the ZOWS activity at elevated pressures and temperatures. The resulting SiO2-coated sheet yielded a STH efficiency of 0.41% at a pressure of 90 kPa and a temperature of 333 K (Fig. 5B). In contrast, a photocatalyst sheet coated with an amorphous TiO2 layer, which is also known to suppress the ORR, did not operate at temperatures above 318 K owing to condensation restricting mass transport across the TiO2 layer. These results show the applicability of non-oxide photocatalysts that are prone to backward reactions under field conditions when appropriate surface modifications are applied. Note that some non-oxide photocatalysts modified with unprotected noble metal cocatalysts have already been reported to achieve STH efficiencies exceeding 1% in ZOWS in the form of a suspension.35,36 Although the applicability of such systems in the form of photocatalyst sheets is interesting, in our experience, such systems will inevitably lose their activity at ambient pressure due to backward reactions. For practical use, it is essential to measure the pressure dependence of ZOWS activity.
Thermodynamically, the LTCA:Mg,Al/Au/BiVO4:Mo system could yield a STH efficiency of approximately 9%, while the presently-achieved efficiency is at most 0.67%. The ZOWS activity could be further improved by refining the synthesis and modification methods for the HEP and OEP. Li and co-workers invented the use of hydrothermally-synthesized BiVO4 exposing anisotropic facets that collect electrons and holes, respectively, under photoexcitation as an OEP.37 The BiVO4 modified with Ir-CoFeOx cocatalysts yielded a STH of 0.6% when combined with ZrO2-modified TaON as the HEP in an aqueous K4Fe(CN)6 solution, which was the highest reported value using suspension systems. The use of such hydrothermally-synthesized BiVO4 was shown to improve the STH efficiency of the LTCA:Mg,Al/Au/BiVO4:Mo sheet system compare to the one using BiVO4:Mo synthesized by solid–liquid reaction, as was demonstrated in our recent work.34
There is also interest in replacing BiVO4 with non-oxide photocatalysts with a narrower bandgap to extend the wavelength range of light available for ZOWS and expand the types of photocatalysts that can be applied as OEPs. A photocatalyst sheet based on Ga-doped LTCA (LTCA:Ga) and LaTiO2N embedded onto Au layer was investigated, but the resultant LTCA:Ga/Au/LaTiO2N sheet exhibited negligible ZOWS activity.38 The Schottky junction at the interface between LTON and Au obviously suppressed the injection of electrons from the OEP to the Au. However, deposition of CoOx onto the LaTiO2N was found to weaken the Schottky barrier and promoted electron injection from LaTiO2N to Au, allowing ZOWS to occur, although the AQY was estimated to be only about 0.04% at 420 nm. It is noteworthy that in a recent study, narrow-bandgap GaN:ZnO synthesized in a sealed evacuated tube and loaded with IrO2 (IrO2/GaN:ZnO) exhibited a relatively negative photoanodic current onset (+0.3 V vs. RHE) compared to CoOx/LaTiO2N (+0.75 V vs. RHE) when studied as a photoanode for photoelectrochemical oxygen evolution.20 Therefore, IrO2/GaN:ZnO would be an interesting candidate of the OEP to replace CoOx/BiVO4:Mo and CoOx/LaTiO2N in the photocatalyst sheet, which may allow to improve the ZOWS performance.
Overall, the systems based on narrow bandgap non-oxide materials have demonstrated lower STH efficiencies at atmospheric pressure than those based on metal oxides, despite the superior light absorption ability of the former. This is due the weaker driving force for forward reactions and susceptibility to backward reactions. Activation and stabilization of narrow bandgap photocatalysts are essential to enable commercialization of photocatalytic solar hydrogen production processes, particularly under outdoor operating conditions. To advance the development of narrow-bandgap OEPs, it is important to precisely control the energy states at the interface between the photocatalyst and the cocatalyst, conductive material, or reaction solution so as to allow facile charge separation and transfer. To this end, development of technologies for tracking the migration of carriers under the reaction environment is important.
6. Summary and future prospects
This perspective reviewed solar-driven hydrogen production using particulate photocatalyst sheets. The STH efficiency achieved with the reported photocatalyst sheets is still lower than those expected based on the light absorption ability of the photocatalysts. From a materials viewpoint, identifying the origin of low AQYs and designing synthesis routes for active narrow-bandgap photocatalysts are vital to allow more excited charge carriers to participate in the hydrogen and oxygen evolution reactions. The STH efficiency is often drastically reduced due to backward reactions at elevated pressures. From a reaction viewpoint, it is important to identify active sites for backward reactions and improve reaction selectivity without sacrificing the inherent activity toward the water splitting reaction. If practical application of solar hydrogen production by OWS is seriously considered, it is necessary to investigate the reaction characteristics in an aqueous solution at atmospheric pressure and elevated temperature.In particulate photocatalyst sheet systems, water splitting performance can be significantly affected by backward reactions and mass transfer of reactants and products. The release of gas bubbles can be a significant concern when OWS is conducted under elevated background pressure, because it would increase the bubble residing time at the sheet surface and thus the opportunity for reverse reaction. Therefore, the identification of active sites and mechanisms for desired and undesired reaction pathways and the understanding of the associated transport phenomena are of particular importance. In the experimental observation, hydrogen and oxygen are probably generated in the narrow cavities between the photocatalyst particles during the water splitting reaction and eventually form submillimetre-sized gas bubbles of oxyhydrogen on the surface of the sheet.16 However, there are still many unsolved phenomena regarding the physical existence of reactant and product molecules. It will be desirable to clarify the mass transport of chemical species and the nucleation/growth of gas bubbles from a chemical engineering and hydrodynamic point of view, taking into account the actual structural and physical properties of the particle layer of photocatalyst sheets. This will enhance the introduction of the reactant (water) and the release of products (hydrogen and oxygen), thereby reducing the probability of reverse reactions under elevated pressure. In addition, visualisation of reduction and oxidation sites on photocatalyst particles has been reported using scanning electrochemical microscopy and single-molecule fluorescence spectroscopy in conjunction with probe molecules.39,40 Such spatially-resolved analytical techniques will be useful in identifying the active sites for backward reactions. Given the insufficient STH efficiencies achieved to date, improvements in charge separation and charge injection within individual photocatalyst particles and composite sheets are still essential. For example, surface photovoltage spectroscopy has been used to determine the facets of photocatalyst particles on which e− and h+ preferentially migrate.41,42 In addition, transient absorption spectroscopy has been used to elucidate carrier dynamics.30 These techniques are expected to be useful in comprehending the timeframe of reactions and the distribution of reaction sites on photocatalytic sheets. Apart from the development of photocatalytic materials and reactors with adequate performance, chemical processes should be carefully designed to balance STH efficiency, cost, and safe operation of renewable hydrogen production systems.
We consider that particulate photocatalyst sheets are promising because they can exhibit OWS activity under such practical conditions with appropriate surface modifications. The technological immaturity of this system offers considerable room for innovation. The authors hope that this contribution will stimulate researchers from diverse fields to collaborate on technical advancements to produce sustainable hydrogen for human society.
Author contribution
Swarnava Nandy: conceptualization, writing original draft, & editing; Takashi Hisatomi: conceptualization, writing & editing; Tsuyoshi Takata: conceptualization, writing; Tohru Setoyama: conceptualization, writing; Kazunari Domen: conceptualization, funding acquisition, writing & editing.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This research work was financially supported by the Artificial Photosynthesis Project (ARPChem) of the New Energy and Industrial Technology Development Organization (NEDO), and by JST, PRESTO, Japan (grant no. JPMJPR20T9).References
- T. Hisatomi and K. Domen, Nat. Catal., 2019, 2, 387–399 CrossRef CAS.
- Z. Wang, C. Li and K. Domen, Chem. Soc. Rev., 2019, 48, 2109–2125 RSC.
- S. Nandy, S. A. Savant and S. Haussener, Chem. Sci., 2021, 12, 9866–9884 RSC.
- K. Domen, S. Naito, M. Soma, T. Onishi and K. Tamaru, J. Chem. Soc. Chem. Commun., 1980, 543–544 RSC.
- G. Segev, J. Kibsgaard, C. Hahn, Z. J. Xu, W.-H. (Sophia) Cheng, T. G. Deutsch, C. Xiang, J. Z. Zhang, L. Hammarström, D. G. Nocera, A. Z. Weber, P. Agbo, T. Hisatomi, F. E. Osterloh, K. Domen, F. F. Abdi, S. Haussener, D. J. Miller, S. Ardo, P. C. McIntyre, T. Hannappel, S. Hu, H. Atwater, J. M. Gregoire, M. Z. Ertem, I. D. Sharp, K.-S. Choi, J. S. Lee, O. Ishitani, J. W. Ager, R. R. Prabhakar, A. T. Bell, S. W. Boettcher, K. Vincent, K. Takanabe, V. Artero, R. Napier, B. R. Cuenya, M. T. M. Koper, R. Van De Krol and F. Houle, J. Phys. D Appl. Phys., 2022, 55, 323003 CrossRef.
- Y. Goto, T. Hisatomi, Q. Wang, T. Higashi, K. Ishikiriyama, T. Maeda, Y. Sakata, S. Okunaka, H. Tokudome, M. Katayama, S. Akiyama, H. Nishiyama, Y. Inoue, T. Takewaki, T. Setoyama, T. Minegishi, T. Takata, T. Yamada and K. Domen, Joule, 2018, 2, 509–520 CrossRef CAS.
- Y. Wang, H. Suzuki, J. Xie, O. Tomita, D. James Martin, M. Higashi, D. Kong, R. Abe and J. Tang, Chem. Rev., 2018, 118, 5201–5241 CrossRef CAS PubMed.
- X. Tao, Y. Zhao, S. Wang, C. Li and R. Li, Chem. Soc. Rev., 2022, 51, 3561–3608 RSC.
- Q. Wang, T. Hisatomi, M. Katayama, T. Takata, T. Minegishi, A. Kudo, T. Yamada and K. Domen, Faraday Discuss., 2017, 197, 491–504 RSC.
- B. S. Pivovar, M. F. Ruth, D. J. Myers and H. N. Dinh, Electrochem. Soc. Interface, 2021, 30, 61 CrossRef.
- T. Ohno, L. Bai, T. Hisatomi, K. Maeda and K. Domen, J. Am. Chem. Soc., 2012, 134, 8254–8259 CrossRef CAS PubMed.
- T. Takata and K. Domen, ACS Energy Lett., 2019, 4, 542–549 CrossRef CAS.
- M. R. Shaner, H. A. Atwater, N. S. Lewis and E. W. McFarland, Energy Environ. Sci., 2016, 9, 2354–2371 RSC.
- Y. Ham, T. Hisatomi, Y. Goto, Y. Moriya, Y. Sakata, A. Yamakata, J. Kubota and K. Domen, J. Mater. Chem. A, 2016, 4, 3027–3033 RSC.
- T. Takata, J. Jiang, Y. Sakata, M. Nakabayashi, N. Shibata, V. Nandal, K. Seki, T. Hisatomi and K. Domen, Nature, 2020, 581, 411–414 CrossRef CAS PubMed.
- H. Nishiyama, T. Yamada, M. Nakabayashi, Y. Maehara, M. Yamaguchi, Y. Kuromiya, Y. Nagatsuma, H. Tokudome, S. Akiyama, T. Watanabe, R. Narushima, S. Okunaka, N. Shibata, T. Takata, T. Hisatomi and K. Domen, Nature, 2021, 598, 304–307 CrossRef CAS PubMed.
- F. Dionigi, P. C. K. Vesborg, T. Pedersen, O. Hansen, S. Dahl, A. Xiong, K. Maeda, K. Domen and I. Chorkendorff, J. Catal., 2012, 292, 26–31 CrossRef CAS.
- A. Xiong, G. Ma, K. Maeda, T. Takata, T. Hisatomi, T. Setoyama, J. Kubota and K. Domen, Catal. Sci. Technol., 2014, 4, 325–328 RSC.
- Z. Li, R. Li, H. Jing, J. Xiao, H. Xie, F. Hong, N. Ta, X. Zhang, J. Zhu and C. Li, Nat. Catal., 2023, 6, 80–88 CrossRef CAS.
- K. Liu, B. Zhang, J. Zhang, W. Lin, J. Wang, Y. Xu, Y. Xiang, T. Hisatomi, K. Domen and G. Ma, ACS Catal., 2022, 12, 14637–14646 CrossRef CAS.
- K. Chen, J. Xiao, J. J. M. Vequizo, T. Hisatomi, Y. Ma, M. Nakabayashi, T. Takata, A. Yamakata, N. Shibata and K. Domen, J. Am. Chem. Soc., 2023, 145, 3839–3843 CrossRef CAS PubMed.
- Q. Wang, M. Nakabayashi, T. Hisatomi, S. Sun, S. Akiyama, Z. Wang, Z. Pan, X. Xiao, T. Watanabe, T. Yamada, N. Shibata, T. Takata and K. Domen, Nat. Mater., 2019, 18, 827–832 CrossRef CAS PubMed.
- J. Xiao, S. Nishimae, J. J. M. Vequizo, M. Nakabayashi, T. Hisatomi, H. Li, L. Lin, N. Shibata, A. Yamakata, Y. Inoue and K. Domen, Angew. Chem., Int. Ed., 2022, 61, e202116573 CrossRef CAS PubMed.
- H. Kato, M. Hori, R. Konta, Y. Shimodaira and A. Kudo, Chem. Lett., 2004, 33, 1348–1349 CrossRef CAS.
- Y. Sasaki, H. Kato and A. Kudo, J. Am. Chem. Soc., 2013, 135, 5441–5449 CrossRef CAS PubMed.
- Q. Wang, Y. Li, T. Hisatomi, M. Nakabayashi, N. Shibata, J. Kubota and K. Domen, J. Catal., 2015, 328, 308–315 CrossRef CAS.
- Q. Wang, T. Hisatomi, Q. Jia, H. Tokudome, M. Zhong, C. Wang, Z. Pan, T. Takata, M. Nakabayashi, N. Shibata, Y. Li, I. D. Sharp, A. Kudo, T. Yamada and K. Domen, Nat. Mater., 2016, 15, 611–615 CrossRef CAS PubMed.
- Q. Wang, T. Hisatomi, Y. Suzuki, Z. Pan, J. Seo, M. Katayama, T. Minegishi, H. Nishiyama, T. Takata, K. Seki, A. Kudo, T. Yamada and K. Domen, J. Am. Chem. Soc., 2017, 139, 1675–1683 CrossRef CAS PubMed.
- Q. Wang, S. Okunaka, H. Tokudome, T. Hisatomi, M. Nakabayashi, N. Shibata, T. Yamada and K. Domen, Joule, 2018, 2, 2667–2680 CrossRef CAS.
- S. Chen, J. J. M. Vequizo, Z. Pan, T. Hisatomi, M. Nakabayashi, L. Lin, Z. Wang, K. Kato, A. Yamakata, N. Shibata, T. Takata, T. Yamada and K. Domen, J. Am. Chem. Soc., 2021, 143, 10633–10641 CrossRef CAS PubMed.
- S. Chen, S. Nandy, J. J. M. Vequizo, T. Hisatomi, M. Nakabayashi, Z. Pan, Q. Xiao, Z. Wang, L. Lin, S. Sun, K. Kato, A. Yamakata, N. Shibata, T. Takata, F. Zhang and K. Domen, ACS Catal., 2023, 13, 3285–3294 CrossRef CAS.
- T. Hisatomi, S. Okamura, J. Liu, Y. Shinohara, K. Ueda, T. Higashi, M. Katayama, T. Minegishi and K. Domen, Energy Environ. Sci., 2015, 8, 3354–3362 RSC.
- Z. Pan, T. Hisatomi, Q. Wang, S. Chen, A. Iwase, M. Nakabayashi, N. Shibata, T. Takata, M. Katayama, T. Minegishi, A. Kudo and K. Domen, Adv. Funct. Mater., 2016, 26, 7011–7019 CrossRef CAS.
- S. Nandy, T. Hisatomi, M. Nakabayashi, H. Li, X. Wang, N. Shibata, T. Takata and K. Domen, Joule, 2023, 7, 1641–1651 CrossRef CAS.
- D. Zhao, Y. Wang, C.-L. Dong, Y.-C. Huang, J. Chen, F. Xue, S. Shen and L. Guo, Nat. Energy, 2021, 6, 388–397 CrossRef CAS.
- Y. Wang, W. Huang, S. Guo, X. Xin, Y. Zhang, P. Guo, S. Tang and X. Li, Adv. Energy Mater., 2021, 11, 2102452 CrossRef CAS.
- Y. Qi, J. Zhang, Y. Kong, Y. Zhao, S. Chen, D. Li, W. Liu, Y. Chen, T. Xie, J. Cui, C. Li, K. Domen and F. Zhang, Nat. Commun., 2022, 13, 484 CrossRef CAS PubMed.
- T. Hisatomi, T. Yamamoto, Q. Wang, T. Nakanishi, T. Higashi, M. Katayama, T. Minegishi and K. Domen, Catal. Sci. Technol., 2018, 8, 3918–3925 RSC.
- G. Askarova, C. Xiao, K. Barman, X. Wang, L. Zhang, F. E. Osterloh and M. V. Mirkin, J. Am. Chem. Soc., 2023, 145, 6526–6534 CrossRef CAS PubMed.
- J. B. Sambur, T.-Y. Chen, E. Choudhary, G. Chen, E. J. Nissen, E. M. Thomas, N. Zou and P. Chen, Nature, 2016, 530, 77–80 CrossRef CAS PubMed.
- R. Chen, J. Zhu, H. An, F. Fan and C. Li, Faraday Discuss., 2017, 198, 473–479 RSC.
- J. Zhu, F. Fan, R. Chen, H. An, Z. Feng and C. Li, Angew. Chem., Int. Ed., 2015, 54, 9111–9114 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |