DOI:
10.1039/D3RA06258A
(Review Article)
RSC Adv., 2023,
13, 35877-35903
Advances in Aβ imaging probes: a comprehensive study of radiolabelled 1,3-diaryl-2-propen-1-ones for Alzheimer's disease: a review
Received
14th September 2023
, Accepted 9th November 2023
First published on 11th December 2023
Abstract
Alzheimer's disease (AD) is a formidable neurodegenerative disorder characterized by cognitive decline, memory impairment and inability to perform everyday tasks. In the pursuit of innovative diagnostic and therapeutic strategies, the synthesis and application of radiolabelled compounds have garnered significant attention. This review delves into the synthesis and biological significance of radiolabelled 1,3-diaryl-2-propen-1-ones, commonly known as chalcones, as Aβ imaging probes for AD. These versatile chalcone derivatives have demonstrated noteworthy potential as radiotracers for visualizing Aβ imaging probes, which are hallmark pathologies of AD. This review encompasses an exploration of chalcone synthesis via diverse methodologies and their biological implications, both as standalone entities and as precursors for intricate natural products. In addition, the pivotal role of advanced imaging techniques, such as single-photon emission computed tomography (SPECT) and positron emission tomography (PET), using various radioisotopes is highlighted. The use of radiopharmaceutical agents, including [18F]FDG, [18F]FMAPO, [11C]6-Me-BTA-1, [124/125I]IBETA, and [64Cu]YW-7 as potent tools for early diagnosis and therapeutic advancement is explored. This review underscores the critical nexus between radiolabelled chalcones and their pivotal role in advancing diagnostic and therapeutic paradigms in AD research. Furthermore, this study encapsulated the role of radiolabelled chalcone emphasizing their prospective implications for drug development and therapeutic interventions. A focal point of paramount significance is the elucidation of Aβ imaging probes and its important role in the combat against AD, with a particular emphasis on their role in facilitating early diagnosis and fostering advancements in therapeutic strategies.
 Sudeep Dhillon | Sudeep Dhillon was born and grew up in Dahola, Jind, Haryana, India. Sudeep Dhillon obtained his bachelor's degree in Non-medical from Panjab University, Chandigarh in 2015 and his Master's in Organic Chemistry from Maharishi Markandeshwar University, Mullana, India in 2017. He perusing his Ph.D. degree from Chaudhary Bansi Lal University, Bhiwani, India with Dr Mayank Kinger, Associate Professor, Chaudhary Bansi Lal University, Bhiwani, India. His current research interests are the Synthesis of new biologically potent heterocycles (anti-cancer, anti-Alzheimer, anti-inflammatory, etc.). |
 Mayank Kinger | Dr Mayank Kinger obtained his bachelor's degree in Medical Science from C. C. S. University, Meerut, India in 2000. He obtained his Master's in Organic Chemistry from C. C. S. University, Meerut, India in 2002. He received his PhD degree from Kurukshetra University, Kurukshetra, India in 2008 under the supervision of Prof. Om Prakash, Emeritus Professor, Kurukshetra University. He was a postdoctoral fellow at Korea Atomic Energy Research Institute, South Korea from 2010 to 2012. Then, he joined M. M. University, Mullana, Ambala, India. In 2018, he joined Chaudhary Bansi Lal University, Haryana as Associate Professor. His current research interests are Synthesis of new biologically potent heterocycles (anti-cancer, anti-Alzheimer, anti-inflammatory, anti-bacterial, anti-fungal agents, etc. |
 Priyanka Rani | Priyanka Rani was born in Bhiwani district, Haryana, India. She received her BSc degree in 2015 from Maharshi Dayanand University, Rohtak, and her MSc degree in 2017 from Baba Mastnath University, Rohtak. Now she continues her study at Chaudhary Bansi Lal University with Dr Mayank Kinger, Associate Professor, Chaudhary Bansi Lal University, Bhiwani, India for research work. Her current research interest focuses on five or six-membered heterocycles having pharmaceutical potential such as anti-diabetic, anti-anxiety, anti-malarial, anti-inflammatory, antifungal etc. |
 Mamta Chahal | Mamta Chahal was born in Jind district, Haryana state, India. She obtained her Bachelor's degree in 2017 and master's degree in organic chemistry from Kurukshetra university, Kurukshetra in 2019. She is perusing her Ph.D. degree with Dr Mayank Kinger, Associate professor, at Chaudhary Bansi Lal University, Haryana, India. Her research area is the synthetic utility of novel heterocycles. |
 Ginna Kumari | Ginna Kumari was born and grew up in Charkhi Dadri district, Haryana, India. She obtained her bachelor's degree in 2013 from Maharshi Dayanand University, Rohtak, and her master's degree in chemistry in 2016 from Panjab University, Chandigarh, India. She is currently working with Dr Mayank Kinger, Department of Chemistry, Chaudhary Bansilal University, Haryana, India. Her research interest is the synthesis of novel heterocycles. |
 Deepak Kumar Aneja | Dr Deepak Kumar Aneja was born in the small village Saniana of District Fatehabad of Haryana State and obtained his early education from the village. He obtained his BSc in 2004 from CRM Jat (PG) College, Hisar and his MSc in 2006 from Kurukshetra University, Kurukshetra, India. He then joined Jubilant Chemsys Ltd, Noida, as Research Associate and worked there for 2 years from 2006 to 2008. Then, he moved back to Kurukshetra University and Joined the research group of Professor Om Prakash for his Ph.D. He submitted his Ph.D. thesis in 2012 and defended in 2013. Meanwhile, he joined GDC Memorial College, Bahal, Bhiwani in 2012 and worked as Assistant Professor for 3 years. In 2015, he joined Chaudhary Bansi Lal University, Haryana as Assistant Professor and is working on Reactivity of hypervalent iodine reagents under different reaction conditions and their importance in different fields of science. |
1. Introduction
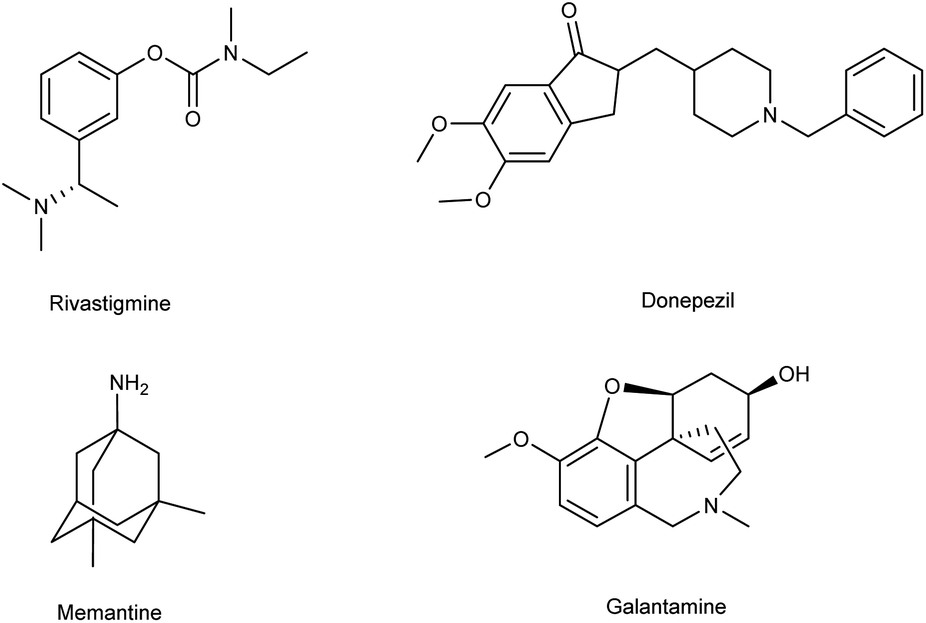
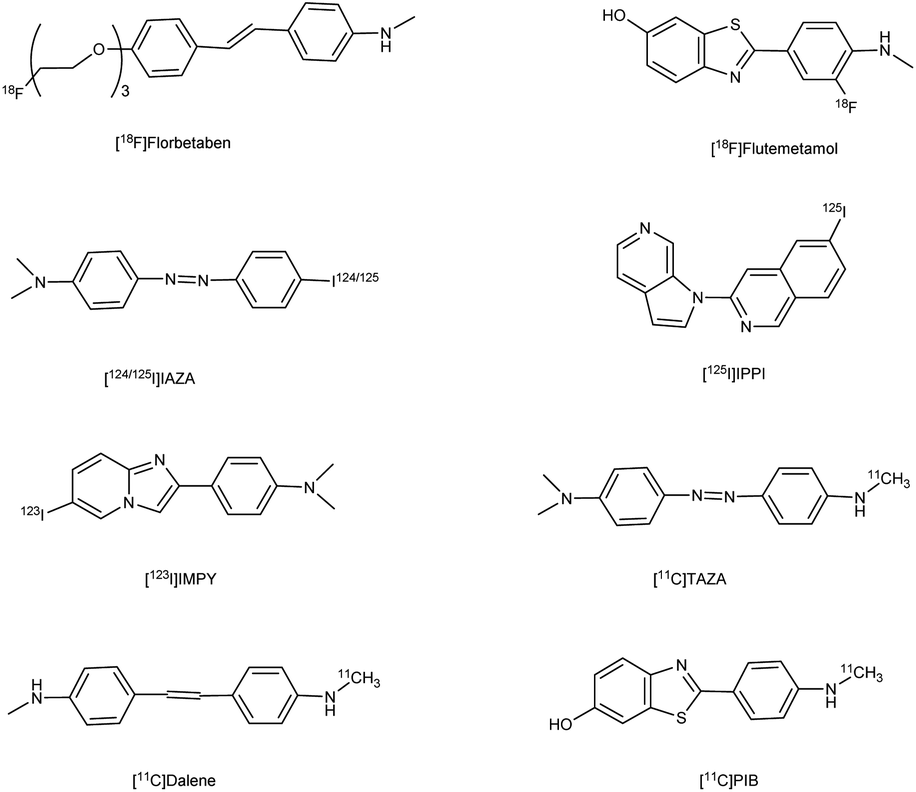
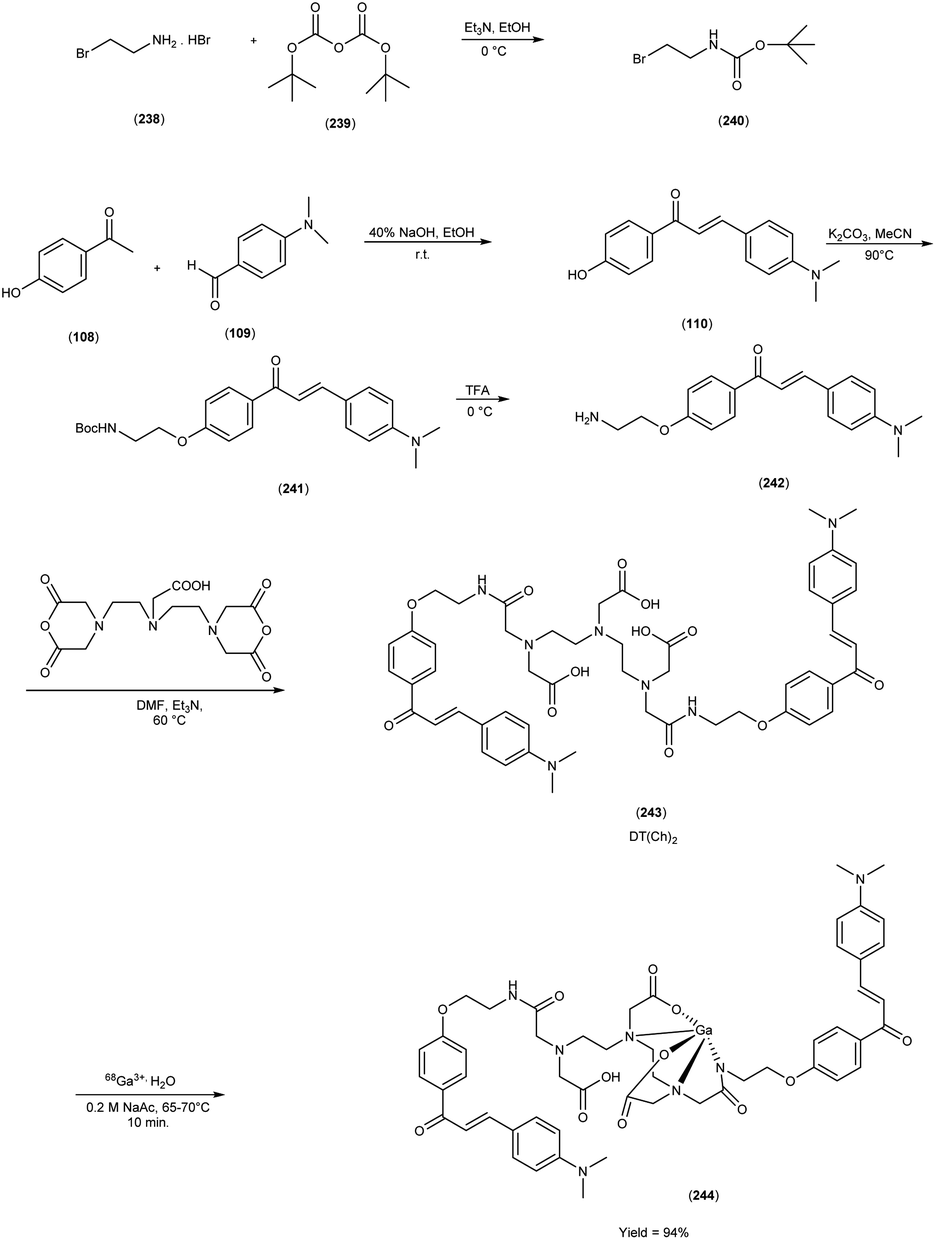
Alzheimer's disease (AD) is a neurological disorder defined by increasing memory loss, cognitive deterioration, language impairment, disorientation, and the inability to perform regular activities.1–3 Extracellular amyloid (Aβ) plaque development, deposition, mitochondrial failure, oxidative stress, neuroinflammation and intracellular neurofibrillary tangles are all symptoms of AD. The pathogenic feature of AD is the production of oligomer aggregates containing amyloid peptides (Aβ40 and Aβ42) by abnormal cleavage of amyloid precursor proteins assisted by α- and γ-secretase in the brain. AD affects the world's aging workforce, and a growing incidence might lead to higher death and disability rates. By 2050, the number of Alzheimer's patients in the U.S. will have grown from 5.8 million to 13.8 million people.4–7 In 1901, Alois Alzheimer, a psychiatrist and neuroanatomist from Germany discovered AD after he treated a 50 year-old lady who was showing signs of intellectual impairment, increasing disorientation and insomnia.8 The medical assessment of her brain in 1906 revealed signs of senility, including plaques and tangles typical of aging. “Alzheimer's Disease” was eventually introduced to describe the disorder.9 Cholinergic, β-amyloid hypotheses and tauopathy have dominated drug development for AD. Recently, other illnesses such as mitochondrial dysfunction and neuroinflammation have been included in the research.10 The cholinesterase inhibitors such as donepezil (DPZ), memantine,11 galantamine,12,13 rivastigmine,14,15 (Fig. 1) and a partial NMDA antagonist have been licensed by the FDA after years of study and breakthroughs in the field of pharmacotherapy. Unfortunately, these pharmaceutical medications can only cure AD symptoms for a short period and cannot halt or stop the disorder from spreading. As a result, more effective treatment drugs that can prevent or reverse disease development should recommended.16 Radiopharmaceuticals17–19 used for the treatment of AD are shown in Fig. 2.
 |
| | Fig. 1 FDA-approved drugs for AD. | |
 |
| | Fig. 2 Radiopharmaceuticals labelled with 18F, 11C, 124/125I, 99Tc for AD. | |
2. Chalcones
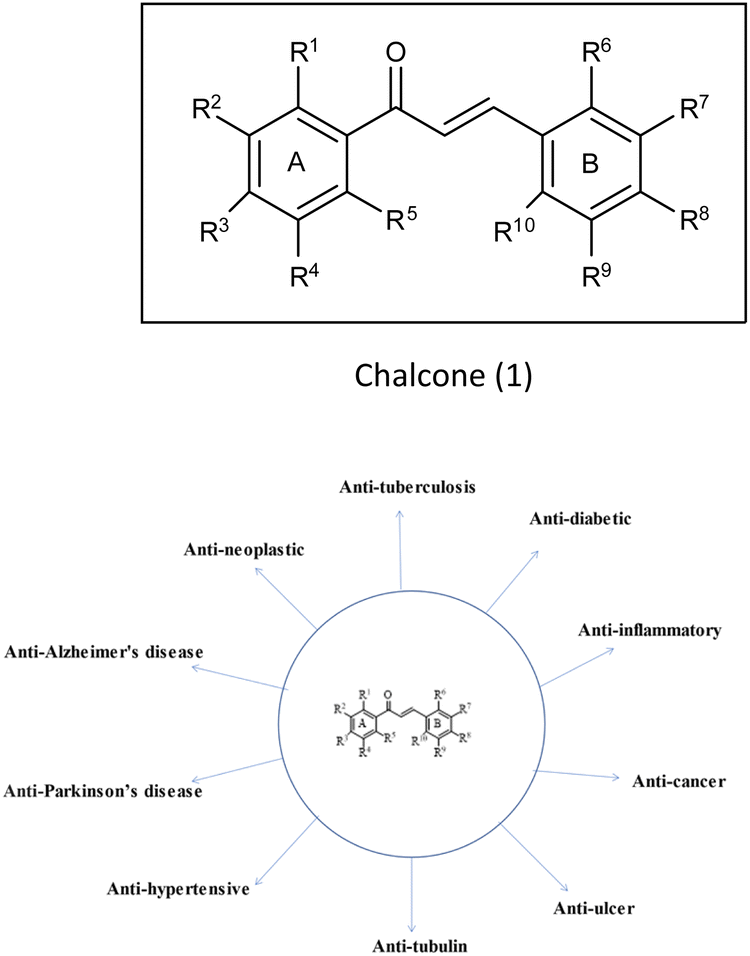
(E)-1,3-Diphenyl-2-propene-1-one,1 commonly known as chalcone, constitutes a distinctive chemical scaffold recurrently encountered in various natural flavonoids and isoflavonoids.20,21 In the nineteenth century, Kostanecki and Tambor22 embarked upon the synthesis of diverse natural chromophores, thereby coining the term “chalcone”. This scaffold has acquired widespread significance within the scientific community due to its open-chain architecture, the abundance of exchangeable hydrogen atoms, and the capacity to yield an entirely novel class of chemical entities through structural modifications.23,24 The synthesis of chalcones is facilitated through various methodologies such as base-catalyzed Claisen–Schmidt condensation, Wittig reaction, Friedel–Crafts acylation and Suzuki coupling reaction,25 etc. This fundamental skeleton1 serves as a versatile framework that can be significantly diversified. Common structural variations include substituting rings A and B with alternative heteroaryl entities and introducing multiple functional groups onto the phenyl ring such as amine, methoxy, hydroxy and halogens (Cl, F, Br), resulting in the enhancement of the biological potential of chalcones.26 Additionally, modifications involving the fusion of ring A with the α-carbon, as well as other alternations enumerated earlier, contribute to the structural diversity. Chalcones play a pivotal role as intermediates in the synthesis of biologically active heterocyclic compounds including isoxazole,27 thiadiazol,28 benzodiazepine,29 pyrazoles,30 pyrimidines,31 benzothiazepines,32 pyrazoline33 etc. The remarkable biological versatility of chalcones is underscored by their engagement in a spectrum of activities encompassing anti-histaminic,34 anti-cancer,35 anti-retroviral,36 anti-tubulin,37 anti-hypertensive,38 anti-oxidant,39 anti-neoplastic,38 anti-ulcer,40 anti-tuberculosis,39 anti-diabetic,41 anti-inflammatory,42 anti-Parkinson's,43 and anti-AD44 (Fig. 3) etc. The profound potential of the chalcone1 has engendered remarkable interest in pharmaceutical research, particularly in the context of central nervous system disorders such as AD.43 Although the structure of these compounds has been explored for its potential biological properties, only a scant number of chalcone-based probes have been investigated as viable radiotracers. Within the context of AD, chalcones and their derivatives have exhibited notable pharmacological potential across multiple targets, notably in the inhibition of Aβ-fibrils aggregation and the activity of pivotal enzymatic systems such as acetylcholinesterase (AChE), butyrylcholinesterase (BuChE) and pseudocholinesterase. These enzymatic systems bear relevance to the development and progression of AD as per the cholinergic hypothesis.4 It is acknowledged that aromatic structures within compounds can impede fibril assembly by disrupting the μ-stacking arrangement within their hydrophobic core.12 Nevertheless, a comprehensive understanding of the nuanced role exerted by functional groups situated on the aryl moieties of chalcones in modulating binding affinity for Aβ-plaques remains a relatively uncharted domain, constituting the focal theme of the present review. A wide variety of radiolabelled chalcones have been studied as a potential building block for anti-AD medication development. Researchers believe that Congo red (CR) and vitamin thioflavin (ThT) have the potential to be used intravenously as probes to detect brain-amyloid plaques. Compounds like [18F]-FDDNP,45,46 [11C] 6-OH-BTA-1,47 and [11C]SB-13,48 have been tested in clinical studies, demonstrating that Aβ-plaques may be seen in the natural human brain.49–51
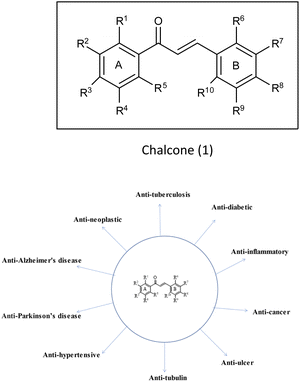 |
| | Fig. 3 Vast biological profile of chalcone. | |
The advent of chalcone-derived Aβ imaging signifies a novel and unexplored domain within the realm of chemical investigation, extending the frontiers of existing knowledge. Chalcones and their analogous emerge as prospective candidates, holding promise for deployment as diagnostic tools or therapeutic entities in the amelioration of AD. This review elucidates the latest strides in the synthesis of radiolabelled chalcones, illuminating their significant contributions in the therapeutic landscape of AD. The organization of this review is structured around five distinct thematic categories, delineating diverse sets of radiotracers harnessed for this purpose.
3. 125I labelled chalcones for AD
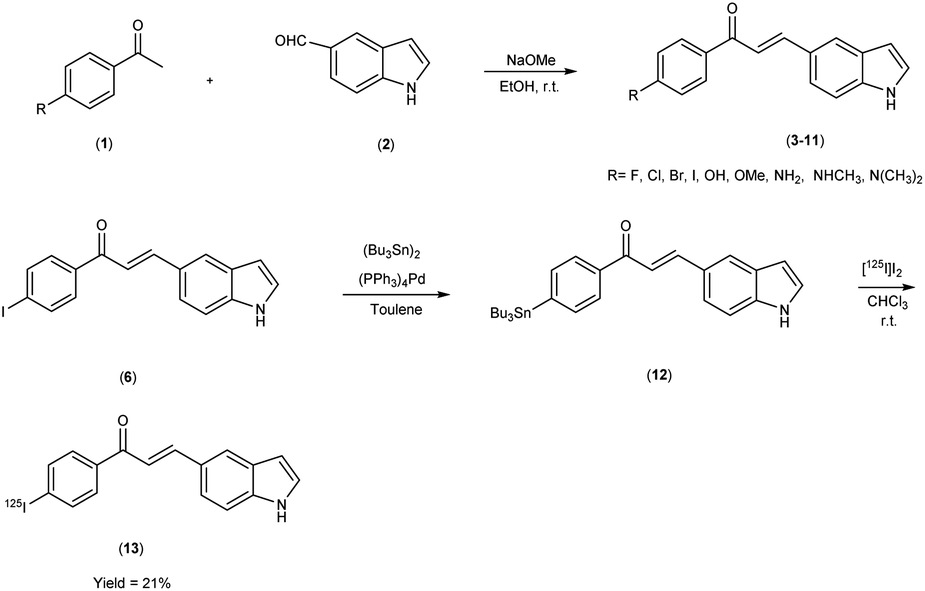
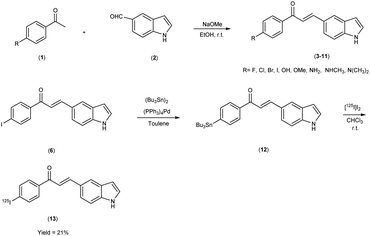
Mengchao Cui and colleagues52 executed the synthesis of chalcone derivatives through a meticulously orchestrated Claisen-condensation reaction, involving substituted acetophenones1 and indole carbaldehyde2 in the presence of a basic catalyst i.e. 28% sodium methoxide (CH3ONa) in ethanol at ambient temperature. To generate the tributyltin precursor,11 a bromo to tributyltin exchange reaction was undertaken, catalyzed by tetrakis(triphenylphosphine)palladium [(PPh3)4Pd]. This synthetic endeavour yielded products across a spectrum from modest to commendable yields. The preparation of the tributyltin precursor material was accomplished via an exchange reaction. The synthetic pathway was then extended to the synthesis of radiolabelled chalcones, employing a strategically designed [125I] iodine-labelled probe. These chalcones were subjected to meticulous biological scrutiny, wherein they underwent competitive binding assays targeting Aβ1–42 aggregates by employing [125I] IMPY as a standard reference. The binding affinities of these compounds displayed a noteworthy range spanning from 4.46 nm to surpassing >1008 nm, contingent upon the specific substitution patterns exhibited on the phenyl ring (Table 1). Notably, the incorporation of halogen moieties, methylated amines and the hydroxy functional group onto the phenyl ring produced augmented binding affinities. Upon the co-introduction of indole derivatives alongside IMPY, the emergence of a shared binding site became evident, suggesting a plausible convergence of binding interactions. Much to our astonishment, DMIC-14 demonstrated a dose-dependent inhibition of the binding of [125I]IMPY. This inhibition exhibited a strong affinity for Aβ1–42 aggregates, as indicated by a high binding constant (Ki = 1.97 nM) (DMIC-14). These results implied that both chalcone and IMPY might target the identical binding site (thioflavin-T) on Aβ aggregates hat possess overlapping binding sites. Nonetheless, empirical investigations involving normal mice unveiled a notably limited in vivo cerebral uptake for [125I]-(E)-3-(1H-indol-5-yl)-1-(4-iodophenyl)prop-2-en-1-one13 (0.41% injected dose per gram of brain tissue at 2 minutes post-administration) (Table 2). The subsequent in vivo experimentation in murine subjects indicated subdued neuroinfiltration of the introduced compounds within brain tissues. The intricate indole-chalcone molecular architecture stands poised for prospective refinements through judicious chemical modifications, with the eventual aim of fashioning a bespoke Aβ-amyloid probe, thereby advancing our investigatory capabilities within the realm of amyloid-associated research (Scheme 1).
Table 1 Inhibition constants (Ki) for binding to Aβ1–42 aggregates versus [125I]IMPY52
| Compound |
R |
Ki (nM) |
| 3 |
4-Fluorophenyl |
35.06 ± 6.21 |
| 4 |
4-Chlorophenyl |
8.43 ± 2.13 |
| 5 |
4-Bromophenyl |
8.96 ± 0.92 |
| 6 |
4-Iodophenyl |
8.22 ± 1.46 |
| 7 |
4-Hydroxyphenyl |
>360 |
| 8 |
4-Methoxyphenyl |
8.52 ± 2.15 |
| 9 |
4-Aminophenyl |
>1008 |
| 10 |
4-Methylaminophenyl |
51.09 ± 7.71 |
| 11 |
4-Diethylaminophenyl |
5.17 ± 0.32 |
| DMIC-14 |
— |
1.97 ± 0.26 |
| IMPY |
— |
10.5 ± 1.0 |
Table 2 Biodistribution in normal ddY mice after injection of radiolabelled compound 13
| Compound |
Organs |
Time after injection (min) |
| 2 min |
15 min |
30 min |
60 min |
120 min |
| 13 |
Blood |
8.85 ± 0.48 |
5.70 ± 0.24 |
4.79 ± 0.53 |
4.15 ± 0.51 |
3.72 ± 0.77 |
| Brain |
0.41 ± 0.02 |
0.29 ± 0.02 |
0.20 ± 0.03 |
0.21 ± 0.05 |
0.13 ± 0.02 |
| Heart |
3.27 ± 0.31 |
2.27 ± 0.25 |
1.72 ± 0.10 |
1.61 ± 0.21 |
1.33 ± 0.26 |
| |
Liver |
2.69 ± 0.22 |
2.21 ± 0.10 |
1.91 ± 0.22 |
11.48 ± 0.25 |
1.43 ± 0.42 |
| Spleen |
3.02 ± 0.48 |
2.96 ± 0.84 |
2.40 ± 0.16 |
2.27 ± 0.34 |
1.89 ± 0.31 |
| Lung |
7.33 ± 0.71 |
4.86 ± 0.17 |
4.20 ± 0.27 |
3.40 ± 0.59 |
3.19 ± 0.63 |
| Kidney |
7.30 ± 1.08 |
4.03 ± 0.12 |
3.26 ± 0.07 |
3.11 ± 0.33 |
2.78 ± 0.19 |
| Stomach |
2.01 ± 0.37 |
1.07 ± 0.28 |
0.85 ± 0.10 |
0.96 ± 0.34 |
1.19 ± 0.52 |
| Intestine |
1.36 ± 0.27 |
2.01 ± 0.43 |
2.32 ± 0.53 |
1.72 ± 0.43 |
2.13 ± 0.56 |
 |
| | Scheme 1 | |
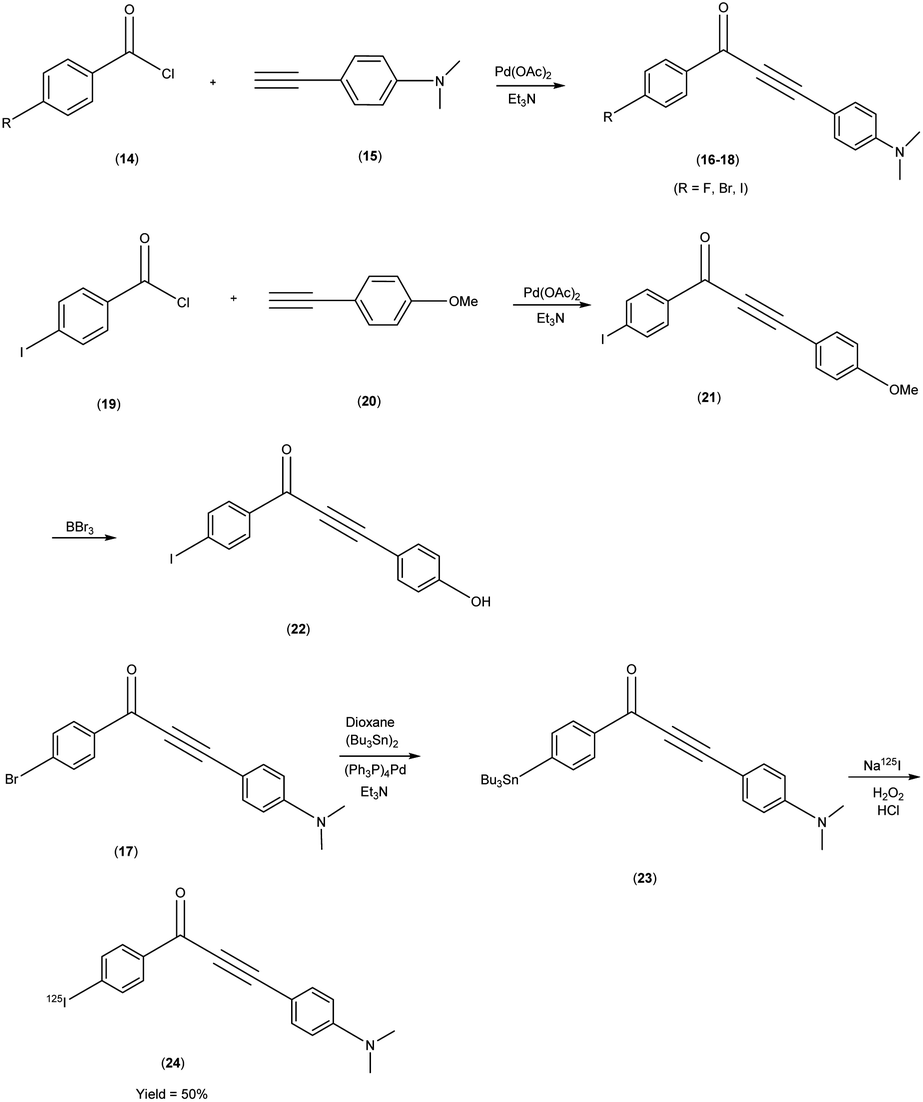
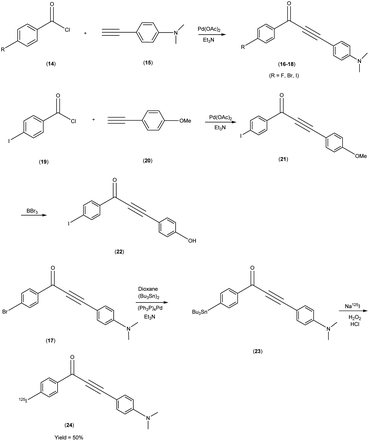
Masahiro Ono and co-researchers53 orchestrated the synthesis of an innovative series of diphenylpropynone (DPP) derivatives through the strategic replacement of double bond inherent in chalcones with a more reactive triple bond. The DPP derivatives16–18 were synthesized through a series of chemical reactions. The initial step involved the coupling of benzoyl chloride, i.e. p-iodobenzoyl chloride, or p-bromobenzoyl chloride, with phenylacetylene(4-ethynyl-N,N-dimethylaniline or p-ethynyl anisole). The reaction was catalyzed by palladium acetate (Pd(OAc)2) and base triethylamine (Et3N) was used. Subsequently, compound 21 was transformed into compound 22 through a demethylation process using boron tribromide (BBr3) in dichloromethane. The tributyltin derivative 23 was prepared from the bromo compound 17 via a bromo to tributyltin exchange reaction catalyzed by palladium (Pd(0)). This tributyltin derivative served as the initial substrate for radioiodination in the synthesis of [125I]24. The novel radioiodinated DPP derivative, denoted as [125I]24, was synthesized through an iododestannylation process employing hydrogen peroxide as the oxidant. This procedure successfully yielded the desired radioiodinated ligand. Notably, the absence of geometric isomerism, a complication often encountered in their double-bonded counterparts, confers a distinct advantage to this novel DPP family. The evaluation of these compounds encompassed their suitability for in vivo imaging applications within in the realm of AD and other neurovegetative disorders. Through a suite of binding assays conducted in vitro, these derivatives revealed a conspicuous and noteworthy affinity toward Aβ1–42 aggregates, as substantiated by ki values spanning the range of 6 nM to 326 nM. Furthermore, the efficacy of these compounds was exemplified through the selective labelling of plaques, which was distinctly observable within brain tissue sections procured from Tg2576 transgenic mice (Table 3). To discern their performance, a comparative analysis was conducted, leveraging the established [125I] IMPY as the standard reference. The radiolabelling of DPP derivative17 was adeptly achieved utilizing [Na125I], followed by meticulous assessment via competitive binding assays. Upon embarking on biodistribution studies in normal murine models, it was observed that in the subsequent biodistribution investigations encompassing normal mice, the radiolabelled derivative [125I] exhibited an appreciable yet measured cerebral uptake (1.55% injected dose per gram of brain tissue at 2 minutes post-administration) and a concomitant progressive reduction in cerebral concentration over time (0.76% injected dose per gram of brain tissue at 60 minutes post-administration) (Table 4). Intriguingly, the non-radioiodinated DPP ligand displayed a discernibly lesser affinity for Aβ plaques when juxtaposed with its radioiodinated DPP ligand counterparts. This finding accentuates the advantageous role of radiolabelling in bolstering the binding affinity of DPP derivatives towards Aβ aggregates (Scheme 2).
Table 3 Inhibition by DPP derivatives of ligand binding to Aβ1–42 aggregates
| Compound |
Ki (nM) |
| 16 |
20.4 ± 1.3 |
| 18 |
6.0 ± 0.15 |
| 21 |
13.7 ± 5.0 |
| 22 |
325.8 ± 13.8 |
| IMPY |
45.6 ± 11.5 |
Table 4 Biodistribution of radioactivity after injection of radiolabelled compound 24 in normal mice
| Compound |
Organ |
Time after injection (min) |
| 2 min |
10 min |
30 min |
60 min |
| 24 |
Blood |
6.19 ± 1.05 |
5.44 ± 0.36 |
3.94 ± 0.46 |
3.07 ± 0.20 |
| Liver |
19.96 ± 2.47 |
15.29 ± 1.81 |
12.02 ± 1.21 |
10.06 ± 1.07 |
| Kidney |
10.45 ± 1.82 |
9.81 ± 1.31 |
8.34 ± 0.77 |
7.75 ± 0.71 |
| Intestine |
2.73 ± 0.79 |
8.44 ± 1.67 |
12.04 ± 1.44 |
14.02 ± 1.77 |
| Spleen |
4.41 ± 0.95 |
5.19 ± 0.93 |
5.47 ± 0.27 |
4.43 ± 1.64 |
| Stomach |
0.75 ± 0.16 |
2.97 ± 2.55 |
2.35 ± 1.38 |
1.46 ± 0.38 |
| Pancreas |
3.90 ± 0.44 |
3.39 ± 0.31 |
2.57 ± 0.30 |
2.09 ± 0.13 |
| Heart |
10.00 ± 1.19 |
7.53 ± 1.14 |
6.03 ± 0.46 |
4.94 ± 0.54 |
| Brain |
1.55 ± 0.26 |
1.23 ± 0.09 |
0.93 ± 0.06 |
0.76 ± 0.07 |
 |
| | Scheme 2 | |
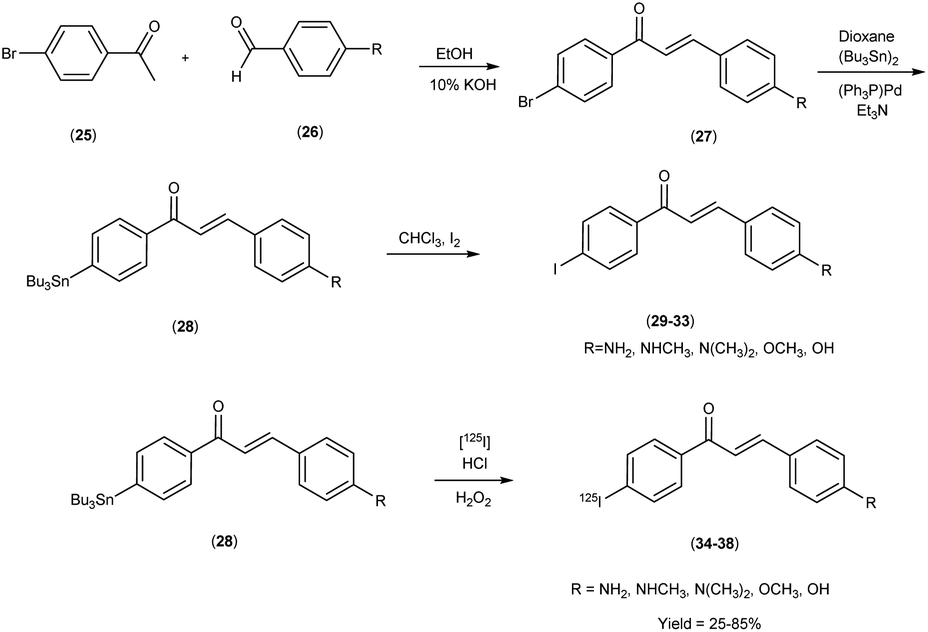
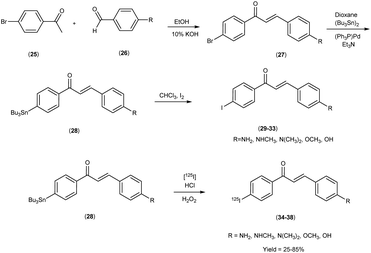
In 2007, Masahiro Ono and co-workers54 revealed the judicious synthesis of chalcones27 through a transformative reaction involving substituted acetophenone25 and carbaldehyde,26 devised within the venerable Claisen condensation framework. The tributyltin derivatives,29 were prepared from their respective bromo compounds through a bromo-to-tributyltin exchange reaction. This transformation was facilitated by palladium in its zero oxidation state, Pd(0). Subsequently, the tributyltin derivatives were readily subjected to a reaction with iodine in chloroform (CHCl3) under ambient conditions, yielding the corresponding iodo derivatives. Additionally, these tributyltin derivatives served as suitable starting materials for radioiodination. The production of novel radioiodinated chalcones was achieved via an iododestannylation reaction, with hydrogen peroxide utilized as the oxidant. This process successfully produced the anticipated radioiodinated ligands (34 to 38) in 25% to 85% yields. These chalcone entities, adorned with radiolabelled, were deftly harnessed as probes to scrutinize the intricate binding dynamics entailed in their interaction with Aβ1–42 aggregates. The span of these architectural scaffolds in the realm of binding experiments is one that spans from the confines of 3 nm to the expanses of 105 nm, thereby encapsulating a diverse panorama of molecular interactions (Table 5). The biodistribution investigations conducted in murine subjects of standard physiological condition after intravenous administration of radioiodinated chalcones34–38 manifested a noteworthy elevation in cerebral uptake (ranging from 2.0 & to 4.7% injected dose per gram) (% ID/g) at the 2 minutes time, concomitant with expeditious elimination from the cerebral region (ranging from 0.2% to 0.6% ID/g at the 30 minutes interval) (Table 6). In a comparative spectroscopic discourse, the resonances of radioiodinated chalcones were elegantly juxtaposed against their non-radioiodinated counterparts. Evidently, the former cohort displayed an elevated predilection for binding to the Aβ plaques, thereby underscoring their heightened affinity towards these pathogenic markers (Scheme 3).
Table 5 Inhibition constants of chalcone derivatives on ligand binding to Aβ1–42 aggregates
| Compound |
R |
Ki (nM) |
| 29 |
Amino |
104.7 ± 12.0 |
| 30 |
Methylamine |
6.3 ± 1.6 |
| 31 |
N-Dimethylamine |
2.9 ± 0.3 |
| 32 |
Methoxy |
6.3 ± 1.7 |
| 33 |
Hydroxy |
21.4 ± 1.4 |
| Thioflavin T |
— |
>10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
| Congo red |
— |
>10000 |
Table 6 Biodistribution of radioactivity after intravenous administration of radiolabelled compounds 34, 35, 36, 37 and 38 in mice
| Compounds |
Organs |
Time after injection (min) |
| 2 min |
10 min |
| 34 |
Blood |
3.62 ± 0.69 |
1.46 ± 0.32 |
| Liver |
7.52 ± 0.72 |
8.16 ± 2.15 |
| Kidney |
6.96 ± 0.43 |
6.34 ± 3.51 |
| Intestine |
1.84 ± 0.32 |
11.27 ± 3.34 |
| Spleen |
1.53 ± 0.37 |
0.60 ± 0.25 |
| Lung |
4.74 ± 1.05 |
1.43 ± 0.23 |
| |
Stomach |
0.87 ± 0.16 |
1.58 ± 0.44 |
| Heart |
4.59 ± 0.54 |
0.84 ± 0.19 |
| Brain |
4.49 ± 0.55 |
0.46 ± 0.07 |
| 35 |
Blood |
1.64 ± 0.47 |
1.47 ± 0.27 |
| Liver |
7.20 ± 1.72 |
8.61 ± 1.13 |
| Kidney |
6.62 ± 1.10 |
11.43 ± 4.02 |
| Intestine |
2.06 ± 0.35 |
10.46 ± 1.71 |
| Spleen |
2.85 ± 0.81 |
0.80 ± 0.28 |
| Lung |
8.30 ± 3.43 |
4.01 ± 0.30 |
| Stomach |
1.02 ± 0.50 |
1.28 ± 0.41 |
| Heart |
4.72 ± 1.00 |
1.08 ± 0.16 |
| Brain |
4.72 ± 1.50 |
0.61 ± 0.11 |
| 36 |
Blood |
1.85 ± 0.40 |
1.40 ± 0.24 |
| Liver |
10.02 ± 0.57 |
11.04 ± 2.42 |
| Kidney |
5.32 ± 0.74 |
11.62 ± 2.06 |
| Intestine |
1.31 ± 0.16 |
10.38 ± 2.37 |
| Spleen |
1.37 ± 0.21 |
0.60 ± 0.08 |
| Lung |
3.40 ± 0.11 |
1.84 ± 0.15 |
| Stomach |
0.96 ± 0.09 |
2.45 ± 0.19 |
| Heart |
3.92 ± 0.36 |
1.18 ± 0.30 |
| Brain |
2.04 ± 0.36 |
0.49 ± 0.08 |
| 37 |
Blood |
1.34 ± 0.20 |
0.46 ± 0.08 |
| Liver |
5.97 ± 0.97 |
4.03 ± 0.72 |
| Kidney |
4.61 ± 0.72 |
3.27 ± 0.28 |
| Intestine |
1.93 ± 0.28 |
10.19 ± 0.51 |
| Spleen |
1.38 ± 0.21 |
0.29 ± 0.07 |
| Lung |
3.81 ± 0.87 |
0.79 ± 0.13 |
| Stomach |
0.9 ± 0.37 |
2.11 ± 2.38 |
| Heart |
3.74 ± 0.92 |
0.63 ± 0.18 |
| Brain |
2.45 ± 0.49 |
0.22 ± 0.08 |
| 38 |
Blood |
1.89 ± 0.13 |
0.57 ± 0.04 |
| Liver |
8.39 ± 1.49 |
6.64 ± 0.46 |
| Kidney |
9.30 ± 0.84 |
3.51 ± 0.28 |
| Intestine |
1.87 ± 0.32 |
12.94 ± 2.43 |
| Spleen |
3.56 ± 0.99 |
1.04 ± 0.25 |
| Lung |
10.04 ± 2.09 |
1.04 ± 0.07 |
| Stomach |
1.22 ± 0.19 |
2.16 ± 0.55 |
| Heart |
5.92 ± 1.32 |
0.49 ± 0.06 |
| Brain |
3.57 ± 0.39 |
0.40 ± 0.02 |
 |
| | Scheme 3 | |
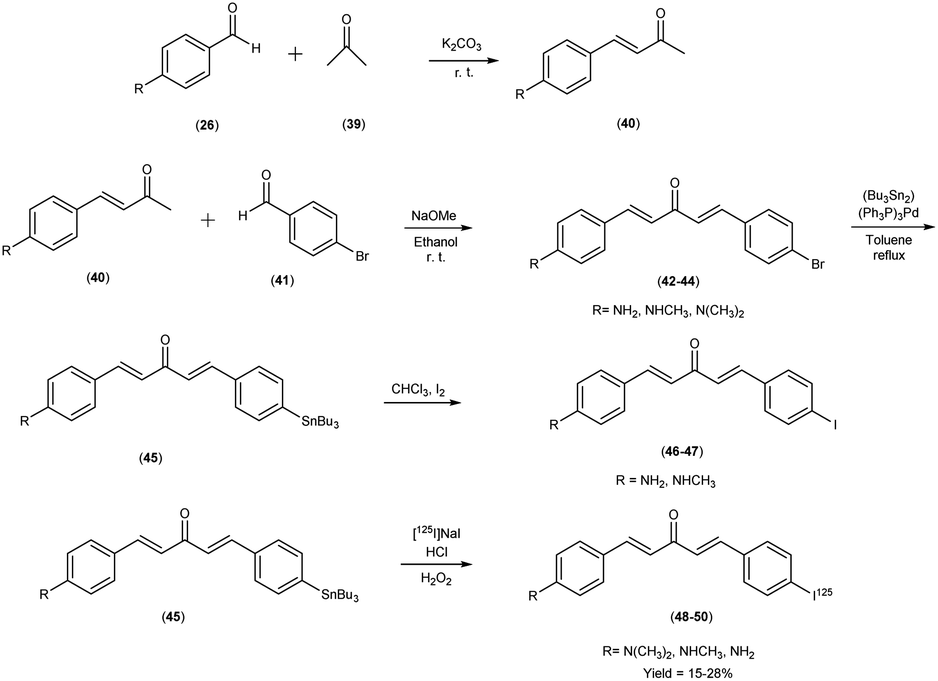
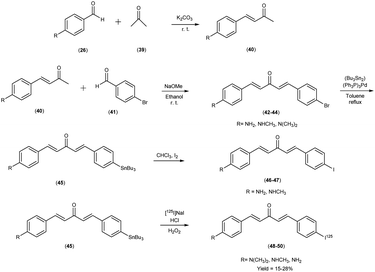
In 2011, Mengchao Cui et al.55 engendered a paradigm shift through the generation of an innovative and heterogeneous assemblage of dibenzylideneactones, which were subsequently subjected to an intricate scrutiny of their affinitive tendencies towards Aβ aggregates across a spectrum of distinctive ligand binding loci. The central and crucial step in this synthetic pathway involved the base-catalyzed Claisen condensation reaction, which was initiated by employing suitably substituted aromatic aldehydes26 in conjunction with aliphatic ketones.39 The tributyltin precursor45 was meticulously prepared from their respective bromo compounds42–44 via a bromo-to-tributyltin exchange reaction. This reaction was efficiently catalyzed by tetrakis(triphenylphosphine)palladium [(Ph3P)4Pd]. The radioiodinated ligands i.e. [125I]48, 49 and 50, were subsequently synthesized from the corresponding tributyltin precursors by means of an iododestannylation reaction. This transformation was conducted with hydrogen peroxide serving as the oxidizing agent, yielding radiochemical yields of 27.6%, 15.3%, and 24.1%, respectively. Finally, the radiochemical identity of [125I]48, 49 and 50 was rigorously verified through HPLC by concurrent injection with their nonradioactive counterparts, thus ensuring the accuracy and reliability of the radiotracer compounds. The introduction of a substituent group at the ortho position resulted in a substantial reduction or complete elimination of binding affinity. Conversely, the para position exhibited remarkable tolerance towards sterically demanding substitutions. During this study, three radioiodinated ligands (compounds 48, 49, and 50) were meticulously synthesized, all of which exhibited exceptionally high affinities for Aβ1–42 aggregates, with binding affinities spanning a range of 0.9 to 7.0 nM (Table 7). A discerning discourse was instigated through a comparative analysis, wherein the venerable [125I] served as the lodestar reference. In the view of the intricate structure–activity relationship (SAR), a cogent narrative emerged wherein the strategic substitution of the phenyl moiety with both electron-donating and electron-withdrawing appendages unveiled a vista of noteworthy compatibility in comparison with Aβ aggregates, thereby etching an indelible imprint upon the binding affinities exhibited by the dibenzylideneacetone substrate. The assessment of binding interactions between radiolabelled tracers (48 to 50) and Aβ plaques within brain tissue sections derived from AD patients or transgenic mice (APP/PS1) was conducted through in vitro autoradiography. As illustrated in Table 8, two of the radioiodinated probes, specifically compounds 48 and 50, demonstrated pronounced affinity for plaques, producing robust signal intensities primarily localized within the cortical region, while maintaining minimal background signal in the white matter of the AD brain sections. The lipophilicity, quantified by the logarithm of the distribution coefficient (logD), for the radiolabelled tracers, was determined under experimental conditions, revealing relatively elevated partition coefficients (logD = 2.97 to 3.66). This observation reflects the inherently lipophilic characteristics of these probes. Furthermore, the thyroidal accumulation of the three radioiodinated probes was observed to attain levels ranging from 14% to 24% of the injected dose per gram of tissue (ID/g) at the 1 hour post-injection time point. This finding is indicative of in vivo deiodination. Intriguingly, these radioiodinated ligand entities showcased a resounding congruence with the Aβ1–42 aggregates, a salient observation borne out by an astute confluence of experimental assays and analytical acumen. Further, refinement through structural exploration culminated in the crystallization of a pivotal insight into the N,N-dimethylamino substitution motif endowed the radiolabelled chalcones with a profound predisposition towards Aβ1–42 aggregates, thereby establishing a cardinal tenet in the realm of molecular recognition. Embarking upon a trajectory of biodistribution inquiries, the radiolabelled compounds exhibited a spirited foray into the cerebral milieu, marked by a robust initial penetration and expeditious clearance. In a poignant juxtaposition, the affinitive ardor of radioiodinated counterparts eclipsed that of their fluorinated brethren, thereby accentuating the heightened affinity engendered by the former compounds (Scheme 4).
Table 7 Inhibition constants (Ki, nM) for binding to aggregates of Aβ1–42 versus [125I]IMPY
| Compound |
R |
Ki (nM) |
| 44 |
Dimethylamino |
2.8 ± 0.5 |
| 46 |
Methyl amino |
2.8 ± 0.5 |
| 47 |
Amino |
7.0 ± 2.2 |
Table 8 Biodistribution of radioactivity after intravenous administration of radiolabelled compounds 48, 49, 50 in mice
| Compounds |
Organs |
Time after injection (min) |
| 2 min |
15 min |
30 min |
60 min |
120 min |
| 48 |
Blood |
11.62 ± 1.22 |
6.58 ± 2.15 |
3.98 ± 0.28 |
2.16 ± 0.55 |
2.29 ± 0.65 |
| Brain |
1.59 ± 0.04 |
1.11 ± 0.28 |
0.63 ± 0.03 |
0.28 ± 0.03 |
0.26 ± 0.07 |
| Heart |
6.42 ± 0.80 |
2.69 ± 0.85 |
1.62 ± 0.20 |
1.03 ± 0.24 |
1.19 ± 0.17 |
| Liver |
44.64 ± 6.19 |
32.80 ± 5.01 |
24.64 ± 2.85 |
11.54 ± 0.60 |
16.47 ± 1.38 |
| Spleen |
11.59 ± 1.10 |
9.94 ± 2.07 |
12.26 ± 3.95 |
6.28 ± 2.87 |
7.34 ± 0.67 |
| Lung |
24.45 ± 1.67 |
12.97 ± 3.05 |
6.91 ± 1.32 |
3.64 ± 0.47 |
3.35 ± 0.12 |
| Kidney |
15.86 ± 1.37 |
22.23 ± 5.52 |
15.63 ± 1.71 |
7.00 ± 2.62 |
8.55 ± 1.98 |
| Stomach |
1.16 ± 0.15 |
1.75 ± 0.15 |
1.85 ± 0.27 |
0.61 ± 0.24 |
0.77 ± 0.29 |
| Intestine |
1.38 ± 0.38 |
7.38 ± 2.75 |
11.62 ± 3.42 |
7.68 ± 2.53 |
17.65 ± 2.91 |
| Thyroid |
11.56 ± 2.61 |
14.93 ± 1.09 |
26.77 ± 1.41 |
23.53 ± 6.25 |
32.22 ± 1.13 |
| 49 |
Blood |
7.48 ± 0.81 |
6.02 ± 1.80 |
4.84 ± 1.40 |
3.35 ± 0.90 |
4.46 ± 0.92 |
| Brain |
4.68 ± 0.25 |
2.62 ± 0.22 |
1.38 ± 0.23 |
0.71 ± 0.06 |
0.54 ± 0.09 |
| Heart |
8.11 ± 1.74 |
4.14 ± 1.36 |
3.29 ± 1.42 |
1.74 ± 0.73 |
1.91 ± 0.60 |
| Liver |
39.22 ± 4.48 |
40.83 ± 4.21 |
23.88 ± 4.21 |
22.63 ± 3.73 |
27.83 ± 2.15 |
| Spleen |
4.57 ± 0.55 |
3.36 ± 0.14 |
2.22 ± 0.94 |
1.52 ± 0.38 |
1.23 ± 0.23 |
| Lung |
9.67 ± 1.12 |
7.42 ± 2.95 |
6.18 ± 2.58 |
3.50 ± 0.70 |
3.90 ± 0.73 |
| Kidney |
14.73 ± 2.23 |
16.49 ± 3.52 |
15.54 ± 5.56 |
10.46 ± 2.92 |
13.43 ± 4.53 |
| Stomach |
3.26 ± 1.37 |
3.82 ± 0.67 |
3.17 ± 0.97 |
3.34 ± 0.93 |
4.49 ± 0.82 |
| Intestine |
3.53 ± 1.19 |
10.33 ± 4.30 |
13.19 ± 5.85 |
23.62 ± 3.87 |
41.29 ± 9.04 |
| Thyroid |
18.20 ± 2.65 |
28.09 ± 4.45 |
27.78 ± 10.74 |
23.96 ± 3.78 |
72.93 ± 10.03 |
| 50 |
Blood |
3.89 ± 0.34 |
4.93 ± 1.91 |
4.12 ± 0.33 |
1.72 ± 0.58 |
2.52 ± 0.19 |
| Brain |
4.56 ± 0.42 |
2.37 ± 0.32 |
1.36 ± 0.19 |
0.54 ± 0.12 |
0.40 ± 0.07 |
| Heart |
7.06 ± 1.37 |
3.45 ± 0.59 |
2.01 ± 0.30 |
0.83 ± 0.17 |
1.11 ± 0.09 |
| Liver |
16.98 ± 2.29 |
29.08 ± 4.93 |
24.17 ± 4.15 |
16.43 ± 2.32 |
17.57 ± 1.63 |
| Spleen |
2.80 ± 0.15 |
2.02 ± 0.41 |
1.79 ± 0.17 |
0.80 ± 0.14 |
1.13 ± 0.39 |
| Lung |
9.25 ± 1.09 |
6.06 ± 1.18 |
4.81 ± 0.43 |
2.43 ± 0.76 |
2.82 ± 0.16 |
| Kidney |
11.20 ± 0.10 |
22.81 ± 3.42 |
16.31 ± 1.81 |
9.00 ± 1.67 |
9.52 ± 0.79 |
| Stomach |
1.37 ± 0.37 |
3.10 ± 2.24 |
3.12 ± 0.92 |
2.12 ± 0.62 |
3.07 ± 0.58 |
| Intestine |
2.27 ± 0.17 |
6.90 ± 2.44 |
10.85 ± 3.68 |
25.97 ± 4.93 |
25.70 ± 1.48 |
| Thyroid |
8.64 ± 2.19 |
8.39 ± 0.51 |
11.49 ± 3.53 |
14.14 ± 4.83 |
21.98 ± 3.75 |
 |
| | Scheme 4 | |
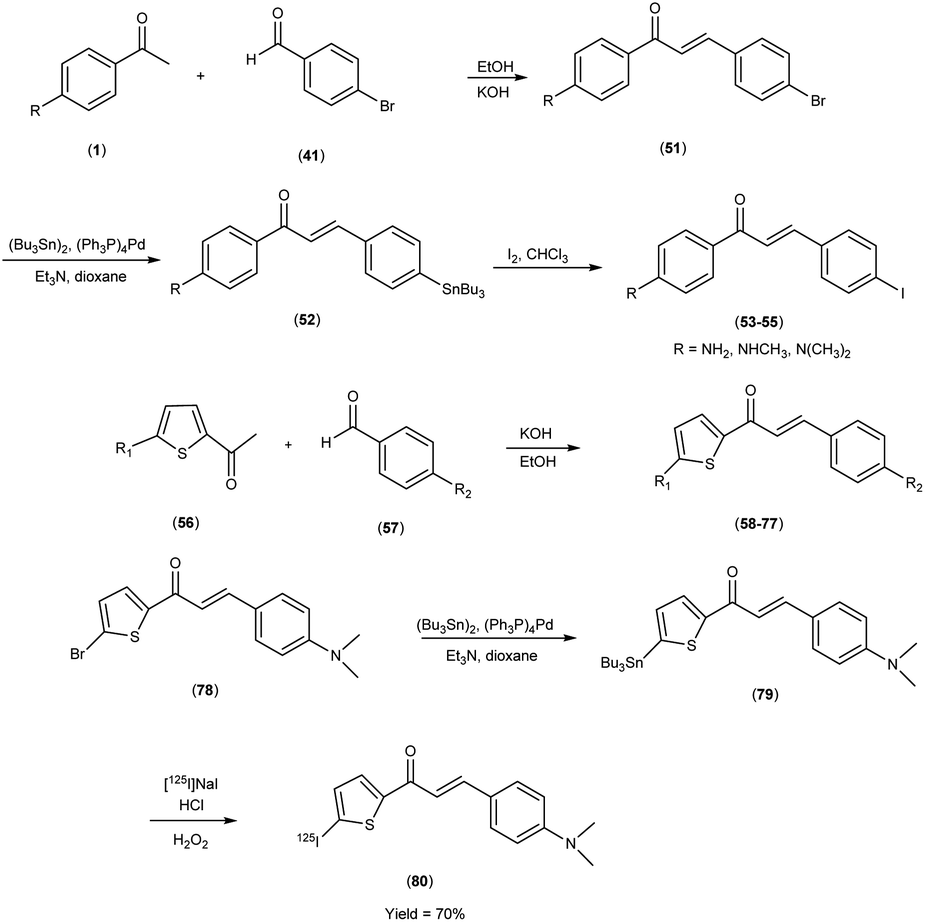
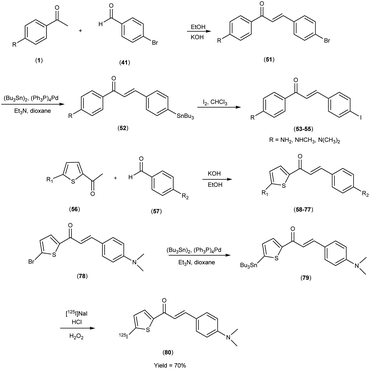
In 2007, Masahiro Ono et al.56 embarked upon the innovative synthesis of chalcone encompassed a base-catalyzed condensation process, wherein appropriately substituted ketones (1 & 56) were coupled with substituted benzaldehydes or heterocyclic aldehydes. Within this procedure, the substituted ketones were treated with the corresponding substituted benzaldehydes41 or heterocyclic aldehydes56 in the presence of a 10% aqueous potassium hydroxide (KOH) solution in ethanol at ambient temperature. This reaction led to the formation of the intended chalcone (51 & 58 to 77). The tin compounds (51 & 78) were derived from their respective bromo compounds (52 & 79). This conversion was facilitated by a bromo-to-tributyltin exchange reaction, catalyzed by palladium in its zero oxidation state, Pd(0). Subsequently, the tributyltin derivative (79) was readily subjected to a reaction with iodine in chloroform under ambient conditions, yielding the corresponding iodo derivatives, 53 to 55. Furthermore, the tributyltin derivative 79 was employed as the starting material for radioiodination in the synthesis of [125I]79. This novel radioiodinated ligand was successfully produced through an iododestannylation reaction, utilizing hydrogen peroxide as the oxidizing agent, ultimately yielding the desired radioiodinated ligand (80). The resulting ensemble of molecular entities, ranging in yield from modest to outstanding, bore witness to the intricate orchestration of this synthetic enterprise. Eager to unveil the intricate nuances of ligand Aβ amyloid interactions, a comprehensive exploration unfolded wherein these newly minted chalcone derivatives assumed the mantle of Aβ-amyloid imaging probes. This experimental saga revealed a pronounced affinity towards the Aβ1–42 aggregates, standing as a poignant testament to the judicious molecular design and synthetic endeavor. During in vitro binding investigations involving Aβ aggregates, a diverse spectrum of Ki values was ascertained, underscoring their structural dependence. Notably, compound 58, which exhibited the most elevated binding affinity towards Aβ aggregates, conspicuously demonstrated specific staining of Aβ-amyloid plaques and cerebrovascular amyloids within the ambit of the in vitro binding assays (Table 9). Collectively, these findings strongly advocate for the imperative pursuit of comprehensive investigations into the utility of the novel radioiodinated compound 80 as a prospective and valuable imaging probe for Aβ-amyloid deposits. The unfolding binding studies, an endeavor of discerning scholarship, unveiled a compelling narrative wherein the radioiodinated chalcones exhibited a heightened proclivity for binding compared to their radioiodinated flavone counterparts. This observation resonates profoundly with the stratagem of molecular modification, resonating with an overarching theme of molecular recognition and affinity. In the context of elucidating the structure–activity relationship (SAR) governing binding affinities towards synthetic Aβ1–42 aggregates, compound [125I](E)-1-(5-iodo-2-thienyl)-3-(4-dimethylaminophenyl)-2-propen-1-one (80) has been found to exhibit the most pronounced binding affinity in the in vitro experiments. Notably, the in vivo investigations involving an AD model murine system revealed the capacity of compound 80 to enable the visualization of Aβ-amyloid plaques within the cerebral tissue. Furthermore, biodistribution studies conducted in healthy mice demonstrated that radiolabelled compound (80) exhibited favorable cerebral uptake, with a measured value of 2.56% ID/g at 2 minutes post-injection, followed by rapid elimination from the cerebral compartment, as evidenced by a diminished level of 0.21% ID/g observed 60 minutes post-injection (Table 10). Against the backdrop of compound 80, the vanguard of radiolabelled moieties, intricate biodistribution scrutiny was pursued, guided by a central tenet in the AD diagnostic the ability of imaging probes to surmount the cerebral barricades and efficaciously retreat from the milieu of affliction. This dualistic capability echoes a cardinal tenet in the quest for superior imaging modalities, thereby etching a profound imprint upon the landscape of neuroimaging diagnostics. Unveiling the cellular dimensions of these chalcone derivatives, the tenor of binding characteristics reverberated with a resonance of significant cellular uptake pertaining to Aβ1–42 aggregates, thereby illuminating the dynamic interplay between molecular design and cellular recognition, thus accentuating the potential utility of these chalcone scaffolds in the pursuit of elucidating the tapestry of Alzheimer's pathology (Scheme 5).
Table 9 Inhibition constants of chalcone and chalcone-like derivatives on ligand binding to Aβ1–42 aggregates
| Compounds |
R1 |
4-Iodophenyl |
Ki (nM) |
| 53 |
4-Aminophenyl 4-iodophenyl |
4-Iodophenyl |
248 ± 56 |
| 54 |
4-Methylaminophenyl |
4-Iodophenyl |
23.9 ± 3.6 |
| 55 |
4-Dimethylaminophenyl |
4-Dimethylaminophenyl |
13.3 ± 1.9 |
| 58 |
5-Iodo-2-thienyl |
Phenyl |
151 ± 16 |
| 59 |
4-Iodophenyl |
2-Furanyl |
908 ± 212 |
| 60 |
4-Iodophenyl |
3-Furanyl |
125 ± 9.2 |
| 61 |
4-Iodophenyl |
2-Thienyl |
102 ± 16 |
| 62 |
4-Iodophenyl |
3-Thienyl |
93 ± 11 |
| 63 |
4-Iodophenyl |
2-Imidazoyl |
797 ± 316 |
| 64 |
4-Iodophenyl |
2-Thiazoyl |
>10000 |
| 65 |
4-Iodophenyl |
5-Dimethylamino-2-furanyl |
1132 ± 344 |
| 66 |
4-Iodophenyl |
5-Dimethylamino-2-thienyl |
113 ± 10 |
| 67 |
4-Iodophenyl |
5-Dimethylamino-2-thienyl |
137 ± 3.4 |
| 68 |
5-Iodo-2-thienyl |
5-Dimethylamino-2-furanyl |
1608 ± 85 |
| 69 |
5-Iodo-2-thienyl |
4-Dimethylaminophenyl |
126 ± 13 |
| 70 |
5-Bromo-2-furanyl |
5-Dimethylamino-2-thienyl |
2648 ± 222 |
| 71 |
5-Bromo-2-furanyl |
5-Dimethylamino-2-furanyl |
>10000 |
| 72 |
5-Bromo-2-furanyl |
4-Aminophenyl |
121 ± 40 |
| 73 |
5-Iodo-2-thienyl |
5-Bromo-2-thienyl |
476 ± 48 |
| 74 |
4-Aminophenyl |
4-Methylaminophenyl |
14.1 ± 0.6 |
| 75 |
5-Iodo-2-thienyl |
5-Bromo-2-thienyl |
198 ± 49 |
| 76 |
4-Methylaminophenyl |
5-Bromo-2-thienyl |
106 ± 7.1 |
| 77 |
4-Dimethylaminophenyl |
5-Bromo-2-thienyl |
248 ± 56 |
| CR |
— |
— |
>10000 |
| ThT |
— |
— |
>10000 |
| AIC |
4-Iodophenyl |
4-Aminophenyl |
105 ± 1.2 |
| IMC |
4-Iodophenyl |
4-Methylaminophenyl |
6.3 ± 1.6 |
| DMIC |
4-Iodophenyl |
4-Dimethylaminophenyl |
2.9 ± 0.3 |
Table 10 Biodistribution of radioactivity after intravenous administration of radiolabelled compound 80 in mice
| Compounds |
Time after injection (min) |
| 2 min |
10 min |
30 min |
60 min |
| 80 |
2.46 ± 0.30 |
0.75 ± 0.31 |
0.31 ± 0.04 |
0.21 ± 0.02 |
 |
| | Scheme 5 | |
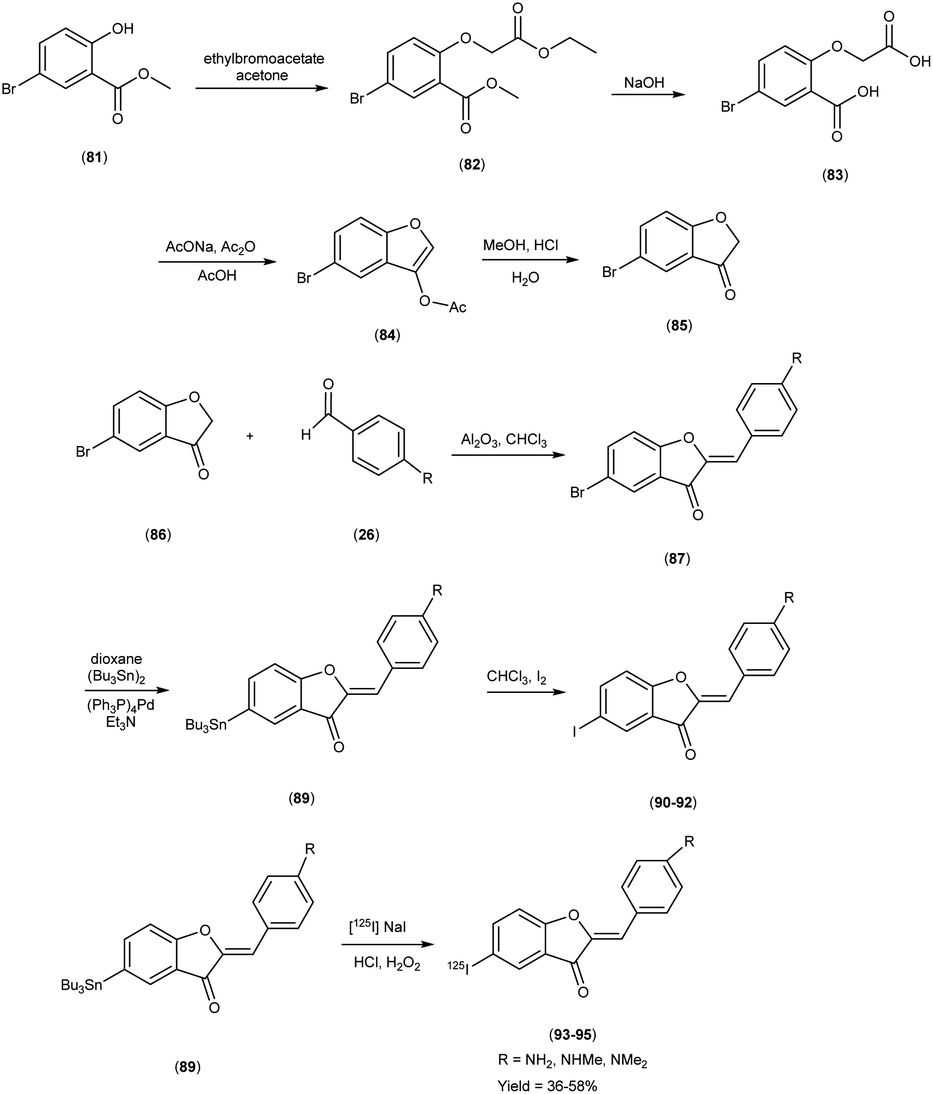
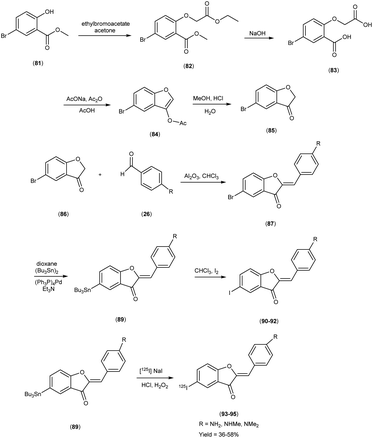
In 2007, Masahiro Ono and colleagues57 orchestrated the synthesis of aurone derivatives, ushering these molecular entities into the crucible of evaluation of Aβ-amyloid plaques inherent in AD pathology. The synthesis of the aurone backbone was accomplished through an aldol reaction i.e. benzofuranones (86) were combined with benzaldehydes26 in the presence of aluminum oxide (Al2O3). Furthermore, the tributyltin derivative (89) was prepared from their corresponding bromo compound (87 & 88), through a bromo-to-tributyltin exchange reaction catalyzed by palladium in its zero oxidation state, Pd(0). Additionally, the tributyltin derivative (89) was a suitable starting material for radioiodination in the synthesis of radioiodinated compounds 93, 94 and 95. The new radioiodinated aurones were successfully generated through an iododestannylation reaction, facilitated by the utilization of hydrogen peroxide as the oxidizing agent. This process yielded the expected radioiodinated ligands, demonstrating the versatility of these derivatives in radiopharmaceutical applications in 36% to 54% yield. In the realm of binding studies, a captivating narrative unfurled wherein the aurone derivatives emerged as avid adherents to Aβ1–42 aggregates. This proclivity for binding resonates as an ode to the strategic synthesis and molecular architecture, elucidating an affinity toward the morbid hallmark's characteristic of AD. Embarking upon an exploration of in vitro manifestations, the aurone derivatives materialized as poignant probes, casting a luminous radiance upon Aβ-amyloid plaques within the confines of AD model mice brains. When conducting in vitro binding investigations employing Aβ1–42 aggregates in the presence of aurone derivatives, notable findings emerged, revealing the manifestation of substantial binding affinities towards Aβ1–42 aggregates. These affinities were quantitatively delineated by Ki values spanning a range of 1.2 to 6.8 nM (Table 11). Upon juxtaposing the Ki values associated with the previously reported radioiodinated flavones, it becomes evident that the Ki values associated with the radioiodinated aurones (93–95) were comparatively lower. This disparity signifies that the radioiodinated aurones exhibit heightened binding affinities towards Aβ-amyloid plaques in comparison to their radioiodinated flavone counterparts. In the context of in vitro plaque labelling endeavors using brain sections obtained from double transgenic mice, it was observed that aurone derivatives exhibited a profound proclivity for vividly staining Aβ-amyloid plaques. The subsequent biodistribution studies conducted in normal mice following intravenous administration of radioiodinated aurones unveiled substantial cerebral uptake, ranging from 1.9% to 4.6% ID/g at 2 minutes post-injection, concomitant with rapid elimination from the cerebral compartment, registering values between 0.11% and 0.26% ID/g at the 60 minutes mark (Table 12). These pharmacokinetic attributes align notably with the prerequisites for effective amyloid imaging agents. The findings gleaned from this investigation collectively propose the potential utility of novel radiolabelled aurones as valuable agents for the detection of Aβ-amyloid plaques in the AD-afflicted brain. Consequently, these commendable pharmacokinetic attributes exhibited by radioiodinated aurones assume pivotal importance in the context of amyloid plaque detection in AD. These biodistribution data further underscore that the novel radioiodinated aurones may possess superior in vivo pharmacokinetic profiles compared to their radioiodinated flavone counterparts, rendering them potentially more suitable for amyloid imaging in the context of AD (Scheme 6).
Table 11 Inhibition constants of aurone derivatives on ligand binding to Aβ1–42 aggregates
| Compound |
Ki (nM) |
| 90 |
2.69 ± 0.16 |
| 91 |
1.24 ± 0.11 |
| 92 |
6.82 ± 0.48 |
Table 12 Biodistribution of radioactivity after intravenous administration of radiolabelled compounds 93, 94 and 95 in mice
| Compounds |
Time after injection (min) |
| 2 |
10 |
30 |
60 |
| 93 |
4.57 ± 0.27 |
1.51 ± 0.17 |
0.49 ± 0.06 |
0.26 ± 0.03 |
| 94 |
3.17 ± 0.45 |
1.22 ± 0.09 |
0.32 ± 0.02 |
0.24 ± 0.03 |
| 95 |
1.89 ± 0.38 |
0.69 ± 0.21 |
0.26 ± 0.04 |
0.11 ± 0.03 |
 |
| | Scheme 6 | |
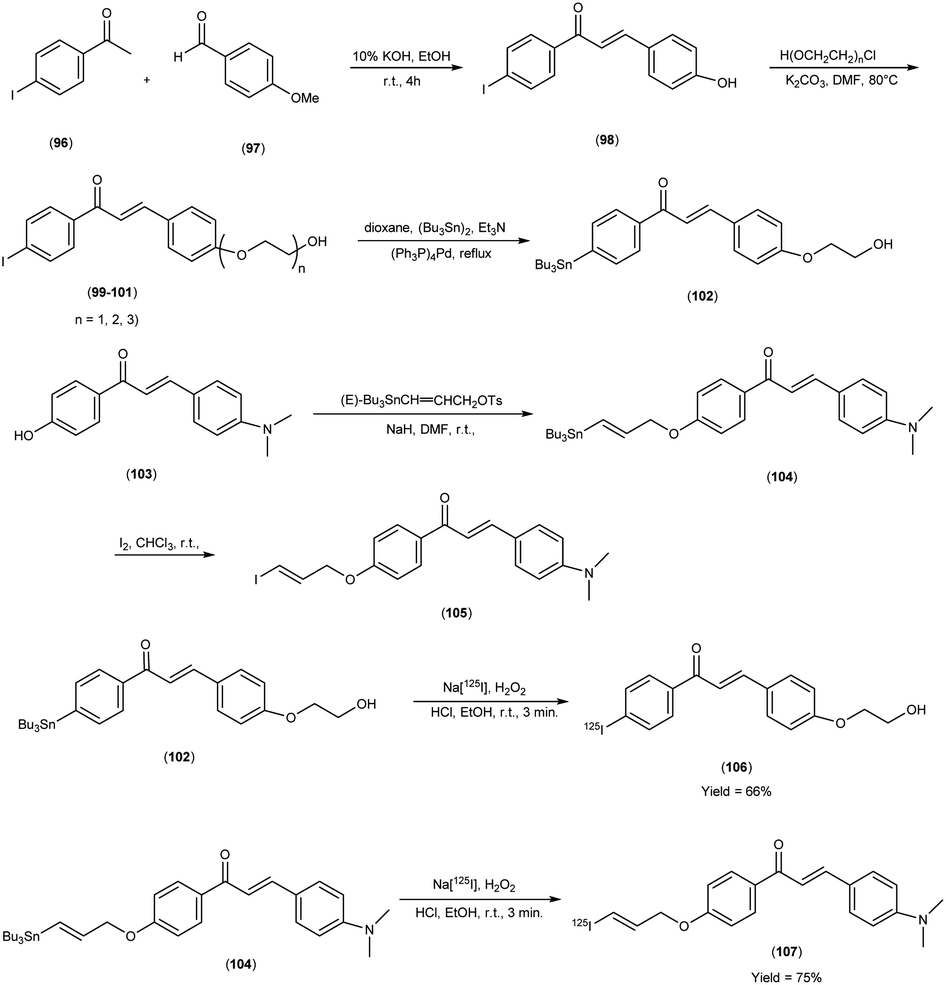
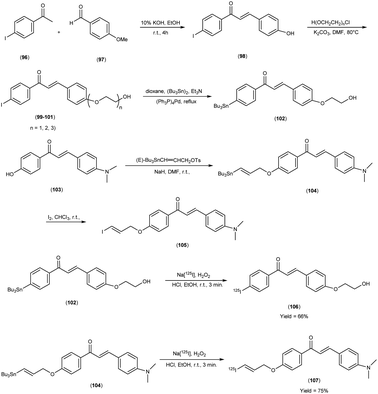
In 2014, Takeshi Fuchigami and colleagues58 synthesized radioiodinated chalcones and characterized them as innovative SPECT imaging agents for Aβ-amyloid plaques. The anticipated chalcone derivatives were synthesized through a series of carefully executed chemical reactions. Initially, 4-iodoacetophenone (96) was subjected to a reaction with 4-methoxybenzaldehyde (97), conducted in the presence of a basic catalyst (10% KOH) in ethanol at room temperature yielded 4-iodo-4-methoxy-chalcone (98) in a quantitative yield. The direct alkylation of compound 98 was then carried out by reacting it with ethylene chlorohydrin, ethylene glycol mono-2-chloroethyl ether, or 2-[2-(2-chloroethoxy)ethoxy]ethanol in the presence of potassium carbonate, conducted in dimethylformamide (DMF). This process provided compounds 99, 100 and 101, with yields of 52%, 49% and 17%, respectively. Subsequently, the tributyltin derivative, denoted as compound 104, was synthesized from the corresponding iodo derivative, via an iodo-to-tributyltin exchange reaction catalyzed by palladium in its zero oxidation state, Pd(0), resulting in a yield of 48%. The p-toluenesulfonate ester of (E)-3-(tri-n-butylstannyl)prop-2-enol was then coupled with phenol 103 to yield the tributyltin derivative 104, with a yield of 45%. The compound 104 underwent reaction with iodine in chloroform, forming the desired compound 105. The radioiodinated chalcones, 106 and 107 were prepared through an iododestannylation reaction, employing hydrogen peroxide as the oxidizing agent. These compounds were obtained with radiochemical yields ranging from 66% to 75%, and they exhibited radiochemical purities exceeding 95% after purification via high-performance liquid chromatography (HPLC). Specifically, the monoethyleneoxy and allyloxy derivatives demonstrated notable affinity towards Aβ1–42 aggregates. Through fluorescence imaging, it was demonstrated that the monoethyleneoxy and allyloxy derivatives prominently labelled thioflavin-S positive antibody-bound plaques within brain slices obtained from Tg2576 transgenic mice. The monoethyleneoxy derivative (99) and allyloxy derivative (105) exhibited notable affinities for Aβ1–42 aggregates, as evidenced by their respective Ki values of 24 and 4.5 nM (Table 13). The evaluation of the inhibition constants (Ki) for compounds 99 to 101 unveiled a discernible influence of the ethyleneoxy group length within the chalcone scaffold on their binding affinities toward Aβ aggregates. The hierarchy in binding affinity for Aβ aggregates was established as follows: monoethyleneoxy derivative 99 (Ki = 24.0 nM) > triethyleneoxy derivative 101 (Ki = 87.8 nM) > diethyleneoxy derivative 100 (Ki = 127.1 nM). The statistical analyses further confirmed the significance of these disparities, with Ki value of compound 99 being markedly lower than both compound 100 (P < 0.001; ANOVA, Bonferroni t-test) and compound 101 (P < 0.001). Additionally, Ki value displayed a statistically significant decrease relative to compound 100 (P < 0.05). In vitro autoradiography assessments unveiled that [125I]106 exhibited negligible accumulation in Aβ plaques within brain sections obtained from Tg2576 mice. Conversely, the distribution pattern of radioiodinated compound 107 closely corresponded with the presence of Aβ plaques, observed both in Tg2576 mouse brain sections and those from AD patient. Notably, biodistribution investigations in normal mice illustrated the favorable in vivo pharmacokinetic attributes of radioiodinated compound 106, with observed values of 4.82% ID/g at 2 minutes and 0.45% ID/g at 60 minutes post-injection. In contrast, [125I]107 demonstrated only modest cerebral uptake (1.62% ID/g at 2 minutes) and exhibited a protracted retention period (0.56% ID/g at 60 minutes) (Table 14). While compound 107 exhibited promising binding properties towards Aβ plaques, it is evident that further structural refinements are warranted to enhance blood–brain barrier permeability and brain clearance. Consequently, the incorporation of the iodoallyloxy group into the chalcone backbone has proven effective in enhancing its affinity for Aβ plaques. This study underscores the potential for further chemical modifications to yield valuable Aβ imaging probes based on the chalcone scaffold (Scheme 7).
Table 13 Inhibition constant (Ki) of chalcone derivatives for Aβ aggregates
| Compounds |
Ki (nM) |
| 99 |
24.0 ± 10.4 |
| 100 |
127.1 ± 27.3 |
| 101 |
87.8 ± 20.3 |
| 105 |
4.5 ± 1.5 |
| DMIC |
10.8 ± 0.9 |
Table 14 Biodistribution of radioactivity after injection of [125I] labelled chalcone derivatives 106 and 107 in normal mice
| Compounds |
Organs |
Time after injection |
| 2 min |
10 min |
30 min |
60 min |
| 106 |
Blood |
3.95 ± 0.30 |
3.01 ± 0.58 |
2.60 ± 0.42 |
2.27 ± 0.60 |
| Liver |
14.76 ± 2.88 |
20.11 ± 4.81 |
18.83 ± 1.20 |
15.57 ± 3.17 |
| Kidney |
10.34 ± 1.72 |
9.05 ± 1.72 |
10.23 ± 1.96 |
9.89 ± 3.07 |
| Intestine |
3.98 ± 0.66 |
10.55 ± 3.41 |
26.39 ± 6.69 |
25.16 ± 6.54 |
| Spleen |
2.69 ± 0.54 |
2.35 ± 0.74 |
1.21 ± 0.10 |
0.81 ± 0.14 |
| Lung |
7.14 ± 1.34 |
4.63 ± 1.37 |
3.21 ± 1.24 |
1.94 ± 0.42 |
| Stomach |
2.22 ± 0.41 |
3.38 ± 2.38 |
4.27 ± 3.43 |
4.01 ± 0.38 |
| Pancreas |
6.12 ± 1.26 |
3.37 ± 1.00 |
1.07 ± 0.44 |
0.90 ± 0.54 |
| Heart |
6.20 ± 1.19 |
3.13 ± 0.88 |
1.54 ± 0.13 |
1.05 ± 0.32 |
| Brain |
4.82 ± 1.19 |
2.86 ± 0.87 |
1.00 ± 0.05 |
0.45 ± 0.09 |
| 107 |
Blood |
2.65 ± 0.21 |
2.29 ± 0.19 |
1.97 ± 0.55 |
2.43 ± 1.18 |
| Liver |
23.21 ± 2.52 |
|
16.91 ± 0.62 |
10.26 ± 0.55 |
| Kidney |
8.98 ± 1.60 |
6.06 ± 0.57 |
4.52 ± 0.59 |
4.17 ± 1.12 |
| Intestine |
1.74 ± 0.59 |
2.38 ± 0.88 |
4.45 ± 1.70 |
6.34 ± 2.57 |
| Spleen |
4.43 ± 1.11 |
5.34 ± 2.03 |
4.68 ± 1.31 |
3.92 ± 1.52 |
| |
Lung |
8.08 ± 1.39 |
4.48 ± 0.38 |
3.16 ± 0.42 |
3.28 ± 1.01 |
| Stomach |
2.49 ± 1.07 |
5.19 ± 2.16 |
8.98 ± 5.05 |
16.44 ± 6.82 |
| Pancreas |
4.02 ± 1.48 |
3.27 ± 0.51 |
2.35 ± 0.63 |
2.14 ± 1.12 |
| Heart |
8.70 ± 1.66 |
3.39 ± 0.38 |
2.20 ± 0.13 |
2.10 ± 0.85 |
| Brain |
1.62 ± 0.30 |
1.63 ± 0.22 |
0.87 ± 0.18 |
0.56 ± 0.16 |
 |
| | Scheme 7 | |
4. 99mTc labelled chalcones for AD
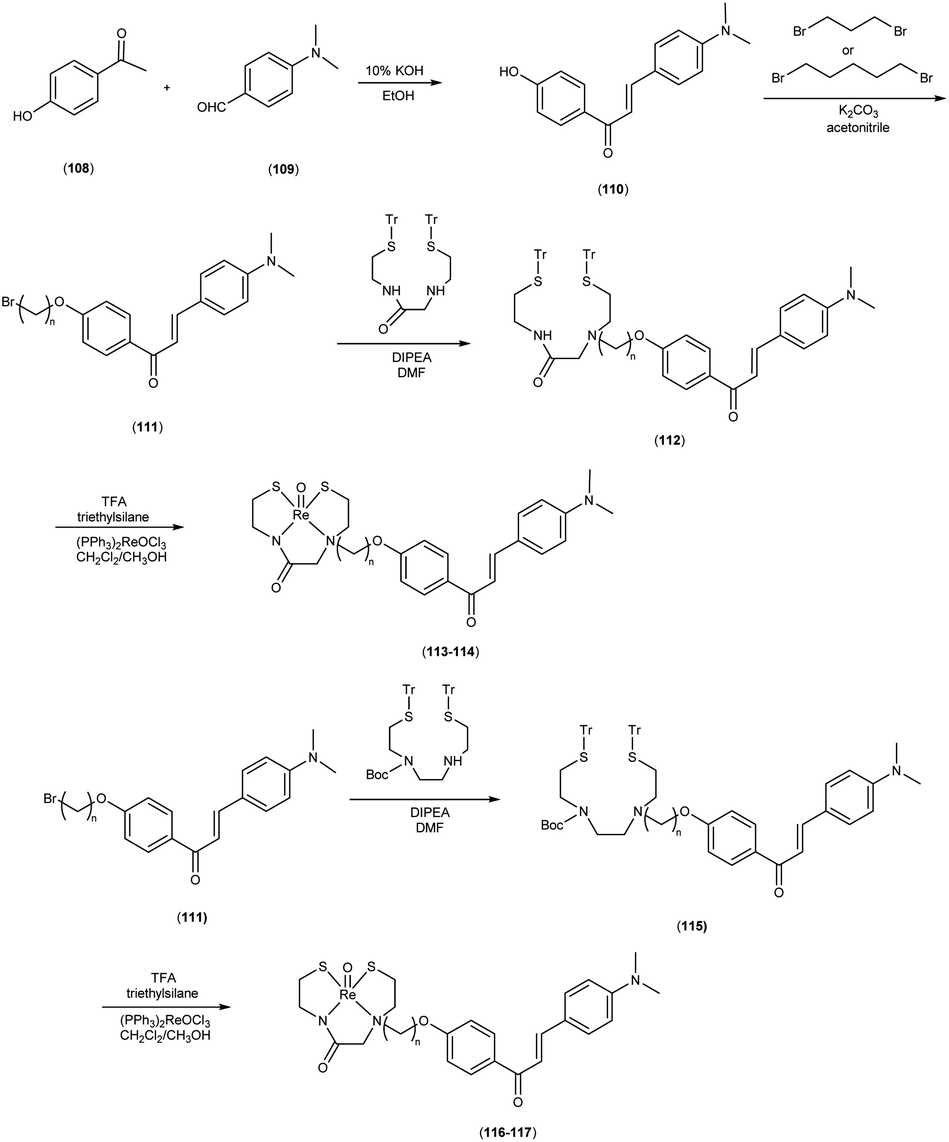
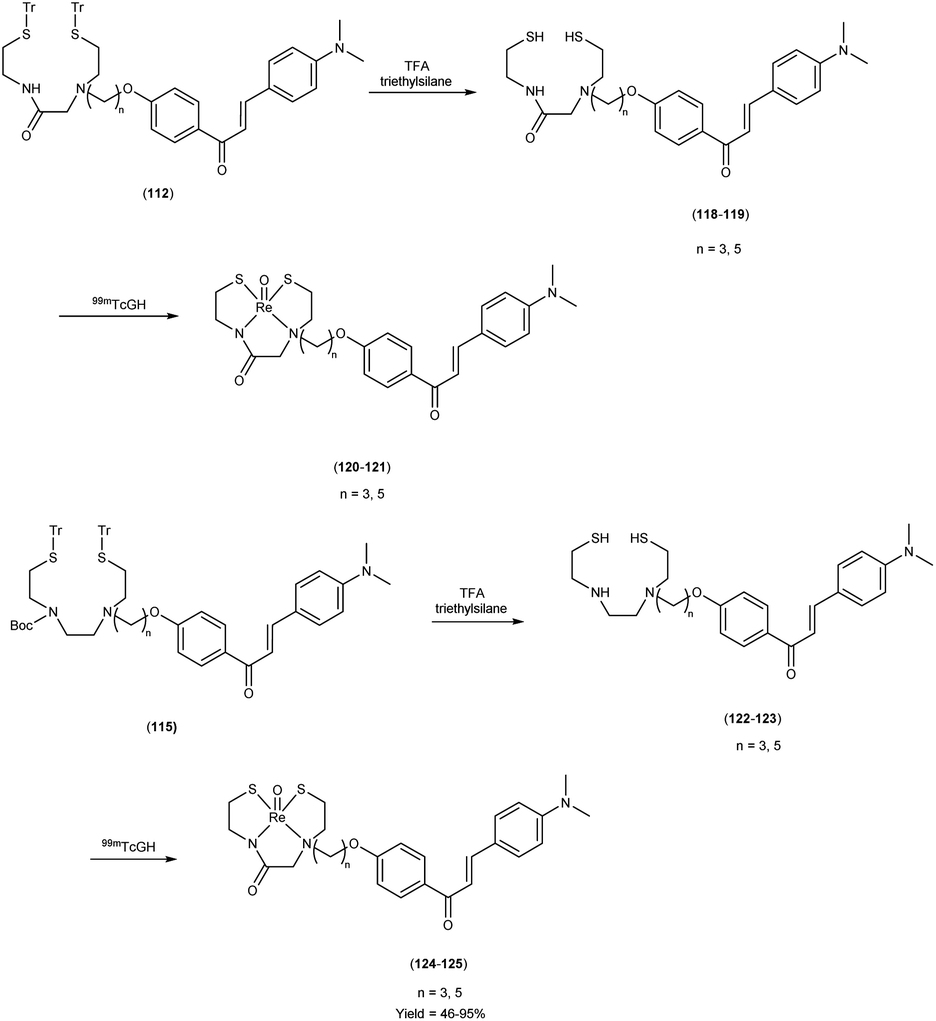
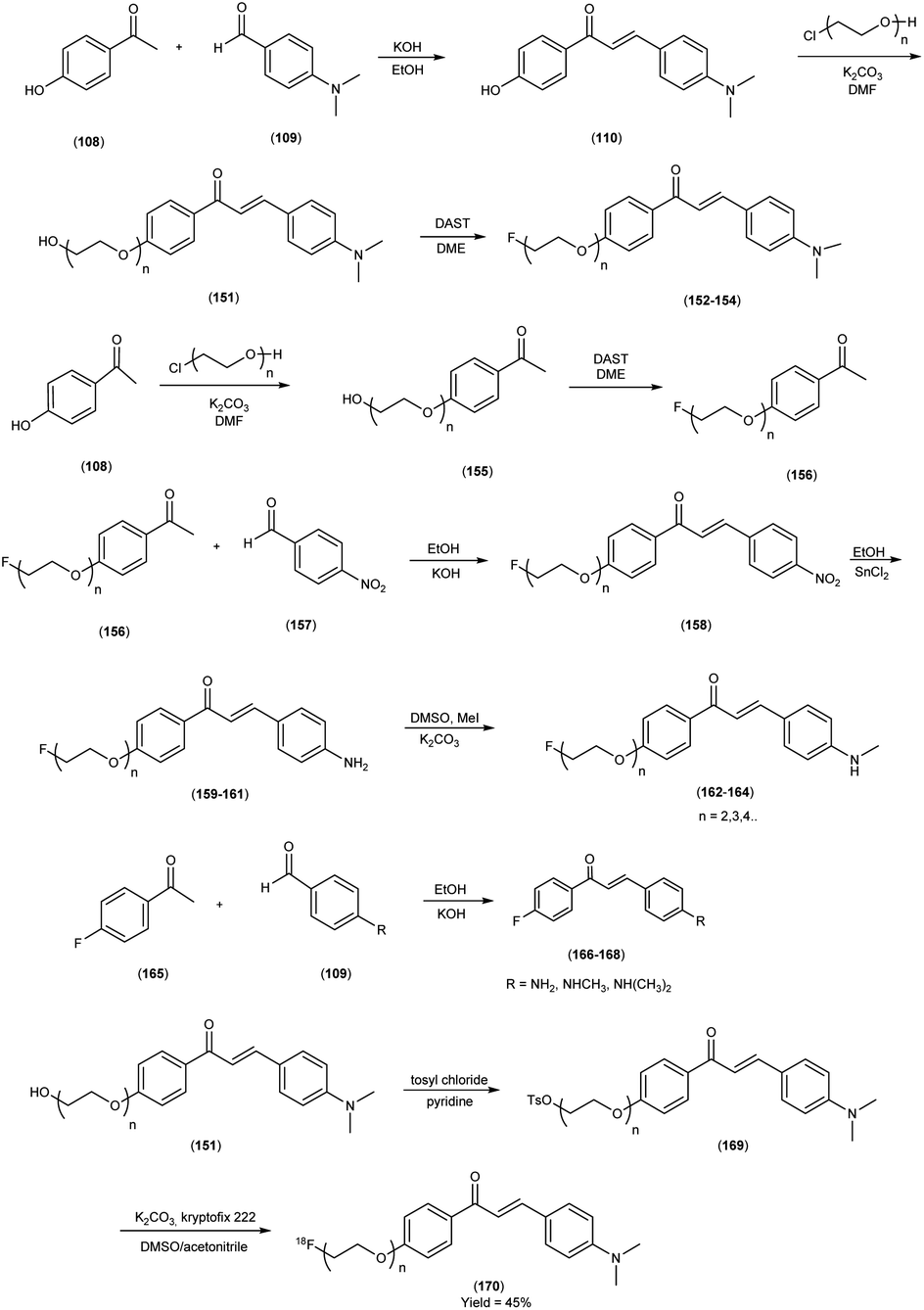
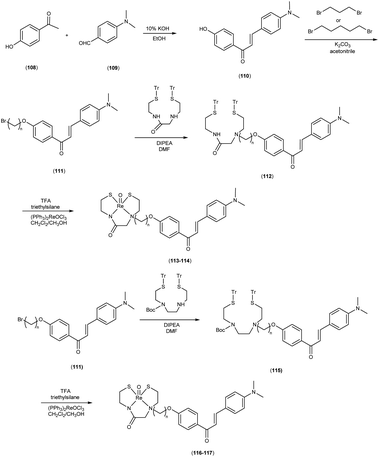
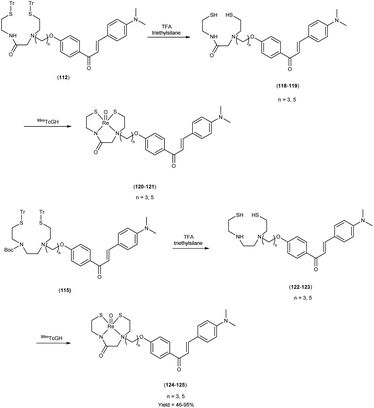
In 2010, Masahiro Ono and colleagues59 reported the exploration into the realm of molecular imaging in AD. Specifically, the authors synthesized and labelled chalcones with technetium-99m (99mTc), subsequently investigating their potential utility as molecular probes for imaging Aβ-amyloid plaques in AD pathology. One of the most widely employed methods for synthesizing chalcones involves the condensation of 4-hydroxyacetophenone (108) with 4-dimethylaminobenzaldehyde (109). This reaction occurred in the presence of a basic catalyst e.g. 10% potassium hydroxide (KOH), conducted in ethanol at room temperature. This reaction resulted in the formation of 4-dimethylamino-4-hydroxy-chalcone (110) with a notable yield of 70%. The subsequent step involved the reaction of dibromo with compound 110, giving rise to two distinct chalcone derivatives (111) both featuring alkyl groups of different lengths (n = 3 or 5). After this step, the compound 111 (n = 5) or 6 (n = 3) was coupled with either Tr-MAMA or Tr-Boc-BAT, leading to the formation of compound 112 (referred to as Tr-MAMA-chalcones or Tr-Boc-BAT). The next set of compounds, specifically 118 and 119 which served as precursors for the labelling with 99mTc, were derived by deprotecting the thiol groups in compounds 122 and 123 respectively. Subsequently, the Re complexes (113 and 114) were synthesized through reactions involving corresponding compounds 112 and 115 respectively, with tetrakis(triphenylphosphine)rhenium(VII) oxide chloride, (PPh3)2ReOCl3. The 99mTc complexes (120, 121, 124, and 125) were prepared through a ligand exchange reaction. This process involved the utilization of the precursor 99mTc-glucoheptonate (GH), leading to the formation of the anticipated 99mTc-labeled complexes. Notably, these synthesized chalcones exhibited enhanced selectivity for targeting Aβ1–42 aggregates, a hallmark of AD pathology. The innovative chalcones analogs, incorporating 99mTc/Re compounds, effectively illuminated amyloid plaques within the brainstem section of experimental animals afflicted with AD. Biodistribution investigation imparts pivotal insights into cerebral uptake, a vital parameter for evaluating the suitability of an imaging probe. An exemplary β-amyloid imaging agent should possess the capability to efficiently breach the blood–brain barrier, thereby facilitating the delivery of an adequate dosage into the cerebral milieu. Simultaneously, it should demonstrate swift clearance from non-pathological brain regions, a factor that significantly contributes to enhancing the signal-to-noise ratio within the AD-afflicted brain. Prior investigations have indicated that the optimal degree of lipophilicity required for effective brain penetration lies within the logP range of 113 to 117, extensive biodistribution studies were also conducted employing normal murine models (Table 15). Remarkably, the chalcones labelled with 99mTc demonstrated a remarkable capacity to traverse the formidable blood–brain barrier, thereby facilitating substantial accumulation within the brain tissue. In the context of this assay, it was ascertained that the proportion of radioactivity associated with nonspecific binding to Aβ1–42 aggregates ranged from 1.9% to 3.2% across the four 99mTc-labelled chalcones. This observation implies that most of the radioactivity is attributed to the occupation of specific binding sites for Aβ aggregates. Furthermore, it was observed that the percentage of radioactivity associated with chalcone binding to these aggregates exhibited a dose-dependent increase contingent upon the Aβ1–42 dosage. Moreover, biodistribution investigations conducted in healthy mice unveiled the commendable in vivo performance of 99mTc-BAT-chalcone, designated as compound 124. This compound exhibited a substantial brain uptake, quantified at 1.48% ID/g, as early as 2 minutes post-administration. Remarkably, compound 124 demonstrated rapid elimination from the cerebral compartment, registering a diminution to 0.17% ID/g after 60 minutes. This rapid clearance attribute aligns favorably with the prerequisites for an ideal imaging agent. Consequently, [99mTc]124 emerges as a promising candidate for the imaging of β-amyloid plaques within the context of AD-afflicted brains (Table 16). This compound exhibited exceptional attributes, notably its heightened absorption profile and rapid clearance from the brain upon administration into the normal meticulous characterization of amyloid plaques in AD pathology (Schemes 8 and 9).
Table 15 HPLC retention times of 99mTc-labelled chalcones and their Re analogues and logP values of 99mTc-labelled chalcones
| 99mTc compound |
Retention time (min) |
Re compound |
Retention time (min) |
Logp of 99mTc compounds |
| 120 |
14.2 |
113 |
13.5 |
2.55 ± 0.19 |
| 121 |
20.3 |
114 |
18.4 |
2.73 ± 0.16 |
| 124 |
9.3 |
116 |
8.6 |
1.51 ± 0.09 |
| 125 |
12.4 |
117 |
11.2 |
2.51 ± 0.05 |
Table 16 Biodistribution of radioactivity after injection of 99mTc-labelled chalcone derivatives in normal mice
| Compounds |
Organs |
Time after injection (min) |
| 2 min |
10 min |
30 min |
60 min |
| 120 |
Blood |
1.85 ± 0.31 |
0.91 ± 0.15 |
0.62 ± 0.16 |
0.28 ± 0.02 |
| Liver |
18.92 ± 2.03 |
24.48 ± 1.34 |
26.63 ± 5.57 |
17.05 ± 1.52 |
| Kidney |
9.45 ± 1.24 |
7.62 ± 3.79 |
9.85 ± 1.35 |
5.25 ± 0.87 |
| Intestine |
4.71 ± 0.63 |
12.45 ± 2.90 |
34.90 ± 3.01 |
36.49 ± 6.04 |
| Spleen |
4.16 ± 0.52 |
3.17 ± 0.52 |
2.37 ± 0.48 |
1.28 ± 0.09 |
| Lung |
16.1 ± 3.34 |
6.59 ± 1.23 |
3.61 ± 1.00 |
1.44 ± 0.17 |
| Stomach |
0.76 ± 0.10 |
1.31 ± 0.15 |
2.06 ± 0.65 |
1.67 ± 0.29 |
| Pancreas |
3.78 ± 0.49 |
5.28 ± 0.33 |
4.71 ± 0.96 |
2.27 ± 0.14 |
| Heart |
11.05 ± 1.99 |
5.10 ± 1.00 |
2.16 ± 0.63 |
0.87 ± 0.22 |
| Brain |
0.22 ± 0.05 |
0.32 ± 0.14 |
0.19 ± 0.030 |
0.11 ± 0.01 |
| 121 |
Blood |
2.49 ± 0.24 |
0.92 ± 0.05 |
0.50 ± 0.11 |
0.35 ± 0.13 |
| Liver |
23.89 ± 2.51 |
24.03 ± 4.51 |
23.18 ± 3.67 |
21.95 ± 4.58 |
| Kidney |
11.26 ± 0.62 |
9.66 ± 0.58 |
7.64 ± 0.88 |
6.39 ± 1.51 |
| Intestine |
6.27 ± 0.31 |
15.99 ± 0.87 |
37.18 ± 2.54 |
54.09 ± 10.94 |
| Spleen |
3.15 ± 0.22 |
1.64 ± 0.33 |
0.69 ± 0.16 |
0.35 ± 0.15 |
| Lung |
15.71 ± 4.59 |
4.54 ± 0.57 |
1.89 ± 0.24 |
1.28 ± 0.49 |
| Stomach |
0.95 ± 0.14 |
1.37 ± 0.19 |
1.77 ± 0.62 |
2.41 ± 0.99 |
| Pancreas |
4.93 ± 0.87 |
3.94 ± 0.84 |
1.71 ± 0.30 |
0.80 ± 0.32 |
| Heart |
13.17 ± 1.42 |
3.03 ± 0.36 |
1.30 ± 0.35 |
0.78 ± 0.10 |
| Brain |
0.78 ± 0.16 |
0.55 ± 0.06 |
0.28 ± 0.09 |
0.16 ± 0.09 |
| 124 |
Blood |
7.84 ± 2.85 |
5.37 ± 2.42 |
1.55 ± 0.62 |
0.41 ± 0.19 |
| Liver |
18.35 ± 1.93 |
24.89 ± 2.27 |
21.29 ± 3.34 |
12.96 ± 3.14 |
| Kidney |
10.50 ± 1.36 |
11.03 ± 2.66 |
9.62 ± 1.54 |
4.65 ± 0.75 |
| Intestine |
3.70 ± 0.55 |
9.60 ± 1.35 |
31.67 ± 4.85 |
41.40 ± 8.54 |
| Spleen |
5.87 ± 2.07 |
4.44 ± 0.69 |
2.71 ± 0.92 |
1.42 ± 0.85 |
| Lung |
16.33 ± 4.74 |
7.87 ± 1.60 |
3.16 ± 0.68 |
4.12 ± 4.25 |
| Stomach |
0.85 ± 0.21 |
1.42 ± 0.28 |
2.29 ± 0.61 |
2.32 ± 0.81 |
| Pancreas |
3.14 ± 1.32 |
5.30 ± 2.14 |
4.64 ± 0.76 |
1.63 ± 0.37 |
| Heart |
13.26 ± 2.46 |
4.96 ± 1.83 |
2.54 ± 0.56 |
1.23 ± 0.28 |
| Brain |
0.62 ± 0.27 |
0.47 ± 0.13 |
0.38 ± 0.11 |
0.16 ± 0.08 |
| 125 |
Blood |
2.81 ± 0.76 |
0.95 ± 0.45 |
0.56 ± 0.30 |
0.29 ± 0.13 |
| Liver |
21.26 ± 2.50 |
27.33 ± 2.45 |
22.08 ± 3.93 |
14.34 ± 0.60 |
| Kidney |
11.21 ± 1.46 |
8.54 ± 0.64 |
4.18 ± 0.52 |
1.92 ± 0.41 |
| Intestine |
6.22 ± 0.40 |
21.95 ± 2.50 |
42.24 ± 3.78 |
53.39 ± 4.78 |
| Spleen |
2.91 ± 0.61 |
2.37 ± 0.55 |
0.74 ± 0.16 |
0.30 ± 0.04 |
| Lung |
10.33 ± 1.80 |
2.47 ± 0.62 |
2.47 ± 0.62 |
0.73 ± 0.27 |
| Stomach |
1.14 ± 0.26 |
1.80 ± 0.22 |
1.93 ± 0.19 |
1.68 ± 0.67 |
| Pancreas |
6.91 ± 2.23 |
4.45 ± 0.54 |
1.44 ± 0.33 |
0.47 ± 0.06 |
| Heart |
11.71 ± 2.13 |
3.01 ± 0.70 |
0.98 ± 0.26 |
0.44 ± 0.10 |
| Brain |
1.48 ± 0.44 |
1.09 ± 0.20 |
0.35 ± 0.14 |
0.17 ± 0.06 |
 |
| | Scheme 8 | |
 |
| | Scheme 9 | |
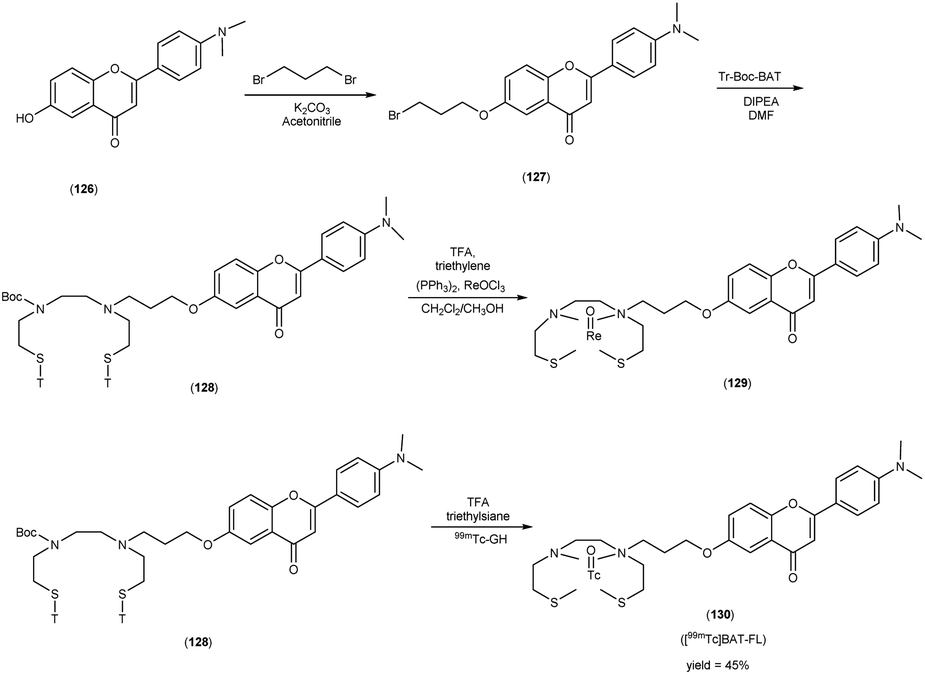
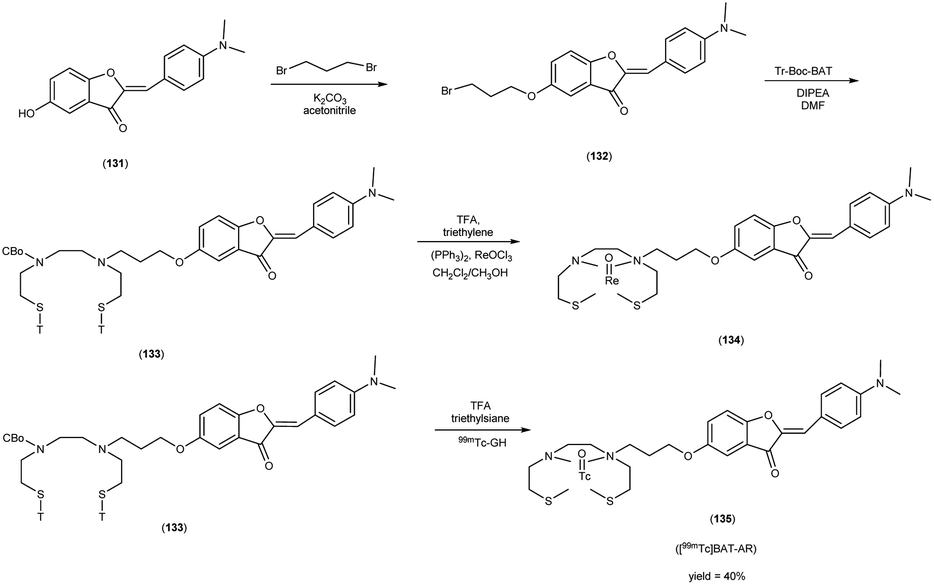
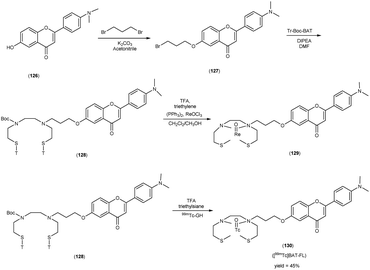
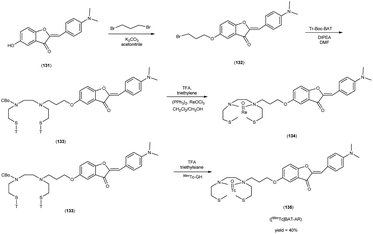
In 2010, Masahiro Ono and collaborators60 synthesized the flavones by using the most useful method known as the Baker–Venkataraman transformation. The 4-hydroxyacetophenone was initially transformed into a benzoyl ester which, upon treatment with a base, underwent conversion into a 1,3-diketone. This diketone was subsequently subjected to an acid-mediated reaction, leading to the synthesis of the desired flavone compound (126). The reaction of dibromopropane with compound 126 resulted in the production of the flavone derivative 127, which included a trimethine group. Subsequently, compound 127 was coupled with Tr-Boc-BAT to generate compound 128, serving as the precursor for the 99mTc/Re reaction. The synthesis of the aurone backbone was achieved through an aldol reaction, involving benzofuranones and benzaldehydes in the presence of aluminum oxide (Al2O3). Specifically, 5-methoxy-3-benzofuranone was reacted with 4-dimethylbenzaldehyde in the presence of Al2O3 in chloroform at room temperature, resulting in the formation of compound 131 with an impressive yield of 92%. The precursor for the subsequent 99mTc/Re reaction, denoted as compound 133, was obtained in a manner analogous to the synthesis of the flavone derivative. Following the deprotection of the thiol groups in compounds 128 and 133 using trifluoroacetic acid (TFA) and triethylsilane, the Re complexes (129 and 134) were synthesized through reactions with tetrakis(triphenylphosphine)rhenium(VII) oxide chloride, (PPh3)2ReOCl3. The corresponding 99mTc complexes, represented as 130 ([99mTc]BAT-FL) and 135 ([99mTc]BAT-AR), were prepared through a ligand exchange reaction employing the precursor compound 99mTc-glucoheptonate (GH) in 40% to 45% yield. In this study, to develop more useful 99mTc imaging agents for the clinical diagnosis of AD, two flavones and aurone derivatives were synthesized with BAT as a chelation ligand. We then evaluated the biological potential of these compounds as probes by testing their affinity for Aβ aggregates and b-amyloid plaques in sections of brain tissue from Tg2576 mice and their uptake by and clearance from the brain in biodistribution experiments using normal mice. The characterization of the complex was conducted through a comparative High-Performance Liquid Chromatography (HPLC) analysis, employing the corresponding rhenium (Re) complexes as reference compounds. The retention times for [99mTc]BAT-FL and [99mTc]BAT-AR, as determined by HPLC with radioactivity detection, were recorded at 11.1 and 16.6 minutes, respectively. In parallel, the retention times of the corresponding Re complexes were measured using HPLC equipped with UV detection, resulting in values of 9.5 and 14.6 minutes, respectively (Table 17). This is the first time 99mTc/Re complexes based on flavone and aurone scaffolds have been proposed as probes for the detection of β-amyloid plaques in the brain. The pertinent 99mTc complexes, namely ([99mTc]BAT-FL) and ([99mTc]BAT-AR), synthesized the precursor 99mTc-glucoheptonate (GH) by employing a ligand exchange methodology. One particularly promising probe, denoted as 99mTc-BATchalcone (n = 3), emerged as a notable contender for the discernment of Aβ-amyloid plaques within the cerebral milieu. In the biodistribution study of the [99mTc]BAT-FL (130) and [99mTc]BAT-AR (135), their measured cerebral uptake, quantified at 0.64% and 0.79% ID/g at the 2 minutes time point following administration, fell below the anticipated levels (Table 18). This confluence of characteristics, comprising an affinity for Aβ-amyloid plaques, efficient cerebral uptake, and prompt clearance, positions compounds 130 and 135 as promising candidates for the detection of Aβ-amyloid plaques within the cerebral milieu. However, it is worth noting that further enhancements are necessitated to augment their cerebral uptake. The outcomes gleaned from this study offer valuable insights to guide the development of 99mTc-labeled probes tailored for the imaging of β-amyloid plaques in the brain (Schemes 10 and 11).
Table 17 HPLC retention times of 99mTc-labeled chalcones and their Re analogues and logP values of 99mTc-labeled chalcones
| Re compound |
Retention time (min) |
99mTc compound |
Retention time (min) |
Logp of 99mTc compounds |
| 129 |
9.5 |
130 |
11.1 |
2.77 ± 0.04 |
| 134 |
14.6 |
135 |
16.6 |
2.23 ± 0.04 |
Table 18 Biodistribution of radioactivity after injection of 99mTc-labelled chalcone derivatives in normal mice
| Compounds |
Organs |
Time after injection (min) |
| 2 min |
10 min |
30 min |
60 min |
| 130 |
Blood |
1.90 ± 0.08 |
0.80 ± 0.16 |
0.41 ± 0.06 |
0.28 ± 0.06 |
| Liver |
19.35 ± 1.30 |
24.75 ± 3.45 |
27.73 ± 3.30 |
24.12 ± 3.08 |
| Kidney |
9.70 ± 0.83 |
5.56 ± 0.84 |
2.38 ± 0.30 |
1.40 ± 0.20 |
| Intestine |
4.54 ± 0.42 |
11.36 ± 1.88 |
26.61 ± 3.93 |
42.67 ± 2.98 |
| Spleen |
3.24 ± 0.61 |
2.21 ± 0.31 |
1.04 ± 0.42 |
0.45 ± 0.07 |
| Lung |
11.42 ± 2.10 |
3.84 ± 0.57 |
1.70 ± 0.24 |
1.07 ± 0.16 |
| Stomach |
0.90 ± 0.15 |
1.36 ± 0.55 |
1.52 ± 0.67 |
2.45 ± 1.04 |
| Pancreas |
4.41 ± 0.29 |
4.31 ± 0.35 |
1.89 ± 0.15 |
0.84 ± 0.17 |
| Heart |
12.00 ± 1.16 |
3.12 ± 0.51 |
0.99 ± 0.18 |
0.44 ± 0.09 |
| Brain |
0.64 ± 0.07 |
0.57 ± 0.14 |
0.36 ± 0.01 |
0.23 ± 0.04 |
| 135 |
Blood |
1.56 ± 0.16 |
0.71 ± 0.07 |
0.35 ± 0.04 |
0.21 ± 0.04 |
| Liver |
17.76 ± 1.51 |
17.77 ± 1.70 |
15.17 ± 0.95 |
12.96 ± 1.48 |
| Kidney |
11.50 ± 0.73 |
8.77 ± 1.15 |
4.83 ± 0.77 |
3.28 ± 1.52 |
| Intestine |
6.78 ± 0.78 |
26.20 ± 2.45 |
46.06 ± 3.17 |
55.33 ± 7.42 |
| Spleen |
2.87 ± 0.30 |
1.92 ± 0.47 |
0.70 ± 0.07 |
0.35 ± 0.15 |
| Lung |
6.10 ± 1.15 |
3.25 ± 0.78 |
1.63 ± 0.42 |
0.85 ± 0.18 |
| Stomach |
1.03 ± 0.13 |
1.63 ± 0.25 |
1.88 ± 0.11 |
1.69 ± 0.49 |
| Pancreas |
5.85 ± 1.09 |
4.20 ± 0.68 |
1.53 ± 0.54 |
0.60 ± 0.30 |
| Heart |
12.30 ± 1.21 |
3.26 ± 0.43 |
1.15 ± 0.30 |
0.40 ± 0.09 |
| Brain |
0.79 ± 0.12 |
0.70 ± 0.05 |
0.27 ± 0.06 |
0.11 ± 0.04 |
 |
| | Scheme 10 | |
 |
| | Scheme 11 | |
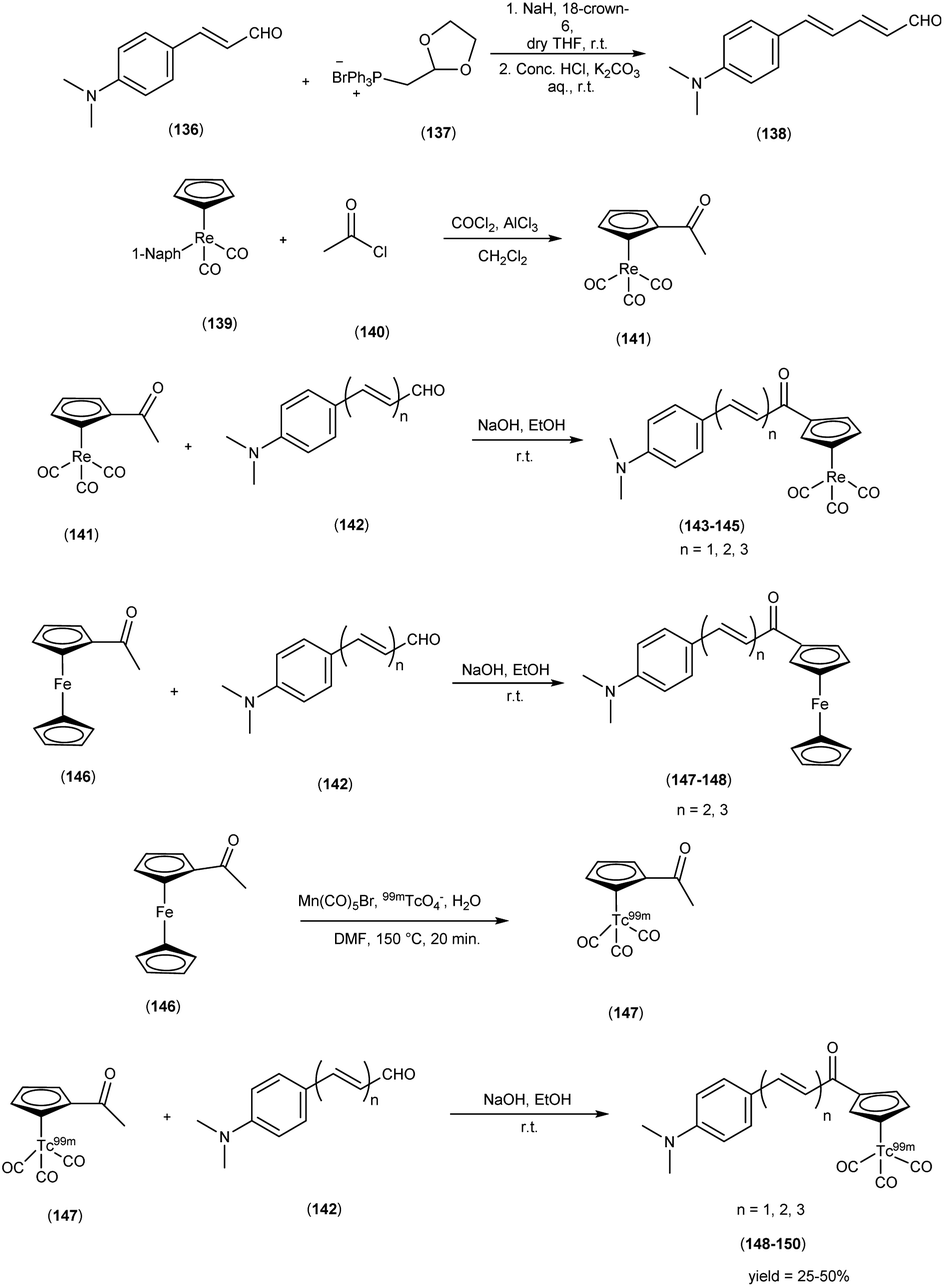
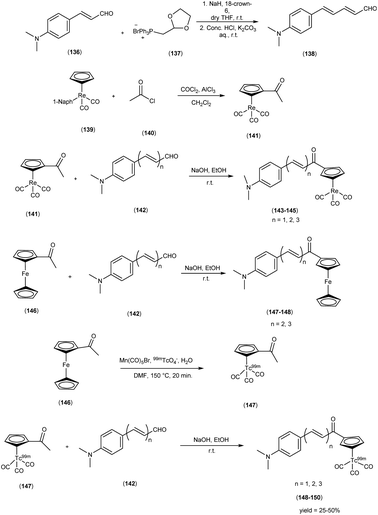
In 2013, Zijing Li and colleagues61 conceived and synthesized innovative chalcone-mimic complexes by introducing a [Cp99mTc(CO)3] core to replace a benzene ring. The synthesis began with the initial acetylation of the cyclopentadienyl group, achieved by the reaction with acetyl chloride in an ice bath. This process produced (acetylcyclopentadienyl)tricarbonylrhenium (141) with an impressive yield of 95%. Subsequently, this complex (141) was subjected to base-catalyzed Claisen condensation with aromatic aldehydes (142), resulting in the formation of the final rhenium complexes (143, 144, and 145). These complexes exhibited varying π conjugation lengths and were synthesized with yields exceeding 90%. It is worth noting that two ferrocene complexes (147 and 148) were also synthesized using the same method, serving as precursors for radiolabelling. To obtain the 99mTc-labelled cyclopentadienyl tricarbonyl complexes 148, 149, and 150, a two-step sequential reaction was employed. In the first step, the compound [CH3COCp99mTc(CO)3] ([99mTc]2) (147) was synthesized through a DLT method conducted at 150 °C for 20 minutes, yielding an average radiochemical yield of 50%. Subsequently, [99mTc]2 was subjected to a base-catalyzed Claisen condensation with the corresponding aldehydes. These complexes have been empirically demonstrated to exhibit a discernible affinity for Aβ-amyloid through both fluorescent staining conducted on brain sections obtained from AD patients and binding assays employing Aβ1–42 aggregates. The assessment of binding affinities, elucidated by Ki values, was contingent upon the extension of the conjugated π system, yielding values within the range of 899 to 108 nM. To provide a quantitative evaluation of the binding affinities of these chalcone-mimic complexes towards Aβ1–42 aggregates, an in vitro inhibition assay was conducted in solution, employing [125I]IMPY as a competing radioligand, adhering to conventional methodologies. The results, as outlined in Table 19, revealed that the three rhenium complexes (143, 144 and 145) effectively inhibited the binding of [125I]IMPY in a dose-dependent manner, with each exhibiting moderate binding affinities towards Aβ1–42 aggregates (Ki = 899 ± 78 nM for 143, Ki = 211 ± 19 nM for 144, and Ki = 108 ± 16 nM for 145). While these values may not be considered optimal, they were nonetheless sufficiently robust for Aβ aggregates, and notably comparable to the values determined for IMPY under the same assay system (Ki = 11.5 ± 2.5 nM). Moreover, these binding affinities align favorably with those reported for chalcone derivatives, where Ki values ranged from 2.9 to >10000 nM as reported. In the context of biodistribution studies, it was observed that all compounds exhibited favorable initial brain uptake and rapid blood clearance. However, a noteworthy trend emerged, where an increase in the conjugation length correlated with a reduction in the initial brain uptake. Specifically, this pattern ranged from 4.10 ± 0.38% ID/g for compound 148 to 1.11 ± 0.34% ID/g for compound 150 (Table 20). It is important to note that these technetium-99m complexes, characterized by their small molecular weight (<500 Da), have been strategically designed through an integrated approach. This collective evidence underscores the potential feasibility of developing a promising 99mTc-labeled agent for the imaging of Aβ plaques within the cerebral milieu. This intricate evaluation affirmed the robust affinity of technetium-99m (99mTc), a radiotracer, demonstrating not only pronounced initial brain uptake but also expeditious clearance kinetics within biodistribution investigations. These ingeniously devised, compact technetium-99m complexes, characterized by their synergistic alludes to the conceivable development of 99mTc-labelled agents that hold the promise of enabling the visualization of Aβ plaques within the cerebral milieu, thus contributing to the burgeoning field of neuroimaging in AD research (Scheme 12).
Table 19 Inhibition constants (Ki, nM) for binding to aggregates of Aβ1–42 versus [125I]IMPY
| Compound |
Ki (nM) |
Compound |
Ki (nM) |
| 143 |
899 ± 78 |
147 |
3.36 ± 0.30 |
| 144 |
211 ± 19 |
148 |
5.08 ± 1.74 |
| 145 |
108 ± 16 |
IMPY |
11.5 ± 2.5 |
Table 20 Biodistribution of [99mTc] in male ICR mice
| Compounds |
Organs |
Time after injection (min) |
| 2 min |
10 min |
30 min |
60 min |
120 min |
| 148 |
Blood |
2.02 ± 0.17 |
0.82 ± 0.05 |
0.51 ± 0.07 |
0.51 ± 0.06 |
0.44 ± 0.08 |
| Brain |
4.10 ± 0.38 |
2.27 ± 0.51 |
0.69 ± 0.09 |
0.50 ± 0.08 |
0.37 ± 0.08 |
| Heart |
11.47 ± 1.65 |
1.87 ± 0.36 |
0.98 ± 0.22 |
0.84 ± 0.16 |
0.55 ± 0.15 |
| Liver |
18.14 ± 1.77 |
24.75 ± 3.38 |
22.43 ± 4.39 |
25.96 ± 2.06 |
25.24 ± 5.17 |
| Spleen |
3.53 ± 0.52 |
1.93 ± 0.30 |
0.81 ± 0.14 |
0.64 ± 0.14 |
0.51 ± 0.15 |
| Lung |
7.39 ± 1.06 |
7.32 ± 0.71 |
5.52 ± 0.49 |
5.38 ± 0.38 |
4.79 ± 0.43 |
| Kidney |
13.36 ± 0.99 |
5.38 ± 0.66 |
3.87 ± 0.79 |
3.17 ± 0.16 |
2.56 ± 0.40 |
| Stomach |
1.69 ± 0.16 |
3.06 ± 0.90 |
2.18 ± 0.39 |
1.38 ± 0.55 |
0.75 ± 0.16 |
| Small pancreas |
6.81 ± 1.57 |
17.98 ± 1.58 |
37.71 ± 7.94 |
34.75 ± 2.26 |
26.22 ± 4.20 |
| 149 |
Blood |
3.80 ± 0.71 |
1.25 ± 0.17 |
0.98 ± 0.49 |
0.64 ± 0.15 |
0.55 ± 0.07 |
| Brain |
2.30 ± 0.27 |
1.85 ± 0.25 |
0.93 ± 0.09 |
0.55 ± 0.08 |
0.49 ± 0.11 |
| Heart |
2.30 ± 0.27 |
1.85 ± 0.25 |
0.93 ± 0.09 |
0.55 ± 0.08 |
0.49 ± 0.11 |
| Liver |
12.94 ± 2.16 |
3.58 ± 0.51 |
1.92 ± 0.26 |
1.36 ± 0.13 |
0.92 ± 0.12 |
| Spleen |
32.64 ± 4.34 |
28.81 ± 4.57 |
34.32 ± 5.35 |
35.93 ± 4.25 |
29.59 ± 5.35 |
| Lung |
4.50 ± 0.91 |
4.34 ± 0.92 |
2.84 ± 0.90 |
1.25 ± 0.23 |
1.00 ± 0.16 |
| Kidney |
9.08 ± 2.20 |
6.18 ± 1.59 |
4.03 ± 0.83 |
3.29 ± 1.45 |
2.74 ± 0.38 |
| Stomach |
1.11 ± 0.24 |
1.28 ± 0.33 |
1.86 ± 0.60 |
1.90 ± 0.22 |
1.38 ± 1.63 |
| Small pancreas |
5.27 ± 0.46 |
15.12 ± 5.32 |
24.42 ± 4.62 |
34.63 ± 4.72 |
29.94 ± 5.47 |
| 150 |
Blood |
13.53 ± 1.37 |
1.01 ± 0.11 |
0.70 ± 0.08 |
0.82 ± 0.06 |
0.98 ± 0.22 |
| Brain |
1.11 ± 0.34 |
0.40 ± 0.05 |
0.38 ± 0.05 |
0.51 ± 0.08 |
0.64 ± 0.11 |
| Heart |
11.48 ± 1.82 |
3.50 ± 0.23 |
2.61 ± 0.28 |
1.70 ± 0.14 |
1.73 ± 0.24 |
| Liver |
52. 40 ± 3.64 |
67.08 ± 3.54 |
57.20 ± 3.09 |
40.49 ± 6.00 |
42.91 ± 3.43 |
| Spleen |
13.14 ± 2.57 |
21.59 ± 2.91 |
20.95 ± 2.72 |
18.63 ± 4.75 |
11.65 ± 4.25 |
| Lung |
31.98 ± 4.58 |
13.89 ± 1.39 |
8.66 ± 0.99 |
9.69 ± 0.52 |
9.18 ± 1.87 |
| Kidney |
7.11 ± 0.80 |
2.83 ± 0.33 |
2.87 ± 0.73 |
2.98 ± 0.36 |
4.12 ± 0.56 |
| Stomach |
0.63 ± 0.10 |
0.50 ± 0.06 |
0.74 ± 0.08 |
0.93 ± 0.12 |
1.15 ± 0.13 |
| Small pancreas |
2.30 ± 0.45 |
3.84 ± 0.48 |
9.85 ± 0.72 |
16.64 ± 2.29 |
14.93 ± 2.70 |
 |
| | Scheme 12 | |
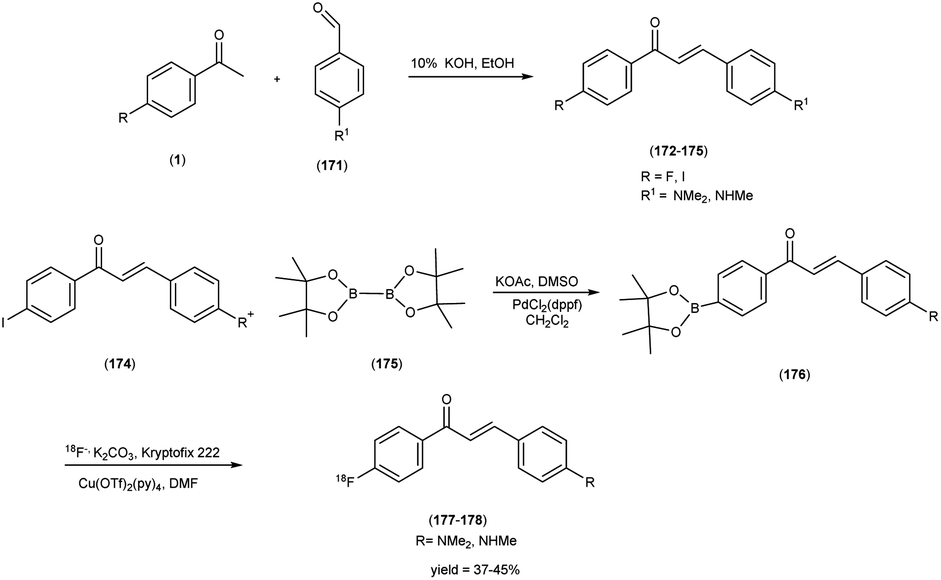
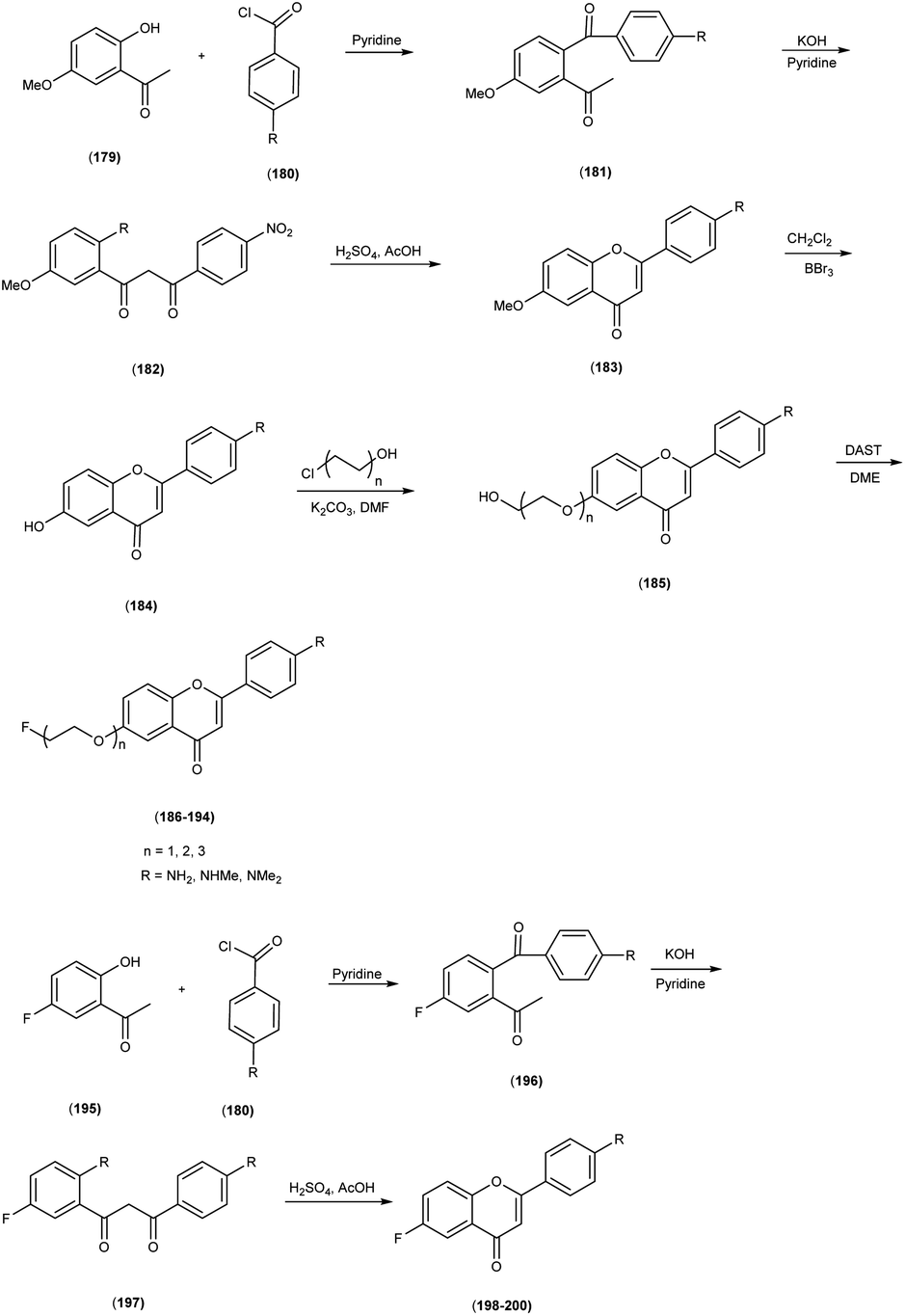
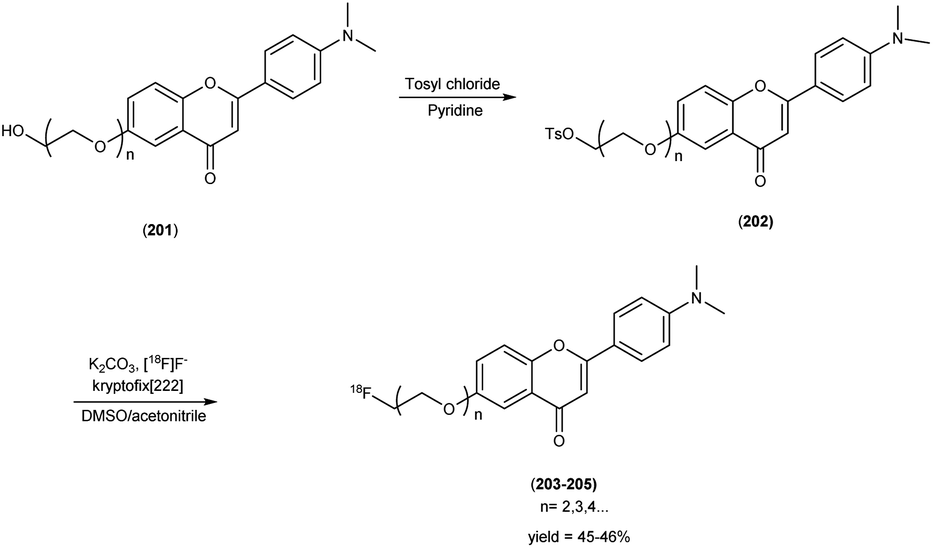
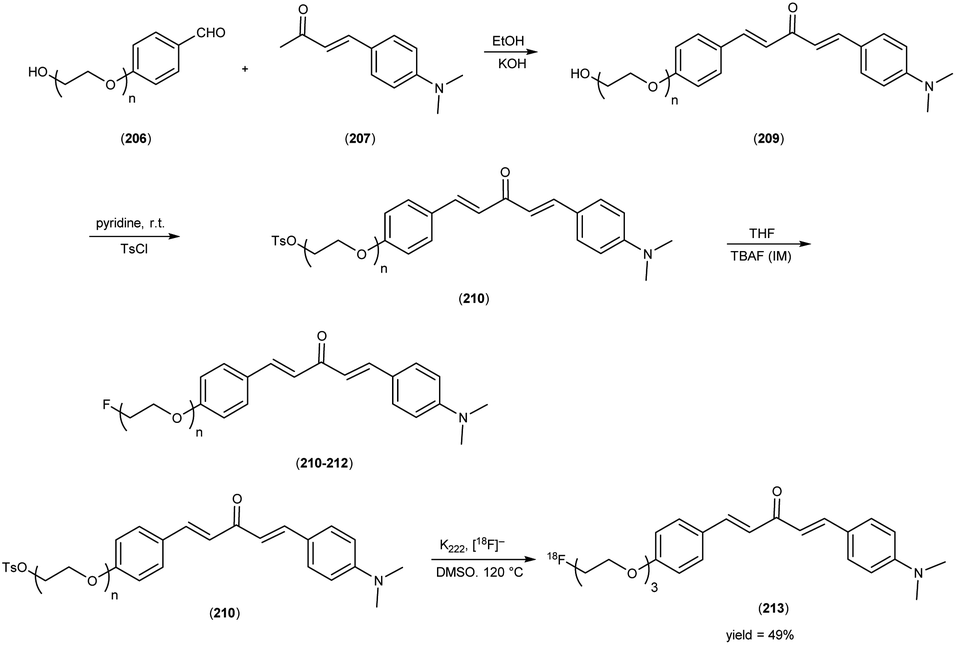
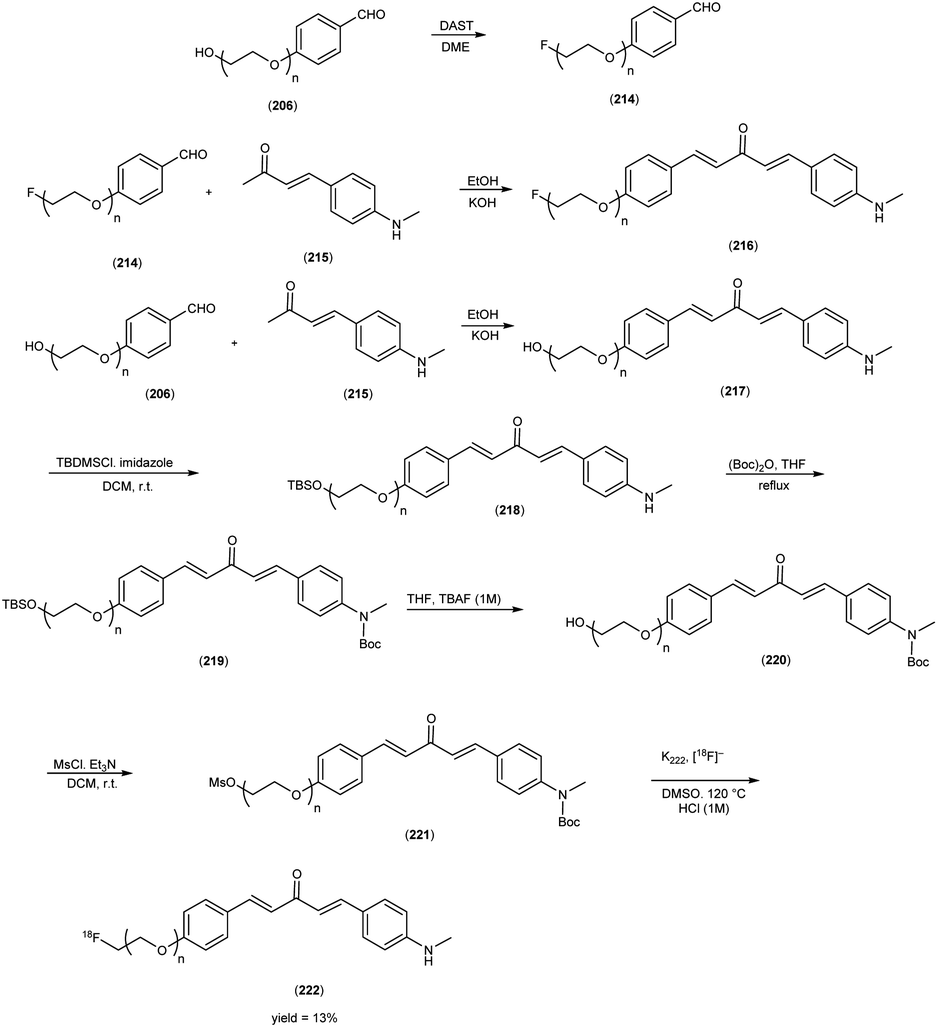
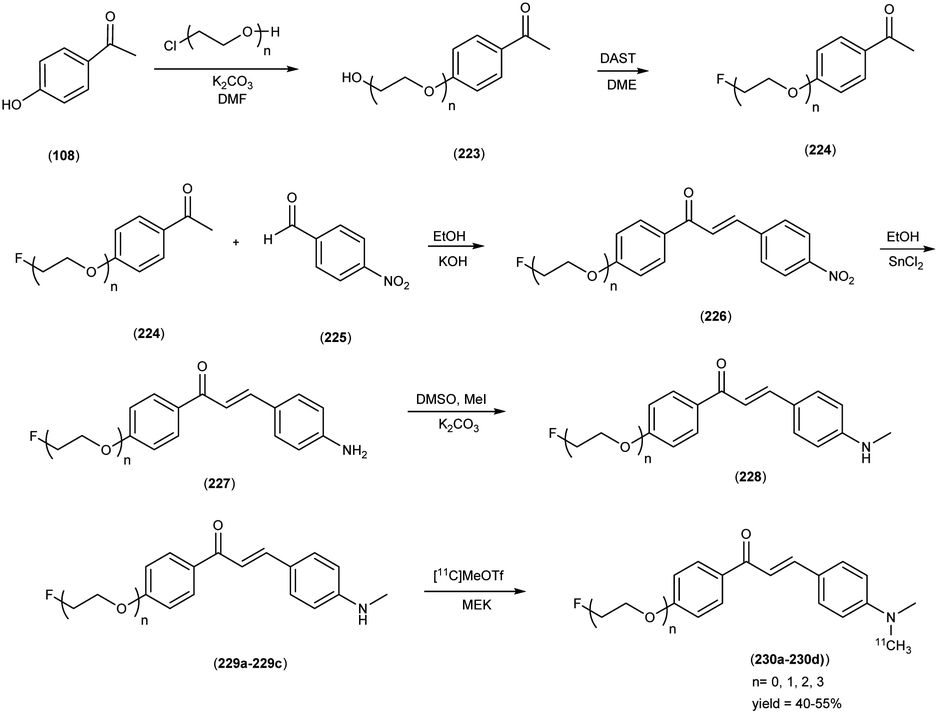
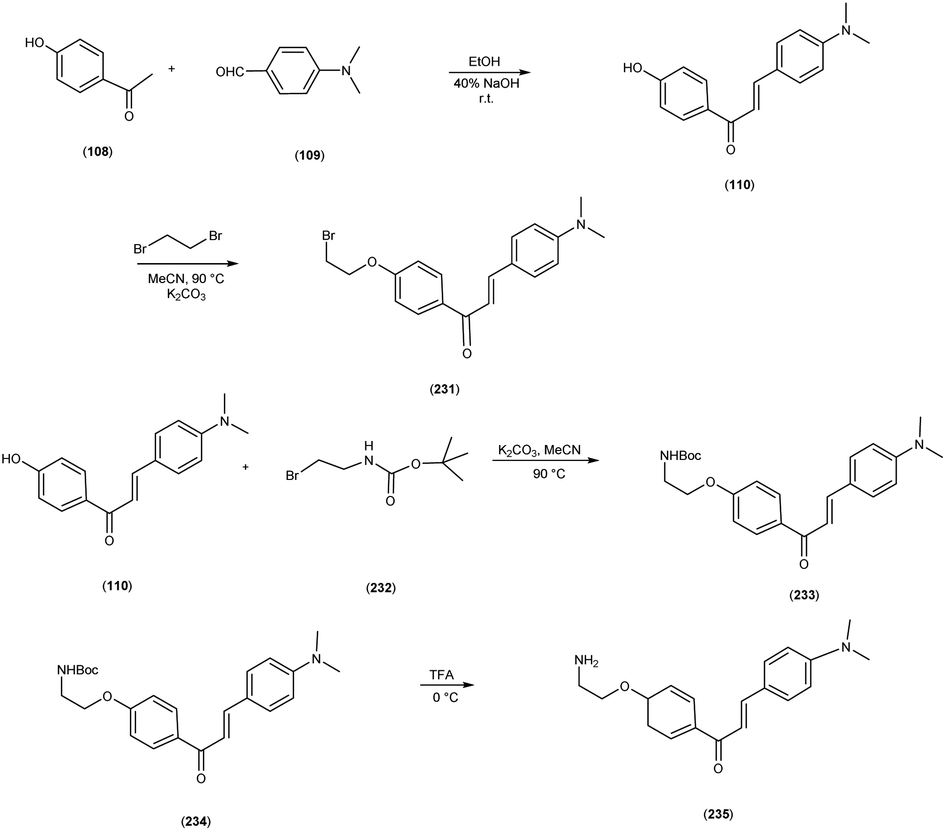
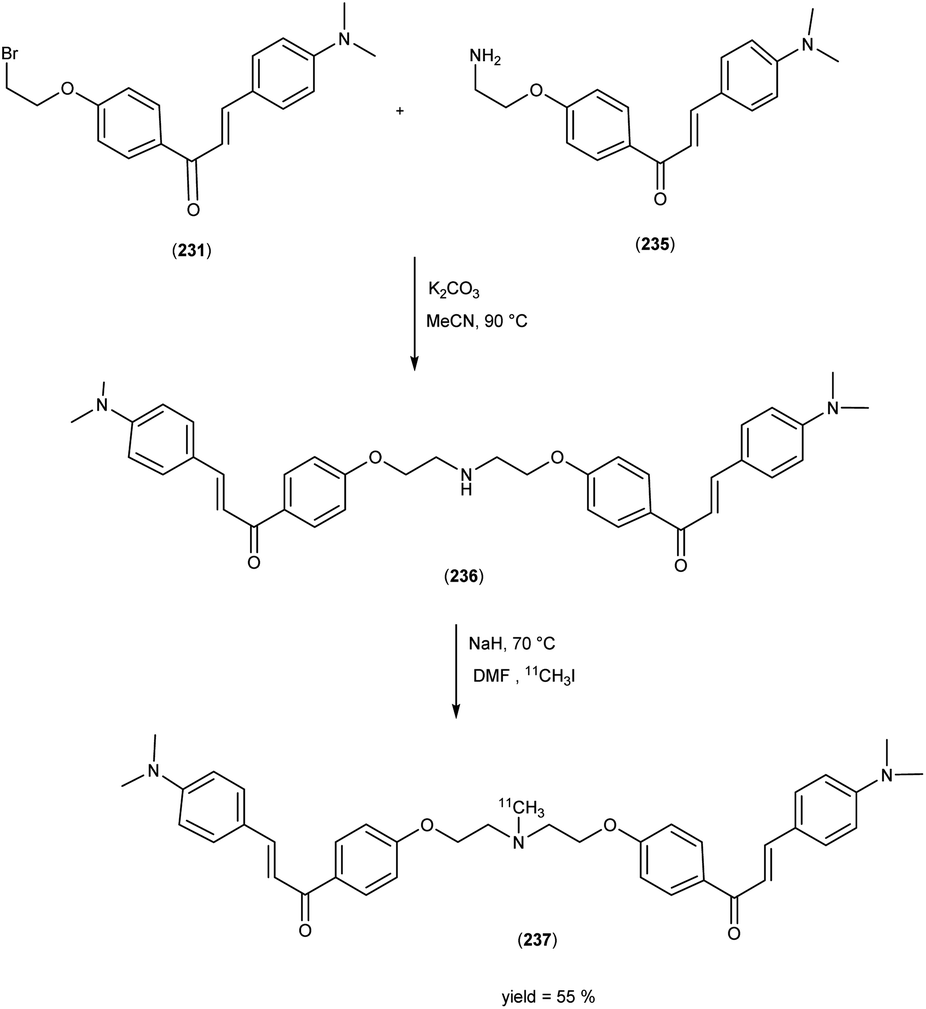
5. 18F labelled chalcones for AD
In 2009, Masahiro Ono and collaborators62 devised and synthesized fluorinated chalcone derivatives with the explicit intention of formulating 18F-labeled probes for deploying positron emission tomography (PET) as an imaging modality for the visualization of Aβ plaques. The most advantageous approach for the synthesis of chalcones was the condensation reaction involving acetophenones (108 & 165) and 4-dimethylaldehyde (109). Within this framework, 4-hydroxy acetophenone or 4-fluoro acetophenone was subjected to a chemical reaction resulting in the formation of 4′-hydroxy-4-dimethylamino-chalcone (110) and 4′-fluoro-4-dimethylamino-chalcone (158), yielding products in quantities of 84.0% and 41.6%, respectively. The fluorination of compounds 151 & 155 to yield compounds 152, 153, 154 and 156 was executed using diethylamino sulfur trifluoride (DAST) after the incorporation of three oligoethylene glycol molecules into the phenolic hydroxyl group. The 18F radiolabelling of tosyl precursor (169) was executed via a nucleophilic displacement reaction with the fluoride anion with a radiochemical yield of 45%. These chalcones were characterized by their incorporation of fluoro-pegylated (FPEG) moieties, synthesized employing the aldol condensation method. These newly synthesized compounds were harnessed for in vitro binding assessments, it was observed that the FPEG chalcone derivatives featuring a dimethylamino group exhibited elevated Ki values, ranging from 20 to 50 nM, in contrast to their counterparts bearing either a monomethylamino or a primary amine group (Table 21). Remarkably, they demonstrated a remarkable proclivity, surpassing monomethyl amine and primary amine groups, signifying a heightened affinity for Aβ binding. Noteworthy among these compounds, [18F](E)-3-(4-dimethylamino)phenyl)-1-(4-(2-((fluoroethoxy)-ethoxy)phenyl)-2-propen-1-one (170) exhibited a pronounced ability to recognize Aβ plaques avidly. Furthermore, compound (170) showcased the attributes of swift cerebral clearance within the brain of normal murine subjects (Table 22). This study not only contributes to the expanding repertoire of potential AD diagnostic tools but also accentuates the potency of these fluoro-pegylated chalcone derivatives in illuminating Aβ-amyloid plaques, setting the stage for further advancements in neuroimaging research (Scheme 13).
Table 21 Inhibition constants (Ki, nM) of compounds for the binding of [125F]DMIC to Aβ1–42 aggregates
| compound |
R1 |
R2 |
Ki (nM) |
| 152 |
FCH2CH2O |
N(CH3)2 |
45.7 ± 7.1 |
| 153 |
F(CH2CH2O)2 |
N(CH3)2 |
20.0 ± 2.5 |
| 154 |
F(CH2CH2O)3 |
N(CH3)2 |
38.9 ± 4.2 |
| 159 |
FCH2CH2O |
NH2 |
678.9 ± 21.7 |
| 160 |
F(CH2CH2O)2 |
NH2 |
1048.0 ± 114.3 |
| 161 |
F(CH2CH2O)3 |
NH2 |
790.0 ± 132.1 |
| 162 |
FCH2CH2O |
NHCH3 |
197.1 ± 58.8 |
| 163 |
F(CH2CH2O)2 |
NHCH3 |
216.4 ± 13.8 |
| 164 |
F(CH2CH2O)3 |
NHCH3 |
470.9 ± 00.4 |
| 166 |
F |
N(CH3)2 |
49.8 ± 6.2 |
| 167 |
F |
NH2 |
663.0 ± 88.3 |
| 168 |
F |
NHCH3 |
234.2 ± 44.0 |
| DMIC |
I |
N(CH3)2 |
13.1 ± 3.0 |
| IMPY |
|
|
28.0 ± 4.1 |
Table 22 Biodistribution of radioactivity after injection of [18F] radiolabelled compound 170 in normal mice
| Compound |
Organs |
Time after injection (min) |
| 2 min |
10 min |
30 min |
60 min |
| 170 |
Blood |
2.09 ± 0.40 |
1.94 ± 0.18 |
2.35 ± 0.33 |
1.87 ± 0.26 |
| Brain |
3.48 ± 0.47 |
1.52 ± 0.03 |
1.08 ± 0.09 |
1.07 ± 0.17 |
| Bone |
1.80 ± 0.31 |
1.76 ± 0.15 |
2.98 ± 0.49 |
3.58 ± 0.41 |
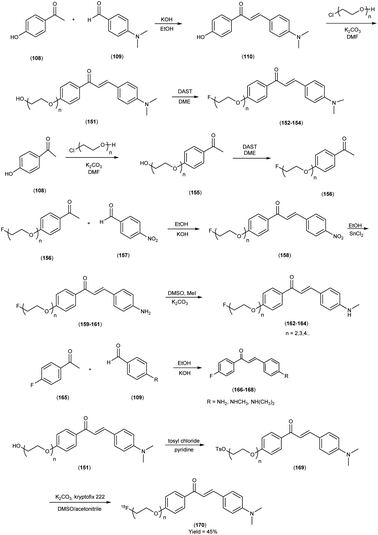 |
| | Scheme 13 | |
In 2018, Sho Kaide and colleagues63 encompassed the synthesis of radio-fluorinated chalcone derivatives, followed by a comprehensive assessment of their potential utility in PET for discerning Aβ-amyloid plaques linked to AD. The synthesis of nonradioactive chalcones (172 to 175) was carried out by the reaction of 4-iodoacetophenone with either 4-dimethylaminobenzaldehyde or 4-methylaminobenzaldehyde in the presence of a basic catalyst (10% KOH) in ethanol at ambient temperature with a yield range of 60% to 63%. To synthesized the precursors necessary for 18F-labelling, boronate derivative 176 was synthesized from compound 174 through coupling reactions with bis(pinacolato)diboron, aided by a base (KOAc) and a palladium catalyst (PdCl2(dppf)CH2Cl2). The synthesized boronate derivative (176) was effectively employed as a precursor in the copper-mediated nucleophilic 18F-labelling process, leading to the successful preparation of radiolabelled compounds 177 and 178. This approach facilitated the direct conversion of iodine moieties, which were originally integrated into the chalcone structure, into fluorine, ultimately yielding the desired radiolabelled compounds 177 and 178. The core strategy employed the transformation of iodine to fluorine as a foundational principle, facilitating the construction of novel radio-fluorinated chalcones designed as PET probes. This ingenious design aimed to foster enhanced specificity in detecting Aβ-amyloid plaques while mitigating undesirable non-specific binding. In the context of in vitro binding investigations employing recombinant Aβ peptides in aggregate form, both [18F]DMFC and [18F]FMC demonstrated notable binding affinities, characterized by Ki values of 4.47 and 6.50 nM, respectively. Furthermore, in the context of in vitro autoradiography (ARG) experiments conducted using brain sections from individuals with AD, [18F]DMFC and [18F]FMC exhibited conspicuous accumulation exclusively within regions abundant in Aβ plaques. This unequivocal pattern of accumulation underscores their discerning recognition of human Aβ plaques in the in vitro setting. The development of these new radio-fluorinated chalcones was executed through a strategic process that entailed the direct conversion of iodine to fluorine, an innovation achieved by implementing 18F-labelling methodologies employing boronated precursors. This transformative approach culminated in the direct incorporation of fluorine-18 into the chalcone scaffold, yielding [18F]4-dimethylamino-4-fluoro-chalcone (177) and [18F]4′-fluoro-4-methylamino-chalcone (178). These novel compounds, representative of the innovation, showcased commendable attributes within in vivo scenarios. In the context of a biodistribution study conducted in healthy mice, both compounds 177 and 178 exhibited notably higher initial brain uptake levels, quantified at 4.43% and 5.47% ID/g, respectively, at the 2 minutes time point following injection. Furthermore, these compounds displayed a substantially more expeditious clearance from the brain, registering values of 0.52% and 0.66% ID/g at the 30 minutes post-injection interval, respectively (Table 23). This contrasted favorably with the performance of a Food and Drug Administration (FDA)-approved Aβ-amyloid imaging agent, [18F]Florbetapir, signifying an enhanced likelihood of detecting Aβ plaques while mitigating non-specific binding in the cerebral milieu. This favourable outcome was attributable to the compound's judiciously designed lipophilicity and molecular dimensions, as realized through the iodine-to-fluorine conversion strategy. Such advancements hold the potential to propel the domain of neuroimaging further, thereby contributing to a more nuanced understanding of Aβ-amyloid plaque distribution within the intricate landscape of the AD-afflicted brain (Scheme 14).
Table 23 Brain uptake of radioactivity after the injection of [18F]chalcones, [125I]chalcones and [ 18F]Florbetapir in normal mice and the 2 min/30 min ratio of radioactivity accumulation
| Compound |
% ID/g brain |
Ratio (2 min/30 min) |
| 2 min |
30 min |
| [18F]DMFC (177) |
4.43 ± 0.45 |
0.52 ± 0.07 |
8.5 |
| [125I]DMIC |
2.43 ± 0.10 |
0.33 ± 0.07 |
7.4 |
| [18F]FMC (178) |
5.47 ± 0.52 |
0.66 ± 0.09 |
8.3 |
| [125I]IMC |
3.52 ± 0.62 |
0.63 ± 0.19 |
5.6 |
| [18F]Florbetapir |
4.90 ± 0.99 |
1.65 ± 0.11 |
3.0 |
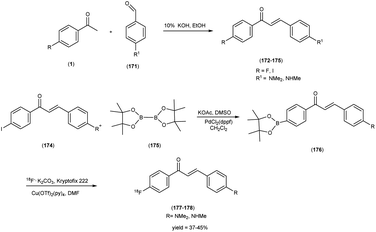 |
| | Scheme 14 | |
In 2009, Masahiro Ono and colleagues64 a compelling endeavor unfolded wherein a series of innovative fluoro-pegylated (FPEG) flavones and their derivatives were synthesized, with the overarching aim of assessing their applicability in unraveling the intricacies of AD development and functionality. In this intricate synthetic process, the initial step involves the reaction of substituted acetophenone (179) with acetyl chloride to synthesize a benzoyl ester (180) denoted as compound 181. Subsequently, this species (181) is subjected to a base-mediated reaction, yielding a 1,3-diketone (182). Further, the treatment of diketone with acid triggers the transformation into the targeted flavone compound denoted as 183. The compound 183 is then subjected to demethylation using BBr3 in CH2Cl2, leading to the formation of compound 184. To synthesize compounds bearing 1–3 ethoxy groups as the polyethylene glycol (PEG) linkage, 2-chlorethanol was coupled with the hydroxyl group of compound 184 to yield compound 185. The fluorinated flavone derivatives, designated as compounds 186 to 194, were successfully prepared by reacting compound 185 with diethylamino sulfur trifluoride (DAST) in dimethoxyethane (DME) or ethylene glycol dimethyl ether. In the final phase of the synthetic process, the synthesis of flavone derivatives (198, 199 and 200) bearing fluorine atoms directly bound to the phenyl group is successfully accomplished. To achieve the desired 18F-labeled FPEG flavones, denoted as compounds 203, 204 and 205, the tosylates 202 serve as the precursors. The free hydroxyl groups of the compound 185 was transformed into tosylate through a reaction with tosyl chloride (TsCl) in the presence of pyridine, resulting in the formation of tosylated compound 202. The synthesized tosylate (202) was combined with [18F]fluoride and potassium carbonate in the presence of Kryptofix 222, all dissolved in dimethyl sulfoxide (DMSO), and heated at 160 °C for a duration of 5 minutes. The meticulous design of these compounds strategically incorporated electron-donating functional moieties, encompassing methoxy, hydroxy, methylamino, dimethylamino and amino groups, all of which play pivotal roles in engendering affinity for Aβ1–42 aggregation, a hallmark of AD pathology. The binding affinities of the FPEG flavone derivatives towards Aβ aggregates displayed a range spanning from 5 to 321 nM (Table 24). Within brain sections obtained from AD model mice, those FPEG flavones featuring a dimethylamino group were observed to robustly stain Aβ-amyloid plaques. To quantitatively assess the affinities of these FPEG flavones for Aβ aggregates, in vitro binding experiments were conducted in solutions employing [125I]DMFV as the ligand. The hierarchy of affinity among the flavone derivatives towards Aβ1–42 aggregates was established as follows: the dimethylamino derivatives (186, 187, 188 and 200) exhibited the highest affinity, followed by the monomethylamino derivatives (190, 193, 194 and 199), and finally, the primary amino derivatives (189, 191, 192 and 198). Notably, this study involved the performance of in vitro plaque labelling on brain slices extracted from Tg2576 mice, revealing that the synthesized FPEG flavones exhibited a pronounced tendency to selectively bind to Aβ-amyloid plaques. Three distinct 18F-labeled FPEG flavones, denoted as [18F]203, [18F]204, and [18F]205, were subjected to comprehensive biodistribution assessments in normal mice (Table 25). Remarkably, all three ligands demonstrated a substantial uptake within the brain, ranging from 2.89% to 4.17% ID/g at the 2 minutes post-injection time point, a level deemed sufficient for effective imaging purposes. Furthermore, these compounds exhibited favorable clearance kinetics, as evidenced by the brain levels of 1.89%, 2.00%, and 1.31% ID/g at the 30 minutes post-injection interval for [18F]203, [18F]204 and [18F]205, respectively. These values corresponded to 45.3%, 56.5%, and 45.3% of the initial peak uptake for radiolabelled compounds 203, 204 and 205 respectively. The noteworthy attributes of rapid initial uptake into the normal brain coupled with swift washout represent highly desirable properties for β-amyloid imaging probes, as they contribute to the attainment of a high signal-to-background ratio. However, it is noteworthy that the free fluoride resulting from this process was not absorbed by brain tissue. Hence, the anticipated interference stemming from this free fluoride is expected to be relatively minimal in the context of brain imaging (Schemes 15 and 16).
Table 24 Inhibition constants (Ki, nM) of compounds for the binding of compound 203 to 205 for Aβ1–42 aggregates
| Compound |
Ki (nM) |
Compound |
Ki (nM) |
| 186 |
5.3 ± 0.8 |
192 |
234.0 ± 60.6 |
| 187 |
14.4 ± 2.5 |
193 |
54.5 ± 10.3 |
| 188 |
19.3 ± 4.0 |
194 |
45.1 ± 5.8 |
| 189 |
234.3 ± 63.5 |
198 |
260.5 ± 43.3 |
| 190 |
99.0 ± 11.8 |
199 |
110.0 ± 47.4 |
| 191 |
321.1 ± 74.4 |
200 |
73.9 ± 5.3 |
Table 25 Biodistribution of 18F-labeled flavones in normal mice
| Compounds |
Organ |
Time after injection (min) |
| 2 min |
10 min |
30 min |
60 min |
| 203 |
Blood |
2.80 ± 0.41 |
2.71 ± 0.13 |
2.53 ± 0.17 |
3.25 ± 0.31 |
| Brain |
4.17 ± 0.77 |
3.62 ± 0.21 |
1.89 ± 0.13 |
2.19 ± 0.18 |
| Bone |
2.02 ± 0.53 |
2.83 ± 0.23 |
4.51 ± 0.55 |
6.21 ± 0.84 |
| 204 |
Blood |
2.09 ± 0.35 |
2.50 ± 0.21 |
2.50 ± 0.21 |
2.94 ± 0.27 |
| Brain |
3.54 ± 0.54 |
2.75 ± 0.21 |
2.00 ± 0.20 |
2.13 ± 0.10 |
| Bone |
1.13 ± 0.22 |
1.65 ± 0.10 |
2.42 ± 0.38 |
3.74 ± 0.30 |
| 205 |
Blood |
2.35 ± 0.54 |
1.50 ± 0.26 |
1.40 ± 0.04 |
1.88 ± 0.08 |
| Brain |
2.89 ± 0.74 |
2.23 ± 0.36 |
1.31 ± 0.14 |
1.37 ± 0.11 |
| Bone |
1.53 ± 0.52 |
2.38 ± 0.39 |
4.06 ± 0.49 |
5.21 ± 0.98 |
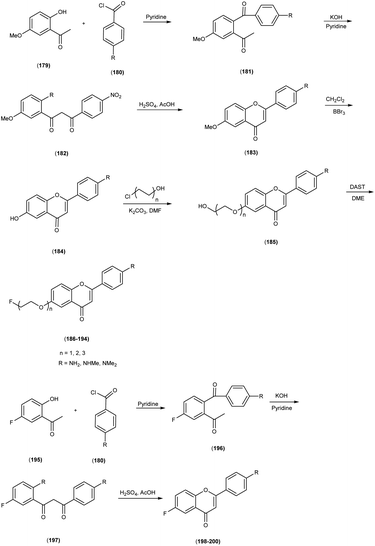 |
| | Scheme 15 | |
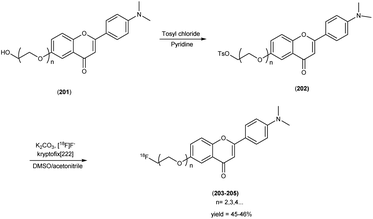 |
| | Scheme 16 | |
Mengchao Cui and colleagues55 orchestrated a transformative shift in paradigm by precisely crafting an innovative and heterogeneous array of dibenzylideneactones. The synthesis of FPEG dibenzylideneacetones, the dimethylated derivative or the monomethylated derivative 206 was subjected to coupling reactions with benzaldehyde modified with polyethylene glycol, resulting in the formation of compound 209. For the dimethylated dibenzylideneacetone series, the hydroxyl groups present in compound 209 was initially protected via tosylation, yielding precursor compound 210. These precursors was subsequently subjected to a reaction with anhydrous tetra-n-butylammonium fluoride (TBAF) at reflux conditions, leading to the formation of non-radioactive FPEG dibenzylideneacetones 210 to 212. In the case of monomethylated dibenzylideneacetone series, compound 86 had its hydroxyl group protected by treatment with tert-butyldimethylsilyl chloride (TBDMSCl), resulting in the TBS-protected compound 218. Further modification involved protecting the methylamino group of compound 219 with butyloxycarbonyl (BOC), yielding compound 220. Following the removal of the TBS-protecting group from compound 219 through treatment with TBAF, the free hydroxyl group was converted into mesylates by reacting with methanesulfonyl chloride in the presence of triethylamine, culminating in the formation of compound 222. To produce the desired 18F-labeled dibenzylideneacetone 213 and 222, the tosylate precursor (210) was mixed with [18F]fluoride and potassium carbonate, with the addition of Kryptofix 222, in dimethyl sulfoxide (DMSO). The mixture was then subjected to heating at 120 °C for 5 minutes to produce compound 213 a radiochemical yield of 49%. In the case of radiolabelled compound 222, the N-BOC-protected mesylate compound 221 served as the precursor. After heating at 120 °C for five minutes, the mixture was exposed to aqueous HCl to remove the N-BOC-protecting group. The crude product was subsequently purified through HPLC, achieving a radiochemical purity greater than 98% and a radiochemical yield of 13%. These compounds were subsequently subjected to a rigorous examination to discern their affinity profiles toward Aβ aggregates across a spectrum of discrete ligand-binding sites. The majority of the synthesized compounds exhibited remarkable affinity for Aβ aggregates, with binding constants (Ki) in the nanomolar (nM) range. The structure–activity relationship (SAR) investigation outlined previously revealed that the incorporation of a substituted group at the ortho position resulted in a diminished or nullified binding interaction. In stark contrast, the para position exhibited remarkable tolerance toward steric bulk substitutions, thereby presenting an enticing prospect for the prospective development of novel radioligands that can be readily labelled for in vivo imaging of Aβ plaques. In the context of PET or SPECT agent development targeted towards Aβ plaques, this observation pertaining to the dibenzylideneacetone scaffold bears significant importance. Notably, the FPEG ligands 210, 212 and 216, distinguished by varying polyethylene glycol unit counts (n = 1 to 3), consistently exhibited exceptional affinity (Ki < 10 nM) (Table 26). Remarkably, the length of the linker moiety did not impart any substantial alteration in the binding characteristics. Intriguingly, augmenting the size of the substituent at the aromatic amino group resulted in a discernible reduction in binding affinity. The lipophilicity (logD) profiles of the radiolabelled tracers [18F]213, and [18F]222 assessed under controlled experimental conditions exhibited relatively elevated partition coefficients (logD = 2.97 to 3.66), indicative of their inherent lipophilic characteristics. Subsequent biodistribution investigations were conducted in healthy murine subjects employing these radiolabelled tracers. As delineated in Table 27, compounds 213 and 222 manifested commendable initial blood–brain barrier (BBB) permeation with remarkable initial cerebral uptake (4.13%, and 5.15% ID/g at 2 minutes, respectively). Additionally, the radiolabelled probes exhibited favorable brain penetration characteristics along with rapid clearance. Notably, these probes unequivocally exhibited a distinctive signal for labelling amyloid plaques, as evidenced in transgenic mouse brain sections and postmortem brain sections from AD patients (Schemes 17 and 18).
Table 26 Inhibition constants (Ki, nM) for binding to aggregates of Aβ1–42 versus [125I]IMPY
| Compound |
R |
Ki (nM) |
| 210 |
Dimethylamino |
5.6 ± 2.5 |
| 211 |
Dimethylamino |
4.4 ± 1.3 |
| 212 |
Dimethylamino |
6.9 ± 1.4 |
| 216 |
Methyl amino |
8.6 ± 1.3 |
| IMPY |
— |
10.5 ± 1.0 |
Table 27 Biodistribution of [18F]213 and [18F]222 in ddY normal mice (% ID/g, mean (SD, n = 4))
| Compounds |
Organ |
Time after injection (min) |
| 2 min |
10 min |
30 min |
60 min |
| 213 |
Brain |
4.13 ± 0.41 |
1.51 ± 0.17 |
1.04 ± 0.23 |
0.90 ± 0.14 |
| Blood |
5.38 ± 0.32 |
3.89 ± 0.36 |
2.46 ± 0.54 |
1.60 ± 0.17 |
| Bone |
2.88 ± 0.37 |
2.42 ± 0.14 |
2.33 ± 0.30 |
2.33 ± 0.30 |
| Liver |
22.3 ± 3.12 |
24.41 ± 3.34 |
14.0 ± 1.94 |
6.88 ± 1.51 |
| Kidney |
12.95 ± 0.79 |
12.77 ± 2.42 |
7.13 ± 2.64 |
3.67 ± 0.85 |
| Spleen |
4.26 ± 1.05 |
6.19 ± 1.06 |
4.46 ± 0.80 |
1.93 ± 0.67 |
| Stomach |
6.28 ± 5.75 |
7.39 ± 1.87 |
6.00 ± 1.14 |
3.98 ± 1.52 |
| Intestine |
4.12 ± 0.48 |
10.53 ± 2.11 |
17.50 ± 4.47 |
21.57 ± 7.37 |
| Lung |
12.30 ± 1.99 |
6.34 ± 0.88 |
3.38 ± 0.43 |
1.86 ± 0.25 |
| Heart |
7.60 ± 1.32 |
3.33 ± 0.35 |
1.90 ± 0.26 |
1.47 ± 0.13 |
| 222 |
Brain |
5.15 ± 0.17 |
1.35 ± 0.19 |
1.35 ± 0.19 |
1.27 ± 0.12 |
| Blood |
7.44 ± 1.22 |
7.99 ± 0.34 |
4.68 ± 0.59 |
3.59 ± 0.93 |
| Bone |
2.97 ± 0.35 |
2.93 ± 0.37 |
2.94 ± 0.91 |
2.54 ± 0.84 |
| Liver |
24.1 ± 5.77 |
30.09 ± 4.43 |
19.24 ± 3.64 |
14.07 ± 1.87 |
| Kidney |
16.48 ± 2.51 |
14.15 ± 0.98 |
11.51 ± 1.76 |
9.43 ± 1.50 |
| Spleen |
5.39 ± 1.79 |
6.62 ± 0.42 |
5.44 ± 1.24 |
5.35 ± 2.06 |
| Stomach |
3.26 ± 0.63 |
2.61 ± 0.18 |
1.93 ± 0.33 |
1.28 ± 0.49 |
| Intestine |
6.58 ± 0.89 |
12.82 ± 3.23 |
24.34 ± 3.83 |
26.26 ± 3.57 |
| Lung |
12.19 ± 2.31 |
11.77 ± 1.94 |
7.43 ± 1.03 |
5.44 ± 1.45 |
| Heart |
10.55 ± 1.10 |
7.64 ± 1.23 |
5.67 ± 0.72 |
5.14 ± 0.88 |
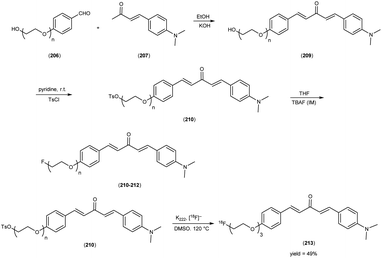 |
| | Scheme 17 | |
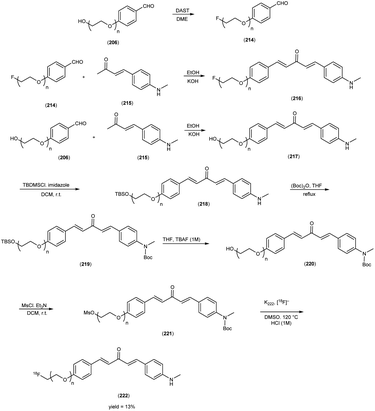 |
| | Scheme 18 | |
6. 11C labelled chalcones for AD
In the scholarly pursuit orchestrated by Mashiro Ono and colleagues,62 a meticulous synthesis endeavor unfolded, involving the preparation of N-nor methyl precursors and subsequent utilization of [11C] methyl triflate for the purpose of synthesizing radiolabelled chalcones tagged with the 11C isotope. In order to synthesize the desired compound (229), fluorination of compound 223 was executed using diethylamino sulfur trifluoride (DAST). Prior to this, the introduction of three oligoethylene glycol moieties into the phenolic hydroxyl group of the compound 108 was carried out by using 1,2 dichloroethane in DMF by using potassium carbonate as a base. The chalcone was synthesized by employing the Claisen–Schmidt condensation reaction of 4-nitrobenzaldehyde (225) and 4-substituted acetophenone (224), engaged within a basic medium. The amino derivatives were readily prepared by reducing compound 226 with SnCl2. Furthermore, the transformation of compound 227 into their respective monomethylamino derivatives (228), was achieved through methylation with CH3I under alkaline conditions. For the synthesis of 11C-labeled chalcones, the starting materials were the N-normethyl precursors (229) and [11C]methyl triflate ([11C]-MeOTf). The radiochemical yields of the final product were found to range from 40% to 55%, and these values were decay corrected to the end of the bombardment. The radiochemical purity of the resulting compounds consistently exceeded 99%, and the specific activity ranged from 22 to 28 GBq μmol−1. This pioneering methodology aimed to endow these chalcones with radiolabelled, facilitating their application as imaging agents. The In vitro binding assays with Aβ1–42 aggregates revealed that FPEG chalcone derivatives featuring a dimethylamino group exhibited notably higher Ki values, ranging from 20 to 50 nM, in comparison to their counterparts containing either a monomethylamino or primary amine group (Table 28). The subsequent phase of inquiry involved conducting biodistribution studies on normal murine subjects, employing four distinct 11C-labelled chalcone compounds. Remarkably, the outcome of these investigations unveiled an expeditious phenomenon whereby the 11C-labelled FPEG chalcones, mere moments after administration, promptly traversed the cerebral barrier. Upon administration of 11C-labeled FPEG chalcones (230a to 230d), the ensuing radioactivity effectively traversed the blood–brain barrier, manifesting a commendable uptake within the range of 3.7% to 6.0% ID/g in the brain at the 2 minutes post-injection time point. This level of uptake was deemed sufficient for enabling the imaging of Aβ plaques within the cerebral environment. Additionally, these compounds exhibited favorable clearance kinetics within the normal brain, as indicated by values of 2.3%, 1.0%, 0.35%, and 1.0% ID/g at the 60 minutes post-injection interval for [11C]230b, [11C]230c, [11C]230d, and [11C]230a, respectively (Table 29). This translocation culminated in their substantial accumulation within the brain tissue, reaching uptake levels that were deemed suitable for the imaging of Aβ plaques. The quantitative range of accumulation spanned from 3.7% to 6.0% injected dose per gram of brain tissue. This accomplishment not only showcases the feasibility of the developed radiolabelling technique but also underscores the potential of these 11C-labelled FPEG chalcones as promising tools for imaging Aβ plaques, thus contributing to the advancement of neuroimaging methodologies with a focus on AD research (Scheme 19).
Table 28 Inhibition constants (Ki, nM) of compounds for the binding of [125I]DMIC to Aβ1–42 aggregates
| Compound |
R1 |
R2 |
Ki (nM) |
| 229 |
FCH2CH2O |
NHCH3 |
197.1 ± 58.8 |
| 229 |
F(CH2CH2O)2 |
NHCH3 |
216.4 ± 13.8 |
| 229 |
F(CH2CH2O)3 |
NHCH3 |
470.9 ± 00.4 |
| DMIC |
I |
N(CH3)2 |
13.1 ± 3.0 |
| IMPY |
|
|
28.0 ± 4.1 |
Table 29 Biodistribution of radioactivity after injection of [11C]230a, [11C]230b, [11C]230c, and [11C]13 in normal mice
| Compounds |
Time after injection |
Organ |
| Blood |
Brain |
| 230a |
2 min |
3.65 ± 0.37 |
6.01 ± 0.61 |
| 10 min |
2.73 ± 0.28 |
3.24 ± 0.39 |
| 30 min |
2.12 ± 0.18 |
2.57 ± 0.26 |
| 60 min |
2.22 ± 0.25 |
2.26 ± 0.41 |
| 230b |
2 min |
3.48 ± 0.56 |
4.73 ± 0.47 |
| 10 min |
2.28 ± 0.84 |
2.23 ± 0.18 |
| 30 min |
2.54 ± 0.96 |
1.14 ± 0.12 |
| 60 min |
1.44 ± 0.36 |
1.00 ± 0.19 |
| 230c |
2 min |
2.44 ± 0.25 |
4.31 ± 0.33 |
| 10 min |
1.52 ± 0.42 |
0.64 ± 0.07 |
| 30 min |
1.01 ± 0.15 |
0.64 ± 0.07 |
| 60 min |
0.68 ± 0.10 |
0.35 ± 0.03 |
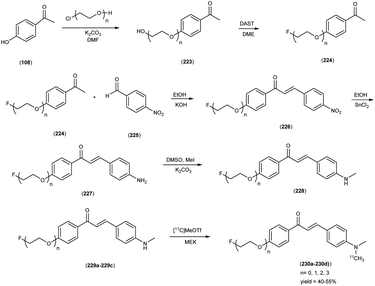 |
| | Scheme 19 | |
In 2018, Kanchan Chauhan and co-authors65 orchestrated a comprehensive investigation, wherein they embarked upon the synthesis of 11C-(chal)2DEA-Me (237), an innovative homodimeric chalcone-based 11C-PET radiotracer. The facile synthetic approach employed in this investigation drew upon classical principles of organic chemistry, incorporating condensation and nucleophilic substitution reactions. Notably, the starting materials, N,N-dimethylbenzaldehyde (109) and 4-hydroxyacetophenone (108), engaged in a Claisen–Schmidt condensation reaction within a basic medium, yielding the E-isomer of chalcone (110). Subsequently, compound 110 was subjected to o-methylation at the hydroxyl position, resulting in the formation of the monomeric derivative, Chal-Me (231). The compound 110 underwent further reaction with 1,2 dibromoethane to produce intermediates (232), which collectively constituted a bivalent ligand bearing C2 symmetry. The compound 235 was generated through the conjugation of bromoethylamine-boc with compound 110, followed by the removal of the boc-protecting group. The culmination of these synthetic steps led to the formation of the dimeric compound, (Chal)2DEA (236). In this study, a wet chemistry methodology was harnessed for radiolabelling, with the radiolabelling process executed in the presence of sodium hydride. The radiolabelling of precursor 236 was accomplished through N–11C-methylation yielding 11C-(Chal)2DEA-Me (237). These radiolabelled tracers were efficiently isolated using reversed-phase high-performance liquid chromatography (HPLC), resulting in high radiochemical yields i.e. 55%, and purity exceeding 98%. The central objective of their endeavor encompassed an in-depth exploration of the radiotracers binding affinity toward Aβ-amyloid plaques, pivotal to the understanding of neurodegenerative disorders. The investigation entailed intricate PET imaging and biodistribution assessments, both of which unveiled the radiotracers judicious brain pharmacokinetics. Remarkably, the bivalent ligand exhibited heightened cerebral outcome underscoring the promising potential of 11C-(chal)2DEA-Me (237) as an adept in vivo imaging tracer of Aβ1–42, accentuating the efficacy of a bivalent approach in augmenting biological responses, particularly for early amyloidosis detection. The utilization of a bivalent approach in the design of radioligands is underscored by a marked and statistically significant enhancement (p < 0.0001) in the binding affinity of compound 237 towards synthetic Aβ1–42 aggregates when compared to its monomeric counterpart, 11C-Chal-Me. This augmentation in binding affinity signifies the efficacy of the bivalent strategy. In the context of PET imaging and biodistribution investigations, both ligands exhibited favorable brain pharmacokinetics, with the bivalent ligand demonstrating higher cerebral uptake. Notably, the analysis of metabolites in brain homogenates derived from healthy ddY mice demonstrated the robust stability of these radiotracers in the cerebral milieu, with over 93% of the tracer remaining intact 30 minutes post-injection (Table 30). Furthermore, both chalcone derivatives exhibited intrinsic fluorescence, and their emission properties underwent substantial alterations upon binding to Aβ1–42. These preliminary findings indicate a promising potential for compound 237 as an in vivo Aβ1–42 imaging tracer. In concert with experimental insights, computational studies corroborated the superior binding affinity and thermodynamic stability of this devised ligand when contrasted with monomeric and conventional ligands. Such initial findings collectively bolster the rationale behind envisioning compound 237 as a prospective in vivo imaging tracer, while concurrently advocating the strategic utility of a bivalent approach in eliciting robust biological responses, thereby offering potential benefits in addressing afflictions as traumatic brain injury (TBI) and AD (Schemes 20 and 21).
Table 30 Percentage of intact tracer in plasma and brain homogenates of ddY mice
| Compounds |
Time after the injection (min) |
Organs |
| Plasma |
Brain |
| 11C-Chal-Me |
10 min |
40.3 |
98.6 |
| 30 min |
25.8 |
94.8 |
| 235 |
10 min |
37.6 |
98.7 |
| 30 min |
23.9 |
93.3 |
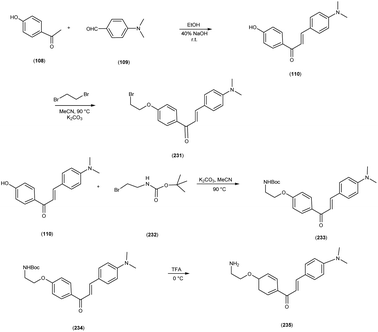 |
| | Scheme 20 | |
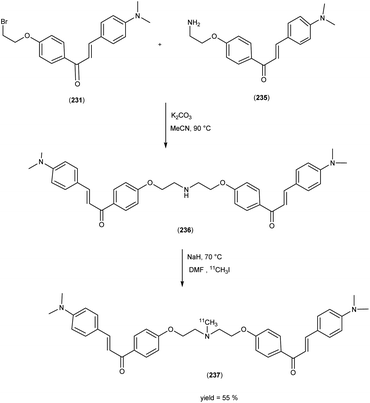 |
| | Scheme 21 | |
7. 68Ga labelled chalcones for AD
In 2014, Kanchan Chauhan and collaborators66 orchestrated a meticulous synthesis endeavor aimed at the production of radiolabelled chalcones, imbued with the potential to serve as PET imaging probes, specifically within the realm of AD. The synthetic protocol employed in this study consisted of a meticulous five-step procedure. To initiate this process, a Claisen–Schmidt condensation reaction was chosen as the method of choice for generating chalcone, specifically (E)-1-(4-hydroxyphenyl)-3-(4-isopropylphenyl)prop-2-en-1-one (110). This intermediate served as the foundational building block for subsequent transformations. The culminating and paramount phase of the synthesis entailed the reaction of diethylenetriamine pentaacetic acid (DTPA) with (E)-1-(4-hydroxyphenyl)-3-(4-isopropylphenyl)prop-2-en-1-one (110), yielding the complex molecular structure denominated as 5,8-bis(carboxymethyl)-13-(4-((E)-3-(4-(dimethylamino)phenyl)acryloyl)phenoxy)-2-(2-(2-(4-((E)3(4(dimethylamino)phenyl)acryloyl)phenoxy)ethylamino)-2-oxoethyl)-10-oxo-2,5,8,11-tetraazatridecane-1-carboxylic acid, abbreviated as DT(Ch)2. It is of noteworthy significance that this intricate chemical transformation was executed with a remarkable 94% yield, denoting a highly efficient synthetic endeavour. This innovative ligand (243) was envisaged as an adept tool for PET-based detection through radiolabelling with the radioisotope 68Ga. In an in vitro binding assay conducted on Aβ1–42 aggregates, it was observed that 68Ga-DT(Ch)2 exhibited a remarkable binding affinity, characterized by an inhibition constant of 4.18 ± 0.62 nM. The fluorescent attributes of this ligand were noteworthy, featuring absorption/emission peaks at 410/540 nM (Table 31). Notably, upon binding to Aβ aggregates, the ligand displayed a distinct blue shift and a notable 5.5-fold enhancement in emission intensity. In the context of blood kinetics, investigations conducted in normal rabbits indicated rapid clearance dynamics, with a half-life (t1/2) of 24 ± 0.08 nM minute for the fast component (t1/2(F)) and 2 hours and 40 ± 0.04 nM minute for the slow component (t1/2(S)). Moreover, ex vivo biodistribution analysis unveiled the blood–brain barrier penetration capabilities of the complex, as evidenced by a brain uptake of 1.24 ± 0.31% ID/g at 2 minutes post-injection, followed by expeditious washout, with negligible activity (0.36% ID/g) remaining at 30 minutes post-injection (Table 32). These initial investigations underscore the potential of the bivalent synthesis approach, as it had a minimal impact on the binding affinity of the compound. Consequently, the synthesized 68Ga-complex, denoted as 68Ga-DT(Ch)2, represents a promising candidate for generator-produced PET imaging probes in the context of AD. The study further delved into the intricate dynamics within the circulatory system of normal rabbits, uncovering the rapid clearance kinetics of the synthesized complex. Importantly, the investigation also disclosed that the strategic bivalent synthesis strategy did not exert any adverse influence on the binding affinity of the resulting compound. This outcome resonates with the binding affinity of the resulting compound (Scheme 22).
Table 31 Inhibition constants (Ki, nM) for ligands binding to Aβ42 aggregates (n = 3)
| Compounds |
Ki (nM) |
| DT(Ch)2 (243) |
21.5 ± 0.94 |
| 69/71Ga-DT(Ch)2 (244) |
4.18 ± 0.62 |
| DMIC |
3.64 ± 0.3 |
| Thioflavin |
T > 6000 |
Table 32 Biodistribution of 68Ga-DT(Ch)2 (244) after tracer injection in normal mice (n = 3)
| Compound |
Organ |
Times after injection |
| 2 min |
10 min |
30 min |
60 min |
| 244 |
Blood |
6.22 ± 1.4 |
5.20 ± 0.81 |
1.69 ± 0.18 |
0.83 ± 0.11 |
| Brain |
1.24 ± 0.31 |
1.06 ± 0.09 |
0.36 ± 0.06 |
0.20 ± 0.01 |
| Heart |
1.34 ± 0.16 |
1.03 ± 0.13 |
0.43 ± 0.12 |
0.36 ± 0.06 |
| Lung |
1.77 ± 0.17 |
3.24 ± 0.27 |
1.54 ± 0.14 |
0.90 ± 0.09 |
| Liver |
11.12 ± 1.02 |
14.67 ± 1.42 |
11.85 ± 1.21 |
6.78 ± 1.08 |
| Spleen |
1.04 ± 0.37 |
2.10 ± 0.31 |
0.67 ± 0.18 |
0.33 ± 0.08 |
| Kidney |
4.86 ± 0.93 |
5.31 ± 0.86 |
2.50 ± 0.57 |
1.28 ± 0.27 |
| Stomach |
0.54 ± 0.13 |
0.82 ± 0.11 |
0.69 ± 0.13 |
0.36 ± 0.07 |
| Intestine |
2.33 ± 0.19 |
3.19 ± 0.15 |
5.98 ± 0.28 |
6.23 ± 0.25 |
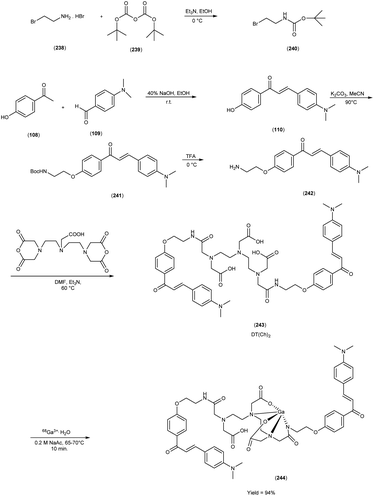 |
| | Scheme 22 | |
8. Current overview and future prospective
The continued exploration of radiolabelled 1,3-diaryl-2propen-1-ones as Aβ imaging probes presents a dynamic trajectory for research endeavors. The future investigations may focus on refining synthetic methodologies to enhance the efficiency and yield of these radiotracers, ultimately facilitating their widespread availability and utilization. The exploration of novel chalcone derivatives with tailored molecular architectures may yield compounds with improved imaging properties, enhanced brain penetration and heightened selectivity for Aβ-amyloid plaques, thereby elevating the precision and accuracy of early AD detection. In the realm of radiochemistry, a diverse array of strategies has been meticulously investigated to enhance the efficacy of radiolabelling procedures. These strategies have predominantly harnessed cutting-edge pathways tailored to the specific attributes of commonly employed radionuclides within nuclear medicine applications. Notably, the radioisotope iodine-125 has been directly introduced onto activated aryl structures, employing well-established methodologies such as iododestannylation reaction. Conversely, the incorporation of fluorine-18 necessitated the utilization of tosylated precursors or more contemporaneously, innovative approaches like the application of aryl boronic precursors. The carbon-11 radiolabelling in its customary execution entailed the reaction between N-nor methyl precursors and [11C]methyl triflate. The utilization of technetium-99m spanned a diverse landscape of chalcone derivatives, involving the bifunctional chelator strategy encompassing MAMA, BAT and DTPA moieties. Alternatively, an integrated approach was pursued, involving the substitution of a section of the chalcone structure with a cyclopentadienyl group. An intriguing synthesis was the generation of homodimer analogs labelled with [68Ga], facilitated through the strategic deployment of the acyclic chelator DTPA. In-depth investigations ensued to comprehend the ramifications of structural modifications and the consequences associated with distinct radionuclide choices on the ultimate affinity of radiotracers for Aβ-amyloid plaques. This multifaceted exploration drew upon an amalgamation of techniques. Conventionally, studies pertaining to affinity and binding assays were conducted on synthetic aggregates. However, there was a parallel endeavor involving in vitro staining utilizing fluorescent chalcone derivatives on brain sections from both murine and human subjects, characterized by plaque expression. Finally, the in vivo biodistribution studies conducted on normal murine models, provided a critical assessment of brain uptake dynamics and radiotracer washout characteristics. The future collaborative efforts among chemists, radiopharmacologists and neuroscientists may lead to the development of innovative radiolabelled chalcones with multi-modal capabilities, enabling simultaneous imaging of various biomarkers associated with AD pathology. These advanced probes could offer comprehensive insights into the disease progression, aiding in elucidating intricate molecular mechanisms and paving the way for more therapeutic interventions.
In parallel, translating the findings of this review into clinical applications holds immense potential. The refinement and validation of radiolabelled chalcones as reliable tools for non-invasive AD diagnosis and patient stratification could expedite the identification of individuals at risk, enabling early intervention and personalized treatment strategies. Additionally, the integration of these imaging probes into larger clinical trials and longitudinal studies may yield critical insights into disease progression, therapeutic efficacy and response to interventions, thereby contributing to the development of novel therapeutic avenues and monitoring tactics. As the field of radiolabelled chalcones for Aβ imaging probes continues to evolve, synergistic collaborations among researchers, clinicians and pharmaceutical industries are likely to propel the translation of these innovative concepts from bench to bedside. The elucidation of the intricate interplay between radiolabelled chalcones and AD pathology, coupled with the clinical validation of their diagnostic and prognostic utility, holds the promise of ushering in a new era of precision medicine in the management of AD.
9. Conclusion
Alzheimer's disease (AD) is a complicated neurological disorder that affects millions of people around the world. In recent decades, after several biochemical and pharmacological studies to decipher the complexity of this degenerative disease, scientists have been able to identify and develop innovative therapeutic agents to control this unpredictable disease. A study on this topic showed that most of the radiopharmaceuticals that reached phase two did not show a narcotic difference and therefore did not reach phase three. However, more than fifty molecules have successfully progressed from phase two to phase three, but only one drug has passed phase three clinical trials since 2003. Interestingly, more than 140 therapeutic agents are currently under investigation at various stages of clinical trials, but it is still unclear how many of them will progress from clinical trials to primary care. In this review, we discussed biosynthesis, the structural significance of radiolabelled chalcones, their therapeutic uses as AD agents, and structure–activity correlation research. Using nuclear imaging techniques such as positron emission tomography (PET) and single photon emission computed tomography (SPECT), radiolabelled chalcones as Aβ imaging agents could lead to in vivo detection of AD. The researchers have primarily focused on the skeletal modification of chalcones to produce new and innovative compounds with a wide range of applications. The reviewed literature endeavored to provide a meaningful overview of radiolabelled chalcones as novel Aβ-imaging probes.
Abbreviations
| PET | Positron emission tomography |
| SPECT | Single-photon emission computed tomography |
| AD | Alzheimer's disease |
| DPP | Diphenylpropynone |
| HPLC | High-performance liquid chromatography |
| IMPY | 6-Iodo-2-(40-dimethylamino-)phenyl-imidazo [1,2]pyridine |
Conflicts of interest
The authors disclose no conflicts of interest.
Acknowledgements
We gratefully acknowledge the University Grant Commission, New Delhi, India, for financial support in the form of a Junior Research Fellowship (JRF) Sudeep Dhillon (Ref. No. 114/(CSIR-UGC NET JUNE) and Mamta Chahal (Ref. No. 2019211610128270) for this research. We are also thankful to the Council of Scientific and Industrial Research (CSIR), New Delhi, India, for providing financial support to Ginna Kumari (Ref. No. 09/1307(16134)/2022-EMR-1). We also acknowledge the Chaudhary Bansi Lal University, Bhiwani for the financial assistance in the form of research project (Grant No. CBLU/DAA/2022/2137). This work was also supported by the National Research Foundation of Korea (NRF) grant (No. 2021R1I1A3059257) funded by the Korea government-MSIT (Ministry of Science and ICT) and Dongguk university Wise campus research Fund.
References
- P. Scheltens, K. Blennow, M. M. B. Breteler, B. de Strooper, G. B. Frisoni, S. Salloway and W. M. Van der Flier, Lancet, 2016, 388, 505–517 CrossRef CAS.
- C. L. Masters, R. Bateman, K. Blennow, C. C. Rowe, R. A. Sperling and J. L. Cummings, Nat. Rev. Dis. Primers, 2015, 1, 15056 CrossRef.
- Y. D. Park, M. Kinger, C. Min, S. Y. Lee, Y. Byun, J. W. Park and J. Jeon, Bioorg. Chem., 2021, 115, 105167 CrossRef CAS.
- P. Thapa, P. Upadhyay, W. Z. Suo, V. Singh, P. Gurung, E. S. Lee and M. Sharma, Bioorg. Chem., 2021, 108, 104681 CrossRef CAS PubMed.
- A. Dement and J. Alzheimer's Ass, Alzheimers. Dement., 2020, 16, 391–460 CrossRef.
- W. Thies and L. Bleiler, Alzheimers. Dement., 2013, 9, 208–245 CrossRef PubMed.
- I. W. Hamley, Chem. Rev., 2012, 112, 5147–5192 CrossRef CAS PubMed.
- T. Athar, K. Al Balushi and S. A. Khan, Mol. Biol. Rep., 2021, 48, 5629–5645 CrossRef CAS PubMed.
- H. Hippius and G. Neundorfer, Dialogues Clin. Neurosci., 2003, 5, 101–108 CrossRef PubMed.
- W. V. Graham, A. Bonito-Oliva and T. P. Sakmar, Annu. Rev. Med., 2017, 68, 413–430 CrossRef CAS.
- S. Matsunaga, T. Kishi, I. Nomura, K. Sakuma, M. Okuya, T. Ikuta and N. Iwata, Expert. Opin. Drug. Saf., 2018, 17, 1053–1061 CrossRef CAS.
- D. Prvulovic, H. Hampel and J. Pantel, Expert Opin. Drug. Metab. Toxicol., 2010, 6, 345–354 CrossRef CAS PubMed.
- M. A. Raskind, Neurologist, 2003, 9, 235–240 CrossRef PubMed.
- G. T. Grossberg, C. Sadowsky and J. T. Olin, Int. J. Clin. Pract., 2010, 64, 651–660 CrossRef CAS.
- K. Articus, M. Baier, F. Tracik, F. Kuhn, U. W. Preussa and A. Kurz, Int. J. Clin. Pract., 2011, 65, 790–796 CrossRef CAS.
- R. Khoury, J. Rajamanickam and G. T. Grossberg, Ther. Adv. Drug Saf., 2018, 9, 171–178 CrossRef CAS PubMed.
- T. T. Reddy, M. H. Iguban, L. L. Melkonyan, J. Shergill, C. Lianga and J. Mukherjee, Molecules, 2023, 28, 865 CrossRef CAS PubMed.
- N. Kumar, V. Kumar, P. Anand, V. Kumar, A. R. Dwivedi and V. Kumar, Bioorg. Med. Chem., 2022, 61, 116742–116771 CrossRef CAS.
- Y. Yang, H. M. Jia and B. L. Liu, Molecules, 2012, 17, 4252–4265 CrossRef CAS.
- K. Barber, P. Mendonca and K. F. Soliman, Brain Sci., 2023, 13, 145 CrossRef CAS.
- C. Simmler, D. C. Lankin, D. Nikolić, R. B. van Breemen and G. F. Pauli, Fitoterapia, 2017, 121, 6–15 CrossRef CAS PubMed.
- D. K. Mahapatra, S. K. Bharti and V. Asati, Eur. J. Med. Chem., 2015, 98, 69–114 CrossRef CAS PubMed.
- P. Singh, A. Anand and V. Kumar, Eur. J. Med. Chem., 2014, 877, 58–77 Search PubMed.
- A. Rammohan, J. S. Reddy, G. Sravya, C. N. Rao and G. V. Zyryanov, Environ. Chem. Lett., 2020, 18, 433–458 CrossRef CAS.
- R. Sasidharan, S. L. Manju, G. Uçar, I. Baysal and B. Mathew, Arch. Pharm., 2016, 349, 627–637 CrossRef CAS.
- X. Q. Wang, L. Y. Zhou, R. X. Tan, G. P. Liang, S. X. Fang, W. Li and Y. P. Chen, Chem. Biodiversity, 2021, 18, 2100341 CrossRef.
- K. C. Gautam and D. P. Singh, Chem. Sci. Trans., 2013, 2, 992–996 CAS.
- A. Singh, V. S. Reddy, K. Santosh, G. B. Kumar, A. B. Shaik, R. Mahesh and S. Kotamraju, MedChemComm, 2014, 5, 1718–1723 RSC.
- N. Kaur and D. Kishore, J. Chem. Sci., 2013, 125, 555–560 CrossRef CAS.
- O. Prakash, A. Kumar, M. Kinger and S. P. Singh, Indian J. Chem., 2006, 45, 456–460 Search PubMed.
- B. M. Sahoo, M. Rajeswari, P. Jnyanaranjan and S. Binayani, Indian J. Pharm. Educ. Res., 2017, 51, 700–706 CrossRef.
- D. M. Lokeshwari, N. D. Rekha, B. Srinivasan, H. K. Vivek and A. K. Kariyappa, Bioorg. Med. Chem. Lett., 2017, 27, 3048–3054 CrossRef CAS PubMed.
- M. Kumar, H. Sushil, P. Jeong and W. Kim Sang, Nat. Prod. J., 2014, 4, 93–114 Search PubMed.
- S. Kantevari, D. Addla, P. K. Bagul, B. Sridhar and S. K. Banerjee, Bioorg. Med. Chem., 2011, 19, 4772–4781 CrossRef CAS PubMed.
- V. R. Suma, R. Sreenivasulu, M. V. B. Rao, M. Subramanyam, M. J. Ahsan, R. Alluri and K. R. M. Rao, Med. Chem. Res., 2020, 29, 1643–1654 CrossRef CAS.
- D. K. Mahapatra, V. Asati and S. K. Bharti, Eur. J. Med. Chem., 2015, 92, 839–865 CrossRef CAS.
- H. Zhang, J. J. Liu, J. Sun, X. H. Yang, T. T. Zhao, X. Lu and H. L. Zhu, Bioorg. Med. Chem. Lett., 2012, 20, 3212–3218 CrossRef CAS.
- A. Detsi, M. Majdalani, C. A. Kontogiorgis, D. Hadjipavlou-Litin and P. Kefalas, Bioorg. Med. Chem., 2009, 17, 8073–8085 CrossRef CAS.
- T. L. B. Ventura, S. D. Calixto, B. D. A. Abrahim-Vieira, A. M. T. D. Souza, M. V. P. Mello, C. R. Rodrigues and M. F. Muzitano, Molecules, 2015, 20, 8072–8093 CrossRef CAS.
- B. Das, S. Samanta, S. Burle and P. C. Behera, Int. J. Pharm. Res., 2015, 7, 1–8 Search PubMed.
- A. Rammohan, B. V. Bhaskar, N. Venkateswarlu, W. Gu and G. V. Zyryanov, Bioorg. Chem., 2020, 95, 103527 CrossRef CAS.
- M. Chen, S. Brøgger Christensen, L. Zhai, M. H. Rasmussen, T. G. Theander, S. Frøkjaer, B. Steffansen, J. Davidsen and A. Kharazmi, J. Infect. Dis., 1997, 176, 1327–1333 CAS.
- R. Sasidharan, S. L. Manju, G. Uçar, I. Baysal and B. Mathew, Arch. Pharm., 2016, 349, 627–637 CrossRef CAS.
- X. Q. Wang, L. Y. Zhou, R. X. Tan, G. P. Liang, S. X. Fang, W. Li and Y. P. Chen, Chem. Biodiversity, 2021, 18, 2100341 CrossRef.
- E. D. Agdeppa, V. Kepe, J. Liu, S. Flores-Torres, N. Satyamurthy, A. Petric, G. M. Cole, G. W. Small, S. C. Huang and J. R. Barrio, J. Neurosci., 2001, 21, 1–5 CrossRef.
- K. Shoghi-Jadid, G. W. Small, E. D. Agdeppa, V. Kepe, L. M. Ercoli, P. Siddarth and J. R. Barrio, Am. J. Geriatr. Psychiatry, 2002, 10, 24–35 CrossRef.
- C. A. Mathis, Y. Wang, D. P. Holt, G. F. Huang, M. L. Debnath and W. E. Klunk, J. Med. Chem., 2003, 46, 2740–2754 CrossRef CAS.
- M. Ono, A. Wilson, J. Nobrega, D. Westaway, P. Verhoeff, Z. P. Zhuang and H. F Kung, Nucl. Med. Biol., 2003, 30, 565–571 CrossRef CAS PubMed.
- C. C. Rowe, U. Ackerman, W. Browne, R. Mulligan, K. L. Pike, G. O'Keefe and V. L. Villemagne, Lancet Neurol.Am. J. Geriatr. Psychiatry, 2008, 7, 129–135 CAS.
- M. Ono, R. Ikeoka, H. Watanabe, H. Kimura, T. Fuchigami, M. Haratake and M. Nakayama, Chem. Neurosci., 2010, 1, 598–607 CrossRef CAS.
- Y. Maya, M. Ono, H. Watanabe, M. Haratake, H. Saji and M. Nakayama, Bioconjugate Chem., 2009, 20, 95–101 CrossRef CAS PubMed.
- M. Cui, M. Ono, H. Kimura, B. L. Liu and H. Saji, Bioorg. Med. Chem. Lett., 2011, 21, 980–982 CrossRef CAS PubMed.
- M. Ono, H. Watanabe, R. Watanabe, M. Haratake, M. Nakayama and H. Saji, Bioorg. Med. Chem. Lett., 2011, 21, 117–120 CrossRef CAS.
- M. Ono, M. Haratake, H. Mori and M. Nakayama, Bioorg. Med. Chem. Lett., 2007, 15, 6802–6809 CrossRef CAS PubMed.
- M. Cui, M. Ono, H. Kimura, B. Liu and H. Saji, J. Med. Chem., 2011, 54, 2225–2240 CrossRef CAS.
- M. Ono, M. Hori, M. Haratake, T. Tomiyama, H. Mori and M. Nakayama, Bioorg. Med. Chem. Lett., 2007, 15, 6388–6396 CrossRef CAS.
- M. Ono, Y. Maya, M. Haratake, K. Ito, H. Mori and M. Nakayama, Biochem. Biophys. Res. Commun., 2007, 36, 116–121 CrossRef.
- T. Fuchigami, Y. Yamashita, M. Haratake, M. Ono, S. Yoshida and M. Nakayama, Bioorg. Med. Chem. Lett., 2014, 22, 2622–2628 CrossRef CAS PubMed.
- M. Ono, R. Ikeoka, H. Watanabe, H. Kimura, T. Fuchigami, M. Haratake and M. Nakayama, Chem. Neurosci., 2010, 1, 598–607 CrossRef CAS.
- M. Ono, R. Ikeoka, H. Watanabe, H. Kimura, T. Fuchigami, M. Haratake and M. Nakayama, Bioorg. Med. Chem. Lett., 2010, 20, 5743–5748 CrossRef CAS.
- Z. Li, M. Cui, J. Dai, X. Wang, P. Yu, Y. Yang and B. Liu, J. Med. Chem., 2013, 56, 471–482 CrossRef CAS PubMed.
- M. Ono, R. Watanabe, H. Kawashima, Y. Cheng, H. Kimura, H. Watanabe and M. Nakayama, J. Med. Chem., 2009, 52, 6394–6401 CrossRef CAS PubMed.
- S. Kaide, M. Ono, H. Watanabe, Y. Shimizu, Y. Nakamoto, K. Togashi and H. Saji, Bioorg. Med. Chem. Lett., 2018, 26, 3352–3358 CrossRef CAS PubMed.
- M. Ono, R. Watanabe, H. Kawashima, T. Kawai, H. Watanabe, M. Haratake and M. Nakayama, Bioorg. Med. Chem. Lett., 2009, 17, 2069–2076 CrossRef CAS.
- K. Chauhan, A. K. Tiwari, N. Chadha, A. Kaul, A. K. Singh and A. Datta, Mol. Pharmaceutics, 2018, 15, 1515–1525 CrossRef CAS.
- K. Chauhan, A. Datta, A. Adhikari, K. Chuttani, A. K. Singh and A. K. Mishra, Org. Biomol. Chem., 2014, 12, 7328–7337 RSC.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Mayank Kinger
a,
Mayank Kinger