
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances of bifunctional catalysts for zinc air batteries with stability considerations: from selecting materials to reconstruction
Wanqi
Tang
ab,
Jiarong
Mai
a,
Lili
Liu
 *a,
Nengfei
Yu
*a,
Lijun
Fu
a,
Yuhui
Chen
a,
Yankai
Liu
c,
Yuping
Wu
*acd and
Teunis
van Ree
e
*a,
Nengfei
Yu
*a,
Lijun
Fu
a,
Yuhui
Chen
a,
Yankai
Liu
c,
Yuping
Wu
*acd and
Teunis
van Ree
e
aState Key Laboratory of Materials-oriented Chemical Engineering, Institute of Advanced Materials (IAM), School of Energy Science and Engineering, Nanjing Tech University, Nanjing 211816, P. R. China. E-mail: liulili@njtech.edu.cn; yunf@njtech.edu.cn; wuyp@fudan.edu.cn
bCollege of Chemical Engineering, Nanjing Tech University, Nanjing 210009, China
cHunan Bolt Power New Energy Co., Ltd, Dianjiangjun Industrial Park, Louxing District, Loudi 417000, Hunan, China
dSchool of Energy and Environment, Southeast University, Nanjing 210096, China
eDepartment of Chemistry, University of Venda, Thohoyandou 0950, South Africa
First published on 19th July 2023
Abstract
With the growing depletion of traditional fossil energy resources and ongoing enhanced awareness of environmental protection, research on electrochemical energy storage techniques like zinc–air batteries is receiving close attention. A significant amount of work on bifunctional catalysts is devoted to improving OER and ORR reaction performance to pave the way for the commercialization of new batteries. Although most traditional energy storage systems perform very well, their durability in practical applications is receiving less attention, with issues such as carbon corrosion, reconstruction during the OER process, and degradation, which can seriously impact long-term use. To be able to design bifunctional materials in a bottom-up approach, a summary of different kinds of carbon materials and transition metal-based materials will be of assistance in selecting a suitable and highly active catalyst from the extensive existing non-precious materials database. Also, the modulation of current carbon materials, aimed at increasing defects and vacancies in carbon and electron distribution in metal–N–C is introduced to attain improved ORR performance of porous materials with fast mass and air transfer. Finally, the reconstruction of catalysts is introduced. The review concludes with comprehensive recommendations for obtaining high-performance and highly-durable catalysts.
 Yuping Wu | Dr Yuping Wu, FRSC, Chair Professor of Southeast University. He got his PhD degree from Institute of Chemistry, CAS, in 1997. In 2003, he was promoted to full professor in Fudan University, China. In 2015 he moved to Nanjing Tech University and shifted to Southeast University in 2021. He has published over 430 papers in peer-reviewed journals with H-index > 96, was awarded the World's Most Influential Minds (2015) by Thomson Reuters, and Albert Nelson Marquis Lifetime Achievement Award (2019), and one of Highly Cited Researchers over the World. He undertook pioneering research work on cutting-edge aqueous batteries, and gel-type and nonporous separators for lithium batteries with high safety, lithium sulfur batteries of high energy density, and hybrid capacitors. |
1. Introduction
The increasing frequency of energy shortages and the deleterious effects of carbon-based fuels, such as environmental pollution and related green-house effects are factors driving the acceleration in the development of secondary energy sources that are mobile, portable, with high-efficiency energy conversion and, most importantly, clean to produce and use. To realize the low-carbon goal and greatly decrease carbon dioxide emissions, cleaner energy sources must be developed. Solar and wind energy and a variety of storage systems such as fuel cells, batteries, and supercapacitors for electricity production and powering of electronic devices have become important issues recently.1 The development of new and clean energy storage techniques is gaining momentum and drives policy-making; hence, highly efficient, safe, functional and easily-commercialized devices are needed to meet the demands of utilization and environmental protection.2Li-ion batteries are increasingly getting involved in our life. In spite of great progress, there still is a great need for higher energy and power density since in some working situations the lithium-ion battery cannot satisfy this need. While some researchers are cautious about the future of this technology, metal–air batteries are growing in importance as possible next-generation devices answering demands for higher performance.3 For example, long-distance traveling of electric vehicles up to 700 km may be hard to achieve with traditional batteries.4 Zinc (Zn), magnesium (Mg), and aluminium (Al)–air batteries are receiving increasing attention, exhibiting safe, low cost and abundant materials. Besides these, Li–air batteries (LABs) show the highest theoretical energy density, rivalling the gasoline engine (13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 W h kg−1), but incomplete cycling and safety issues are hindering wide usage.4,5 Therefore, zinc–air batteries are an appropriate choice for both achieving capacity and safety (compared to, e.g., Na).6 In the following paragraphs, articles published recently focusing on the catalyst for zinc–air batteries will be discussed.
000 W h kg−1), but incomplete cycling and safety issues are hindering wide usage.4,5 Therefore, zinc–air batteries are an appropriate choice for both achieving capacity and safety (compared to, e.g., Na).6 In the following paragraphs, articles published recently focusing on the catalyst for zinc–air batteries will be discussed.
Unlike metal-ion batteries and supercapacitors, secondary metal–air batteries involve two reversible reactions on the air cathode surface, viz. the oxygen evolution reaction (OER) and oxygen reduction reaction (ORR), corresponding to the charge and discharge processes, respectively. The developing voltage gap between catalyst and metal has the potential to meet the demands for both high current density and stable discharge voltage.7 However, this technology usually is associated with problems with dendrite formation and efficiency loss in the electrolyte, especially in the alkaline electrolyte, hindering the long-term use of metal–air batteries.3,8
Secondary metal–air batteries are highly sensitive to the concentrations of reactants and rate of electron transfer, which on a macroscopic scale correspond to current density and stability. The reaction rate is limited by the concentrations of the reactants and the electron transfer rate. During operation, the low transfer rate and the unstable electrode–electrolyte interfaces limit long-term use, which should be considered when balancing the requirements for real working conditions.9 Therefore, for commercial application of metal–air batteries, research is not only aimed at improving catalyst ability but also focusing on synthesis of new electrode materials that support long-term electrode stability with morphology control or other techniques.10,11
Among the applications of metal–air batteries, the user-friendly zinc–air battery (ZAB) system is attractive as a substitute for Li-ion-based batteries. Zinc–air batteries and Li–air batteries are regarded as promising and viable energy systems with advanced technology that will improve upon the existing engineering process and theoretical performance limits of Li batteries.12 A typical ZAB usually consists of a zinc anode, electrolyte, separator, and bifunctional air cathode. During charging and discharging, cycling between Zn-ion and Zn metal, the Zn metal interacts with the electrolyte.13 Zn–air batteries can also be characterized as all-solid or aqueous according to the electrolyte phase. The OER and ORR reactions at the air electrode match the generation and consumption of O2 mainly from the oxygen dissolved in the electrolyte and outside air (in theory). For the ORR reaction during discharge, atmospheric O2 diffuses through the air cathode, which usually consists of three compressed layers (gas diffusion layer, current collector, and electrocatalyst), simultaneously accepting electrons from the cathode to form hydroxide ions in an alkaline enviroment.14 In the OER process, Zn is deposited from the electrolyte, while the air cathode generates O2 bubbles. This causes a mechanical breakdown of the catalyst by the airflow and massive corrosion of carbon or catalyst when the charging voltage reaches ∼2.0 V. Some researchers separate the OER and ORR reactions by adding two different catalysts that work when charging and discharging, respectively.
As three-phase reactions, the OER and ORR reactions are strongly catalyzed by state-of-the-art precious metals such as Pd, Pt, Ir, or Au from the point of view of activity and durability. High prices and lack of resources are regarded as barriers to the large-scale application of noble metals.15 Although remarkably, 95% of fuel cells are applying Pt as an efficient catalyst,16 transition metals still have the potential for substituting the noble metals. In recent years, more and more researchers have attempted to improve the properties of catalysts based on transition metals such as Ni, Co, Fe, and Mn exhibiting bifunctional catalytic ability for the OER and ORR reactions. Carbon-based materials like graphene and N-doped carbon nanotubes (CNTs) and non-metal-doped oxides such as spinel and perovskites have been introduced to improve the performance of ZABs. Many metal-based materials deposited on highly conductive nanosheets like MXenes,17 porphyrin-based metal–organic frameworks,18 and MOF/PCP-based materials,19 like non-thermal plasma,20 are also used to modify the performance of ZABs.21
Many reviews have focused on mechanism analysis and new ZAB systems to give advice on how to rationally select catalysts.22 Some are proposing new techniques of catalyst optimization for better electrochemical performance, like interfacial engineering23 and advanced architecture design.24 In this discussion we categorize modification into three types: (1) higher conductivity (like carbon materials), (2) catalytic ability (lower adsorption energy with hetero-doping), and (3) larger reactive area (structure and geometrical optimization). The attempts made to meet the goals of achieving a high-performance cell with longer stability, are shown schematically in Fig. 1.
 | ||
| Fig. 1 Schematic illustration of key processes catalyzed by bifunctional materials. | ||
2. OER and ORR reactions on air cathode of ZABs
The zinc–air battery was first studied in 1878, and a few years later a gas diffusion electrode with a porous carbon layer on a nickel current collector was investigated, also called the Walker Wilkins battery.25 Currently, some commercialized zinc–air cells can be found in many systems like remote communications, seismic telemetry, and navigator signalling.But this primary battery cannot be used on a large scale because it cannot be recharged. Zinc–air batteries have been studied intensively from 1975 to 2000, and as Li-ion battery technology matured, many international companies, such as EOS, Fluid Energy, and ZincNyx have joined the field with excellent research work.25 However, even though the commercial foundation of ZABs has been laid at the lab level – showing potential for extended use and good performance in applications where Li-ion batteries are less useful – their commercial use is still hampered by the limited performance at high current density and over the long term. Therefore, researchers are focusing on extended aqueous/flexible/combined ZABs with high performance and safer configuration than Li-based batteries, as more and more demand arises for advanced wearable electronics and super-thin devices.
The metal redox reaction takes place at the zinc foil, which is an excellent metal source just below Li in terms of power density per mass, and should support the capacity of the battery and sustain the voltage for a long time. However, because zinc suffers from many problems,26–29 the low efficiency of this metal electrode limits its use. As a result, electrolytes, separators, and also cell configurations are studied to remove these barriers. In this review, we focus on the air electrode, which provides a possible interface for the OER and ORR reactions.
The oxygen evolution reaction presumably contributes to water electrolysis and has been studied by many researchers seeking to unravel the complicated pathways and mechanisms of water electrolysis. In a typical alkaline electrolyte, the intermediates playing important roles in these mechanisms arise as follows:
| M + OH− → M–OH + e− | (1) |
| M–OH + OH− → M–O + H2O + e− | (2) |
| M–O + OH− → M–OOH + e− | (3) |
| M–OOH + OH− → M + H2O + O2 + e− | (4) |
M–OH, M–O, and M–OOH represent the intermediates formed at the active site for reaction, and M–O corresponds to the reactive metal–O bonds. While these single-electron half-reactions take place during oxygen evolution, the interface is unstable due to two mechanisms – lattice oxidation mechanism (LOM), where lattice oxygen participates in the reaction, and adsorbate evolution mechanism (AEM), where the lattice oxygen is not involved. By deeply studying these two mechanisms, the degrading mechanisms of different catalysts for the OER are now better understood. Also, the stability of ZABs using metal as a catalyst is strongly influenced by the OER performance in an alkaline or flexible battery. Hence, the development of efficient and robust OER catalysts is urgently needed to meet the demand for rechargeable ZABs.30
For the reverse mechanism, two major reaction pathways are widely accepted. One is a direct four-step single-electron pathway producing hydroxide, representing a highly efficient way for reduction of O2 in ZABs; while the other is an indirect two-electron pathway that produces peroxide. Over recent years several active sites have been proposed by some researchers when highly conductive carbon or N-doped carbon-based materials are introduced;31 however, most intermediate half-reactions can be represented as follows:
| M + H2O + O2 + e− → M–OOH + OH− | (5) |
| M–OOH + e− → M–O + OH− | (6) |
| M–O + H2O + e− → M–OH + OH− | (7) |
| M–OH + e− → M + OH− | (8) |
As formulated, oxygen is absorbed by the metal in the presence of water, and one electron contributes to formation of M–OOH while another electron is transferred to form M–O. After this transfer, M–O gains an electron and a proton to form M–OH, and a final electron transfer reduces this product back to the initial M with formation of a hydroxide ion.32 In this half-reaction series, the rates of generation and transformation of the intermediates play a vital role in the reaction kinetics. To obtain high reaction rates and capacity, the binding energy between surface-active sites and intermediates should be optimized.
For the OER reaction, Ru, RuO2, Ir, and IrO2 are known as benchmark catalysts for the long-term oxygen evolution process in alkaline or acidic electrolyte.33 The corrosion resistance during reaction leads to higher stability than that of other metal-based oxides.34 More and more research is dedicated to highly-stable resources in both catalytic performance and crystal structure to unravel the mechanism of the OER reaction on precious metals.35–38
For the discharge process, which is an ORR reaction on an air cathode, Pt is considered to be the material that best balances activity and stability, and is regarded as the ideal material for a 4-electron transfer reaction. This precious metal is usually mixed with carbon, exhibiting high ORR performance in both acidic and alkaline electrolytes.39
To estimate more objectively the ORR performance at laboratory level, a rotating disk electrode (RDE) with a three-electrode system is normally used, removing diffusion influence to some extent.40 As the ORR process is actually co-affected by surface reactions and mass transfer, the half-wave potential (E1/2) – the potential needed to reach half of the limiting current density, obtained from the LSV curve – is a widely accepted indicator (although it may not be comprehensive) for evaluating this ORR performance, which may differ little as diffusion ability changes.41,42
Recently, due to high electrolysis demands, as in PEMFCs, the Pt/C electrode has been modified to increase the operation time by avoiding carbon corrosion, Pt dissolution, active site leaching, Ostwald ripening, and agglomeration of nanoparticles.43 Alloys, nanostructured material, and loading on other carbon-based materials are approaches used to enhance the coordination process and stabilize the crystal structure for the ORR reaction.
Before considering the substitution of these precious metals with more abundant metals and carbonaceous materials to find suitable catalytic materials, it is crucial to understand the mechanism of the ORR process. Numerous early reports have mentioned that the first electron transfer steps are the rate-determining steps (RDS) for the ORR, which means the O2 adsorption process is the limiting process. However, recently it has been pointed out that the intermediate steps are regarded as RDS processes, which means that the adsorption and desorption of M–O, M–OH, and M–OOH determine the reaction rate. Norskov et al. introduced a method to calculate the free energy of all intermediates using density functional theory (DFT) calculations, based on the potential of the electrode to determine the adsorption energy. The results show that electron transfer is impossible when oxygen is adsorbed strongly on the Pt(111) face,44,45 but by lowering the activation energy needed, the reaction would proceed. Volcano plots were used to calculate the adsorption energy for removal of O and OH species, showing that Pt and Pd provide the best potential for effective catalytic performance.
D-band theory was also introduced by Nørskov for further understanding of the intermediates and reaction rate. The adsorption energy of ORR intermediates on the active site of the catalyst directly affects the reaction activity. Nørskov et al. used DFT to study the adsorption energy of oxygen intermediates at an early stage. There exists a unique scaling relationship between the free energies of the oxygen intermediates. Briefly, the d-band theory enables comparison of the oxygen–metal bond on one transition metal surface, and the strength of coupling of 2p states of oxygen and d states of metal. Therefore, a fast reaction should result in an upward shift of the catalyst's Fermi level of the strength or energy of bonds, leading to less filling, and a suitable bond can be formed. For a precious metal such as Pt, the Fermi level can also be modulated by alloying, surface modulation, and varying the structure of the catalyst.46,47
As the active site of metal cations influences the multi-valence characteristics of the working performance of transition metals, the modification of a bifunctional catalyst for the previous two reactions is mostly based on optimization of the geometry and structure for (1) more active reaction sites (reaction rate), (2) doping, tailoring crystal structure and metal-bonds for higher catalytic performance (better onset potential), and (3) combination and added carbon base for higher conductivity (more electron transfer). Carbon-based materials and transition metals-based materials are common catalysts widely used in energy storage devices.
However, the evaluation of a catalyst is based on its electrochemical performance, which is influenced by primary modifications like doping and surface decoration. As the OER and ORR reactions are interfacial reactions that may change crystal structure with the higher voltage and high-alkaline environment in ZABs, rational catalyst design and selection should also consider reconstruction of functional interfaces with highly stable structures. Studies on materials and the reconstruction process help build an understanding of the relationship between stability and performance.
3. Metal-free carbon material
Before being applied in ZABs, carbon materials with their high conductivity have long been considered as highly active support or base for many materials.48,49 Over the last two decades, most carbon materials have been modulated and applied to fuel cells after treatment avoiding their hydrophilicity, but they are seldom used in alkaline electrolysis or zinc–air batteries, because of their extensive corrosion during the OER reaction32,50Due to the stability problems of the OER reaction experienced with precious metals like Pt and Pd, carbon materials such as CNTs and graphene are considered rational substitutes and ideal ORR reaction catalysts with low extraction and processing costs.51,52 High-dispersion structures with large active areas and fast reactant transfer channels enlarge the interface for higher activity, while simultaneously the low resistance facilitates electron transfer. Heteroatom doping of carbon materials has been widely recognized as an efficient method that can significantly improve ORR activity compared to pristine carbon materials.53–55
More recently, the preparation of metal-free materials has concentrated on the synthesis methodology for achieving highly strained regions in sp2 networks; functional and activated nanostructures (O atoms for example); heteroatom doping (N, B alternating electron and active sites); and active sites at defects, vacancy pentagons/heptagons, and edges.54,56 In this section, we concentrate on the origin of 4-electron transfer in metal-free materials, and their high stability in ZABs.
3.1 N-doped graphenes and CNTs
By modification of carbon nanomaterials using various innovative strategies, including surface functionalization,57–59 geometric structuring,60–62 and heteroatom doping,63–65 the OER and ORR reactions can be improved by rational heteroatom doping with B, N, Si, P, and S. N-doped carbonaceous materials are widely used in zinc–air batteries without any added metal.66,67 However, the performance of N-doped graphene68 and N-doped CNTs is strongly affected by their source and processing.In a typical graphene synthesis process, as shown in Fig. 2a, melamine and L-cysteine are mixed in a mass ratio of 4:1 to form a homogeneous precursor that is heated at 600 °C to form C3N4 nanosheets doped with L-cysteine. Then, carbonization and pyrolysis at 800–1000 °C transform the previously formed nanosheets into a 3D interconnected carbon network, with the –C–S–C– bond playing an important role during carbonization. The result is an N-doped graphene named N-GRW that has good OER and ORR characteristics, with a half-wave potential of 0.84 V (vs. RHE) and 0.92 V (vs. RHE) onset potential, and 360 mV overpotential for the OER process. RRDE measurements confirmed a low yield (below 5%) of H2O2 and a high electron transfer number reaching 3.95. This high performance is ascribed to the graphitic N and pyridinic N in the graphene.69 The Mott–Schottky effect accelerates electron transfer on the catalyst, which is either p-type or n-type. As shown by X-ray absorption near edge structure (XANES) spectroscopy of researched intermediates, the quaternary N with n-type doping, rather than the p-type doping by pyridinic N, is responsible for the ORR. Furthermore, the fast adsorption of OOH* and O* intermediates on carbon atoms next to the pyridinic N is responsible for the OER reaction (Fig. 2a). As discussed above, from another angle, doping quaternary N atoms in graphene provides electrons to the π-conjugated system (n-type doping), leading to increased nucleophile strength for the adjacent carbon rings Cd− to enhance O2 adsorption (because O2 has high densities of O lone pair electrons Od+), and hence accelerating the ORR.70,71
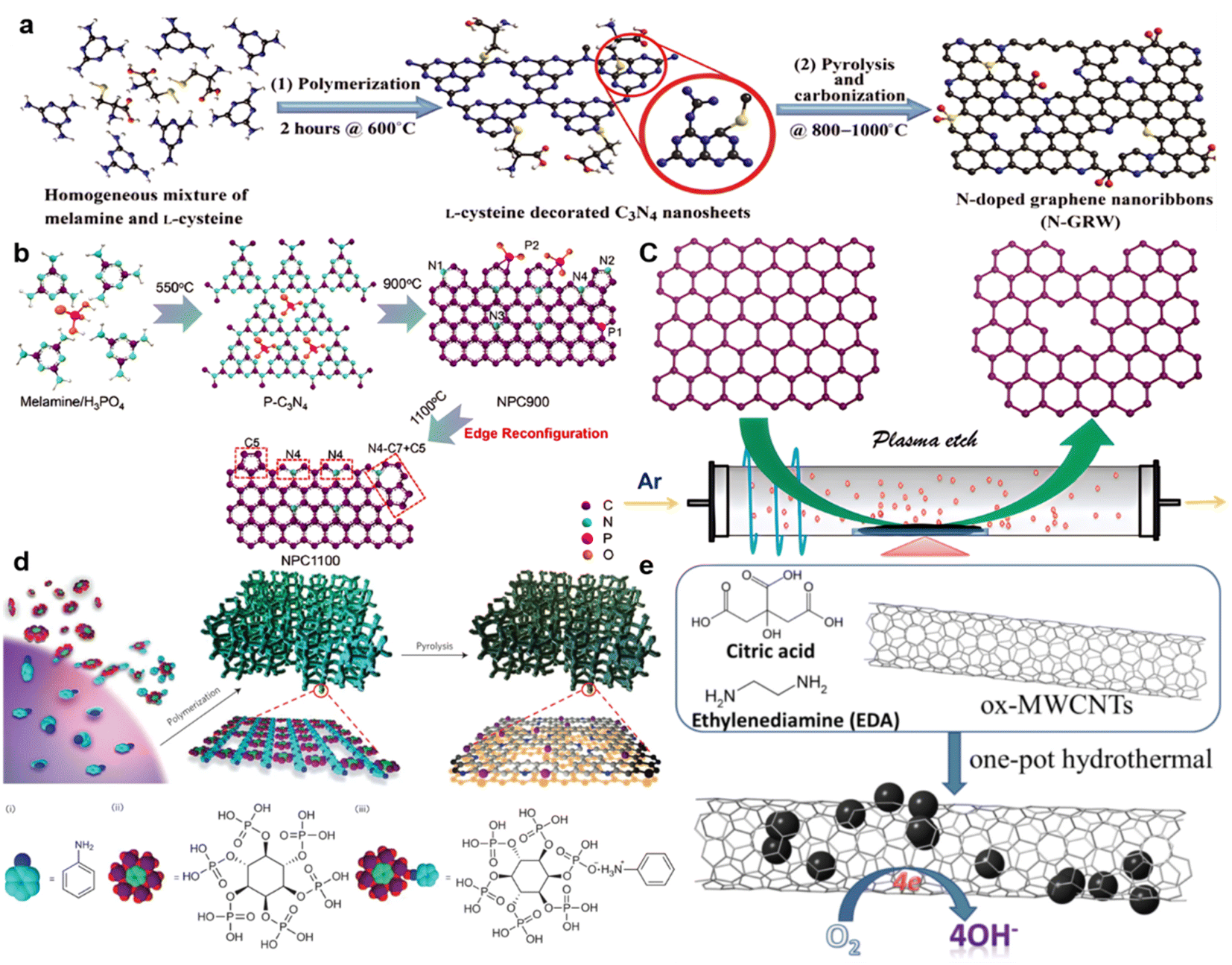 | ||
| Fig. 2 Graphenes and CNTs doped by N. Configuration refinement assisted by (a) S and N, (reproduced from ref. 69 with permission from AAAS, CC BY-NC) (b) P, (reproduced from ref. 73 with permission from Nature) (c) Ar plasma, (reproduced from ref. 74 with permission from the Royal Society of Chemistry). (d) PANi, (reproduced from ref. 78 with permission Copyright ©2015, Nature) and (e) CQDs. (Reproduced from ref. 95 with permission Copyright © 2019 Elsevier). | ||
Also, the defects and edges in the graphene are regarded as active sites for OER and ORR reactions.72 Edge-rich graphene usually exhibits high ORR reactivity. Zigzags in the graphene exhibit different performances before and after N doping. The edge effect also can be enhanced by P doping by the reconfiguration effect, when N4 bonds can be oriented by heating P–C bonds. Consequently, electron transfer numbers over the same potential range are improved from 3.48–3.57 for samples treated at 900 °C, to 3.75–3.94 for samples reconstituted at 1100 °C.73 As shown in Fig. 2b, S and P doping in the precursor (mostly not found in the final product) can also enhance the coupling effect at the edges and inside the graphene structure. After treatment at 1100 °C (the high temperature removing S and P), the final carbon materials are rich in holes and edges on the graphene structure at S and P vacancies. With an Ar plasma (Fig. 2c), the graphite oxide formed by thermal expansion has a voluminous, fluffy structure and the few-layer carbon nanotubes and graphite obtained were also enhanced with more edges, despite the absence of dopants.74 As a result, in 0.1 M KOH, the plasma-treated edge-rich material (P-G) exhibited a potential of 0.912 V (vs. RHE), which is more positive than that of pristine G (0.806 V), with an electron-transfer number of 3.85.
Polymers are widely used as precursors for N-doped materials.75,76 Unlike most of the other synthetic polymers, conducting polymers possess robust conjugated structures that are excellent candidates to produce carbon-based materials, due to their ample carbonization and architecture-maintaining capacity.77 As shown in Fig. 2d, a N,P-doped carbon foam with a large surface area of ∼1663 m2 g−1 that was formed after treatment at 1000 °C of a PANi hydrogel co-doped with nitrogen and phosphorus, exhibited a positive onset potential of 0.94 V versus the reversible hydrogen electrode (RHE) and a half-wave potential of 0.85 V versus RHE in 0.1 M KOH.78 Precursors like PANI-PPy nanofiber,79 PANI with MWCNTs,80 CMP on a SiO2 template,81 crosslinked fluoropolymers,82 and rationally controlled asymmetric sidechain conjugated polymers,83 combined with fast processing such as polymer dehalogenation,84 exemplify synthesis processes for high-active-area carbon materials.85
Besides graphene materials, carbon nanotubes are also a suitable form of carbon with good electron transfer and channels for quick mass transfer.86 Similarly, high nitrogen doping content modifies the charge distribution in the carbon rings, leading to increased C 1s binding energy and inhibition of carbon corrosion during the OER. Thus, N-doped CNTs should exhibit efficient OER and ORR reactions.87 The advantages of this material include improved mass transfer of reagents/products, enhanced electronic conductivity, and higher resistance to corrosion.30,88
Several reviews have described the characteristics of active sites obtained by new strategies and N doping on CNTs and graphene nanoribbons (GNRs), and also other materials like carbon nanofibers,89 carbon nanoribbons derived from MWCNTs,79,90 and N-doped hybrids.72,91 Some new methods have been introduced that link the N doping effects on carbon materials. Several fluorescent carbonaceous nanomaterials such as graphene quantum dots (GQDs), polymer dots (PDs), and nanodiamond QDs have been reported.92–94 Nitrogen-doped CQDs (N-doped CQDs)95 have been introduced to enhance the ORR activity, as shown in Fig. 2e, and highly N-doped CQDs are located on ox-MWCNTs, similar to N,P co-doped CQDs, with electron transfer numbers reaching 3.8, proving a highly catalytically active surface. Synthesis of COF/COP/MOF precursors,96 carbon materials derived from ionic liquid precursors, and solvothermal/hydrothermal synthesis has contributed to a better understanding of carbon materials.97
3.2 Heterodoping of N-doped carbon materials
Doping of carbon materials is not limited to N-doping to enhance ORR reactions; B, P, and S are regarded as ideal heteroatoms to improve the N-doping effects for achieving higher yielding ORR reactions.98 The ORR and OER activities of CNTs can be boosted considerably by an effective oxygen-induced surface electron density modulation strategy.99For co-doping with N and B,100 the introduction of boron by adding a boron source before or during pyrolysis of the carbon material is generally done, as shown in Fig. 3a. Introducing oxidation or reduction processes on pretreated carbon material to form stable bonds, is also commonly done. For example, after the oxidation of graphene with H2SO4/KMnO4 and reduction with NH3–BH3, the graphene edge sites contained more heteroatomic dopants, and the catalyst ORR activity was enhanced. However, harsh oxidation also increased the number of permanent defects in the catalyst, reducing the number of electron pathways.101 Using the zeolitic imidazolate framework (ZIF) ZIF-8 as a template for NaBH4 reduction, the boron is introduced into a box-like carbon structure, as shown in Fig. 3b.102 Apart from traditional pyrolysis, a CVD process could also be introduced in this process, with B located on the graphene103 or CNTs104 to obtain optimized three-coordinate structures. These exhibit sp2-like hybridization to form B–C bonds, contributing to the higher electronegativity of carbon with respect to boron.
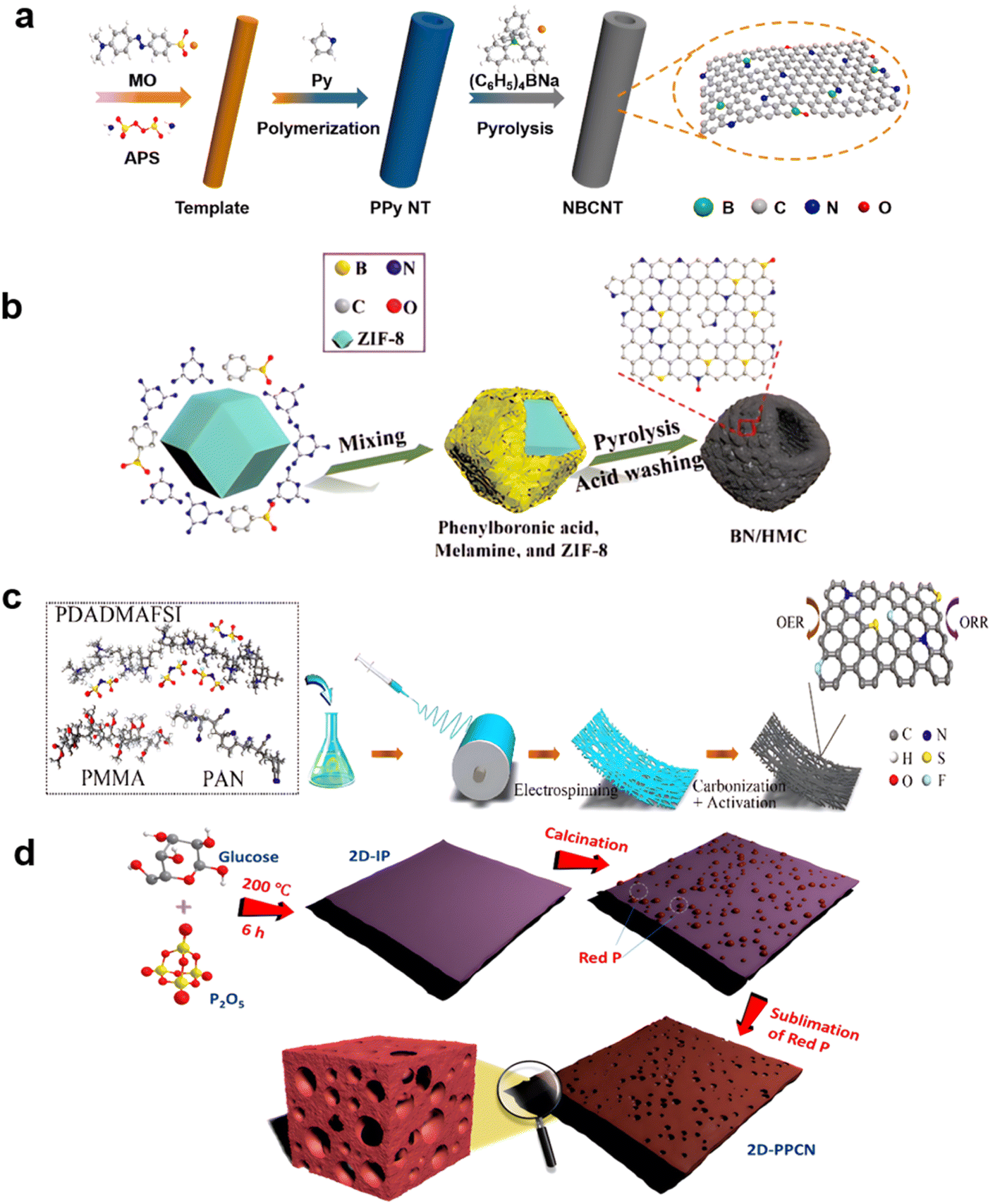 | ||
| Fig. 3 Methods for introducing S and P into carbon materials: (a) (C6H5)4NB on polymers, (reproduced from ref. 100 with permission. Copyright © 2021 Elsevier) (b) NaBH4 on ZIF-8, (reproduced from ref. 102 with permission. Copyright © 2021, American Chemical Society) (c) electrospun with PDADMAFSI, (reproduced from ref. 105 with permission. Copyright © 2021 Elsevier) (d) red P. (reproduced from ref. 107 with permission. Copyright © 2018, American Chemical Society). | ||
More complicated doping with three or more non-metal elements doped on carbon nanofibers was used to functionalize good bifunctional materials in zinc–air or lithium–air batteries.105,106 As shown in Fig. 3c, N, F, and S co-doped nanosheets have been produced by electro-spinning polyacrylonitrile (PAN)/polymerized ionic liquid (PIL) nanofibers, resulting in a positive half-wave potential (0.91 V) for the ORR and a low overpotential (380 mV at 10 mA cm−2). N, F, and S tri-doping also improved electrochemical properties, especially the OER reaction.105 When phosphorus was added before calcination, large-sized two-dimensional phosphorus-doped carbon nanosheets (2D-PPCN) with tunable porosity were synthesized by combining P2O5 with a common saccharide as a template. The efficient production resulted in a 20–35 nm-thick 2D morphology, considerable heteroatom P doping, and excellent tunable porosity. 2D-PPCN-2/6 had a half-wave potential (E1/2) of 0.85 V and an overpotential of 365 mV after iR correction,107 at a current density of 10 mA cm−2 (Fig. 3d).
3.3 Synthesis of porous carbon materials
The evaporation method generated 3D highly porous heteroatom-doped ultrathin carbon nanosheet networks during pyrolysis. Based on the low evaporation temperature of Zn, the precursor of ZIF-8 obtained by solid grinding and water mixing exhibited two kinds of porous structures that are widely used in carbon materials.108 Also, as shown in Fig. 4a, N-doped ultrathin carbon nanosheets with ultrahigh specific surface area (1793 m2 g−1) were obtained by carbonizing a uniform mixture of citric acid and NH4Cl at 1000 °C. This product was rich in inside edge defects and exhibited low overpotential and robust stability for the ORR, OER, and HER.109 Hard-template-like silicon bases are used to generate hollow structures.110,111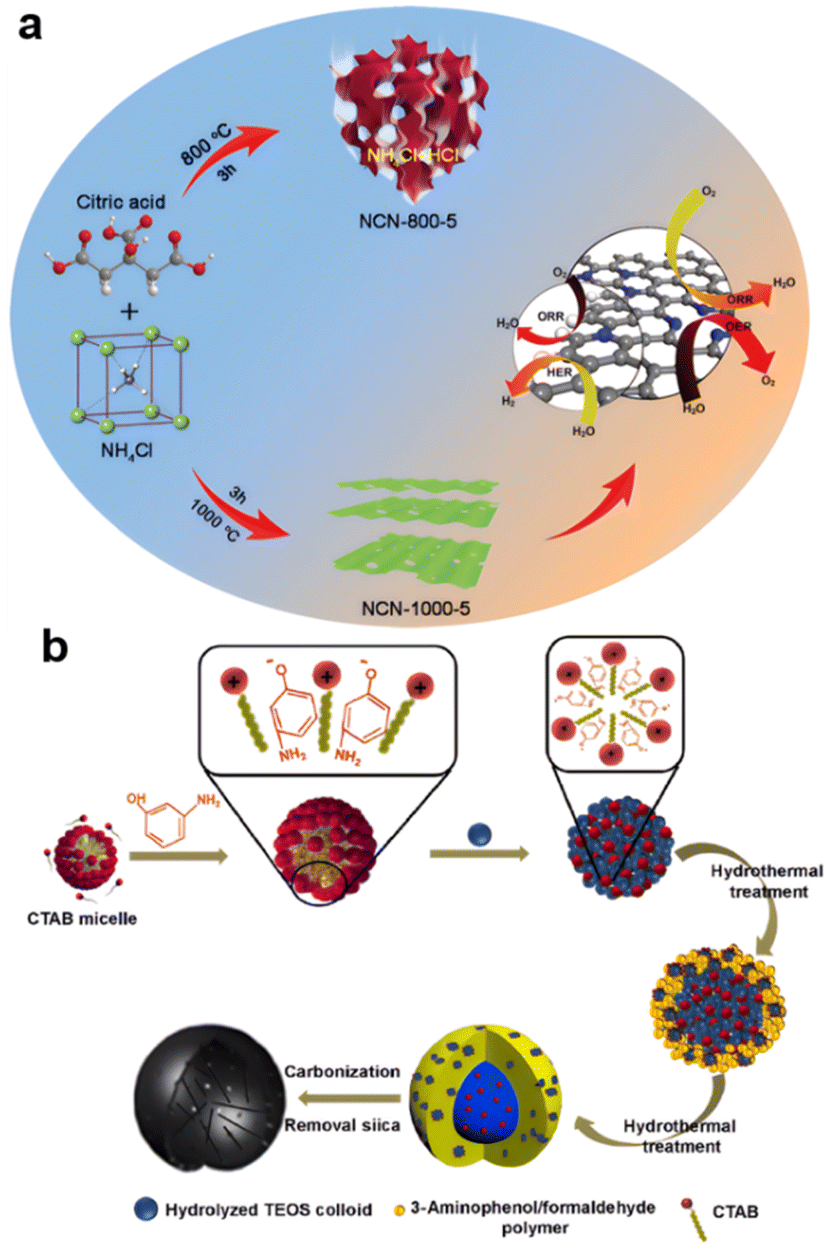 | ||
| Fig. 4 Templates and evaporation method generating porous (a) NH4Cl (reproduced from ref. 109 with permission. Copyright © 2021 Elsevier) (b) silicon and CTAB assisted (reproduced from ref. 112 with permission. Copyright © 2020, Nature). | ||
As shown in Fig. 4b, mesoporous silica nanospheres were formed by self-assembly of silicate oligomers obtained by hydrolysis of TEOS and CTAB micelles, because the hydrolysis and condensation rate of TEOS was faster than the polymerization rate of 3-aminophenol/formaldehyde in the presence of ammonia. As a result, N-doped carbon nanocages (N-CCs) with a porous self-supported architecture and high specific surface area were synthesized.112
Electrospinning and hydrogels113 are also improved techniques for designing porous structures from precursors, starting with highly dispersed nanocarbon fibers, unlike synthesis from hard templates. Electrospun nanomaterials with outstanding chemical stability and structural diversity are promising electrocatalysts for the ORR114–116 As shown in Fig. 5a, inside NPCNF-O, β-cyclodextrin acts as a pore inducer and oxygen regulator. Benefitting from the large specific surface area and synergistic effect of N, O co-doping, the NPCNF-O catalyst exhibits superior ORR (E1/2 = 0.85 V vs. reversible hydrogen electrode (RHE)) and OER (Ej=10 = 1.556 V vs. RHE) activities with excellent stability.91
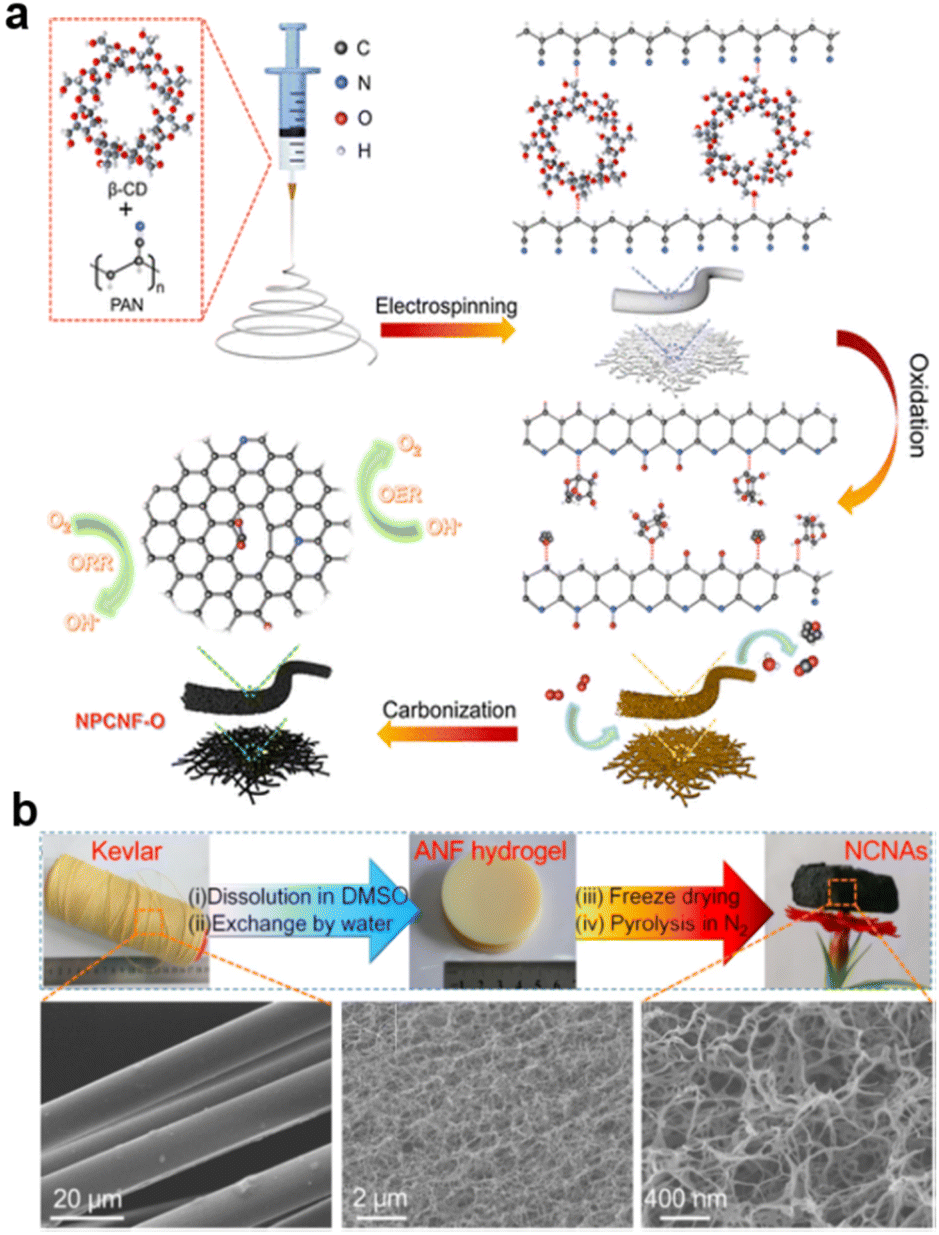 | ||
| Fig. 5 Obtaining highly porous carbon materials by (a) Electrospinning (reproduced from ref. 91 with permission. Copyright © 2022, American Chemical Society) and (b) hydrogel (reproduced from ref. 119 with permission. Copyright © 2018 Elsevier). | ||
Besides being used as electrolyte,117,118 nanofiber aerogels derived from commercial aramid have been prepared using a facile and cost-effective process based on solvent exchange followed by freeze-drying and pyrolysis under a N2 atmosphere. Derived from porous aramid nanofiber (ANF) hydrogel, as illustrated in Fig. 5b, the as-prepared catalyst exhibits high electrocatalytic activity in the oxygen reduction reaction with a half-wave potential estimated to be 0.91 V (vs. RHE) in 0.1 M KOH.119
3.4 Stability of carbon materials
Carbonaceous materials play an important role in zinc–air batteries; however, the effects of conductive carbon materials on the cycling stability of ZABs are usually overlooked.120As shown in Table 1, metal-free carbon materials exhibit similar stability in the alkaline electrolyte at least at 10 mA cm−2 for the ORR reaction, which is close to that of benchmark materials like Pt/C for ORR and Ir/C for the OER process. Most doped carbon materials are known to have long-term stability in the ORR reaction, but there is insufficient data for the OER process. Results differ case-by-case, due to the high-potential carbon corrosion in a highly alkaline environment while charging and discharging at a stable current density or voltage step.
| Catalyst | Electrode | Electrolyte | OER stability | ORR stability | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|
| Time | Potential drop | Potential (or current density) | Time | Potential drop | Potential (or current density) | ||||
| N-GRW | 900 rpm | 1 M KOH | 24 h | 9% | 360 mV | 12 h | 10% | @0.7 V | 69 |
| ZnS/C-1100 | 0.1 M KOH | 10 h | 5.5% | @0.789 V | 128 | ||||
| NCNAS | 1600 rpm | 12 h | 10% | 119 | |||||
| N,P–HCNF-8 | 900 rpm | 0.1 M KOH | 20 h | 9% | @0.7 V | 129 | |||
| NPCNF-O | 1600 rpm | 0.1 M KOH | Slight change | Slight change | 91 | ||||
| DAPHPNC-1020 | 0.1 M KOH | 24 h | 10.9% | 24 h | 14.9% | 130 | |||
| NPCNSS-1000 | 7500 s | 17.6% | @0.8 V (vs. RHE) | 131 | |||||
| MESO/MICRO-POPD | 1600 rpm | 0.1 M KOH | 10000 cycle |
10 mV (half-wave) | 10 mV s−1 | 132 | |||
| NOC-1000-1 | 0.1 M KOH | 24 h | 13.37% | @0.5 V | 133 | ||||
| N-GDY-900 | 1 M KOH | 5000 cycle | 9% | 100 mV s−1 | 134 | ||||
| D/G-HASP | 10 h | 0.1% (overpotential) | 10 mA cm−2 | 10000 cycle |
17 mV (half-wave) | 135 | |||
| PEMAC@CNTS-90 | 1 M KOH | 30000 s |
7% | 5 mA cm−2 | |||||
| ZG-NC-1000 | 0.1 M KOH | 30000 s |
7.7% | @0.7 V | 136 | ||||
| NFS-CNF-0.5-1000 | 1600 rpm | 0.1 M KOH | 30000 s | 23% | @1.6 V (vs. RHE) | 60000 s |
8% | @0.5 V (vs. RHE) | 137 |
| NPSC-DNA-2 | 1600 rpm | 1 M KOH | 10 h | 15% | 105 | ||||
| NBCNT-10 | 1600 rpm | 0.1 M KOH | 12 h | 7.4% | @0.2 V (vs. RHE) | 138 | |||
| N-S-C-2 | 0.1 M KOH | 10000 cycle |
9 mV (half-wave) | ADT (0.6–1.0 V vs. RHE, 100 mV s−1) | 100 | ||||
| CNDA900 | 400 rpm | 0.1 M KOH | 4 h | 12.06% | @0.40 V (vs. RHE) | 139 | |||
| CNT@NSCF | 400 rpm | 0.1 M KOH | 2000 cycles | Small drop | OER LSV | 20000 s |
5.65% | 140 | |
| BP-100 | 0.1 M KOH | 20000 s |
9.7% | @−0.25 V | 141 | ||||
| A-N,S-G | 1 M KOH | 10 h | Steady | @10 mA cm−2 | 142 | ||||
| CNS | 0.1 M KOH | 15000 s |
∼14% | 143 | |||||
| BNF-LCFS | 0.1 KOH | 2000 cycle | 3 mV | @10 mA cm−2 | 40000 s |
15% | @0.665 V (vs. RHE) | 144 | |
| GNP-900 | 1600 rpm | 1 M KOH | 5000 cycle | 13 mV | @10 mA cm−2 | 5000 cycle | 15.4 mV (half-wave) | 106 | |
| PCN–CFP | 0.1 M KOH | 30 h | 6.6% | @1.63 V | 30 h | 2.2% | @0.40 V | 145 | |
| G-C3N4NS–CNT | 0.1 M KOH | 10 h | 13.3% | @1.54 V | 146 | ||||
| NGM | 0.1 M KOH | 8 h | 1.5% | @0.60 V | 59 | ||||
| NCF | 1600 rpm | 0.1 M KOH | 24 h | 11.1% | @0.7 V | 147 | |||
| HPC(MV-C-PN) | 0.1 M KOH | 10 h | No drop | 148 | |||||
The carbon corrosion in both alkaline and acid electrolytes provides a high barrier for application in water electrolysis,121 PEMFCs,122 and zinc–air batteries. The equilibrium potential of carbon is 0.207 V vs. the reversible hydrogen electrode (RHE), making it thermodynamically unstable at potentials higher than the required 1.23 V vs. RHE necessary for the oxygen evolution reaction (OER).123 Real-time monitoring of electrochemical carbon corrosion in alkaline media has been recorded, as shown in Fig. 6a and b.124 An obvious peak proving the relationship between charging-oxidation and CO generation can be observed.125 Oxidative attack of ORR intermediates, demethylation, protonation/neutralization, and micropore flooding are the most commonly observed degradation mechanisms.126
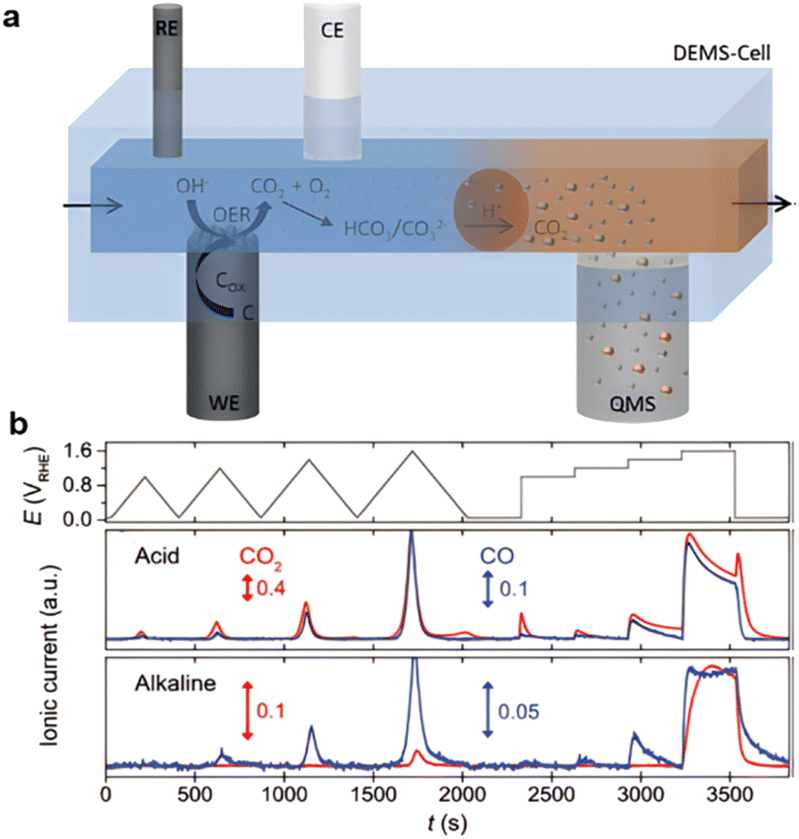 | ||
| Fig. 6 Observation of high corrosion of carbon during oxidization: (a) DEDMs system, (reproduced from ref. 124 with permission. Copyright © 2020 Wiley-VCH) (b) ionic peak of CO. (Reproduced from ref. 125 with permission from the Royal Society of Chemistry). | ||
We display the results for some metal-free carbon materials in Table 1. The stability measured of the rotating-disk electrode (RDE) in a weakly alkaline solution may not imply the same stability in a highly corrosive environment in ZABs. Interestingly, carbon materials with a higher intrinsic OER activity show lower durability and this has been tentatively attributed to the formation of active (e.g., radical) OER intermediates, leading to chemical degradation of carbon materials.122,127
As shown in Table 1, several authors have studied the ORR stability by means of chronopotentiometry. The accelerated durability test (ADT) recorded the linear scan voltammetry (LSV) change over up to 30 hours or 10000 cycles, finding up to 10% long-term degradation of the ORR reaction. This is still far away from cycling for 300 hours, of which 150 hours are discharging, not to mention the OER process. N,P-doped carbon materials, enabling high performance in the OER reaction, and also increasing OER stability, are ideal for long durability. Nitrogen atoms in the carbon materials are redistributed after the reaction.149 To be more specific, highly graphitized platelet-type carbon nanofibers (pCNFs) are tolerant to electrochemical corrosion under oxygen evolution reaction (OER) conditions in a concentrated KOH electrolyte.150 More reports reveal the use of metals to improve OER performance, for example using nickel nanoparticles on rGO151 or NixB coating to decrease the corrosion during OER testing in an alkaline environment.152 This may be attributed to the transition metal working as reaction active sites.
4. Transition metal-based materials
Besides the precious metal and carbon materials with good OER and ORR performance, mentioned in the preceding paragraphs, transition metals have shown higher stability than other materials.153 Many recent publications have described the lower corrosiveness leading to enhanced conductivity and high stability attained by rational modification of transition-metal-based materials used in aqueous alkaline electrolytes. The bi-functional oxygen reaction rates compare favorably to those of the precious metal benchmark catalysts, and the transition metal-based materials are therefore often the first choice as functional zinc–air electrode materials to substitute precious metals. Most of the reported catalysts are metal oxides, but other compounds such as nitrides and sulfides have also been shown to exhibit bi-functional activity.1544.1 Transition metal oxides
The performance of transition metal oxides in the OER in the alkaline electrolyte is usually excellent and stable, promising better long-term use than carbon-based materials; however, the ORR performance of transition-metal-based materials is not as good as that of noble metals like Pt/C. The ORR reaction mechanisms on the metal oxides are different. The surface cations of transition metal oxides coordinate with the oxygen of H2O and at the same time, the hydrogen atoms of H2O are distributed on the catalyst surface. The protonation of surface oxygen ligands is charge-compensated by the reduction of metal cations such as Mn4+, Co3+, and Fe3+. The M–OH intermediates also react further in end-on and side-on reactions.42 The transition metal oxides apparently are also involved in strong metal-to-oxygen covalent interactions, established by the overlapping of the π-orbitals of O2 and dz−2 orbitals of the transition metal.155 In 2011, the Shao-Horn group calculated the volcano activities of the ORR caused by moderate eg sigma-orbital filling with intermediate-strength bonding.156 Surface lattice oxygen was oxidized while the oxygen adsorbate was reduced, highlighting the crucial role of functional transition metal redox in ORR as described by Mueller et al.157Thus, multivalent transition metals like Ni, Co, Fe, and Mn are regarded as the metals of choice for higher OER and ORR activity. Commercialization efforts concentrate on the improvement of transition metal oxides, especially of the manganese oxides (MnO2, Mn2O3, Mn3O4, etc.) that are known to be amongst the most investigated and utilized single metal oxide oxygen catalysts due to their affordability and environmental benignity.158 The higher oxidation state of Mn leads to a favorable OER reaction rate, but due to the decreased activity in the ORR and lower 4-electron transfer rate, the manganese oxides do not perform as well as the noble metals.159
Hence, for application in zinc–air batteries, the improvement of MnO2 starts with improving the ORR activity. Crystal structure modifications introducing more oxygen vacancies, with no foreign additives, were introduced into manganese oxide crystals by simple heat treatment under an air and Ar atmosphere, with Mn3O4 and MnOOH forming the lattice. Forming oxygen-nonstoichiometric oxides by heating at 350–400 °C for about two hours increased the electron transfer number from 3.5 to 3.7 and improved the stability. The formation energy of OOH* on β-MnO2 (110) decreased from 3.09 eV to 0.96 and 0.55 eV by introducing one or two O2c vacancies, respectively (Fig. 7a).160 Secondly, it is also necessary to increase the conductivity. By linking the Mn to NCNTs, the MnO2–NCNT had 220 mV overpotential in 0.1 M KOH for the OER process and higher electron transfer ability, with lower ohmic loss compared to MnO2 and almost 50% lower catalyst resistance.161 Similarly, materials combining metal and carbon like the MnO2–NCNT linked material obtained by the synproportionation of manganese(II) nitrate with potassium permanganate (s-MnOx/MWCNTox) or from the incipient moisture impregnated manganese(II) nitrate (i-MnOx/MWCNTox) perform better in the OER. Modifications that partially achieve higher oxidation states like Mn4+ without changing the reductive carbon base enhance the catalyst's performance in the OER reaction.162
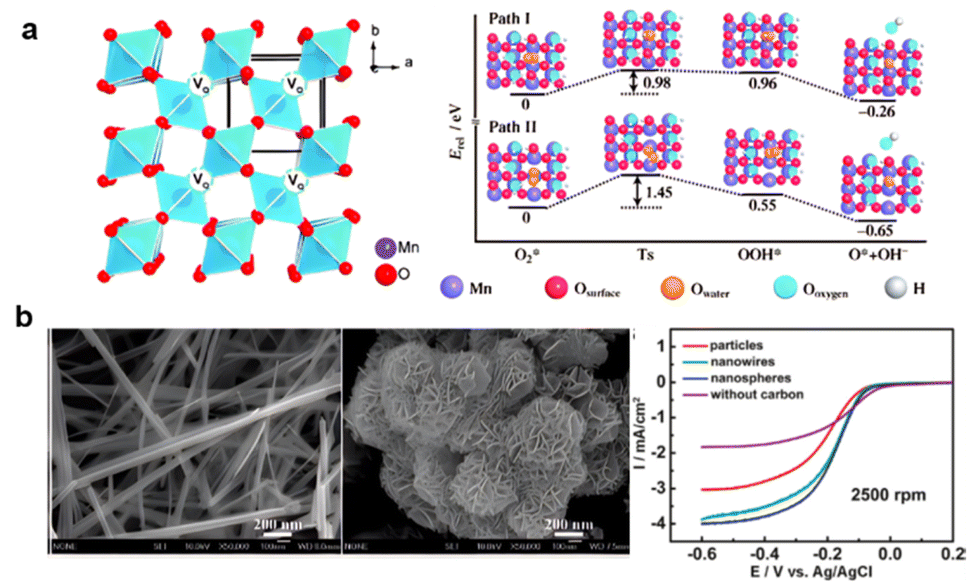 | ||
| Fig. 7 (a) Oxygen induced MnO2 (reproduced from ref. 160 with permission. Copyright © 2013 Wiley-VCH) (b) morphology control of MnO2 and ORR performance (reproduced rom ref. 163 with permission. Copyright © 2010, American Chemical Society). | ||
More active sites can also be obtained by morphology control of MnO2. As shown in Fig. 7b, nanocrystals and nanoplatletes of α-MnO2 can increase the electron transfer number to 3.8, which is only 3.6 for other nanoparticles. To further increase conductivity for O2 adsorption, HO2− chemical-transforming on active sites and ionic and electron transport, MnO2-NWs@Ni-NPs are also recommended.163,164 More complicated methods such as N–C doping in polymer-derived Mn–Fe–N–C SACs further increase Mn-based material activities,165 while CeO2 could help stabilize Mn valency.166,167 Notably, MnO2 is one of the few materials that can be employed in various fields of EESSs: alkaline batteries, supercapacitors, aqueous rechargeable lithium-ion batteries, and metal–air batteries. Yet, the technology still entails bottlenecks and is not yet ready for commercialization.168
Similarly, modifications of other oxide catalysts like MnO2 are categorized by the crystal structure and the original phase. Strategies to enhance the electrocatalytic performance of manganese oxides include doping with cations, coating with metals, and integrating conductive nanostructures.92,159,169–171
Spinels and perovskites are typical structures of transition metal materials with highly stable and active edge and active site structures. Spinel oxides with a typical formula of AB2O4 (A = Li, Mn, Zn, Cd, Co, Cu, Ni, Mg, Fe, Ca, Ge, Ba, etc.; B = Al, Cr, Mn, Fe, Co, Ni, Ga, In, Mo, etc.) have attracted much interest for application in electrocatalysis and other energy-related processes.172 In an AB2O4 spinel, A occupies the tetrahedral centers, whereas B occupies the octahedrally coordinated centers, which can be described as ATd(B2)OhO4 with the two sites influencing the catalytic performance via different mechanisms.
Cations at octahedral and tetrahedral sites in spinel oxides are believed to have different functions toward oxygen electrocatalysis. For example, when a tetrahedral Co in Co3O4 was replaced by Zn, the resulting ZnCo2O4 had OER activity similar to that of Co3O4.154,173 Besides increasing activity from CoAl2O4 to ZnCo2O4 to Co3O4, heating the CoAl2O4 increased the Co octahedral occupancy, resulting in better OER performance. On the other hand, the tetrahedral cations do not contribute significantly to the OER, based on the eg orbital theory.174 As shown in Fig. 8a and b, Xu's and Shao-Horn's groups consolidated the role of electron orbital filling in metal oxide catalysis, and by analyzing MnxCo3−xO4 (x = 2, 2.5, 3), LixMn2O4 (x = 0.7, 1), XCo2O4 (X = Co, Ni, Zn), and XFe2O4 (X = Mn, Co, Ni), found that the eg occupancy of the octahedral cation site plays an important role in metal oxide catalysis, where a descriptor study on the ORR/OER of spinel oxides was presented by the volcano shape eg filling. The volcano profiles result from the eg electron-governed rate-limiting steps and binding strength of reaction intermediates.175 The edge-sharing [Mn–O6] and [Co–O6] octahedra in the spinel lattice allow the Mn–O–Co super exchange interaction via the edge oxygen, and thus the eg occupancy of active Mn can be regulated by varying the Mn/Co ratio176 or Mn/Fe ratio.177 Besides cation doping, the spin states of the Co sites also play an important role in the OER reaction. In spinel zinc–cobalt oxide (not considered as a superior catalyst for the electrochemical oxygen evolution reaction), in the t2g6eg0 configuration, the existence of low-spin (LS) state cobalt cations hinders the OER activity and gives rise to a purely localized electronic structure, exhibiting poor binding affinity for the key reaction intermediate and lowering the OER activity. High-spin (HS) state cobalt cations in thermally controlled spinel ZnCo2O4 have been found to propagate a spin channel that promotes spin-selected charge transport across the Fermi level during the OER (not working for Co3O4) and generates more favorable orbitals for the adsorption of intermediates *OOH178 (Fig. 8c).
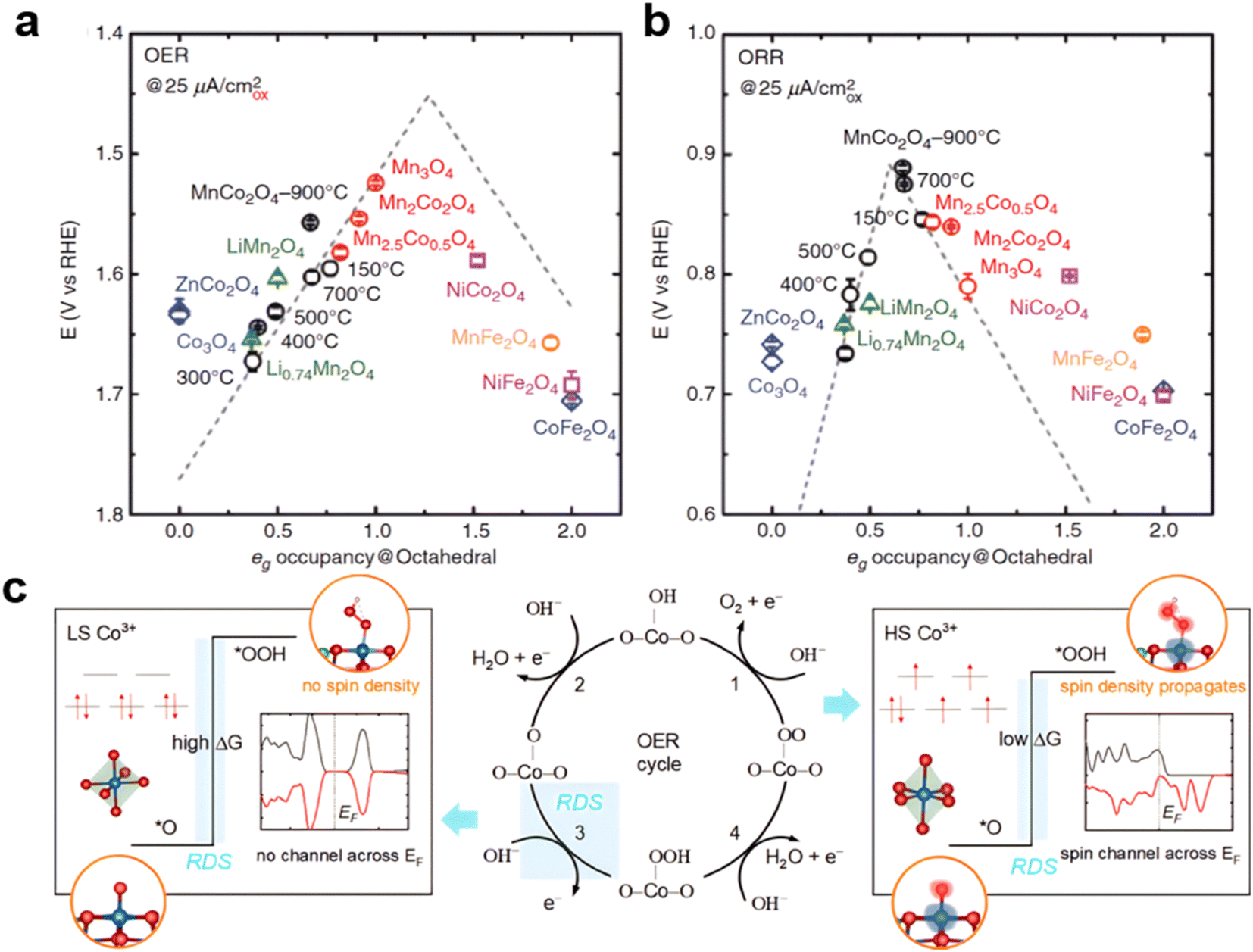 | ||
| Fig. 8 (a) and (b) Volcano relationship between eg filling and OER/ORR performance. (reproduced from ref. 175 with permission. Copyright © 2017 Wiley-VCH). (c) High and low spin of Co and OER RDS. (Reproduced from ref. 178 with permission. Copyright © 2021 Wiley-VCH). | ||
Thus, for a potentially bifunctional material, cation doping, anion doping, and introducing vacancies are typical ways to influence the crystal structure, which increases the intrinsic activity of the metal lattice in the spinels.7,179 In recent years, spinel catalysts have also been created using new approaches such as solid-, solution-,and vapor-phase methods.180
Cation doping is a widely used method for improving the performance of spinels. In addition to single-metal oxides, the rational substitution of a functional metal could efficiently change the physical property of spinels and the activity at interfaces.
In this regard, the incorporation of Ni,82,181,182 Zn,183,184 and Mn185 in cobalt-based spinel oxides has been demonstrated to boost the ORR and OER performance. Wu et al. substituted half of the [Co3+]Oh in Co3O4 by Fe3+, which turned the normal spinel into an inverse spinel structure. The coexistence of Co3+ and Fe3+ in the crystal led to a dissimilarity effect and polarization of octahedral cations, which regulates the adsorption energy of oxygen species and the bond length of adsorbed O2, resulting in superior ORR activity.186 A three-dimensional ordered mesoporous Co3O4 decorated with Mg has been synthesized using PS beads as a hard template and the synthesized MgxCo3−xO4 (3DOM MgxCo3−xO4) displayed a great improvement of the ORR. Substituting Mg2+ in the tetrahedral sites led to more Co3+ occupying octahedral sites and a considerable decrease in the energy level of the d band center (εd).187
Instead of bimetal doping, ternary doping of the functional cations in Co0.5Ni0.5Mn2O4 is also possible.188 The metal can also be changed by a magnetic field. For example, FeCo2O4 nanofiber was annealed in a magnetic field of 2500 Oe, producing a right-shifted half-wave potential of 20 mV for the ORR and a left-shifted overpotential of 60 mV at 10 mV cm−2 for the OER as compared with its non-treated counterpart. Calculations showed that the improvement is caused by the shift close to the Fermi level of the d-band centers (εd) of Co-3d and Fe-3d in the tetrahedral and octahedral sites of the FeCo2O4-M nanofibers.189
Anion doping is also a useful strategy to improve the electrocatalytic activity of metal oxides, and the substitution of O by N, P, S, and Se has been investigated a great deal. Anion doping can lead to increased electronic conductivity.190,191 Derived from a Co-PBA derived Co3O4 nanobox, treated with Na2S at 120 °C for 36 hours, the Co3O4/CoS microbox heterostructure, as shown in Fig. 9a, exhibited a great increase in stability with 98.9% retention of current density after 50 hours CA measurement at 0.4 V in 0.1 M KOH and improved ORR performance with electron transfer number rising from 3.37 to 3.99 as a result of S doping. At the same time, the OER performance of these materials is also increased as evidenced by EIS spectra recorded at a constant potential of 1.65 V (vs. RHE) by a decrease in charge transfer resistance from 290.4 (Co3O4) ohms to 30.8 ohms (Co3O4/CoS).192 The P–O group in a P-doped Co3O4 has been studied using an on-chip micro-device. The coupled P–O groups effectively promote the metal–oxygen covalence of newly-formed oxides, accelerating electron transfer between the active metallic center and oxygen adsorbates, thus leading to enhanced electrocatalytic activity.193 On MnO2 nanorods, coating Mn2+ and Co2+ by TA treatment produced MnS2Ox/MnCo2S4. The low OER activity of MnO2 was greatly increased, getting closer to the fast-charging needs of the zinc–air battery.194 (Fig. 9b).
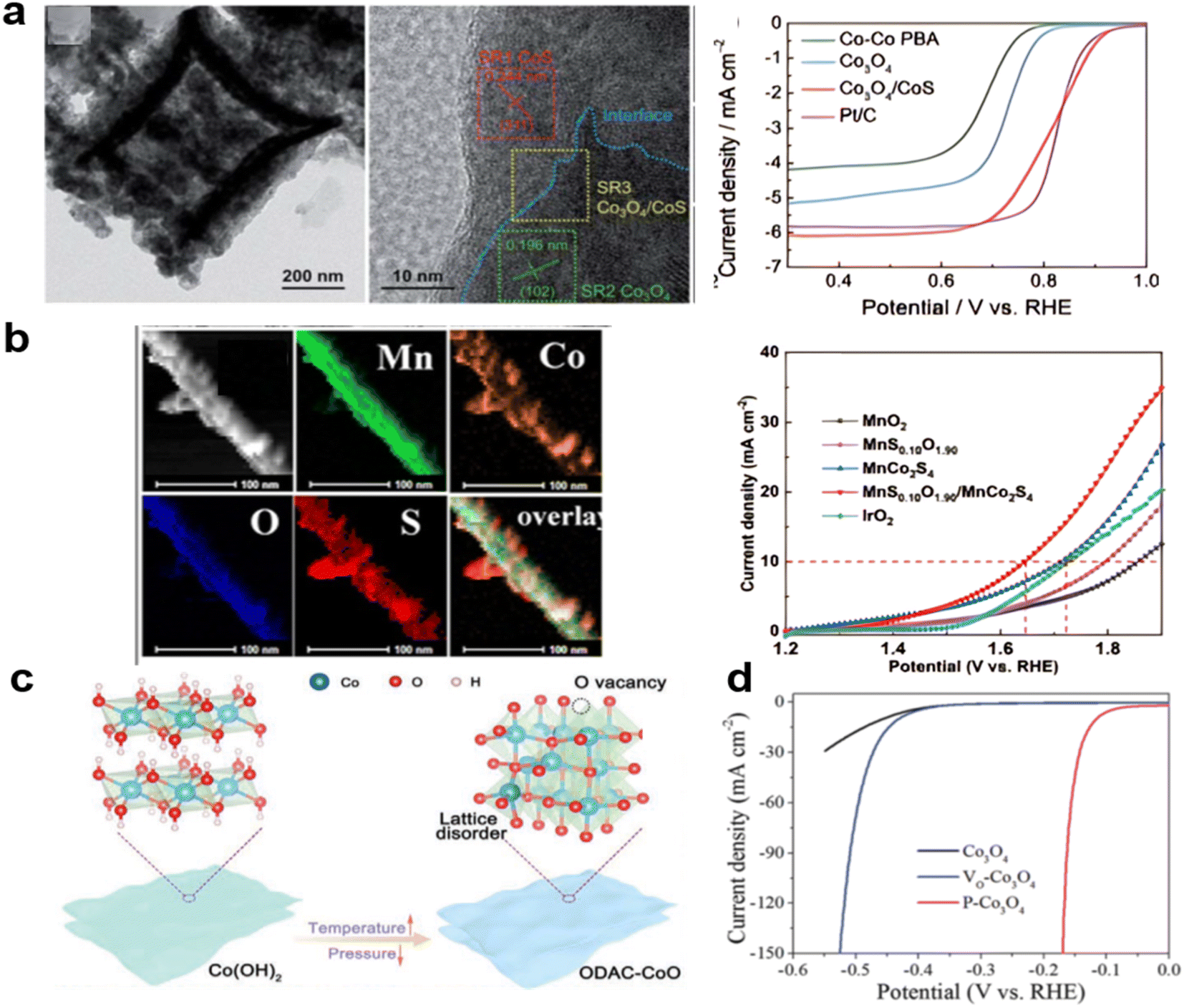 | ||
| Fig. 9 Heterodoped spinels for high performance: (a) S on Co3O4 derived from PBA, (reproduced from ref. 192 with permission from the Royal Society of Chemistry) (b) S on MnCo2O4 needles, (reproduced from ref. 194 with permission. Copyright © 2021 Elsevier) (c) O vacancies introduced by vacuum heating, (reproduced from ref. 198 with permission. Copyright ©2021 Wiley-VCH) (d) P by plasma treatment (reproduced from ref. 200 with permission from the Royal Society of Chemistry). | ||
Increasing the vacancies in spinels with good ORR activity such as Co3O4 tends to improve the OER.195,196 New methods that are widely used are plasma treatment with multi-function: surface atom doping or reconstructing, introducing vacancies or defects, partially reducing or oxidizing surfaces, and increasing the porosity or roughness.197 It has been shown that vacancies can be generated by heating in a vacuum. As shown in Fig. 9c, the vacuum-calcination strategy was utilized to convert Co(OH)2 into oxygen-defective amorphous-crystalline CoO (namely ODAC-CoO) nanosheets with dramatically enhanced ORR and OER electrocatalytic activities.198 This method can be widely used in LDH-derived materials for high porosity and more vacancies. For low and high states of transition metals, Wang et al. used quasi-operando X-ray photoelectron spectroscopy (XPS) and operando X-ray absorption fine structure (XAFS) on the Co3O4 for the OER reaction to confirm that with ample vacancies, the Co2+ to Co3+ process could pre-oxidize the metal to generate Co–OOH⋅ sites for the OER, facilitating the deprotonation process at a lower potential.199 Also, the crystal can be further enhanced by Ar plasma doping and P doping. OER performance was improved in 1 M KOH with higher Co3+ content, showing better ability than direct Ar-plasma treated Co3O4, obtaining more vacancies and higher conductivity, suggesting that the favored catalytic ability of P-Co3O4 is dominated by the Co2+ (Td) site. (Fig. 9d).200.
Besides the influence on the crystal, morphology control also has a great influence on mass transport and accessibility of active sites. As the metal oxides have a low number of active sites exposed to electrolytes, gas, and ions, the performance, especially the ORR performance is sensitive to the morphology. As a simple example, different morphologies of NiCo2O4 were compared and synthesized with a SiO2 template, a P-123 template, and template-free. The Co and Ni valences here exhibited widely different values.201
As shown in Fig. 10, the different added non-carbon catalysts for the ORR reaction produced different morphologies, with the P-123-derived sample exhibiting a unique nanoneedle structure significantly affecting both OER and ORR.201
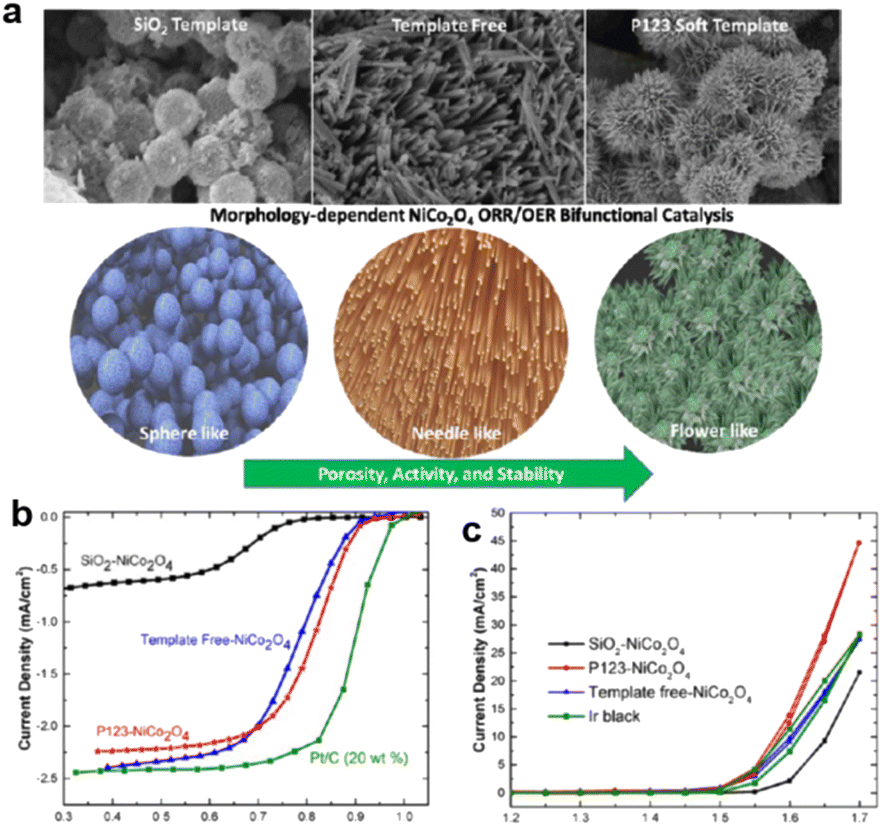 | ||
| Fig. 10 (a) Morphology and (b) OER and (c) ORR performance of NiCo2O4 formed by SiO2 templated, P123 templated, and template free synthesis.201 (Reproduced from ref. 201 with permission. Copyright © 2017, American Chemical Society). | ||
A Fe-containing MOF, Fe4(Fe(CN)6)3 (Prussian Blue, PB) cubes-derived material used to form the porous hollow spinel AFe2O4 (A = Zn, Ni, Co) has been proposed as a good precursor for spinels as energy storage material. Using the self-sacrificial template strategy described in the article, high energy-conversion active materials can be obtained without influencing the box structure.202 Similarly, doping with hierarchical hollow nanoplates (NPs) composed of ultrathin Co3O4 nanosheets doped with 13 different metal atoms, using a Lewis acid etching and metal species coordination process, has been reported. As shown in Fig. 11a, the nitrates of the foreign metals are adsorbed on the previous ZIF configuration and together transformed to a spinel in the calcination process.203 The weakly acidic solution can result in a gentler etching process, cation exchange, before subsequent thermal oxidation processes similar to tannic acid (TA)204 or controlled solutions. Also, on a self-supported system on carbon cloth, as shown in Fig. 11b, a N-confined Co3O4 can be synthesized with nanorod morphology, with N doping and special box structures derived from ZIF-67.205
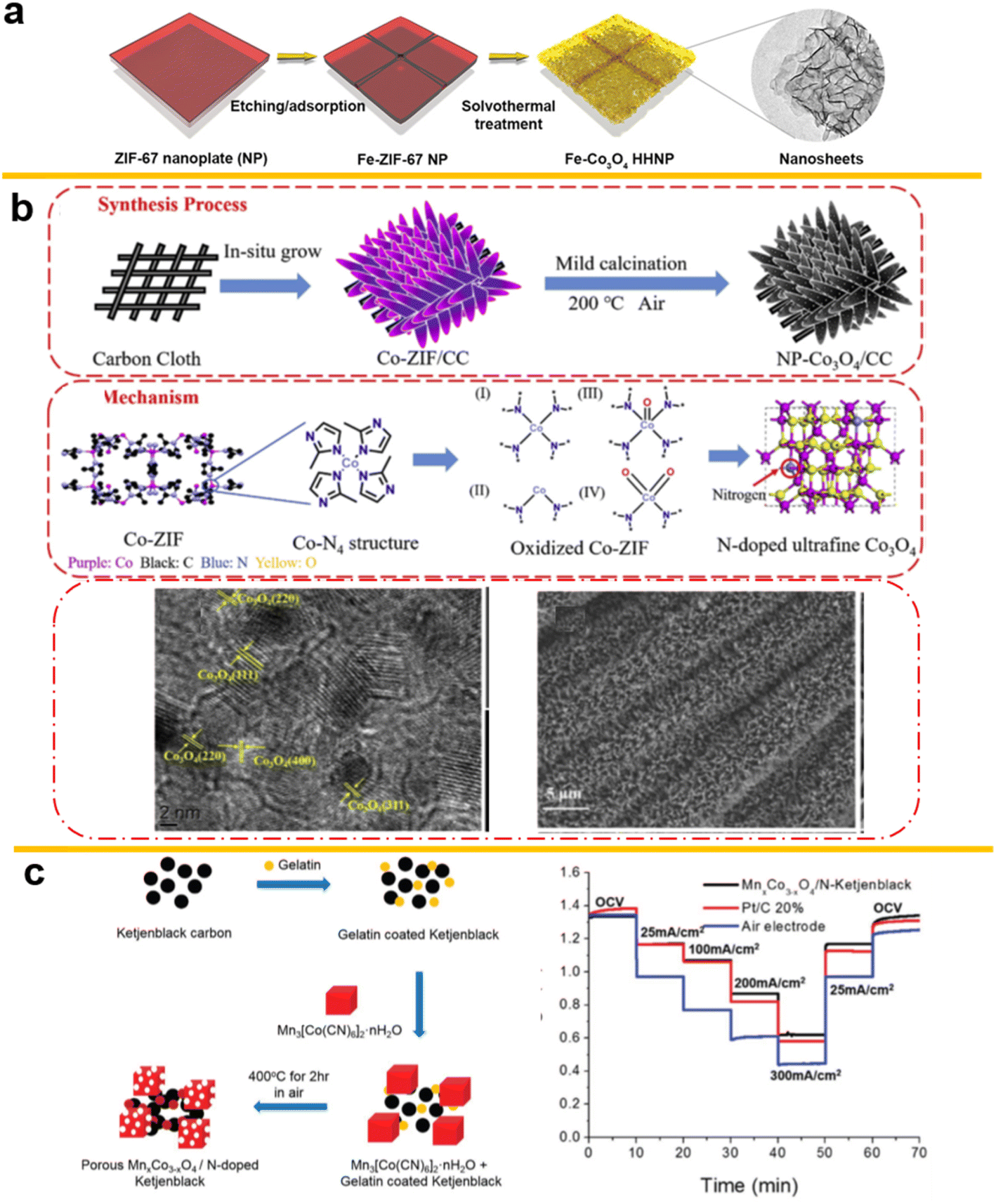 | ||
| Fig. 11 (a) Metal on Co3O4 derived from ZIF-67, (reproduced from ref. 203 with permission Copyright © 2020 from Songlin Zhang et al., published by Wiley) (b) N-doped Co3O4 on Carbon Cloth, (reproduced from ref. 205 with permission Copyright © 2020 Elsevier) (c) MnxCo3−xO4 derived from Prussian blue with high stability in ZABs. (Reproduced from ref. 206 with permission. Copyright © 2016 Wiley-VCH). | ||
In 2016, the Cui group used the Prussian Blue analogue Mn3[Co(CN)6]2·nH2O and gelatin-coated Ketjen black decomposed at 400 °C to form nitrogen-doped carbon material and porous spinel oxides as template (Fig. 11c). The resulting cell displayed the stability of these MOF-derived oxides and the importance of higher transfer ability206 in 6 M KOH rather than 0.1 M KOH with 10 times lower transferability at ambient O2 concentrations in 6 M KOH rather than 0.1 M KOH where the environment RDE test is usually done.207
High reduction performance has been achieved using some new methods. By surface reduction with 5% Li, the La0.6Sr0.4Co0.2Fe0.8O3−δ (LSCF) ORR performance was greatly enhanced, with the half-wave potential increasing from 0.62 V for pristine material to 0.72 V versus RHE. The conductivity of the lithium-reduced LSCF nanopowders increased greatly, with the electron transfer number increasing from 3.71 with the pristine material to 3.91. Improved oxygen vacancies at the surface play an important role in OER and ORR reactions (Fig. 12a).211 Potassium-substituted LaMnO3, in an alkaline environment, performs excellently, with improved kinetics and activity of the ORR (half-wave potential of 0.78 V), OER (potential at the current density of 10 mA cm−2 is 1.66 V) and battery performance (no visible sign of degradation even after 1000 charge–discharge cycles).212
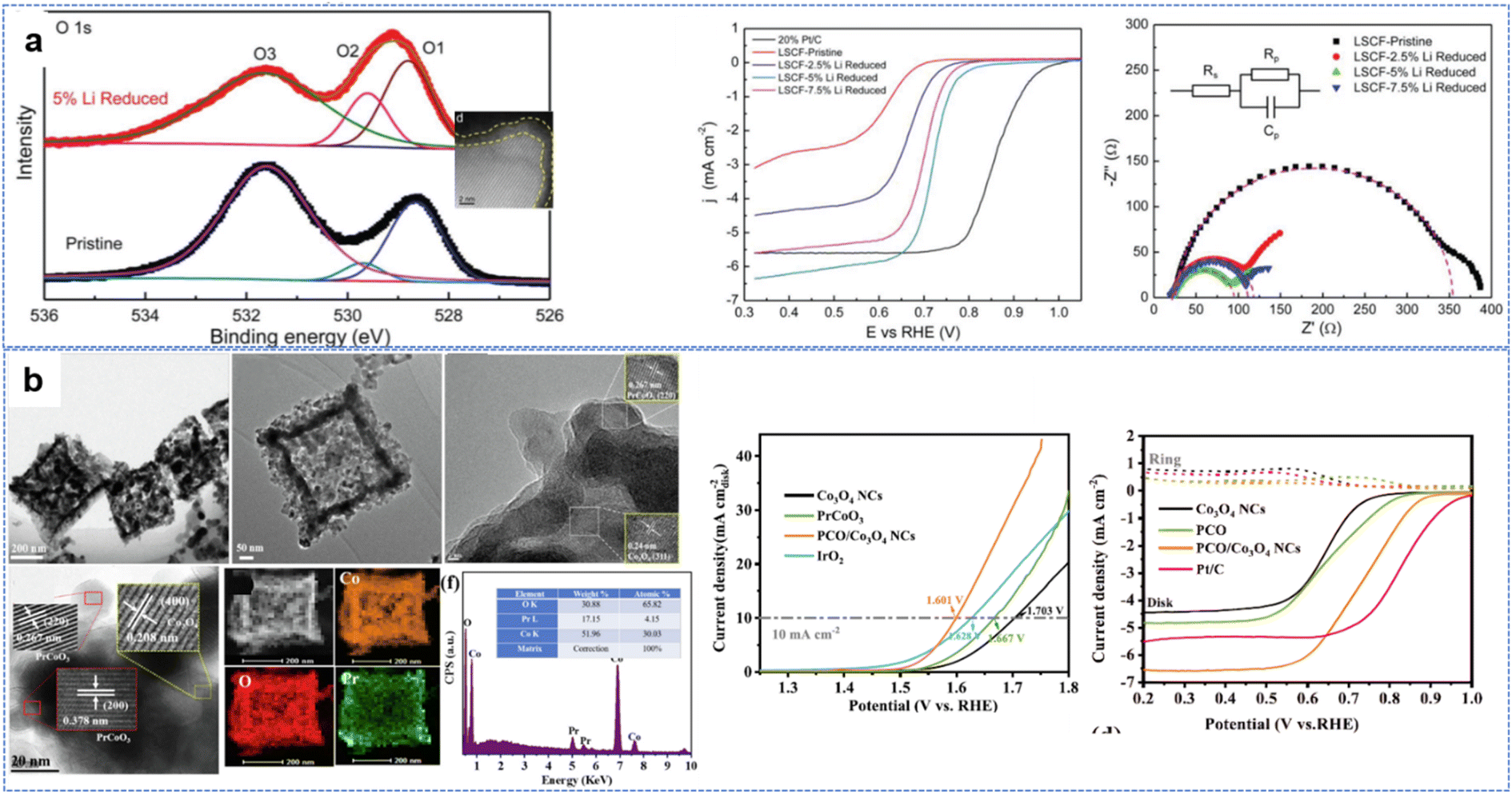 | ||
| Fig. 12 Perovskites used for OER and ORR reactions: (a) Li reduced LSCF, (reproduced from ref. 211 with permission Copyright © 2018 Wiley-VCH) (b) P doped PCO/Co3O4 heterojunction from ZIF-67. (Reproduced from ref. 214 with permission Copyright © 2022 Elsevier). | ||
The yolk–shell structure of the perovskite LaCo1−xFexO3 showing improved ORR performance and possessing an amorphous CoOOH layer during the reaction has proved the vital role of the new interface for reaction sites.213 As shown in Fig. 12b, when composite perovskite/spinel heterojunction PrCoO3/Co3O4 nanocages obtained by Pr3+ adsorption on ZIF-67 boxes were calcined in air, edge-sharing interfaces (where activities are high) of spinel and perovskite with rich oxygen vacancies.214 This PCO/Co3O4 NC catalyst had a low overpotential of 371 mV in 0.1 M KOH at 10 mA cm−2 for the OER and a remarkable decrease of the half-wave potential to 0.72 V for the ORR. Similar perovskite and spinel heterojunctions greatly increased the OER performance of spinels and the ORR performance of perovskites215,216 Dual phases of Co3O4 and LaCoO3 are also similar to this heterojunction with the electron transfer number (n) for Co3O4@LaCoO3 estimated to be 4.0.216 S and P doping217 help increase conductivity, enhancing the bifunctional performance.
Also, increased conductivity is obtained by combining two oxides, such as CoxSey nanosheets on ZnCo2O4 sub-micro/nanospheres with high ORR performance and inside doping like NiCoSe Se-doped spinel.190 Oxides modified with S and P to form CoB,224 plasma-treated NiS,225 CoS nanorods,226 CoS@C,227 Co8S9 (ref. 228) and Co2P,229 scalable COP from MOF,230 NiCoP,231 and CoO/CoP232etc. are also widely used in zinc–air batteries.
4.2 Metal–N–C (transition metals)
Compared to low conductivity and high peroxide selectivity, loading metal materials in the carbon network layer is a more rational design for high activity, resulting in a more complete reaction and higher speed of electron transfer in the ORR, exposing more metal active sites to the alkaline electrolyte. By mixing carbonaceous materials and metal precursors, the metal atoms that are regarded as highly active sites for both OER and ORR are receiving electrons and ions, featuring a low loading of the amount of metal on the carbon substrate. This can be mitigated by introducing heteroatoms into the carbon network, especially nitrogen atoms, which provide an additional electron in the valence shell for higher binding energy.233 For example, with heterojunctions like Ni3N/Co2N, a unique flower-like nickel-cobalt layered double hydroxide (N–NiCo-LDH), redistributes electrons at the Ni3N/Co2N heterojunction interface favoring the energy barrier for OERs and thus, improve the electrocatalytic activity.234The electron transfer number of the ORR reaction is an important index for estimating the extent of the ORR reaction. According to the low selectivity for formation of H2O2, which is an undesired product of the ORR reaction, materials like metal–N/C rationally control the adsorption and desorption of intermediates in superficial structures on the metal–carbon surfaces without losing activity.235 For different metal–N materials, a study taking advantage of the well-defined structure of the tetramethoxyphenylporphyrin (TMP P) unit, showed that the 2-electron transfer ORR reaction at the M–N4 site (M = Mn, Fe, Co, Ni, Cu) is most efficient for Fe while Co had the lowest activity.236
The use of M–N/C catalysts as NPMCs started in the 1960s with the discovery of the ORR activity of cobalt phthalocyanine under alkaline conditions.237 In recent years, in addition to macrocycles, a series of low-cost nitrogen precursors such as polyacrylonitrile (PAN), polypyrrole (PPy), polyaniline (PANI), ethylenediamine (EN), and cyanamide (CA) were utilized in the preparation of M–N/C catalysts by doping carbon supports with these metal-coordinating polymers.238
In this section, we discuss the common metal–N–C materials for zinc–air batteries with special reference to Co and Fe, as well as a special kind of single-atom catalyst, describing the activity and morphology of these kinds of material.
With its very high performance, Co–N–C is suitable for use in a zinc–air battery with high durability and high electron transfer number.241 CoN3 active sites dispersed on N-doped graphitic carbon nanosheets enable Zn–air batteries with ultrahigh durability for over 6000 cycles (∼2000 h). The Zn–air battery based on CoSA/NC catalyst showed a high peak power density of 255 mW cm−2242 (Fig. 13a). The synergistic effects of Co–N moieties and Co metallic nanoparticles encapsulated in an N-doped carbon matrix enabled an overall oxygen redox activity (OER/ORR) of 0.72 V in 0.1 M KOH.243 Gas transfer through the Co–N–C's porous structure can also be enhanced by morphology control. A two-step carbonization method has been reported for the synthesis of a Co–N doped coal tar pitch (CTP) carbon-based electrocatalyst that simultaneously had a hierarchical pore structure, a high degree of local order, and an efficient Co–Nx active structure.244
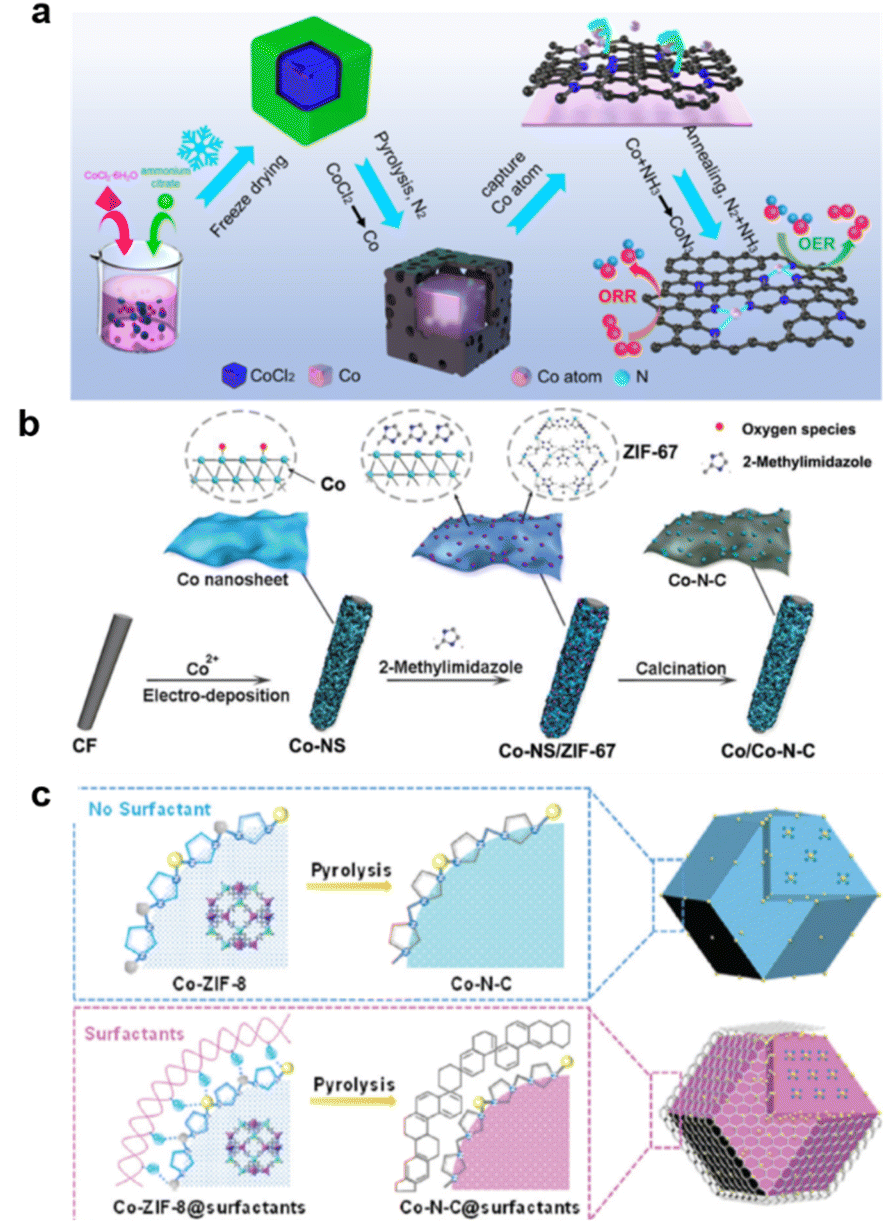 | ||
| Fig. 13 Typical process for Co–N–C formation: (a) pyrolysis and annealing, (reproduced from ref. 242 with permission Copyright © 2022 Elsevier) (b) ZIF-67 coupling with electro spin, (reproduced from ref. 245 with permission Copyright © 2019 Wiley-VCH) (c) surfactant assisted. (Reproduced from ref. 247 with permission from the Royal Society of Chemistry). | ||
For a more porous structure, the electrodeposition of Co–N–C nanosheets on carbon felts (Co/Co–N–C) has been reported. Wavelet transforms extended X-ray absorption fine spectroscopy (WT-EXAFS) and X-ray photoelectron spectroscopy confirmed the formation of Co (mainly Co0) and the Co–N–C (mainly Co2+ and Co3+) structure. Co–Co and Co–N bonds existing in Co/Co–N–C could facilitate the ORR, whereas the Co–O bond is beneficial for the OER (Fig. 13b).245
Another way to achieve a highly active surface is to combine the supramolecular coordination polymers Co(II)-adenine (CoA) with polyacrylonitrile (PAN)/cellulose acetate (CA) in a controlled electrospinning process to synthesize nanofibers (CoA@NFs).246 Surfactants also affect the synthesis process,247 influencing the formation of precursors and forming a protective layer during pyrolysis (Fig. 13c). Furthermore, introduced zinc can regulate the electronic structures of electrocatalysts and construct bimetallic active sites (N–Co/N–Zn species), facilitating the adsorption of reaction intermediates and further enhancing electrocatalytic performance.248
Furthermore, the synthesis methodologies can be classified as the pyrolytic strategy, deposition, and other strategies.250 Moreover, pyrolysis always causes agglomeration, which can be reduced by electrospinning technology combined with a pyrolytic process in an NH3 atmosphere.254
Typically, except for MOF-derived material,255,256 the Fe must first coordinate with a C, N precursor with stable bonds. For example, the 11,11′-bis(dipyrido[3,2-a:2′,3′-c]phenazinyl) (bidppz) ligand was selected for the formation of a nitrogen-rich iron-coordinated coordination polymer (Fe-bidppz) that forms a self-supporting catalyst containing high concentrations of nitrogen and iron doping obtained by pyrolysis (800 °C) as shown in Fig. 14a.238 Similarly, melamine was introduced as an added carbon source to support the growth of Fe–N/Fe sites on the nanotubes257 and CNTs by PPy.249 Other methods like metal co-doping could also change the Fe–N–C performance. As shown in Fig. 14b and c, by the introduction of Mn, the Fe spin in the FeIII sites of metal–N–C materials is regulated more efficiently by both spin-state transition and electronic modulation, rendering excellent ORR performance and good durability of Fe, Mn/N–C in both alkaline and acidic media (half-wave potential 0.928 V in 0.1 M KOH), outperforming Pt/C.258 The etching process of Fe3+ on a copper foil and then adding 2-MeIM to achieve enclosed ZIF-8, followed by pyrolysis, easily produced Cu@Fe–N–C, especially with Cu foil supported, with bimetallic active sites, large surface area, high nitrogen doping level, and conductive carbon frameworks. This material showed excellent ORR performance, with a 50 mV higher half-wave potential of 0.892 V.259
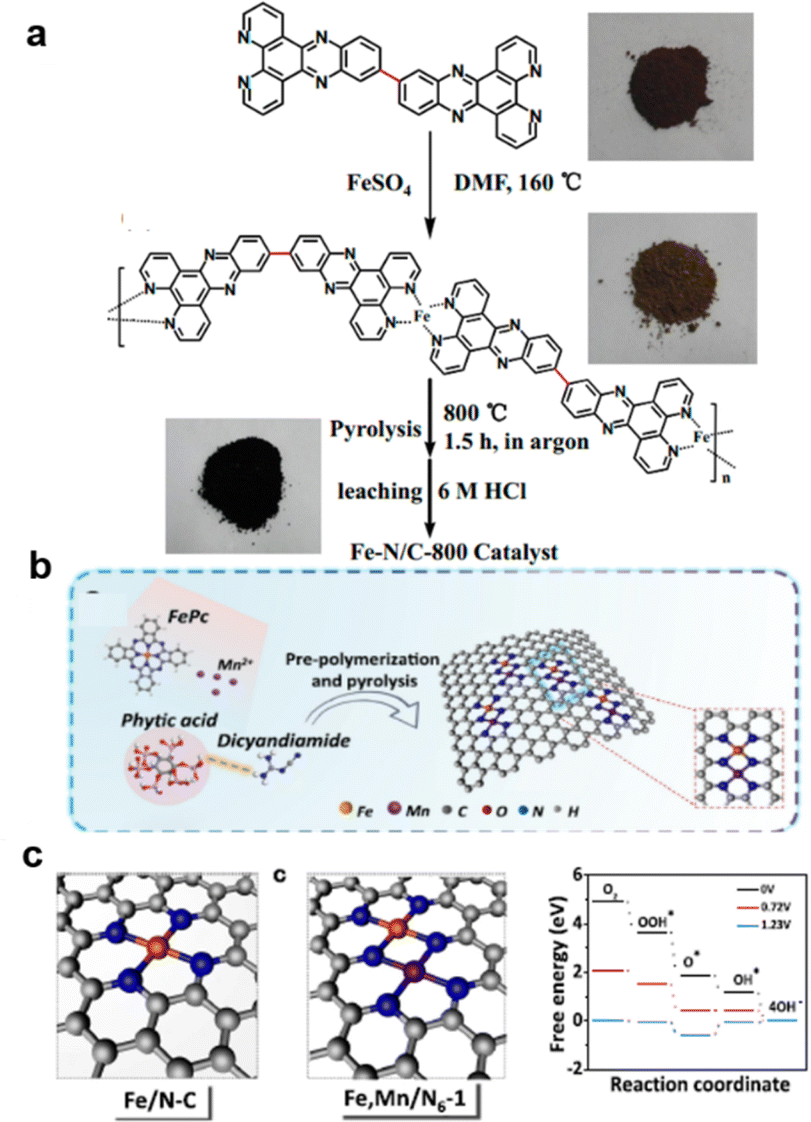 | ||
| Fig. 14 Typical way for Improving Fe–N–C: (a) coupled with N polymers for higher N coupled, (reproduced from ref. 238 with permission. Copyright © 2014, American Chemical Society) (b) and (c) Mn and Fe dual metal enhancing reaction coordination. (Reproduced from ref. 258 with permission. Copyright © 2021 CC-BY). | ||
A second or third carbon could be substituted by S or P to influence the electron distribution on the center metal coupled with N and C.262 For example, in Fig. 15a,263 with abundant tubular channels, Fe-SAs@NCTCs exhibits excellent ORR performances with high E1/2 of 0.91 V (vs. RHE) in 0.1 M KOH by rationally controlling the active sites(SACs-CN).
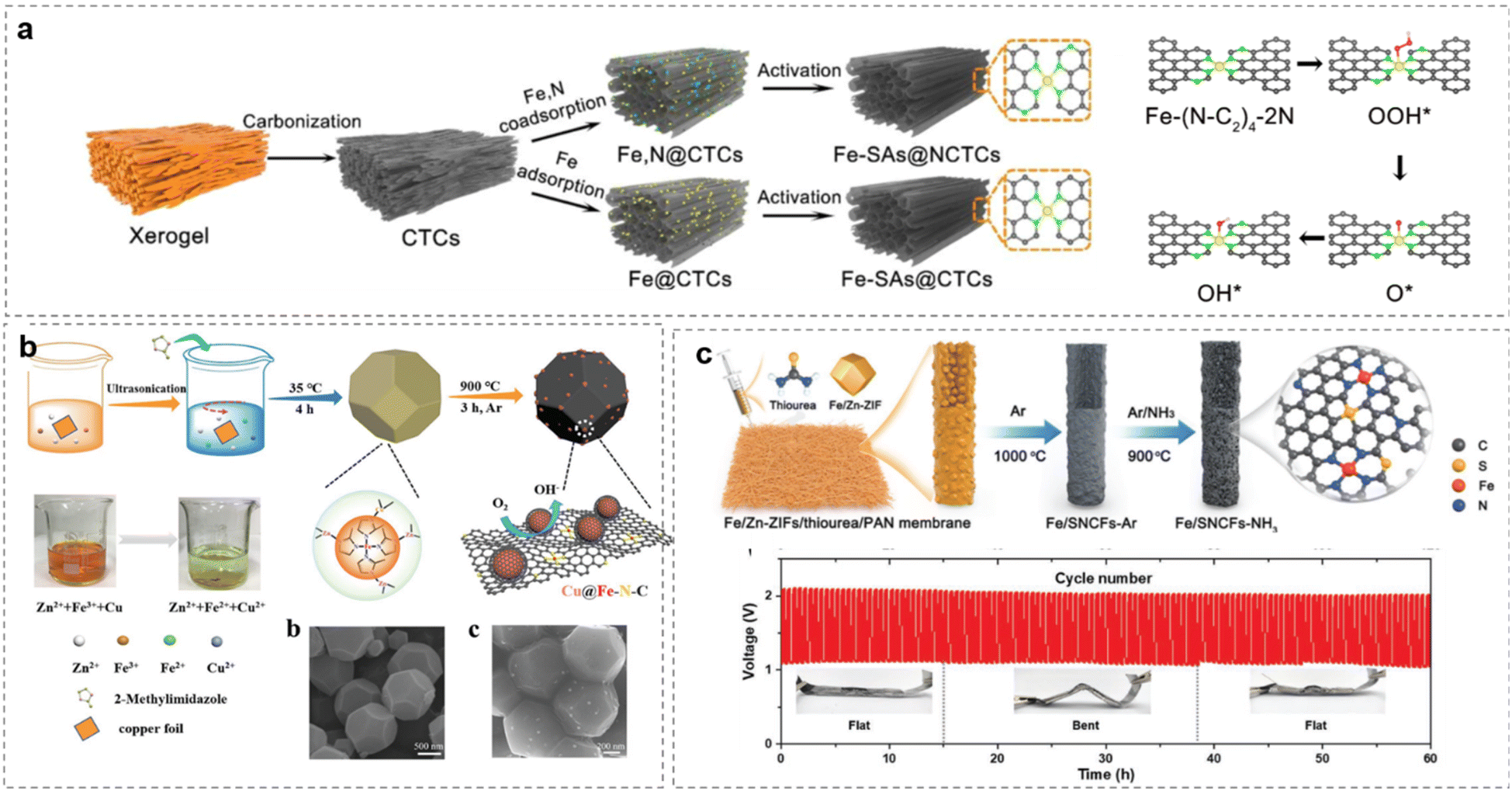 | ||
| Fig. 15 Enhancing Fe-SACs by (a) N-controlling, (reproduced from ref. 263 with permission. Copyright © 2022 Elsevier) (b) Cu coupling from ZIF-8, (reproduced from ref. 259 with permission. Copyright © 2018 Wiley-VCH) (c) separating on PAN Membrane. (Reproduced from ref. 254 with permission. Copyright © 2021 Wiley-VCH). | ||
Doping by black P has shown that Co–N4–bPC (ηORR = 0.31 V; ηOER = 0.22 V), Rh–N4–bPC (ηORR = 0.33 V; ηOER = 0.62 V), and Ir–N4–bPC (ηORR = 0.21 V; ηOER = 0.21 V) are promising candidates as bifunctional catalysts for both the ORR and OER and are comparable or superior to TM–N4–graphene in terms of overpotential.264 Boron also can be introduced into the second surrounding sites. Boron (B)-doped Co–N–C active sites confined in hierarchical porous carbon sheets (denoted as Co–N, B-CSs) were obtained by a soft template self-assembly pyrolysis method. Significantly, the introduced B element provides an electron-deficient site that can activate electron transfer around the Co–N–C sites, strengthen the interaction with oxygenated species, and thus accelerate reaction kinetics in the 4e− processed ORR and OER.265
Choosing the right supports for SACs is important for electrolyte infiltration and electron transfer.266 The large specific surface area exposes a high concentration of Fe–N4/C sites embedded in the carbon matrix. Again, as mentioned before, the Cu@Fe–N–C has a well-defined morphology of truncated rhombic dodecahedra, with excellent durability and methanol tolerance in both acidic and alkaline solutions, which makes it one of the best Pt-free catalysts reported to date for the ORR (Fig. 15b).259
Modulation of local atomic configurations by sulfur doping of a Fe/SNCFs-NH3 catalyst leads to excellent ORR and enhanced OER activities. The liquid-state ZABs utilizing Fe/SNCFs-NH3 catalyst as air cathode (Fig. 15c) deliver a high peak power density of 255.84 mW cm−2 and long-term cycle durability over 1000 h.254
Besides Fe and Co, which are common SACs and metal–N–C choices for high-ability bifunctional materials for ZABs, the metal source can be changed to Ce267 or CuN3.268 Ultrahigh loading of Zn–N–C results in activity and stability comparable to Fe–N–C.269 Pt–CoO has also been introduced to modulate Co SACs.270 The Ni or Fe single atom has been demonstrated to be coordinated with four N atoms by the formation of a Ni–N4 or Fe–N4 planar configuration on the inner and outer walls of graphene hollow nanospheres (GHSs).271,272 To prevent sintering, it is essential to introduce appropriate supports to optimize the local coordination environment and electronic properties, as well as induce strong metal-support interactions (SMSI).266
4.3 Synthesis of porous hybrids
From the viewpoint of designing good bifunctional materials, the supporting materials should be porous and able to accelerate the transfer of air and matter. The inner-Helmholtz plane (IHP) and the outer-Helmholtz plane (OHP) exist in the bilayer structure at the electrode/electrolyte interface. In an alkaline medium, water will be a source of protons, and alkali metals will fill the OHP. Then, after the formation of (O2˙−)ads, proton transfer not only occurs in the solvation shells of O2ads, and (O2˙−)ads but may occur also from water molecules (adsorbed on the electrode surface). The presence of hydroxyl species facilitates the transfer of electrons from the outer sphere.42 Hence, the kinetics of mass transfer at the catalytic layer is very important as it determines the reaction rate, especially of the ORR.As mentioned before, the carbon-supported materials, besides inside defects and edges provided by N doping, could act as bifunctional materials, where the coupling N or C could separate and increase the metal active sites for bifunctional reaction; this has resulted in the great bifunctional activity of low-loading oxides or metal composites on the carbon material. For example, N-doped B–N273 and MXene-like Al etched Ti3AlC2 (ref. 274) nanosheets with high conductivity were found to be stable in a corrosive environment and to be a potential support for loading catalyst. Coupled metallic hybrids of NiFe layered double hydroxide nanosheet/Ti3C2 MXene quantum dots were deposited on a nitrogen-doped graphene surface (LDH/MQD/NG) for high-performance flexible Zn–air batteries (ZABs), as shown in Fig. 16a.275 The electronic and chemical coupling of LDH/MQD/NG modulates the local electronic and surface structure of the active LDH to provide metallic conductivity and abundant active sites. Consequently, the ZABs have significantly improved bifunctional activity, with a power density of 113.8 mW cm−2 and excellent cycle stability over 150 h at 5 mA cm−2, while the kinetics and electrochemical performance are improved significantly by a change in the valence states of Ni and Fe, M–OH vibration, flat band potential, carrier density, metallic conductivity, and Fermi energy level.275 The graphene and CNTs are highlighted based on the widely commercialized application in other areas with a layered and separated structure for high air and mass transfer when in contact with an electrolyte.
 | ||
| Fig. 16 Enabling high porosity: (a) LDHs on MQDs and NG (reproduced from ref. 275 with permission from X. Han et al. published by UESTC and John Wiley. CC BY-NC 3.0) (b) sponge CNTs and graphene.5 (Reproduced from ref. 279 with permission from the Royal Society of Chemistry). | ||
After choosing supports, the OER active layer is loaded on a graphene-like substrate. For example, the LDH precursor of spinels like MnCo2O4,276 ZnCo2O4,277 or Cu–Co bimetallic oxide quantum dots278 are loaded on CNTs or graphene. The oxides-CNTs/graphene form a hollow structure with reaction interface-conductivity channels supporting a high reaction rate. Together with carbon from bottom-up-synthesis, NiCo2O4 nanoarrays on the CNT sponge are derived from a melamine sponge to regulate the holes for air transfer between CNTs, Fig. 16b.279
Controlling the combination of CNTs and oxides is also challenging. After being treated with NH3 for reduction of core–shell CoFe2O4/NiFe2O4 using cellulose as the precursor, the transition metal nitride/CNT hybrid Fe2Ni2N/Co@NCNT exhibited an extremely low voltage gap with ΔE (ΔE = Ej=10 − E1/2) = 0.71 V in O2-saturated 1 M KOH solution.
Also, after 30 charges at a current density of 20 mA cm−2 over 1 h cycles, the in situ-XAFS of the air electrode showed very little difference between charging and discharging, by analyzing the Fe coordination environment, as shown in Fig. 17a and b, implying high stability of this catalyst.280 This points to the stability of metal–N–C on typical CNTs during charging and discharging in typical ZABs.
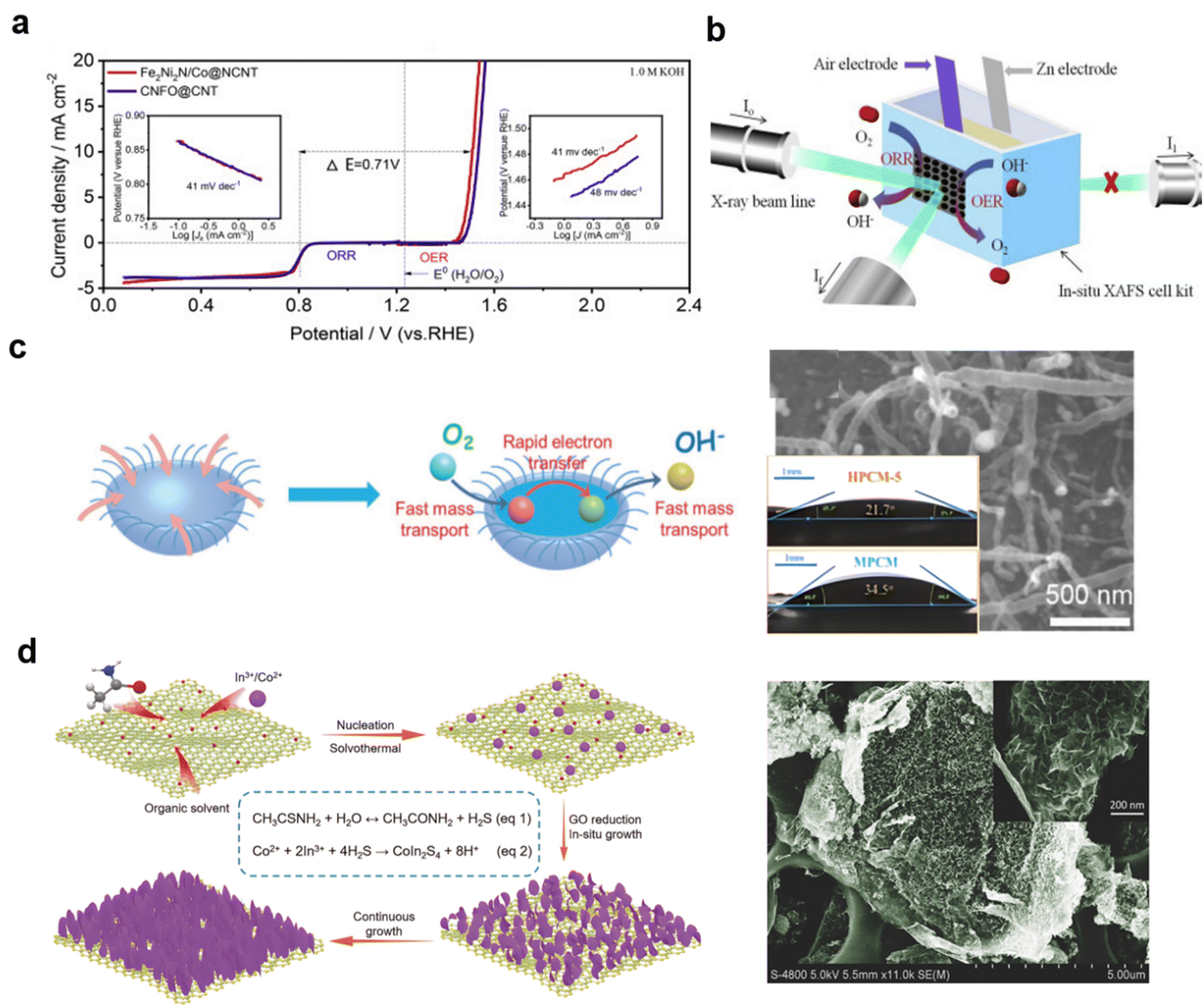 | ||
| Fig. 17 Combining transition metal materials on CNTs with graphene: (a) and (b) Fe2Ni2N/Co@NCNT with in situ XAFS for ZABs, (reproduced from ref. 280 with permission. Copyright © 2019 Elsevier) (c) jellyfish-like CNTs with Mott–Schottky junction, (reproduced from ref. 282 with permission Copyright © 2020 Wiley-VCH) (d) thiospinel CoIn2S4 reduced on GO. (reproduced from ref. 284 with permission. Copyright © 2018 Wiley-VCH). | ||
Apart from adding the CNTs from an external source, in situ-grown CNTs are also widely used as efficient supports. Highly active CNTs can be obtained by treatment at 650 °C, and grown between metals. Oxidative thermal scission of the CNTs and oxidation of Co and Mn nanoparticles inside them, produced spinel Mn–Co oxide nanoparticles partially embedded in the nanotubes.281 A novel jellyfish-like Mott–Schottky type electrocatalyst has been developed to obtain fast electron transfer and obtain fast electron transfer, and to better understand how the porous structure enhances mass transport during the ORR process. Thus, mass transport of oxygen species was improved, because of two effects: the electrolyte streaming effect caused by the tentacle-like carbon nanotubes; and the higher chance for effective collisions in the half-open cavity (Fig. 17c).282
Similarly, Co2S on graphene can be formed also by thermolysis of a Co–thiourea complex in specially made Swagelok cells at 400 and 500 °C, resulting in pure CoS2.283 Also, the indium-based ternary thiospinel CoIn2S4 can be formed during the reduction of GO. As shown in Fig. 17d, the separated nanosheets help to build a large specific surface area on the GO of 102 m2 g−1 and a total pore volume of 0.2 cm3 g−1.284
Formation of the porous structure can be assisted by using templates or special conditions. For example, a ternary nanoalloy-based oxygen electrocatalyst, Ni46Co40Fe14 nanoalloy, with a size distribution of 30–60 nm, dispersed in a carbon matrix, could be formed by the sol–gel method by directly injecting benzene into a reactor at 400 °C at 2.5 MPa (supercritical reaction) followed by heat treatment at 900 °C.285
As shown in Fig. 18a, Fe, Co-SA/CS (single atom, carbon-supported) on nitrogen-doped hollow carbon nanospheres derived from Zn-ZIF for generating SACs and polystyrene to form spherical structures, were formed as a thin layer.286 Carbon templates like PVDF,169 P123,139 and hydrogel have been reported as precursors for generating porous templates. MOF-derived samples exhibit a remaining porous structure that accelerates mass transfer.287 Also, as shown in Fig. 18b,288 ZIF and MPSA have been used to synthesize CoP for controlling the Co valence on graphene. In samples of Co@NC, Co2P@NPC, Co2P/CoP@NPC, and CoP@NPC by ratio control of ZIF and MPSA, as-synthesized Co2P@NPC exhibited the best bifunctional ORR/OER activity among the CoxP@NPC analogs.
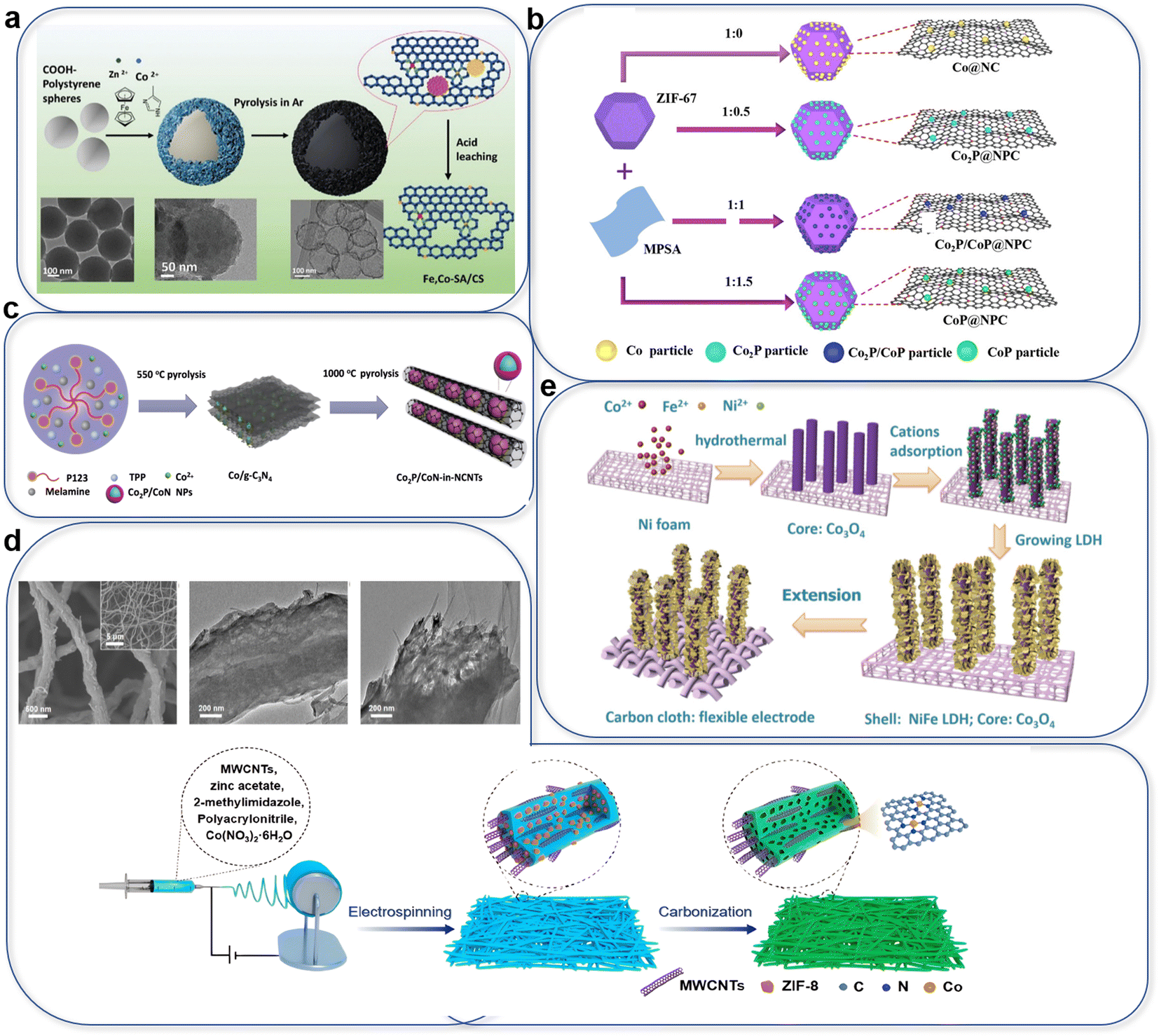 | ||
| Fig. 18 Special applications enabling highly dispersed and porous structures: (a) carbon-sphere template, (reproduced from ref. 286 with permission. Copyright © 2020 Wiley-VCH) (b) ratio control of MPSA and ZIF precursor, (reproduced from ref. 288 with permission. Copyright © 2021 Zhengzhou University). (c) P123 and TPP soft-template. (Reproduced from ref. 289 with permission. Copyright © 2018 Wiley-VCH). (d) Electrospinning by PAN and MWCNTs, (reproduced from ref. 290 with permission. Copyright © 2022, American Chemical Society). (e) Self-growth of Co3O4 on carbon cloth. (Reproduced from ref. 291 with permission. Copyright © 2019, American Chemical Society). | ||
Using the soft template P123 to control the Co, Co2P/CoN particles derived from C3N4 were wrapped in NCNTs, controlling the growth of CNTs. (Fig. 18c).289 By electro-spinning on a self-supported template, the catalyst can be loaded easily on carbon paper or an assembled support for application as an air cathode. The high dispersion enabled high electron and mass transfer on nanorods or nanoneedles. By combining MWCNTs, a ZIF-8-derived Co SAC is stable for 600 hours in a binder-free zinc–air battery at 10 mA cm−2 (Fig. 18d).290 Self-supported NiFe-LDH on Co3O4 can be loaded on carbon cloth for higher activity and flexibility (Fig. 18e).291
5. Reconstruction
Compared with zinc dendrite formation, electrolyte concentration loss, and failure of flexible cells, the air electrode performance may not be the main obstacle for sustainable use, but the ability to achieve low-mass loading and high current density at the air electrode still needs to be improved so that the cycle life can be lengthened before commercialization becomes feasible. RDE measurement removes the effects of diffusion and long-time corrosion in an air-abundant, stable electrolyte in the usually weakly alkaline environment.Real-life working conditions in the electrolyte may differ, and therefore, to prevent the efficiency from decreasing, it is important to monitor the structural, crystal, and catalyst changes during and after the OER/ORR reactions and cycling in ZABs. Two key factors are worth pointing out: one is that the potential dynamics-driven methodology applied in ORR or OER measurements is different from the continuous galvanostatic technique for battery operation, and the other is that alkaline electrolytes used in Zn–air batteries are far more concentrated, so that a lower electrochemical barrier is required for valence variation of the main elements.
As mentioned before, PGM-based electrodes, which exhibit excellent stability in the OER and ORR compared with other materials, are made of expensive and scarce metals and, although ideal, are difficult to commercialize successfully in OER and ORR reactions. The high stability originates from the high active surface area and thermal stability during the reactions.292,293 To replace the expensive metals with transition-metal-based materials while maintaining high stability and high efficiency, remains a great challenge, as the OER and ORR are three-phase reactions taking place on the catalyst surface. The catalyst is usually a nano- or micro-sized material with a complex reaction mechanism occurring in the conducting layer and diffusion layer, which determines the efficiency of the three-phase reactions. To obtain a high current density, which corresponds to a high reaction rate, a higher voltage should be applied on the electrode compared to measurement on RDE, when the reactant and catalyst interface are not abundant. Thus, the stability is highly dependent on the interactions in high current density locations, and changes during long-term cycling.
The OER process commonly proceeds at a high positive potential of more than 1.4 V. At such high potential, (sub)surface atoms on an electrocatalyst tend to be oxidized. The surface sites are therefore dynamic in nature, driving in situ changes and reconstruction.294 The reconstruction involves changes in one or more forms of the composition, phase(s), micro/nanostructures, and crystallinity, at the surface/subsurface of the pre-catalyst. These reconstruction features can be reversible or irreversible depending on the reaction and service conditions, as shown in Fig. 19a.294
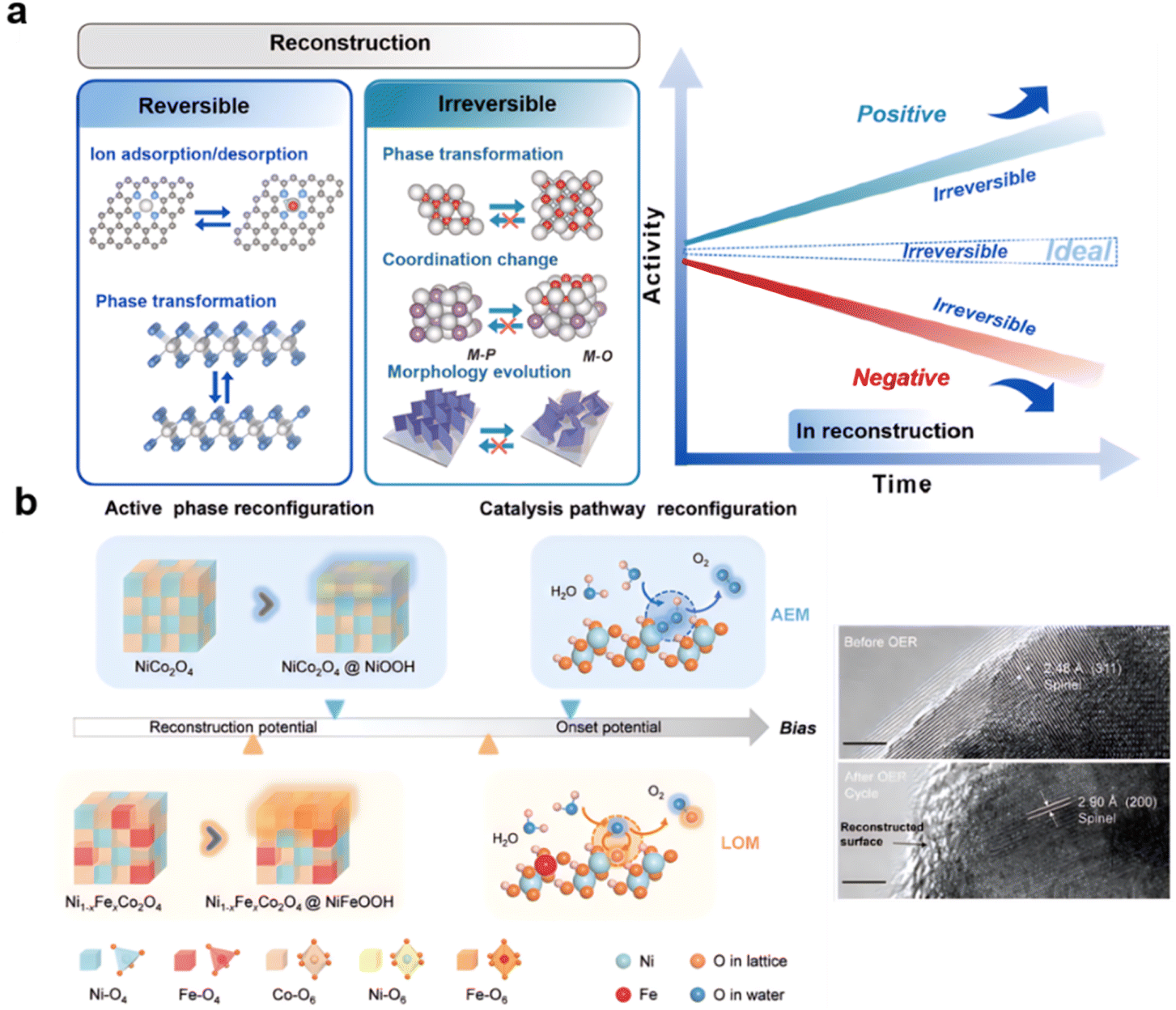 | ||
| Fig. 19 (a) Diagram of different types of reconstruction and durability, (reproduced from ref. 294 with permission. Copyright © 2021 from John Wang et al.) (b) surface reconstruction of NiCo2O4 and Fe doped NiCo2O4. (Reproduced from ref. 299 with permission. Copyright © 2022 Wiley-VCH). | ||
Much research has concentrated on the reconstruction behavior during the OER process. For morphology, the self-reconstruction of Co into Co3O4 and CoOOH during the OER process exhibits higher adsorption energy for OH− and presents an exothermic reaction in the OER process.295 Testing 10 wt% to 60 wt% loading of FeCo in a ZAB at 100 mA cm−2 on the cathode area, cycling between 60 min of discharging and 60 min of charging, the optimal loading was found to be 40 wt%, with both CoFe2O4 and CoO nanocrystals detected. The OER catalytic activity was the limiting factor for the ZAB's cyclability.296
During the OER on NiCo-LDH, the low-crystallinity films were transformed from a local spinel structure to amorphous CoO at an anode potential of 0.2 V vs. Ag/AgCl and then to the active structure NiOOH-h-CoO2 at 0.3 V vs. Ag/AgCl. The initial transformation from spinel to an a-CoO structure is irreversible; however, the subsequent conversion to NiOOH-h-CoO2 is reversible. Incorporation of Ni into this active structure improved the OER activity of NiCoOxHy catalysts, which was increased by 100% compared to when Ni was coordinated in a spinel structure.297 At the same time, the double-exchange-induced conductivity was enhanced by another metal on the nickel-based oxyhydroxides, following a complicated dual-metal engaged mechanism during reconstruction.298 The pathway evolution and structure evolution of the model catalyst spinel NiCo2O4 during water oxidation has been studied, and doping with, for instance, Fe enabled optimization of this reconfiguration, to finally establish a lifetime dynamic structure–performance correlation and obtain outstanding catalysis activity (Fig. 19b).299
5.1 Reconstruction of metal–N–C
Materials based on metal–N–C are considered to be good substitution materials with high ORR rates. The stability of metal–N–C is attracting attention, as the economic-efficiency can be increased when activity and reaction efficiency last longer. While non-precious metal-based catalysts (NPMCs) have emerged as highly promising alternatives, but they tend to degrade quickly under the harsh operating conditions of typical OER devices.300 While the metal–N–C is not stable under working conditions, and operando XAFS analyses imply that initially formed Cu–N4 could be transformed to Cu–N3/Cu-NC under working conditions.301 N-doping also influences stability. For example, N1-OMCHS shows remarkably high stability due to the modification of N-quaternary to N-pyridinic species, while the N-pyrrolic species are oxidized to N-quaternary species at N2-OMCHS.149 For a MOF like Prussian Blue Analog (PBA), it has been found that the partially amorphous nickel hydroxide after activation acts as an active species, and the wrapping of the shell facilitates the transformation of PBA and protects the integrity of the materials.302Reports on reconstruction of metal–N–C in a ZABs level are rare. Deng et al. presents a deep understanding of cycling process. Deng et al. used a cation-carving process in which Fe2+ was added to attack a Co-based nano-cuboid precursor to obtain a hollow structure material denoted (Co,Fe)3N_R, as shown in Fig. 20. EDS mapping then confirmed the composition as (Co0.56Fe0.44)3N.303 The author pointed out that the galvanostatic system for the battery is different from the potential-dynamic-driven method for OER and ORR, so that it was necessary to measure the charging and discharging processes in situ. When applied to a ZAB, the first and following cycles exhibited big differences with several cycles of “activation” as shown in Fig. 20, first discharging at 158 mW cm−2 and charging 225, and then discharging at 234 mW cm−2. The following cycles were stable with a similar voltage gap between charge and discharge cycles. The WAXs (Fig. 20) and SXRD validate the new phase which was shown to be a hexagonal CoOOH or (Co,Fe)OOH mixture. EPR was used to determine the generated phase. During the charging phase of g ≈ 5, Co2+ ions were generated, while for g = 2, which exhibited a maximum in (Co,Fe)3N_1C, the fluctuation is considered to be due to formation of temporary Co4+ and oxygen vacancies. The maturation process was also recorded using XANES. In conflict with the XAS result, a higher state than CoOOH was observed, implying that the valence state of Co continues to increase after the maturation process and light reconstruction, even after only two cycles.
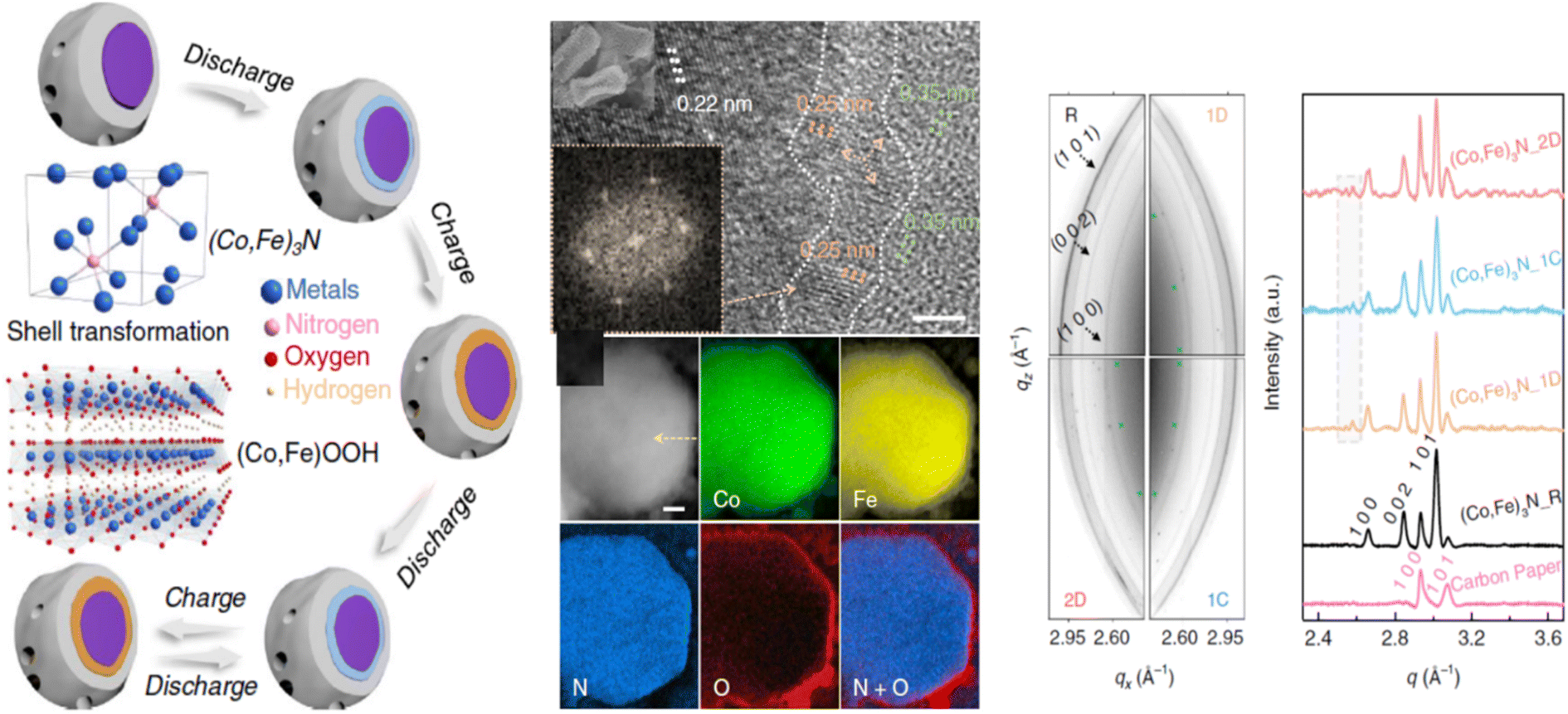 | ||
| Fig. 20 Dynamic electrocatalyst with current-driven oxyhydroxide shell of (Co,Fe)3N. (Reproduced from ref. 303 with permission. Copyright © from Y. Deng et al.). | ||
The shell and bulk Co do not show the same electrochemical accessibility between XAS and EPR. Another noticeable evolution is the gradual fading of the pre-edge peak at 7712 eV, implying a weakening nitride feature; the continuously increasing oxidation state of Co causes increased Co occupation of dipole-forbidden octahedral sites (described by Deng et al.) in the oxyhydroxide. Hence, CoII is observed in the oxyhydroxide shell upon discharge, while CoIV exists after the battery is charged. Also, a slight right shift and intensity increase of the Co–N peak is found in (Co,Fe)3N_1D, while the intensity of the peak of the Co-metal shell is reduced. EXAFS was also used to identify the increased Co–O coordination number suggested by the increased Co–O/N. Starting with two major peaks at 1.4 and 2.1 Å, a slight right shift takes place with the first discharge of Co–N and shows an overlap of Co–N with the Co–O shell at 1.4 Å. A new peak at 2.5 Å corresponds to the formation of oxyhydroxides during discharge; when charging, the high valence of Co constrains the distance between neighbouring metal or oxygen in the corresponding peaks. A clear mid-layer can be seen on the XANES contour map due to reflecting O-rich and N-deficient boundaries, while more Fe occurs between the carbon coated layer and core Co–N–C material. The peak energy position and the intensity ratio of L3/(L2 + L3), commonly stated as the L3 branch ratio are the main criteria of Co and Fe L-edge EELS. As a clear difference in shell and bulk regions, Fe shows higher intensity in the first discharge and similar intensity and similar ratio to Co in the second discharge corresponding to increasing Co concentration in the generated oxyhydroxide during maturation. The prediction on shell-bulk-type configuration and periodic valence swings of surficial Co between CoII and CoIV during cycling is confirmed. The author points out that, interestingly, the generated intermediate phase was a hexagonal crystal different from the amorphous feature from the previous report. Thus, Co plays an important role in the (Co,Fe)OOH oxyhydroxide during battery cycling.303
Until recently, the main approach to improve the stability of metal–N–C and SACs has been to form strong bonds between the metal atoms and support stabilizing the single metal atoms. However, the appropriate electron transfer between isolated metal atoms and the coordination ligands could also suppress the adverse structural evolution of SACs, which leads to the degradation of SACs. Recent advances in stable SACs have been in terms of metal and support materials, synthetic strategies, and catalytic stability in electrocatalysis.304
5.2 Reconstruction of transition metal-based oxides, and ramifications
As many transition-metal materials have shown, the OER process is involved in surface reconstruction and generates oxyhydroxide. The surface layer introducing a new phase into the reaction usually exhibits a different performance from the original surface layer, especially under high current density, high voltage, long-term, and high-pH environments. The new interface with the amorphous layer and original chemicals could synergistically catalyse OER and ORR processes, based on metal valence and change of coordination state, influencing the adsorption and desorption energy needed of intermediates that determine reaction rate, and also change the structure and even compounds of origin. The non-metal elements are doped to increase the oxide's conductivity and also increase stability. But during the reaction, the crystal morphology also changes, such as Co3O4 spinel,305 NiMoO4,306 and CoFe alloy doped with P on graphene.307For the metal phosphides, dephosphorization during the OER process accompanying surface oxidation has been widely reported. As shown in Fig. 21a, Co/Co2P heterojunctions on N-doped carbon nanotubes provide higher bifunctional activity with Egap at 0.68 V. However, high-potential operation can result in irreversible structural damage to the amorphous carbon materials. After the OER process (1.1–2.1 V vs. RHE), Co/Co2P@NCNTs and Co@NCNTs electrodes demonstrated a greatly enhanced stability. The Co oxyhydroxides are formed during cycling from the Co2+/Co3+ couple and Co3+/Co4+ couple with mean state at 3+. Spectra of the N K-edge reveal the transformation from πN–C to graphitic-N after the ADT test, which confirms the restructuring of N active sites, which may not decrease performance in ORR the reaction,308 similarly on MnCoP,309 NiCoP,231 and NiS.310
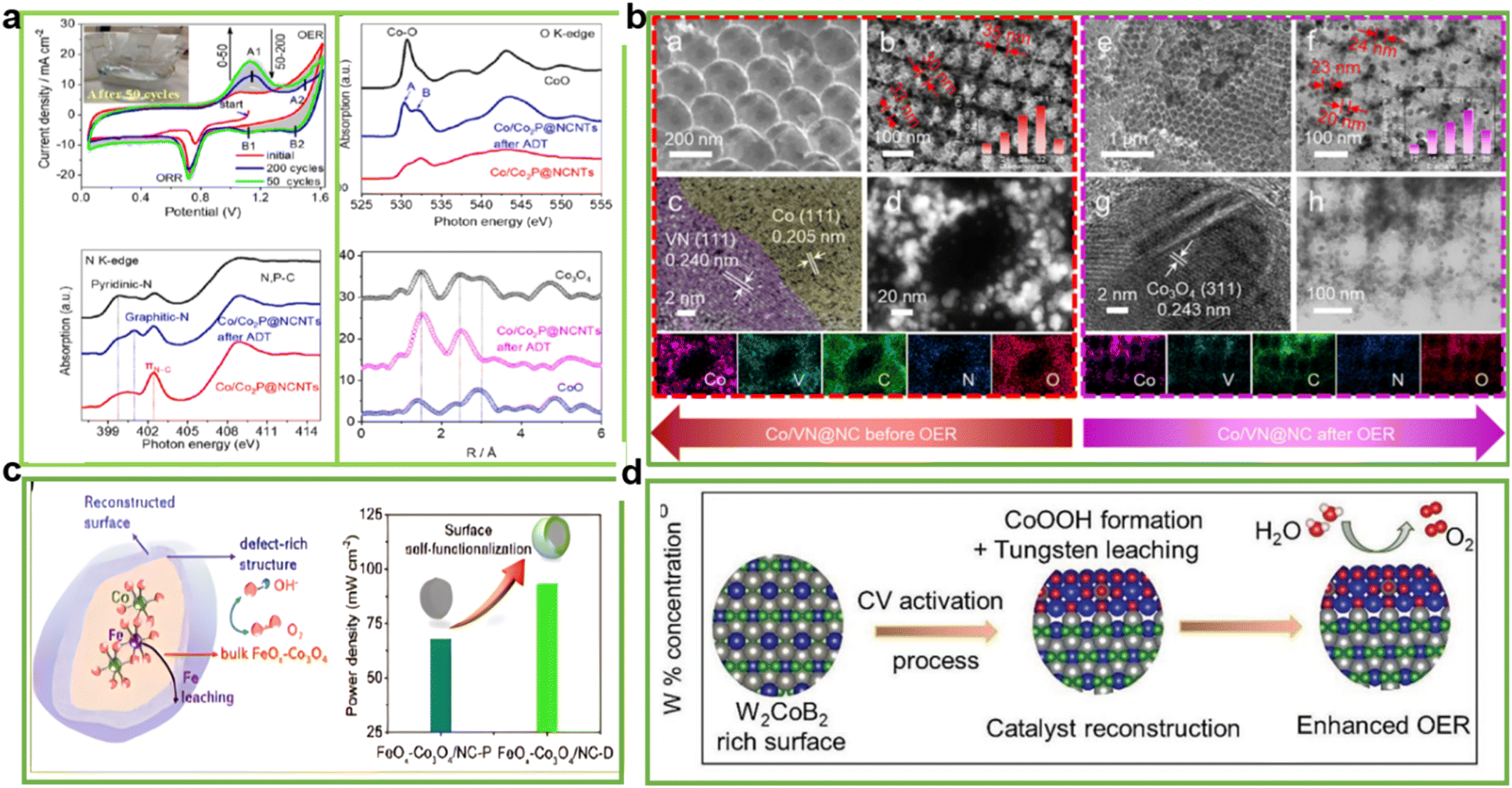 | ||
| Fig. 21 Reconstruction process of (a) Co2P, (reproduced from ref. 308 with permission. Copyright © 2021, American Chemical Society). (b) Co/VN, (reproduced from ref. 295 with permission. Copyright © 2021 published by Elsevier) (c) FeOx–Co3O4 (reproduced from ref. 314 with permission. Copyright © 2022, American Chemical Society). and (d) W2CoB2 (reproduced from ref. 315 with permission. Copyright © 2022 Wiley-VCH). | ||
Morphology also changed during the reaction of Co/VN with three-dimensionally ordered macroporous architecture. VN worked as a sacrificial promoter to accelerate the conversion of Co into CoOOH and Co3O4, which are the true active components during the OER process (Fig. 21b).295 Further analysis of the size-dependent structural transformation during the OER of CoOOH, showed that electron redistribution and oxyl radical (M–O˙) formation within the surface Co3+–O6 units is triggered as a predominant surface-terminating motif, contrasting the long-standing view of high-valence metal ions driving the OER, and leading to a fundamental understanding of the oxygen-evolving near-surface chemistry. Also, S-doped CoS was transformed to CoSx at 0.5 mA cm−2 after 12.5 min treatment, and CoSx crystallization to CoOOH is initiated on the surface of the electrocatalysts with a morphology change via the Co(OH)2 intermediate during the OER measurement, where CoOOH is confirmed as the actual active species.311,312
The reconstruction process enhances the faces and generates vacancies, thereby accelerating the reaction. Activation of the MnCo2O4 (MnCo2O4-A) surface structure with a high Mn/Co tetrahedron occupancy, displays excellent ORR performance via the four-electron pathway with an ultrahigh onset potential and half-wave potential of 0.78 and 0.92 V, respectively, ideal mass activity (MA), and turnover frequency (TOF) values.313 Similarly, the Fe3+ leaching will increase the oxygen vacancies, at the same time increase performance, but if this continues, the danger of crystal destruction is real (Fig. 21c).314
Similarly, a newly formed layer was also detected during the OER reaction on WxCoBx. The bifunctional material for zinc–air or water oxidation underwent Co3+ formation during the CV test. The OER and ORR reactions were enhanced by the formation of an amorphous layer of (oxy)-hydroxide. Theoretical calculations provided an insight into the energy needed to form *OOH from *O due to its large energy barrier, which is therefore the rate-determining step (RDS) for the OER process. The new CoOOH–W2CoB2 interface provides a lower energy barrier than the original phase, and Co and B in W2CoB2 are easy to oxidize according to a Bader charge analysis. Pronounced electron redistribution is observed throughout the CoOOH–W2CoB2 system caused by the more conductive electronic structure of W2CoB2, which is beneficial for providing more electrons to the CoOOH layer (Fig. 21d).315
6. Conclusion and perspectives
6.1 Rational design of bifunctional materials
Based on the foregoing analysis of selected bifunctional materials, apart from considering performance, stability is also an important index for long-time usage. Flexible and rechargeable batteries are different from traditional batteries.316 The procedure for rationally designing OER and ORR catalysts can be separated into four steps.Highly porous materials are also desirable for highly conductive materials with multi-channels to transport air and reactants. Carbon-based materials combined with transition metal compounds, like MOF with porous structure,319 modulated to form porous and multi-channel structures, are very important in the highly alkaline environments with low oxygen levels needed for longtime cycling in the electrolyte in both solid and aqueous zinc–air batteries. The “lung model” helps build air channels for high gas mass transfer rates (Fig. 22a).320 The porous structure of carbon-based materials can be derived from polymers or MOF materials, while some hard-templated models, commonly with high N contents, are also useful. Electro-spun or self-supported321 materials are also widely used to control the morphology. Finally, the ideal product is recommended to be a functional material combining OER and ORR catalysts with highly porous structure and highly uniform dispersion. As shown in Fig. 22b, simply combining Co/NC and LDH could reach extremely high performance, pointing to the possibility of such design routes.322
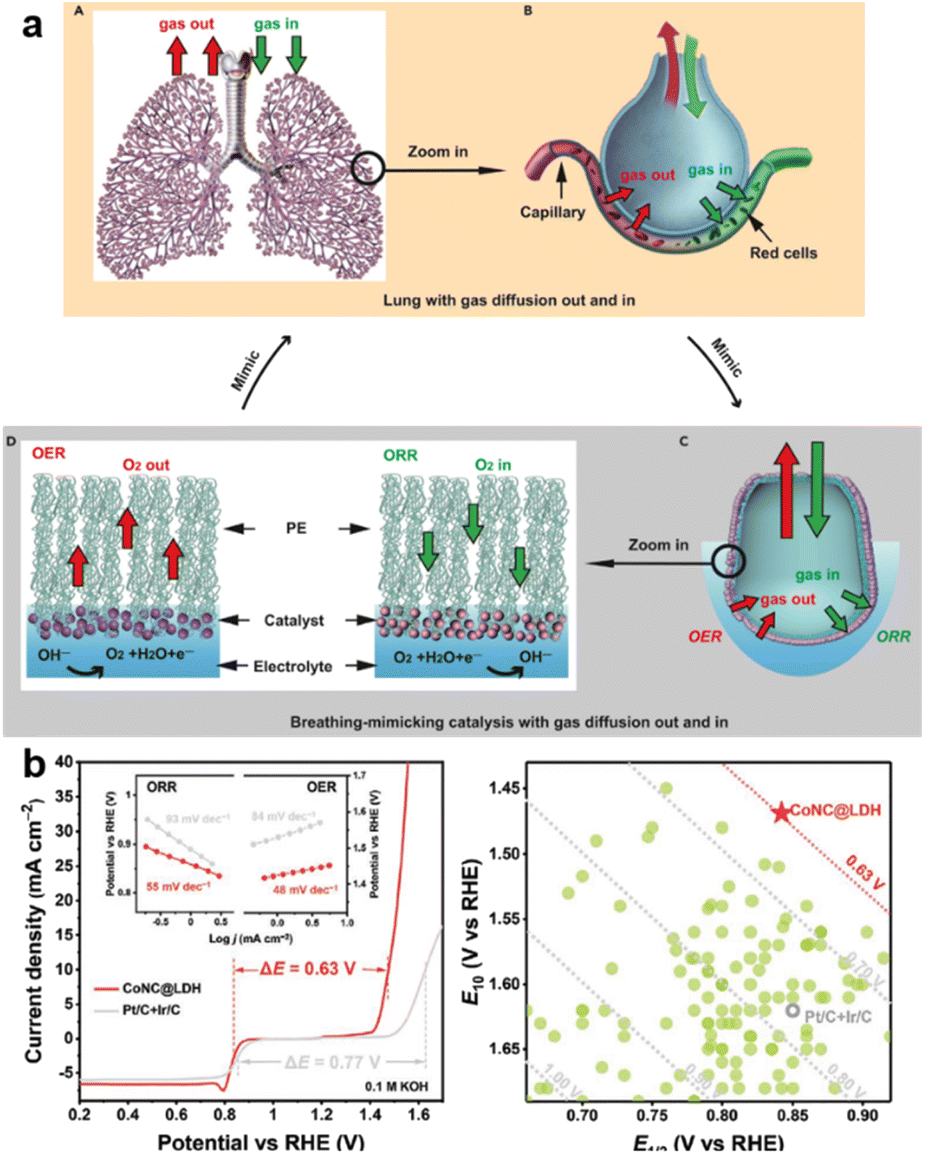 | ||
| Fig. 22 (a)“Lung-inspired” air cathode (reproduced from ref. 320 with permission. Copyright © 2021 Wiley-VCH) (b) CoNC@LDH with extremely high performance. (reproduced from ref. 322 with permission. Copyright © 2021 Wiley-VCH). | ||
More and more techniques will help us understand the reconstruction process during the OER and ORR processes. Operando techniques have provided insightful information on the underlying mechanism of a metal-based catalyst. It is expected that if the same information is attainable for MFC systems, great advances in MFC technology can be achieved.323
Also, besides synthesizing catalysts from synthetic chemical products, the source of the catalyst could be environmentally friendly and easily-sourced. Biomass324 or recycled carbon-based materials can be combined with C3N4 (ref. 325) as carbon-based precursor to synthesize bifunctional materials.
6.2 Stability considerations
As mentioned before, the stability of carbon in the alkaline electrolyte during the OER process is still a big problem for metal-based materials. However, these are not the main limitations of the zinc–air batteries, based on the complicated structure for assembly. The zinc are also unstable in some aqueous electrolyte.For an aqueous battery at ambient temperature, the electrolyte326 and zinc dendrites27 often cause a loss of efficiency, so the design of the cell is very important, while MnO2 is a good material for low-current density use.28 Moreover, the mechanical stability of an air electrode that generates high-volume bubbles will cause the catalyst layer to fail. Selection of the right binder materials and changes in the assembly process may ultimately help solve this problem in aqueous or flexible batteries.
The reconstruction process for both oxides and metal–N–C is determined by the reaction rate of the OER reaction while the combined interface will accelerate the ORR reaction during the charging and discharging processes. That means, for example, if LDHs are used in the charging and discharging processes, the stability of the materials may not be good. However, the good news is that when applying the same current density for the OER reaction, a high-performance catalyst will normally need a lower potential, which is low enough that it will not affect the crystal's stability. So when choosing bifunctional materials, the intended use and desired stability strongly affect the catalyst choice for either quick discharge or long-term stability (i.e., better, longer).
An ideal bifunctional battery catalyst for air electrodes should include: a thin and highly active oxyhydroxide shell that serves as the primary activity contributor responsible for catalyzing the oxygen reaction in battery cycling; and a highly conductive bulk material to maintain the electrochemical kinetics.303 Therefore, compared to direct loading of a highly active oxyhydroxide on carbon materials, rational design with highly-dispersed oxides or metal–N–C on carbon materials with strong bonds will allow the generated reaction sites to benefit from the carbon supports and have no limitation on the ORR. (Also, the stability of the carbon will be enhanced).
6.3 Outlook
Based on the increasing volume of articles and increasing research on zinc–air battery air-cathode catalysts, the use of carbon-based materials and N doping allows the performance of catalysts to reach higher efficiencies than traditional MnO2. However, the processes and techniques for newly made materials have not yet reached maturity with lower costs and higher stability.Notably, the traditional electrochemical test results suggest that electrochemical tests conducted at low current densities may be inadequate in predicting practical battery cycling performance296 or performance at low temperature.327 More advanced monitoring of the efficiency loss during cycling in the real high-alkalinity electrolyte will help estimate the relationship between the crystal structure of functional material and conditions like mass, potential, and current density.22 By defect engineering, heteroatom doping, and vacancy construction, more efficient catalysts may be produced for different electrocatalytic reactions. More and more new, high-potential materials like MOFs are being designed that can be used in many energy-conversion materials,328 and carbon-free functional catalysts like graphene and CNTs are finding wide application in different areas.
We have categorized 100 reports on zinc–air batteries with performance of different bifunctional materials. Fig. 23 portrays the peak power density and potential for charging @ 50 mA cm−2 and discharging @ 100 mA cm−2. Although most reported peak power densities of zinc–air batteries are between 50 and 200 mW cm−2, and only a few excellent results have reached levels higher than 250 mW cm−2, by rational design and advanced energy storage research, it should be possible to achieve better and more active electrocatalysts with high stability and longer battery cycle time.
 | ||
| Fig. 23 Performance of different type of catalysts in the zinc–air battery according to reports.91,100,105,106,128,130,131,133–135,141,144,148,187,189,192,194,198,212,214,216,225,226,240,242,246,248,253–255,257,258,263,267,287,289,290,295,308,314,322,329–385 | ||
No rational relationship between performance and the type of catalyst could be observed, pointing to more important requirements in upgrading equipment for higher ability (Fig. 23).91,100,105,106,128,130,131,133–135,141,144,148,187,189,192,194,198,212,214,216,225,226,240,242,246,248,253–255,257,258,263,267,287,289,290,295,308,314,322,329–385
As a backup battery, higher actual specific energy is expected to meet the needs of wearable devices; zinc–air batteries need a flexible substrate and high-power output.
Therefore, follow-up ZAB research directions should look into compounding with other materials with the objective to increase the number of active sites, and especially to prepare atom-dispersed electrocatalysts.
Author contributions
Wanqi Tang: writing, review, and editing; Jiarong Mai: writing and review; Lili Liu: review and editing; Nengfei Yu: review and editing; Lijun Fu: review and editing; Yuhui Chen: review and editing; Yankai Liu: review; Yuping Wu: supervision, writing-review and editing, funding acquisition; and Teunis van Ree: writing, review, and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support of National Key R & D Program of China (2021YFB2400400), NSFC Key Project (52131306 and 52122209), Project on Carbon Emission Peak and Neutrality of Jiangsu Province (BE2022031-4) and the University of Venda Research and Innovation Directorate is gratefully acknowledged.Notes and references
- M. Xu, J. Qu and M. Li, Sustainability, 2022, 14, 10014 CrossRef.
- R. Schlogl, ChemSusChem, 2010, 3, 209–222 CrossRef PubMed.
- Y. Li and J. Lu, ACS Energy Lett., 2017, 2, 1370–1377 CrossRef CAS.
- Q. Xia, Y. Zhai, L. Zhao, J. Wang, D. Li, L. Zhang and J. Zhang, Energy Mater., 2022, 2, 200015 Search PubMed.
- M. Balaish, A. Kraytsberg and Y. Ein-Eli, Phys. Chem. Chem. Phys., 2014, 16, 2801–2822 RSC.
- P. Gu, M. Zheng, Q. Zhao, X. Xiao, H. Xue and H. Pang, J. Mater. Chem. A, 2017, 5, 7651–7666 RSC.
- Y. Dai, J. Yu, C. Cheng, P. Tan and M. Ni, Chem. Eng. J., 2020, 397, 125516 CrossRef CAS.
- Y. Li, M. Gong, Y. Liang, J. Feng, J. E. Kim, H. Wang, G. Hong, B. Zhang and H. Dai, Nat. Commun., 2013, 4, 1805 CrossRef PubMed.
- K. Tang, H. Hu, Y. Xiong, L. Chen, J. Zhang, C. Yuan and M. Wu, Angew. Chem., Int. Ed. Engl., 2022, 61, e202202671 CAS.
- X. Chen, Z. Zhou, H. E. Karahan, Q. Shao, L. Wei and Y. Chen, Small, 2018, 14, e1801929 CrossRef PubMed.
- D. Yang, L. Zhang, X. Yan and X. Yao, Small Methods, 2017, 1, 1700209 CrossRef.
- J. Sun, C. Liu, X. Song, J. Zhang, Y. Liu, L. Liang, R. Jiang and C. Yuan, Appl. Phys. Rev., 2022, 9, 031301 CAS.
- J. Chang, G. Wang and Y. Yang, Small Sci., 2021, 1, 2100044 CrossRef CAS.
- J. Wu, B. Liu, X. Fan, J. Ding, X. Han, Y. Deng, W. Hu and C. Zhong, Carbon Energy, 2020, 2, 370–386 CrossRef CAS.
- Z. Huang, J. Wang, Y. Peng, C. Jung, A. Fisher and X. Wang, Adv. Energy Mater., 2017, 7, 1700544 CrossRef.
- M. Tang, S. Zhang and S. Chen, Chem. Soc. Rev., 2022, 51, 1529–1546 RSC.
- R. Yoo, E. Pranada, D. Johnson, Z. Qiao and A. Djire, J. Electrochem. Soc., 2022, 169, 063513 CrossRef CAS.
- B. Yao, Y. He, S. Wang, H. Sun and X. Liu, Int. J. Mol. Sci., 2022, 23, 6036 CrossRef CAS PubMed.
- L. Tang, Q. Xu, Y. Zhang, W. Chen and M. Wu, Electrochem. Energy Rev., 2022, 5, 32–81 CrossRef CAS.
- Z. Ye, L. Zhao, A. Nikiforov, J. M. Giraudon, Y. Chen, J. Wang and X. Tu, Adv. Colloid Interface Sci., 2022, 308, 102755 CrossRef CAS PubMed.
- X. Liu, G. Zhang, L. Wang and H. Fu, Small, 2021, 17, e2006766 CrossRef PubMed.
- S. Lee, J. Choi, M. Kim, J. Park, M. Park and J. Cho, Chem. Sci., 2022, 13, 6159–6180 RSC.
- M. Luo, W. Sun, B. B. Xu, H. Pan and Y. Jiang, Adv. Energy Mater., 2020, 11, 2002762 CrossRef.
- J. Pan, Y. Y. Xu, H. Yang, Z. Dong, H. Liu and B. Y. Xia, Adv. Sci., 2018, 5, 1700691 CrossRef PubMed.
- J. Liu, C. Zhao, J. Wang, D. Ren, B. Li and Q. Zhang, Energy Environ. Sci., 2022, 15, 4542–4553 RSC.
- Y. Deng, R. Liang, G. Jiang, Y. Jiang, A. Yu and Z. Chen, ACS Energy Lett., 2020, 5, 1665–1675 CrossRef CAS.
- M. Wu, G. Zhang, H. Yang, X. Liu, M. Dubois, M. A. Gauthier and S. Sun, InfoMat, 2021, 4, 12265 Search PubMed.
- Y. Zhang, Z. Chen, H. Qiu, W. Yang, Z. Zhao, J. Zhao and G. Cui, NPG Asia Mater., 2020, 12, 4 CrossRef CAS.
- Z. Zhao, X. Fan, J. Ding, W. Hu, C. Zhong and J. Lu, ACS Energy Lett., 2019, 4, 2259–2270 CrossRef CAS.
- Q. Li, R. Cao, J. Cho and G. Wu, Adv. Energy Mater., 2014, 4, 1301415 CrossRef.
- L. Yang, J. Shui, L. Du, Y. Shao, J. Liu, L. Dai and Z. Hu, Adv. Mater., 2019, 31, e1804799 CrossRef PubMed.
- C. X. Zhao, J. N. Liu, J. Wang, D. Ren, B. Q. Li and Q. Zhang, Chem. Soc. Rev., 2021, 50, 7745–7778 RSC.
- Y. Nie, L. Li and Z. Wei, Chem. Soc. Rev., 2015, 44, 2168–2201 RSC.
- S. Cherevko, S. Geiger, O. Kasian, N. Kulyk, J. Grote, A. Savan, B. R. Shrestha, S. Merzlikin, B. Breitbach, A. Ludwig and K. J. J. Mayrhofer, Catal. Today, 2016, 262, 170–180 CrossRef CAS.
- M. E. Lyons and S. Floquet, Phys. Chem. Chem. Phys., 2011, 13, 5314–5335 RSC.
- J. Cheng, H. Zhang, G. Chen and Y. Zhang, Electrochim. Acta, 2009, 54, 6250–6256 CrossRef CAS.
- E. Antolini, ACS Catal., 2014, 4, 1426–1440 CrossRef CAS.
- Y. E. Roginskaya, T. V. Varlamova, M. D. Goldstein, I. D. Belova, B. S. Galyamov, R. R. Shifrina, V. A. Shepelin and V. N. Fateev, Mater. Chem. Phys., 1991, 30, 101–113 CrossRef CAS.
- J. Zhao, C. Fu, K. Ye, Z. Liang, F. Jiang, S. Shen, X. Zhao, L. Ma, Z. Shadike, X. Wang, J. Zhang and K. Jiang, Nat. Commun., 2022, 13, 685 CrossRef CAS PubMed.
- J. Scholz, M. Risch, K. A. Stoerzinger, G. Wartner, Y. Shao-Horn and C. Jooss, J. Phys. Chem. C, 2016, 120, 27746–27756 CrossRef CAS.
- J. Wang, C. Zhao, J. Liu, D. Ren, B. Li, J. Huang and Q. Zhang, Nano Mater. Sci., 2021, 3, 313–318 CrossRef CAS.
- X. Ge, A. Sumboja, D. Wuu, T. An, B. Li, F. W. T. Goh, T. S. A. Hor, Y. Zong and Z. Liu, ACS Catal., 2015, 5, 4643–4667 CrossRef CAS.
- J. Zhang, Y. Yuan, L. Gao, G. Zeng, M. Li and H. Huang, Adv. Mater., 2021, 33, e2006494 CrossRef PubMed.
- V. Tripković, E. Skúlason, S. Siahrostami, J. K. Nørskov and J. Rossmeisl, Electrochim. Acta, 2010, 55, 7975–7981 CrossRef.
- V. Stamenkovic, B. S. Mun, K. J. Mayrhofer, P. N. Ross, N. M. Markovic, J. Rossmeisl, J. Greeley and J. K. Norskov, Angew. Chem., Int. Ed., 2006, 45, 2897–2901 CrossRef CAS PubMed.
- Y. Bing, H. Liu, L. Zhang, D. Ghosh and J. Zhang, Chem. Soc. Rev., 2010, 39, 2184–2202 RSC.
- V. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. Mayrhofer, C. A. Lucas, G. Wang, P. N. Ross and N. M. Markovic, Nat. Mater., 2007, 6, 241–247 CrossRef CAS PubMed.
- L. Chen, X. Xu, W. Yang and J. Jia, Chin. Chem. Lett., 2020, 31, 626–634 CrossRef CAS.
- S. Wu, X. Xu, Y. Ren, X. Guo, H. Sun and G. Zhou, Ionics, 2021, 28, 1017–1036 CrossRef.
- R. Paul, L. Zhu, H. Chen, J. Qu and L. Dai, Adv. Mater., 2019, 31, e1806403 CrossRef PubMed.
- D. Wang and D. Su, Energy Environ. Sci., 2014, 7, 576–591 RSC.
- L. Wang, Z. Liu and J. Zhang, Nanoscale, 2022, 14, 13473–13489 RSC.
- D. Yan, Y. Li, J. Huo, R. Chen, L. Dai and S. Wang, Adv. Mater., 2017, 29, 1606459 CrossRef PubMed.
- R. Paul, Q. Dai, C. Hu and L. Dai, Carbon Energy, 2019, 1, 19–31 CrossRef.
- A. Parkash, N. Solangi, T. H. Seehar, G. Zhang, M. Akram and S. Ali, ECS J Solid State Sci. Technol., 2022, 11 Search PubMed.
- J. Ortiz-Medina, Z. Wang, R. Cruz-Silva, A. Morelos-Gomez, F. Wang, X. Yao, M. Terrones and M. Endo, Adv. Mater., 2019, 31, e1805717 CrossRef PubMed.
- M. Steenackers, A. M. Gigler, N. Zhang, F. Deubel, M. Seifert, L. H. Hess, C. H. Lim, K. P. Loh, J. A. Garrido, R. Jordan, M. Stutzmann and I. D. Sharp, J. Am. Chem. Soc., 2011, 133, 10490–10498 CrossRef CAS PubMed.
- S. Kang, S. Park, D. Kim, S. Y. Park, R. S. Ruoff and H. Lee, Adv. Funct. Mater., 2011, 21, 108–112 CrossRef CAS.
- T. Ma, S. Dai, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed. Engl., 2014, 53, 7281–7285 CrossRef CAS PubMed.
- L. Jiao, X. Wang, G. Diankov, H. Wang and H. Dai, Nat. Nanotechnol., 2010, 5, 321–325 CrossRef CAS PubMed.
- D. Yu, K. Goh, H. Wang, L. Wei, W. Jiang, Q. Zhang, L. Dai and Y. Chen, Nat. Nanotechnol., 2014, 9, 555–562 CrossRef CAS PubMed.
- L. Ci, L. Song, D. Jariwala, A. L. ElÃ-as, W. Gao, M. Terrones and P. M. Ajayan, Adv. Mater., 2009, 21, 4487–4491 CrossRef CAS.
- X. H. Li and M. Antonietti, Angew. Chem., Int. Ed., 2013, 52, 4572–4576 CrossRef CAS PubMed.
- J. Duan, S. Chen, M. Jaroniec and S. Z. Qiao, ACS Catal., 2015, 5, 5207–5234 CrossRef CAS.
- J. Liang, Y. Jiao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed. Engl., 2012, 51, 11496–11500 CrossRef CAS PubMed.
- Y. Arafat, M. R. Azhar, Y. Zhong, M. O. Tadé and Z. Shao, Mater. Res. Bull., 2021, 140 Search PubMed.
- B. Wu, H. Meng, D. M. Morales, F. Zeng, J. Zhu, B. Wang, M. Risch, Z. J. Xu and T. Petit, Adv. Funct. Mater., 2022, 32, 2204137 CrossRef CAS.
- H. Wang, C. Tang, C. X. Zhao, J. Q. Huang and Q. Zhang, Adv. Funct. Mater., 2022, 32, 1803329 Search PubMed.
- H. Yang, J. Miao, S. Hung, J. Chen, H. Tao, X. Wang, L. Zhang, R. Chen, J. Gao, H. M. Chen, L. Dai and B. Liu, Sci. Adv., 2016, 2, e1501122 CrossRef PubMed.
- Z. Luo, S. Lim, Z. Tian, J. Shang, L. Lai, B. MacDonald, C. Fu, Z. Shen, T. Yu and J. Lin, J. Mater. Chem., 2011, 21, 8038–8044 RSC.
- Y. Okamoto, Appl. Surf. Sci., 2009, 256, 335–341 CrossRef CAS.
- T. Xiang, Z. Wu, Z. Sun, C. Cheng, W. Wang, Z. Liu, J. Yang and B. Li, J. Colloid Interface Sci., 2022, 610, 486–494 CrossRef CAS PubMed.
- Q. Lai, K. Wei, Z. Tang, X. Liu, J. Zheng and Y. Liang, J. Mater. Sci., 2021, 56, 19577–19588 CrossRef CAS.
- L. Tao, Q. Wang, S. Dou, Z. Ma, J. Huo, S. Wang and L. Dai, Chem. Commun., 2016, 52, 2764–2767 RSC.
- J. Barrio, M. Volokh and M. Shalom, J. Mater. Chem. A, 2020, 8, 11075–11116 RSC.
- D. Sharma and B. K. Satapathy, Polym. Rev., 2021, 62, 439–484 CrossRef.
- W. Li, C. Wang and X. Lu, Coord. Chem. Rev., 2022, 464 Search PubMed.
- J. Zhang, Z. Zhao, Z. Xia and L. Dai, Nat. Nanotechnol., 2015, 10, 444–452 CrossRef CAS PubMed.
- J. He, Y. He, Y. Fan, B. Zhang, Y. Du, J. Wang and P. Xu, Carbon, 2017, 124, 630–636 CrossRef CAS.
- G. Wu, N. H. Mack, W. Gao, S. Ma, R. Zhong, J. Han, J. K. Baldwin and P. Zelenay, ACS Nano, 2012, 6, 9764–9776 CrossRef CAS PubMed.
- H. Sun, P. Zhou, X. Ye, J. Wang, Z. Tian, Z. Zhu, C. Ma, W. Liang and A. Li, J. Colloid Interface Sci., 2022, 617, 11–19 CrossRef CAS PubMed.
- H. Li, M. R. Gadinski, Y. Huang, L. Ren, Y. Zhou, D. Ai, Z. Han, B. Yao and Q. Wang, Energy Environ. Sci., 2020, 13, 1279–1286 RSC.
- D. Li, B. Wang, X. Long, W. Xu, Y. Xia, D. Yang and X. Yao, Angew. Chem., Int. Ed. Engl., 2021, 60, 26483–26488 CrossRef CAS PubMed.
- Y. Chang, G. Zhang, B. Han, H. Li, C. Hu, Y. Pang, Z. Chang and X. Sun, ACS Appl. Mater. Interfaces, 2017, 9, 29753–29759 CrossRef CAS PubMed.
- E. Gottlieb, K. Matyjaszewski and T. Kowalewski, Adv. Mater., 2019, 31, e1804626 CrossRef PubMed.
- J. Sheng and Y. Li, ACS Appl. Mater. Interfaces, 2022, 14, 20455–20462 CrossRef CAS PubMed.
- R. Ma, G. Lin, Y. Zhou, Q. Liu, T. Zhang, G. Shan, M. Yang and J. Wang, npj Comput. Mater., 2019, 5, 78 CrossRef.
- H. Yao, Y. Zhao, N. Yang, W. Hao, H. Zhao, S. Li, J. Zhu, L. Shen and W. Fang, J. Energy Chem., 2022, 65, 141–148 CrossRef CAS.
- F. Zhang, P. C. Sherrell, W. Luo, J. Chen, W. Li, J. Yang and M. Zhu, Adv. Sci., 2021, 8, e2102859 CrossRef PubMed.
- J. Jiang, J. Turnbull, W. Lu, P. Boguslawski and J. Bernholc, J. Chem. Phys., 2012, 136, 014702 CrossRef PubMed.
- F. Qiang, J. Feng, H. Wang, J. Yu, J. Shi, M. Huang, Z. Shi, S. Liu, P. Li and L. Dong, ACS Catal., 2022, 12, 4002–4015 CrossRef CAS.
- S. A. Shaik, S. Sengupta, R. S. Varma, M. B. Gawande and A. Goswami, ACS Sustainable Chem. Eng., 2020, 9, 3–49 CrossRef.
- R. Rabeya, S. Mahalingam, A. Manap, M. Satgunam, M. Akhtaruzzaman and C. H. Chia, Int. J. Quantum Chem., 2022, 122 Search PubMed.
- Y. Han, D. Tang, Y. Yang, C. Li, W. Kong, H. Huang, Y. Liu and Z. Kang, Nanoscale, 2015, 7, 5955–5962 RSC.
- Y. Huang and W. Liao, Appl. Surf. Sci., 2019, 495 CAS.
- Q. Pang, L. Yang and Q. Li, Small Struct., 2022, 3, 202100203 Search PubMed.
- D. Liu, L. Dai, X. Lin, J. F. Chen, J. Zhang, X. Feng, K. Mullen, X. Zhu and S. Dai, Adv. Mater., 2019, 31, e1804863 CrossRef PubMed.
- J. P. Paraknowitsch and A. Thomas, Energy Environ. Sci., 2013, 6, 2839–2855 RSC.
- F. Yang, X. Liu, H. Zhang, J. Zhou, J. Jiang and X. Lu, Energy Stor. Mater., 2020, 30, 138–145 Search PubMed.
- P. Wei, X. Li, Z. He, X. Sun, Q. Liang, Z. Wang, C. Fang, Q. Li, H. Yang, J. Han and Y. Huang, Chem. Eng. J., 2021, 422, 130134 CrossRef CAS.
- G. Kang, S. Lee, D. C. Lee, C. W. Yoon and H. Joh, Curr. Appl. Phys., 2020, 20, 456–461 CrossRef.
- X. Chen, Y. Xie, Y. Shao, K. Shen and Y. Li, ACS Appl. Mater. Interfaces, 2021, 13, 42598–42604 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, L. Ge, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed. Engl., 2013, 52, 3110–3116 CrossRef CAS PubMed.
- L. Yang, S. Jiang, Y. Zhao, L. Zhu, S. Chen, X. Wang, Q. Wu, J. Ma, Y. Ma and Z. Hu, Angew. Chem., Int. Ed. Engl., 2011, 50, 7132–7135 CrossRef CAS PubMed.
- H. Li, T. A. Ha, S. Jiang, C. Pozo-Gonzalo, X. Wang, J. Fang, P. C. Howlett and X. Wang, Electrochim. Acta, 2021, 377, 138089 CrossRef CAS.
- Y. Wang, R. Gan, S. Zhao, W. Ma, X. Zhang, Y. Song, C. Ma and J. Shi, Appl. Surf. Sci., 2022, 598, 153891 CrossRef CAS.
- W. Lei, Y. Deng, G. Li, Z. P. Cano, X. Wang, D. Luo, Y. Liu, D. Wang and Z. Chen, ACS Catal., 2018, 8, 2464–2472 CrossRef CAS.
- T. Liu, S. Feng, J. Huo, Q. Li, C. Xie and S. Wang, ChemCatChem, 2018, 10, 4562–4568 CrossRef CAS.
- H. Jiang, J. Gu, X. Zheng, M. Liu, X. Qiu, L. Wang, W. Li, Z. Chen, X. Ji and J. Li, Energy Environ. Sci., 2019, 12, 322–333 RSC.
- Z. Pei, H. Li, Y. Huang, Q. Xue, Y. Huang, M. Zhu, Z. Wang and C. Zhi, Energy Environ. Sci., 2017, 10, 742–749 RSC.
- Z. Xu, Z. Ma, K. Dong, J. Liang, L. Zhang, Y. Luo, Q. Liu, J. You, Z. Feng, D. Ma, Y. Wang and X. Sun, Chem. Commun., 2022, 58, 5025–5028 RSC.
- N. Jia, Q. Weng, Y. Shi, X. Shi, X. Chen, P. Chen, Z. An and Y. Chen, Nano Res., 2018, 11, 1905–1916 CrossRef CAS.
- P. Zhang, K. Wang, P. Pei, Y. Zuo, M. Wei, X. Liu, Y. Xiao and J. Xiong, Mater. Today Chem., 2021, 21, 100538 CrossRef CAS.
- Z. Xu, H. Zhao, J. Liang, Y. Wang, T. Li, Y. Luo, X. Shi, S. Lu, Z. Feng, Q. Wu and X. Sun, Mater. Today Phys., 2020, 15, 100280 CrossRef.
- C. Ding and Z. Qiao, CrystEngComm, 2022, 24, 6289–6296 RSC.
- G. Nie, Z. Zhang, T. Wang, C. Wang and Z. Kou, ACS Appl. Mater. Interfaces, 2021, 13, 37961–37978 CrossRef CAS PubMed.
- L. Ma, S. Chen, D. Wang, Q. Yang, F. Mo, G. Liang, N. Li, H. Zhang, J. A. Zapien and C. Zhi, Adv. Energy Mater., 2019, 9, 1803046 CrossRef.
- K. Lu, T. Jiang, H. Hu and M. Wu, Front. Chem., 2020, 8, 546728 CrossRef CAS PubMed.
- G. Ren, Q. Chen, J. Zheng, B. Huang and Y. Qian, J. Electroanal. Chem., 2018, 829, 177–183 CrossRef CAS.
- Z. Zhao, W. Yu, W. Shang, Y. He, Y. Ma, Z. Zhang and P. Tan, Int. J. Energy Res., 2022, 46, 7694–7703 CrossRef CAS.
- M. Chatenet, B. G. Pollet, D. R. Dekel, F. Dionigi, J. Deseure, P. Millet, R. D. Braatz, M. Z. Bazant, M. Eikerling, I. Staffell, P. Balcombe, Y. Shao-Horn and H. Schafer, Chem. Soc. Rev., 2022, 51, 4583–4762 RSC.
- J. Zhao, Z. Tu and S. H. Chan, J. Power Sources, 2021, 488, 229434 CrossRef CAS.
- S. Maass, F. Finsterwalder, G. Frank, R. Hartmann and C. Merten, J. Power Sources, 2008, 176, 444–451 CrossRef CAS.
- S. Moller, S. Barwe, J. Masa, D. Wintrich, S. Seisel, H. Baltruschat and W. Schuhmann, Angew. Chem., Int. Ed. Engl., 2020, 59, 1585–1589 CrossRef PubMed.
- S. G. Ji, H. Kim, W. H. Lee, H. Oh and C. H. Choi, J. Mater. Chem. A, 2021, 9, 19834–19839 RSC.
- H. Singh, S. Zhuang, B. Ingis, B. B. Nunna and E. S. Lee, Carbon, 2019, 151, 160–174 CrossRef CAS.
- I. S. Filimonenkov, C. Bouillet, G. Kéranguéven, P. A. Simonov, G. A. Tsirlina and E. R. Savinova, Electrochim. Acta, 2019, 321 Search PubMed.
- T. Liu, X. J. Du, S. Li, Q. L. Wu, Q. Guo, Z. Z. Liu, J. P. Zhao and F. C. Liu, Nanoscale, 2022, 14, 14248–14254 RSC.
- Y. Gao, Z. Xiao, D. Kong, R. Iqbal, Q. Yang and L. Zhi, Nano Energy, 2019, 64, 103879 CrossRef CAS.
- F. Gui, Q. Jin, D. Xiao, X. Xu, Q. Tan, D. Yang, B. Li, P. Ming, C. Zhang, Z. Chen, S. Siahrostami and Q. Xiao, Small, 2022, 18, e2105928 CrossRef PubMed.
- P. Jia, J. Zhang, G. Xia, Z. Yu, J. Sun and X. Ji, Polymers, 2022, 14, 2581 CrossRef CAS PubMed.
- H. W. Liang, X. Zhuang, S. Bruller, X. Feng and K. Mullen, Nat. Commun., 2014, 5, 4973 CrossRef CAS PubMed.
- Q. Xie, W. Si, Y. Shen, Z. Wang and H. Uyama, Nanoscale, 2021, 13, 16296–16306 RSC.
- T. Lu, X. Hu, J. He, R. Li, J. Gao, Q. Lv, Z. Yang, S. Cui and C. Huang, Nano Energy, 2021, 85 Search PubMed.
- G. Yasin, S. Ibrahim, S. Ibraheem, S. Ali, R. Iqbal, A. Kumar, M. Tabish, Y. Slimani, T. A. Nguyen, H. Xu and W. Zhao, J. Mater. Chem. A, 2021, 9, 18222–18230 RSC.
- Y. Zhang, X. Fan, J. Jian, D. Yu, Z. Zhang and L. Dai, Energy Environ. Sci., 2017, 10, 2312–2317 RSC.
- F. Liu and B. He, Electrochim. Acta, 2020, 362, 137225 CrossRef CAS.
- Q. Yu, J. Wang, H. Li, R. Li, S. Zeng, R. Li, Q. Yao, H. Chen, K. Qu and Y. Zheng, Chem. Eng. J., 2022, 429, 132102 CrossRef CAS.
- X. Ding, F. Li, Q. Cao, H. Wu, Y. Qin, L. Yang, T. Wang, X. Zheng and C. Wang, Chem. Eng. J., 2022, 429, 132469 CrossRef CAS.
- B. J. Ferraz, B. Li, Z. Guo, C. Blackman and Z. Liu, ChemElectroChem, 2021, 8, 3954–3961 CrossRef CAS.
- X. Wang, G. Li, Z. Lu, S. Cao, C. Hao, S. Wang and G. Sun, Catal. Sci. Technol., 2022, 12, 181–191 RSC.
- J. Liu, P. Song and W. Xu, Carbon, 2017, 115, 763–772 CrossRef CAS.
- X. Li, X. Duan, C. Han, X. Fan, Y. Li, F. Zhang, G. Zhang, W. Peng and S. Wang, Carbon, 2019, 148, 540–549 CrossRef CAS.
- S. Cao, W. Shang, G. Li, Z. Lu, X. Wang, Y. Yan, C. Hao, S. Wang and G. Sun, Carbon, 2021, 184, 127–135 CrossRef CAS.
- Z. Liu, M. Wang, X. Luo, S. Li, S. Li, Q. Zhou, W. Xu and R. Wu, Appl. Surf. Sci., 2021, 544, 148912 CrossRef CAS.
- T. Ma, J. Ran, S. Dai, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed. Engl., 2015, 54, 4646–4650 CrossRef CAS PubMed.
- C. Tang, H. Wang, X. Chen, B. Q. Li, T. Z. Hou, B. S. Zhang, Q. Zhang, M. M. Titirici and F. Wei, Adv. Mater., 2016, 28, 6845 CrossRef CAS PubMed.
- L. Zhang, T. Gu, K. Lu, L. Zhou, D. S. Li and R. Wang, Adv. Funct. Mater., 2021, 31, 2103187 CrossRef CAS.
- J. C. Carrillo-Rodríguez, A. M. Garay-Tapia, B. Escobar-Morales, J. Escorcia-García, M. T. Ochoa-Lara, F. J. Rodríguez-Varela and I. L. Alonso-Lemus, Int. J. Hydrogen Energy, 2021, 46, 26087–26100 CrossRef.
- Y. Sato, N. Yamada, S. Kitano, D. Kowalski, Y. Aoki and H. Habazaki, J. Mater. Chem. A, 2022, 10, 8208–8217 RSC.
- C. Madan, L. Sharma, S. Mukerjee and A. Halder, Int. J. Hydrogen Energy, 2022, 47, 22738–22751 CrossRef CAS.
- S. Möller, S. Barwe, S. Dieckhöfer, J. Masa, C. Andronescu and W. Schuhmann, ChemElectroChem, 2020, 7, 2680–2686 CrossRef.
- Y. Sun, G. Chen, S. Xi and Z. J. Xu, ACS Catal., 2021, 11, 13947–13954 CrossRef CAS.
- J. O. Olowoyo and R. J. Kriek, Small, 2022, 18, e2203125 CrossRef PubMed.
- E. Yeager, J. Mol. Catal., 1986, 38, 5–25 CrossRef CAS.
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and Y. Shao-Horn, Nat. Chem., 2011, 3, 546–550 CrossRef CAS PubMed.
- D. N. Mueller, M. L. Machala, H. Bluhm and W. C. Chueh, Nat. Commun., 2015, 6, 6097 CrossRef CAS PubMed.
- D. U. Lee, P. Xu, Z. P. Cano, A. G. Kashkooli, M. G. Park and Z. Chen, J. Mater. Chem. A, 2016, 4, 7107–7134 RSC.
- K. Zhang, X. Han, Z. Hu, X. Zhang, Z. Tao and J. Chen, Chem. Soc. Rev., 2015, 44, 699–728 RSC.
- F. Cheng, T. Zhang, Y. Zhang, J. Du, X. Han and J. Chen, Angew. Chem., Int. Ed. Engl., 2013, 52, 2474–2477 CrossRef CAS PubMed.
- Z. Chen, A. Yu, R. Ahmed, H. Wang, H. Li and Z. Chen, Electrochim. Acta, 2012, 69, 295–300 CrossRef CAS.
- K. Mette, A. Bergmann, J.-P. Tessonnier, M. Hävecker, L. Yao, T. Ressler, R. Schlögl, P. Strasser and M. Behrens, ChemCatChem, 2012, 4, 851–862 CrossRef CAS.
- F. Cheng, Y. Su, J. Liang, Z. Tao and J. Chen, Chem. Mater., 2009, 22, 898–905 CrossRef.
- J. Wu, J. Xue, F. Huang, N. Zhang, C. Tao and X. Fan, J. Alloys Compd., 2022, 907, 164520 CrossRef CAS.
- S. Ding, Z. Lyu, E. Sarnello, M. Xu, L. Fang, H. Tian, S. E. Karcher, T. Li, X. Pan, J. McCloy, G. Ding, Q. Zhang, Q. Shi, D. Du, J. Li, X. Zhang and Y. Lin, J. Mater. Chem. A, 2022, 10, 5981–5989 RSC.
- B. Chutia, N. Hussain, P. Puzari, D. Jampaiah, S. K. Bhargava, E. V. Matus, I. Z. Ismagilov, M. Kerzhentsev and P. Bharali, Energy Fuels, 2021, 35, 10756–10769 CrossRef CAS.
- M. Sridharan and T. Maiyalagan, J. Mater. Sci.: Mater. Electron., 2022, 33, 9538–9548 CrossRef CAS.
- J. Shin, J. K. Seo, R. Yaylian, A. Huang and Y. S. Meng, Int. Mater. Rev., 2019, 65, 356–387 CrossRef.
- N. Jia, T. Yang, S. Shi, X. Chen, Z. An, Y. Chen, S. Yin and P. Chen, ACS Sustainable Chem. Eng., 2020, 8, 2883–2891 CrossRef CAS.
- T. N. Lambert, D. J. Davis, W. Lu, S. J. Limmer, P. G. Kotula, A. Thuli, M. Hungate, G. Ruan, Z. Jin and J. M. Tour, Chem. Commun., 2012, 48, 7931–7933 RSC.
- D. W. Wang, F. Li, L. C. Yin, X. Lu, Z. G. Chen, I. R. Gentle, G. Q. Lu and H. M. Cheng, Chemistry, 2012, 18, 5345–5351 CrossRef CAS PubMed.
- X. Liu, X. Cui, K. Dastafkan, H. Wang, C. Tang, C. Zhao, A. Chen, C. He, M. Han and Q. Zhang, J. Energy Chem., 2021, 53, 290–302 CrossRef CAS.
- Y. Zhou, S. Xi, J. Wang, S. Sun, C. Wei, Z. Feng, Y. Du and Z. J. Xu, ACS Catal., 2017, 8, 673–677 CrossRef.
- S. Sun, Y. Sun, Y. Zhou, S. Xi, X. Ren, B. Huang, H. Liao, L. P. Wang, Y. Du and Z. J. Xu, Angew. Chem., Int. Ed. Engl., 2019, 58, 6042–6047 CrossRef CAS PubMed.
- C. Wei, Z. Feng, G. G. Scherer, J. Barber, Y. Shao-Horn and Z. J. Xu, Adv. Mater., 2017, 29, 1606800 CrossRef PubMed.
- Y. Zhou, S. Sun, S. Xi, Y. Duan, T. Sritharan, Y. Du and Z. J. Xu, Adv. Mater., 2018, 30, 1705407 CrossRef PubMed.
- Y. Zhou, Y. Du, S. Xi and Z. J. Xu, Electrocatalysis, 2017, 9, 287–292 CrossRef.
- Y. Sun, X. Ren, S. Sun, Z. Liu, S. Xi and Z. J. Xu, Angew. Chem., Int. Ed. Engl., 2021, 60, 14536–14544 CrossRef CAS PubMed.
- Y. Chen, J. K. Seo, Y. Sun, T. A. Wynn, M. Olguin, M. Zhang, J. Wang, S. Xi, Y. Du, K. Yuan, W. Chen, A. C. Fisher, M. Wang, Z. Feng, J. Gracia, L. Huang, S. Du, H. J. Gao, Y. S. Meng and Z. J. Xu, Nat. Commun., 2022, 13, 5510 CrossRef CAS PubMed.
- Q. Zhao, Z. Yan, C. Chen and J. Chen, Chem. Rev., 2017, 117, 10121–10211 CrossRef CAS PubMed.
- J. Béjar, L. Álvarez-Contreras, J. Ledesma-García, N. Arjona and L. G. Arriaga, J. Electroanal. Chem., 2019, 847, 113190 CrossRef.
- M. Prabu, K. Ketpang and S. Shanmugam, Nanoscale, 2014, 6, 3173–3181 RSC.
- V. Rajic, I. Stojkovic Simatovic, L. Veselinovic, J. B. Cavor, M. Novakovic, M. Popovic, S. D. Skapin, M. Mojovic, S. Stojadinovic, V. Rac, I. J. Castvan and S. Markovic, Phys. Chem. Chem. Phys., 2020, 22, 22078–22095 RSC.
- J. Bao, Z. Wang, W. Liu, L. Xu, F. Lei, J. Xie, Y. Zhao, Y. Huang, M. Guan and H. Li, J. Alloys Compd., 2018, 764, 565–573 CrossRef CAS.
- M. Prabu, P. Ramakrishnan and S. Shanmugam, Electrochem. Commun., 2014, 41, 59–63 CrossRef CAS.
- G. Wu, J. Wang, W. Ding, Y. Nie, L. Li, X. Qi, S. Chen and Z. Wei, Angew. Chem., Int. Ed. Engl., 2016, 55, 1340–1344 CrossRef CAS PubMed.
- Y. Zhang, Z. Zhang, G. Jiang, A. H. Mamaghani, S. Sy, R. Gao, Y. Jiang, Y. Deng, Z. Bai, L. Yang, A. Yu and Z. Chen, Nano Energy, 2022, 100 Search PubMed.
- S. Kosasang, H. Gatemala, N. Ma, P. Chomkhuntod and M. Sawangphruk, Batteries Supercaps, 2020, 3, 631–637 CrossRef CAS.
- Z. Zhang, J. Li, J. Qian, Z. Li, L. Jia, D. Gao and D. Xue, Small, 2022, 18, e2104248 CrossRef PubMed.
- Z. Qian, Y. Chen, Z. Tang, Z. Liu, X. Wang, Y. Tian and W. Gao, Nano-Micro Lett., 2019, 11, 28 CrossRef PubMed.
- Y. Zhu, X. Liu, S. Jin, H. Chen, W. Lee, M. Liu and Y. Chen, J. Mater. Chem. A, 2019, 7, 5875–5897 RSC.
- K. Min, S. Kim, E. Lee, G. Yoo, H. C. Ham, S. E. Shim, D. Lim and S.-H. Baeck, J. Mater. Chem. A, 2021, 9, 17344–17352 RSC.
- X. Zhou, X. Liao, X. Pan, M. Yan, L. He, P. Wu, Y. Zhao, W. Luo and L. Mai, Nano Energy, 2021, 83 CAS.
- K. Wang, Z. Wang, Y. Liu, J. Liu, Z. Cui, X. Zhang, F. Ciucci and Z. Tang, Chem. Eng. J., 2022, 427 Search PubMed.
- B. Mu, X. Zhang, Y. Zhang, P. Lu, J. Hao and J. Zhang, RSC Adv., 2022, 12, 9821–9827 RSC.
- K. Xiang, Z. Xu, T. Qu, Z. Tian, Y. Zhang, Y. Wang, M. Xie, X. Guo, W. Ding and X. Guo, Chem. Commun., 2017, 53, 12410–12413 RSC.
- J. Tang, C. Su and Z. Shao, Energy Technol., 2022, 10, 2200235 CrossRef CAS.
- Y. Tian, X. Liu, L. Xu, D. Yuan, Y. Dou, J. Qiu, H. Li, J. Ma, Y. Wang, D. Su and S. Zhang, Adv. Funct. Mater., 2021, 31, 2101239 CrossRef CAS.
- Z. Xiao, Y. C. Huang, C. L. Dong, C. Xie, Z. Liu, S. Du, W. Chen, D. Yan, L. Tao, Z. Shu, G. Zhang, H. Duan, Y. Wang, Y. Zou, R. Chen and S. Wang, J. Am. Chem. Soc., 2020, 142, 12087–12095 CrossRef CAS PubMed.
- Z. Xiao, Y. Wang, Y.-C. Huang, Z. Wei, C.-L. Dong, J. Ma, S. Shen, Y. Li and S. Wang, Energy Environ. Sci., 2017, 10, 2563–2569 RSC.
- S. V. Devaguptapu, S. Hwang, S. Karakalos, S. Zhao, S. Gupta, D. Su, H. Xu and G. Wu, ACS Appl. Mater. Interfaces, 2017, 9, 44567–44578 CrossRef CAS PubMed.
- H. Yu, H. Fan, B. Yadian, H. Tan, W. Liu, H. H. Hng, Y. Huang and Q. Yan, ACS Appl. Mater. Interfaces, 2015, 7, 26751–26757 CrossRef CAS PubMed.
- S. L. Zhang, B. Y. Guan, X. F. Lu, S. Xi, Y. Du and X. W. D. Lou, Adv. Mater., 2020, 32, e2002235 CrossRef PubMed.
- Y. Huang, S. L. Zhang, X. F. Lu, Z. P. Wu, D. Luan and X. W. D. Lou, Angew. Chem., Int. Ed. Engl., 2021, 60, 11841–11846 CrossRef CAS PubMed.
- X. Wang, Z. Liao, Y. Fu, C. Neumann, A. Turchanin, G. Nam, E. Zschech, J. Cho, J. Zhang and X. Feng, Energy Stor. Mater., 2020, 26, 157–164 Search PubMed.
- J. Lee, G. Nam, J. Sun, S. Higashi, H.-W. Lee, S. Lee, W. Chen, Y. Cui and J. Cho, Adv. Energy Mater., 2016, 6, 1601052 CrossRef.
- R. E. Davis, G. L. Horvath and C. W. Tobias, Electrochim. Acta, 1967, 12, 287–297 CrossRef CAS.
- Y. Matsumoto, H. Yoneyama and H. Tamura, J. Electroanal. Chem. Interfacial Electrochem., 1977, 79, 319–326 CrossRef CAS.
- B. Li, C. Tang, H. F. Wang, X. L. Zhu and Q. Zhang, Sci. Adv., 2016, 2, e1600495 CrossRef PubMed.
- J. I. Jung, H. Y. Jeong, M. G. Kim, G. Nam, J. Park and J. Cho, Adv. Mater., 2015, 27, 266–271 CrossRef CAS PubMed.
- G. Ou, C. Yang, Y. Liang, N. Hussain, B. Ge, K. Huang, Y. Xu, H. Wei, R. Zhang and H. Wu, Small Methods, 2019, 3, 1800279 CrossRef.
- V. Kotha, I. Karajagi, P. C. Ghosh and L. S. Panchakarla, ACS Appl. Energy Mater., 2022, 5, 7297–7307 CrossRef CAS.
- B. Bao, Y. Liu, M. Sun, B. Huang, Y. Hu, P. Da, D. Ji, P. Xi and C. H. Yan, Small, 2022, 18, e2201131 CrossRef PubMed.
- B. He, Y. Deng, H. Wang, R. Wang, J. Jin, Y. Gong and L. Zhao, J. Colloid Interface Sci., 2022, 625, 502–511 CrossRef CAS PubMed.
- X. Wang, Z. Pan, X. Chu, K. Huang, Y. Cong, R. Cao, R. Sarangi, L. Li, G. Li and S. Feng, Angew. Chem., Int. Ed. Engl., 2019, 58, 11720–11725 CrossRef CAS PubMed.
- J. Ran, J.-F. Wu, Y. Hu, M. Shakouri, B. Xia and D. Gao, J. Mater. Chem. A, 2022, 10, 1506–1513 RSC.
- J. Ran, T. Wang, J. Zhang, Y. Liu, C. Xu, S. Xi and D. Gao, Chem. Mater., 2020, 32, 3439–3446 CrossRef CAS.
- S. Sun, X. Jin, B. Cong, X. Zhou, W. Hong and G. Chen, J. Catal., 2019, 379, 1–9 CrossRef CAS.
- J. Zhang, X. Bai, T. Wang, W. Xiao, P. Xi, J. Wang, D. Gao and J. Wang, Nano-Micro Lett., 2019, 11, 2 CrossRef CAS PubMed.
- X. Gong, H. Zhong, L. A. Estudillo-Wong, N. Alonso-Vante, Y. Feng and D. Li, J. Energy Chem., 2022, 74, 376–386 CrossRef CAS.
- C.-X. Zhao, B.-Q. Li, M. Zhao, J.-N. Liu, L.-D. Zhao, X. Chen and Q. Zhang, Energy Environ. Sci., 2020, 13, 1711–1716 RSC.
- H. Wang, C. Tang, B. Wang, B. Q. Li and Q. Zhang, Adv. Mater., 2017, 29, 1702327 CrossRef PubMed.
- B. Wang, C. Tang, H. F. Wang, B. Q. Li, X. Cui and Q. Zhang, Small Methods, 2018, 2, 1800055 CrossRef.
- J. Masa, P. Weide, D. Peeters, I. Sinev, W. Xia, Z. Sun, C. Somsen, M. Muhler and W. Schuhmann, Adv. Energy Mater., 2016, 6, 1502313 CrossRef.
- Z. Shao, Q. Zhu, Y. Sun, Y. Zhang, Y. Jiang, S. Deng, W. Zhang, K. Huang and S. Feng, Adv. Mater., 2022, 34, e2110172 CrossRef PubMed.
- Y. Wang, X. Wu, X. Jiang, X. Wu, Y. Tang, D. Sun and G. Fu, Chem. Eng. J., 2022, 434 Search PubMed.
- B. Chen, R. Li, G. Ma, X. Gou, Y. Zhu and Y. Xia, Nanoscale, 2015, 7, 20674–20684 RSC.
- C. Hu, J. Liu, J. Wang, W. She, J. Xiao, J. Xi, Z. Bai and S. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 33124–33134 CrossRef CAS PubMed.
- H. Liu, J. Guan, S. Yang, Y. Yu, R. Shao, Z. Zhang, M. Dou, F. Wang and Q. Xu, Adv. Mater., 2020, 32, e2003649 CrossRef PubMed.
- Z. Wang, K. Shen, L. Chen and Y. Li, Sci. China: Chem., 2021, 65, 619–629 CrossRef.
- H. Wang, C. Li, J. An, Y. Zhuang and S. Tao, J. Mater. Chem. A, 2021, 9, 18421–18430 RSC.
- W. Jin, J. Chen, B. Liu, J. Hu, Z. Wu, W. Cai and G. Fu, Small, 2019, 15, e1904210 CrossRef PubMed.
- X. Li, H. Rong, J. Zhang, D. Wang and Y. Li, Nano Res., 2020, 13, 1842–1855 CrossRef CAS.
- Y. Wen, J. Qi, P. Wei, X. Kang and X. Li, J. Mater. Chem. A, 2021, 9, 10260–10269 RSC.
- X. Guo, S. Lin, J. Gu, S. Zhang, Z. Chen and S. Huang, ACS Catal., 2019, 9, 11042–11054 CrossRef CAS.
- K. Dong, J. Liang, Y. Ren, Y. Wang, Z. Xu, L. Yue, T. Li, Q. Liu, Y. Luo, Y. Liu, S. Gao, M. S. Hamdy, Q. Li, D. Ma and X. Sun, J. Mater. Chem. A, 2021, 9, 26019–26027 RSC.
- R. Jasinski, Nature, 1964, 201, 1212–1213 CrossRef CAS.
- L. Lin, Q. Zhu and A. W. Xu, J. Am. Chem. Soc., 2014, 136, 11027–11033 CrossRef CAS PubMed.
- C. W. B. Bezerra, L. Zhang, K. Lee, H. Liu, A. L. B. Marques, E. P. Marques, H. Wang and J. Zhang, Electrochim. Acta, 2008, 53, 4937–4951 CrossRef CAS.
- Y. Zhu, Z. Zhang, Z. Lei, Y. Tan, W. Wu, S. Mu and N. Cheng, Carbon, 2020, 167, 188–195 CrossRef CAS.
- S. Chen, T. Luo, X. Li, K. Chen, J. Fu, K. Liu, C. Cai, Q. Wang, H. Li, Y. Chen, C. Ma, L. Zhu, Y. R. Lu, T. S. Chan, M. Zhu, E. Cortes and M. Liu, J. Am. Chem. Soc., 2022, 144, 14505–14516 CrossRef CAS PubMed.
- P. Li, H. Wang, X. Tan, W. Hu, M. Huang, J. Shi, J. Chen, S. Liu, Z. Shi and Z. Li, Appl. Catal., B, 2022, 316 Search PubMed.
- D. Lyu, Y. Du, S. Huang, B. Y. Mollamahale, X. Zhang, S. W. Hasan, F. Yu, S. Wang, Z. Q. Tian and P. K. Shen, ACS Appl. Mater. Interfaces, 2019, 11, 39809–39819 CrossRef CAS PubMed.
- J. Zhang, T. Zhang, J. Ma, Z. Wang, J. Liu and X. Gong, Carbon, 2021, 172, 556–568 CrossRef CAS.
- P. Yu, L. Wang, F. Sun, Y. Xie, X. Liu, J. Ma, X. Wang, C. Tian, J. Li and H. Fu, Adv. Mater., 2019, 31, e1901666 CrossRef PubMed.
- K. Gao, M. Shen, C. Duan, C. Xiong, L. Dai, W. Zhao, W. Lu, S. Ding and Y. Ni, ACS Sustainable Chem. Eng., 2021, 9, 17068–17077 CrossRef CAS.
- Y. He, S. Hwang, D. A. Cullen, M. A. Uddin, L. Langhorst, B. Li, S. Karakalos, A. J. Kropf, E. C. Wegener, J. Sokolowski, M. Chen, D. Myers, D. Su, K. L. More, G. Wang, S. Litster and G. Wu, Energy Environ. Sci., 2019, 12, 250–260 RSC.
- L. Xu, D. Deng, Y. Tian, H. Li, J. Qian, J. Wu and H. Li, Chem. Eng. J., 2021, 408, 127321 CrossRef CAS.
- J. Li, Z. Yang, D. Tang, L. Zhang, P.-X. Hou, S. Zhao, C. Liu, M. Cheng, G. Li, F. Zhang and H. Cheng, NPG Asia Mater., 2018, 10, e461 CrossRef CAS.
- Y. Wang, L. Wang and H. Fu, Sci. China Mater., 2022, 65, 1701–1722 CrossRef CAS.
- C. Shao, L. Wu, Y. Wang, K. Qu, H. Chu, L. Sun, J. Ye, B. Li and X. Wang, Appl. Catal., B, 2022, 316, 121607 CrossRef CAS.
- R. Ma, J. Wang, Y. Tang and J. Wang, J. Phys. Chem. Lett., 2022, 13, 168–174 CrossRef CAS PubMed.
- Z. Mei, S. Cai, G. Zhao, Q. Jing, X. Sheng, J. Jiang and H. Guo, Energy Stor. Mater., 2022, 50, 12–20 Search PubMed.
- L. Yang, X. Zhang, L. Yu, J. Hou, Z. Zhou and R. Lv, Adv. Mater., 2022, 34, e2105410 CrossRef PubMed.
- F. Liu, L. Shi, S. Song, K. Ge, X. Zhang, Y. Guo and D. Liu, Small, 2021, 17, e2102425 CrossRef PubMed.
- W. Zhai, S. Huang, C. Lu, X. Tang, L. Li, B. Huang, T. Hu, K. Yuan, X. Zhuang and Y. Chen, Small, 2022, 18 Search PubMed.
- C. Yang, S. Shang, Q. Gu, J. Shang and X.-y. Li, J. Energy Chem., 2022, 66, 306–313 CrossRef CAS.
- G. Yang, J. Zhu, P. Yuan, Y. Hu, G. Qu, B. A. Lu, X. Xue, H. Yin, W. Cheng, J. Cheng, W. Xu, J. Li, J. Hu, S. Mu and J. N. Zhang, Nat. Commun., 2021, 12, 1734 CrossRef CAS PubMed.
- Z. Wang, H. Jin, T. Meng, K. Liao, W. Meng, J. Yang, D. He, Y. Xiong and S. Mu, Adv. Funct. Mater., 2018, 28, 1802596 CrossRef.
- Y. Mu, T. Wang, J. Zhang, C. Meng, Y. Zhang and Z. Kou, Electrochem. Energy Rev., 2021, 5, 145–186 CrossRef.
- Z. Zhang, S. Yang, R. Jiang, T. Sheng, C. Shi, Y. Chen and L. Wang, Nano Res., 2022, 15, 8928–8935 CrossRef CAS.
- J. Liu, J. Xiao, B. Luo, E. Tian and G. I. N. Waterhouse, Chem. Eng. J., 2022, 427, 132038 CrossRef CAS.
- F. Lu, K. Fan, L. Cui, B. Li, Y. Yang, L. Zong and L. Wang, Appl. Catal., B, 2022, 313, 121464 CrossRef CAS.
- D. Chen, Z. Chen, Z. Lu, X. Zhang, J. Tang and C. V. Singh, Nanoscale, 2020, 12, 18721–18732 RSC.
- Y. Guo, P. Yuan, J. Zhang, Y. Hu, I. S. Amiinu, X. Wang, J. Zhou, H. Xia, Z. Song, Q. Xu and S. Mu, ACS Nano, 2018, 12, 1894–1901 CrossRef CAS PubMed.
- H. Xu, Y. Zhao, Q. Wang, G. He and H. Chen, Coord. Chem. Rev., 2022, 451, 214261 CrossRef CAS.
- J. Li, X. Qin, F. Xiao, C. Liang, M. Xu, Y. Meng, E. Sarnello, L. Fang, T. Li, S. Ding, Z. Lyu, S. Zhu, X. Pan, P. X. Hou, C. Liu, Y. Lin and M. Shao, Nano Lett., 2021, 21, 4508–4515 CrossRef CAS PubMed.
- J. Tang, Z. Zeng, H. Liang, Z. Wang, W. Nong, Z. Yang, C. Qi, Z. Qiao, Y. Li and C. Wang, ACS Omega, 2022, 7, 19794–19803 CrossRef CAS PubMed.
- J. Li, S. Chen, N. Yang, M. Deng, S. Ibraheem, J. Deng, J. Li, L. Li and Z. Wei, Angew. Chem., Int. Ed. Engl., 2019, 58, 7035–7039 CrossRef CAS PubMed.
- G. W. Sievers, A. W. Jensen, J. Quinson, A. Zana, F. Bizzotto, M. Oezaslan, A. Dworzak, J. J. K. Kirkensgaard, T. E. L. Smitshuysen, S. Kadkhodazadeh, M. Juelsholt, K. M. O. Jensen, K. Anklam, H. Wan, J. Schafer, K. Cepe, M. Escudero-Escribano, J. Rossmeisl, A. Quade, V. Bruser and M. Arenz, Nat. Mater., 2021, 20, 208–213 CrossRef CAS PubMed.
- J. Chen, H. Li, C. Fan, Q. Meng, Y. Tang, X. Qiu, G. Fu and T. Ma, Adv. Mater., 2020, 32, e2003134 CrossRef PubMed.
- F. Zheng, Y. Ji, H. Dong, C. Liu, S. Chen and Y. Li, J. Phys. Chem. C, 2021, 126, 30–39 CrossRef.
- H. Liu, X. H. Zhang, Y. X. Li, X. Li, C. K. Dong, D. Y. Wu, C. C. Tang, S. L. Chou, F. Fang and X. W. Du, Adv. Energy Mater., 2020, 10, 1902521 CrossRef CAS.
- X. Xie, Y. Xue, L. Li, S. Chen, Y. Nie, W. Ding and Z. Wei, Nanoscale, 2014, 6, 11035–11040 RSC.
- X. Han, N. Li, P. Xiong, M. G. Jung, Y. Kang, Q. Dou, Q. Liu, J. Y. Lee and H. S. Park, InfoMat, 2021, 3, 1134–1144 CrossRef CAS.
- S. G. Chandrappa, P. Moni, G. Karkera and A. S. Prakash, Nanoscale Adv., 2019, 1, 2392–2399 RSC.
- Z. Pu, Q. Liu, C. Tang, A. M. Asiri, A. H. Qusti, A. O. Al-Youbi and X. Sun, J. Power Sources, 2014, 257, 170–173 CrossRef CAS.
- H. Cheng, M.-L. Li, C.-Y. Su, N. Li and Z.-Q. Liu, Adv. Funct. Mater., 2017, 27, 1701833 CrossRef.
- P. K. Gangadharan, S. N. Bhange, N. Kabeer, R. Illathvalappil and S. Kurungot, Nanoscale Adv., 2019, 1, 3243–3251 RSC.
- M. Wu, G. Zhang, J. Qiao, N. Chen, W. Chen and S. Sun, Nano Energy, 2019, 61, 86–95 CrossRef CAS.
- A. Zhao, J. Masa, W. Xia, A. Maljusch, M. G. Willinger, G. Clavel, K. Xie, R. Schlogl, W. Schuhmann and M. Muhler, J. Am. Chem. Soc., 2014, 136, 7551–7554 CrossRef CAS PubMed.
- Z. Sun, Y. Wang, L. Zhang, H. Wu, Y. Jin, Y. Li, Y. Shi, T. Zhu, H. Mao, J. Liu, C. Xiao and S. Ding, Adv. Funct. Mater., 2020, 30 CAS.
- P. Ganesan, M. Prabu, J. Sanetuntikul and S. Shanmugam, ACS Catal., 2015, 5, 3625–3637 CrossRef CAS.
- G. Fu, J. Wang, Y. Chen, Y. Liu, Y. Tang, J. B. Goodenough and J.-M. Lee, Adv. Energy Mater., 2018, 8 CAS.
- G. Nam, Y. Son, S. O. Park, W. C. Jeon, H. Jang, J. Park, S. Chae, Y. Yoo, J. Ryu, M. G. Kim, S. K. Kwak and J. Cho, Adv. Mater., 2018, 30, e1803372 CrossRef PubMed.
- V. Jose, H. Hu, E. Edison, W. Manalastas Jr, H. Ren, P. Kidkhunthod, S. Sreejith, A. Jayakumar, J. M. V. Nsanzimana, M. Srinivasan, J. Choi and J.-M. Lee, Small Methods, 2021, 5, 2000751 CrossRef CAS PubMed.
- Z. Lei, Y. Tan, Z. Zhang, W. Wu, N. Cheng, R. Chen, S. Mu and X. Sun, Nano Res., 2020, 14, 868–878 CrossRef.
- Y. Guo, P. Yuan, J. Zhang, H. Xia, F. Cheng, M. Zhou, J. Li, Y. Qiao, S. Mu and Q. Xu, Adv. Funct. Mater., 2018, 28, 1805641 CrossRef.
- Q. Shi, Q. Liu, Y. Zheng, Y. Dong, L. Wang, H. Liu and W. Yang, Energy Environ. Mater., 2021, 5, 515–523 CrossRef.
- Y. Han, H. Duan, C. Zhou, H. Meng, Q. Jiang, B. Wang, W. Yan and R. Zhang, Nano Lett., 2022, 22, 2497–2505 CrossRef CAS PubMed.
- X. Guo, X. Hu, D. Wu, C. Jing, W. Liu, Z. Ren, Q. Zhao, X. Jiang, C. Xu, Y. Zhang and N. Hu, ACS Appl. Mater. Interfaces, 2019, 11, 21506–21514 CrossRef CAS PubMed.
- B. Geboes, J. Ustarroz, K. Sentosun, H. Vanrompay, A. Hubin, S. Bals and T. Breugelmans, ACS Catal., 2016, 6, 5856–5864 CrossRef CAS.
- Y. Chung, D. Y. Chung, N. Jung, H. Y. Park, S. J. Yoo, J. H. Jang and Y.-E. Sung, J. Phys. Chem. C, 2014, 118, 9939–9945 CrossRef CAS.
- Z. Kou, X. Li, L. Zhang, W. Zang, X. Gao and J. Wang, Small Sci., 2021, 1, 2100011 CrossRef CAS.
- X. Zhao, Q. Han, J. Li, X. Du, G. Liu, Y. Wang, L. Wu and Z. Chen, Chem. Eng. J., 2022, 433, 133509 CrossRef CAS.
- W. J. Sim, M. T. Nguyen, Z. Huang, S. Kheawhom, C. Wattanakit and T. Yonezawa, Nanoscale, 2022, 14, 8012–8022 RSC.
- Z. Chen, L. Cai, X. Yang, C. Kronawitter, L. Guo, S. Shen and B. E. Koel, ACS Catal., 2018, 8, 1238–1247 CrossRef CAS.
- B. Tian, H. Shin, S. Liu, M. Fei, Z. Mu, C. Liu, Y. Pan, Y. Sun, W. A. Goddard 3rd and M. Ding, Angew. Chem., Int. Ed. Engl., 2021, 60, 16448–16456 CrossRef CAS PubMed.
- Y. Sun, J. Wu, Y. Xie, X. Wang, K. Ma, Z. Tian, Z. Zhang, Q. Liao, W. Zheng, Z. Kang and Y. Zhang, Adv. Funct. Mater., 2022, 32, 2207116 CrossRef CAS.
- X. Xie, L. Du, L. Yan, S. Park, Y. Qiu, J. Sokolowski, W. Wang and Y. Shao, Adv. Funct. Mater., 2022, 32, 2110036 CrossRef CAS.
- G. Xing, M. Tong, P. Yu, L. Wang, G. Zhang, C. Tian and H. Fu, Angew. Chem., Int. Ed. Engl., 2022, 61, e202211098 CAS.
- H. Zhang, P. Li, S. Chen, F. Xie and D. J. Riley, Adv. Funct. Mater., 2021, 31, 2106835 CrossRef CAS.
- Y. P. Deng, Y. Jiang, R. Liang, S. J. Zhang, D. Luo, Y. Hu, X. Wang, J. T. Li, A. Yu and Z. Chen, Nat. Commun., 2020, 11, 1952 CrossRef CAS PubMed.
- H. Hu, J. Wang, P. Tao, C. Song, W. Shang, T. Deng and J. Wu, J. Mater. Chem. A, 2022, 10, 5835–5849 RSC.
- Z. Hu, L. Hao, F. Quan and R. Guo, Catal. Sci. Technol., 2022, 12, 436–461 RSC.
- R. N. Durr, P. Maltoni, H. Tian, B. Jousselme, L. Hammarstrom and T. Edvinsson, ACS Nano, 2021, 15, 13504–13515 CrossRef CAS PubMed.
- J.-T. Ren, Y.-D. Ying, Y.-P. Liu, W. Li and Z.-Y. Yuan, J. Energy Chem., 2022, 71, 619–630 CrossRef CAS.
- M. Wu, G. Zhang, N. Chen, Y. Hu, T. Regier, D. Rawach and S. Sun, ACS Energy Lett., 2021, 6, 1153–1161 CrossRef CAS.
- L. An, Y. Hu, J. Y. Li, J. M. Zhu, M. Z. Sun, B. L. Huang, P. X. Xi and C. H. Yan, Adv. Mater., 2022, 34, e2202874 CrossRef PubMed.
- B. Yan, D. Krishnamurthy, C. H. Hendon, S. Deshpande, Y. Surendranath and V. Viswanathan, Joule, 2017, 1, 600–612 CrossRef CAS.
- F. T. Haase, A. Bergmann, T. E. Jones, J. Timoshenko, A. Herzog, H. S. Jeon, C. Rettenmaier and B. R. Cuenya, Nat. Energy, 2022, 7, 765–773 CrossRef CAS.
- H. Wang, S. Hung, H. Chen, T. Chan, H. Chen and B. Liu, J. Am. Chem. Soc., 2016, 138, 36–39 CrossRef CAS PubMed.
- L. An, Y. Hu, J. Li, J. Zhu, M. Sun, B. Huang, P. Xi and C. H. Yan, Adv. Mater., 2022, 34, e2202874 CrossRef PubMed.
- C. Weng, X. Lv, J. Ren, Y. Wang, W. Tian, L. Gao, H. Wang and Z. Yuan, ACS Sustainable Chem. Eng., 2022, 10, 6456–6465 CrossRef CAS.
- A. Saad, Y. Gao, A. Ramiere, T. Chu, G. Yasin, Y. Wu, S. Ibraheem, M. Wang, H. Guo, P. Tsiakaras and X. Cai, Small, 2022, 18, e2201067 CrossRef PubMed.
- X. Wang, X. Yang, H. Liu, T. Han, J. Hu, H. Li and G. Wu, Small Struct., 2021, 3, 2100103 CrossRef.
- M. Fan, L. Cui, X. He and X. Zou, Small Methods, 2022, 6, e2200855 CrossRef PubMed.
- C. Liu, Q. Li, W. Kang, W. Lei, X. Wang, C. Lu and M. Naebe, J. Mater. Chem. A, 2022, 10, 10–49 RSC.
- Y. Zhong, X. Xu, W. Wang and Z. Shao, Batteries Supercaps, 2018, 2, 272–289 CrossRef.
- J. Li, Y. Zhu, W. Chen, Z. Lu, J. Xu, A. Pei, Y. Peng, X. Zheng, Z. Zhang, S. Chu and Y. Cui, Joule, 2019, 3, 557–569 CrossRef CAS.
- P. Wang, T. Jia and B. Wang, J. Electrochem. Soc., 2020, 167 CAS.
- C. X. Zhao, J. N. Liu, J. Wang, D. Ren, J. Yu, X. Chen, B. Q. Li and Q. Zhang, Adv. Mater., 2021, 33, e2008606 CrossRef PubMed.
- Y. Yuan, M. Li, Z. Bai, G. Jiang, B. Liu, T. Wu, Z. Chen, K. Amine and J. Lu, Adv. Mater., 2019, 31, e1805609 CrossRef PubMed.
- S. S. Sekhon, J. Lee and J.-S. Park, J. Energy Chem., 2022, 65, 149–172 CrossRef CAS.
- A. Hayat, M. Sohail, J. Ali Shah Syed, A. G. Al-Sehemi, M. H. Mohammed, A. A. Al-Ghamdi, T. A. Taha, H. Salem AlSalem, A. M. Alenad, M. A. Amin, A. Palamanit, C. Liu, W. I. Nawawi, M. Tariq Saeed Chani and M. Muzibur Rahman, Chem. Rec., 2022, 22, e202100310 CAS.
- Z. Pei, L. Ding, C. Wang, Q. Meng, Z. Yuan, Z. Zhou, S. Zhao and Y. Chen, Energy Environ. Sci., 2021, 14, 4926–4935 RSC.
- C. Zhao, J. Liu, N. Yao, J. Wang, D. Ren, X. Chen, B. Q. Li and Q. Zhang, Angew. Chem., Int. Ed. Engl., 2021, 60, 15281–15285 CrossRef CAS PubMed.
- X. Lu, Y. Fang, D. Luan and X. W. D. Lou, Nano Lett., 2021, 21, 1555–1565 CrossRef CAS PubMed.
- G. Zhou, G. Liu, X. Liu, Q. Yu, H. Mao, Z. Xiao and L. Wang, Adv. Funct. Mater., 2021, 32, 2107608 CrossRef.
- L. Zhong, Q. Huang, J. Ding, Y. Guo, X. Wang, L. Chai, T.-T. Li, Y. Hu, J. Qian and S. Huang, J. Power Sources, 2021, 492, 229632 CrossRef CAS.
- N. Manna, S. K. Singh, M. Kurian, A. Torris and S. Kurungot, ACS Appl. Energy Mater., 2022, 5, 8756–8768 CrossRef CAS.
- X. Liu, Q. Yin, C. Dai, G. Li, J. Lian, Y. Zhao, S. Yang and H. Li, ACS Sustainable Chem. Eng., 2021, 9, 5345–5355 CrossRef CAS.
- D. Wang, B. Li, X. Tao, S. Rao, J. Li, W. Wang, J. Yang and Y. Zhou, J. Electroanal. Chem., 2021, 898, 115627 CrossRef CAS.
- S. Das, S. Bhattacharjee, S. Mondal, S. Dutta, N. Bothra, S. K. Pati and S. Bhattacharyya, ACS Sustainable Chem. Eng., 2021, 9, 14868–14880 CrossRef CAS.
- H. Sun, M. Wang, S. Zhang, S. Liu, X. Shen, T. Qian, X. Niu, J. Xiong and C. Yan, Adv. Funct. Mater., 2020, 31 CAS.
- Y. Niu, X. Teng, S. Gong, X. Liu, M. Xu and Z. Chen, Energy Stor. Mater., 2021, 43, 42–52 Search PubMed.
- Y. Dai, J. Yu, J. Wang, Z. Shao, D. Guan, Y. C. Huang and M. Ni, Adv. Funct. Mater., 2022, 32, 2111989 CrossRef CAS.
- B. Wang, P. Zhao, J. Feng, D. Chen, Y. Huang, L. Sui, H. Dong, S. Ma, L. Dong and L. Yu, J. Colloid Interface Sci., 2021, 588, 184–195 CrossRef CAS PubMed.
- Q. Lu, X. Zou, Y. Bu, K. Liao, W. Zhou and Z. Shao, Small, 2022, 18, e2105604 CrossRef PubMed.
- J. Zhang, J. Chen, Y. Luo, Y. Chen, Y. Luo, C. Zhang, Y. Xue, H. Liu, G. Wang and R. Wang, J. Mater. Chem. A, 2021, 9, 5556–5565 RSC.
- H. Pan, X. Huang, Z. Lu, Z. Zhang, B. An, D. Wu, T. Wang, X. Chen and F. Cheng, Chem. Eng. J., 2021, 419 Search PubMed.
- N. Yu, H. Chen, J. Kuang, K. Bao, W. Yan, J. Ye, Z. Yang, Q. Huang, Y. Wu and S. Sun, Nano Res., 2022, 15, 7209–7219 CrossRef CAS.
- X. Liu, S. Peng, X. Li, C. Liu, J. Zeng, X. Qi and T. Liang, J. Electrochem. Soc., 2021, 168, 090514 CrossRef CAS.
- F. Zhang, L. Chen, Y. Zhang, Y. Peng, X. Luo, Y. Xu and Y. Shi, Chem. Eng. J., 2022, 447, 137490 CrossRef CAS.
- C. Shao, S. Zhuang, H. Zhang, Q. Jiang, X. Xu, J. Ye, B. Li and X. Wang, Small, 2021, 17, e2006178 CrossRef PubMed.
- L. Xie, X. P. Zhang, B. Zhao, P. Li, J. Qi, X. Guo, B. Wang, H. Lei, W. Zhang, U. P. Apfel and R. Cao, Angew. Chem., Int. Ed. Engl., 2021, 60, 7576–7581 CrossRef CAS PubMed.
- Y. Zhang, W. Xiao, Y. Yin, D. Z. Peng, H. Wang, M. Zhou, Z. Hou, Y. Liu and B. He, New J. Chem., 2022, 46, 17032–17039 RSC.
- L. Song, H. Fan, T. Wang, T. Xiang, M. Zhang, C. Hu, W. Zhou and J. He, Energy Stor. Mater., 2021, 43, 365–374 Search PubMed.
- S. Chang, H. Zhang and Z. Zhang, J. Energy Chem., 2021, 56, 64–71 CrossRef CAS.
- N. Li, L. Liu, K. Wang, J. Niu, Z. Zhang, M. Dou and F. Wang, Chemistry, 2021, 27, 10987–10997 CrossRef CAS PubMed.
- Z. Meng, G. Zhu, J. Wu, R. Wang, T. Tian, H. Tang, R. Luo, D. Ye, R. Zhang, F. Kwofie, Y. Cheng and H. Tang, Mater. Today Energy, 2022, 24, 100935 CrossRef CAS.
- H. Xu, D. Wang, P. Yang, L. Du, X. Lu, R. Li, L. Liu, J. Zhang and M. An, Appl. Catal., B, 2022, 305, 121040 CrossRef CAS.
- R. Zhao, H. Wang, X. Zhang, J. Liu, G. Du and T. Chen, Langmuir, 2022, 38, 11372–11381 CrossRef CAS PubMed.
- M. Wang, L. Cao, X. Du, Y. Zhang, F. Jin, M. Zhang, Z. Li and K. Su, ACS Appl. Mater. Interfaces, 2022, 14, 25427–25438 CrossRef CAS PubMed.
- K. Li, R. Cheng, Q. Xue, P. Meng, T. Zhao, M. Jiang, M. Guo, H. Li and C. Fu, Chem. Eng. J., 2022, 450, 137991 CrossRef CAS.
- X. Wang, R. K. M. Raghupathy, C. J. Querebillo, Z. Liao, D. Li, K. Lin, M. Hantusch, Z. Sofer, B. Li, E. Zschech, I. M. Weidinger, T. D. Kuhne, H. Mirhosseini, M. Yu and X. Feng, Adv. Mater., 2021, 33, e2008752 CrossRef PubMed.
- C. Chen, H. Su, L.-N. Lu, Y.-S. Hong, Y. Chen, K. Xiao, T. Ouyang, Y. Qin and Z.-Q. Liu, Chem. Eng. J., 2021, 408, 127814 CrossRef CAS.
- X. Qiao, Y. Deng, X. Cao, J. Wu, H. Guo, W. Xiao and S. Liao, Catalysts, 2022, 12, 1023 CrossRef CAS.
- C. Jin, H. Deng, J. Zhang, Y. Hao and J. Liu, Chem. Eng. J., 2022, 434, 134617 CrossRef CAS.
- X. Chen, J. Pu, X. Hu, Y. Yao, Y. Dou, J. Jiang and W. Zhang, Small, 2022, 18, e2200578 CrossRef PubMed.
- Y. Wang, N. Xu, R. He, L. Peng, D. Cai and J. Qiao, Appl. Catal., B, 2021, 285, 119811 CrossRef CAS.
- Y. Jiang, Y. P. Deng, R. Liang, N. Chen, G. King, A. Yu and Z. Chen, J. Am. Chem. Soc., 2022, 144, 4783–4791 CrossRef CAS PubMed.
- H. Lei, Q. Zhang, Z. Liang, H. Guo, Y. Wang, H. Lv, X. Li, W. Zhang, U. P. Apfel and R. Cao, Angew. Chem., Int. Ed. Engl., 2022, 61, e202201104 CAS.
- Z. Liang, H. Guo, G. Zhou, K. Guo, B. Wang, H. Lei, W. Zhang, H. Zheng, U. P. Apfel and R. Cao, Angew. Chem., Int. Ed. Engl., 2021, 60, 8472–8476 CrossRef CAS PubMed.
- S. Liang, Z.-D. Wang, Z.-F. Guo, X.-Y. Chen, S.-Q. Li, B.-D. Wang, G.-L. Lu, H. Sun, Z.-N. Liu and H.-Y. Zang, New J. Chem., 2021, 45, 3947–3953 RSC.
- Y. Liao, H. Chen, C. Ou, L. Bao, R. Li and H. Liu, J. Electroanal. Chem., 2022, 921, 116560 CrossRef CAS.
- Y. Peng, F. Zhang, Y. Zhang, X. Luo, L. Chen and Y. Shi, Dalton Trans., 2022, 51, 12630–12640 RSC.
- J. Yin, J. Jin, H. Liu, B. Huang, M. Lu, J. Li, H. Liu, H. Zhang, Y. Peng, P. Xi and C. H. Yan, Adv. Mater., 2020, 32, e2001651 CrossRef PubMed.
- T. Hu, Z. Jiang, Z. Fu and Z.-J. Jiang, J. Mater. Chem. A, 2022, 10, 8739–8750 RSC.
- H. Zheng, Y. Zhang, J. Long, R. Li and X. Gou, J. Electrochem. Soc., 2020, 167, 084516 CrossRef CAS.
- X. Yang, X. Zheng, H. Li, B. Luo, Y. He, Y. Yao, H. Zhou, Z. Yan, Y. Kuang and Z. Huang, Adv. Funct. Mater., 2022, 32, 202200397 Search PubMed.
- Z. Zhang, H. Zhang, Y. Hou, P. Liu, X. Hao, Y. Liu, B. Xu and J. Guo, J. Mater. Chem. A, 2022, 10, 13013–13020 RSC.
- C. Li, Y. Wu, M. Fu, X. Zhao, S. Zhai, Y. Yan, L. Zhang and X. Zhang, ACS Appl. Energy Mater., 2022, 5, 4340–4350 CrossRef CAS.
- W. Zhai, S. Huang, C. Lu, X. Tang, L. Li, B. Huang, T. Hu, K. Yuan, X. Zhuang and Y. Chen, Small, 2022, 18, e2107225 CrossRef PubMed.
- C. Lai, X. Liu, C. Cao, Y. Wang, Y. Yin, T. Liang and D. D. Dionysiou, Carbon, 2021, 173, 715–723 CrossRef CAS.
- C. Zhou, X. Chen, S. Liu, Y. Han, H. Meng, Q. Jiang, S. Zhao, F. Wei, J. Sun, T. Tan and R. Zhang, J. Am. Chem. Soc., 2022, 144, 2694–2704 CrossRef CAS PubMed.
- M. Christy, H. Rajan, H. Lee, I. Rabani, S. M. Koo and S. C. Yi, ACS Appl. Energy Mater., 2021, 4, 1876–1886 CrossRef CAS.
- Y. Zhao, X. Wang, X. Guo, D. Cheng, H. Zhou, N. Saito and T. Fan, Carbon, 2021, 184, 609–617 CrossRef CAS.
- Y. Gao, D. Kong, F. Cao, S. Teng, T. Liang, B. Luo, B. Wang, Q.-H. Yang and L. Zhi, Nano Res., 2022, 15, 7959–7967 CrossRef CAS.
- Y. Ma, W. Shang, W. Yu, X. Chen, W. Xia, C. Wang and P. Tan, Energy Fuels, 2021, 35, 14188–14196 CrossRef CAS.
- H. Begum, M. S. Ahmed and S. Jung, J. Mater. Chem. A, 2021, 9, 9644–9654 RSC.
- J. Ding, D. Wu, S. Huang, C. Lu, Y. Chen, J. Zhang, L. Zhang, J. Li, C. Ke, D. Tranca, E. Kymakis and X. Zhuang, Nanoscale, 2021, 13, 13249–13255 RSC.
- A. Wang, C. Zhao, M. Yu and W. Wang, Appl. Catal., B, 2021, 281, 119514 CrossRef CAS.
- C. Shao, L. Wu, H. Zhang, Q. Jiang, X. Xu, Y. Wang, S. Zhuang, H. Chu, L. Sun, J. Ye, B. Li and X. Wang, Adv. Funct. Mater., 2021, 31, 2100833 CrossRef CAS.
- W. Zhang, X. Zhao, W. Niu, H. Yu, T. Wan, G. Liu, D. Zhang and Y. Wang, Nanotechnology, 2021, 33, 065409 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2023 |