
Nanoarchitected graphene/copper oxide nanoparticles/MoS2 ternary thin films as highly efficient electrodes for aqueous sodium-ion batteries†
Maria K.
Ramos
a,
Gustavo
Martins
a,
Luiz H.
Marcolino-Junior
 a,
Márcio F.
Bergamini
a,
Marcela M.
Oliveira
b and
Aldo J. G.
Zarbin
*a
a,
Márcio F.
Bergamini
a,
Marcela M.
Oliveira
b and
Aldo J. G.
Zarbin
*a
aDepartment of Chemistry, Federal University of Paraná (UFPR), CP 19032, 81531-980, Curitiba, PR, Brazil. E-mail: aldozarbin@ufpr.br; Tel: +55 41 3361-1565
bDepartment of Chemistry and Biology, Technological Federal University of Paraná (UTFPR), Curitiba, PR, Brazil
First published on 20th September 2023
Abstract
Sodium-ion batteries (SIBs) operating in aqueous electrolyte are an emerging technology that promises to be safer, cheaper, more sustainable and more efficient than their lithium-based counterparts. One of the great challenges associated with this technology is the development of advanced materials with high specific capacity to be used as electrodes. Herein, we describe an ingenious strategy to prepare unprecedented tri-component nanoarchitected thin films with superior performance when applied as anodes in aqueous SIBs. Taking advantage of the broadness and versatility of the liquid–liquid interfacial route, three transparent nanocomposite films comprising graphene, molybdenum sulphide and copper oxide nanoparticles have been prepared. The samples were characterized using several techniques, and the results demonstrated that depending on the specific experimental strategy, different nanoarchitectures are achieved, resulting in different and improved properties. An astonishing capacity of 1377 mA h g−1 at 0.1 A g−1 and a degree of recovery of 100% were observed for the film in which the interactions among the components were optimized. This is among the highest capacity values reported in the literature and demonstrates the potential of these tri-component materials to be used as anodes in aqueous sodium-ion batteries.
 Aldo J. G. Zarbin | The arrival of Materials Horizons in 2014 brought a new perspective to the Materials Science community, and it has established a reputation for quality and become a source to find the most exciting breakthroughs in the field. I have had the honor and responsibility of being part of the Materials Horizons Advisory Board since 2017, and of having published a review on the technique developed in our group for preparing thin films at liquid interfaces in 2021. We are very happy to contribute this paper for the celebration of a decade of huge success for Materials Horizons. It is time to celebrate its 10th anniversary and to wish it a long life. |
New conceptsThis work describes unprecedented materials, prepared by an ingenious methodology, with the aim of application in an emergent and promising technology. Tri-component nanoarchitected materials comprising graphene, molybdenum disulfide and copper oxide nanoparticles were prepared and processed as thin and transparent films at the interface between immiscible liquids. This innovative preparation technique allows different experimental arrangements, producing three outstanding samples in which the distribution of the components (or the nanoarchitecture of the films) is different, resulting in different electrochemical properties. The performance of the materials as electrodes in aqueous sodium-ion batteries was evaluated, resulting in an impressive specific capacity due to several factors that are directly dependent on the synthetic pathway, among them: (i) the sum of the capacities of the individual components; (ii) the individual characteristics of each component, and (iii) the synergism resulting from the nanoarchitected structure. |
1. Introduction
Novel, unique, and synergistic functionalities arise from materials prepared through the arrangement of different nanostructures in a specific configuration, producing the so-called nanoarchitected nanocomposites.1 Based on the chosen constituents, bi-, three- or multi-component materials can be designed, presenting improved mechanical, optical, electrical, chemical and electrochemical properties resulting from the specific interactions among the individual nano-objects.1,2 One of the major challenges related to the real application of these nanoarchitected materials concerns their processability, particularly as thin films.3 Traditional techniques for thin film synthesis and/or processing (such as dip-coating,4 spin-coating,5 vapor deposition techniques,6,7 sol–gel processes,8 hydrothermal routes,9 and layer-by-layer deposition,10 among others2) are not successful with multicomponent materials prepared in nanoarchitected configurations, and the situation is even worse if transparent and flexible films are desired.2,11 Thereby, it is a huge scientific and technological challenge to prepare and process nanoarchitected materials as thin and transparent films that can be deposited over any solid surface or directly integrated into devices in a reliable and reproducible way.Our research group developed a straightforward technique for both synthesizing and processing multi-component materials as thin, transparent and homogeneous films.12 The so-called liquid–liquid interfacial route (LLIR) is based on the self-assembly of solid materials at the interface between immiscible liquids, resulting in a continuous network that behaves like a malleable film and is easily transferred over any kind of solid substrate.12 One of the many advantages of the LLIR is the possibility of synthesizing compounds in the course of the process, which means that desirable materials can be obtained already processed in a one-pot and single-step procedure. Also, due to the intrinsic characteristics of the LLIR, unprecedented multi-component thin films can be prepared by adjusting the experimental conditions, which could result in different nanoarchitectures. Numerous nanoarchitected thin films have been prepared by us through this technique, based on the mixture of low-dimensional materials, polymers and inorganic compounds,13–15 and applied in fields such as sensors,16,17 photovoltaics,18,19 batteries,20–22 supercapacitors,23 and catalysis,24 among others.
Energy storage device technology can be positively impacted by nanoarchitected thin films,25,26 including novel sustainable, inexpensive and safe devices, for example, aqueous sodium-ion batteries (SIBs). Currently, lithium-ion batteries (LIBs) are a well-established and widely used energy storage device. Nonetheless, this technology presents some drawbacks:27–29 traditional electrolytes for LIBs are flammable and volatile solvents;30 the growth of lithium dendrites during the charge/discharge cycles can connect the electrodes and generate a short circuit;31–33 and lithium resources have a limited and poor global distribution.34,35 Alternatively, SIBs are emerging as a compelling energy storage device, as they are potentially low-cost, sodium is abundant and widely globally distributed, and sodium mining is accomplished using a cleaner process.36 Additionally, replacing the flammable and environmentally impactful organic solvents currently used in LIBs with water is a straightforward way towards sustainable and safe devices.37
Despite the fact that SIBs work in a similar way to LIBs (which could favor the faster development of SIBs by extrapolating and adapting the mature knowledge gained from LIBs), important differences related to the intercalation/deintercalation of these ions are observed, which makes imperative the development of advanced materials with high specific capacity to be used as electrodes in SIBs.33,38,39 For aqueous SIBs, the challenge is even greater, as the intercalation/deintercalation process involves the hydrated cation and the electrochemical window of the aqueous electrolyte is narrow.40,41 Electrode materials based on different nanostructures have been recently prepared for this specific purpose.42–45
The goal of this work is to demonstrate the potential of the LLIR to produce unique tri-component thin-film nanoarchitected materials to be used as high-capacity electrodes in aqueous SIBs. By combining molybdenum disulfide (MoS2), copper oxide nanoparticles (CuxO-NPs) and graphene, synergistic and improved properties arising from the specific nanoarchitecture are obtained, and the resulting thin-film material presents one of the highest specific capacities ever reported for this kind of application. MoS2 is a bidimensional (2D) material with a large interlayer distance (0.62 nm)46,47 and pseudocapacitive properties, which allows surface redox reaction and the reversible intercalation/deintercalation of different ions.48–50 The theoretical specific capacity of MoS2 is 669 mA h g−1,51–53 but even higher values can be reached by controlling its morphology or preparing MoS2-based nanocomposites.45,54,55 The volume changes of MoS2 during ion intercalation/deintercalation (which causes mechanical stress, re-stacking and structure degradation),35,50 as well as its inherently low electronic conductivity,48,49 are characteristics that decrease the experimental capacity of this material, and which can be improved by preparing suitable nanocomposites.56
Copper oxides (Cu2O and CuO) have attracted great attention as electrodes in metal-ion batteries due to their theoretical specific capacity, modular morphology, easy production and low toxicity.57–67 CuO exhibits a higher theoretical specific capacity (674 mA h g−1) compared to Cu2O (369 mA h g−1), but interestingly, some properties of the mixture of these two copper oxides, such as electrochemical reactivity and electrical/ionic conductivity, have been demonstrated to be higher than those of the individual components.68,69 However, Cu2O/CuO presents several drawbacks for use as electrodes in SIBs, including significant volume changes during conversion reactions (over 228%),70,71 low energy density, low power density, and poor stability.72,73 Once again, the preparation of nanocomposites with suitable materials, such as graphene, is one way to overcome these limitations.45,56,74,75
Graphene is a promising material for use as electrodes in metal-ion batteries. The structure of graphene, which is characterized by a one-atom-thick bidimensional material of sp2-hybridized carbon atoms (the stacking of which forms graphite)76–78 can theoretically host metal ions on both sides of the sheet, resulting in a theoretical capacity twice that of graphite (744 vs. 372 mA h g−1, respectively).78 Thus, preparing nanocomposites comprising graphene, MoS2 and CuxO nanoparticles could result in a material in which the three components are expected to contribute to the electrode capacity, and the interactions among them can result in synergistic properties to further increase this capacity.
We recently demonstrated the preparation of thin and transparent bi-component films between MoS2/carbon nanotubes and graphene/CuxO nanoparticles through the LLIR.56,75 Herein, unprecedented tri-component nanoarchitected materials combining MoS2, graphene and CuxO nanoparticles are introduced. The huge potential of the LLIR to prepare these complexes and multi-component materials is demonstrated for three different samples based on three different experimental approaches, which results in different nanoarchitectures and different properties. Finally, the use of these films as anodes in aqueous SIBs is demonstrated, and the results are among the highest performances ever reported for this kind of application.
2. Methodology
2.1. Chemicals
Ammonium molybdate ((NH4)6Mo7O24·4H2O, Vetec), ammonium sulfide ((NH4)2S, solution 20%, Vetec), sulfuric acid (H2SO4, 98%, Anidrol), acetonitrile (gradient grade for liquid chromatography, 99.9% LiChrosolv, Merck), graphite (90%, Graflake, Nacional de Grafite), potassium permanganate (KMnO4, synth, 99%), hydrochloric acid (HCl, 37% Panreac), sodium nitrate (NaNO3, Vetec), hydrogen peroxide aqueous solution (H2O2, 30%, Vetec), toluene (99.9% Sigma Aldrich or 99% Neon), copper nitrate (Cu(NO3)2, Vetec), sodium borohydride (NaBH4, 98%, Acros Organic), and sodium chloride (NaCl, Neon) were used as received. The solutions and dispersions were prepared with deionized water using a Milli-Q ultra-pure water purification system with R = 18.2 MΩ cm.2.2. Synthesis, exfoliation and film preparation of MoS2
MoS2 was synthesized and exfoliated according our previous report.79 The thin films of neat MoS2 were prepared through the LLIR according the procedure summarized in Fig. 1a:79 5 mL of an acetonitrile dispersion of MoS2 (0.14 mg mL−1) was added to a 50 mL round-bottom flask and maintained under magnetic stirring at 2500 rpm. Then, 20 mL of ultrapure water and 20 mL of toluene were added to the system, and the resulting biphasic system was magnetically stirred for 12 h. After interruption of the stirring, a homogeneous, continuous and self-assembled film of MoS2 was obtained at the liquid–liquid interface. The film was transferred to a glass Becker containing the substrate (glass, quartz, Si, FTO/glass electrodes) at the bottom and deposited by pulling the substrates across the liquid/liquid interface. The film was dried at 200 °C for 4 h before characterization.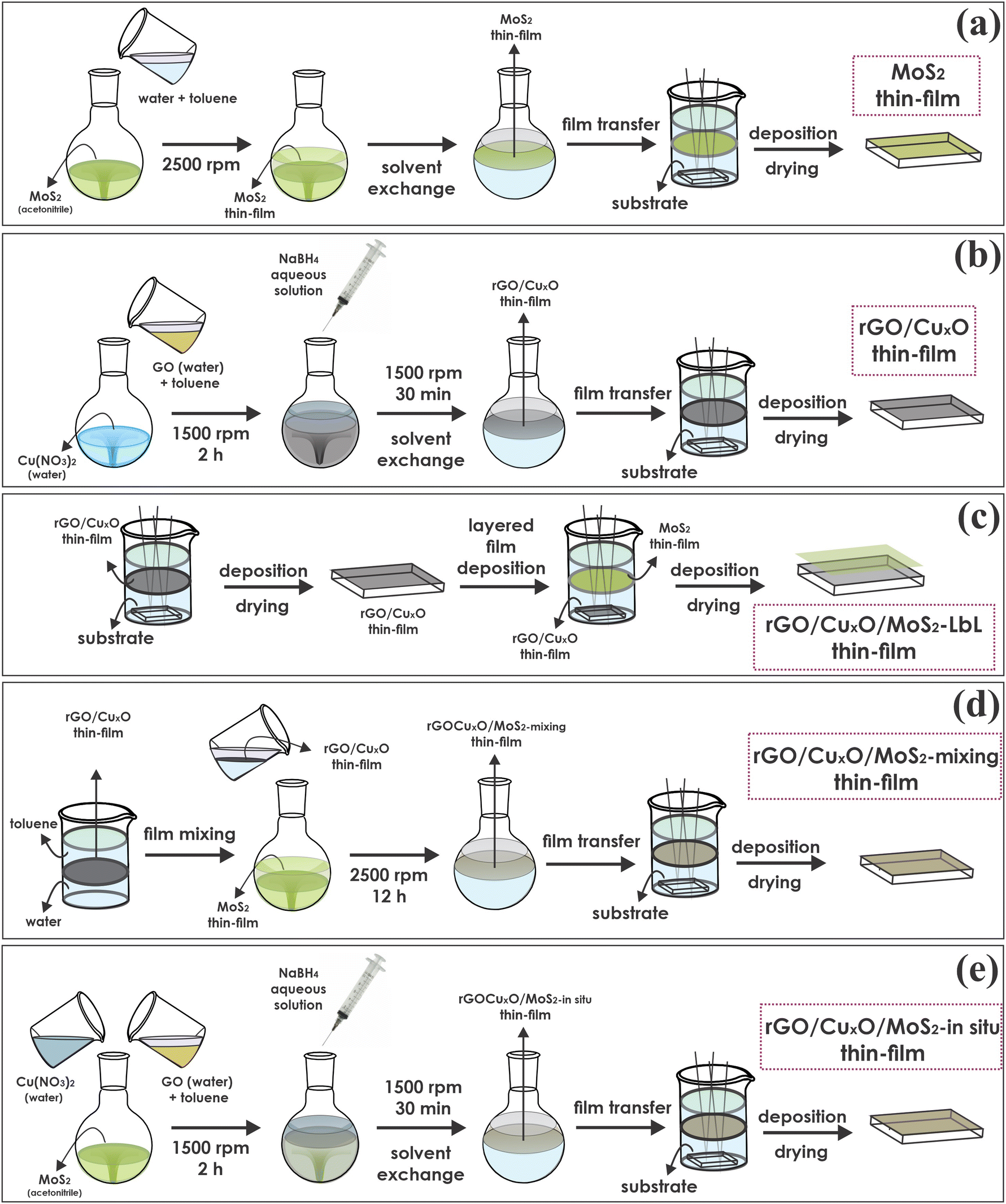 | ||
| Fig. 1 Schematic representation of the general steps for the thin film preparation: (a) MoS2; (b) rGO/CuxO or rGO; (c) rGO/MoS2-LbL and rGO/CuxO/MoS2-LbL; (d) rGO/MoS2-mixing and rGO/CuxO/MoS2-mixing; (e) rGO/MoS2-in situ and rGO/CuxO/MoS2-in situ. | ||
2.3. Preparation of the graphene/CuxO nanoparticles thin films
The graphene/copper oxide nanoparticles thin films were prepared according a methodology previously reported by us, as summarized in Fig. 1b.75 The method involves the simultaneous reduction of graphene oxide (GO) and Cu2+ in a water/toluene biphasic system in air. GO was prepared using a modified Hummers’ method.12 Then, 8 mL of a 0.3 mg mL−1 Cu(NO3)2 aqueous solution, 4 mL of an aqueous dispersion of GO (0.15 mg mL−1) and 18 mL of water were mixed in a 50 mL round bottom flask, resulting in a total volume of 30 mL for the aqueous phase. Afterwards, 20 mL of toluene was added to the flask to obtain the liquid–liquid interface. The entire system was stirred for two hours at 1500 rpm. After this time, 3 mL of an aqueous NaBH4 solution (40 mg mL−1) were added under stirring, and the reaction was performed for 30 minutes. Subsequently, the stirring was turned off, and a dark gray thin film was observed at the liquid–liquid interface, which was characterized by reduced graphene oxide/CuxO nanoparticles (hereafter referred as rGO/CuxO thin film). The aqueous phase was exchanged for fresh ultra-pure water (by removing the aqueous phase using a Pasteur pipette and adding a novel portion of 30 mL of water), and the system was stirred for a few minutes. This procedure was done to remove any soluble impurities remaining in the system, and it was repeated 10 times. Subsequently, the same procedure was carried out with toluene. As a control, the same procedure was also performed without copper nitrate, aiming for the preparation of a neat rGO film. Both films were deposited over suitable substrates as described earlier in Section 2.2.2.4. Thin films of nanocomposites comprising rGO, CuxO nanoparticles and MoS2
Starting from the LLIR, three different approaches were adopted to prepare the tri-component films, as described in the following sections and schematically represented in Fig. 1c, d and e. The strategies will be referred to here as the layer-by-layer (Fig. 1c), mixing (Fig. 1d) and in situ synthesized (Fig. 1e) strategies.2.5. Characterization
X-ray diffraction (XRD) was performed using a Shimadzu diffractometer (XRD-6000) with CuKα radiation (λ = 1.5418 Å) and a thin-film accessory. Raman spectra were acquired using a confocal Raman spectrometer (WITec Alpha 300R) with a 532 nm laser line at 0.9 mW of power, with 30 accumulations of 10 s of acquisition time. UV-Vis spectra were obtained using a Shimadzu UV-2450 spectrophotometer directly on the films deposited on quartz substrates, using air as a reference. The scanning electron microscopy (SEM) images were obtained using a Mira FEG-SEM (Tescan) with an accelerating voltage of 10 kV coupled to an EDS detector (Oxford Instruments) for elemental analysis. For the transmission electronic microscopy (TEM) images, the films were deposited over copper grids covered with a hollow carbon thin film, and the images were collected using a JEOL 120 kV instrument at 100 kV. High-resolution transmission electronic microscopy (HRTEM) images were acquired using a JEOL JEM F200 device with a resolution of 0.1 nm, and the EDS-type elemental chemical analysis was obtained using an SDD detector with an area of 100 mm2 in a scanning mode (STEM). Electrochemical measurements were performed with an Autolab potentiostat operated via GPES and NOVA 1.11 software, using a conventional three-electrode cell with a Pt wire as a counter electrode, Ag/AgCl (3.0 mol L−1) as the reference electrode and the thin films deposited over ITO as the working electrode. Cyclic voltammetry (CV) was performed from −0.2 V to 0.4 V at different scan rates (5 mV s−1 to 50 mV s−1) in an aqueous solution of 0.1 mol L−1 NaCl. The galvanostatic charge/discharge (GCD) measurements were performed using NaCl aqueous solution (0.1 mol L−1) as the electrolyte in the range of −0.2 V to 0.4 V (vs. Ag/AgCl) at five current densities (0.1, 0.25, 0.5, 0.75, 1 and 2 A g−1), and evaluation of the materials for 200 or 1000 consecutive cycles was performed at a current density of 2 A g−1. The cyclic voltammograms were integrated to obtain the approximate electroactive mass of the components in each film: MoS2 (9.35 × 10−8 g cm−2), rGO/CuxO (3.41 × 10−7 g cm−2), rGO/MoS2-LbL (5.19 × 10−8 g cm−2), rGO/CuxO/MoS2-LbL (2.89 × 10−7 g cm−2), rGO/MoS2-mixing (5.07 × 10−8 g cm−2), rGO/CuxO/MoS2-mixing (1.58 × 10−7 g cm−2), rGO/MoS2-in situ (5.39 × 10−8 g cm−2) and rGO/CuxO/MoS2-in situ (2.06 × 10−7 g cm−2). Electrochemical impedance spectroscopy (EIS) was performed in 0.1 mol L−1 aqueous NaCl solution at −0.1 V with an amplitude of 10 mV and frequency of 10 MHz to 10 kHz. Electrochemical quartz crystal microbalance (EQCM) measurements were performed using an Autolab 128n in EQCM mode and the software nova 2.15, with a gold circular electrode (3 mm radius) coupled to a quartz crystal, a Pt wire as a counter electrode, and Ag/AgCl (3.0 mol L−1) as the reference electrode, in 0.1 mol L−1 aqueous solution of NaCl in the range of −1.0 to 0.6 V at 10 mV s−1.3. Results
The main challenge presented in this work was to obtain the unprecedented tri-component materials directly processed as transparent films, and to demonstrate their efficiency as electrodes in aqueous SIBs. Beyond describing a nanoarchitected material that had never been reported before, this work also demonstrates the potential and broadness of the LLIR to achieve these sophisticated materials in different nanoarchitectures, which will result in different properties, as will be demonstrated in the following.Nine different films have been prepared, consisting of three control samples (MoS2, rGO, and rGO/CuxO), along with six nanocomposites generated through three different approaches. The first strategy involved producing the films separately and subsequently depositing the most conductive film (rGO or rGO/CuxO) onto the substrate, followed by the deposition of the MoS2 film above the previous one, in a layer-by-layer (LbL) method (samples rGO/MoS2-LbL and rGO/CuxO/MoS2-LbL). The second method involved the preparation of each individual film at a liquid–liquid interface, followed by the combination (mixing) of those films at the liquid–liquid interface before deposition over the substrates, resulting in the samples rGO/MoS2-mixing and rGO/CuxO/MoS2-mixing. Finally, the third method involved the chemical synthesis of the rGO/CuxO in-situ at the liquid–liquid interface in the presence of a MoS2 dispersion in a one-step approach, resulting in the rGO/MoS2-in situ and rGO/CuxO/MoS2-in situ films.
Digital photographs of the films deposited over planar quartz substrates are shown in Fig. 2a. The high homogeneity, transparency, and optical quality of the films are evident. The MoS2 film has a characteristic orange-yellowish hue, while the rGO and rGO/CuxO films appear gray. Due to the multilayer deposition method, darker films were obtained for the layer-by-layer films, compared to the those synthesized using the mixing and in situ strategies.
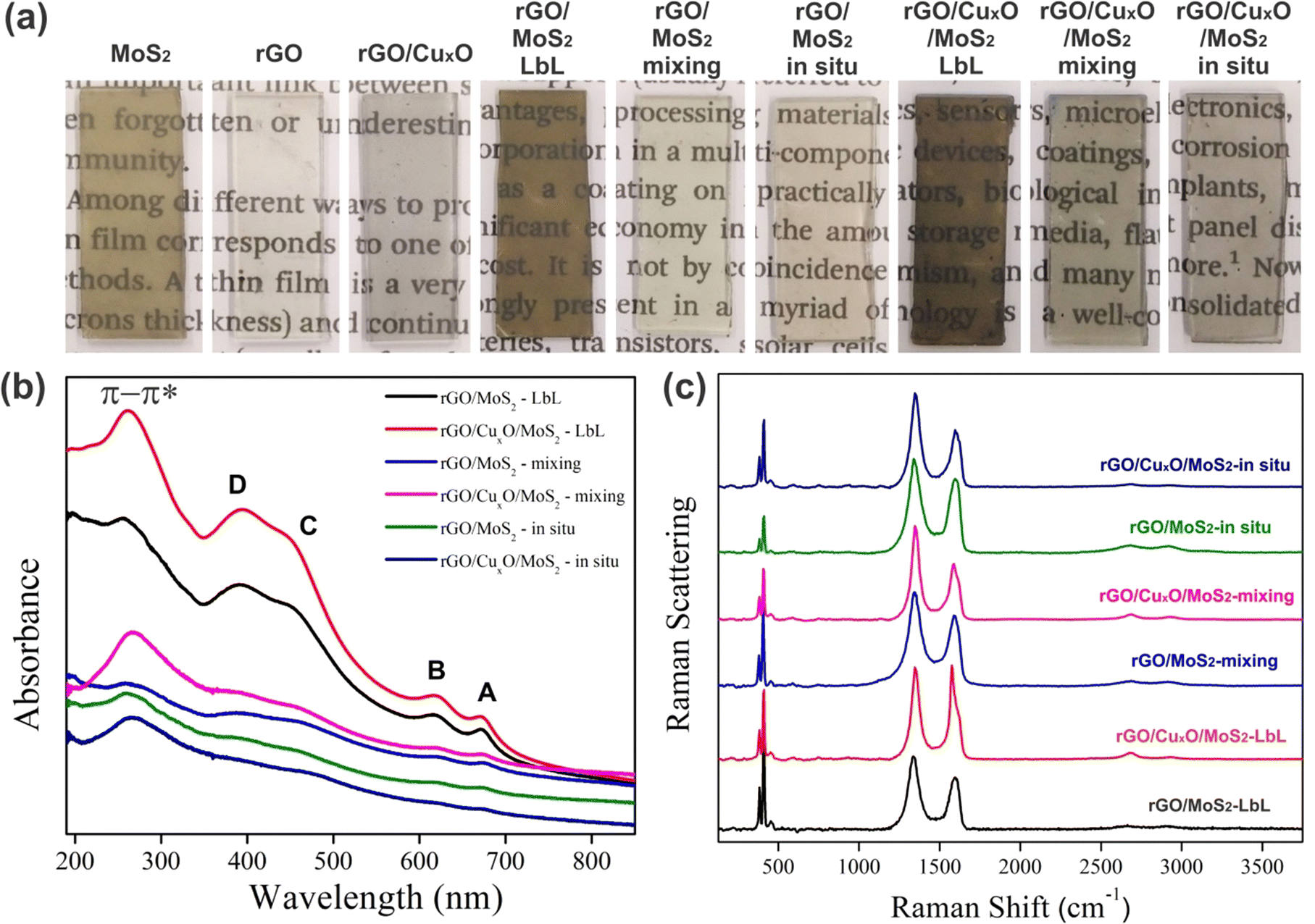 | ||
| Fig. 2 Digital images of the films deposited over glass (a); UV-Vis (b) and Raman (c) (λ = 532 nm) spectra of the different nanocomposites. | ||
Fig. 2b and Fig. S1a (ESI†) present the UV-Vis spectra of the samples deposited over planar quartz substrates. The main absorption bands are detailed in Table S1 (ESI†). The MoS2 spectrum (Fig. S1a, ESI†) presents the characteristic A and B bands at 673 nm and 621 nm, respectively, due to excitonic transitions in the valence band related to excitons at point K of the Brillouin zone, and the bands C and D at 453 nm and 392 nm due to the excitonic transitions from the valence to the conduction band of the semiconductor.80–82 The spectrum of rGO/CuxO (Fig. S1a, ESI†) shows a single band at 270 nm attributed to the π–π* transition in graphene,75,83 confirming the reduction of GO to rGO in this sample. This band is also observed in the rGO sample at 259 nm. It is well known that the energy of the π–π* transition in rGO samples is dependent on the effectiveness of the GO to rGO reduction (which is directly related to the sp2 backbone restoration), and occurs at lower energy for the most-reduced samples.84,85 The occurrence of this band at 259 nm for rGO and 270 nm for rGO/CuxO indicates that the GO to rGO reduction was more effective in the rGO/CuxO sample compared to in the neat rGO. Furthermore, another band at 305 nm can be seen in the neat rGO spectrum (Fig. S1, ESI†) due to the n–π* transitions involving residual oxygenated functional groups that were not fully removed during the reaction, which provides further evidence that the GO to rGO reduction was not as effective in the neat rGO reaction as in the rGO/CuxO one.86 Taking into account the fact that the amounts of GO and NaBH4 are exactly the same in both reactions, as well as the fact that the reducing agent was shared with the Cu2+ cations during the synthesis of rGO/CuxO (meaning less reducing agent available to GO), we would expect the opposite. This apparent paradox can be explained by considering that the nascent CuxO nanoparticles catalyze the GO to rGO reduction, promoting more-reduced rGO after 30 min of reaction. The difference in the energy of the UV-Vis band was not observed for the same reactions conducted for 24 h (data not shown), corroborating this hypothesis. No bands due to the CuxO nanoparticles were detected in the rGO/CuxO spectrum.
The spectra of the nanocomposites (Fig. 2b) show all the MoS2 and graphene bands described earlier, corroborating the presence of both components in the samples. Some shifts in the MoS2 bands were observed in the spectra of the in situ synthesized samples (Table S1, ESI†), mainly in the band-gap-related C and D bands, which may be indicative of changes in the electronic structure of MoS2 due to the interactions among the components.
The Raman spectra of the films are presented in Fig. 2c and Fig. S1b, S2 (ESI†), and a summary of the main features is presented in Table S2 (ESI†). The MoS2 film (Fig. S1b, ESI†) presents two characteristic bands at 384 and 412 cm−1 due to the E12g and A1g modes, respectively.87–92 The graphene bands are readily observable in the rGO and rGO/CuxO samples:93–100 the G band at 1588 cm−1 (due to the E2g mode); the D band at 1352 cm−1 (resulting from a symmetry break due to structural defects or partially disordered graphitic domains); the D′ band at 1620 cm−1 (also due to disorder or defects in the structure, usually observed in well-reduced rGO samples in which the sp2 structure was efficiently rebuilt after the reduction process); the 2D band at 2683 cm−1 (attributed to the structural organization of graphene in the two-dimensional plane); and the D + G band at 2940 cm−1 (a combinatory mode). Comparing the rGO and rGO/CuxO spectra, there is a slight shift in the band positions (Table S2, ESI†), the bands are narrower in the rGO/CuxO spectrum, and the D′ band does not appear in the rGO spectrum. All these features are spectral evidence that the rGO is more reduced in the rGO/CuxO than in the rGO films,87,95–97 corroborating the data obtained from UV-Vis spectroscopy.
All the MoS2 and graphene bands discussed earlier can be found in the spectra of the nanocomposites presented in Fig. 2c and Fig. S2 (ESI†), including the evidence for more-reduced rGO in the samples containing the CuxO nanoparticles. The ratio between the intensities of the D and G band (the ID/IG ratio) was calculated (Table S2, ESI†), and a significant increase in ID/IG was observed for all the CuxO-containing materials, as expected for more effectively reduced rGO.92,98 The chemical interactions among the components can also be responsible for the increase in the ID/IG ratio observed for the nanocomposite samples.94,99–101
Fig. S3a (ESI†) shows the X-ray diffraction patterns of the thin films. MoS2 and all the MoS2-containing films present an intense peak due to the (002) planes of 2H-MoS2 at 6.15 Å (JCPDS 37–1492), which is enlarged in comparison to that of bulk MoS2, evidencing the presence of few-layer MoS2.102 The other two less-intense peaks of MoS2 expected to be seen in this range (specifically, the (100) and (103) peaks) are not observable in the patterns presented in Fig. S3a (ESI†) due to the specificity of the MoS2 sample used in this work (characterized as few-layer, polycrystalline and exhibiting a small grain size of 64 nm × 42 nm)79 and due to the data being collected directly from the thin films using a grazing angle accessory. A large and low-intensity signal at 3.85 Å can be seen in the profile of the rGO and rGO/CuxO films due to the irregular stacking of the rGO sheets as a result of their re-organization in the solid state after the reduction process.103 The copper oxide nanoparticle signal appears between 25 and 45° (in 2θ). An expanded view of this region can be seen in Fig. S3b (ESI†), demonstrating that the samples are constituted of a mixture of CuO and Cu2O.75
In order to identify the nature of the copper oxide produced in the rGO/CuxO/MoS2-in situ tri-component sample, several syntheses were carried out, and the film was collected from the liquid–liquid interface with the aim of accumulating material to acquire a conventional X-ray diffractogram profile. The result is shown in Fig. S3c (ESI†); the material is characterized by the presence of the (002) peak of MoS2 at d = 6.15 Å (JCPDS 37–1492); the broader and poorly defined (002) peak of the rGO at 3.97 Å; three peaks attributed to Cu2O at 3.01 Å (110), 2.46 Å (111) and 2.13 Å (200) (JCPDS 74-1230); and four peaks due to CuO at 2.75 Å (110), 2.52 Å (111), 2.32 Å (200) and 1.87 Å (202) (JCPDS 72-0629). It is interesting to note that although the rGO/CuxO and rGO/CuxO/MoS2-in situ samples were prepared in exactly the same way (except, of course, that the latter was prepared in the presence of dispersed MoS2), the former has predominantly CuO and the latter a mixture of Cu2O (predominant) and CuO. These findings suggest that MoS2 is not just a spectator during the synthetic procedure, and someway affects the reaction pathway.
Fig. 3a–i (and Fig. S4–S8, ESI†) show the scanning electron microscopy images of the films. The MoS2 thin film (Fig. 3a and Fig. S4a–c, ESI†) is composed of a continuous network of small platelets (average size of 64 nm × 42 nm),56,79 while rGO (Fig. 3b and Fig. S4d–f, ESI†) consists of large interconnected graphene sheets (∼1 μm × 500 nm) with clear edges and folds.93 Notably, this rGO morphology can also be seen in the sample rGO/CuxO (Fig. 3c and Fig. S4g–i, ESI†), together with cubic-shaped CuxO nanoparticles75 (average lateral size of 120 nm) distributed over, between and wrapped by the graphene sheets. These three morphological profiles appear on different nanoarchitectures in the bi- and tri-component nanocomposite films: (i) in both rGO/MoS2-LbL (Fig. 3d and Fig. S5a–c, ESI†) and rGO/CuxO/MoS2-LbL (Fig. 3g and Fig. S5d–f, ESI†), the morphology observed is consistent with films that have been sequentially deposited, one on top of another, in which the morphology of individual films can be detected; (ii) in contrast to the –LbL samples, both the rGO/MoS2-mixing (Fig. 3e and Fig. S6a–c, ESI†) and rGO/CuxO/MoS2-mixing (Fig. 3h and S6d–e) films exhibit closer interaction among the components. The MoS2 plates are sandwiched between graphene sheets, creating a more intimate contact between the materials; (iii) a similar nanoarchitecture was observed in both the rGO/MoS2-in situ (Fig. 3f and Fig. S7a–c, ESI†) and rGO/CuxO/MoS2-in situ (Fig. 3i and Fig. S7d–f, ESI†) films, which were characterized by very close and intimate contact among the three components, but the CuxO NPs were larger in size (approximately ∼200 nm), demonstrating that the presence of MoS2 during the synthesis also affects the final particle size. Images using a backscattered electron (BSE) detector were collected for the rGO/CuxO/MoS2-in situ sample (Fig. S8, ESI†). In the BSE images, the regions containing heavy elements (high Z values) appear brighter, facilitating the identification of the large amount and good distribution of copper oxide nanoparticles and MoS2 nanoplatelets.
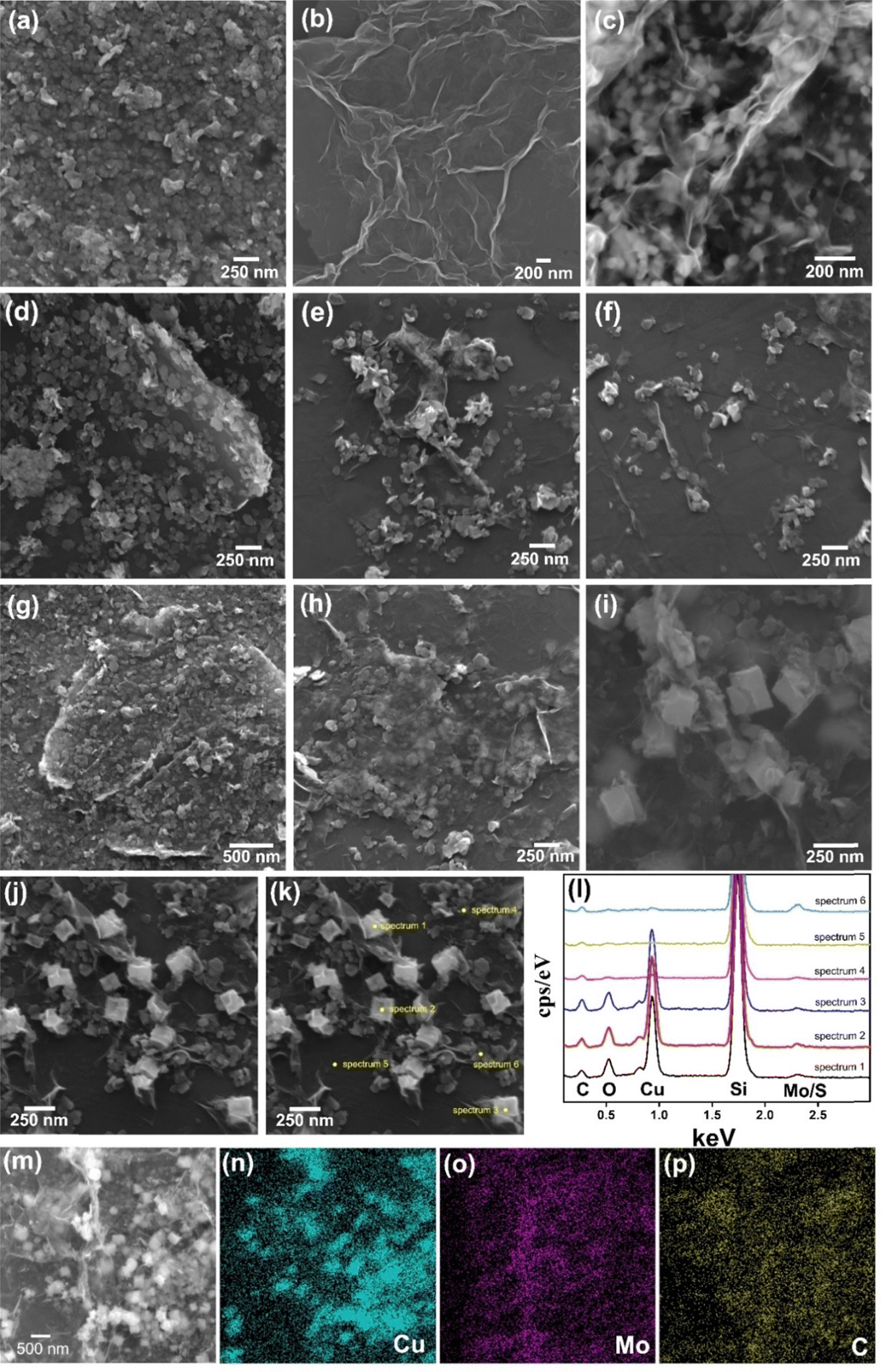 | ||
| Fig. 3 Scanning electron microscopy images of MoS2 (a); rGO (b); rGO/CuxO (c); rGO/MoS2-LbL (d); rGO/MoS2-mixing (e); rGO/MoS2-in situ (f); rGO/CuxO/MoS2-LbL (g); rGO/CuxO/MoS2-mixing (h); and rGO/CuxO/MoS2-in situ (i). Energy dispersive spectroscopy data (j–p) of rGO/CuxO/MoS2-in situ, including point analysis (j–l) and mapping (m–p). | ||
Energy dispersive spectroscopy (EDS) analysis (Fig. 3j–p and Fig. S9, ESI†) provided information on the elemental composition of the materials. The survey spectra of all the nanocomposite films (Fig. S9, ESI†) show the presence of C, Mo, and S (the Si signal is from the substrate). In addition, Cu and O were also detected in the samples containing CuxO nanoparticles, as expected. Fig. 3j–l present the spatially resolved spectra, and Fig. 3m–p show the elemental mapping obtained for the rGO/CuxO/MoS2-in situ sample. Analysis of these data revealed that the cubic particles of this sample are composed of Cu2O/CuO, as shown by the intense Cu and O signals in the point analysis spectra of these cubes (Fig. 3l) and by the Cu mapping (Fig. 3n). The MoS2 plates were found to be homogeneously distributed throughout the sample (Fig. 3o).
Fig. 4 and Fig. S10–S16 (ESI†) show the transmission electron microscopy (TEM) images collected for the samples rGO, MoS2, rGO/CuxO, rGO/MoS2-in situ and rGO/CuxO/MoS2-in situ. Graphene (Fig. S10, ESI†) has large, flat sheets with small layers, as well as some characteristic folds. On the other hand, MoS2 (Fig. S11, ESI†) presents a very small two-dimensional plate (64 nm × 42 nm, as previously shown by SEM), structure with the stacking of a few layers (3 to 10 layers, Fig. S12, ESI†). The rGO/MoS2-in situ sample (Fig. 4a–b and Fig. S13, ESI†) has MoS2 plates homogeneously distributed across large graphene sheets. The rGO/CuxO (Fig. 4c–f and Fig. S14, ESI†) sample, in addition to the cubic morphology seen via SEM in regions with large amounts of particles, also shows very small (∼10 nm) and mostly spherical particles distributed throughout the graphene sheets. The presence of CuO (with a crystallographic plane d200 = 0.32 nm) and Cu2O (with a crystallographic plane d111 = 0.24 nm) in rGO/CuxO was confirmed through high-resolution images and FFT (fast Fourier transform) analysis, as shown in Fig. S15 (ESI†). EDS analysis obtained using STEM (Fig. 4g–j) of the rGO/CuxO sample show Cu and O on the darker and geometric-shape particles in Fig. 4g, again corroborating the CuxO composition of those nanoparticles, and a homogeneous distribution of C matching the graphene-like morphology.
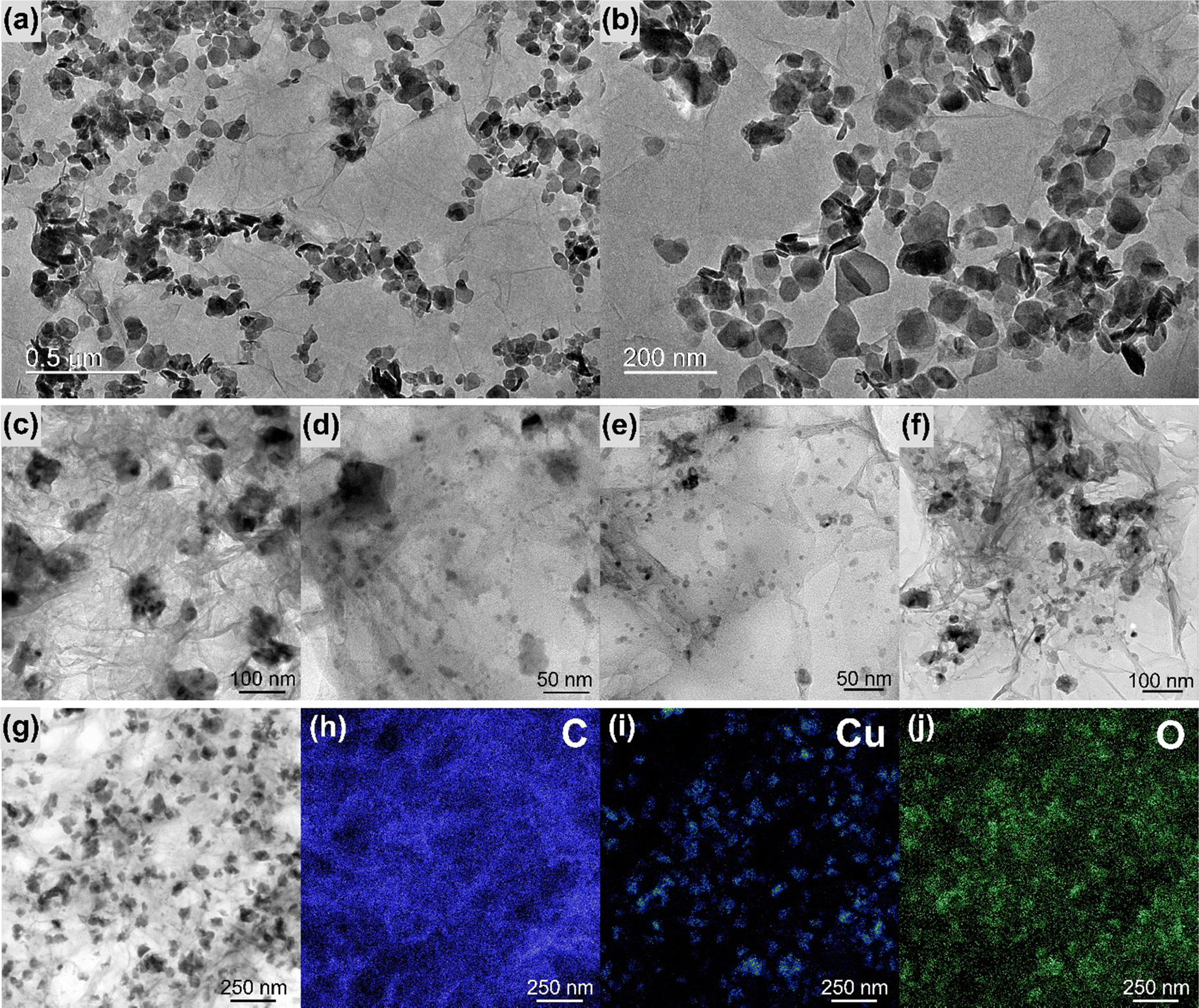 | ||
| Fig. 4 Transmission electron microscopy images of the samples rGO/MoS2 (a) and (b) and rGO/CuxO (c)–(f). Energy dispersive spectroscopy mapping of rGO/CuxO (g)–(j). | ||
The TEM and HRTEM images of the sample rGO/CuxO/MoS2-in situ (Fig. 5 and Fig. S16, ESI†) reveal details regarding the nanoarchitected way in which the three components are organized. As can be seen, the film is supported by interconnected graphene sheets on which the MoS2 and the CuxO NPs are homogeneously distributed (Fig. 5a, b and Fig. S16, ESI†). Looking closer, as in the two different regions illustrated in Fig. 5c and d, it can be noted that the three components are in very intimate contact with no segregation or predominance of individual components. This finding highlights the hypothesis that graphene and MoS2 can act as nucleation and growth sites for the CuxO nanoparticles. Looking even closer (Fig. 5e and f), it can be seen that in addition to the cubic-shaped ones, the sample is full of very small CuxO nanoparticles (1 to 10 nm) spread over the entire film. Fig. 5f elucidates the intimate contact between these small CuxO nanoparticles, the MoS2 (the fringes of the few-layer MoS2 can be detected mixed with the CuxO NPs) and the graphene.
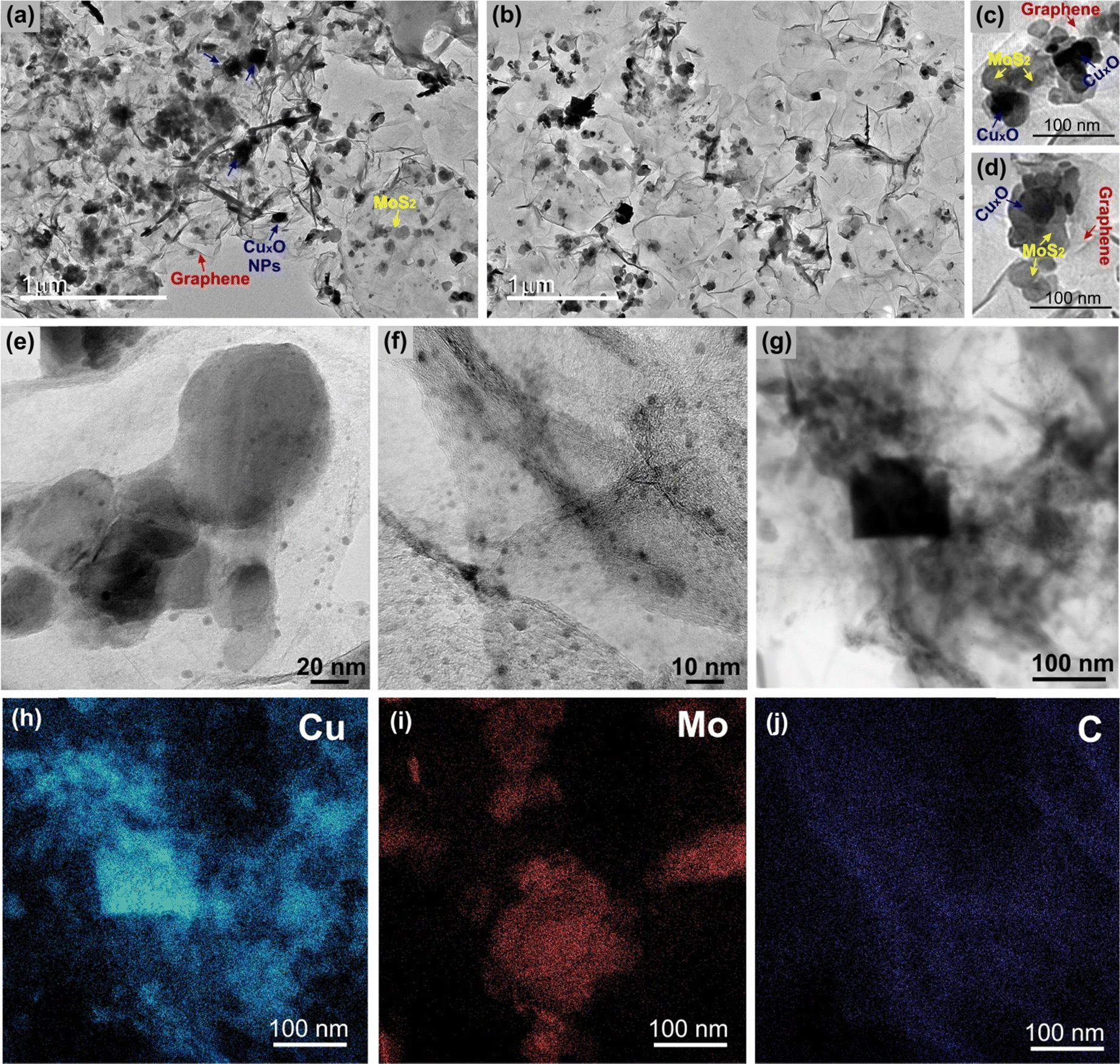 | ||
| Fig. 5 Transmission electron microscopy images (a)–(g) and energy dispersive spectroscopy mapping images (h)–(j) of the rGO/CuxO/MoS2-in situ sample. | ||
The EDS elemental mapping (Fig. 5h–j) of the region shown in Fig. 5g clearly shows the distribution of the elements and corroborates the identification of each component in the film.
The electrochemical performance of the nanocomposites deposited over ITO/glass was investigated using cyclic voltammetry (CV), galvanostatic charge/discharge curves (GCD), an electrochemical quartz crystal microbalance (EQCM) and electrochemical impedance spectroscopy (EIS), carried out in NaCl aqueous solution as electrolyte. Fig. 6 shows the CVs of the nanocomposites and the control samples at 5 mV s−1 within the potential range of −0.2 V to 0.4 V in an aqueous solution of 0.1 mol L−1 NaCl. Fig. S17 (ESI†) shows the CV data at different scan speeds (5 to 50 mV s−1). Pseudo-capacitive behavior was observed in the MoS2 thin-film (Fig. 6a), with a redox pair at an E1/2 of 0.14 V. Two mechanisms are predicted in the literature to explain this profile:104–106 the first one describes the pseudo-capacitive behavior from the faradaic process in which Na+ ions are intercalated into the lamellar structure of MoS2 (eqn (1)), and the second one is related to the non-faradaic process due to the formation of an electrical double layer due to the adsorption of cations onto the MoS2 surface.
| MoS2 + xNa+ + xe− ↔ NaxMoS2 (x < 1) | (1) |
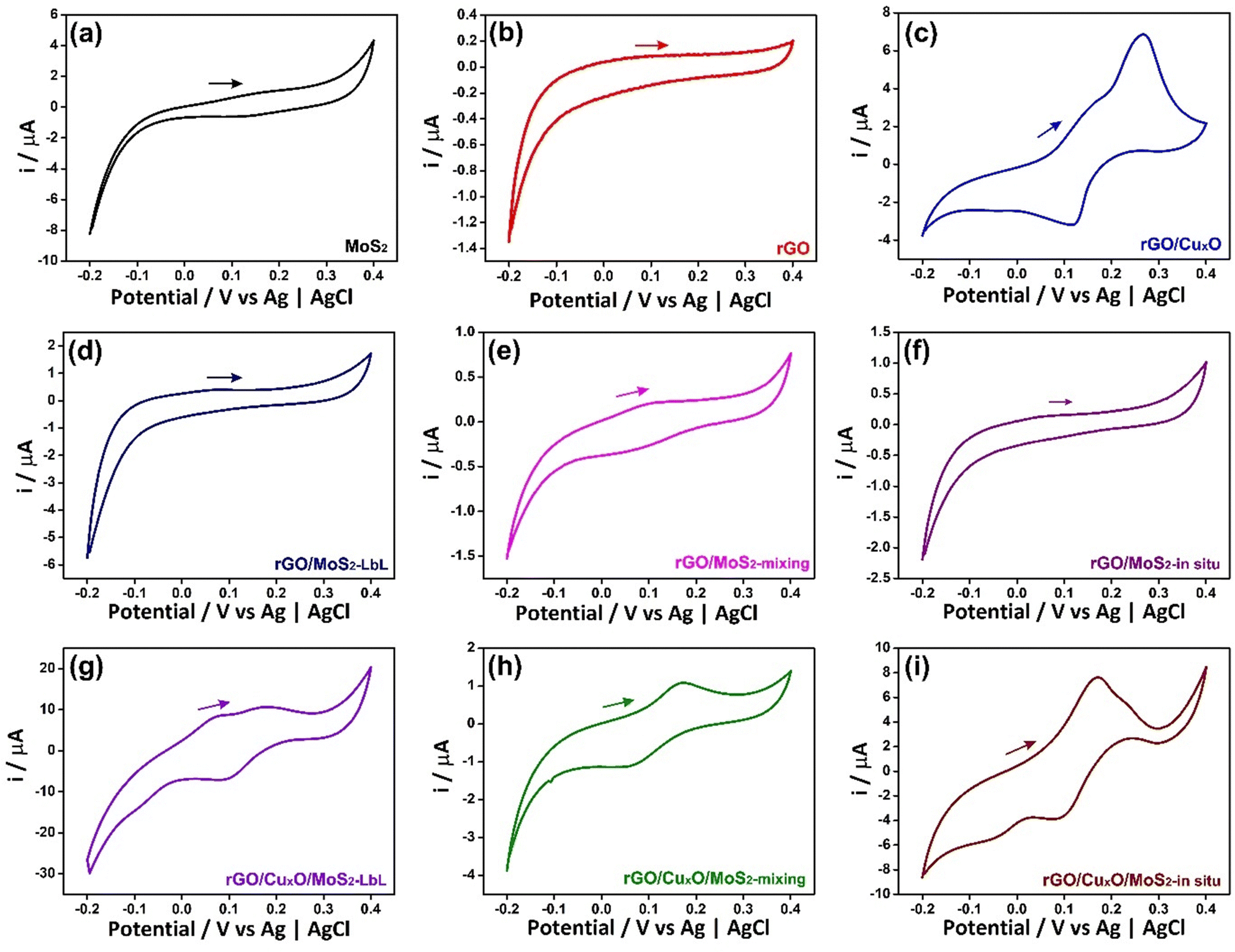 | ||
| Fig. 6 Cyclic voltammograms of the thin films: (a) MoS2; (b) rGO; (c) rGO/CuxO; (d) rGO/MoS2-LbL; (e) rGO/MoS2-mixing; (f) rGO/MoS2-in situ; (g) rGO/CuxO/MoS2-LbL; (h) rGO/CuxO/MoS2-mixing; (i) rGO/CuxO/MoS2-in situ. Electrolyte: 0.1 mol L−1 NaCl aqueous solution. Scan speed of 50 mV s−1. | ||
The CV profile of rGO is typical of a capacitive material related to the occurrence of an electric double-layer, as can be seen in Fig. 6b. The nanocomposites between rGO and MoS2 exhibit similar behavior, showing a faradaic process at E1/2 = 0.1 V (attributed to MoS2) and a rectangular profile coherent with the rGO. The faradaic process is more defined in the CV curve of rGO/MoS2-mixing and occurs at lower potential than in the other samples (E1/2 = 0.06 V) due to the improved contact between the two components, as described earlier.
The cyclic voltammogram of rGO/CuxO exhibits two well-defined redox pairs, a predominant one at E1/2 = 0.19 V (0.26 V/0.12 V), corresponding to the Cu+/Cu2+ process, and a less-intense one at 0.16 V/−0.05 V, attributed to the Cu+/Cu2+ oxidation pair of small copper oxide nanoparticles with different species/sizes/shapes in the film. As previously described, this sample contains two species of copper oxide, with CuO being present in greater quantity and Cu2O in smaller quantity. These species have different sizes and shapes that are consistent with the processes observed in the film. Furthermore, CV experiments were conducted on rGO/CuxO in different electrolytes (aqueous solutions of NaCl, NaNO3, and Na2SO4), and the CV profile obtained in NaNO3 (Fig. S18, ESI†) indicates several well-defined oxidation processes that are consistent with the presence of Cu2O/CuO nanoparticles. Literature reports of these materials mainly focus on organic electrolytes that are not within our working range. Other systems have been studied in alkaline aqueous electrolytes, and, although the processes may be slightly different, they occur in the same potential range as our work, and these studies have previously demonstrated the Cu+/Cu2+ process in this range.107–109
The tri-component materials present significant differences in their CV profiles due to the different ways in which they are nanoarchitected. The rGO/CuxO/MoS2-LbL sample shows an oxidation process at 0.07 V attributed to MoS2 and another redox pair at 0.2 V/0.09 V attributed to Cu+/Cu2+ processes; the rGO/CuxO/MoS2-mixing film exhibits a single redox process at E1/2 = 0.11 V (0.17 V/0.05 V), which has a profile similar to that of the rGO/MoS2-mixing control sample. This process is attributed to the combined processes of both materials (MoS2/CuxO) due to their close contact, as discussed earlier. The rGO/CuxO/MoS2-in situ sample exhibits two processes: a redox pair at 0.17 V/−0.06 V and another at 0.23 V/0.08 V. These processes are comparable to those observed in the rGO/CuxO control sample and can be attributed to the Cu+/Cu2+ redox process, although the profiles are shifted in comparison to the rGO/CuxO control sample due to the larger amount of Cu2O relative to CuO in this sample. Also, the redox process attributed to MoS2 (eqn (1)) at the same potential should contribute to the observed intensity of the redox pair. The oxidation process becomes noticeably broader as the scanning speed of the cyclic voltammograms increases (Fig. S17I, ESI†), indicating the occurrence of two processes in the same region, possibly originating from CuxO nanoparticles and MoS2.
Fig. S19 (ESI†) displays the Nyquist plots derived from the EIS data of all the nanocomposite films. The rGO film closely resembles an ideal capacitor according to its curve profile and CV measurements; when the system kinetics are straightforward, current limitation occurs due to mass transfer phenomena, resulting in a more-linear and less-distinct semicircular region that corresponds to the sodium diffusion process within the electrode.110–112 In contrast, the neat MoS2 is characterized by an evident semi-circle at high frequency, corresponding to the charge transfer resistance, indicating that the electrochemical process is governed by charge transfer phenomena between the active material and the electrolyte.111 The diameter of the semi-circle is directly proportional to the charge transfer resistance and inversely proportional to the rate of heterogeneous charge transfer. The incorporation of CuxO NPs or MoS2 into graphene to give the bi-component material leads to the appearance of the semi-circle in the high-frequency region, and the tri-component materials exhibit smaller semi-circle diameter. Notably, the rGO/CuxO/MoS2-in situ film possesses the lowest charge transfer resistance, which implies enhanced conductivity.113 This is consistent with the superior specific capacity results of these samples, as will be discussed further.
The electrochemical quartz crystal microbalance (EQCM) is a convenient tool for monitoring mass changes during the electrochemical evaluation of electrode materials. It provides direct information that allows the understanding of various surface processes related to sensors, capacitors, batteries, and other electrochemical devices.114–117Fig. 7 and Fig. S20 (ESI†) show the CV profiles recorded in NaCl 0.1 mol L−1 and the respective EQCM frequency responses recorded simultaneously for the samples MoS2, rGO/CuxO and rGO/CuxO/MoS2-in situ deposited on a gold/quartz electrode. The slight variations observed in the voltametric behavior of these films compared to those presented earlier are due to the working electrode utilized (films deposited over gold/quartz) as well as to the wider potential range selected, which was determined by EQCM stabilization (a feat that was unattainable within a smaller range). In Fig. 7a, the CV obtained for MoS2 reveals two redox pairs with E1/2 values of −0.24 V and 0.25 V, respectively, which are ascribed to the pseudocapacitive processes discussed earlier. Thomas et al.115 presented the electrodeposition technique for obtaining MoS2 using EQCM under different working conditions than those used in this study, but observed a similar electrochemical profile.115 As shown in the EQCM results for MoS2 (Fig. 7a), a slight increase in mass occurs near a potential of 0.2 V (1) during the reduction process, followed by a significant increase starting at −0.3 V (2). During oxidation, the mass begins to decrease at around −0.3 V (3), and another drop is observed at 0.2 V (4). The suggested mechanism, which is schematically represented in eqn (1), is based on Na+ cation intercalation during the reduction of the electrode material, which involves the filling of the electrode with cations, leading to a mass increase. Conversely, cation deintercalation occurs during the oxidation process when Na+ cations are removed from the electrode material, resulting in a decrease in mass.116 This is exactly what can be seen in Fig. 7a for the MoS2 film: the mass variations happen in the same region as the charge changes (redox processes), suggesting an intercalation/deintercalation process within the material. Notably, the deintercalation at 0.2 V (4) is in close proximity to the discharge potential, as discussed in the subsequent GCD analyses.
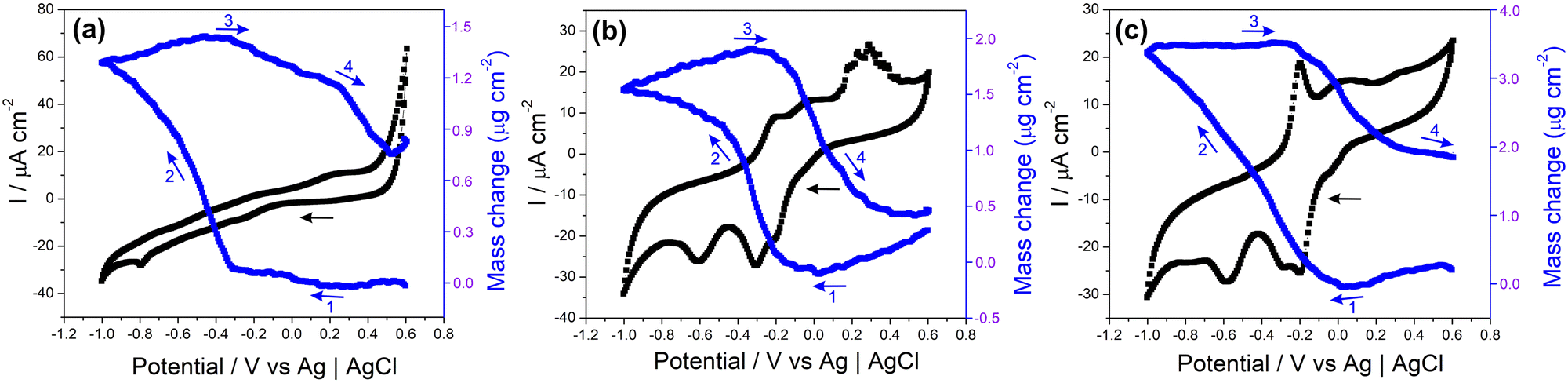 | ||
| Fig. 7 Cyclic voltammograms (black) and evolution of the film mass (blue) during the third cycle (arrows indicate the scanning direction) for the MoS2 (a), rGO/CuxO (b) and rGO/CuxO/MoS2-in situ (c) films. Conditions: Gold/quartz substrate, NaCl electrolyte (0.1 mol L−1) and scan rate 10 mV s−1. | ||
In both the rGO/CuxO (Fig. 7b) and rGO/CuxO/MoS2-in situ (Fig. 7c) samples, there is a similar mass change profile with a steady mass increase starting at 0.0 V (1) and continuing up to −0.4 V for rGO/CuxO and up to −1.0 V for rGO/CuxO/MoS2-in situ (2). In fact, the mass change profile of the tri-component material appears to be a mixture between the profiles of MoS2 (Fig. 7a) and rGO/CuxO (Fig. 7b), indicating the contribution of all the materials through the entire process. During oxidation, the mass starts to decrease near −0.3 V (3) and remains constant up to −0.2 V for rGO/CuxO and 0.6 V for rGO/CuxO/MoS2-in situ (4). Furthermore, the differences in mass obtained during the CV cycles reveal increases in mass of 8.1, 11.3 and 13.5% for the rGO/CuxO, MoS2 and rGO/CuxO/MoS2-in situ films, respectively, indicating that the ternary film has the greatest intercalation capacity for Na+ ions, corroborating the EIS data discussed earlier and supporting the performance of this material as electrodes, as will be discussed next.
The performance of the nanoarchitected thin films as anodes for aqueous sodium-ion batteries was analyzed based on the CV profiles of each sample. GCD curves were obtained in 0.1 mol L−1 aqueous solution of NaCl in the potential window between −0.2 and 0.4 V (Fig. 8). Currents of 0.1, 0.25, 0.5, 0.75, 1 and 2 A g−1 (Fig. S21, ESI†) were applied, and the specific capacities (SC) were calculated (Table 1). Fig. 8a and b displays the GCD profile of the materials operating at a current of 0.1 A g−1. The GCD curves exhibit a plateau between 0.1 to 0.2 V, indicating the intercalation of Na+ in the thin films, as previously shown in the CV curves (Fig. 6) and EQCM measurements (Fig. 7). Fig. 8c and d show the variation of the specific capacity according the number of CD cycles, demonstrating the decrease in the storage capacity of the materials as the current density increases, which is the expected profile for batteries due to the internal resistance of the materials, which limits the speed at which cations can migrate in and out of the structure.
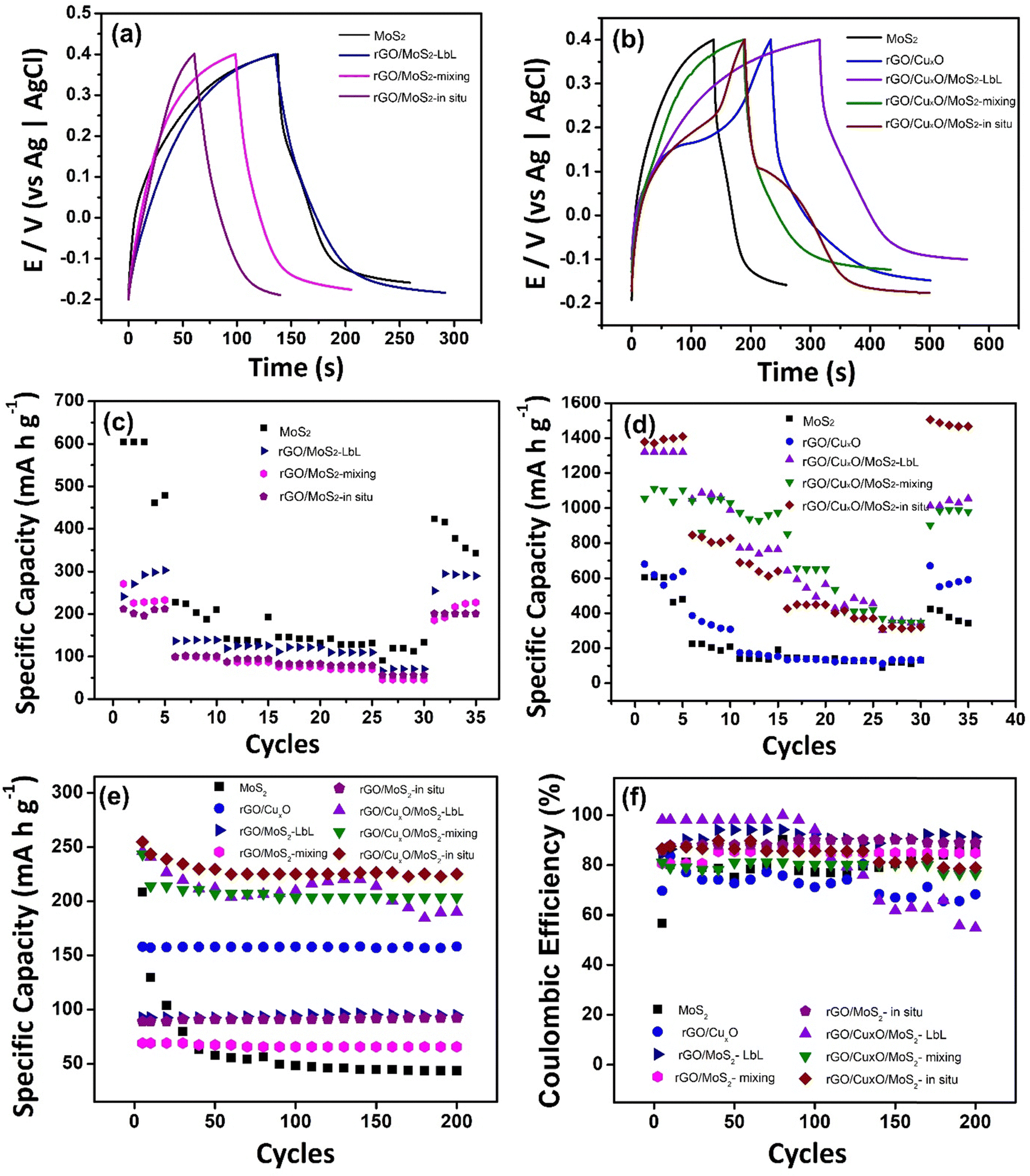 | ||
| Fig. 8 Charge and discharge curves (a) and (b); specific capacity of the discharge process (c) and (d) at different current densities (0.1, 0.25, 0.5, 0.75, 1 and 2 A g−1); specific capacity measured during 200 cycles at 2 A g−1 (e) and Coulombic efficiency (f) of the nanocomposites in 0.1 mol L−1 NaCl aqueous solution and the potential range of -0.2 to 0.4 V. | ||
| Sample | Capacity (mA h g−1) | Retention rate (%) | Coulombic efficiency (%) |
|---|---|---|---|
| MoS2 | 551 | 70 | 88 |
| rGO/CuxO | 620 | 95 | 68 |
| rGO/MoS2-LbL | 280 | 100 | 92 |
| rGO/CuxO/MoS2-LbL | 1321 | 78 | 85 |
| rGO/MoS2-mixing | 238 | 88 | 89 |
| rGO/CuxO/MoS2-mixing | 1056 | 90 | 55 |
| rGO/MoS2-in situ | 206 | 98 | 77 |
| rGO/CuxO/MoS2-in situ | 1377 | 100 | 80 |
The MoS2 film exhibited an impressive SC value (551 mA h g−1) that was significantly higher than the values obtained for this same film in KCl aqueous solution (276 mA h g−1),56 suggesting that hydrated Na+ is more easily intercalated than hydrated K+. This high capacity was not retained in the rGO-based MoS2 binary composites, in which the SC values were 280 mA h g−1 (rGO/MoS2-LbL), 238 mA h g−1 (rGO/MoS2-mixing) and 206 mA h g−1 (rGO/MoS2-in situ). The SC for the pristine rGO film was not determined, as it does not exhibit a redox process. The copper-oxide-nanoparticle–based materials, however, present impressive electrochemical performance, with rGO/CuxO having an SC value of 620 mA h g−1. In addition to the redox process attributed to copper oxide NPs, this result is also explained by the presence of the more-conductive rGO in this sample (due to the more effective reduction of GO to rGO, as discussed earlier), which is expected to improve the electrical double layer. Regarding the nanoarchitected tri-component structures, the findings are particularly noteworthy: (i) rGO/CuxO/MoS2-LbL presents an SC of 1321 mA h g−1, which represents a 240%, 213% and 472% increase compared to those of MoS2, rGO/CuxO and rGO/MoS2-LbL, respectively; (ii) rGO/CuxO/MoS2-mixing presents an SC of 1056 mA h g−1, which is 192%, 170% and 444% higher than those of MoS2, rGO/CuxO and rGO/MoS2-mixing, respectively; (iii) rGO/CuxO/MoS2-in situ presents an impressive SC of 1377 mA h g−1, which represents a 250%, 222% and 668% increase compared to those of MoS2 rGO/CuxO and rGO/MoS2-in situ respectively. These data are summarized in Table 1.
Fig. 8e presents the specific capacity measured during 200 charge/discharge cycles at 2 A g−1. The MoS2 film exhibits instability during the first 50 cycles, which can be attributed to the volume change and structural breakage that occurs in this material during the GCD processes. When MoS2 is combined with rGO or rGO/CuxO, greater stability is observed during cycling. In the films generated using rGO/CuxO, a negligible loss of stability is observed in the initial 10 cycles, which can be ascribed to the adjustment of the CuO/MoS2 in the structure during the process of Na+ intercalation/de-intercalation. The only film that exhibits a high loss of stability during cycling is the rGO/CuxO/MoS2-LbL film, in which the contact among the components is not as close as in the other films. The coulombic efficiency of the materials, as determined from 200 charge/discharge cycles (as presented in Fig. 8f and Table 1), ranges between 70% and 90%. The rGO/CuxO/MoS2-LbL film is an exception, showing low stability as previously discussed. The performance of the rGO/CuxO/MoS2-in situ thin film was followed over 1000 consecutive charge/discharge cycles at elevated current (Fig.S22, ESI†), and it demonstrated a stability of 50%.
The SC values of the tri-component films obtained in this study are comparable to the remarkable values reported in the literature. These results are attributed to several factors: (i) the high charge storage capacity of the three materials used in this study; (ii) the improved conductivity achieved through the use of reduced and better-structured graphene; (iii) the formation of nanocomposites that prevent the restacking of 2D materials and accommodate the volume changes of the MoS2 and CuxO nanoparticles; (iv) the employment of nanoarchitected structures that possess high surface area and homogeneity; (v) the intrinsic characteristics of each of the tri-component structures obtained in the samples, e.g., the small flakes of the few-layer MoS2, the coexistence of Cu2O/CuO, and the very small CuxO nanoparticles dispersed through the entire sample; and (vi) the synergistic effect among the three components due to the intimate contact among them, which is directly dependent on the preparation method. In the best sample, rGO/CuxO/MoS2-in situ, each of the three components is in intimate contact as a result of the nanoarchitected way in which they are structured. The graphene sheets contribute to the overall performance, allowing cation adsorption and playing a crucial role in enhancing the conductivity (as observed by EIS) and the resistance to mechanical stress of both MoS2 and CuxO NPs during cycling. The insertion of Na+ into the electrode occurs through the interaction with the three components, while the excellent electrical conductivity of graphene facilitates efficient charge transfer during the intercalation process, contributing to the overall performance and stability of the electrode.
Table 2 presents a summarized comparison between the results reported in this work with those using different MoS2/graphene composites and some ternary combinations (MoS2/graphene/other) applied to energy storage. The full table is available in the ESI† (Table S3). It can be noted that the majority of the data presented in the literature is based on LIBs and organic electrolytes, which makes our results for SIBs in aqueous electrolyte even more impressive. Finally, it is important to point out that this is the first report of a ternary material comprising rGO, MoS2 and CuxO nanoparticles prepared as a thin and transparent film. Similar ternary combinations have been reported only in two very recent papers by Selvamani et al. (2020)118 and (2021),119 which were prepared as powders for alkaline supercapacitor applications.
| Sample | Electrode | Cation | Solvent | Current density | Specific capacity or capacitance | Retention rate | Ref. |
|---|---|---|---|---|---|---|---|
| rGO/CuxO/MoSs-LbL | Anode | Na+ | Aqueous | 100 mA g−1 | 1321 mA h g−1 | 78% | This work |
| rGO/CuxO/MoS2-mixing | Anode | Na+ | Aqueous | 100 mA g−1 | 1056 mA h g−1 | 90% | This work |
| rGO/CuxO/MoS2-in situ | Anode | Na+ | Aqueous | 100 mA g−1 | 1377 mA h g−1 | 100% | This work |
| MoS2/G | Anode | Li+ | Organic | 100 mA g−1 | 1902 mA h g−1 | 76.45% | 120 |
| MoS2/rGO | Anode | Li+ | Organic | 100 mA g−1 | 1289 mA h g−1 | 77% | 49 |
| MoS2/G | Anode | Li+ | Organic | 1 A g−1 | 1897 mA h g−1 | 91% | 52 |
| MoS2/Gra | Anode | Li+ | Organic | 100 mA g−1 | 1145 mA h g−1 | 88% | 53 |
| MoS2/rGO | Anode | Li+ | Organic | 100 mA g−1 | 1140 mA h g−1 | 94% | 27 |
| N-GRs/MoS2 | Anode | Li+ | Organic | 100 mA g−1 | 1151 mA h g−1 | 86% | 45 |
| MoS2-rGO | Anode | Li+ | Organic | 0.05 A g−1 | 1102 mA h g−1 | 74% | 55 |
| (MoS2)-graphene | Anode | Li+ | Organic | 100 mA g−1 | 1300 mA h g−1 | 93% | 121 |
| MoS2/rGO | Anode | Li+ | Organic | 100 mA g−1 | 1180 mA h g−1 | 94% | 46 |
| MoS2/Gr/PAni | Anode | Li+ | Organic | 200 mA g−1 | 785 mA h g−1 | 82.3% | 122 |
| MoO2@MoS2/rGO | Anode | Li+ | Organic | 100 mA g−1 | 604 mA h g−1 | 90.3% | 29 |
| CNTs/S@MoS2/G | Cathode | Li+ | Organic | 0.1 C | 1537 mA h g−1 | 78.3% | 123 |
| CNTs/S@MoS2/Gr | Cathode | Li+ | Organic | 0.1 C | 1537 mA h g−1 | 78.3% | 123 |
| MS/MO/CNT/G | Anode | Li+ | Organic | 100 mA g−1 | 640 mA h g−1 | 78.5% | 124 |
| PEDOT/MoS2/Gr | Anode | Li+ | Organic | — | 1143.7 F g−1 | 73.3% | 125 |
| CuO/MoS2/rGO | Anode | KOH | Aqueous | 1 A g−1 | 1445 F g−1 | 91% | 119 |
4. Conclusion
The results presented and discussed in this work represent significant advances in the search for novel electrode materials for aqueous sodium ion batteries. The superlative performance of ternary materials specifically designed for this application was demonstrated for the first time, in which the individual response of each component was synergistically potentiated by the unique nanoarchitected structure in which the materials were prepared. Equally relevantly, the potential of immiscible liquid interfaces to both prepare and process sophisticated materials in a controlled way has also been demonstrated. None of the widely known techniques for thin film deposition could be used to prepare graphene/MoS2/copper oxide nanoparticle tri-component thin films materials like those reported here. However, through the wide range of experimental variations provided by LLIR, three ternary films have been obtained, in which the specific interactions among the components, and consequently the electrochemical response of the final materials, could be controlled.The remarkable architectures of these materials place them among the best reported so far for use as electrodes in aqueous sodium-ion batteries. Faradaic mechanisms due to MoS2 and Cu2O/CuO have been observed and identified in the three-component films, while graphene improves the conductivity and makes a non-faradaic contribution to the electrode capacity. The insertion/de-insertion of cations into the electrodes was followed using a quartz microbalance and found to coincide with the charge changes. These ternary materials demonstrate notable specific capacity enhancements, ranging from 200% to 600% in comparison to their respective control films. The best results, with a specific capacity of 1377 mA h g−1 and a recovery rate of 100%, were obtained for the sample rGO/CuxO/MoS2-in situ, which is the sample that was prepared with the best, most-improved and most-intimate interactions among the components.
Finally, it is important to note that the unique nanostructures of the ternary materials presented here make them promising candidates for several other technological applications, such as electrochemical sensors or catalysts. Also, the ease with which the films can be deposited over ordinary substrates, such as plastics, will enable their use in flexible devices, and the high optical quality exhibited by these films opens great avenues toward applications in which transparency is required, such as optical devices, photovoltaics and transparent batteries.
Author contributions
Maria K. Ramos: conceptualization, visualization, methodology, formal analysis, writing – original draft, writing - review & editing; Gustavo Martins: methodology, formal analysis, EQCM data curation; Márcio F. Bergamini: methodology, formal analysis, EQCM data curation; Luiz H. Marcolino-Juniors: methodology, formal analysis, EQCM data curation; Marcela Mohallem Oliveira: methodology, TEM data curation; formal analysis; Aldo J. G. Zarbin: conceptualization, funding acquisition, methodology, project administration, resources, supervision, validation, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support of CAPES, CNPq, INCT-Nanocarbon and INCT-NanoVida. We kindly thank CME-UFPR for the TEM and HRTEM images and for the Raman spectra. M.K.R. thanks CAPES for the fellowship.References
- B. Ates, S. Koytepe, A. Ulu, C. Gurses and V. K. Thakur, Chem. Rev., 2020, 120, 9304–9362 CrossRef CAS PubMed.
- E. Omanovi, A. Badnjevi, A. Kazlagi and M. Hajlovac, Health Technol., 2020, 51–59 Search PubMed.
- A. T. Lawal, Biosens. Bioelectron., 2019, 141, 111384 CrossRef CAS PubMed.
- W. Qing, F. Liu, H. Yao, S. Sun, C. Chen and W. Zhang, Adv. Colloid Interface Sci., 2020, 282, 102207 CrossRef CAS PubMed.
- E. Benrezgua, B. Deghfel, A. Zoukel, W. J. Basirun, R. Amari, A. Boukhari, M. K. Yaakob, S. Kheawhom and A. A. Mohamad, J. Mol. Struct., 2022, 1267, 133639 CrossRef CAS.
- E. Lee, S. G. Lee, W. H. Lee, H. C. Lee, N. N. Nguyen, M. S. Yoo and K. Cho, Chem. Mater., 2020, 32, 4544–4552 CrossRef CAS.
- C. Ding, L. Wang, W. Zhou, D. Wang, Y. Du and G. Wen, Chem. Eng. J., 2018, 353, 340–349 CrossRef CAS.
- Y. Aparna, K. V. Rao and P. S. Subbarao, J. Nano- Electron. Phys., 2012, 4, 4–7 Search PubMed.
- M. Liu, C. Yu, X. Liu, P. Wang and Y. Chen, Beilstein Arch., 2021, 1, 20215 Search PubMed.
- K. Ariga, J. P. Hill and Q. Ji, Phys. Chem. Chem. Phys., 2007, 9, 2319–2340 RSC.
- E. T. Thostenson, C. Li and T. Chou, Compos. Sci. Technol., 2005, 65, 491–516 CrossRef CAS.
- A. J. G. Zarbin, Mater. Horizons, 2021, 8, 1409–1432 RSC.
- C. M. Ferreira, M. K. Ramos and A. J. G. Zarbin, Eur. J. Inorg. Chem., 2021, 3373–3384 CrossRef CAS.
- A. Schmidt, M. K. Ramos, C. S. Pinto, A. F. Pereira, V. H. R. Souza and A. J. G. Zarbin, Electrochem. Commun., 2022, 134, 107183 CrossRef CAS.
- C. S. Pinto, V. H. R. Souza, A. Schmidt and A. J. G. Zarbin, Synth. Met., 2023, 293, 117259 CrossRef CAS.
- F. M. Morawski, G. Martins, M. K. Ramos, A. J. G. Zarbin, L. Blanes, M. F. Bergamini and L. H. Marcolino-Junior, Anal. Chim. Acta, 2023, 1258, 341169 CrossRef PubMed.
- L. S. M. Alves, M. F. F. D. Neves, L. Benatto, M. K. Ramos, M. Eising, C. K. B. Q. M. De Oliveira, A. J. G. Zarbin and L. S. Roman, IEEE Sens. J., 2023, 23, 1845–1853 CAS.
- R. V. Salvatierra, C. E. Cava, L. S. Roman and A. J. G. Zarbin, Adv. Funct. Mater., 2013, 23, 1490–1499 CrossRef CAS.
- R. V. Salvatierra, C. E. Cava, L. S. Roman, M. M. Oliveira and A. J. G. Zarbin, Chem. Commun., 2016, 52, 1629–1632 RSC.
- S. Husmann, A. J. G. Zarbin and R. A. W. Dryfe, Electrochim. Acta, 2020, 349, 136243 CrossRef CAS.
- L. C. Lopes, S. Husmann and A. J. G. Zarbin, Electrochim. Acta, 2020, 345, 136199 CrossRef CAS.
- S. Husmann, M. K. Ramos and A. J. G. Zarbin, Electrochim. Acta, 2022, 422, 140548 CrossRef CAS.
- V. H. R. Souza, M. M. Oliveira and A. J. G. Zarbin, J. Power Sources, 2014, 260, 34–42 CrossRef.
- L. Hostert, E. G. C. Neiva, A. J. G. Zarbin and E. S. Orth, J. Mater. Chem. A, 2018, 6, 22226–22233 RSC.
- W. H. Khoh, B. H. Wee and J. D. Hong, Colloids Surf., A, 2019, 581, 123815 CrossRef CAS.
- F. Chen, Z. Chen, J. Zhou, X. Fan, X. Xu, Z. Liang, L. Zhan, L. Ma and X. Zhou, Appl. Surf. Sci., 2021, 541, 148497 CrossRef CAS.
- N. Lingappan and D. J. Kang, Electrochim. Acta, 2016, 193, 128–136 CrossRef CAS.
- M. Choi, S. K. Koppala, D. Yoon, J. Hwang, S. M. Kim and J. Kim, J. Power Sources, 2016, 309, 202–211 CrossRef CAS.
- Y. Luo, X. Ding, X. Ma, D. Liu, H. Fu and X. Xiong, Electrochim. Acta, 2021, 388, 138612 CrossRef CAS.
- Y. Ge, C. Pozo-Gonzalo, Y. Zhao, X. Jia, R. Kerr, C. Wang, P. C. Howlett and G. G. Wallace, Chem. Commun., 2018, 54, 5338–5341 RSC.
- D. Lisbona and T. Snee, Process Saf. Environ. Prot., 2011, 89, 434–442 CrossRef CAS.
- Q. Wang, P. Ping, X. Zhao, G. Chu, J. Sun and C. Chen, J. Power Sources, 2012, 208, 210–224 CrossRef CAS.
- J. R. González, R. Alcántara, J. L. Tirado, A. J. Fielding and R. A. W. Dryfe, Chem. Mater., 2017, 29, 5886–5895 CrossRef.
- S. Anwer, Y. Huang, B. Li, B. Govindan, K. Liao, W. J. Cantwell, F. Wu, R. Chen and L. Zheng, ACS Appl. Mater. Interfaces, 2019, 11, 22323–22331 CrossRef CAS PubMed.
- H. Li, X. Wen, F. Shao, S. Xu, C. Zhou, Y. Zhang, H. Wei and N. Hu, J. Alloys Compd., 2021, 877, 160280 CrossRef CAS.
- L. Zhao, T. Zhang, W. Li, T. Li, L. Zhang, X. Zhang and Z. Wang, Engineering DOI:10.1016/j.eng.2021.08.032.
- B. E. Lebrouhi, S. Baghi, B. Lamrani, E. Schall and T. Kousksou, J. Energy Storage, 2022, 55, 105471 CrossRef.
- J. Deng, C. Zeng, C. Ma, J. Fold von Bülow, L. Zhang, D. Deng, Z. Tian and X. Bao, Mater. Today Energy, 2018, 8, 151–156 CrossRef.
- J. Li, H. Tao, Y. Zhang and X. Yang, J. Electrochem. Soc., 2019, 166, A3685–A3692 CrossRef CAS.
- D. Chao, W. Zhou, F. Xie, C. Ye, H. Li, M. Jaroniec and S.-Z. Qiao, Sci. Adv., 2020, 6, eaba4098 CrossRef CAS PubMed.
- N. Alias and A. A. Mohamad, J. Power Sources, 2015, 274, 237–251 CrossRef CAS.
- S. Mateti, M. M. Rahman, P. Cizek and Y. Chen, RSC Adv., 2020, 10, 12754–12758 RSC.
- B. Cui, X. Cai, W. Wang, P. Saha and G. Wang, J. Energy Chem., 2022, 66, 91–99 CrossRef CAS.
- L. Xu, Z. Jiao, P. Hu, Y. Wang, Y. Wang and H. Zhang, ChemElectroChem, 2016, 3, 1503–1512 CrossRef CAS.
- Z. Xiao, L. Sheng, L. Jiang, Y. Zhao, M. Jiang, X. Zhang, M. Zhang, J. Shi, Y. Lin and Z. Fan, Chem. Eng. J., 2021, 408, 127269 CrossRef CAS.
- G. Liu, Y. Feng, Y. Li, M. Qin, H. An, W. Hu and W. Feng, Part. Part. Syst. Charact., 2015, 32, 489–497 CrossRef CAS.
- W. Qiu, J. Jiao, J. Xia, H. Zhong and L. Chen, RSC Adv., 2014, 4, 50529–50535 RSC.
- J. Chao, L. Yang, H. Zhang, J. Liu, R. Hu and M. Zhu, J. Power Sources, 2020, 450, 227680 CrossRef CAS.
- Z. Yu, J. Ye, W. Chen, S. Xu and F. Huang, Carbon, 2017, 114, 125–133 CrossRef CAS.
- Y. Huang, J. Zou, L. Luo, Z. Zhao, H. Liu, Y. Huang, A. Ren and Z. Wang, J. Mater. Sci., 2022, 57, 1246–1260 CrossRef CAS.
- J. Su, Y. Pei, Z. Yang and X. Wang, RSC Adv., 2014, 4, 43183–43188 RSC.
- M. Yang, S. Ko, J. S. Im and B. G. Choi, J. Power Sources, 2015, 288, 76–81 CrossRef CAS.
- Y. Liu, X. He, D. Hanlon, A. Harvey, J. N. Coleman and Y. Li, ACS Nano, 2016, 10, 8821–8828 CrossRef CAS PubMed.
- T. Wang, G. Zhao, C. Sun, L. Zhang, Y. Wu, X. Hao and Y. Shao, Adv. Mater. Interfaces, 2017, 4, 1–8 Search PubMed.
- M. Choi, J. Hwang, H. Setiadi, W. Chang and J. Kim, J. Supercrit. Fluids, 2017, 127, 81–89 CrossRef CAS.
- A. Schmidt, M. K. Ramos, C. M. Ferreira, B. A. Braz and A. J. G. Zarbin, Electrochim. Acta, 2021, 387, 138500 CrossRef CAS.
- G. Zhang, X. Xiao, B. Li, P. Gu, H. Xue and H. Pang, J. Mater. Chem. A, 2017, 5, 8155–8186 RSC.
- A. L. Brisse, P. Stevens, G. Toussaint, O. Crosnier and T. Brousse, Electrochim. Acta, 2018, 279, 161–167 CrossRef CAS.
- Z. Luo, L. Fu, J. Zhu, W. Yang, D. Li and L. Zhou, J. Power Sources, 2020, 448, 227569 CrossRef CAS.
- W. Chen, W. Zhang, L. Chen, L. Zeng and M. Wei, J. Alloys Compd., 2017, 723, 172–178 CrossRef CAS.
- R. Gusain, P. Kumar, O. P. Sharma, S. L. Jain and O. P. Khatri, Appl. Catal., B, 2016, 181, 352–362 CrossRef CAS.
- F. Xu, M. Deng, G. Li, S. Chen and L. Wang, Electrochim. Acta, 2013, 88, 59–65 CrossRef CAS.
- M. Rashad, M. Rüsing, G. Berth, K. Lischka and A. Pawlis, J. Nanomater., 2013, 2013, 1–6 CrossRef.
- C. Sarkar and S. K. Dolui, RSC Adv., 2015, 5, 60763–60769 RSC.
- S. Sun, X. Zhang, Q. Yang, S. Liang, X. Zhang and Z. Yang, Prog. Mater. Sci., 2018, 96, 111–173 CrossRef CAS.
- J. Singh and M. Rawat, J. Bioelectron. Nanotechnol., 2016, 1, 1–9 Search PubMed.
- S. S. Sawant, A. D. Bhagwat and C. M. Mahajan, J. Nano- Electron. Phys., 2016, 8, 1–5 Search PubMed.
- C. Xu, K. V. Manukyan, R. A. Adams, V. G. Pol, P. Chen and A. Varma, Carbon, 2019, 142, 51–59 CrossRef CAS.
- Y. T. Xu, Y. Guo, C. Li, X. Y. Zhou, M. C. Tucker, X. Z. Fu, R. Sun and C. P. Wong, Nano Energy, 2015, 11, 38–47 CrossRef CAS.
- G. Wang, Y. Sui, M. Zhang, F. Du and B. Zou, J. Alloys Compd., 2019, 774, 668–676 CrossRef CAS.
- L. Zhang, Q. Li, H. Xue and H. Pang, ChemSusChem, 2018, 11, 1581–1599 CrossRef CAS PubMed.
- M. Zheng, H. Tang, L. Li, Q. Hu, L. Zhang, H. Xue and H. Pang, Adv. Sci., 2018, 5, 1700592 CrossRef PubMed.
- S. Mukherjee, Z. Ren and G. Singh, Nano-Micro Lett., 2018, 10, 1–27 CrossRef PubMed.
- V. O. Koroteev, S. G. Stolyarova, A. A. Kotsun, E. Modin, A. A. Makarova, Y. V. Shubin, P. E. Plyusnin, A. V. Okotrub and L. G. Bulusheva, Carbon, 2021, 173, 194–204 CrossRef CAS.
- M. K. Ramos and A. J. G. Zarbin, Appl. Surf. Sci., 2020, 515, 146000 CrossRef CAS.
- X. J. Lee, B. Y. Z. Hiew, K. C. Lai, L. Y. Lee, S. Gan, S. Thangalazhy-Gopakumar and S. Rigby, J. Taiwan Inst. Chem. Eng., 2019, 98, 163–180 CrossRef CAS.
- N. Kumar, R. Salehiyan, V. Chauke, O. Joseph Botlhoko, K. Setshedi, M. Scriba, M. Masukume and S. Sinha Ray, FlatChem, 2021, 27, 100224 CrossRef CAS.
- B. Wang, T. Ruan, Y. Chen, F. Jin, L. Peng, Y. Zhou, D. Wang and S. Dou, Energy Storage Mater., 2020, 24, 22–51 CrossRef.
- A. Schmidt and A. J. G. Zarbin, J. Colloid Interface Sci., 2019, 554, 80–90 CrossRef CAS PubMed.
- A. Molina-Sánchez, K. Hummer and L. Wirtz, Surf. Sci. Rep., 2015, 70, 554–586 CrossRef.
- L. Muscuso, S. Cravanzola, F. Cesano, D. Scarano and A. Zecchina, J. Phys. Chem. C, 2015, 119, 3791–3801 CrossRef CAS.
- R. Coehoorn, C. Haas and R. A. De Groot, Phys. Rev. B: Condens. Matter Mater. Phys., 1987, 35, 6203–6206 CrossRef CAS PubMed.
- W. Cheung, M. Patel, Y. Ma, Y. Chen, Q. Xie, J. V. Lockard, Y. Gao and H. He, Chem. Sci., 2016, 7, 5192–5199 RSC.
- S. Pei and H. M. Cheng, Carbon, 2012, 50, 3210–3228 CrossRef CAS.
- J. Zhang, H. Yang, G. Shen, P. Cheng, J. Zhang and S. Guo, Chem. Commun., 2010, 46, 1112–1114 RSC.
- F. T. Johra, J. W. Lee and W. G. Jung, J. Ind. Eng. Chem., 2014, 20, 2883–2887 CrossRef CAS.
- M. D. Patel, E. Cha, N. Choudhary, C. Kang, W. Lee, J. Y. Hwang and W. Choi, Nanotechnology, 2016, 27, 1–10 Search PubMed.
- Y. Yang, X. Chuan, J. Li, F. Liu and A. Li, Int. J. Electrochem. Sci., 2020, 15, 6052–6059 CrossRef CAS.
- S. García-Dalí, J. I. Paredes, J. M. Munuera, S. Villar-Rodil, A. Adawy, A. Martínez-Alonso and J. M. D. Tascón, ACS Appl. Mater. Interfaces, 2019, 11, 36991–37003 CrossRef.
- Y. Pan, L. Gong, X. Cheng, Y. Zhou, Y. Fu, J. Feng, H. Ahmed and H. Zhang, ACS Nano, 2020, 14, 5917–5925 CrossRef CAS PubMed.
- M. Allen, V. Tung and R. Kaner, Am. Chem. Soc., 2010, 110, 132–145 CAS.
- H. Mehl, C. F. Matos, E. G. C. Neiva, S. H. Domingues and A. J. G. Zarbin, Quim. Nova, 2014, 37, 1639–1645 CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- Y. T. Xu, Y. Guo, L. X. Song, K. Zhang, M. M. F. Yuen, X. Z. Fu, R. Sun and C. P. Wong, RSC Adv., 2014, 4, 58005–58010 RSC.
- W. Huang, C. Li, L. Gao, Y. Zhang, Y. Wang, Z. N. Huang, T. Chen, L. Hu and H. Zhang, J. Mater. Chem. C, 2020, 8, 1172–1197 RSC.
- T. M. N. Nguyen, V. D. Vuong, M. T. Phong and T. Van Le, J. Nanomater., 2019, 2019, 16–19 Search PubMed.
- J. Li, Y. Hou, X. Gao, D. Guan, Y. Xie, J. Chen and C. Yuan, Nano Energy, 2015, 16, 10–18 CrossRef CAS.
- T. Xu and L. Sun, Defects in Advanced Electronic Materials and Novel Low Dimensional Structures, 2018, vol. 5, pp. 137–160 Search PubMed.
- M. Guo, Y. Zhao, F. Zhang, L. Xu, H. Yang, X. Song and Y. Bu, RSC Adv., 2016, 6, 50587–50594 RSC.
- M. Nasrollahzadeh, F. Babaei, P. Fakhri and B. Jaleh, RSC Adv., 2015, 5, 10782–10789 RSC.
- Y. Yang, Z.-H. Lu, Y. Hu, Z. Zhang, W. Shi, X. Chen and T. Wang, RSC Adv., 2014, 4, 13749–13752 RSC.
- H. S. S. Ramakrishna Matte, A. Gomathi, A. K. Manna, D. J. Late, R. Datta, S. K. Pati and C. N. R. Rao, Angew. Chem., Int. Ed., 2010, 49, 4059–4062 CrossRef PubMed.
- H. Ju, S. Hun, S. Choi and H. Lee, Mater. Lett., 2010, 64, 357–360 CrossRef CAS.
- A. Ramadoss, T. Kim, G.-S. Kim and S. J. Kim, New J. Chem., 2014, 38, 2379–2385 RSC.
- P. Iamprasertkun, W. Hirunpinyopas, A. M. Tripathi, M. A. Bissett and R. A. W. Dryfe, Electrochim. Acta, 2019, 307, 176–187 CrossRef CAS.
- Y.-X. Wang, S.-L. Chou, D. Wexler, H.-K. Liu and S.-X. Dou, Chem. – A Eur. J., 2014, 20, 9607–9612 CrossRef CAS PubMed.
- K. Wang, X. Dong, C. Zhao, X. Qian and Y. Xu, Electrochim. Acta, 2015, 152, 433–442 CrossRef CAS.
- J. J. Ruan, Y. Q. Huo and B. Hu, Electrochim. Acta, 2016, 215, 108–113 CrossRef CAS.
- P. Subalakshmi, M. Ganesan and A. Sivashanmugam, Mater. Des., 2017, 119, 104–112 CrossRef CAS.
- A. C. Lazanas and M. I. Prodromidis, ACS Meas. Sci. Au, 2023, 3, 162–193 CrossRef CAS PubMed.
- S. H. Choi, Y. N. Ko, J. K. Lee and Y. C. Kang, Adv. Funct. Mater., 2015, 25, 1780–1788 CrossRef CAS.
- S. Sarwar, S. Karamat, A. Saleem Bhatti, M. Kadri Aydinol, A. Oral and M. U. Hassan, Chem. Phys. Lett., 2021, 781, 138969 CrossRef CAS.
- T. Wang, C. Jia, B. Wang and P. Yang, J. Alloys Compd., 2020, 813, 152211 CrossRef CAS.
- X. Zhao, H. Gao, Y. Hou, L. Gbologah, X. Zeng and Y. Wang, J. Electroanal. Chem., 2022, 904, 115936 CrossRef CAS.
- S. Thomas, D. E. Smith, V. K. Greenacre, Y. J. Noori, L. Hector, C. H. K. De Groot, G. Reid and P. N. Bartlett, J. Electhochem. Soc., 2020, 167, 106511 CrossRef CAS.
- S. Yamagata, I. Takahara, M. Wang, T. Mizoguchi and S. Yagi, J. Alloys Compd., 2020, 846, 156469 CrossRef CAS.
- Y. Ji, Z. Yin, Z. Yang, Y. Deng and H. Chen, Chem. Soc. Rev., 2021, 50, 10743–10763 RSC.
- P. S. Selvamani, J. J. Vijaya, L. J. Kennedy, B. Saravanakumar and M. Bououdina, Mater. Lett., 2020, 275, 1–5 CrossRef.
- P. S. Selvamani, J. J. Vijaya, L. J. Kennedy, B. Saravanakumar, M. Bououdina and J. R. Rajabathar, Synth. Met., 2021, 278, 116843 CrossRef CAS.
- J. Wang, X. Zhao, Y. Fu and X. Wang, Appl. Surf. Sci., 2017, 399, 237–244 CrossRef CAS.
- S. Kalluri, K. H. Seng, Z. Guo, A. Du, K. Konstantinov, H. K. Liu and S. X. Dou, Sci. Rep., 2015, 5, 1–8 Search PubMed.
- S. Han, Y. Ai, Y. Tang, J. Jiang and D. Wu, RSC Adv., 2015, 5, 96660–96664 RSC.
- X. Fang and M. Zhang, Ionics, 2021, 27, 3875–3885 CrossRef CAS.
- J. Li, S. Du, H. Tao and X. Yang, Ionics, 2021, 27, 75–84 CrossRef CAS.
- D. Sarmah and A. Kumar, J. Energy Storage, 2021, 35, 102289 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3mh00982c |
| This journal is © The Royal Society of Chemistry 2023 |