
Programmable precise kinetic control over crystal phase, size, and equilibrium in spontaneous metathesis reaction for Cs–Pb–Br nanostructure patterns at room temperature†
Marek
Piotrowski‡
 a,
Zhongsheng
Ge‡
a,
Yixi
Wang
a,
Anil Kumar
Bandela
*b and
Udayabhaskararao
Thumu
*a
a,
Zhongsheng
Ge‡
a,
Yixi
Wang
a,
Anil Kumar
Bandela
*b and
Udayabhaskararao
Thumu
*a
aInstitute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China, Chengdu 610054, China. E-mail: uday@uestc.edu.cn
bDepartment of Chemistry, Ben Gurion University of the Negev Beer, Sheva 84105, Israel. E-mail: bandela@post.bgu.ac.il
First published on 11th October 2022
Abstract
Growth kinetics involved in spontaneous random clustering of perovskite precursors to a particular cesium–lead–bromide (Cs–Pb–Br) nanocrystal (NC) is a poorly understood phenomenon and its spectroscopic investigation is highly challenging. There is scarcely any method that has been optimized yet in which perovskites and their related NCs of a particular size can be grown, viewed, or tuned to another by reaction handling. Here, for the first time, we shed light on the largely overlooked process of growth kinetics of these transformations throughout the reaction trajectory of anionic [PbBrx]n− crystallization dictated by Cs+ cation and report a simple and direct approach to control the metathesis reaction between two precursors (specifically Cs+- and PbBr2-associated oligomeric complexes) in one solvent at room temperature to monitor the NC growth characteristics in a stepwise manner even in the early stages of nucleation. Altering the molar ratio of the two precursors up to a factor of 10 leads to the formation of three prominent phases (CsPbBr3, Cs4PbBr6, CsBr) as dictated by Cs+ to trigger distinct morphological forms (nanobelts, nanoplatelets, rhombohedral NCs, pseudo-rhombic NCs, spherical CsBr NCs, cubic CsBr NCs) including a transient phase that is formed out of linearly self-assembled CsPbBr3 clusters. Our results pave the way towards understanding spontaneous crystallization to develop well-defined, hassle-free routes for diverse perovskite NCs in a simple yet controlled manner.
Introduction
Spontaneous chemical reactions at room temperature (RT) yielding excellent materials of unique properties for a wide range of applications in electronics and optoelectronics are much sought and greatly regarded for the advancement of materials research. In view of easy device processing methods, reactions that do not require any external energy source are desirable to obtain well-controlled materials at the nanoscale by varying the stoichiometry of the precursors.1 It is not conventional to employ such spontaneous reaction processes for controlling the shape, size, and crystal structures of noble metal nanoparticles and quantum dots.2 In contrast to metal and semiconductor nanosystems,3 cesium lead halide perovskite nanocrystals (NCs) and their derivatives exhibit unique, spontaneous reaction tendencies.4,5 Cesium lead halide perovskite NCs have a predominantly ionic lattice, so the reactions between oligomeric Cs+, Pb2+, and X− precursors simply follow the metathesis route, i.e., the redistribution of cations and anions in their respective complexes by the scission and regeneration of Cs–X and Pb–X bonds followed by the stabilization of the surface at the nanoscale by charged ligands (R–NH3+ and R–COO−).6,7 Recently, a few chemical equilibria have been suggested for the inter-phase transformations of CsPb2Br5, CsPbBr3, and Cs4PbBr6 in the presence of external stimuli such as polarity, chelating agents, or thermal influence.8–15 In addition, most of the systematic studies focused on thermodynamic control of these NCs at specific reaction conditions such as chemical nature of the ligand, temperature, use of a different source of halide, type of mixing, additives, etc.16–23 Despite progress in thermodynamics, such as stable phase-controlled synthesis and structural transformations for shape- and size-controlled NCs, the growth kinetics mechanism for perovskite NCs is largely overlooked and is of increasing interest through systematic investigation of reactions,7 including robotic synthesis.24 During a spontaneous reaction, a controlled equilibrium can be achieved to obtain targeted perovskite NCs,25–29 for instance, nanoplatelets,25,30 quantum dots,26,31 nanobelts,32–35 and nanosheets36via particular precursor ratios and selective ligands. However, it is anticipated that the best way to gain deep insight into kinetically stable intermediate states and their fine-tuning is to carefully study the reaction process at RT that could potentially nullify the dissipation of thermal energy, which otherwise could cause polar molecule byproducts, ligand destabilization, oriented self-assembly, phase transformations, etc. In this regard, herein, we demonstrate a systematic approach to understanding the direct influence of Cs+ ion diffusion kinetics on the metathesis reaction that takes place among anionic [PbBrx]n− and Cs+ complexes towards the formation of perovskite NCs at RT in a non-polar solvent. In other words, this work aims not to study the products themselves or other variations in reaction conditions but rather to mainly focus on understanding the growth kinetics of these transformations throughout the reaction trajectory of anionic [PbBrx]n− crystallization with Cs+ cation. In a typical experiment, a microliter quantity (2–250 μL range) of precursor Cs-oleate (PCs-ol) was titrated against a fixed PbBr2 (PPb-br) complexed with oleic acid (OA) and oleylamine (OLAM) in octadecene (ODE) in 2 mL hexane at RT in a 5 mL vial and shaken at 900 rpm (see a schematic illustration in Fig. 1 and ESI† for the detailed synthesis protocols). Because the conditions used in the original hot injection method are widely employed and still a primarily implemented strategy, the PCs-ol and PPb-br used in this protocol are the same as those in the original synthesis route.4 In our approach, experiments were specially designed with a fixed volume of hexane as a solvent and tiny amounts of PCs-ol and PPb-br because such an approach not only provides the feasibility to vary the precursor stoichiometry ratios without abrupt changes in the concentrations but also provides a simple way to characterize the resultant NCs in solution without further dilutions or the necessity to conduct post-synthesis purification methods. At these reaction conditions, firstly, we investigated in detail the growth kinetics during titration of PCs-ol against PPb-br; then, studies pertaining to concentration influence to produce kinetically favored crystal states; and finally, experiments that deal with the role of polarity in distorting the reaction memory.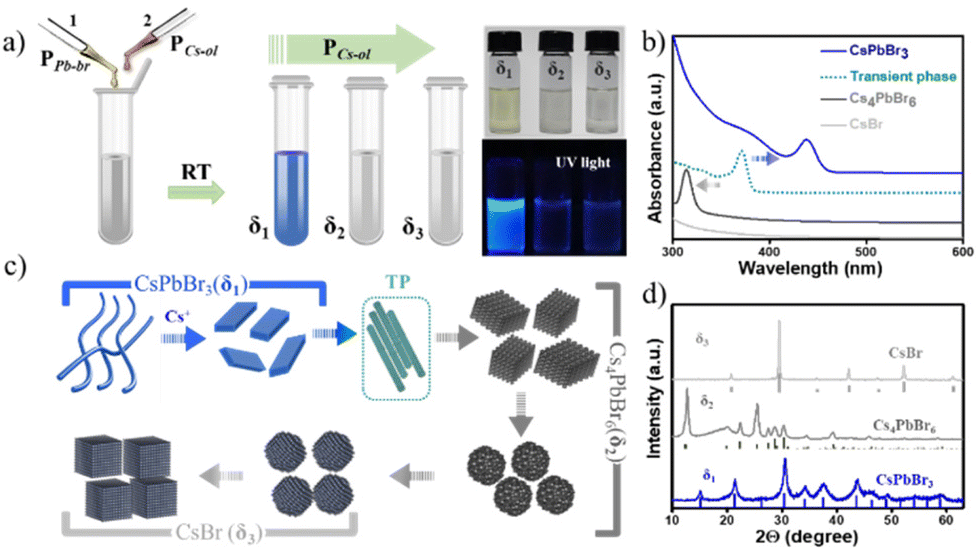 | ||
| Fig. 1 Schematic illustration of the RT synthesis of Cs–(Pb)–Br nanostructures at different amounts of PCs-ol titrated against PPb-br (the amount of PCs-ol increased from 2 μL to 250 μL, δ1 to δ3) in hexane (a) and absorption spectra (b) of solutions collected from reaction vials corresponding to four main regions (δ1−δ3) with three types of crystal phases and a transient phase. (c) Schematic representation of Cs+-concentration-triggered growth of three prominent phases and their distinct morphologies – nanobelts, nanoplatelets, rhombohedral NCs, pseudo-rhombic NCs, spherical CsBr NCs, cubic CsBr NCs including a transient phase. (d) Powder X-ray diffraction patterns of three selected samples in the three regions of δ1, δ2, and δ3. The vertical bars indicate the ICSD reference patterns of CsPbBr3, Cs4PbBr6, and CsBr NCs. | ||
Results and discussion
In this study, a solution of PCs-ol is directly titrated against PPb-br, which is in stark contrast to the regular methods, and we observed that the initial milky white CsOA turned to a clear solution immediately after mixing both the precursors, and no sedimentation was found. In order to assess the reaction kinetics, initially, about 30 titrations were carried at infinitesimally small (1 μL) increments of PCs-ol, specifically 2, 3, 4, 5, …, 30 μL. In these regions, the spectral changes are very sensitive to the amounts of PCs-ol whereas changes are less sensitive to further addition. Therefore, subsequent titrations are performed at each 10 μL increment of PCs-ol from 40 to 250 μL. The PPb-br reacts with PCs-ol during titration and keeps on proliferating from one phase to another. Thus, overall, around 50 titrations and their repetitions at various reaction times were performed. Each sample was carefully studied by spectroscopy and microscopy analysis and a conclusion was arrived at to divide these precursor ratios into three categories: δ1 = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.03 to 1:1, δ2 = 1:1 to 1:5, and δ3 = 1:6 to 1:10 (S2-Table 1†), where δ = mole ratio precursors (PPb-br/PCs-ol). The colorless precursor solutions turned to yellowish or pale green in the δ1 region, and the solutions turned to colorless with further incremental additions of PCs-ol in δ2 and δ3 regions (Fig. 1a and Fig. S1†). Optical absorption spectra of the NCs at δ1, δ2, and δ3 exhibit characteristic absorption peaks at 420 nm and 313 nm, corresponding to two monolayers of CsPbBr324 and Cs4PbBr6,37,38 respectively, whereas a featureless spectrum at δ3 corresponds to CsBr NCs (Fig. 1b). In between δ1 and δ2, there is another product not related to any of these crystal phases whose absorption is around 370 nm attributed to transient phase (TP) of linear assemblies of CsPbBr3 clusters between CsPbBr3 and Cs4PbBr6. Fig. 1c is a schematic illustration of the formation of three prominent phases (CsPbBr3, Cs4PbBr6, and CsBr) as dictated by Cs+ to trigger distinct morphologies (nanobelts, nanoplatelets, rhombohedral NCs, pseudo-rhombic NCs, spherical CsBr NCs, cubic CsBr NCs). The next section deals with the in-depth study of these systematic absorption changes with respect to the minor changes of PCs-ol against PPb-br. The ratios (δ) and the resulting crystal phase, shape, and sizes are detailed in ESI (S2-Table 1†).
:0.03 to 1:1, δ2 = 1:1 to 1:5, and δ3 = 1:6 to 1:10 (S2-Table 1†), where δ = mole ratio precursors (PPb-br/PCs-ol). The colorless precursor solutions turned to yellowish or pale green in the δ1 region, and the solutions turned to colorless with further incremental additions of PCs-ol in δ2 and δ3 regions (Fig. 1a and Fig. S1†). Optical absorption spectra of the NCs at δ1, δ2, and δ3 exhibit characteristic absorption peaks at 420 nm and 313 nm, corresponding to two monolayers of CsPbBr324 and Cs4PbBr6,37,38 respectively, whereas a featureless spectrum at δ3 corresponds to CsBr NCs (Fig. 1b). In between δ1 and δ2, there is another product not related to any of these crystal phases whose absorption is around 370 nm attributed to transient phase (TP) of linear assemblies of CsPbBr3 clusters between CsPbBr3 and Cs4PbBr6. Fig. 1c is a schematic illustration of the formation of three prominent phases (CsPbBr3, Cs4PbBr6, and CsBr) as dictated by Cs+ to trigger distinct morphologies (nanobelts, nanoplatelets, rhombohedral NCs, pseudo-rhombic NCs, spherical CsBr NCs, cubic CsBr NCs). The next section deals with the in-depth study of these systematic absorption changes with respect to the minor changes of PCs-ol against PPb-br. The ratios (δ) and the resulting crystal phase, shape, and sizes are detailed in ESI (S2-Table 1†).
The structural conformation of the samples was studied by analyzing powder X-ray diffraction (XRD) patterns of the selected samples in the three regions (Fig. 1d), where all peak positions observed represent CsPbBr3, Cs4PbBr6, and CsBr crystal phases. Also, mixed phases are observed in the regions where the borders of one phase to another phase are overlapped (Fig. S2†). The stoichiometry ratios of the pristine nanostructures were further confirmed by HAADF-STEM EDS analysis of pure and mixed regions. The atomic percentages values of Cs:Pb:Br of 1:1:2.6, 4:1:5 and 1:0.06:1.1 are in good agreement with those of the three phases CsPbBr3, Cs4PbBr6, and CsBr, respectively (Fig. S3 and S4†).
Some of the representative spectra are compared to follow the absorption changes in a stepwise manner that clearly showcase the phase transformations going from δ1 to δ3 regions (Fig. 2a). In δ1, initially an addition of 2 μL of PCs-ol against PPb-br evoked a rapid spectral change with the appearance of an absorption band at 390 nm, which red shifts to 420 nm over time in a span of 15 min (Fig. S5†). These two bands correspond to single- and two-monolayered CsPbBr3 nanostructures, respectively.24,36 Later on, the changes were slowed down, indicating the formation of a fairly stable phase of CsPbBr3 NBs. Thus, the major changes are (i) appearance of a new absorption band at 390 nm, (ii) appearance of a shoulder at ca. 420 nm, (iii) rise of the shoulder to a strong absorption band at 420 nm at the expense of decreasing absorption at 390 nm (that is, the transformation of single- to two-monolayered CsPbBr3 nanostructures). Remarkably, the spectral changes are accompanied by the appearance of isosbestic points at 408 and 380 nm (Fig. S5†), indicating a stoichiometric conversion of single-layer (390 nm) to two-monolayered nanostructures (420 nm). These isosbestic points are clearly seen with an increase in the Cs+ amount until the titrations of 12 μL of PCs-ol to PPb-br. A few more increments later, the isosbestic features are distorted and finally vanished beyond certain additions of PCs-ol. The presence of distorted isosbestic features is due to the two simultaneous growth and shrinkage transformations which significantly lead to blue as well as red shifts in the absorption spectra with the increase in the presence of PCs-ol (Fig. S6–S9†). In Fig. 2a (in the δ1 region), the absorption was collected after 4 hours of reaction, showing the band at 430 nm.
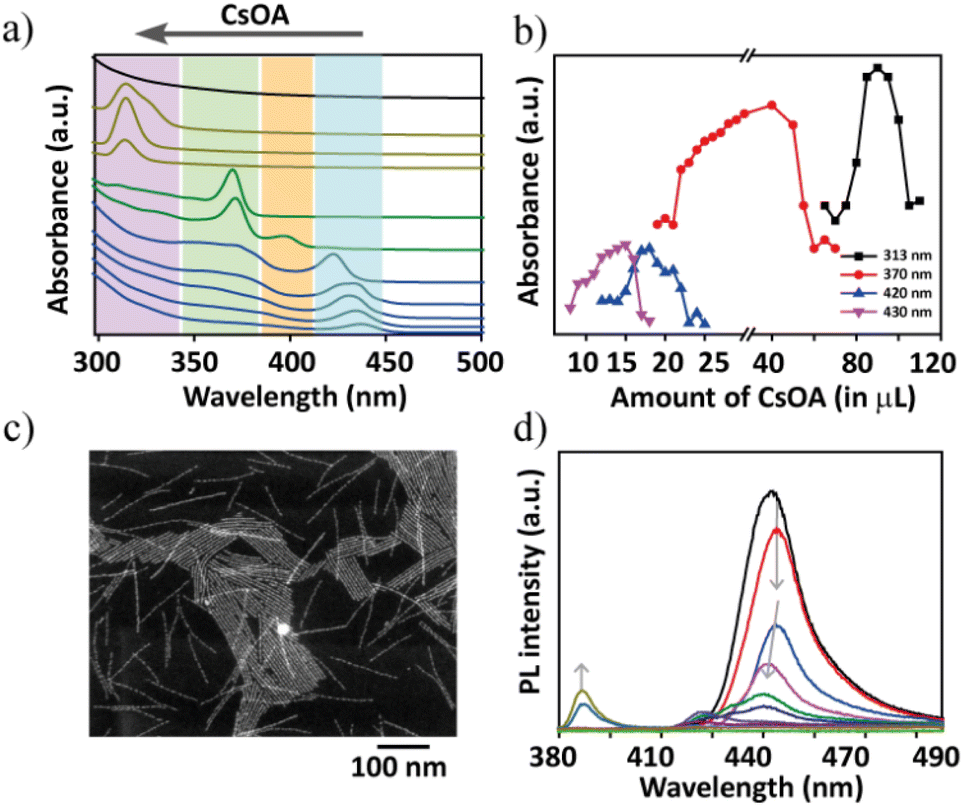 | ||
| Fig. 2 (a) Absorption spectra of the transformation of Cs–(Pb)–Br nanostructures from CsPbBr3 to Cs4PbBr6 NCs to CsBr NCs via the intermediate transient phase. The systematic trend of characteristic peak positions is labelled with different colours to represent the four major bands at ≈430 nm, 420 nm, 370 nm, and 313 nm. The upper black trace is featureless, corresponding to CsBr NCs. The top arrow indicates the incremental addition of PCs-ol to the reaction. Titrations were carried out at RT and a constant concentration of the host, PPb-br. (b) The appearance of characteristic absorption bands (≈430 nm, purple; 400 nm, blue; 370 nm, red; 317 nm, black) and their gradual progress during the titration of PCs-ol from 2 μL to 200 μL. (c) STEM image of the intermediate product formed between CsPbBr3 and Cs4PbBr6 phases corresponding to the absorption at 370 nm, which persisted for a long titration period from 20 to 50 μL of PCs-ol addition (red trace in b). (d) The changes in PL intensity and the PL wavelength with respect to the titrations of (b). | ||
Interestingly, overall, this band at 430 nm undergoes a gradual blue shift (Fig. 2a, Fig. S6–S8†) during incremental addition of PCs-ol from 2 to 20 μL. The spectrum gradually loses its original absorption structure with a blue shift from 430 nm to 400 nm and then to an entirely new spectrum with absorption at 370 nm (Fig. 2a and Fig. S9†). Afterwards, an era of new region δ2 begins with the emergence of zero-dimensional Cs4PbBr6 NCs as a dominating product with a clear characteristic absorption band at 313 nm (Fig. 2a and Fig. S10†). This crystal phase remained from the titration of 50 μL to 120 μL of PCs-ol against PPb-br. Similar to the above-mentioned δ1 condition, a rapid reaction change in the absorption fine structure (Fig. S11†), that is, the appearance of a broad peak composed of the absorption at 313 nm and 320 nm from Cs4PbBr6 and [PbBr4]2−, respectively, was seen immediately after mixing the precursors at δ2 (Fig. S10,† 100 μL to 180 μL of PCs-ol). Beyond these, a new region δ3 occurred where the absorption is nearly featureless, corresponding to CsBr NCs (Fig. 2a, black trace; and Fig. S12†). As one might have expected, no further changes in the absorption spectra were observed when δ3 = 1:20 equivalent of PCs-ol was added.
Fig. 2b unambiguously indicates the appearance of four prominent absorption bands during the course of titration of PCs-ol from 2 to 200 μL. It is evident from the data that the characteristic CsPbBr3 nanostructure absorption at 435 nm could occur only at the lower Cs+ additions (2 to 11 μL); later, it undergoes a clear shift from 435 nm to 420 nm from around 12 μL addition of PCs-ol. This absorption band further blue shifts to 370 nm with a small band at 400 nm for the subsequent incremental additions of PCs-ol (also refer to Fig. S13†). The blue shift of the CsPbBr3 absorption peaks with an increased amount of Cs+ is clearly indicative of the size reduction in the nanostructure. The emergence of the absorption peak at 370 nm with a shoulder peak at 320 nm is not so clearly observed or identified in Cs–Pb–Br literature to date, even for single-monolayer sizes. The absorption band of one-monolayer 2D (OLAM)2PbBr4 sheet is at 398 nm, as seen in the literature.36 We ruled out the other possible compositions to characterize this intermediate phase such as CsPb2Br5 and PbBr2 NCs in the reaction medium because their absorption peaks are supposed to be around 320 to 350 nm, respectively.24 In our studies, we observed that this absorption band was relatively stable without any sign of degradation for about 24 hours (Fig. S9†) and also persistent for a more extended titration volume from 26 μL to 70 μL of PCs-ol against PPb-br signifying it is a long-standing transient state. In view of their emission (see the next section), we attributed this phase to being made up of CsPbBr3 clusters. In TEM, defined structures related to this transient state are composed of a linear arrangement of CsPbBr3 clusters into ultra-thin structures (Fig. S14†). However, the HADDF-STEM image shows the presence of ultra-thin nanoplatelets (NPs) of bright contrast (Fig. 2c and Fig. S15†) and the morphology suggests linear assembled structures. In a set of reports, it was shown that nanoclusters39 and particles40 could assemble in a one-dimensional fashion under the impact of electrostatic interactions. Thus, it is possible that these NPs are intermediate state structures, and we attribute these structures to CsPbBr3 clusters that assembled into ultra-thin NBs. However, more detailed studies are required to understand the origin of the optical properties of this structure. From 70 μL to 120 μL, the Cs4PbBr6 NC phase dominates with a maximum yield at 90 μL addition of PCs-ol. Beyond 120 μL to 200 μL, the reactions show a weak absorption at 313 nm and 320 nm. These peaks are inextricably linked to [PbBr6]4− and the peak intensities are interchangeable over time. This scenario implies that these Cs4PbBr6 NCs and [PbBr6]4− are in chemical equilibria.41
Photoluminescence (PL) studies of these reactions further confirm the blue shift in the emission with incremental Cs+ additions. In addition to the blue shift, the emission intensity drastically reduced for the single-layer species reminiscent of nanoplatelets, and such poor emission properties are attributed to the increased surface defects at this atomic scale of perovskites. At 30 μL of (PCs-ol) a new and weak emission at 380 nm appeared corresponding to the intermediate transient species (λabs of 370 nm). The results of PL experiments indicate that the influence of stoichiometry on the shape and crystal structure transformations results in the change of NC emission properties. Fig. 2d shows the PL results with an increase in the concentration of Cs+ ions. At molar ratios δ = 1:0.25 – δ = 1:1, nanobelts emit cyan blue at 420 nm. In the following stages, Cs+ increase results in the appearance of a very weak emission at 400 nm corresponding to one-monolayer-thick nanosheets, and, next, the appearance of a weak emission at 380 nm. As expected for other phases Cs4PbBr6 and CsBr, we have not observed any luminescent features in all NC systems at values δ ≤ 1 (Fig. 2d).
The TEM measurements of the spontaneous reaction solution at the lower δ1 region show formation of atomically thin lamellar nanoplatelets (NPs), which correlate to the one-monolayer thickness (Fig. S16†). These structures subsequently arrange into two-monolayer perovskite structures within several minutes and further grow to nanobelts of length 1–3 μm and width ∼6 nm (Fig. 3a). At moderate δ1, shortening and thinning of NBs take place, resulting in NPs of 20–30 nm in length and ∼3 nm in width (Fig. 3b, inset). At the lower δ2 region, a different behavior has been observed for Cs-rich conditions, where the surface plane is mainly ended with Cs+ ions. At this ratio, we started to observe the appearance of additional phases in the solution along with CsPbBr3 NPs, and they are ∼100 nm-sized rhombohedral Cs4PbBr6 NCs (Fig. S17†). At this stage, the absorption spectra showed the presence of mixed peaks centered at 400 nm (weak), 370 nm, and 313 nm as well. At moderate δ2 values, NPs disappear while 100–150 nm-sized Cs4PbBr6 rhombohedral NCs are exclusively seen (Fig. 3c). On further increase in δ2, the size of the rhombohedral Cs4PbBr6 NCs decreases, and apparently ∼20 nm spherical Cs4PbBr6 NCs have emerged (Fig. 3d). Remarkably, these are highly monodisperse, and they form large self-assembled monolayers of hexagonally packed sheets upon drop-casting of the as-made sample in hexane on a TEM grid (Fig. S18†).
 | ||
| Fig. 3 TEM images collected at reaction condition δ1, corresponding to CsPbBr3 NBs (a) and nanoplatelets (b). (c and d) TEM images collected for reaction condition δ2, related to two different sized Cs4PbBr6 structures. (e and f) TEM images collected for reaction condition δ3, related to spherical and cube-shaped CsBr NC structures. (g, h and i) TEM images of the NCs with two different packings (HCP and CCP) during the reaction progress while transferring reaction conditions from δ2 to δ3. Scale bars are 100 nm in all the regular images and 10 nm for the image in the inset of (b). | ||
Now one important question arises: What happens if the Pb2+ ratio further reduces in our reaction conditions? Intending to answer this question, we decided to further increase the amount of Cs+, which eventually decreases the Pb2+ availability for the reaction. At lower δ3, a mixed phase consisting of spherical particles (≈12 nm) with two types of packing tendencies has been observed (Fig. S19 and S20†). Even though all NCs formed are spherical, they are arranged in three different packings through self-assembly upon solvent evaporation. The three different packings are CCP, HCP, and mixed quasi-crystalline packing, which implies the presence of two types of NC structures (Fig. 4 and 5a–c). The XRD data confirm the presence of both Cs4PbBr6 and CsBr NCs (Fig. 5d). The results of HRTEM studies in Fig. 4 are in agreement with the assigned crystal phase compositions. The lattice spacings of 0.68 nm, 0.4 nm, and 0.33 nm correspond to (100), (110), and (131) of the Cs4PbBr6 phase, respectively. The lattice spacings of 0.43 nm and 0.3 nm correspond to (100) and (110) of the CsBr phase, respectively. Typically, the single perovskite NCs assemblies lead to a simple cubic packing following the cube-shaped morphology. In addition, Kovalenko et al. explored other complex factors such as shape and steric influences for designing functional materials formed via this NC self-assembly.42 We propose that the presence of the unique packing pattern of CsBr and Cs4PbBr6 could result from different ligand structures around the spherical NCs. This kind of dynamic change in the arrangement has also been observed in soft-matter materials, such as polymers. Recently, Pradhan et al. found such packing differences in pseudo-spherical CsPbBr3 NCs due to the change in their facets.19,43 Such unique self-assembly arrangements are seen even from the initial stages, since the reaction process is also consistently rapid even at δ2 and δ3 conditions as confirmed in TEM, with the formation of tiny spherical Cs4PbBr6 (≈6 nm) and CsBr NCs (≈3 nm) (Fig. 3g–i and Fig. S21–S24†). Even though both the NCs are spherical, their arrangement is unique, i.e., HCP for Cs4PbBr6 and CCP for CsBr seeds (Fig. 3g). At moderate δ3, the spherical CsBr NCs (≈15 nm) were the dominating fraction and arranged in CCP fashion (Fig. 3e–h). At higher δ3 = 1:8 to 1:10, with a further increase of the Cs+ precursor amount in the reaction, the tendency to form cube-shaped CsBr (≈20 nm) is completely dominant (Fig. 3f–i). It is worth noting that the residual amount of Pb2+ could not suppress the formation of Pb2+-free CsBr structures. This observation is justifiable based on two reasons: (i) the relative reduction of Pb2+ in comparison to other ions (Cs+ and Br−); (ii) an increase in the amount of OA in solution could bind Pb2+ and is not involved in the reaction. Increasing the amount of PCs-ol adds both Cs+ as well as OA into the reaction medium; however, the amount of OA is very little (1 μL PCs-ol contains 0.0625 μL of OA; overall 2.5 μL OA in 40 mL octadecane in a typical synthesis; details in ESI†) which did not alter the reaction products as confirmed through the experiments maintaining at constant OA (Fig. S25†). The absorption profiles show the same trend in the presence and absence of small additions of OA along with PCs-ol, reflecting the similar characteristic crystal phase irrespective of minute changes in the OA amounts. Consequently, these minute changes in ligand concentrations alter the morphology of a particular phase as discussed below.
 | ||
| Fig. 4 (a and b) HAADF-STEM image of the 100 nm Cs4PbBr6 NCs prepared at the lower δ2 region. (c and d) TEM images of the ≈15 nm Cs4PbBr6 NCs as self-assembled monolayers prepared at higher δ2 regions. (e) TEM image of isolated Cs4PbBr6 NCs and their HRTEM analysis. TEM images of cubic (f) and spherical (h) CsBr NCs and their respective HRTEM images (g and i). The lattice spacings of 0.68 nm, 0.4 nm, and 0.33 nm correspond to (100), (110), and (131) of the Cs4PbBr6 phase, respectively. The lattice spacings of 0.43 nm and 0.3 correspond to (100) and (110) of the CsBr phase, respectively. | ||
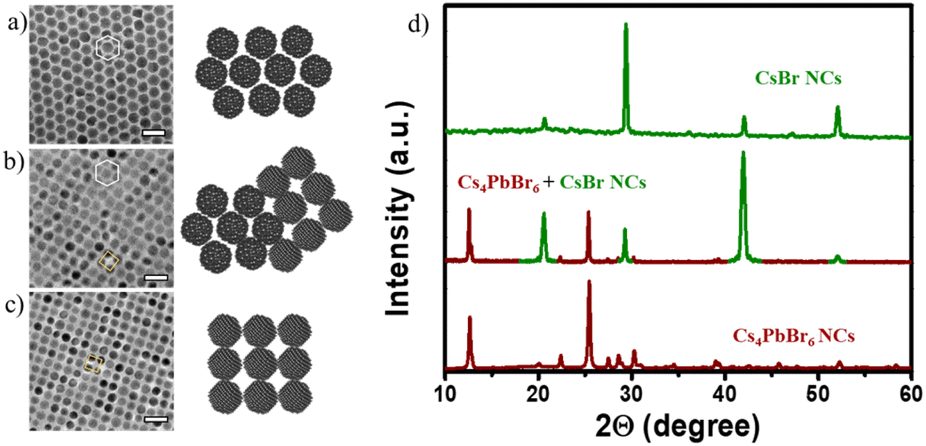 | ||
| Fig. 5 (a–c) TEM images of NCs with two different packings (hexagonal close packing, HCP, and cubic close packing, CCP) while the reaction condition was transferring from δ2 to δ3. Scale bars are 20 nm in all the images. (d) Powder XRD patterns of the samples in the regions of δ2 and δ3 corresponding to Cs4PbBr6 and CsBr NCs along with their mixed region. | ||
At the same condition, soft-edged cube-shaped CsBr NCs were formed upon the addition of small (1–3 μL) quantities of OA. Typically, OA supports the formation of sharp edges of NCs, whereas OLAM suppresses the sharp edges leading to spherical NCs (Fig. S26 and S27†). Further increase in the Cs+ ions in δ3 (1:10) lowers the Pb2+ amounts to result in the formation of sharp-edged highly monodisperse CsBr nanocubes readily undergoing square packed assembly on TEM grids into 200 nm to 1 μm square-shaped sheets (Fig. S28†). Beyond this δ (1:10) value, formation of larger and polydisperse CsBr nanocubes has been observed (Fig. S29†). Overall, in these δ1 to δ3 regions, we demonstrated that size, shape, structure, and surface anisotropy could be kinetically controlled solely based on the amount of Cs+ ions. Moreover, we believe such dynamic changes in the packing patterns, hexagonal close packing or face-centered cubic packing, based on the type of morphology, size, and crystal structure of NCs even from their nucleation stages, provide simple traditional microscopic crystal structure and uniqueness from others of the same size.
Another important issue is dealing with the chemical equilibria which could be reversible to either side of the reaction providing requisite chemical inputs. To crosscheck our hypothesis, first, CsPbBr3 NBs were created by subjecting the titration reaction at δ1 conditions whose characteristic absorption peak at 430 nm is shown in Fig. 6a. These pre-preformed NBs were subjected to post-modification through forward titration reaction by adding the required amount of PCs-ol. Here, the forward titration reaction indicates that first the NBs are formed at δ1 conditions, and next the reaction conditions are shifted to δ2 by providing additional δ1. During this process, the absorption peak shows a clear blue shift from 430 nm to 313 nm (Fig. 6a), the TEM images corresponding to the obvious structural and morphology changes from CsPbBr3 to Cs4PbBr6 (Fig. 6b). After 12 hours of reaction time, there are pure Cs4PbBr6 NCs, and such are the conditions originally seen at initial δ2 condition. In another case, at first, the Cs4PbBr6 NCs mixed with [PbBr6]4− were created by titrating at δ2 conditions. This reaction was subjected to backward titration conditions by adding the required amount of PPb-br leading to the appearance of a new absorption peak at 370 nm (Fig. 6c). TEM images show the transformation of spherical Cs4PbBr6 NCs to CsPbBr3 clustered NBs (Fig. 5d), such conditions being originally seen at higher δ1. Such reversible equilibrium is not successful in the case of δ2 to δ3. This could explain the influence of chemical equilibrium on the size and morphology of the NCs (discussed in the next section).
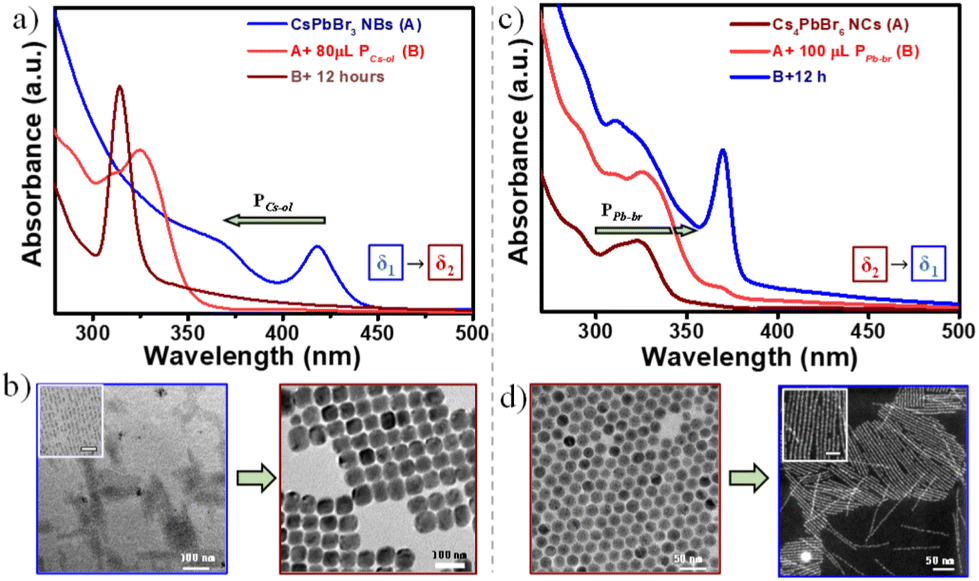 | ||
| Fig. 6 (a) Absorption spectral studies of reaction, with the deliberate shifting of reaction conditions via introducing additional PCs-ol to the already prepared CsPbBr3 NBs at δ1 condition. Initially, NB structures are made by titrating 5 μL of PCs-ol with 100 μL of PPb-br in 2 mL hexane (A), its characteristic absorption spectrum of 430 nm being represented by a blue trace. At this point, 80 μL PCs-ol was added (B) in order to shift the reaction toward the formation of Cs4PbBr6 NCs, confirmed by the peak at 313 nm (brown trace) upon 12 h of reaction. (c) Absorption spectral studies of reaction, with deliberate reversing of the reaction conditions via introducing additional PPb-br to the already stabilized Cs4PbBr6 NC system. Initially, Cs4PbBr6 NC structures are made at δ2 conditions by titrating 80 μL of PCs-ol with 100 μL of PPb-br in 2 mL hexane (A), the characteristic absorption peak being at 313 nm (brown trace). At this point, 100 μL PPb-br was added (B) in order to shift the reaction backward to the formation of CsPbBr3 clustered NWs, confirmed by the peak at 370 nm upon 12 h of reaction. (b–d) TEM and STEM images representing the morphology changes during the forward and backward titration reactions. Scale bars of the insets are 20 nm. | ||
The crystalline products at different δ1, δ2, and δ3 conditions can be explained in the following way. The sources of Pb2+ and Br− ions are apparently from PbBr2, so a fixed amount of PPb-br results in the equivalent amount of Pb2+ and doubles the amount of Br− concerning incremental addition of Cs+. The δ1 region conditions are similar to a typical synthesis of CsPbBr3 NCs in terms of precursor ratio where the amount of Pb2+ used for the synthesis is higher than that of Cs+ in the reaction; hence Pb-oleate and PbBr-oleate are the expected byproducts. In the case of the lower Cs+ addition side of δ1, we observed the formation of NBs, and higher Cs+ addition leads to nanoplatelets. The CsPbBr3 NB formation can be explained by the cooperative effect of stoichiometry, and micelle-based linearly assembled Pb+, Br− complexed OA, and OLAM ligand interactions; the first one dictates CsPbBr3 (Fig. 7a), and the latter directs the NB morphology. With a further increase of the Cs+ amount, the competitive force between the interconnection among [PbBr6]4− octahedra versus Cs+ separated octahedra. In this situation, the NBs become shortened and turn to NPs as seen in TEM images and blue shift in the absorption. When the incremental addition of Cs+ reaches δ2, the second factor dominates that pushes the crystallization toward the crystal structure where octahedra are separated by Cs+ ions for the formation of Cs4PbBr6 (Fig. 7b). At this condition, instead of preferring the three-dimensional growth while maintaining the CsPbBr3 phase, a phase transformation occurs and results in the formation of Cs4PbBr6 NCs. Interestingly, beyond the δ = 1:6 region, the transformation to CsBr NCs indicates that the residual Pb2+ is not enough to participate in the reaction; hence it could no longer influence the reaction. However, in the presence of an excess amount of Cs+, cesium rapidly reacts with bromine (double the concentration of Pb2+), causing the nucleation of CsBr NCs (Fig. 7c). Importantly, the concentration of Cs+ ions creates competitive forces which alter the growth kinetics determining the size and shape of particular NCs (NBs vs. NPs; 100 nm vs. 20 nm Cs4PbBr6 NCs; or spherical vs. cubic CsBr NCs). Few assumptions are expected for the cause of the size variation in these NCs. As shown in earlier sections, there is a dynamic equilibrium among CsPbBr3 and Cs4PbBr6 systems. At increased Cs+ concentrations, CsPbBr3 NB equilibrium shifts to Cs4PbBr6 NCs, limiting the dissolution of the resulting crystal phase and resulting in the larger sizes of Cs4PbBr6 NCs. Whereas the tendency of balanced equilibrium among Cs4PbBr6 and CsBr systems in the middle of δ2 and δ3 regions prefers competition among the crystal phases at rapid formation, more dissolution results in smaller NCs. Finally, at the higher δ3 region (dominated by Cs+ concentrations, the solubility of Pb2+ in the presence of OA), equilibrium shifts toward the formation of CsBr NCs and results in larger CsBr NCs. However, for an in-depth understanding of mechanistic insights, further qualitative studies are required on this system.
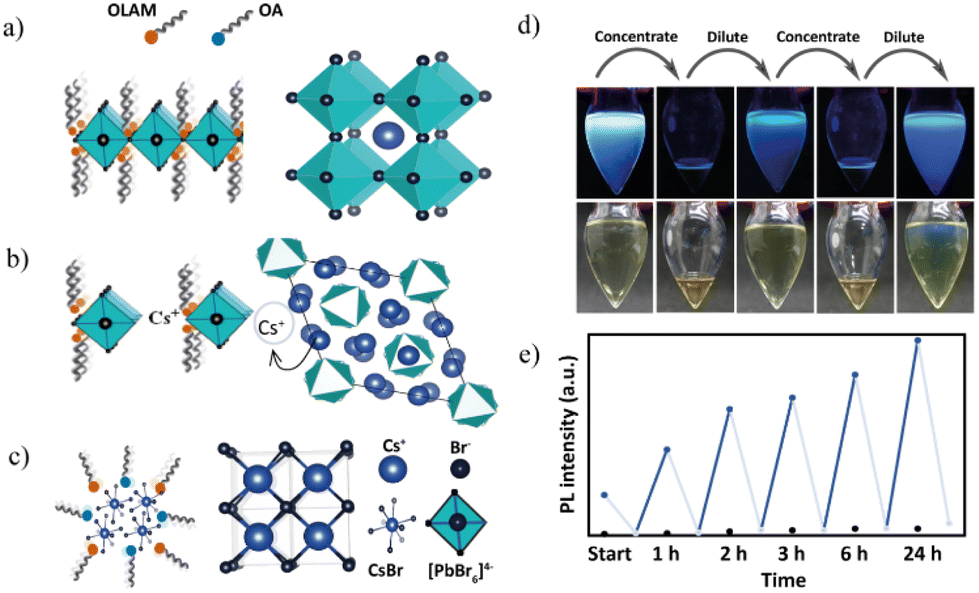 | ||
| Fig. 7 (a–c) Schematic representation of the effect of stoichiometry on the final crystal structures. At δ1, the attachment of (PbBr6)− octahedra predominantly occurs; at δ2, the Cs4PbBr6 structure where the octahedra are separated by the Cs+ ions is predominant; at δ3, the structures are influenced by Cs+ and Br−, and, as a result, the final products are CsBr structures. (d) Photographs of the solutions (NBs) for the cyclic experiment in visible (bottom) and UV light (top) and (e) the corresponding PL experiment. The black dots represent a control experiment having two precursors (without hexane solvent). | ||
To better understand the kinetic control over the crystallization process, as a proof of concept, we studied the impact of dilution at δ1 as this region exhibits visible changes (Fig. 7d). At δ1 = 1:0.25, the prerequisite amounts of PCs-ol to the host PPb-br at initial concentration lead to ultrathin NB formation. PL emission spectra of these NBs were collected at two different constant concentrations during the reaction progress over 24 hours (refer to ESI for experimental details†). The rising blue lines in Fig. 7e indicate original solutions during reaction progress at 1 h, 2 h, 3 h, 6 h, and 24 h. Falling gray lines in Fig. 7e represent the quenching of PL intensity between the reaction progress subjected to solvent evaporation, thereby increasing the concentration. The PL intensities of concentrated sample reaction time intervals are at 0.5 h, 1.5 h, 2.5 h, 3.5 h, 6.5 h, and 24.5 h. We expect that at the increased concentrations, the stoichiometry of NCs could be altered as a result of changes in the local concentration of Cs+ due to the disturbance to ligand dynamics associated with modified electrostatic interactions.
The PL emission of the sample is quenched at the concentrated condition after hexane evaporation, whereas upon re-dilution, it regains its PL emission. Blue dots imply PL emission at regular dilutions, and grey ones indicate the concentrated conditions. This procedure was repeated several times in a period of over 24 hours, and the results of cyclic quenching and recovery of PL emission are presented in Fig. 7e. As a control experiment, samples without hexane solvent and with hexane undisturbed were prepared, and their PL intensity was measured at the same time intervals. The results are compared in Fig. S30–S33,† and show that there is no or ill-defined reaction (no signs of CsPbBr3 NBs) for the sample without hexane. The undisturbed reaction shows a gradual PL emission but a relatively lower intensity than that in the cyclic experiment. Interestingly, based on the PL emission results of the cyclic experiment, the reaction “remembers” the original reaction path with a quicker resurgence of NB formation at every dilution step. In addition, kinetically stable products trapped at suitable dilution predominated over spontaneous aggregation directed by stoichiometry. Our results on the study of the effect of concentration align with the findings that dilution is the prerequisite to producing luminescent nanobelts. Some of the recently reported NB syntheses are two-step processes, where, at first, the formation of non-luminescence nanostructures occurs. Next, in the second step, the redissolution of non-luminescence species in a solvent to the desired dilution results in the formation of nanobelts. From our experiment, one can conclude that the reaction can be controlled via suitable concentrations (a direct synthetic protocol) without any need for stepwise synthesis or post-synthetic purifications.32
Finally, to study the effect of polarity on the reaction process, first, we studied the effect of aging of CsPbBr3 NB phase without quenching it by subjection to washing procedures. Fig. 8a highlights that continuing the reaction for 24 hours has a dramatic impact on PL intensity and peak position as the emission grows strongly and exhibits a uniform redshift from 420 nm to 465 nm over time. This redshift reflects the NB diameter associated with quantum confinement. Moreover, these reactions could be stopped at any time by the addition of antisolvent, e.g., methyl acetate. The PL results are in good agreement with the TEM observations, which confirm the growth of ultrathin nanobelts (∼3 nm) to NBs (∼6 nm) to thicker NBs (∼9 nm), as can be seen from Fig. 8b–e. The formation of nanobelts is quite spontaneous, so the provided precursors were immediately consumed, leading to the formation of the ultrathin NBs. Therefore, we assume that the formation of thicker NBs occurred through the fusing mechanism given in Fig. 8e. Importantly, in our experimental conditions, either side of excess host PPb-br or titrant PCs-ol, could not produce cube-shaped CsPbBr3 NCs. Few reports show the formation of such CsPbBr3 NCs at RT using ligands other than OA and OLAM,16,44 wherein the ligands themselves provide sufficient polar and acidic strength to change the dynamics toward the different sizes and morphologies. Since such conditions were not provided in our reaction system, there is less likelihood of the formation of CsPbBr3 nanocubes. To prove the same, we performed experiments with the addition of ethanol (a polar solvent), and, as expected, we observed the formation of cubic CsPbBr3 instantaneously (Fig. S34†). As prepared highly emissive CsPbBr3 was stable after washing with methyl acetate, centrifuging, and redispersing the pellet in hexane. The addition of microliter quantities of ethanol to the δ = 1:0.25 conditions leads to a redshift of the PL positions with respective quantum-confined nanostructures. The shift is higher (100 nm) at higher amounts of ethanol and lower (30 nm) at 5 μL of ethanol in comparison to that of the nanobelts formed in the absence of polar reagents. Fig. 8f shows photographs of such quantum-confined species and their respective PL emissions. This can be explained that ethanol does not allow sufficient passivation of ligands on the nanobelts’ surface; they tend to grow upon oriented self-assembly to next-level quantum confined structures (Fig. 8g). The thermal and polar influences could aid ligand-destabilization-induced crystallization for the formation of CsPbBr3 NCs.45 These observations can also support the formation of other nanostructures in a solution depending on the polar medium's strength and amount. The growth kinetics of the reaction is highly influenced by the polarity of the solvent, which could significantly alter the reaction pathway.
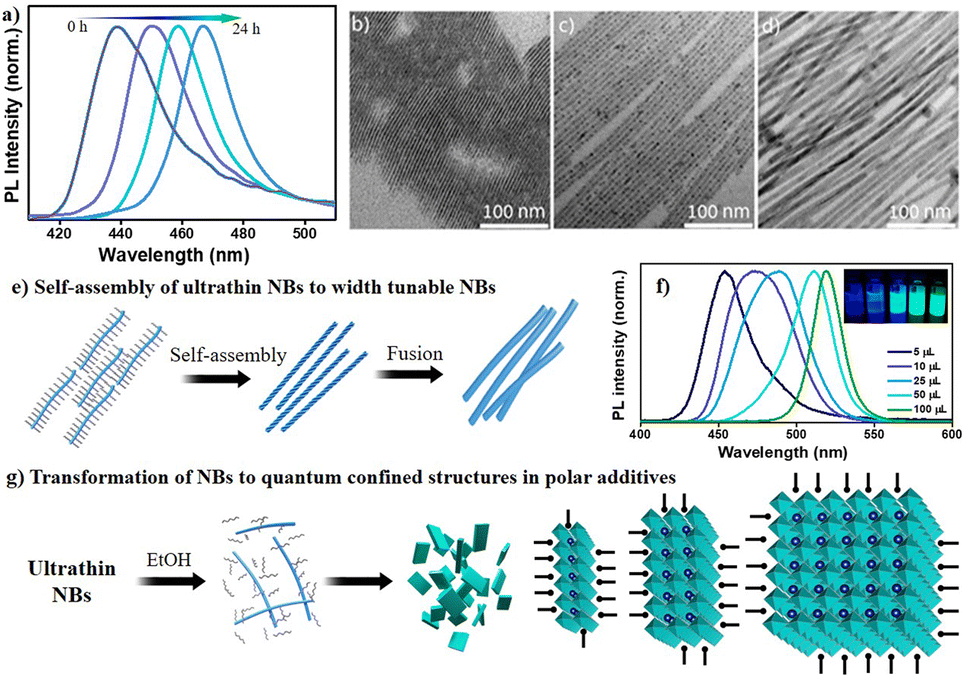 | ||
| Fig. 8 (a) PL emission of ultrathin NBs upon the continuation of reaction for several hours; corresponding TEM images are presented in (b) to (d). (e) Schematic illustration of the RT growth profile of NBs from lamellar-type Cs–Pb–Br structures to 9 nm NBs. (f) PL emission of ethanol-induced growth of NBs to NCs and corresponding photographs under UV light. (g) Schematic illustration of the transformation of NBs in the presence of ethanol resulting in ligand destabilization on the NB surface and followed by proactive growth. | ||
Conclusions
In summary, we have demonstrated the growth kinetics of transformations throughout the reaction path of anionic [PbBrx]n− crystallization with Cs+ as governed by the supply of Cs+ to offer a landscape for diverse cesium–(lead)–bromide NCs at RT in a non-polar solvent. In the process, we provide new insights into the transformation of the crystalline phase and the habit of kinetically stable states. By increasing the Cs+ concentration with respect to PbBr2, we observed the formation of CsPbBr3 NBs with uniform diameters and thickness (δ1), followed by larger rhombohedral and spherical Cs4PbBr6 (δ2), and finally spherical and cubic CsBr NCs (δ3). This study offers possible reasons behind the size and shape control of kinetically stable phases at suitable concentrations and packing patterns of the self-assembled states, for example, the CsPbBr3 nanobelt scission to nanoplatelets, size variation in Cs4PbBr6 NCs, and effect of polarity, dilution, etc. Depending on the crystal nature, they can form large HCP and CCP self-assembled monolayers on solid surfaces, even from the stage of NC seeds. Additionally, this facile method for self-pattern assemblies from these as-synthesized NCs could contribute to the application of single and binary NC superlattices for super-fluorescence. This study highlights that the precise size control of NCs is possible and indeed related to the availability of Cs+ and its dynamic exchange to mediate or guide the crystal habit and growth kinetics. Overall, our findings highlight the concepts of rapid crystallization, shifting the equilibrium to a designated path by chemical input as an internal cue (an unprecedented approach for Cs–Pb–Br metathesis reaction at room temperature) and ultimately controlling the growth kinetics of the perovskite NC systems.Author contributions
UT conceived the project. UT, MP, and AKB designed the experiments. MP, ZG, and YW performed the experiments. MP, UT, and AKB analyzed the data. AKB and UT co-wrote the manuscript and all the authors approved the manuscript. MP and ZG contributed equally.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC Grant No. 22050410280) and UESTC grants. MP and YW would like to acknowledge the University of Electronic Science and Technology of China (UESTC) and Institute of Fundamental and Frontier Sciences (IFFS) for a post-doctoral fellowship. ZG acknowledges the UESTC for a Ph.D Grant. We thank the State Key Laboratory of Electronic Thin Films and Integrated Devices at the UESTC for support.References
- Q. A. Akkerman, G. Rainò, M. V. Kovalenko and L. Manna, Nat. Mater., 2018, 17, 394–405 CrossRef CAS PubMed.
- Y. Xia, Y. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60–103 CrossRef CAS PubMed.
- F. Wang, V. N. Richards, S. P. Shields and W. E. Buhro, Chem. Mater., 2014, 26, 5–21 CrossRef CAS.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed.
- J. Shamsi, A. S. Urban, M. Imran, L. De Trizio and L. Manna, Chem. Rev., 2019, 119, 3296–3348 CrossRef CAS PubMed.
- G. Nedelcu, L. Protesescu, S. Yakunin, M. I. Bodnarchuk, M. J. Grotevent and M. V. Kovalenko, Nano Lett., 2015, 15, 5635–5640 CrossRef CAS PubMed.
- G. Almeida, L. Goldoni, Q. Akkerman, Z. Dang, A. H. Khan, S. Marras, I. Moreels and L. Manna, ACS Nano, 2018, 12, 1704–1711 CrossRef CAS PubMed.
- S. Toso, D. Baranov and L. Manna, ACS Energy Lett., 2020, 5, 3409–3414 CrossRef CAS.
- S. Bera, R. K. Behera, S. D. Adhikari, A. K. Guria and N. Pradhan, J. Phys. Chem. Lett., 2021, 12, 11824–11833 CrossRef CAS PubMed.
- L. Wu, H. Hu, Y. Xu, S. Jiang, M. Chen, Q. Zhong, D. Yang, Q. Liu, Y. Zhao, B. Sun, Q. Zhang and Y. Yin, Nano Lett., 2017, 17, 5799–5804 CrossRef CAS PubMed.
- F. Palazon, C. Urso, L. De Trizio, Q. Akkerman, S. Marras, F. Locardi, I. Nelli, M. Ferretti, M. Prato and L. Manna, ACS Energy Lett., 2017, 2, 2445–2448 CrossRef CAS PubMed.
- J.-R. Wen, B. J. Roman, F. A. Rodriguez Ortiz, N. Mireles Villegas, N. Porcellino and M. Sheldon, Chem. Mater., 2019, 31, 8551–8557 CrossRef CAS.
- J. Shamsi, Z. Dang, P. Ijaz, A. L. Abdelhady, G. Bertoni, I. Moreels and L. Manna, Chem. Mater., 2018, 30, 79–83 CrossRef CAS PubMed.
- T. Udayabhaskararao, L. Houben, H. Cohen, M. Menahem, I. Pinkas, L. Avram, T. Wolf, A. Teitelboim, M. Leskes, O. Yaffe, D. Oron and M. Kazes, Chem. Mater., 2018, 30, 84–93 CrossRef CAS.
- S. K. Balakrishnan and P. V. Kamat, Chem. Mater., 2018, 30, 74–78 CrossRef CAS.
- A. Pan, B. He, X. Fan, Z. Liu, J. J. Urban, A. P. Alivisatos, L. He and Y. Liu, ACS Nano, 2016, 10, 7943–7954 CrossRef CAS PubMed.
- S. Sun, D. Yuan, Y. Xu, A. Wang and Z. Deng, ACS Nano, 2016, 10, 3648–3657 CrossRef CAS PubMed.
- B. A. Koscher, Z. Nett and A. P. Alivisatos, ACS Nano, 2019, 13, 11825–11833 CrossRef CAS PubMed.
- S. Bera, R. K. Behera and N. Pradhan, J. Am. Chem. Soc., 2020, 142, 20865–20874 CrossRef CAS PubMed.
- C. Otero-Martínez, D. García-Lojo, I. Pastoriza-Santos, J. Pérez-Juste and L. Polavarapu, Angew. Chem., Int. Ed., 2021, 60, 26677–26684 CrossRef PubMed.
- Q. A. Akkerman, S. G. Motti, A. R. Srimath Kandada, E. Mosconi, V. D'Innocenzo, G. Bertoni, S. Marras, B. A. Kamino, L. Miranda, F. De Angelis, A. Petrozza, M. Prato and L. Manna, J. Am. Chem. Soc., 2016, 138, 1010–1016 CrossRef CAS PubMed.
- L. Peng, A. Dutta, R. Xie, W. Yang and N. Pradhan, ACS Energy Lett., 2018, 3, 2014–2020 CrossRef CAS.
- Y. Wang, H. Zhao, M. Piotrowski, X. Han, Z. Ge, L. Dong, C. Wang, S. K. Pinisetty, P. K. Balguri, A. K. Bandela and U. Thumu, Micromachines, 2022, 13, 1318 CrossRef PubMed.
- J. C. Dahl, X. Wang, X. Huang, E. M. Chan and A. P. Alivisatos, J. Am. Chem. Soc., 2020, 142, 11915–11926 CrossRef CAS PubMed.
- H. Huang, Y. Li, Y. Tong, E.-P. Yao, M. W. Feil, A. F. Richter, M. Döblinger, A. L. Rogach, J. Feldmann and L. Polavarapu, Angew. Chem., Int. Ed., 2019, 58, 16558–16562 CrossRef CAS PubMed.
- F. Zhang, H. Zhong, C. Chen, X.-g. Wu, X. Hu, H. Huang, J. Han, B. Zou and Y. Dong, ACS Nano, 2015, 9, 4533–4542 CrossRef CAS PubMed.
- Y. Dong, T. Qiao, D. Kim, D. Parobek, D. Rossi and D. H. Son, Nano Lett., 2018, 18, 3716–3722 CrossRef CAS PubMed.
- H. Yang, Y. Zhang, J. Pan, J. Yin, O. M. Bakr and O. F. Mohammed, Chem. Mater., 2017, 29, 8978–8982 CrossRef CAS.
- A. A. M. Brown, P. Vashishtha, T. J. N. Hooper, Y. F. Ng, G. V. Nutan, Y. Fang, D. Giovanni, J. N. Tey, L. Jiang, B. Damodaran, T. C. Sum, S. H. Pu, S. G. Mhaisalkar and N. Mathews, Chem. Mater., 2021, 33, 2387–2397 CrossRef CAS.
- C. Otero-Martínez, J. Ye, J. Sung, I. Pastoriza-Santos, J. Pérez-Juste, Z. Xia, A. Rao, R. L. Z. Hoye and L. Polavarapu, Adv. Mater., 2022, 34, 2107105 CrossRef PubMed.
- X. Li, Y. Wu, S. Zhang, B. Cai, Y. Gu, J. Song and H. Zeng, Adv. Funct. Mater., 2016, 26, 2435–2445 CrossRef CAS.
- Y. Xu, Q. Zhang, L. Lv, W. Han, G. Wu, D. Yang and A. Dong, Nanoscale, 2017, 9, 17248–17253 RSC.
- Y. Liu, M. Guo, S. Dong, X. Jiao, T. Wang and D. Chen, J. Mater. Chem. C, 2018, 6, 7797–7802 RSC.
- A. Kostopoulou, M. Sygletou, K. Brintakis, A. Lappas and E. Stratakis, Nanoscale, 2017, 9, 18202–18207 RSC.
- E. H. Massasa, R. Strassberg, A. Vurgaft, Y. Kauffmann, N. Cohen and Y. Bekenstein, Nano Lett., 2021, 21, 5564–5571 CrossRef CAS PubMed.
- M. C. Weidman, M. Seitz, S. D. Stranks and W. A. Tisdale, ACS Nano, 2016, 10, 7830–7839 CrossRef CAS PubMed.
- Z. Liu, Y. Bekenstein, X. Ye, S. C. Nguyen, J. Swabeck, D. Zhang, S.-T. Lee, P. Yang, W. Ma and A. P. Alivisatos, J. Am. Chem. Soc., 2017, 139, 5309–5312 CrossRef CAS PubMed.
- Q. A. Akkerman, S. Park, E. Radicchi, F. Nunzi, E. Mosconi, F. De Angelis, R. Brescia, P. Rastogi, M. Prato and L. Manna, Nano Lett., 2017, 17, 1924–1930 CrossRef CAS PubMed.
- Y. Yang, J. T. Lee, T. Liyanage and R. Sardar, J. Am. Chem. Soc., 2019, 141, 1526–1536 CAS.
- A. Pan, M. Jurow, Y. Zhao, F. Qiu, Y. Liu, J. Yang, J. J. Urban, L. He and Y. Liu, Nanoscale, 2017, 9, 17688–17693 RSC.
- J. Hui, Y. Jiang, Ö. Ö. Gökçinar, J. Tang, Q. Yu, M. Zhang and K. Yu, Chem. Mater., 2020, 32, 4574–4583 CrossRef CAS.
- I. Cherniukh, G. Rainò, T. Stöferle, M. Burian, A. Travesset, D. Naumenko, H. Amenitsch, R. Erni, R. F. Mahrt, M. I. Bodnarchuk and M. V. Kovalenko, Nature, 2021, 593, 535–542 CrossRef CAS PubMed.
- B. Hudait, S. K. Dutta and N. Pradhan, ACS Energy Lett., 2020, 5, 650–656 CrossRef CAS.
- J. Zhao, S. Cao, Z. Li and N. Ma, Chem. Mater., 2018, 30, 6737–6743 CrossRef CAS.
- B. J. Bohn, Y. Tong, M. Gramlich, M. L. Lai, M. Döblinger, K. Wang, R. L. Z. Hoye, P. Müller-Buschbaum, S. D. Stranks, A. S. Urban, L. Polavarapu and J. Feldmann, Nano Lett., 2018, 18, 5231–5238 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: General information protocols of the titration conditions and 34 supplementary figures. See DOI: https://doi.org/10.1039/d2nr04102b |
| ‡ M. P. and Z. G. contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |