
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUncovering selective and active Ga surface sites in gallia–alumina mixed-oxide propane dehydrogenation catalysts by dynamic nuclear polarization surface enhanced NMR spectroscopy†
Pedro
Castro-Fernández
 a,
Monu
Kaushik
b,
Zhuoran
Wang
b,
Deni
Mance
c,
Evgenia
Kountoupi
a,
Elena
Willinger
a,
Paula M.
Abdala
a,
Christophe
Copéret
c,
Anne
Lesage
*b,
Alexey
Fedorov
*a and
Christoph R.
Müller
*a
a,
Monu
Kaushik
b,
Zhuoran
Wang
b,
Deni
Mance
c,
Evgenia
Kountoupi
a,
Elena
Willinger
a,
Paula M.
Abdala
a,
Christophe
Copéret
c,
Anne
Lesage
*b,
Alexey
Fedorov
*a and
Christoph R.
Müller
*a
aDepartment of Mechanical and Process Engineering, ETH Zürich, CH-8092, Zürich, Switzerland. E-mail: fedoroal@ethz.ch; muelchri@ethz.ch
bHigh-Field NMR Center of Lyon, CNRS, ENS Lyon, Université Lyon1, UMR 5082, F-69100, Villeurbanne, France. E-mail: Anne.Lesage@ens-lyon.fr
cDepartment of Chemistry and Applied Biosciences, ETH Zürich, CH-8093, Zürich, Switzerland
First published on 12th November 2021
Abstract
Gallia–alumina (Ga,Al)2O3(x![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :y) spinel-type solid solution nanoparticle catalysts for propane dehydrogenation (PDH) were prepared with four nominal Ga:Al atomic ratios (1:6, 1:3, 3:1, 1:0) using a colloidal synthesis approach. The structure, coordination environment and distribution of Ga and Al sites in these materials were investigated by X-ray diffraction, X-ray absorption spectroscopy (Ga K-edge) as well as 27Al and 71Ga solid state nuclear magnetic resonance. The surface acidity (Lewis or Brønsted) was probed using infrared spectroscopy with pyridine and 2,6-dimethylpyridine probe molecules, complemented by element-specific insights (Ga or Al) from dynamic nuclear polarization surface enhanced cross-polarization magic angle spinning 15N{27Al} and 15N{71Ga} J coupling mediated heteronuclear multiple quantum correlation NMR experiments using 15N-labelled pyridine as a probe molecule. The latter approach provides unique insights into the nature and relative strength of the surface acid sites as it allows to distinguish contributions from Al and Ga sites to the overall surface acidity of mixed (Ga,Al)2O3 oxides. Notably, we demonstrate that (Ga,Al)2O3 catalysts with a high Al content show a greater relative abundance of four-coordinated Ga sites and a greater relative fraction of weak/medium Ga-based surface Lewis acid sites, which correlates with superior propene selectivity, Ga-based activity, and stability in PDH (due to lower coking). In contrast, (Ga,Al)2O3 catalysts with a lower Al content feature a higher fraction of six-coordinated Ga sites, as well as more abundant Ga-based strong surface Lewis acid sites, which deactivate through coking. Overall, the results show that the relative abundance and strength of Ga-based surface Lewis acid sites can be tuned by optimizing the bulk Ga:Al atomic ratio, thus providing an effective measure for a rational control of the catalyst performance.
:y) spinel-type solid solution nanoparticle catalysts for propane dehydrogenation (PDH) were prepared with four nominal Ga:Al atomic ratios (1:6, 1:3, 3:1, 1:0) using a colloidal synthesis approach. The structure, coordination environment and distribution of Ga and Al sites in these materials were investigated by X-ray diffraction, X-ray absorption spectroscopy (Ga K-edge) as well as 27Al and 71Ga solid state nuclear magnetic resonance. The surface acidity (Lewis or Brønsted) was probed using infrared spectroscopy with pyridine and 2,6-dimethylpyridine probe molecules, complemented by element-specific insights (Ga or Al) from dynamic nuclear polarization surface enhanced cross-polarization magic angle spinning 15N{27Al} and 15N{71Ga} J coupling mediated heteronuclear multiple quantum correlation NMR experiments using 15N-labelled pyridine as a probe molecule. The latter approach provides unique insights into the nature and relative strength of the surface acid sites as it allows to distinguish contributions from Al and Ga sites to the overall surface acidity of mixed (Ga,Al)2O3 oxides. Notably, we demonstrate that (Ga,Al)2O3 catalysts with a high Al content show a greater relative abundance of four-coordinated Ga sites and a greater relative fraction of weak/medium Ga-based surface Lewis acid sites, which correlates with superior propene selectivity, Ga-based activity, and stability in PDH (due to lower coking). In contrast, (Ga,Al)2O3 catalysts with a lower Al content feature a higher fraction of six-coordinated Ga sites, as well as more abundant Ga-based strong surface Lewis acid sites, which deactivate through coking. Overall, the results show that the relative abundance and strength of Ga-based surface Lewis acid sites can be tuned by optimizing the bulk Ga:Al atomic ratio, thus providing an effective measure for a rational control of the catalyst performance.
Introduction
The growing demand for propene has motivated its on-purpose production through the propane dehydrogenation reaction (PDH, Scheme 1A). However, catalysts used at the industrial scale, i.e. CrOx/Al2O3 (Catofin process) or PtSn/Al2O3 (Oleflex process), still suffer from shortcomings, such as toxicity concerns associated with the use of Cr6+ and the high cost of Pt.1,2 Catalysts based on gallium oxide (gallia) have been considered as possible alternatives to CrOx and PtSn-based PDH catalysts.1,2 More recently, a PtGa/Al2O3 catalyst has been developed for the use in the FCDh (fluidized catalytic dehydrogenation) process, which allows energy usage (and therefore the carbon footprint) of PDH to be reduced.3 However, the role of platinum in PtGa catalysts is currently debated, ranging from a promoter of coordinately unsaturated Ga3+ active sites,4 to the active phase itself, such as site-isolated Pt atoms in metallic PtGa alloyed nanoparticles.5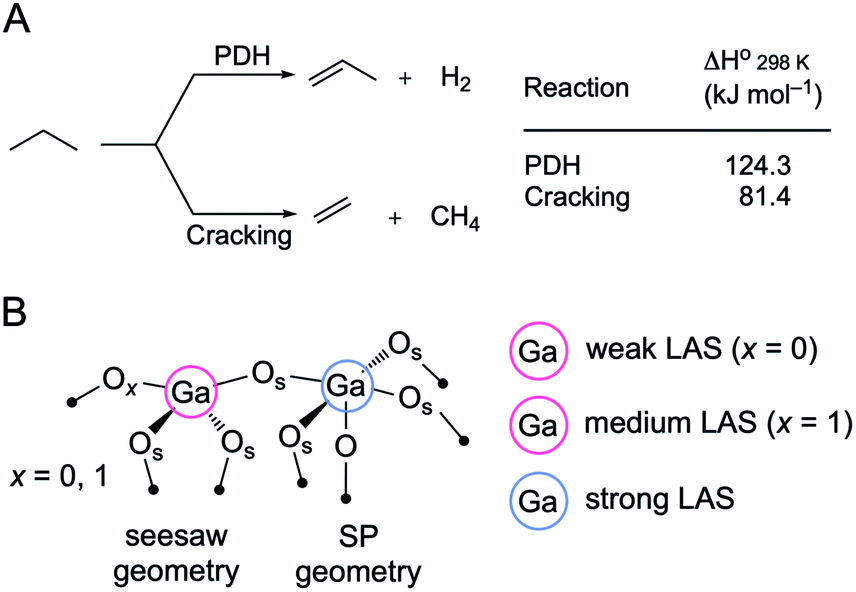 | ||
| Scheme 1 (A) Competing propane dehydrogenation (PDH) and propane cracking reactions. (B) A possible coordination geometry for a GaIV–O–GaVI surface linkage with the attribution of Lewis acidity strength according to the 15N chemical shift of bound pyridine probe molecule.12 SP stands for square pyramidal. | ||
In this context, the atomic-scale understanding of active sites in Ga2O3-based PDH catalysts is essential.6–8 It has been argued that the active sites in gallia catalysts are tetracoordinated (GaIV) Lewis acidic Ga3+ surface sites,1,9 associated with weak Lewis acidity.10,11 In β-Ga2O3, weak Lewis acid sites (LAS, assessed using pyridine as a probe molecule) active in PDH have been attributed to tricoordinated Ga sites (GaIII) with a neighboring oxygen vacancy (Vo, when x = 0 in Scheme 1B).12 In contrast, stronger LAS have been associated with an accelerated catalyst deactivation through coking and linked to pentacoordinated Ga surface sites (GaV, likely with square pyramidal (SP) geometry, formed when surface termination contains Ga in octahedral positions, Scheme 1B).12 In addition to coking, another undesired side reaction that competes with PDH is the cracking of propane, which forms methane and ethene (Scheme 1A). Cracking could be caused by either Lewis or Brønsted acidity.13 PDH and cracking reactions proceed on different sites in Ga2O3 catalysts since the rate of propene formation and propene selectivity decrease with time on stream (TOS) but the selectivity to cracking products and their rate of formation is stable with TOS.12
One strategy to control the distribution of bulk GaIV and GaVI sites, and thereby presumably also influence the coordination environment of surface Ga sites, is to exploit (Ga,Al)2O3 spinel-type solid solutions as alkane dehydrogenation catalysts.6,9,14 In these materials, the bulk GaIV:GaVI ratio can be varied since Al atoms preferentially occupy octahedral positions in the defect spinel-type structure of Ga2O3,15 increasing thereby the relative fraction of GaIV sites (i.e., the proposed active sites). For instance, gallia–alumina mixed oxides prepared by coprecipitation demonstrated an improved activity and stability in PDH and lower coke-related deactivation relative to the γ-Ga2O3 benchmark.9 The reduced deactivation and increased activity might be related to the higher relative fraction of weak LAS in (Ga,Al)2O3 (as assessed by NH3-TPD) and linked to a higher relative density of undercoordinated Ga sites, such as putative GaIII sites discussed above.9,12 Note that the alkane dehydrogenation activity of various phases of Al2O3 (α-, δ-, γ-, θ-) is generally low and requires a pre-treatment with CO or H2 at high temperature (600 °C) to become considerable.16,17 Due to their limited activity, Al2O3 catalysts are typically tested in PDH at substantially higher temperatures (600–630 °C) relative to Ga2O3 catalysts (550 °C).16,18
DFT studies proposed that doping (100) and (110) γ-Al2O3 surfaces with Ga could lead to Al-rich gallia–alumina solid solutions with an increased dehydrogenation activity.19,20 The presence of Ga sites has been suggested to increase the activity of such surfaces by lowering the C–H activation barriers of the kinetically favored concerted alkane dehydrogenation pathway.19 In addition, the increase in PDH activity of the Ga-doped (110) γ-Al2O3 surface was attributed to vicinal AlIII and GaIV sites.20 However, an experimental determination of the coordination geometry, Lewis acidity and performance in PDH (i.e., activity, selectivity and stability) of Ga surface sites in the presence of Al surface sites on the surface of mixed (Ga,Al)2O3 catalysts is highly challenging.
Dynamic nuclear polarization surface enhanced NMR spectroscopy (DNP SENS) has emerged over the last decade as a unique approach to probe the surface structure of active sites in catalytic materials.21 For instance, it has been recently demonstrated that unique structural information could be obtained on the nature and relative strength of acid sites in alumina or silica-alumina materials from 15N DNP enhanced NMR spectroscopy using 15N-labelled pyridine as a probe molecule.22–24
In this work, we prepare nanocrystalline (Ga,Al)2O3 solid solution nanoparticles (NPs) via a colloidal route from Ga3+ and Al3+ acetylacetonate precursors and oleylamine. Al-rich (Ga,Al)2O3(1:6) produces the least amount of coke (and deactivates least with TOS), while (Ga,Al)2O3(1:3) is the most active catalyst (initial Ga-based activity). The variation of the PDH performance of (Ga,Al)2O3 NP catalysts is related to their surface acidity, assessed by Fourier transform infrared (FTIR) studies of adsorbed pyridine and 2,6-dimethylpyridine probe molecules, in addition to element-specific insights from a combination of DNP SENS experiments, namely cross-polarization magic angle spinning (CPMAS), 15N{27Al} and 15N{71Ga} J coupling mediated heteronuclear multiple quantum correlation (J-HMQC) experiments, using adsorbed 15N pyridine. These studies provide atomic level insights on the coordination geometry and Lewis acidity of surface sites, allowing one to categorize individual contributions of Al and Ga Lewis sites. In addition, we correlate element-specific Al and Ga Lewis acidity to the distribution of coordination environments of bulk Ga (GaIV and GaVI) and Al (AlIV, AlV and AlVI) sites obtained from fittings of 27Al and 71Ga solid-state NMR spectra. The improved propene selectivity, stability and Ga-based activity of Al-rich catalysts is related to an increased relative abundance of surface GaIV–O–AlVI linkages. These active sites feature a decreased Lewis acidity of Ga atoms relative to the Lewis acidity of Ga atoms in gallia-only or (Ga,Al)2O3 materials with low Al content, which is directly related to their high Ga-based catalytic activity, propene selectivity and stability in PDH.
Results
Synthesis and characterization
Colloidal solutions of gallia–alumina nanoparticles were prepared by heating Ga(acac)3 and Al(acac)3 precursors in oleylamine (200 °C, 7 h), using four nominal Ga:Al molar ratios, i.e. 1:6, 1:3, 3:1 and 1:0, the latter composition provided an Al-free γ-Ga2O3 NPs benchmark (Fig. 1).25 The synthesis of colloidal Al2O3 NPs by the same method was unsuccessful (i.e., no material could be isolated). The diameters of the as-synthesized NPs as determined by TEM were below 5 nm for all compositions (particle agglomeration prevents a more precise assessment of the particle size distribution, see Fig. 1 for a representative TEM image). After washing (see ESI† for details), the toluene colloidal solutions were dried and calcined (650 °C, 2 h) to give (Ga,Al)2O3(x:y) NPs, where x:y denotes the nominal molar Ga:Al ratio used.
 | ||
| Fig. 1 (A) Synthesis of (Ga,Al)2O3(x:y) catalysts, where the x:y index indicates the nominal Ga:Al molar ratio used for the synthesis (1:6, 1:3, 3:1, 1:0). (B) A representative TEM image of as-prepared (Ga,Al)2O3(1:3) nanoparticles in toluene colloidal solution. | ||
The specific surface areas and pore diameters of the prepared materials were determined by applying the Brunauer–Emmett–Teller (BET) method and Barrett–Joyner–Halenda (BJH) model to the N2 physisorption data, respectively (Table S1†). The addition of Al(acac)3 in the synthesis of (Ga,Al)2O3(x:y) NPs leads to a notable increase of the BET surface area of the calcined materials, that is from 98 m2 g−1 for Ga2O3 NPs (x:y = 1:0) to ca. 245–286 m2 g−1 for Al-containing (Ga,Al)2O3(x:y) NPs; an increase of the BJH pore diameter accompanies the increase of the BET surface area (Table S1†).
Inductively coupled plasma – optical emission spectroscopy (ICP-OES) measurements were performed to compare the experimental atomic Ga:Al ratios to the nominal compositions used (Table S1†). The ICP-OES determined Ga:Al ratio is closest to the nominal ratio in (Ga,Al)2O3(1:6) (1:5.6), and are ca. 2:1 and 1:2 for (Ga,Al)2O3(3:1) and (Ga,Al)2O3(1:3), respectively.
The X-ray powder diffractograms of the (Ga,Al)2O3(x:y) materials display peaks of the γ-phase (cubic spinel-type structure, Fig. S1†).26 We have recently reported that calcination of γ-Ga2O3 NPs, prepared by a colloidal route from Ga(acac)3 and oleylamine, induces a phase transition of γ-Ga2O3 to the thermodynamically stable monoclinic β-Ga2O3 phase. This transition starts already at ca. 300 °C in the local environment of the Ga atoms, while the transformation of the periodic structure sets in at ca. 550 °C.27 Therefore, we assessed to which extent calcined (Ga,Al)2O3(x:y) materials (650 °C, 2 h) have undergone this spinel-type-to-monoclinic phase transition. While the gallia-only material (Ga,Al)2O3(1:0) shows clearly peaks of the β-Ga2O3 phase (marked by red symbols in Fig. S1†), the X-ray diffraction (XRD) patterns of Al-containing (Ga,Al)2O3 nanoparticles are consistent with a cubic spinel-type structure with no apparent transformation to the monoclinic phase (Fig. S1†), i.e. Al atoms stabilize notably the spinel structure.9,28–31 A linear increase of the lattice parameter (a) is observed with increasing Ga at% in (Ga,Al)2O3(x:y) materials (Fig. S1†), explained by the lower atomic radius of Al compared to that of Ga (125 pm and 130 pm, respectively).32 Using the Scherrer equation,33 we estimated the average crystallite sizes in (Ga,Al)2O3(x:y) NPs and observed a decrease of the average crystallite size when Al is introduced into the structure of γ-Ga2O3, i.e. from ca. 5 nm in Ga-only (Ga,Al)2O3(1:0) to 2–3 nm in Al-containing materials (Table S1†). Overall, the XRD data is consistent with the formation of a solid solution with a spinel-type structure in all gallia–alumina materials.31
Ga K-edge X-ray absorption near edge structure (XANES) spectra of (Ga,Al)2O3(x:y) materials provide information about the relative abundances of GaIV and GaVI sites in the prepared materials. The features at ca. 10375 eV and 10380 eV are related to GaIV and GaVI sites, respectively.34 Consistent with previous studies,31 increasing the content of Al atoms in (Ga,Al)2O3(x:y) materials leads to a higher relative fraction of GaIV sites, as seen from the increased intensity of the white line feature of the GaIV sites (and the correspondingly decreased intensities of GaVI features), in particular for (Ga,Al)2O3(1:6) and (Ga,Al)2O3(1:3) (Fig. 2A). The local environment around Ga, was investigated by the extended X-ray absorption fine structure (EXAFS), exhibiting two distinguishable peaks due to Ga–O and Ga–Ga/Al coordination shells (Fig. 2B). The corresponding fittings provided average coordination numbers (CN) and average interatomic distances. We modeled the EXAFS data using one average Ga–O shell and two Ga–Ga/Al subshells. In general, shorter average Ga–O distances and lower average Ga–O coordination numbers can be related to a higher fraction of GaIV sites. The fitting results presented in Table S2 and Fig. S2† show a decrease in the average Ga–O distances and coordination numbers with increasing Al loadings in the (Ga,Al)2O3(x:y) materials, consistent with the corresponding XANES data. In particular, the average Ga–O distance decreases from 1.91(1) to 1.89(1), 1.86(1) and 1.84(1) Å for (Ga,Al)2O3(x:y) NPs with increasing nominal Al content from 1:0 to 3:1, 1:3 and 1:6 (Table S2†).
 | ||
| Fig. 2 Structural characterization of the calcined (Ga,Al)2O3(x:y) materials. (A) Ga K-edge XANES spectra, and (B) Fourier transform of the EXAFS data of (Ga,Al)2O3(x:y) NPs. Captions in (A) indicate features corresponding to GaIV and GaVI sites and captions of the peaks in (B) indicate the Ga–O and Ga–Ga/Al coordination sphere. Panels (C) and (D) show 27Al and 71Ga MAS NMR spectra, respectively, obtained at 700 MHz in 1.3 mm rotors spun at 50 kHz. (E) ADF-STEM images of (Ga,Al)2O3(1:3) showing terminating rows of octahedral cation sites. In (F), the region in the white box features a spinel-type structure oriented along the [100]-zone axis (FFT pattern is shown in the top right inset), with arrows indicating Ga atoms in tetrahedral and octahedral crystallographic positions. In (G), the image shows a (Ga,Al)2O3(1:6) particle, also oriented along the [100]-zone axis, where the arrows indicate preferential occupation of tetrahedral sites by Ga. The spinel model in (H) shows Ga occupying both tetrahedral and octahedral positions, as in the case of (Ga,Al)2O3(1:3). (I) Shows an ADF-STEM model for the spinel structure along the [100]-zone axis, where Ga and Al occupy tetrahedral and octahedral positions, respectively. | ||
The relative distributions of Ga and Al sites between tetrahedral and octahedral geometries were then assessed quantitatively using 27Al and 71Ga magic-angle spinning nuclear magnetic resonance spectroscopy (MAS NMR). 27Al MAS NMR spectra feature asymmetric peaks due to distorted AlIV and AlVI sites centered at ca. 70 and 15 ppm, respectively, although an additional minor peak at ca. 35 ppm, attributed to AlV sites, appears in Al-rich (Ga,Al)2O3(1:6) (Fig. 2C).3571Ga MAS NMR spectra show two peaks located at ca. 150 and 30 ppm, associated with distorted GaIV and GaVI sites, respectively (Fig. 2D).31 Quantification of the relative ratios, average isotropic chemical shift (![[small delta, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0c5.gif) iso), distribution of isotropic chemical shift (Δδiso) and average quadrupolar coupling constant (
iso), distribution of isotropic chemical shift (Δδiso) and average quadrupolar coupling constant (![[C with combining macron]](https://www.rsc.org/images/entities/i_char_0043_0304.gif) Q) of the Ga and Al sites in the materials was performed using the Gaussian isotropic model (GIM) or Czjzek model implemented in the DMFit software (Fig. S3, Tables S3 and S4†).36–38 In the case of 71Ga, the quadrupolar interaction is significantly larger than the spinning speed, and the computation takes into account the latter to reproduce the many spinning sidebands observed for this nuclei. The relative abundance of GaIV sites in the prepared materials increases with Al content, i.e. from 57% in (Ga,Al)2O3(3:1) to 72% in (Ga,Al)2O3(1:3) and 86% and (Ga,Al)2O3(1:6) (Fig. S3, Table S3†). The highest amount of AlVI sites (92%) is found in (Ga,Al)2O3(3:1), and this amount decreases to 88% and 66% for (Ga,Al)2O3(1:3) and (Ga,Al)2O3(1:6), i.e. with increasing Al content. In Al-rich (Ga,Al)2O3(1:6), the fitted fraction of AlIV sites is 22%, and decreases to 12% and 8% in (Ga,Al)2O3(1:3) and (Ga,Al)2O3(3:1), respectively (Fig. S3, Table S4†). The fitted fraction of AlV sites in (Ga,Al)2O3(1:6) is 12%. This value appears to be slightly higher than the AlV values reported for γ-Al2O3 (5–10%).39 Residual amounts of AlV sites (<10%) could be possibly present in (Ga,Al)2O3(1:3), but their low intensity and high intrinsic error in the fit (due to strongly overlapping contributions) does not allow to accurately quantify them. Besides a slight decrease in the chemical shifts with increasing Al content, no notable changes could be observed neither in the average chemical shifts nor in the average quadrupolar couplings between the different (Ga,Al)2O3 materials (Fig. S3, Table S4†). The spectra of (Ga,Al)2O3(1:0) are complicated by the coexistence of γ and β polymorphs, and no robust simulations could be performed in this case. The β polymorph is nevertheless known to show both GaVI and GaIV environments with δiso = 40 and 200 ppm and CQ = 13.4 and 17.5 MHz, respectively.40
Q) of the Ga and Al sites in the materials was performed using the Gaussian isotropic model (GIM) or Czjzek model implemented in the DMFit software (Fig. S3, Tables S3 and S4†).36–38 In the case of 71Ga, the quadrupolar interaction is significantly larger than the spinning speed, and the computation takes into account the latter to reproduce the many spinning sidebands observed for this nuclei. The relative abundance of GaIV sites in the prepared materials increases with Al content, i.e. from 57% in (Ga,Al)2O3(3:1) to 72% in (Ga,Al)2O3(1:3) and 86% and (Ga,Al)2O3(1:6) (Fig. S3, Table S3†). The highest amount of AlVI sites (92%) is found in (Ga,Al)2O3(3:1), and this amount decreases to 88% and 66% for (Ga,Al)2O3(1:3) and (Ga,Al)2O3(1:6), i.e. with increasing Al content. In Al-rich (Ga,Al)2O3(1:6), the fitted fraction of AlIV sites is 22%, and decreases to 12% and 8% in (Ga,Al)2O3(1:3) and (Ga,Al)2O3(3:1), respectively (Fig. S3, Table S4†). The fitted fraction of AlV sites in (Ga,Al)2O3(1:6) is 12%. This value appears to be slightly higher than the AlV values reported for γ-Al2O3 (5–10%).39 Residual amounts of AlV sites (<10%) could be possibly present in (Ga,Al)2O3(1:3), but their low intensity and high intrinsic error in the fit (due to strongly overlapping contributions) does not allow to accurately quantify them. Besides a slight decrease in the chemical shifts with increasing Al content, no notable changes could be observed neither in the average chemical shifts nor in the average quadrupolar couplings between the different (Ga,Al)2O3 materials (Fig. S3, Table S4†). The spectra of (Ga,Al)2O3(1:0) are complicated by the coexistence of γ and β polymorphs, and no robust simulations could be performed in this case. The β polymorph is nevertheless known to show both GaVI and GaIV environments with δiso = 40 and 200 ppm and CQ = 13.4 and 17.5 MHz, respectively.40
Annular dark-field scanning transmission electron microscopy imaging (ADF-STEM) was carried out to obtain insight into the distribution of Ga and Al sites (distinguished by a brighter contrast for Ga atoms relative to Al atoms) on the surface of (Ga,Al)2O3(1:3) and (Ga,Al)2O3(1:6) nanoparticles. While the particles are often found to be poorly crystalline (Fig. S4†), we observe that the terminating surfaces have an enhanced contrast over the bulk (marked by arrows in Fig. 2E, corresponding to (Ga,Al)2O3(1:3)). For γ-Al2O3, it has been suggested that this enhanced contrast is due to aluminum-terminated (111) and (100) planes.41,42 This literature result is similar to what we have reported for γ-Ga2O3 nanoparticles, which feature Ga-terminated (111) facets in solely octahedral positions.27 In a mixed (Ga,Al)2O3 oxide, the enhanced contrast at the particle edge could be due to a mixture of Ga and Al cations. However, given that Al atoms feature lower line profile intensities relative to Ga atoms, the intensity line profile suggests that the particle edge contains mainly aluminum cations (Fig. S5†).
Based on ADF-STEM images of (Ga,Al)2O3(1:3) particles (boxed region in Fig. 2F) and (Ga,Al)2O3(1:6) (Fig. 2G), we can observe an atomic arrangement that is characteristic for a spinel-type structure (Fig. 2H) oriented along the [100]-zone axis (FFT pattern in the top right inset in Fig. 2F). The spinel tetrahedral and octahedral positions are distinguishable along this zone axis. The non-spinel positions are not shown due to their low occupancies and, therefore, low contribution to the contrast of the ADF-STEM image. The analysis of the ADF-STEM image shows that Ga atoms (bright dots) can be found in both tetrahedral and octahedral positions (Fig. 2F). In (Ga,Al)2O3(1:6), Ga atoms are found to occupy mainly tetrahedral positions (marked by arrows in Fig. 2G), which is consistent with the NMR and X-ray absorption spectroscopy (XAS) results discussed above. Fig. 2I shows an ADF-STEM model for the spinel structure oriented along the [100] zone axis, built on the assumption that Ga occupies solely tetrahedral sites, while Al occupies octahedral sites. A clear resemblance is observed between the model in Fig. 2I and the experimental ADF image shown in Fig. 2G, confirming the preferential Ga occupancies of tetrahedral sites in (Ga,Al)2O3(1:6). An additional discussion of the results of the electron energy loss spectroscopy (EELS) and X-ray photoelectron spectroscopy (XPS) is provided in the ESI (Fig. S13 and S14, respectively†).
Catalytic tests and coke deposition
Catalytic tests were performed at 550 °C and at a weight hourly space velociy (WHSV) = 7.2 h−1 using 10% propane in N2. The changes of the Ga weight-normalized (ICP based) rates of formation of propene and cracking products (methane and ethene) as well as the selectivity to propene with time on stream are plotted in Fig. 3. The activity of the tested catalysts is normalized by the Ga content determined by ICP-OES measurements (mol C3H6 mol Ga−1 h−1) because, as we have discussed above, the activity of unsupported Al-based catalysts in PDH becomes significant only at higher temperatures (600–630 °C) and requires a CO pre-treatment;16,17 both of those requirements are not available in our experiments. Therefore, we consider Al sites inactive when tested in the present conditions. Table S5† summarizes the propane conversions and propene selectivities after 4 and 144 min TOS. The amount of coke deposited by the catalysts (determined in a separate thermogravimetric analysis (TGA) experiment) is presented as weight gain (%) normalized by either BET surface area or Ga wt% (Table S5†). Surface area-normalized activities (based on the surface area of the calcined NPs and expressed in μmol C3H6 m−2 h−1) are also presented in Fig. S6† and summarized in Table S6.†The initial Ga-normalized activity after 4 min TOS decreases as following: (Ga,Al)2O3(1:3) > (Ga,Al)2O3(1:6) > (Ga,Al)2O3(3:1) > (Ga,Al)2O3(1:0) (4.48, 3.41, 2.24 and 1.21 mol C3H6 mol Ga−1 h−1, respectively, Fig. 3A). While all catalysts deactivate, the activity of (Ga,Al)2O3(1:6) increases first to 4.51 mol C3H6 mol Ga−1 h−1 within 24 min TOS, before deactivation also sets in for this catalyst. After 144 min of TOS, the catalysts have deactivated to varying extents, i.e. the decrease of activity is less pronounced for the Ga-poor materials (Ga,Al)2O3(1:6) and (Ga,Al)2O3(1:3) (decrease to 3.32 and 2.67 mol C3H6 mol Ga−1 h−1, respectively) and more noticeable for the Ga-rich materials (Ga,Al)2O3(3:1) and especially (Ga,Al)2O3(1:0) (decrease to 0.6 and 0.2 mol C3H6 mol Ga−1 h−1, respectively). When comparing the productivity, (Ga,Al)2O3(1:3) also outperforms the other three catalysts, showing productivities of 0.80 and 0.47 g C3H6 gcat h−1 after 4 and 144 min TOS, respectively (Fig. S7, Table S6†).
The initial surface area-normalized activity shows a different activity trend, i.e. (Ga,Al)2O3(1:0) > (Ga,Al)2O3(3:1) > (Ga,Al)2O3(1:3) > (Ga,Al)2O3(1:6), (125, 77, 66 and 39 μmol C3H6 m−2 h−1, respectively, Fig. S6, Table S6†), which is explained by decreasing the surface density of active sites (Ga atoms) with increasing Al content. Therefore, comparing the catalysts' activity after normalization by their Ga content (as presented in Fig. 3) may reflect the intrinsic activities of the active sites more precisely, at least for (Ga,Al)2O3(1:3), (Ga,Al)2O3(1:6) and (Ga,Al)2O3(3:1) catalysts that feature similar specific surface areas (Table S1†). Noteworthy, the surface area-normalized PDH activity and propene selectivity of the bulk γ-Al2O3 reference catalyst are poor, i.e. ca. 8 μmol C3H6 m−2 h−1, ca. 15 times lower than the initial areal activity of (Ga,Al)2O3(1:0), and 42%, respectively (Fig. S8†).
Interestingly, while the selectivity to propene declines with TOS from 87% to 62% for (Ga,Al)2O3(1:0) and from 76% to 68% for Ga-rich (Ga,Al)2O3(3:1) (after a ca. 20 min initial increase of propene selectivity for this catalyst), the propene selectivity is high and stable at ca. 86–90% for the Al-rich (Ga,Al)2O3(1:3) and (Ga,Al)2O3(1:6) materials (Fig. 3B, Table S5†). (Ga,Al)2O3(1:0), and the Al-rich catalysts (Ga,Al)2O3(1:3) and (Ga,Al)2O3(1:6), crack propane to methane and ethene with similar stable activities of ca. 0.1 mol (C1 + C2=) mol Ga−1 h−1. (Ga,Al)2O3(3:1) shows an initial cracking activity of ca. 0.4 mol (C1 + C2=) mol Ga−1 h−1 that decreases within the first ca. 24 min TOS and then stabilizes at ca. 0.16 mol (C1 + C2=) mol Ga−1 h−1 (Fig. 3C).
In situ TGA measurements under reaction conditions show that (Ga,Al)2O3(1:0) deposits higher amounts of coke per surface area relative to the other catalysts studied and that the amount of deposited coke decreases with the increasing Al content (Table S5†). In contrast, the reference γ-Al2O3 material shows no detectable coke deposition. If the amount of coke is normalized per Ga content, (Ga,Al)2O3(1:3) deposits the highest amounts of carbon while Al-rich (Ga,Al)2O3(1:6) deposits the lowest amounts of carbon.
Acidity of surface sites by Py-FTIR
To probe the nature of the surface acid sites (i.e., Brønsted vs. Lewis), their strengths and distributions, a Fourier-transform infrared spectroscopy study was performed using pyridine as a probe molecule (Py-FTIR). Pyridine was adsorbed at room temperature on self-supporting pellets of the catalysts that had been previously outgassed at 500 °C under ca. 10−5 mbar for 2 h, followed by pyridine desorption at room temperature, 100, 200 and 300 °C.43–47 The spectra and description of results obtained are presented in the ESI (Fig. S9–S11. Table S7†). In brief, we observe that bands ascribed to Py on weak LAS are more abundant in Al-rich (Ga,Al)2O3(1:6) and (Ga,Al)2O3(1:3) NPs. Strong LAS are present in all four (Ga,Al)2O3 catalysts, but their relative fraction is lowest in Al-rich (Ga,Al)2O3(1:6). Experiments were also performed using 2,6-dimethylpyridine as the probe molecule, but these experiments did not yield additional insights relative to the Py-FTIR results (Fig. S12†).
Acidity of surface sites by DNP SENS of adsorbed 15N pyridine
In order to obtain further insight into the strength, the distribution and the nature of LAS and BAS on the surface of (Ga,Al)2O3 NPs, dynamic nuclear polarization surface enhanced NMR spectroscopy48 of adsorbed 15N-labelled pyridine was applied. This surface selective spectroscopy has been recently shown to be a powerful approach to characterize acid surface sites because the 15N chemical shifts are sensitive to the nature and strength of pyridine adsorption onto Lewis or Brønsted surface sites.12,22,23,49DNP enhanced 15N{1H} CPMAS spectra were acquired for (Ga,Al)2O3(1:6), (Ga,Al)2O3(1:3), (Ga,Al)2O3(3:1) and (Ga,Al)2O3(1:0) (Fig. 4A). Three major peaks resonating at ca. 240, 265 and 281 ppm are identified from the CPMAS spectrum of (Ga,Al)2O3(1:0), which are assigned to strong, medium and weak gallium LAS, respectively (Fig. 4A).12 A minor peak is also observed at ca. 305 ppm corresponding to pyridine adsorbed on weak BAS. It should be noted that the peak at 281 ppm has been commonly linked to the presence of weak or mild BAS, in particular in silica- and silica-alumina supported materials with abundant surface silanols; however, we have recently shown, based on DFT modeling and chemical shift calculations on gallia materials, that a peak at this chemical shift is related to weak LAS rather than to weak or mild BAS (i.e., GaOH sites).12 In the CPMAS spectrum of (Ga,Al)2O3(3:1) the three mentioned LAS peaks are observed as well, while the minor peak at ca. 305 ppm disappears, suggesting that the amount of weak BAS associated with Ga is reduced when Al is introduced. Deconvolution of the CPMAS spectra of (Ga,Al)2O3(1:3) and (Ga,Al)2O3(1:6) clearly demonstrates that increasing further the content of Al results in the appearance of an additional strong LAS peak resonating at ca. 234–235 ppm (Fig. 4A; Table S8†). This emerging site is likely related to surface Al atoms. The CPMAS spectrum of (Ga,Al)2O3(1:6) shows the minor peak at ca. 305 ppm again, corresponding to weak BAS. Due to the high content of Al in this material, this weak BAS probably relates to aluminols instead of ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) GaOH sites. By comparing the CPMAS spectrum of (Ga,Al)2O3(3:1) with that of (Ga,Al)2O3(1:0), we observe that the intensity of weak LAS peak at ca. 281 ppm decreases with the incorporation of Al (from 14% to 11–9%, see Table S8†). Varying further the ratio between Ga and Al in a large range (i.e., from 3:1 to 1:6 nominal ratio) does not significantly change the relative amount of weak LAS (ca. 10%). In parallel, we observe clearly that the intensity of the strong LAS peak at around 241 ppm decreases with increasing Al content.
GaOH sites. By comparing the CPMAS spectrum of (Ga,Al)2O3(3:1) with that of (Ga,Al)2O3(1:0), we observe that the intensity of weak LAS peak at ca. 281 ppm decreases with the incorporation of Al (from 14% to 11–9%, see Table S8†). Varying further the ratio between Ga and Al in a large range (i.e., from 3:1 to 1:6 nominal ratio) does not significantly change the relative amount of weak LAS (ca. 10%). In parallel, we observe clearly that the intensity of the strong LAS peak at around 241 ppm decreases with increasing Al content.
 | ||
| Fig. 3 Results of the PDH catalytic tests, i.e. Ga weight-normalized activity (A), selectivity to propene (B), and formation rates to cracking products (combined rates to ethene and methane), (C) for the (Ga,Al)2O3 catalysts. WHSV = 7.2 h−1, T = 550 °C. Initial and final conversions and selectivities are presented in Table S5,† while surface area-normalized, Ga-weight normalized activities and productivities are in Table S6.† | ||
Element-specific (Ga or Al) acidity of surface sites
In the materials containing both Ga and Al, the surface acidity can be associated with either Ga or Al atoms. To further characterize the nature of the LAS observed in the 15N{1H} CPMAS spectra, 1D 15N{27Al} J-HMQC spectra were acquired for (Ga,Al)2O3(1:6), (Ga,Al)2O3(1:3) and (Ga,Al)2O3(3:1) (see Fig. 4B). This experiment relies on the existence of a sizeable J coupling between 15N and 27Al spin pair and therefore can be used to probe Al-based LAS; the one-bond J(15N–27Al) coupling value increasing with Lewis acidity. We first note that the signal-to-noise ratio of the 15N{27Al} J-HMQC spectrum of (Ga,Al)2O3(3:1) is relatively low due to the low Al content. Nevertheless, the spectrum clearly shows that the aluminum Lewis acid sites contribute to the resonances observed in the 260–280 ppm chemical shift range. Strong aluminum LAS peaks are also identified at 234 ppm in the spectra of (Ga,Al)2O3(1:6) and (Ga,Al)2O3(1:3), with relative intensities around 19 and 20%, respectively (Fig. 4B; Table S9†). This peak is hardly detectable in the spectrum of (Ga,Al)2O3(3:1). These results agree well with what we observe in the corresponding CPMAS spectra (Fig. 4A), i.e. confirming that the resonance at around 234 ppm relates exclusively to strong Al-based LAS. The deconvolution also shows the presence of a weak resonance at ca. 242 ppm in these two spectra. The intensity of this peak increases with the content of Al, which is contrary to its behavior in the CPMAS spectra of Fig. 4A. In addition, there is a difference between the full widths at half maximum (FWHM, in ppm) of this resonance in (Ga,Al)2O3(1:6) and (Ga,Al)2O3(1:3), decreasing from ca. 15 and 15.6, respectively, in the CPMAS spectra to around 9.3 ppm in 15N{27Al} J-HMQC spectra (see Tables S8 and S9†). This implies that the resonance at around 242 ppm has contributions both from Ga and Al Lewis acid sites and that the relative amount of Ga-based strong LAS decreases when the content of Al is increased. The Al-based medium LAS peak that resonates at ca. 263–264 ppm is slightly shifted upfield (by 1 to 3 ppm) with respect to the chemical shift extracted from the CPMAS spectra, and its linewidth increases with Al content, viz. from a FWHM of 16 ppm in (Ga,Al)2O3(3:1) to 20 and 23 ppm. This is in contrast to what is observed for the medium LAS peak in the CPMAS spectra, where the linewidth remains within 26.4 and 23.3 ppm in all materials (Table S8†). These two observations indicate that both Ga- and Al-based medium LAS contribute to the shifts in the 260–265 ppm range. Finally, the peak resonating at ca. 275–277 ppm can be assigned to Al-based weak LAS. The relative intensity of this peak decreases when the Al content increases, i.e. from 28% in (Ga,Al)2O3(3:1) to 16% in (Ga,Al)2O3(1:3) and 6% in (Ga,Al)2O3(1:6). In the corresponding CPMAS spectra, the weak LAS peak appears at 282 ppm and its intensity remains between 9–11% in the Ga–Al materials. This indicates that weak LAS also have contributions from both Ga and Al. A 1D 15N{71Ga} J-HMQC spectrum of (Ga,Al)2O3(3:1) (natural abundance of 71Ga is 30.8%) has also been recorded and is presented in Fig. 4C. While the signal-to-noise ratio does not allow for the unambiguous observation of the strong gallium Lewis acid sites at around 240 ppm in (Ga,Al)2O3(3:1), we observe clearly a broad resonance in the 260–280 ppm range, with a maximum at ca. 276 ppm, which confirms the presence of Ga-based weak LAS at this chemical shift (and rules out the assignment of this peak to Py on weak/mild BAS).
Discussion
Our study shows that an increasing Al content leads to higher amounts of GaIV sites in (Ga,Al)2O3 nanoparticle catalysts, influencing thereby the distribution and properties of the surface sites and in turn the catalytic performance of the materials. In particular, an increased selectivity and stability was reported for (Ga,Al)2O3 catalysts with high Al contents, i.e. with a high fraction of GaIV sites.9 Results of the present study further advance our understanding of (Ga,Al)2O3 catalysts by disentangling element-specific contributions of Ga or Al sites to the overall Lewis acidity and by correlating these new insights to catalytic properties and the relative populations of bulk GaIV and GaVI sites as well as AlIV, AlV and AlVI sites. Our experimental results are graphically summarized in Fig. 5 and discussed in detail below. | ||
| Fig. 4 Deconvoluted CPMAS (A) and 15N{27Al} J-HMQC (B) spectra of (Ga,Al)2O3 materials. The peak intensities are normalized with respect to the most intense peak in each spectrum. Peak maxima and fitting results are summarized in Tables S8 and S9.†15N-Py was desorbed at 100 °C. (C) Comparison of 15N{71Ga} J-HMQC spectrum with CPMAS and 15N{27Al} J-HMQC spectra of (Ga,Al)2O3(3:1). The red box marks the feature at ca. 276 ppm attributed to Py on weak Ga LAS. Panel (D) shows a sketch explaining the observed variation of Lewis acidity of Ga–O–Ga and Ga–O–Al linkages with increasing substitution of Ga atoms in octahedral positions by Al atoms. | ||
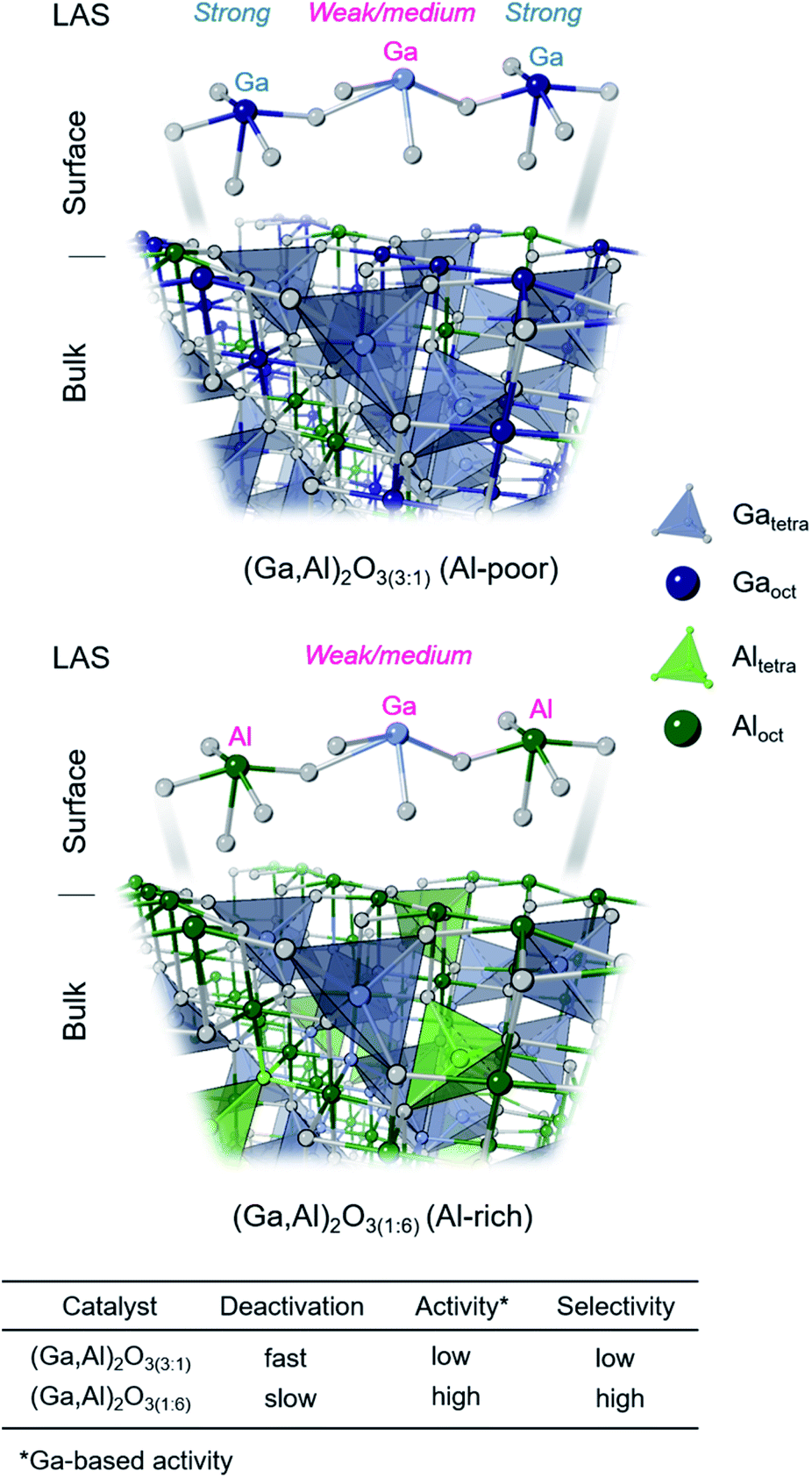 | ||
| Fig. 5 Schematic representation of the distribution of bulk metal sites (MIV and MVI, M = Ga,Al) in (Ga,Al)2O3(3:1) and (Ga,Al)2O3(1:6) catalysts as well as the representative surface sites in these materials. The nature of surface metal sites and their Lewis acidity is linked to their performances in PDH. | ||
We observe that in contrast to the Ga-only benchmark (Ga,Al)2O3(1:0), which transforms after calcination at 650 °C partially into the monoclinic β-phase, Al-containing (Ga,Al)2O3 materials retain their cubic spinel-type structure (γ-phase) characteristic of the as-prepared (Ga,Al)2O3 NPs. The addition of Al decreases the average crystallite size of (Ga,Al)2O3 NPs (from ca. 5 nm nm in (Ga,Al)2O3(1:0) to ca. 2–3 nm in (Ga,Al)2O3(3:1), (Ga,Al)2O3(1:3) and (Ga,Al)2O3(1:6)) and increases the specific surface area of the Al-containing materials, by ca. 2.5–2.9 times (note that the molecular weight of Al is ca. 2.6 times lower than that of Ga). Ga K-edge XAS, 27Al and 71Ga MAS NMR data confirm that increasing the fraction of Al in the materials leads to a higher fraction of GaIV sites, with Al cations found hexa- (major), tetra- (minor) and penta-coordinated (minor, only in Al-rich (Ga,Al)2O3(1:3) and (Ga,Al)2O3(1:6) materials). Interestingly, the (Ga,Al)2O3(1:6) catalyst demonstrates the highest Ga-based activity and selectivity in PDH, as well as a reduced deactivation (26% after 144 min TOS, relative to the highest activity after 24 min TOS). This contrasts with the Ga-rich catalysts (Ga,Al)2O3(3:1) and (Ga,Al)2O3(1:0), that deactivate strongly with time on stream (by 76% and 81% after 144 min TOS). This order of deactivation correlates with the amount of deposited coke determined by in situ TGA experiments (normalized per catalysts' surface area), suggesting therefore that the catalysts deactivate by coking (Table S5†). Therefore, coking can be related to Ga surface atoms in SP geometry that display strong Lewis acidity (such surface sites are derived from bulk GaVI sites, Scheme 1B and Fig. 5).
15N{27Al} J-HMQC DNP SENS experiments suggest that Ga–O–Ga linkages are replaced by Ga–O–Al linkages not only in the bulk but also on the surface of the catalysts studied in this work (Fig. 4D). More abundant Ga–O–Al surface linkages correlate with more abundant weak Lewis acidity observed by Py-FTIR in (Ga,Al)2O3(1:3) and (Ga,Al)2O3(1:6) catalysts. In particular, (Ga,Al)2O3(1:6) has the highest fraction of GaIV sites among the materials studied in this work (and the lowest fraction of GaVI sites), features weak LAS (Py bands at ca. 1606 and 1597 cm−1), shows a high Ga-weight normalized activity (similar to (Ga,Al)2O3(1:3) catalyst), low deactivation with TOS, stable selectivity to propene at ca. 85%, and deposits the lowest amounts of coke during in situ TGA experiments. Yet, these Al-rich catalysts still contain strong LAS, evidenced by a band at ca. 1618 cm−1 at Tdes = 200 °C in Py-FTIR (Fig. S10†), and deposit low amounts of coke, likely related to the presence of remaining low amounts of Ga-based strong LAS. Comparison of CPMAS, 15N{27Al} and 15N{71Ga} J-HMQC spectra allows to refine the conclusions of the Py-FTIR study and deconvolute the contributions of Lewis acid sites linked with Ga- and Al-based LAS. In line with the FTIR results, CPMAS spectra in Fig. 4A and fittings in Table S8† show that the relative fraction of Ga-based strong LAS associated with the peak at 240–242 ppm decreases from 23% and 24% in Ga-rich (Ga,Al)2O3(1:0) and (Ga,Al)2O3(3:1) catalysts to 15% and 7% in Ga-poor (Ga,Al)2O3(1:3) and (Ga,Al)2O3(1:6) catalysts. However, 15N{27Al} J-HMQC spectra reveal that the peak at ca. 242 ppm in CPMAS spectra contains contributions from Al-based strong LAS, fitted to 4% in both (Ga,Al)2O3(1:3) and (Ga,Al)2O3(1:6) (Fig. 4B, Table S9†), which means that the actual amounts of Ga-based strong LAS in (Ga,Al)2O3(1:3) and (Ga,Al)2O3(1:6) catalysts is less than 15% and 7%. This suggests that (Ga,Al)2O3(1:6), and to a slightly lesser extent (Ga,Al)2O3(1:3), have a strongly diminished fraction of Ga-based strong LAS as compared to (Ga,Al)2O3(1:0) and (Ga,Al)2O3(3:1) catalysts. Deconvolution of the strong LAS peaks in CPMAS spectra shows that the broadening of the peak at 240–242 ppm observed in Al-rich (Ga,Al)2O3(1:3) and (Ga,Al)2O3(1:6) is due to the contribution of a more shielded peak resonating at 234 ppm, which indicates the emergence of new, stronger LAS in these two materials. The presence of the peak at 234 ppm in the 15N{27Al} J-HMQC spectra allows us to assign this more shielded peak to Py on Al-based strong LAS (Fig. 4B). Noteworthy, the presence of strong Al-based LAS in (Ga,Al)2O3(1:6) (as well as the presence of mostly weak/mild Ga-based LAS in this material) does not lead to coking, which is in contrast to other (Ga,Al)2O3 catalysts studied here that contain strong Ga-based LAS and produce coke. This result suggests that strong Al-based LAS are not involved in coking.
While the relative fraction of Py on Al-based medium LAS associated with the peak at 263–264 ppm changes in the 15N{27Al} J-HMQC spectra from 65% in (Ga,Al)2O3(3:1) to 60% and 71% in (Ga,Al)2O3(1:3) and (Ga,Al)2O3(1:6), the respective fraction of Py on weak Al-based LAS (peak at 275–277 ppm) decreases constantly, i.e. from 28% in (Ga,Al)2O3(3:1) to 16% in (Ga,Al)2O3(1:3) and to 6% in (Ga,Al)2O3(1:6). That being said, the fittings of the CPMAS spectra, which capture contributions from Al-based and Ga-based LAS, reveal that the fraction of weak LAS (centered at 281–282 ppm) remains stable at around 10% in Al-containing (Ga,Al)2O3(3:1), (Ga,Al)2O3(1:3) and (Ga,Al)2O3(1:6) catalysts, and is higher at 14% only in (Ga,Al)2O3(1:0). These results suggest that in Al-containing catalysts, the relative fraction of Ga-based weak LAS increases with the increasing Al content. Importantly, the presence of the weak LAS peak resonating at 276 ppm in the 15N{71Ga} J-HMQC spectrum of (Ga,Al)2O3(3:1) demonstrates unequivocally the attribution of this peak to weak Ga-based LAS sites rather than to weak/medium BAS sites. We have reported previously that the higher catalytic activity of β-Ga2O3 is linked to a notably higher relative fraction of weak LAS in this material relative to the γ-Ga2O3 polymorph, identified by the 15N peak at 281 ppm.12 Therefore, we explain the higher relative fraction of weak LAS in (Ga,Al)2O3(1:0) as compared to the Al-containing (Ga,Al)2O3 catalysts by the presence of the β-Ga2O3 phase in (Ga,Al)2O3(1:0), which follows from the XRD results in Fig. S1.† Indeed, the presence of Al stabilizes notably the γ-spinel-type structure of the (Ga,Al)2O3 catalysts against the transformation to the β-polymorph, and the β-Ga2O3 phase is not observed in Al-containing (Ga,Al)2O3 catalysts.
CPMAS and 15N{27Al} J-HMQC experiments reveal a fundamental divergence of the Lewis acidity strength related to the respective AlIV/GaIV (and AlVI/GaVI) sites (Fig. 4A and B). We discussed above that Al atoms preferentially occupy octahedral sites in the (Ga,Al)2O3 materials, but with increasing Al content, also tetrahedral sites become occupied and comprise 8, 12 and 22% in (Ga,Al)2O3(3:1), (Ga,Al)2O3(1:3) and (Ga,Al)2O3(1:6), respectively, according to the quantitative fittings of the 27Al NMR spectra. Although these values reflect the quantity of bulk AlIV sites, a qualitative correlation is found with the relative amount of strong Al-based LAS, fitted to 7% of the total area in (Ga,Al)2O3(3:1) but increasing to ca. 20–25% in (Ga,Al)2O3(1:3) and (Ga,Al)2O3(1:6) (Table S9†). In line with these results, the Al-poor (Ga,Al)2O3(3:1) catalyst contains 92% of bulk AlVI sites and features a fitted area of weak/medium LAS peaks totaling to 93%. This data allows us to associate strong Al-based Lewis acidity in (Ga,Al)2O3 catalysts with AlIV sites and weak/medium Lewis acidity with Al surface sites in SP geometry (Scheme 1B), consistent with the previous data on γ-Al2O3.22 Analysis of CPMAS results discussed above displays the opposite correlation (relative to Al sites) between the coordination geometry and Lewis acidity of Ga LAS in γ-(Ga,Al)2O3 spinels. Specifically, an increasing amount of bulk GaIV sites (57, 72 and 86% in (Ga,Al)2O3(3:1), (Ga,Al)2O3(1:3) and (Ga,Al)2O3(1:6), respectively) correlates with the increasing fraction of weak/medium LAS. In turn, the respectively decreasing amount of bulk GaVI sites correlates with a decreasing fraction of strong Ga LAS. Therefore, strong Ga-based Lewis acidity is associated with Ga LAS in GaVI surface termination positions, which likely yields Ga surface sites in square pyramidal geometry, and weak/medium Lewis acidity with GaIV positions, providing GaIV and GaIII (next to a Vo site) surface sites.12 The underlying physical reason why SP surface Ga sites are strong LAS in Ga2O3 and γ-(Ga,Al)2O3 materials is currently unclear, but is consistent with reported DFT calculations.12
We can now relate results of DNP SENS experiments to the catalytic performance of the studied materials. The high and stable selectivity to propene displayed by (Ga,Al)2O3(1:3) and (Ga,Al)2O3(1:6) (at ca. 90 and 85%, respectively) and the high activity (Ga-content normalized) of these catalysts is associated with the low relative amounts of strong Ga-based LAS and high relative amounts of weak and medium Ga-based LAS assigned to tetra- or tricoordinated Ga surface sites. When the relative amount of weak/mild Ga LAS decreases and the relative amount of strong Ga LAS increases, as in (Ga,Al)2O3(3:1), the activity and propene selectivity decline. Interestingly, while Ga sites in octahedral positions at surface termination (GaV surface sites with SP geometry, Scheme 1B) can be linked to deactivation by coking,12 the current results show that surface Al sites in these positions do not coke significantly; yet, strong Al-based LAS (AlIV) do not coke either. However, in addition to deactivation by coking, we cannot exclude that strongly bound surface hydrides contribute to the observed deactivation.
We also note that another parameter changing alongside the Al loading in (Ga,Al)2O3 catalysts is the oxygen basicity, which is linked with the electron density on the oxygen atom.50 For instance, oxygen basicity has been shown to increase with the increasing content of the framework Al sites in zeolites.51 Therefore, it is likely that the basicity of surface oxygen atoms is higher in Al-rich (Ga,Al)2O3 catalysts, which is beneficial for the proton transfer step during the C–H activation of propane;52 a higher electron density on the oxygen atoms can also decrease Lewis acidity of Ga in Ga–O–Al surface linkages. The progress in understanding the relation between oxygen basicity and Lewis acidity in Ga-based PDH catalysts will be reported in due course.
A control experiment with γ-Al2O3 (SBET = 100 m2 g−1) showed a surface area-normalized activity that is ca. 15 times lower than the initial activity of (Ga,Al)2O3(1:0), and it showed a poor propene selectivity of 42%, consistent with previous reports.30,53 No coke deposition could be detected in this material by TGA. While a higher activity of alumina for PDH was reported to require a high temperature CO pre-treatment,16,18,54,55 such pre-treatment was not performed in the present work. Crystalline alumina-based catalysts also require higher reaction temperatures (600–630 °C), as was already mentioned above. Thus, the activity trends of (Ga,Al)2O3 catalysts in this work are related to Ga sites, that are influenced by the coordination geometry of both Ga and Al atoms, and the replacement on the surface of Ga–O–Ga linkages in Ga-rich materials by Ga–O–Al linkages in Al-rich materials (Fig. 4D).20 High resolution TEM imaging of (Ga,Al)2O3(1:3) nanoparticles showed that the atomic termination rows contain AlVI cations.
Overall, the experimental results suggest that the high catalytic activity, stability and selectivity in PDH is related to the weak/mild Lewis acidity of Ga-based active sites, presumably residing in GaIV–O–AlVI surface linkages, that is when Ga and Al are in tetrahedral and octahedral positions in atomic termination rows, respectively (Fig. 4D). The replacement of Ga by Al in (Ga,Al)2O3 spinel-type solid solutions attenuates the relative fraction of strong Lewis acidity of Ga-based sites and increases the relative fraction of weak and mild Ga-based sites. Thus, weak and mild Lewis acidity is associated with Ga atoms that produce little coke and yield high propene selectivity, and that are found in tetrahedral atomic termination rows with tetra- or tricoordinated (if in the vicinity of a Vo site) geometry of Ga sites. Since the presence of strong Al-based LAS in Al-rich (Ga,Al)2O3 catalysts does not induce coking, it is the strongly Lewis acidic Ga sites found in GaVI atomic termination rows that are most likely responsible for deactivation by coking.
Conclusions
We have described how control over the Ga:Al atomic ratio in (Ga,Al)2O3 spinel-type solid solutions allows one to improve the catalytic performance (activity, selectivity, stability) of these materials in the dehydrogenation of propane. The presence of Al atoms in (Ga,Al)2O3 nanoparticles stabilizes the spinel-type structure, leads to a higher relative fraction of bulk and surface GaIV sites and, for Al-rich compositions, provides a relatively high fraction of weak/mild Ga-based surface Lewis acid sites. These weak/mild Ga LAS that suffer less from catalyst deactivation (owing to decreased coke deposition during PDH) while retaining a high activity and selectivity are ascribed to GaIV–O–AlVI surface linkages. (Ga,Al)2O3 catalysts that are rich in Ga display a higher presence of bulk GaVI sites, a higher relative fraction of stronger Ga-based surface LAS and this correlates with a faster deactivation by coke deposition.
Data availability
Data are available within the article or in the ESI file.†Author contributions
P. C.-F. and A. F. conceived the research project and planned the experimental work. P. C.-F. prepared materials, characterized and tested the catalysts, and analyzed the data. M. K. and Z. W. planned and performed DNP SENS solid-state NMR experiments, supervised by A. L., analysed and described results. D. M. performed 27Al and 71Ga solid-state NMR experiments. E. K. performed part of Py-FTIR experiments. E. W. performed ADF-STEM imaging and EELS characterization. P. M. A. supervised XAS experiments and data analysis. The data were discussed among all co-authors. P. C.-F. and A. F. wrote the first draft that was edited by all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement No. 819573) for partial funding of this work. Financial support from the ANR-17-CE29-0006-01 contract is gratefully acknowledged. M. K. acknowledges financial support from the Deutsche Forschungsgemeinschaft (KA 5221/1-1) and L'oréal-UNESCO foundation. We also acknowledge the Scientific Centre for Optical and Electron Microscopy (ScopeM) of ETH Zürich for providing access to electron microscopes. The Swiss Norwegian Beamlines (SNBL) at European Synchrotron Facilities (ESRF) is acknowledged for provision of beamtime. Dr Pierre Florian (University of Orléans) is thanked for valuable advice on fitting and interpreting of 27Al and 71Ga MAS NMR spectra. We are also grateful to Dr Agnieszka Kierzkowska (ETH Zürich) for carrying out ICP-OES measurements. E. K. thanks ETH Zürich for financial support (ETH-40 19-2).Notes and references
- J. J. H. B. Sattler, J. Ruiz-Martinez, E. Santillan-Jimenez and B. M. Weckhuysen, Chem. Rev., 2014, 114, 10613–10653 CrossRef CAS PubMed.
- S. Chen, X. Chang, G. Sun, T. Zhang, Y. Xu, Y. Wang, C. Pei and J. Gong, Chem. Soc. Rev., 2021, 50, 3315–3354 RSC.
- M. Monai, M. Gambino, S. Wannakao and B. M. Weckhuysen, Chem. Soc. Rev., 2021, 50, 11503–11529 RSC.
- J. J. H. B. Sattler, I. D. Gonzalez-Jimenez, L. Luo, B. A. Stears, A. Malek, D. G. Barton, B. A. Kilos, M. P. Kaminsky, T. W. G. M. Verhoeven, E. J. Koers, M. Baldus and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2014, 53, 9251–9256 CrossRef CAS PubMed.
- P.-A. Payard, L. Rochlitz, K. Searles, L. Foppa, B. Leuthold, O. V. Safonova, A. Comas-Vives and C. Copéret, JACS Au, 2021, 1, 1445–1458 CrossRef CAS PubMed.
- T. Otroshchenko, G. Jiang, V. A. Kondratenko, U. Rodemerck and E. V. Kondratenko, Chem. Soc. Rev., 2021, 50, 473–527 RSC.
- C. Copéret, F. Allouche, K. W. Chan, M. P. Conley, M. F. Delley, A. Fedorov, I. B. Moroz, V. Mougel, M. Pucino, K. Searles, K. Yamamoto and P. A. Zhizhko, Angew. Chem., Int. Ed., 2018, 57, 6398–6440 CrossRef PubMed.
- S. R. Docherty, L. Rochlitz, P. A. Payard and C. Copéret, Chem. Soc. Rev., 2021, 50, 5806–5822 RSC.
- M. Chen, J. Xu, F. Z. Su, Y. M. Liu, Y. Cao, H. Y. He and K. N. Fan, J. Catal., 2008, 256, 293–300 CrossRef CAS.
- N. S. Nesterenko, O. A. Ponomoreva, V. V. Yuschenko, I. I. Ivanova, F. Testa, F. Di Renzo and F. Fajula, Appl. Catal., A, 2003, 254, 261–272 CrossRef CAS.
- P. Jiang, H. Fu, H. Ma, W. Qian, H. Zhang and W. Ying, Catal. Lett., 2021, 151, 1894–1901 CrossRef CAS.
- P. Castro-Fernández, D. Mance, C. Liu, I. B. Moroz, P. M. Abdala, E. A. Pidko, C. Copéret, A. Fedorov and C. R. Müller, ACS Catal., 2021, 11, 907–924 CrossRef.
- N. Rane, M. Kersbulck, R. A. van Santen and E. J. M. Hensen, Microporous Mesoporous Mater., 2008, 110, 279–291 CrossRef CAS.
- H. Xiao, J. Zhang, P. Wang, X. Wang, F. Pang, Z. Zhang and Y. Tan, Catal. Sci. Technol., 2016, 6, 5183–5195 RSC.
- D. S. Cook, J. E. Hooper, D. M. Dawson, J. M. Fisher, D. Thompsett, S. E. Ashbrook and R. I. Walton, Inorg. Chem., 2020, 59, 3805–3816 CrossRef CAS PubMed.
- D. Zhao, H. Lund, U. Rodemerck, D. Linke, G. Jiang and E. V Kondratenko, Catal. Sci. Technol., 2021, 11, 1386–1394 RSC.
- Z. Xie, Z. Li, P. Tang, Y. Song, Z. Zhao, L. Kong, X. Fan and X. Xiao, J. Catal., 2021, 397, 172–182 CrossRef CAS.
- P. Wang, Z. Xu, T. Wang, Y. Yue, X. Bao and H. Zhu, Catal. Sci. Technol., 2020, 10, 3537–3541 RSC.
- M. Abdelgaid, J. Dean and G. Mpourmpakis, Catal. Sci. Technol., 2020, 10, 7194–7202 RSC.
- S. P. Batchu, H. Wang, W. Chen, W. Zheng, S. Caratzoulas, R. F. Lobo and D. G. Vlachos, ACS Catal., 2021, 11, 1380–1391 CrossRef CAS.
- T. Kobayashi, F. A. Perras, I. I. Slowing, A. D. Sadow and M. Pruski, ACS Catal., 2015, 5, 7055–7062 CrossRef CAS.
- I. B. Moroz, K. Larmier, W.-C. Liao and C. Copéret, J. Phys. Chem. C, 2018, 122, 10871–10882 CrossRef CAS.
- I. B. Moroz, A. Lund, M. Kaushik, L. Severy, D. Gajan, A. Fedorov, A. Lesage and C. Copéret, ACS Catal., 2019, 9, 7476–7485 CrossRef CAS.
- M. Kaushik, C. Leroy, Z. Chen, D. Gajan, E. Willinger, C. R. Müller, F. Fayon, D. Massiot, A. Fedorov, C. Copéret, A. Lesage and P. Florian, Chem. Mater., 2021, 33, 3335–3348 CrossRef CAS.
- T. Wang, S. S. Farvid, M. Abulikemu and P. V. Radovanovic, J. Am. Chem. Soc., 2010, 132, 9250–9252 CrossRef CAS.
- S. J. Pearton, J. Yang, P. H. Cary, F. Ren, J. Kim, M. J. Tadjer and M. A. Mastro, Appl. Phys. Rev., 2018, 5, 011301 Search PubMed.
- P. Castro-Fernández, M. V. Blanco, R. Verel, E. Willinger, A. Fedorov, P. M. Abdala and C. R. Müller, J. Phys. Chem. C, 2020, 124, 20578–20588 CrossRef.
- M. Hirano, K. Sakoda, K. Souma, H. Nishimoto, K. Jinno and Y. Hirose, Ceram. Int., 2015, 41, 14285–14292 CrossRef CAS.
- T. Watanabe, Y. Miki, T. Masuda, H. Deguchi, H. Kanai, S. Hosokawa, K. Wada and M. Inoue, Ceram. Int., 2011, 37, 3183–3192 CrossRef CAS.
- M. Chen, J. Xu, Y. Liu, Y. Cao, H. He and J. Zhuang, Catal. Lett., 2008, 124, 369–375 CrossRef CAS.
- C. Otero Areán, M. Rodríguez Delgado, V. Montouillout and D. Massiot, Z. Anorg. Allg. Chem., 2005, 631, 2121–2126 CrossRef.
- J. C. Slater, J. Chem. Phys., 1964, 41, 3199–3204 CrossRef CAS.
- P. Scherrer and P. Debye, Nachr. Ges. Wiss. Goettingen, Math.-Phys. Kl., 1918, 2, 101–120 Search PubMed.
- K. Nishi, K. Shimizu, M. Takamatsu, H. Yoshida, A. Satsuma, T. Tanaka, S. Yoshida and T. Hattori, J. Phys. Chem. B, 1998, 102, 10190–10195 CrossRef CAS.
- D. Lee, N. T. Duong, O. Lafon and G. De Paëpe, J. Phys. Chem. C, 2014, 118, 25065–25076 CrossRef CAS.
- D. Massiot, F. Fayon, M. Capron, I. King, S. Le Calvé, G. Hoatson, I. King, B. Alonso, J.-O. Durand, B. Bujoli, Z. Gan and G. Hoatson, Magn. Reson. Chem., 2002, 40, 70–76 CrossRef CAS.
- J. B. d'Espinose de Lacaillerie, C. Fretigny and D. Massiot, J. Magn. Reson., 2008, 192, 244–251 CrossRef PubMed.
- G. Le Caër and R. A. Brand, J. Phys.: Condens. Matter, 1998, 10, 10715–10774 CrossRef.
- C. V. Chandran, E. Breynaert, J. A. Martens, C. E. A. Kirschhock, F. Taulelle and S. Radhakrishnan, Chem. Soc. Rev., 2018, 48, 134–156 RSC.
- D. Massiot, I. Farnan, N. Gautier, D. Trumeau, A. Trokiner and J. Pierre, Solid State Nucl. Magn. Reson., 1995, 4, 241–248 CrossRef CAS PubMed.
- D. A. Jefferson, Philos. Trans. R. Soc., A, 2000, 358, 2683–2692 CrossRef CAS.
- Y. Rozita, R. Brydson and A. J. Scott, J. Phys.: Conf. Ser., 2010, 241, 012096 CrossRef.
- T. Barzetti, E. Selli, D. Moscotti and L. Forni, J. Chem. Soc., Faraday Trans., 1996, 92, 1401–1407 RSC.
- E. P. Parry, J. Catal., 1963, 2, 371–379 CrossRef CAS.
- J. A. Lercher, C. Gründling and G. Eder-Mirth, Catal. Today, 1996, 27, 353–376 CrossRef CAS.
- M. C. Kung and H. H. Kung, Catal. Rev., 1985, 27, 425–460 CrossRef.
- X. Liu and R. E. Truitt, J. Am. Chem. Soc., 1997, 119, 9856–9860 CrossRef CAS.
- P. Berruyer, L. Emsley and A. Lesage, eMagRes, 2018, 7, 93–104 CAS.
- G. E. Maciel, J. F. Haw, I. S. Chuang, B. L. Hawkins, T. A. Early, D. R. McKay and L. Petrakis, J. Am. Chem. Soc., 1983, 105, 5529–5535 CrossRef CAS.
- D. Barthomeuf, Catal. Rev. - Sci. Eng., 1996, 38, 521–612 CrossRef.
- R. Heidler, G. O. A. Janssens, W. J. Mortier and R. A. Schoonheydt, J. Phys. Chem., 1996, 100, 19728–19734 CrossRef CAS.
- C. Copéret, Chem. Rev., 2010, 110, 656–680 CrossRef PubMed.
- K. Nakagawa, M. Okamura, N. Ikenaga, T. Suzuki and T. Kobayashi, Chem. Commun., 1998, 3, 1025–1026 RSC.
- U. Rodemerck, E. V. Kondratenko, T. Otroshchenko and D. Linke, Chem. Commun., 2016, 52, 12222–12225 RSC.
- M. Dixit, P. Kostetskyy and G. Mpourmpakis, ACS Catal., 2018, 8, 11570–11578 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: experimental procedures, XAS, MAS NMR, DNP, J-HMQC, HAADF-TEM, catalytic tests, Py-FTIR, 2,6-DMPY FTIR, EELS, XPS, BET, BJH, ICP-OES, TGA, crystallite size, and catalytic equations. See DOI: 10.1039/d1sc05381g |
| This journal is © The Royal Society of Chemistry 2021 |