
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA brief review on solid lipid nanoparticles: part and parcel of contemporary drug delivery systems
Yongtao Duana,
Abhishek Dharb,
Chetan Patelc,
Mehul Khimanic,
Swarnali Neogib,
Prolay Sharmab,
Nadavala Siva Kumar d and
Rohit L. Vekariya*ef
d and
Rohit L. Vekariya*ef
aHenan Provincial Key Laboratory of Children's Genetics and Metabolic Diseases, Children's Hospital Affiliated to Zhengzhou University, Zhengzhou University, Henan 450018, China
bDepartment of Instrumentation & Electronics Engineering, Jadavpur University, Kolkata 700106, India
cSchool of Sciences, P P Savani University, NH-8, GETCO, Near Biltech, Village: Dhamdod, Kosamba, Dist., Surat 394125, Gujarat, India
dDepartment of Chemical Engineering, King Saud University, P.O. Box 800, Riyadh 11421, Saudi Arabia
eDepartment for Management of Science and Technology Development, Ton Duc Thang University, Ho Chi Minh City, Vietnam
fFaculty of Applied Sciences, Ton Duc Thang University, Ho Chi Minh City, Vietnam. E-mail: rohit.vekariya@tdtu.edu.vn
First published on 17th July 2020
Abstract
Drug delivery technology has a wide spectrum, which is continuously being upgraded at a stupendous speed. Different fabricated nanoparticles and drugs possessing low solubility and poor pharmacokinetic profiles are the two major substances extensively delivered to target sites. Among the colloidal carriers, nanolipid dispersions (liposomes, deformable liposomes, virosomes, ethosomes, and solid lipid nanoparticles) are ideal delivery systems with the advantages of biodegradation and nontoxicity. Among them, nano-structured lipid carriers and solid lipid nanoparticles (SLNs) are dominant, which can be modified to exhibit various advantages, compared to liposomes and polymeric nanoparticles. Nano-structured lipid carriers and SLNs are non-biotoxic since they are biodegradable. Besides, they are highly stable. Their (nano-structured lipid carriers and SLNs) morphology, structural characteristics, ingredients used for preparation, techniques for their production, and characterization using various methods are discussed in this review. Also, although nano-structured lipid carriers and SLNs are based on lipids and surfactants, the effect of these two matrixes to build excipients is also discussed together with their pharmacological significance with novel theranostic approaches, stability and storage.
Introduction
With the development of technology in the last two decades, the particle size of materials ranges from the micro- to nano-scale. The reduction in the particle size of materials at the nanometer scale increases their overall surface area by several orders of magnitude. Particles with a size in the range of 1 nm to 1000 nm are known as nanoparticles. The word “nano” can be easily defined, but it covers numerous areas of application. Fig. 1 represents several nano-based systems composed of different types of materials, which can be utilized as nanocarriers.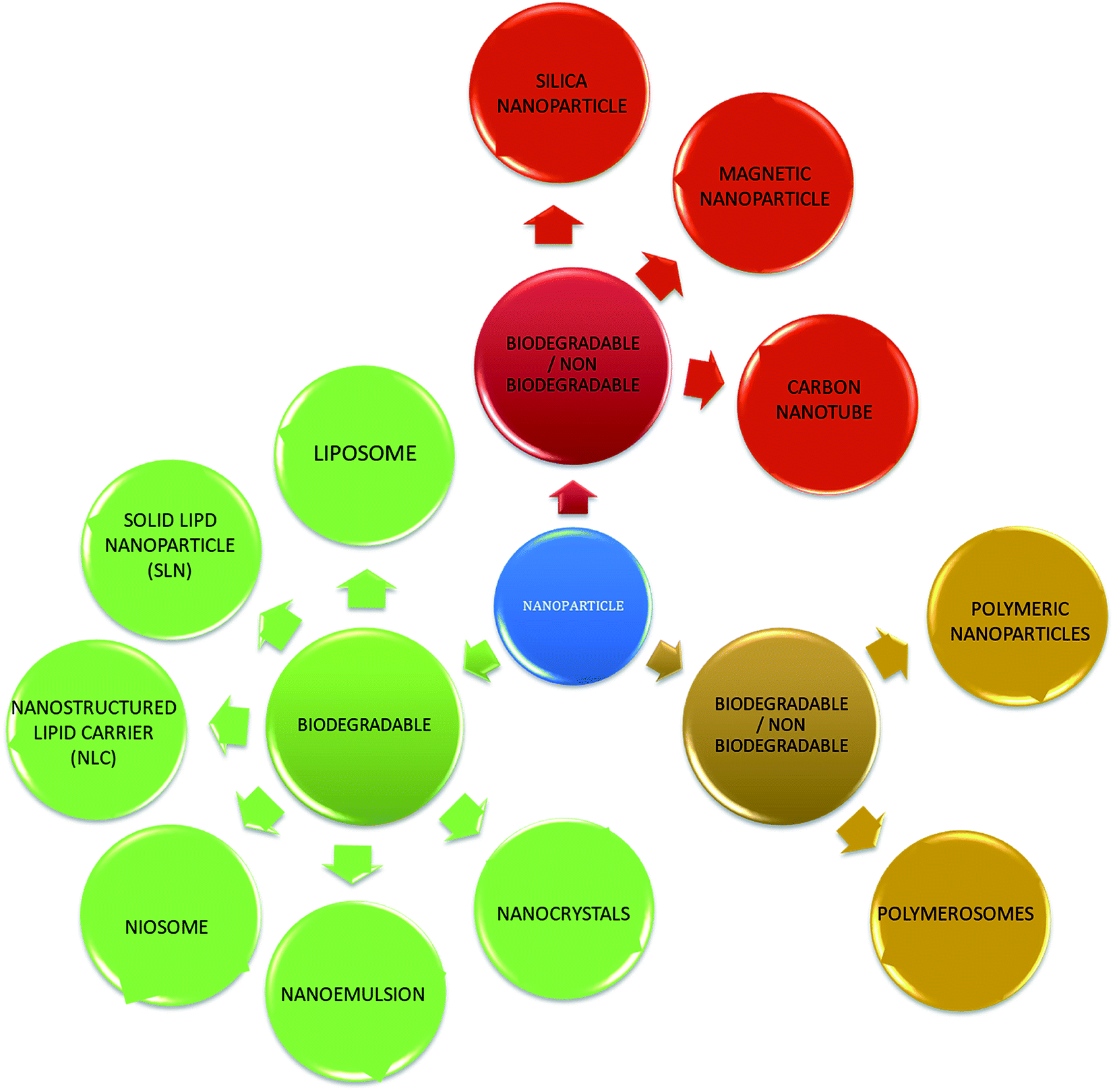 | ||
| Fig. 1 Schematic diagram of the different types of nanoparticles. | ||
However, nanomaterials with excellent biodegradability and biocompatibility are considered to be the best vehicles for drug delivery systems in biomedical applications. Currently, scientists and researchers are focused on discovering new methods/routes to control the pharmacokinetics (ADME), pharmacodynamics, non-specific toxicity, immunogenicity, biorecognition, and drug efficacy of drugs. These new strategies are often called novel drug delivery systems (NDDS) and are based on interdisciplinary approaches that combine polymer science, pharmaceutics, bioconjugate chemistry, and molecular biology. Some of the different approaches for novel drug delivery include transdermal patches, sustained and controlled release by polymeric and magnetic control, liposomes, hydrogels, implants, microspheres, erythrocytes, and nanoparticles. Nanoparticular drug delivery systems are a successful approach in the treatment of chronic human diseases, which have excellent function in satisfying the biopharmaceutical and pharmacological considerations. The emergence of nanotechnology and the growing capabilities of functional proteomics, genomics, and bioinformatics combined with combinatorial chemistry have driven scientists to become more enthusiastic to express their technical expertise to discover, invent and explore novel approaches for drug delivery systems through new techniques. Novel drug delivery systems remain the foundation to deliver drugs having complications that cannot be minimized by conventional drug delivery systems, where the therapeutic effectiveness of drugs depends on their pharmacokinetics and site of administration. Pharmacokinetics are also based on physico-chemical properties such as solubility, crystallinity, toxicity, and HLB value. After understanding the biopharmaceutics and pharmacokinetics, the administration route, absorptive surface area, and transportation of drugs in the body are the key points for their absorption and distribution. Furthermore, metabolism and elimination depend on the aforementioned properties.1 The formulation design has a major impact on the effective delivery of the active pharmaceutical ingredient (API), and thus all the above parameters are crucial challenges. Drugs based on the HLB scale are categorized into two classes, hydrophilic and lipophilic molecules. Lipophilic molecules exhibit very poor solubility, and depending on this, they produce a great challenge to design safe, efficacious, and cost-effective drug delivery systems and have been a source of frustration for pharmaceutical scientists.2 Lipophilic molecules allow the design of formulations for hydrophobic drug molecules, and despite all the problems confronted by pharmaceutical scientists, the current solid lipid nanoparticles are the result of their great effort. Traditionally, lipid-based novel drug delivery systems have focused on the delivery of lipophilic molecules, but recently, lipoid drug delivery systems have received attention due to their inherent properties such as biocompatibility, self-assembly capabilities, ability to cross the blood brain barrier, particle size variability and finally cost effectiveness, making lipid-based delivery systems much more attractive.3 Lipid-based nanoparticles can also be subcategorized as follows in Chart 1.
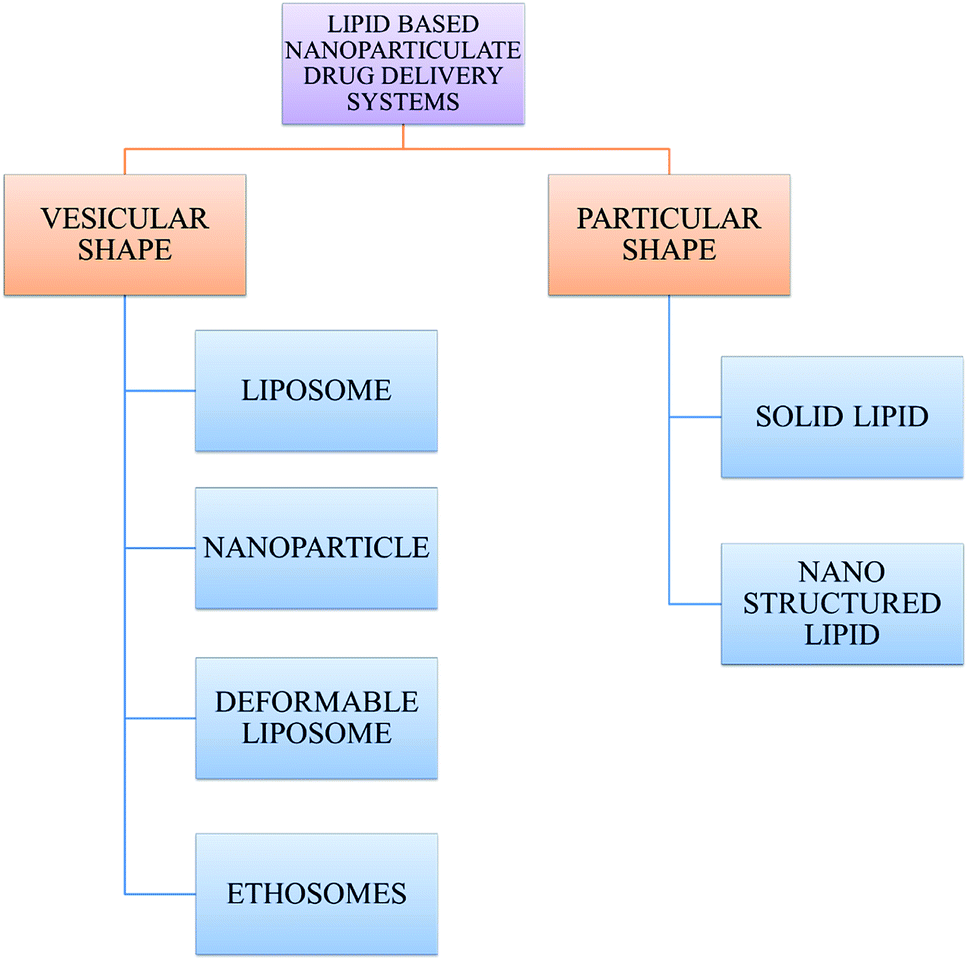 | ||
| Chart 1 Classification of lipid-based nanoparticle drug delivery systems. | ||
Over the past few years, nanomaterials have emerged as drug carriers. Liposomes are important biological molecules, which have been used for many years, but currently, there are various alternative molecules. Niosomes are one of the promising economical alternatives to liposomes. Niosomes are highly stable and slightly more leaky than liposomes. The size of niosomes decreases substantially upon freezing in liquid nitrogen and subsequent thawing, as evident by cryo-EM and dynamic light scattering. The successful delivery of drugs through nanoparticles depends on their ability to penetrate barriers, continuously release drugs and their stability. However, the scarcity of regulatory approved polymers, i.e. the Food and Drug Administration (FDA), and their expensive costs have limited their clinical application.4 Thus, to overcome these limitations, scientists and researchers have proposed lipids as alternative carriers. These lipid-based nanoparticles are known as solid lipid nanoparticles (SLNs), which have attracted worldwide interest due to their advantages (Table 1).5
| Issue | Advantages of SLNs over liposomes | Advantages of SLNs over polymeric nanoparticles |
|---|---|---|
| Avoidance of organic solvents | Avoidance of organic solvents when desired | Avoidance of organic solvents when desired |
| Preparation and reproducibility | Excellent reproducibility and feasible large-scale production | Excellent reproducibility and feasible large-scale production with cost-effective high-pressure homogenization method as the preparation method6 |
| Stability | Increased stability of the active ingredient because of the rigid core lipid matrix8 | Increased product stability of about 3 years7 |
| Biodegradability | Both liposomes and SLNs are biodegradable | Lipids of SLNs are physiological and biodegradable, and hence have better biocompatibility and sterilization. On the other hand, polymeric nanoparticles may accumulate undesirably in the liver, spleen etc.9 |
| Binding, entrapment and release | SLNs impose greater entrapment efficiency for hydrophobic drugs (since they do not contain an aqueous core with lipid bilayer like liposomes) | Drug delivery is extremely site specific for SLNs, whereas polymeric nanoparticles may produce non-specific drug delivery or show unpredictable release towards siRNAs10 |
| Ability to allow controlled release (similar to polymeric nanoparticles) and drug targeting by coating/attaching ligands to SLNs12 | 11 |
Solid lipid nanoparticle overview
For lipid and lipid-based drug delivery systems, phospholipids are an important constituent because of their various properties, such as amphiphilic nature, biocompatibility and multifunctionality. However, liposomes, lipospheres, and microsimulation carrier systems have many drawbacks such as their complicated production method, low percentage entrapment efficiency (% EE), difficult large-scale manufacture, and thus the SLN delivery system has emerged.13,14 SLNs are commonly spherical in shape with a diameter in the range of 50 to 1000 nm. The key ingredients of SLN formulations include lipids, which are in the solid state at room temperature, emulsifiers and sometimes a mixture of both, active pharmaceutical ingredients (APIs) and an adequate solvent system (Fig. 2). Nanocarrier-based drug delivery systems can be subcategorized in many aspects depending on the route of administration, degree of degradability, etc. The route of administration includes nanoparticles for parenteral administration, oral administration, ocular administration, and topical administration, and nanoparticles for protein peptide delivery. Nanocarrier systems can also be subcategorized based on the degree of their degradability as follows.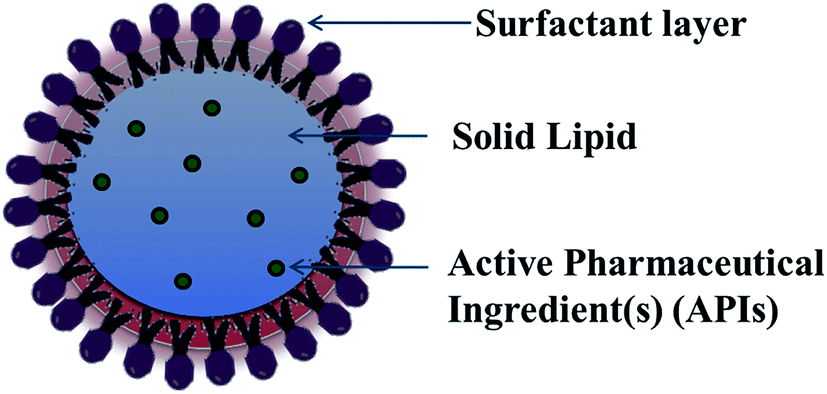 | ||
| Fig. 2 Schematic presentation of the complete structure of solid lipid nanoparticles. | ||
An ideal nanoparticulate drug delivery system must contain the following characteristics:
(1) Maximum drug bioavailability.
(2) Tissue targeting.
(3) Controlled release kinetics.
(4) Minimal immune response.
(5) Ability to deliver traditionally difficult drugs such as lipophiles, amphiphiles and biomolecules.
(6) Sufficient drug loading capacity.
(7) Good patient compliance.
Solid lipid nanoparticles have changed the dimension of drug delivery by combining all the advantageous characteristics of polymeric nanoparticles, liposomes and microemulsion.15 All the properties of lipid nanoparticles are upgraded with surface modification, better pharmacokinetic acceptability, formation of inclusion complexes, improved stability pattern and incorporation of chemotherapeutic agents. SLNs are appropriate for intravenous applications because of their effortless dispersion in solution, which are aqueous or aqueous-surfactant. Nanoparticles undergo phagocytic uptake,16 and thus by surface modification, their phagocytic uptake can be minimized.17 A pharmacokinetic study also showed a good increase in the of concentration doxorubicin in with solid lipid nanoparticles compared with conventional commercial drug solutions, and it was found that the drug concentrations were higher in the lungs, spleen and brain of rats.18 In drug delivery technology, cyclodextrin is used as a complex agent, which can be used to increase aqueous solubility, bioavailability and improve the physicochemical properties of drugs by forming inclusion complexes. The incorporation of these inclusion complexes into solid lipid nanoparticles increases their release profile compared to solid lipid nanoparticles without cyclodextrin.19 Furthermore, the stability pattern of solid lipid nanoparticles (SLNs) is more attractive than that of other nanoparticulate formulations. Aqueous SLNs can be stored for up to 3 years or longer, and their gelling tendency due to long term storage and light exposure can be stabilized by inhibiting the transitions by lipid modification.20 The major aim of solid lipid nanoparticles (SLN) in terms of drug delivery is to enhance the bioavailability and efficacy of drugs, and control the non-specific toxicity, immunogenicity, pharmacokinetics and pharmacodynamics of drugs. This review focuses on the potential of SLNs in various types of chemotherapy such as cancer, where conventional chemotherapy is hindered by different obstacles such as drug resistance, low specificity and poor stability of chemotherapeutic compounds.9 These issues may be partly overcome by encapsulating drugs as SLNs. The new generations of SLN such as nanostructured lipid carriers (NLC), lipid drug conjugates (LDC), polymeric lipid hybrid nanoparticles (PLN), and long-circulating SLNs, improve the role of SLNs as versatile drug carriers for various types of chemotherapy, and treatment of parasitic infections and tuberculosis.14,17 Cell line studies have shown that SLNs can be easily internalized and may be designed as surrogate colloidal drug carriers for the administration of chemotherapeutic agents, especially for the treatment of malignant melanoma and colorectal cancer.21 Besides their antitumor activities, SLNs are also capable of hindering the adhesive interactions between cancerous cells (resulting from human breast, prostate cancers, melanoma, etc.) with the cells present on human umbilical vein endothelium.22 Furthermore, since SLNs are based on nontoxic and non-irritating materials, they are ideal for use in topical formulations.23 Accordingly, there has been extensive research on the topical applications of SLNs (containing lipids such as glyceryl palmitostearate and glyceryl behenate) to treat several skin diseases since SLNs adhere strongly because of their greater surface area as a result of their smaller sizes.24,25 The coenzyme Q10 penetrated the stratum corneum more effectively as SLNs in comparison with liquid paraffin and isopropanol.26 The extent of drug release was higher and more rapid for SLNs of Compritol®(Retinol-loaded) compared to conventional carriers.27,28 Also, SLNs were found to be significant vehicles for numerous sunscreen agents.29,30
The delivery of genetic material via nanotechnology is now gaining significant attention. Cationically modified SLNs can effectively deliver DNA to binding sites, where the transfection efficiency and cytotoxicity are also very low.31 Furthermore, solid lipid nanoparticles (SLNs) and nanostructured lipid carriers (NLCs) have been considered as effective and safe alternatives to potentially treat both genetic and non-genetic diseases. Lipid nanoparticles (LNs) easily overcome the main biological barriers for cell transfection, including degradation by nucleases, cell internalization, intracellular trafficking, and selective targeting to a specific cell type. SLNs and NLCs can effectively be used for gene therapy, and the treatment of ocular diseases, infectious diseases, and lysosomal storage disorders. SLNs and NLCs have been established to be very effective in the topical delivery of antifungals such as clotrimazole and ketoconazol. Various studies have shown that because of several factors such as stability, complete release, and low toxicity, SLNs can also be considered as new potential vehicles for the pulmonary delivery of antitubercular drugs.32
Claus-Michael Lehr and co-workers showed that a two-tail cationic lipid had a greater transfection efficiency than a one-tail cationic lipid, and concluded that higher tolerability and transfection efficiency can be achieved with SLNs.33 Ocular drug delivery is one of the most critical drug delivery technologies, which is still lacking regarding sensitivity. Accordingly, since SLNs contain no inflammatory lipid material, they may be suitable for ocular drug delivery. Tobramycin was incorporated in SLNs and compared to a reference eye drop, showing a 1.5-fold and 8-fold increase in Cmax and tmax value with respect to the reference solution. SLNs show occlusive properties and UV blocking potential, which are ideal for cosmetic preparation, resulting in excellent skin hydration.34,35 Thus, SLNs are interesting for drug delivery, where they mostly cover all the sites for drug delivery and have numerous applications with respect to the route of administration. Furthermore, stability-related issues are not a major problem, and drugs, proteins and peptides can also be deliverable to the target site. Thus, SLNs are potential carriers for bioactive materials.
Principle of lipid nanoparticle formulation36
General ingredients
SLNs are comprised of a phospholipid-coated solid hydrophobic core matrix (containing the hydrophobic tails of the phospholipid section) (Fig. 2). Also, SLNs consist mainly of solid lipid(s), emulsifiers together with APIs such as drugs, genes, DNA, plasmid, and proteins. The lipids utilized in the formation of SLNs are surfactant stabilized, and thus solid at both physiological and room temperature. Depending on their structure, lipids are mainly divided into fatty acids, fatty esters, fatty alcohols, triglycerides, and partial glycerides. Ionic and nonionic polymers (Pluronic® such as F-68 and F127), surfactants, and organic salts are used as emulsifiers. However, their physicochemical characteristics also affect the behavior of the corresponding SLNs in both in vivo and in vitro release. The formation of colloidal nanoparticles depends on the interfacial tension and surface tension between two liquids. Thus, the main principle for the formation of solid lipid nanoparticles is the adhesive forces between two liquids. Normally, the interfacial tension between two liquids is less than their surface tension because of the weaker adhesive forces compared to that with gas. Molecules at the interface constitute surface free energy of interfacial tension, while they undergo agitation and form a spherical system to minimize the surface free energy.To increase the surface of the dispersed particles, the amount of work needed to be done is as follows:
| W = γ × ΔA |
Surfactant selection also based on the HLB scale, as described by Griffin, where a high value denotes hydrophilic molecule and low value indicates a hydrophobic molecule.
In the case of non-ionic surfactants whose hydrophilic portion is only polyoxyethylene
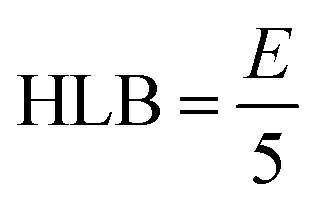
In the case of polyhydric alcohol fatty acid esters
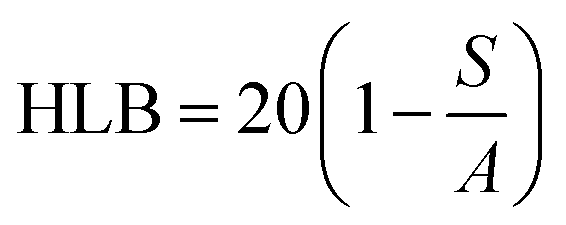
SLNs are very similar to emulsions, where solid lipids are used as a substitute for the oil phase and melted and mixed with the aqueous phase. Agitation at high speed is applied to this mixture, which results in the formation of fine droplets of dispersed phase in the dispersion medium. By adding a surfactant as a third substance, the interfacial tension between the two liquids is reduced, thereby is also reducing the surface energy, and stable SLNs are formed.
Surfactant literally means ‘surface-active agent’. Surfactants lower the surface tension between the contact surface of two or more substances existing in the same or different physical states. Surfactants enhance the drug loading capacity and stability of SLNs. For example, CPC, Poloxamer 407 and Tween-80 are widely used surfactants to increase the efficacy of SLNs during drug delivery.
Techniques for the fabrication of SLNs
Preparation method
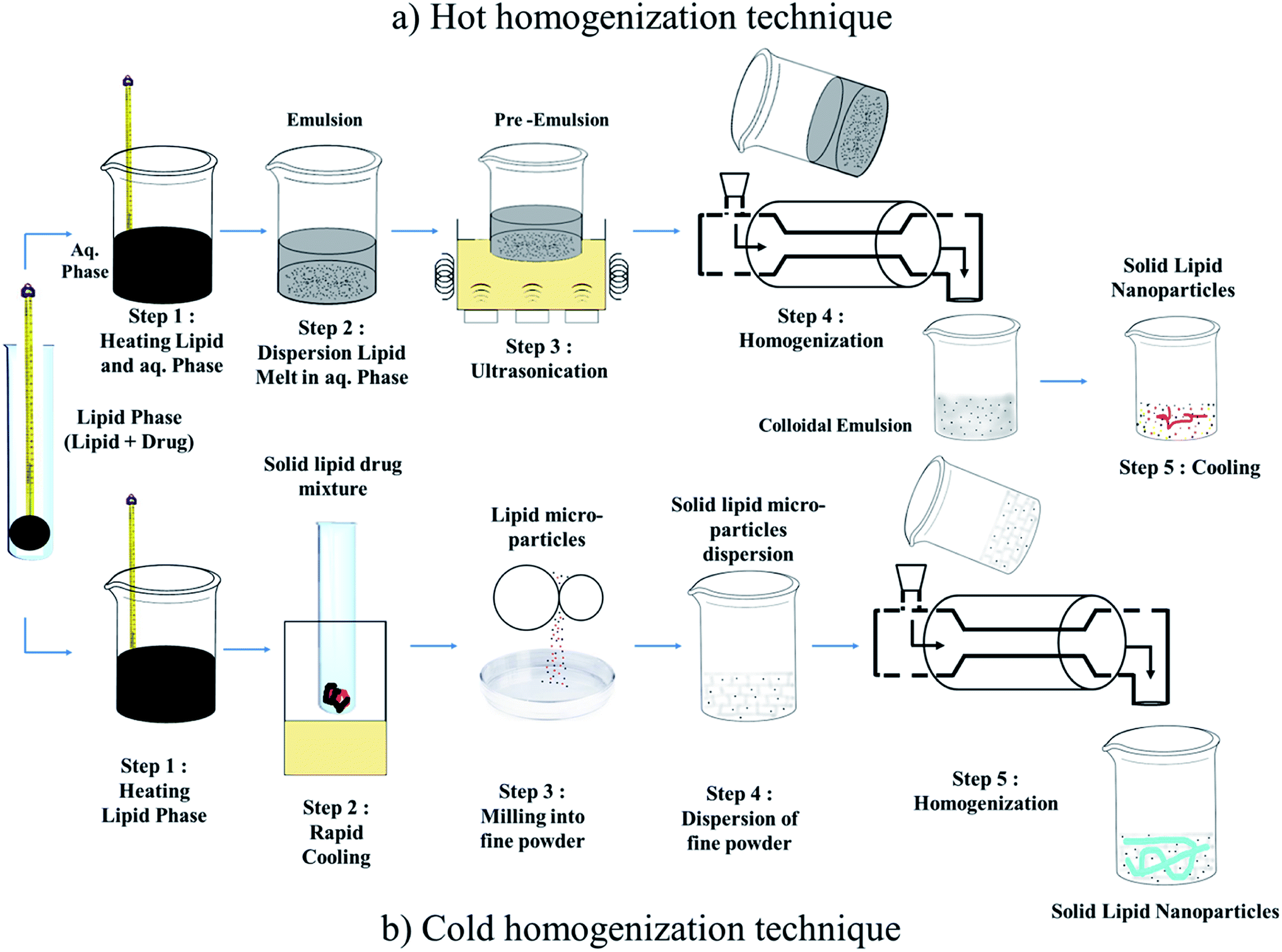 | ||
| Fig. 3 Homogenization technique: (a) Hot homogenization technique and (b) Cold homogenization technique. | ||
 | ||
| Chart 2 Preparation of solid lipid nanoparticles by oil/water (o/w) microemulsion method. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 kDa (Chart 4).
000 kDa (Chart 4).
 | ||
| Chart 3 Solvent-emulsification diffusion technique for the synthesis of solid lipid nanoparticles. | ||
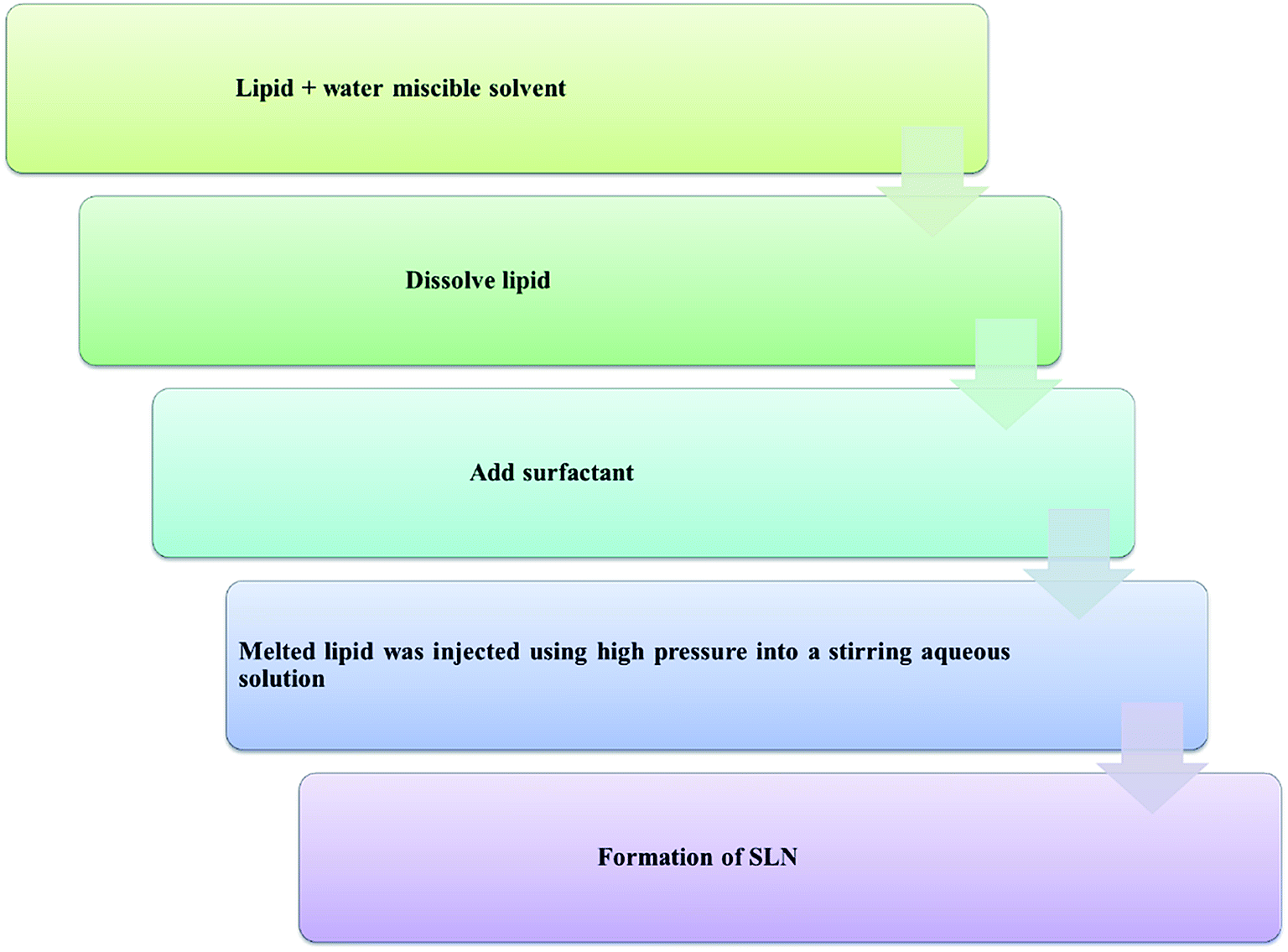 | ||
| Chart 4 Solvent injection method for the synthesis of solid lipid nanoparticles. | ||
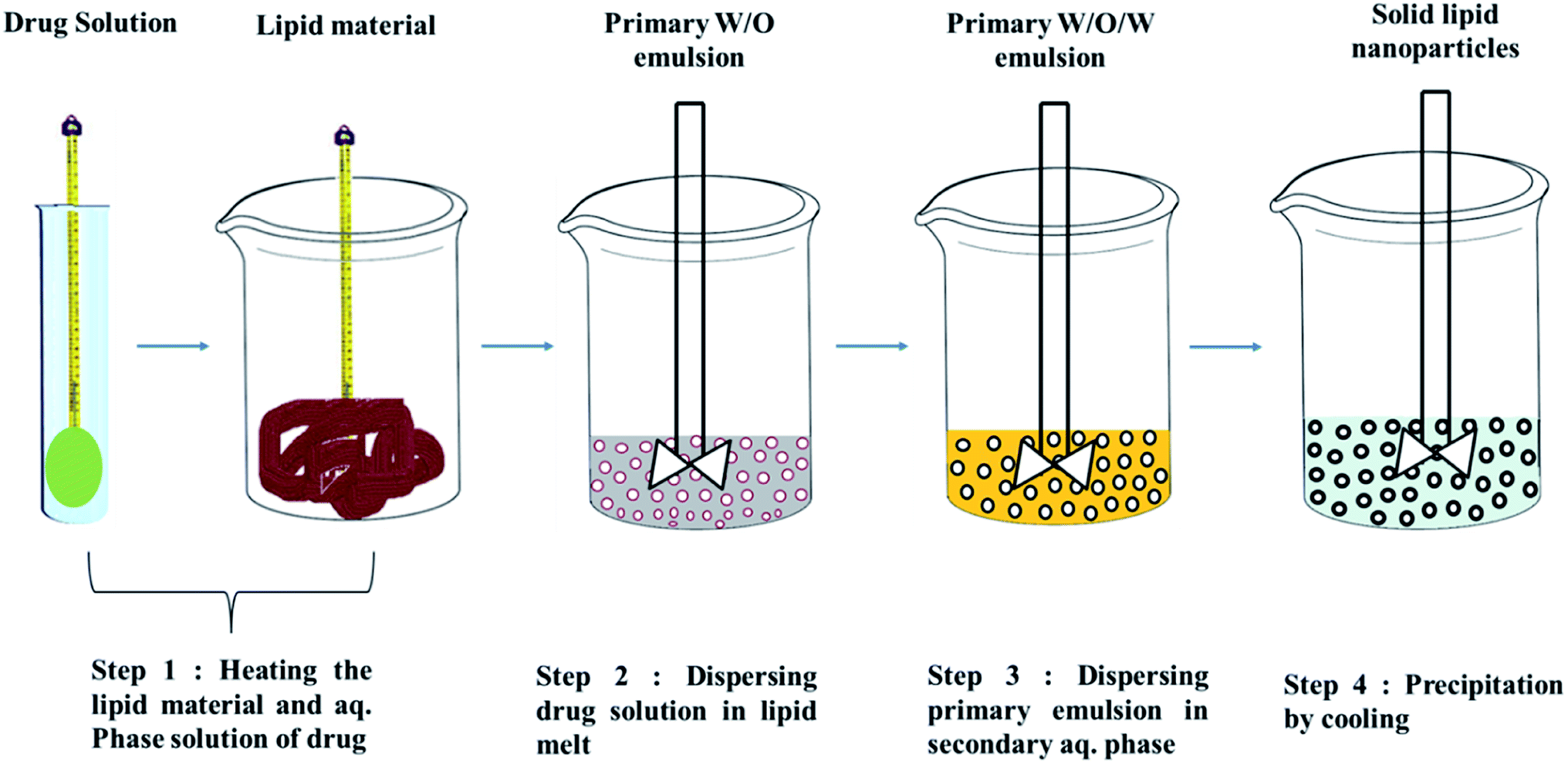 | ||
| Fig. 4 w/o/w double emulsion technique for the preparation of solid lipid nanoparticles. | ||
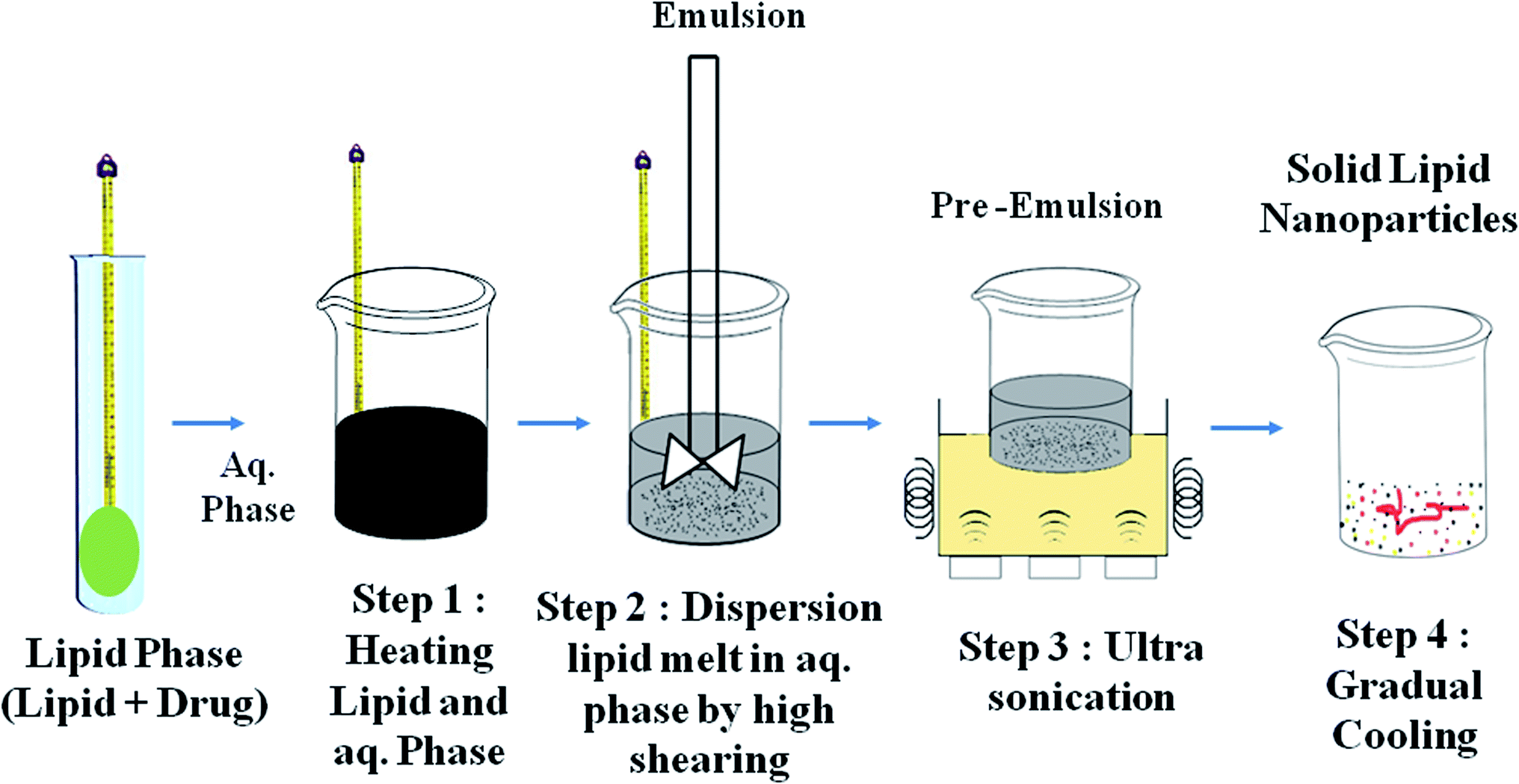 | ||
| Fig. 5 Ultrasonication technique for the preparation of solid lipid nanoparticles. | ||
Some other advanced techniques have also been introduced to formulate SLNs.
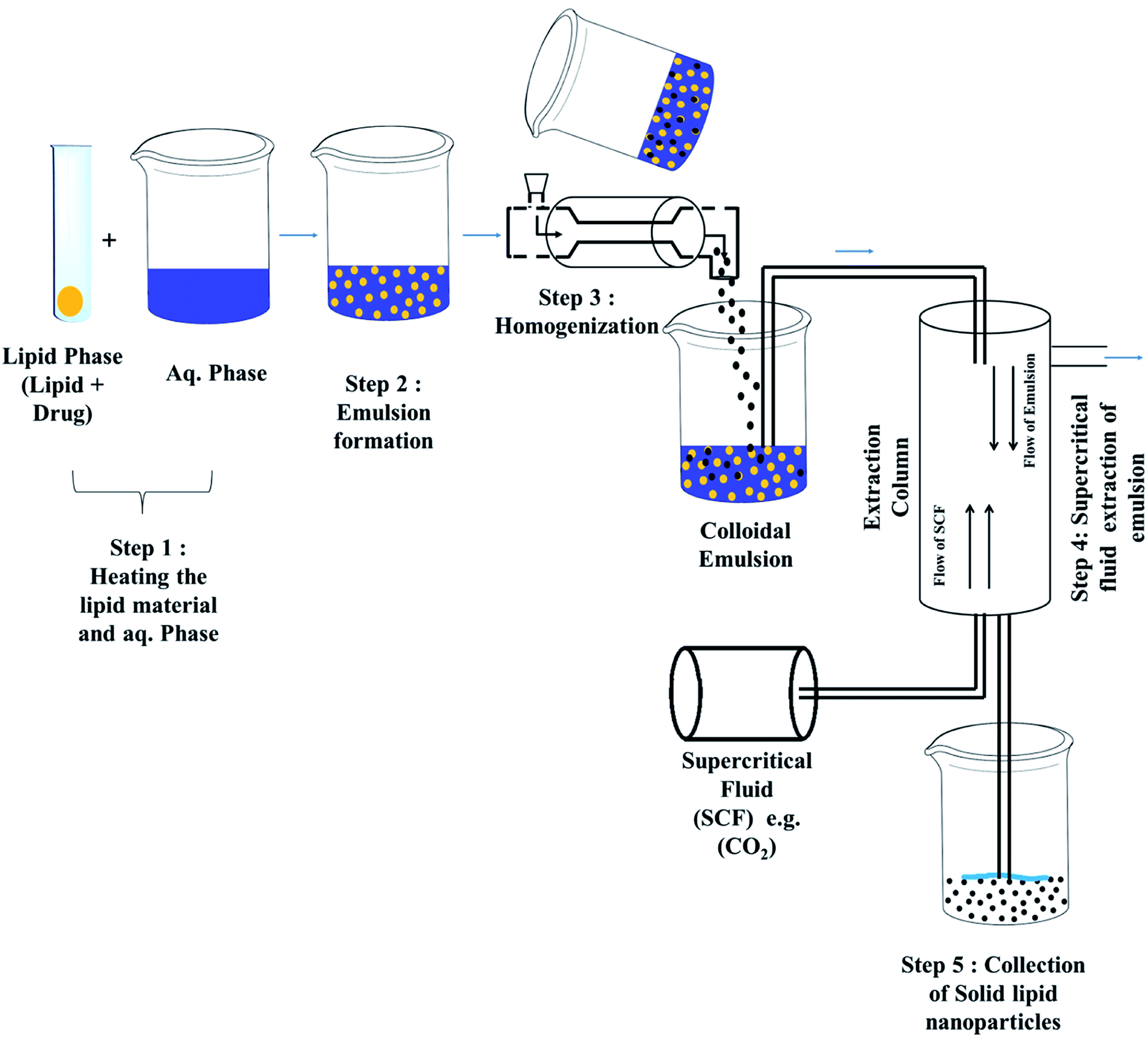 | ||
| Fig. 6 Super critical fluid technique for the preparation of solid lipid nanoparticles. | ||
The conversion of liquid lipid nanoparticles into a solid plays a pivotal role in enhancing the stability and safe storage of drug delivery systems. Besides spray drying, lyophilization is also suitable for converting nanolipid dispersions into dry, solid particles. Among these techniques, spray drying is cost-effective and can be used beneficially for large-scale purposes. Spray drying of lipid nanoparticles is a very sensitive process since low melting temperature lipids are used in the formulation. Some studies36,41 demonstrated the use of an organic solvent to reduce the processing temperature and facilitate the drying of heat-sensitive materials. The removal of organic solvents from the lipid nanoparticle matrix again requires exposure to high temperatures, which is not always advisable.
Effect of lipids and surfactants
The process for the production of SLNs is not responsible for any chemical instability. Obviously, the concentration of lipid used may be the special consideration that can alter the stability. It is reported that the maximum lipid degradation is around 10% over 2 years of storage, and SLNs prepared with triglycerides are more stable than SLNs prepared with mono and diglycerides.82 The melting point of the lipid is also a point of discussion regarding the particle size distribution.83 Thus, for the preparation of SLNs, lipids that do not undergo hydrolyzation with the aqueous phase should be chosen, and for SLNs prepared from natural lipids, the addition of preservatives can stabilize the microbial contamination.84 Amphiphilic molecules such as surfactants and block copolymers are used as stabilizing agents, emulsifier, and co-emulsifiers in the preparation of SLNs. Some examples including phospholipids (tricaprin),85 ethylene oxide or propylene oxide copolymers (poloxamer 188 or Pluronic® 68),86 sorbitan ethylene oxide or propylene oxide copolymers (Tween 80 and Tween 20),87,88 bile salts (sodium taurocholate)89 and others are listed in Table 2.| Drug | Lipid | Surfactant/emulsifier | Co-Surfactant | Method for preparation of SLNs | Techniques for characterization of SLNs | Size (nm) | Ref. |
|---|---|---|---|---|---|---|---|
| Amphotericin B | Compritol® ATO 888, Precirol ATO 5 and stearic acid, | Pluronic® F-68, Pluronic® F-127, | Solvent diffusion method | DLS, DSC, zeta potential | 111–415.8 | 37 | |
| Compritol® ATO 888 (glycerylbehenate), glycerylpalmitostearate (Precirol® ATO 5), medium chain triglyceride | Tween 20, Pluronic® F-127, Cremophor RH40, polyoxyethylene (40) stearate (Myrj 52) | HPH | DLS, zeta potential, HPLC, TEM, FTIR, DSC, PXRD, 1H NMR | 90–260 | 38 | ||
| Baclofen | Stearic acid | Epikuron 200 (92% phosphatidylcholine) | Propionic acid, butyric acid, and sodium taurocholate | Multiple (w/o/w) warm, microemulsion | DLS | 161.4 | 39 |
| BuspironeHCl | Cetyl alcohol, Spermaceti | Pluronic® F-68, Tween 80 | Emulsification-evaporation followed by ultrasonication | DLS | 86–123 | 40 | |
| Camptothecin | Soybean lecithin, stearic acid | Pluronic® F-68, Tween 80 | Glycerol, PEG 400, PPG | Hot HPH | TEM | 196.8 | 41 |
| Carvedilol | Stearic acid | Pluronic® F-68 | Sodium taurocholate and ethanol | Microemulsion | TEM, DLS | 120–200 and 600–800 | 42 |
| Clozapine | Trimyristin, tripalmitin, tristearin, soy phosphatidylcholine | Pluronic® F-68 | Ultrasonication method | DLS, zeta potential | 96.7 ± 3.8 to 163.3 ± 0.7 | 43 | |
| Crypto-Tanshinone | Glycerylmonostearate, Compritol 888 ATO | Soy lecithin, Tween 80, sodium dehydrocholate | Ultrasonic and high-pressure homogenization method | TEM, DLS, DSC | 121.4 ± 6.3 and 137.5 ± 7.1 | 44 | |
| Curcumin | Compritol 888 ATO | Soy lecithin, Tween 80 | Microemulsion | DLS, TEM | 134.6, 40–120 | 45 | |
| Tristearin | Polyoxyethylene (10) stearyl ether (Brij®S10), polyoxyethylene (100) stearyl ether (Brij® S100) | Oil-in-water emulsion technique | PCS, zeta potential | 111–350 | 45 | ||
| Cyclosporine A | Imwitor® 900 | Tagat®S, sodium cholate | HPH, hot HPH | DLS | 157, 143 | 46 and 47 | |
| Diazepam | Compritol 888 ATO, Imwitor® 900 | Pluronic® F-68, Tween 80 | Ultrasound techniques modified high-shear homogenization and | TEM | <500 | 48 | |
| Doxorubicin hydrochloride | Glycerylcaprate | Polyethylene glycol 660 hydrox-ystearate (Solutol®HS15) | Ultrasonic homogenization | DLS, zeta potential, DSC | 199 | 49 | |
| Fenofibrate | Vitamin E TPGS, Vitamin E 6–100 | Hot HPH | DLS | 58 | 50 | ||
| Hydrocortisone | Precirol® ATO 5, Compritol® 888 ATO, Rylo TM MG 14 Pharma, Dynasan® 114 Dynasan® 118, Tegin® 4100 | Tween 80 | Hot high pressure homogenization | DLS, DSC | 150–220 | 51 | |
| Ibuprofen | Trilaurin, tripalmitin, stearic acid | Pluronic®F127, sodium taurocholate | Solvent-free high-pressure homogenization (HPH) | DLS, X-ray powder diffraction, DSC, AFM | 111–121 (empty SLN) 175–189 (loaded sample) | 52 | |
| Idarubicin | Stearic acid | Epikuron 200 (soy phosphatidylcholine 95%) | Taurocholate sodium salt | Microemulsion | PCS, 90 PLUS | 80 ± 10((loaded sample)) | 53 |
| Emulsifying wax | Polyoxyl 20-stearyl ether (Brij 78), D-alpha-tocopheryl polyethylene glycol succinate (vitamin E TPGS),DSPE-PEG3000 | Sodium taurodeoxycholate (STDC), sodium tetradecylsulfate (STS) | PCS, Zetasizer nano Z | 94.4 (blank), 80–104 (loaded sample) | 54 | ||
| Ketoprofen | Beeswax and carnauba wax | Tween 80, egg lecithin | Microemulsion technique | PCS, DSC | 65–250 (loaded sample) | 55 | |
| Lopinavir | Compritol 888 ATO (glycerylbehenate) | Pluronic®F127 | Hot homogenization, ultrasonication | DLS, zeta potential, HPLC, DSC, WAXS, AFM | 230 | 56 | |
| Lovastatin | Triglyceride, and phosphatidylcholine 95% | Pluronic®F68 | Hot homogenization ultrasonication | DLS, HPLC, DSC, PXRD, LC-MS/MS | 60–119 | 57 | |
| Methotrexate | Stearic acid, monostearin, tristearin, and Compritol 888 ATO | L-α-Soya lecithin, and Sephadex G-50 | Solvent diffusion method | DLS, zeta potential, TEM | 120–167 | 58 | |
| Nevirapine | Steric acid, Compritol 888 ATO | Dimethyldioctadecyl ammonium bromide (DODAB), Tween 80, Lecithin | 1-Butanol | Microemulsion | DLS, zeta potential, field emission scanning electron microscopy (FE-SEM), DSC | 153.1 | 59 |
| Nitrendipine | triglyceride and phosphatidylcholine | Pluronic®F68 | Hot homogenization ultrasonication method | DLS, zeta potential, scanning electron microscopy (SEM) | 110–140 | 60 | |
| Octadecylamine-fluorescein isothiocyanate | Stearic acid | Otcadecylamine, polyethylene glycol monostearate (PEG2000-SA) | Solvent diffusion | DLS, zeta potential | 203 | 61 | |
| Pentoxifylline | Stearic acid, cetyl alcohol, soy lecithin, | Tween 20, Pluronic F®68 | Homogenization followed by the ultrasonication | DLS, zeta-potential | 255–4000 | 62 | |
| Praziquantel | Hydrogenated castor oil | Poly vinyl alcohol (PVA) | Hot homogenization and ultrasonication | DLS, zeta-potential, SEM | 344.0 | 63 | |
| Puerarin | Monostearin, and soy lecithin | Pluronic F®68 | Solvent injection method | DLS and zeta-potential | 160 | 64 | |
| Quercetin | Glycerylmonostearat, soy lecithin | Tween-80 and PEG 400 | Emulsification-solidification | DLS, zeta-potential, TEM | 65 | ||
| Rifampicin | Stearic acid | PVA | Emulsion-solvent diffusion | 66 | |||
| Tobramycin | Stearic acid | Epikuron 200 | Sodium taurocholate | Microemulsion | DLS, TEM | 70–100 | 67 |
| Vinpocetine | Glycerylmonostearat, soy lecithin, polyoxyethylene hydrogenated castor oil | Tween 80 | Ultrasonic-solvent emulsification | DLS, TEM | 70–170 | 68 |
The general mechanism of all these surfactants is to reduce the interfacial tension between the lipid and aqueous phase by applying their amphiphilic nature. Previous studies have shown that the use of surfactant together with a co-surfactant is likely to result in a smaller particle size.90 Recrystallization of the lipid phase results in the rapid growth of particle size, and thereby the long-term stability of the aqueous SLN dispersion is reduced.91 The surfactant structure and interaction between lipid molecules are also responsible for the crystallization process,92 and thus the impact of the lipid and surfactant with or without a co-surfactant and their concentration are significant.
SLN characterization
Physical and chemical characterization are also required after the preparation of SLNs. Due to the particle size, complexity and dynamic nature of the delivery system, the characterization of SLNs is a serious challenge. The parameters needed to evaluate SLNs include particle size, zeta potential, degree of crystallinity, drug release, entrapment efficiency (% EE) and surface morphology. Particle size, polydispersity index and charge analysis can be measured by photon correlation spectroscopy (PCS), dynamic light scattering (DLS) and quasi-elastic light scattering (QELS).93 The main advantage of these techniques is that they are not time-consuming, with speedy analysis and high sensitivity.94 The crystallinity of lipid or polymorphic modifications can be analyzed via differential scanning calorimetric analysis (DSC).95 The crystallinity within nanoparticles is measured by the function of the glass and melting point temperature associated with the enthalpies. Nuclear magnetic resonance (NMR) can also be used to determine the size and qualitative nature of nanoparticles. Changes in their chemical shift are related to the molecular dynamics, which provide information about the physicochemical state of the constituents inside the nanoparticles. Electron microscopy is an advanced technique that can offer a direct way of observing nanoparticles. The size, surface topography, stability and structural changes of SLNs with time can be better investigated using scanning electron microscopy (SEM) and transmission electron microscopy (TEM). However, cryo-microscopic analysis involves rapid freezing, and thus the specimen is preserved in its hydrated state. Cryoelectron microscopy (cryo-EM) such as cryo-TEM and cryo-FESEM provides 3D images of stable frozen-hydrated particles.96 Atomic force microscopy (AFM) is more advanced than TEM and SEM. This method allows atomic-level resolution to be accessed together with size, colloidal attraction and resistance to deformation, making AFM an important tool. The surface distribution of surfactant molecules, bio-conjugation confirmation in case of cationic SLNs, and functionalization of nanoparticles can be estimated by X-ray photoelectron spectroscopy (XPS).97,98 SLN entrapment can be measured by either centrifugation or micro-centrifugation techniques. The samples are centrifuged at high rpm, and the amount of free compound is determined by UV-Visible spectroscopy or high-performance liquid chromatography in a clear supernatant.99–101 The drug loading and release profile or release kinetics of SLNs depend on the crystalline state and melting behavior of the lipid.102Pharmacological performance of SLNs
Nanoparticles for drug delivery or nanotechnology-induced drug delivery systems are going to be the most innovative and crucial cornerstones in the pharmaceutical research area with a great economic impact.103 Gradually, novel SLNs will be widely accepted pharmaceutical carriers for drug delivery to a specific site with increasing interest and improved pharmacokinetic profiles compared to traditional drug delivery.104 Targeted dug delivery, oral administration, topical administration, cosmetics, intravenous administration, protein peptide delivery and ocular delivery are the areas covered by SLNs.84,105–113 Targeting the brain for the successful delivery of pharmaceutical actives is a challenging part of NDDS since 98% of drugs cannot cross the blood brain barrier (BBB).114 Accordingly, SLNs demonstrate a potential approach due to their lipid behavior and effective nanometer size range for targeted drug delivery.Stability issue and storage conditions of SLNs
It has already been reported that SLNs are stable for more than three years. The stability of SLNs is mainly associated with their lipid material, surfactant concentration, and temperature optimization during their preparation. Thus, all these parameters should be considered for their stability and storage. Triglycerides undergo α (alpha), β (beta) and β′ (beta prime) crystal modification during their preparation and storage.115 The kinetics of their polymorphic transitions largely depend on their chain length, where the crystallization process is slower for longer chain than shorter chain triglycerides.116 Sometimes SLNs undergo gel formation, and their gelling tendency strongly depends upon β′ modification due to exposure to light, temperature and shear force.7 Also, the size of the particles can vary because of exposure to light.117In a study, SLNs were exposed to various destabilizing factors, and it was found that gelation occurred and their zeta potential decreased.118 However, SLNs have several stability issues and the drug may be hydrolyzed in aqueous dispersion. Thus, drying is a necessary option for the prolonged storage of SLNs. Freeze drying, spray drying, and lyophilization are techniques for drying. Recently, the electrospray method was employed to prepare SLNs, where a dry SLN powder was obtained directly.119 The formulation of SLNs in a powder form, which may be loaded into pellets, capsules, or tablets, makes these materials highly advantageous for drug delivery. On the other hand, the applications of SLN formulations may be restricted due to their uncontrolled particle growth through coagulation or agglomeration, generating very swift “burst release” of the drug.112 SLNs possess perfect crystal lipid matrices, which carry the loaded drug in its molecular form between fatty acid chains.76,83 The formation and uncontrolled, unwanted enhancement of the crystal structure during both the production and storage of SLNs often result in the release of the loaded drug solution, which is a huge drawback of SLNs.98
Applications in drug delivery
SLNs have been widely applied for various medical applications due to their flexible surface topology and versatile properties (Table 2).Novel theranostic approach
Recently, the emerging trends of nanoparticulate drug delivery systems include nanotherapeutics with diagnostic imaging on the same platform based on image-guided drug delivery to track the pharmacokinetics and biodistribution of the therapeutic agent in real time. Thus, the in vivo theranostics approach can be a great dimensional change in drug delivery systems and diagnostic imaging.120 Image-guided drug delivery systems include the combination of disease diagnosis and therapy, bio-distribution tracking, drug distribution at the target site, drug response prediction, drug efficacy, monitoring and quantification.121 Nanotheranostics or drug delivery together with diagnostic imaging using nanoparticles has a great impact on localizing the target site, and the disease-specific targeting of active pharmaceutical ingredients can also be monitored.122 Since nanoparticles possess dimensions similar to that of various biomolecules such as DNA, RNA, and proteins, they can play a crucial role in surgery together with drug delivery and imaging.123 Nowadays, polymeric nanoparticles, liposomal drug delivery systems, dendrimers and silica nanoparticles are used in the theranostics approach.124–128 Furthermore, pH-triggered nanoparticles, and magnetic and photo-responsive theranosomes are also included in image-guided drug delivery systems for cancer therapy.129,130 Also, SLNs inserted with prostacyclin (PGI2) can be used for the image-guided treatment of atherosclerosis by inhibiting platelet aggregation.131Lipid vesicles can be used as a theranostics platform for non-invasive drug delivery and imaging, and since SLNs are lipid nanovesicles, they have potential for application in the theranostic approach.
Conclusion
Solid lipid nanoparticles are colloidal dispersions with modified properties of other nanoparticles such as microemulsions, suspensions, liposomes and polymeric nanoparticles. The major problems encountered with nanoparticles can be successively avoided using SLNs, and finally a chemically stable and physiologically suitable drug delivery system can be achieved with less limitations. Only their gelation tendency seems to be the main problem, but nanostructured lipid carriers are a possible way to overcome this problem. In addition, the active component, i.e. the drug, may be degraded during their production based on the hot homogenization method because of the generated heat and stress. Thus, choosing an appropriate production method is crucial. Several other difficulties such as particle size, coexistence of various colloidal forms, different shapes and drug ejection from the lipid matrix also need to be addressed.132 The various well-established methods for the bulk production of the SLN matrix and its characterization were discussed. Drugs with physicochemical incompatibility, lower pharmacokinetic profile, and thermolabile drugs can be delivered to the target site via SLNs. Protein and peptide delivery with a higher degree of efficiency and lower toxicity can also be achieved with SLNs. Thus, the addition of the theranostics approach with SLNs can take therapeutics and diagnostics in a new direction.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by National Natural Science Foundation of China (Project No. 81903623), the China Postdoctoral Science Foundation (No. 2019M652586), and the Postdoctoral Research Grant in Henan Province (Project No. 19030008).References
- M. J. Mycek, R. A. Harvey and P. C. Champe, Pharmacology, Lippincott Williams & Wilkins, Philadelphia, P.A., 2000 Search PubMed.
- R. H. Muller, K. Mader and S. Gohla, Eur. J. Pharm. Biopharm., 2000, 50, 161–177 CrossRef CAS PubMed.
- S. S. Salunkhe, N. M. Bhatia and M. S. Bhatia, Drug Deliv., 2016, 23, 1306–1316 CAS.
- U. Scheffel, B. A. Rhodes, T. K. Natajaran and H. N. Wagner, J. Nucl. Med., 1970, 13, 498–503 Search PubMed.
- M. Jumaa and B. W. Muller, Eur. J. Pharm. Sci., 2000, 9, 285–290 CrossRef CAS PubMed.
- S. H. Gohla and A. Dingler, Pharmazie, 2001, 56, 61–63 CAS.
- C. Freitas and R. H. Müller, Eur. J. Pharm. Biopharm., 1999, 47, 125–132 CrossRef CAS PubMed.
- P. R. Lockman, M. O. Oyewumi, J. M. Koziara, K. E. Roder, R. J. Mumper and D. D. Allen, J. Controlled Release, 2003, 93, 271–282 CrossRef CAS PubMed.
- C. Schwarz, J. Microencapsul., 1999, 16, 205–213 CrossRef CAS PubMed.
- R. Kammari, N. G. Das and S. K. Das, Emerging Nanotechnologies for Diagnostics, Drug Delivery and Medical Devices Micro and Nano Technologies, 2017, pp. 105–144 Search PubMed.
- W. Mehnert and K. Mader, Adv. Drug Delivery Rev., 2001, 47, 165–196 CrossRef CAS PubMed.
- W. Mehnert and K. Mader, Adv. Drug Deliv. Rev., 2001, 47, 165–196 CrossRef CAS PubMed.
- S. B. Lim, A. Banerjee and H. Önyüksel, J. Controlled Release, 2012, 163, 34–45 CrossRef CAS PubMed.
- R. S. Dhakad, R. K. Tekade and N. K. Jain, Curr. Drug Delivery, 2013, 10, 477–491 CrossRef CAS PubMed.
- S. C. Yang, L. F. Lu, Y. Cai, J. B. Zhu, B. W. Liang and C. Z. Yang, J. Controlled Release, 1999, 59(3), 299–307 CrossRef CAS PubMed.
- H. H. Gustafson, D. Holt-Casper, D. W. Grainger and H. Ghandehari, Nano Today, 2015, 10, 487–510 CrossRef CAS PubMed.
- C. Bocca, O. Caputo, R. Cavalli, L. Gabriel, A. Miglietta and M. R. Gasco, Int. J. Pharm., 1998, 175(2), 185–193 CrossRef CAS.
- G. P. Zara, R. Cavalli, A. Fundaro, A. Bargoni, O. Caputo and M. R. Gasco, Pharmacol. Res., 1999, 40, 281–286 CrossRef CAS PubMed.
- R. Cavalli, E. Peira, O. Caputo and M. R. Gasco, Int. J. Pharm., 1999, 182, 59–69 CrossRef CAS PubMed.
- C. Freitas and R. H. Müller, Eur. J. Pharm. Biopharm., 1999, 47, 125–132 CrossRef CAS PubMed.
- A. Miglietta, R. Cavalli, C. Bocca, L. Gabriel and M. R. Gasco, Int. J. Pharm., 2000, 210, 61–67 CrossRef CAS PubMed.
- C. Dianzani, G. P. Zara, G. Maina, P. Pettazzoni, S. Pizzimenti, F. Rossi, C. L. Gigliotti, E. S. Ciamporcero, M. Daga and G. Barrera, Drug delivery nanoparticles in skin cancers, BioMed Res. Int., 2014, 895–986 Search PubMed.
- V. Jenning, A. Gysler, M. Schäfer-Korting and S. H. Gohla, Eur. J. Pharm. Biopharm., 2000, 49, 211–218 CrossRef CAS PubMed.
- C. C. Muller-Goymann, Eur. J. Pharm. Biopharm., 2004, 58, 343–356 CrossRef CAS PubMed.
- M. Schafer-Korting, W. Mehnert and H.-C. Korting, Adv. Drug Delivery Rev., 2007, 59, 427–443 CrossRef PubMed.
- R. H. Muller, M. Radtke and S. A. Wissing, Adv. Drug Delivery Rev., 2002, 54, S131–S155 CrossRef CAS PubMed.
- V. Jenning, A. Gysler, M. Schafer-Korting and S. H. Gohla, Eur. J. Pharm. Biopharm., 2000, 49, 211–218 CrossRef CAS PubMed.
- V. Jenning, M. Schafer-Korting and S. Gohla, J. Controlled Release, 2000, 66, 115–126 CrossRef CAS PubMed.
- S. A. Wissing and R. H. Muller, Adv. Drug Delivery Rev., 2001, 23, 233–243 CAS.
- S. A. Wissing, A. Lippacher and R. H. Muller, J. Cosmet. Sci., 2001, 52, 313–323 CAS.
- C. Olbrich, U. Bakowsky, C. M. Lehr, R. H. Müller and C. Kneuer, J. Controlled Release, 2001, 77, 345–355 CrossRef CAS.
- D. P. Gaspar, F. Vasco, L. M. D. Goncalves, P. Taboad, C. Remunan-Lopez and A. Almeida, Int. J. Pharm., 2016, 497, 199–209 CrossRef CAS PubMed.
- K. Tabatt, M. Sameti, C. Olbrich, R. H. Müller and C. M. Lehr, Eur. J. Pharm. Biopharm., 2004, 57, 155–162 CrossRef CAS PubMed.
- S. A. Wissing and R. H. Muller, Int. J. Pharm., 2003, 254, 65–68 CrossRef CAS PubMed.
- S. A. Wissing and R. H. Muller, J. Pharmacokinet. Biopharm., 2003, 56, 67–72 CrossRef CAS.
- S. Kumar and J. K. Randhawa, Mater. Sci. Eng., C, 2013, 33, 1842–1852 CrossRef CAS PubMed.
- D. Butani, C. Yewale and A. Misra, Colloids Surf., B, 2016, 139, 17–24 CrossRef CAS PubMed.
- P. Jansook, Z. Fulop and G. C. Ritthidej, Drug Dev. Ind. Pharm., 2019, 45, 560–567 CrossRef CAS PubMed.
- L. Priano, G. P. Zara, N. El-Assawy, S. Cattaldo, E. Muntoni, E. Milano, L. Serpe, C. Musicanti, C. Pérot and M. R. Gasco, Eur. J. Pharm. Biopharm., 2011, 79, 135–141 CrossRef CAS PubMed.
- J. Varshosaz, M. Tabbakhian and M. Y. Mohammadi, J. Liposome Res., 2010, 20, 286–296 CrossRef CAS PubMed.
- S. Yang, J. Zhu, Y. Lu, B. Liang and C. Yang, Pharm. Res., 1999, 16, 751–757 CrossRef CAS PubMed.
- B. Sanjula, F. M. Shah, A. Javed and A. Alka, J. Drug Target., 2009, 17, 249–256 CrossRef CAS PubMed.
- K. Manjunath and V. Venkateswarlu, J. Controlled Release, 2005, 107, 215–228 CrossRef CAS PubMed.
- L. Hu, Q. Xing, J. Meng and C. Shang, AAPS PharmSciTech, 2010, 11, 582–587 CrossRef CAS PubMed.
- V. Kakkar, S. Singh, D. Singla and I. P. Kaur, Mol. Nutr. Food Res., 2011, 55, 495–503 CrossRef CAS PubMed.
- R. Muller, S. Runge, V. Ravelli, W. Mehnert, A. Thunemann and E. Souto, Int. J. Pharm., 2006, 317, 82–89 CrossRef CAS PubMed.
- R. Muller, S. Runge, V. Ravelli, A. Thunemann, W. Mehnert and E. Souto, Eur. J. Pharm. Biopharm., 2008, 68, 535–544 CrossRef CAS PubMed.
- G. Abdelbary and R. H. Fahmy, AAPS PharmSciTech, 2009, 10, 211–219 CrossRef CAS PubMed.
- R. K. Subedi, K. W. Kang and H.-K. Choi, Eur. J. Pharm. Sci., 2009, 37, 508–513 CrossRef CAS PubMed.
- A. Hanafy, H. Spahn-Langguth, G. Vergnault, P. Grenier, M. T. Grozdanis, T. Lenhardt and P. Langguth, Adv. Drug Delivery Rev., 2007, 59, 419–426 CrossRef CAS PubMed.
- L. B. Jensen, E. Magnussson, L. Gunnarsson, C. Vermehren, H. M. Nielsen and K. Petersson, Int. J. Pharm., 2010, 390, 53–60 CrossRef CAS PubMed.
- S. G. Potta, S. Minemi, R. K. Nukala, C. Peinado, D. A. Lamprou, A. Urquhart and D. Douroumis, J. Microencapsul., 2011, 28, 74–81 CrossRef CAS PubMed.
- G. P. Zara, A. Bargoni, R. Cavalli, A. Fundarò, D. Vighetto and M. R. Gasco, J. Pharm. Sci., 2002, 91, 1324–1333 CrossRef CAS PubMed.
- P. Ma, X. Dong, C. L. Swadley, A. Gupte, M. Leggas, H. C. Ledebur and R. J. Mumper, J. Biomed. Nanotechnol., 2009, 5, 151–161 CrossRef CAS PubMed.
- S. Kheradmandnia, E. Vasheghani-Farahani, M. Nosrati and F. Atyabi, Nanomed. Nanotechnol. Biol. Med., 2010, 6, 753–759 CrossRef CAS PubMed.
- M. A. Alex, A. Chacko, S. Jose and E. Souto, Eur. J. Pharm. Sci., 2011, 42, 11–18 CrossRef PubMed.
- G. Suresh, K. Manjunath, V. Venkateswarlu and V. Satyanarayana, AAPS PharmSciTech, 2007, 8, E162–E170 CrossRef PubMed.
- R. Paliwal, S. Rai, B. Vaidya, K. Khatri, A. K. Goyal, N. Mishra, A. Mehta and S. P. Vyas, Nanomed. Nanotechnol. Biol. Med., 2009, 5, 184–191 CrossRef CAS PubMed.
- Y.-C. Kuo and J.-F. Chung, Colloids Surf., B, 2011, 83, 299–306 CrossRef CAS PubMed.
- K. Manjunath and V. Venkateswarlu, J. Drug Targeting, 2006, 14, 632–645 CrossRef CAS PubMed.
- H. Yuan, J. Chen, Y.-Z. Du, F.-Q. Hu, S. Zeng and H.-L. Zhao, Colloids Surf., B, 2007, 58, 157–164 CrossRef CAS PubMed.
- J. Varshosaz, M. Minayian and E. Moazen, J. Liposome Res., 2010, 20, 115–123 CrossRef CAS.
- S. Xie, B. Pan, M. Wang, L. Zhu, F. Wang, Z. Dong, X. Wang and W. Zhou, Nanomedicine, 2010, 5, 693–701 CrossRef CAS PubMed.
- C.-F. Luo, M. Yuan, M.-S. Chen, S.-M. Liu, L. Zhu, B.-Y. Huang, X.-W. Liu and W. Xiong, Int. J. Pharm., 2011, 410, 138–144 CrossRef CAS PubMed.
- H. Li, X. Zhao, Y. Ma, G. Zhai, L. Li and H. Lou, J. Controlled Release, 2009, 133, 238–244 CrossRef CAS PubMed.
- R. Pandey, S. Sharma and G. Khuller, Tuberculosis, 2005, 85, 415–420 CrossRef CAS PubMed.
- R. Cavalli, A. Bargoni, V. Podio, E. Muntoni, G. P. Zara and M. R. Gasco, J. Pharm. Sci., 2003, 92, 1085–1094 CrossRef CAS PubMed.
- Y. Luo, D. Chen, L. Ren, X. Zhao and J. Qin, J. Controlled Release, 2006, 114, 53–59 CrossRef CAS PubMed.
- A. R. Gardouh, S. Gad, H. M. Ghonaim and M. M. Ghorab, Br. J. Pharm. Res., 2013, 3, 326–346 CrossRef.
- C. Schwarz, W. Mehnert, J. Lucks and R. Müller, J. Controlled Release, 1994, 30, 83–96 CrossRef CAS.
- M. O. Emeje, E. I. Akpabio, I. C. Obidike and S. I. Ofoefule, Nanotechnology in Drug Delivery. INTECH Open Access Publisher, 2012 Search PubMed.
- A. Waghmare, N. Grampurohit, M. Gadhave, D. Gaikwad and S. Jadhav, Int. Res. J. Pharm., 2012, 3, 100–107 CAS.
- C. Shah, V. Shah and U. Upadhyay, Curr. Pharma Res., 2011, 1, 351–368 CrossRef.
- M. Trotta, F. Debernardi and O. Caputo, Int. J. Pharm., 2003, 257, 153–160 CrossRef CAS PubMed.
- M. A. Schubert and C. C. Müller-Goymann, Eur. J. Pharm. Biopharm., 2003, 55, 125–131 CrossRef CAS PubMed.
- Y. W. Naguib, B. L. Rodriguez, X. Li, S. D. Hursting, R. O. Williams III and Z. Cui, Mol. Pharmaceutics, 2014, 11, 1239–1249 CrossRef CAS PubMed.
- H. Svilenov and C. Tzachev, Solid lipid nanoparticles–apromising drug delivery system, Nanomedicine, 2014, 187–237 Search PubMed.
- Y. J. Chen, R. X. Jin, Y. Q. Zhou, J. Zeng, H. Zhang and Q. R. Feng, China J. Chin. Mater. Med., 2006, 31(5), 376–379 CAS.
- C. Charcosset, A. El-Harati and V. Fessi, J. Controlled Release, 2005, 108, 112–120 CrossRef CAS PubMed.
- R. Sridhar and S. Ramakrishna, Biomatter, 2013, 3, e24281 CrossRef PubMed.
- F. S. Abdel-Salam, S. A. Elkheshen, A. A. Mahmoud and H. O. Ammar, Bull. Fac. Pharm. Cairo Univ., 2016, 54, 1–7 CrossRef.
- A. Radomska-Soukharev, Adv. Drug Delivery Rev., 2007, 59, 411–418 CrossRef CAS PubMed.
- B. Siekmann and K. Westesen, Pharm. Pharmacol. Lett., 1992, 1, 123–126 CAS.
- T. Skotland, K. Sagini, K. Sandvig and A. Llorente, Adv. Drug Delivery Rev., 2020 DOI:10.1016/j.addr.2020.03.002.
- F. Castelli, C. Puglia, M. G. Sarpietro, L. Rizza and F. Bonina, Int. J. Pharm., 2005, 304, 231–238 CrossRef CAS PubMed.
- C. Schwarz and W. Mehnert, Int. J. Pharm., 1997, 157, 171–179 CrossRef CAS PubMed.
- J. Liu, W. Hu, H. Chen, Q. Ni, H. Xu and X. Yang, Int. J. Pharm., 2007, 328, 191–195 CrossRef CAS PubMed.
- T. S. Awad, T. Helgason, J. Weiss, E. A. Decker and D. J. McClements, Cryst. Growth Des., 2009, 9, 3405–3411 CrossRef CAS.
- R. Cavalli, M. R. Gasco, P. Chetoni, S. Burgalassi and M. F. Saettone, Int. J. Pharm., 2002, 238, 241–245 CrossRef CAS PubMed.
- R. Cavalli, Pharmazie, 1998, 53, 392–396 CAS.
- Z. Muhlen, Feste Lipid Nanopartikel mit prolongierter Wirkstoffliberation. Herstellung, Langzeitstabilitat, Charakterisierung, Freiset zungsverhalten und -mechanismen. PhD thesis, Freie Universitat Berlin, 1996 – from 1992-2 ref. no 2.
- A. Kovacevic, S. Savic, G. Vuleta, R. H. Müller and C. M. Keck, Int. J. Pharm., 2011, 406, 163–172 CrossRef CAS PubMed.
- M. Alexander and D. G. Dalgleish, Food Biophys., 2006, 1, 2–13 CrossRef.
- S. Mukherjee, S. Ray and R. S. Thakur, Indian J. Pharm. Sci., 2009, 71, 349 CrossRef CAS PubMed.
- P. J. Dale, J. Kijlstra and B. Vincent, Langmuir, 2005, 21, 12250–12256 CrossRef CAS PubMed.
- A. Saupe, K. C. Gordon and T. Rades, Int. J. Pharm., 2006, 314, 56–62 CrossRef CAS PubMed.
- V. K. Venishetty, R. Samala, R. Komuravelli, M. Kuncha, R. Sistla and P. V. Diwan, Nanomed. Nanotechnol. Biol. Med., 2013, 9, 388–397 CrossRef CAS PubMed.
- V. K. Venishetty, R. Komuravelli, M. Kuncha, R. Sistla and P. V. Diwan, Nanomed. Nanotechnol. Biol. Med., 2013, 9, 111–121 CrossRef CAS PubMed.
- N. K. Jain and R. K. Tekade, Drug Deliv., 2013, 373–409 CAS.
- J. Kayat, V. Gajbhiye, R. K. Tekade and N. K. Jain, Nanomedicine, 2011, 7, 40–49 CrossRef CAS PubMed.
- R. K. Tekade, T. Dutta, V. Gajbhiye and N. K. Jain, J. Microencapsul., 2009, 26, 287–296 CrossRef CAS PubMed.
- A. Zur Muhlen, C. Schwarz and W. Mehnert, Eur. J. Pharm. Biopharm., 1998, 45, 149–155 CrossRef CAS PubMed.
- S. K. Sahoo and V. Labhasetwar, Drug discovery Today, 2003, 8, 1112–1120 CrossRef CAS PubMed.
- A. Fundaro, R. Cavalli, A. Bargoni, D. Vighetto, G. P. Zara, G. Paolo and M. R. Gasco, Pharmacol. Res., 2000, 42, 337–343 CrossRef CAS PubMed.
- S. Martins, I. Tho, I. Reimold, G. Fricker, E. Souto, D. Ferreira and M. Brandl, Int. J. Pharm., 2012, 439, 49–62 CrossRef CAS PubMed.
- C. Zhang, C. Gu, F. Peng, W. Liu, J. Wan, H. Xu and X. Yang, Molecules, 2013, 18, 13340–13356 CrossRef CAS PubMed.
- J. Liu, W. Hu, H. Chen, Q. Ni, H. Xu and X. Yang, Int. J. Pharm., 2007, 328, 191–195 CrossRef CAS PubMed.
- S. A. Wissing and R. H. Muller, Int. J. Pharm., 2003, 254, 65–68 CrossRef CAS PubMed.
- C. Carrillo, N. Sanchez-Hernandez, E. Garcia-Montoya, P. Perez-Lozano, J. M. Sune-Negre, J. R. Tico, C. Sune and M. Minarro, Eur. J. Pharm. Sci., 2013, 49, 157–165 CrossRef CAS PubMed.
- A. Del Pozo-Rodríguez, D. Delgado, M. Á. Solinís, J. L. Pedraz, E. Echevarría, J. M. Rodríguez and A. R. Gascon, Int. J. Pharmacother., 2010, 385, 157–162 CrossRef PubMed.
- M. D. Joshi and R. H. Müller, Eur. J. Pharm. Biopharm., 2009, 71, 161–172 CrossRef CAS PubMed.
- R. Cortesi, M. Campioni, L. Ravani, M. Drechsler, M. Pinotti and E. Esposito, N. Biotech., 2014, 31, 44–54 CrossRef CAS PubMed.
- J. F. Fangueiroa, T. Andreania, M. A. Egead, M. L. Garciad, S. B. Souto, A. M. Silva and E. B. Soutoa, Int. J. Pharmacother., 2014, 461, 64–73 CrossRef PubMed.
- W. M. Pardridge, Alzheimer's Dement, 2009, 5, 427–432 CrossRef PubMed.
- V. Luzzati, A. Tardieu and T. Gulik-Krzywicki, Nature, 1968, 217, 1028–1030 CrossRef CAS PubMed.
- H. Bunjes, K. Westesen and M. H. Koch, Int. J. Pharm., 1996, 129, 159–173 CrossRef CAS.
- R. H. Muller, et al., Die Pharmazeutische Industrie, 1997, 59(7), 614–619 CAS.
- C. Freitas and R. H. Müller, Int. J. Pharm., 1998, 168, 221–229 CrossRef CAS.
- Y. Wu, B. Yu, A. Jackson, W. Zha, L. J. Lee and B. E. Wyslouzil, Mol. Pharmaceutics, 2009, 6, 1371–1379 CrossRef CAS PubMed.
- H. Yi-Ping and K. W. Leong, Nanoscale, 2010, 2, 60–68 RSC.
- T. Lammers, F. Kiessling, W. E. Hennink and G. Storm, Mol. Pharmaceutics, 2010, 7, 1899–1912 CrossRef CAS PubMed.
- T. Lammers, S. Aime, W. E. Hennink, G. Storm and F. Kiessling, Acc. Chem. Res., 2011, 44, 1029–1038 CrossRef CAS PubMed.
- H. E. Gendelman, et al., Nanomed. Nanotechnol. Biol. Med., 2015, 11, 751–767 CrossRef CAS PubMed.
- B. T. Luk, R. H. Fang and L. Zhang, Theranostics, 2011, 2, 1117–1126 CrossRef PubMed.
- S. Li, B. Goins, L. Zhang and A. Bao, Bioconjugate Chem., 2012, 23, 1322–1332 CrossRef CAS PubMed.
- N. T. Chen, S. H. Cheng, J. S. Souris, C. T. Chen, C. Y. Mou and L. W. Lo, J. Mater. Chem. B, 2013, 3128–3135 RSC.
- T. Krasia-Christoforou and T. K. Georgiou, J. Mater. Chem. B, 2013, 24, 3002–3025 RSC.
- S. T. Lo, A. Kumar, J. T. Hsieh and X. Sun, Mol. Pharmaceutics, 2013, 10, 793–812 CrossRef CAS PubMed.
- A. K. Silva, J. Kolosnjaj-Tabi, S. Bonneau, I. Marangon, N. Boggetto, K. Aubertin and C. Wilhelm, ACS Nano, 2013, 7, 4954–4966 CrossRef CAS PubMed.
- H. Chen, T. Moore, B. Qi, D. C. Colvin, E. K. Jelen, D. A. Hitchcock and J. N. Anker, ACS Nano, 2013, 7, 1178–1187 CrossRef CAS PubMed.
- K. Oumzil, M. A. Ramin, C. Lorenzato, A. Hémadou, J. Laroche, M. J. Jacobin-Valat, S. Mornet, C. E. Roy, T. Kauss, K. Gaudin, G. Clofent-Sanchez and P. Barthelemy, Bioconjugate Chem., 2016, 27, 569–575 CrossRef CAS PubMed.
- W. Mehnert and K. Meader, Adv. Drug Deliv. Rev., 2001, 47, 165–196 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |