
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-assembly of Janus Au:Fe3O4 branched nanoparticles. From organized clusters to stimuli-responsive nanogel suprastructures†
Javier
Reguera
 *abc,
Tatjana
Flora
de,
Naomi
Winckelmans
f,
José C.
Rodríguez-Cabello
de and
Sara
Bals
f
*abc,
Tatjana
Flora
de,
Naomi
Winckelmans
f,
José C.
Rodríguez-Cabello
de and
Sara
Bals
f
aBCMaterials, Basque Center for Materials, Applications and Nanostructures, UPV/EHU Science Park, 48940 Leioa, Spain. E-mail: javier.reguera@bcmaterials.net
bCIC biomaGUNE, Paseo de Miramón 182, 20014 Donostia-San Sebastián, Spain
cIkerbasque, Basque Foundation for Science, 48013 Bilbao, Spain
dBIOFORGE Lab, University of Valladolid, Edificio Lucia, Paseo de Belén 19, 47011 Valladolid, Spain
eBiomedical Research Networking Center in Bioengineering Biomaterials and Nanomedicine, Ciber-BBN, Spain
fEMAT – University of Antwerp, Groenenborgerlaan 171, B-2020 Antwerp, Belgium
First published on 22nd April 2020
Abstract
Janus nanoparticles offer enormous possibilities through a binary selective functionalization and dual properties. Their self-assembly has attracted strong interest due to their potential as building blocks to obtain molecular colloids, supracrystals and well-organized nanostructures that can lead to new functionalities. However, this self-assembly has been focused on relatively simple symmetrical morphologies, while for complex nanostructures this process has been unexplored. Here, we study the assembly of plasmonic-magnetic Janus nanoparticles with a branched (nanostar) – sphere morphology. The branched morphology enhances their plasmonic properties in the near-infrared region and therefore their applicability, but at the same time constrains their self-assembly capabilities to obtain more organized or functional suprastructures. We describe the self-assembly of these nanoparticles after amphiphilic functionalization. The role of the nanoparticle branching, as well as the size of the polymer-coating, is explored. We show how the use of large molecular weight stabilizing polymers can overcome the anisotropy of the nanoparticles producing a change in the morphology from small clusters to larger quasi-cylindrical nanostructures. Finally, the Janus nanoparticles are functionalized with a thermo-responsive elastin-like recombinamer. These nanoparticles undergo reversible self-assembly in the presence of free polymer giving rise to nanoparticle-stabilized nanogel-like structures with controlled size, providing the possibility to expand their applicability to multi-stimuli controlled self-assembly.
Introduction
Janus nanoparticles exhibit a surface composed of two chemically or physically different domains or regions. They offer a unique broken symmetry combined with an ability to impart molecule-like directionality by their chemical anisotropy at a nanometer scale, which makes them exceptional candidates for a large number of applications. For instance, thanks to their high interfacial activity in comparison with their homogeneous counterparts, they could be used to form thermodynamically stable emulsions,1–4 with high applicability in cosmetics, waste-water treatments, heterogeneous catalysis or energy among others.5–8 Also, in biomedicine, they have been proposed in applications ranging from biosensing9 and biomimicking10 to cell-membrane penetration elements.11,12 Other uses include nanomotors and nanojets, electronic displays, anti-reflective surfaces, etc.13–17One remarkable use of Janus nanoparticles is as building blocks for nanoparticle self-assembly, enabling an advance in fields such as molecular colloids or supracrystals.16,18,19 Therefore, a huge effort has been dedicated to the synthesis of novel Janus nanoparticles with a variety of morphologies and high uniformity, as well as their use in the generation of novel self-assembled suprastructures. Among many examples, spherical micelles have been formed with spherical soft Janus nanoparticles,20,21 stacked sandwich structures with disc-shapes,22 worm-like structures with hard amphiphilic Janus SiO2 spheres,23 and pH-dependent aggregates with Janus dipolar hard spheres.24 More complex morphologies such as sphere–sphere (also called the snowman shape) have shown a wealth of assembled architectures. Honciuc et al. have reported how polymeric snowman nanoparticles with different Janus ratios and lobe separations could give rise to mono-walled capsules, spherical micelles, planar bilayers or capsules with random orientation nanoparticles.25,26 Snowman hard Au–SiO2 nanoparticles have been used to produce small clusters,27 while Au–Fe3O4 nanoparticles have been employed for the formation of clusters and double- and mono-layer vesicles.28,29
All these reported studies have been directed towards relatively simple and highly symmetric Janus nanoparticles. Thus, there is a lack of understanding of the self-assembly of more complex Janus nanoparticles, such as those with irregular shapes or those exhibiting hybrid soft–hard materials. Advances in their self-assembly will improve our answers to the old but still highly relevant question of “How far can we push chemical self-assembly?”.30 For this, Janus Au–Fe3O4 nanoparticles have been tested, further modifying the Au part into a branched morphology and varying the polymer surface. This morphology has the advantage of presenting dual superparamagnetic properties and a plasmonic response in the near infrared (NIR) region making it highly interesting in biosensing, multimodal imaging, or dual magnetic-plasmonic hyperthermia among others.31–33 Several stimuli have been tested to trigger the directed self-assembly of these nanoparticles. First, solvent exchange was performed by adding water, which acts as a bad solvent for the hydrophobic nanoparticle side, to a stabilized nanoparticle solution. The water caused the hydrophobic aggregation of the corresponding region, thereby driving the assembly. We show that the molecular weight of the hydrophobic polymer has dramatic effects on the assembly of the morphology decreasing the effect of the nanoparticle anisotropy at high molecular weights. Second, temperature variation was applied as a stimulus to nanoparticles with one side stabilized with elastin-like recombinamers (ELR). ELRs are smart polymers that exhibit a fast and reversible hydrophobic aggregation upon the increase of temperature above their lower critical solution temperature (LCST), also known as inverse temperature transition.34,35 ELRs have shown a high tunability in their LCST and can be easily modified to respond to a wide range of stimuli, such as light, pH, redox potential, ionic strength, etc.34–37 Furthermore, given the recombinant nature of ELRs, a blend of different structural protein domains or enhanced functionalities such as bioactive sequences can be easily incorporated into their backbone.38,39 In this case, the addition of a free ELR to the solution of nanoparticles, functionalized with both a hydrophilic and a thermo-responsive ELR, allowed the size control of the assemblies upon the change of temperature. Finally, given the thermal transduction properties of these nanoparticles, alternating magnetic and electromagnetic stimuli could also be applied. As an example, we show how the photothermal transduction, by irradiating at NIR with a relatively low concentration and dose, can be used to increase the temperature over the LCST and trigger the assembly.
Results and discussion
Janus nanoparticles composed of two epitaxially bond nanomaterials, Au nanostars and Fe3O4 nanospheres, were synthesized as previously reported (see the ESI†).32 Two versions of those nanoparticles were obtained sharing the same F3O4 part with a diameter of 16 nm, and Au nanostars with two different sizes (hereafter called nanoparticles (1) and (2)). Nanoparticles (1) have Au nanostars smaller than the Fe3O4 lobe (dequivalent ∼11 nm) and short tips. Nanoparticles (2) have Au-nanostars bigger than the Fe3O4 part (dequivalent ∼23 nm) and longer tips (see Fig. 1B and ESI Fig. S1†). By ICP (see the ESI) the volume ratio corresponds to an Au/Fe3O4 volume ratio of 0.36 and 1.31 for nanoparticles (1) and (2) respectively. The nanoparticles were synthesized with polyvinylpyrrolidone (PVP) as a weak capping agent. This facilitates ligand exchange with molecules containing groups such as thiols for the Au surface or catechols and silanes for Fe3O4. The Janus nanoparticles were modified to obtain amphiphiles in a two-step selective functionalization (see Fig. 1) following a previously reported protocol.40 In the first step, ligand exchange with thiol-terminated polystyrene (with two molecular weights 2 kg mol−1 and 20 kg mol−1) was followed by UV-Vis with a red shift of the Au plasmon band due to the change in the refractive index (ESI, Fig. S2†). Once the ligand exchange was produced, catechol-terminated polyethylene glycol was added to functionalize the Fe3O4 surface in a second step, minimizing in this way the catechol-terminated molecule adhesion to the Au surface. | ||
| Fig. 1 (A) Schematic representation of the different steps of nanoparticle functionalization for two sets of nanoparticles (1 and 2). (B) TEM images corresponding to the initial heterodimer seeds and after the growth of Au nanostars with two different sizes. (1) Size of Fe3O4 > size of Au and (2) size of Au > size of Fe3O4. (C) UV-Vis of the nanoparticles shown in (B). | ||
Polystyrene with a low molecular weight (2 kg mol−1) is a small polymer or oligomer of only ∼19 monomers, which corresponds to a maximum length of ∼5 nm (for a completely stretched polymer chain). Thus, this polymer is expected to form a thin hydrophobic layer on the Au surface in a way that the assembly is highly dictated by the Au morphology. On the other side, the PEG with a molecular weight of 5 kg mol−1 is expected to form a more expanded hydrophilic region. If nanoparticles are in a suitable solvent for both sides, such as DMF or THF, and a small quantity of water is added (10%) to the system, the nanoparticles self-assemble into small clusters of 2 to 4 nanoparticles with the hydrophobic side facing the interior and the hydrophilic side facing the exterior (Fig. 2). The slow assembly process can be followed by UV-Vis where a red shift is produced over approximately two hours (ESI, Fig. S3†). Interestingly, similar small clusters were formed for nanoparticles with the two different sizes of Au nanostars (nanoparticles 1 and 2), which were only slightly higher for nanoparticles with bigger stars (Fig. 2C.1 and C.2). TEM images and 3D reconstructions obtained by electron tomography of the clusters show that nanoparticles are, in fact, arranged quite differently (Fig. 2B.1 and B.2). For small nanostars, the hydrophilic Fe3O4 tends to be located on opposite sides to maximize the hydrophilic surface of the cluster. However, for bigger nanostars, the longer tips impose big constrains on the cluster. They interdigitate to maximize the hydrophobic contact while the hydrophilic Fe3O4 exterior cannot be organized in opposite directions, but still conferring enough colloidal stability to the cluster in water. Note here that when simple nanostars are functionalized with thiol-terminated PEG they do not exhibit any aggregation while when this functionalization is performed with thiol-terminated PS they completely separate from the solution sedimenting and sticking to the surface of the solution vial.
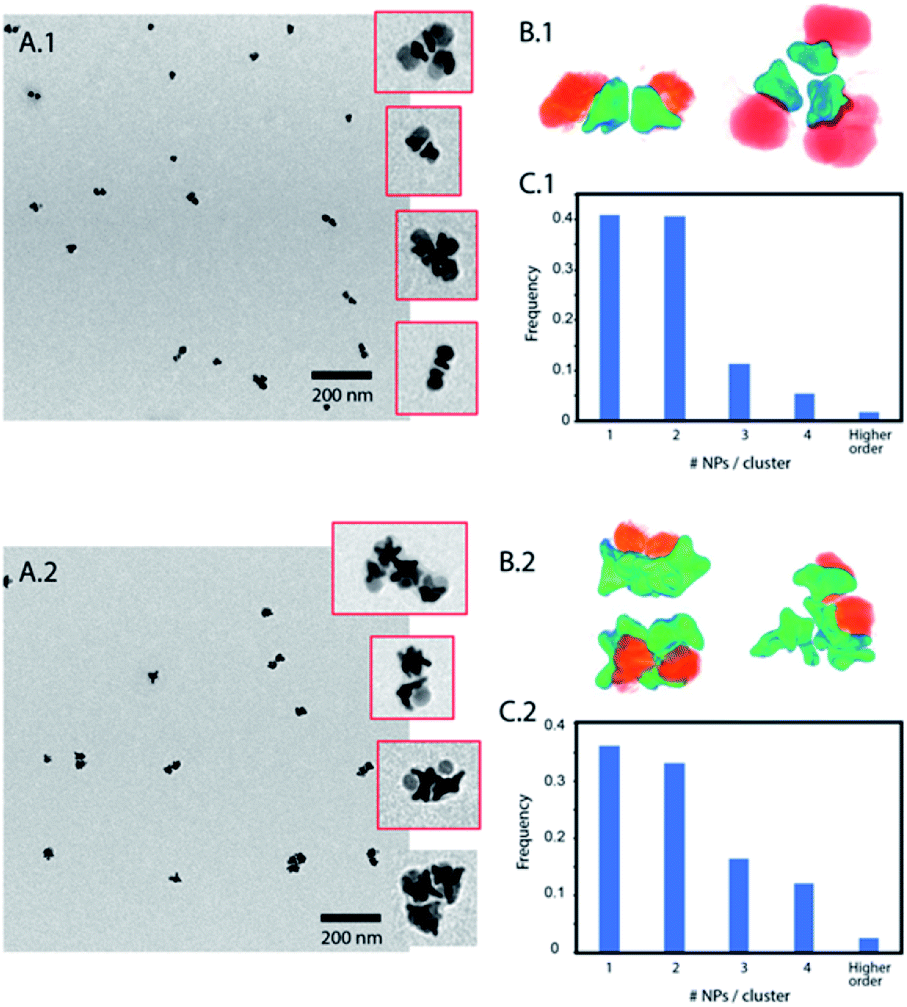 | ||
| Fig. 2 (A) TEM images of assemblies of nanoparticle amphiphiles with short PS. Insets show some of those clusters. (B) Electron tomography showing the cluster hydrophobic core formed by Au nanostars and the hydrophilic exterior formed by hydrophilic Fe3O4. (C) Quantification of cluster sizes according to the number of nanoparticles per cluster, observed in the TEM images. (A.1–C.1) correspond to nanoparticles (1) and (A.2–C.2) to nanoparticles (2). | ||
To overcome the limitations of nanostar irregular shapes, a hydrophobic polymer with a higher molecular weight was used to form a bigger hydrophobic “cushion” and soften the Au branched morphology. Therefore, we modified the Au surface with thiol-terminated PS, with 20 kg mol−1 molecular weight, and performed a similar experiment. The hydrophobic association was much faster than in the previous case (a few minutes) and the assemblies tended to sediment with time (completely sedimented after one day). After purification and dispersion in water, the TEM images showed much bigger assemblies (Fig. 3). They had an elongated morphology with a hydrophobic interior and one-nanoparticle-layer thickness shell. The inset in Fig. 3A shows the side of one of these assemblies where, as expected, the exterior is formed by the Fe3O4 hydrophilic parts (light grey). For the nanoparticles inside the structure, the orientation preference is not obvious, possibly due to the branching of the nanoparticles and the collapse during the drying process. The thickness of those micelles was around 120 ± 25 nm for nanoparticles with small nanostars. Assuming a nanoparticle size of 20–25 nm, this corresponds to an interior diameter of 70–80 nm that could be due to the formation of a second nanoparticle layer, as previously described for dumbbell-like nanoparticles,28,29 or due to the presence of non-bonded polystyrene in solution. The nanoparticles with a bigger nanostar size gave rise to similar structures and, as expected for these bigger nanostars and containing more PS per nanoparticle, the assemblies grew to larger sizes with average diameters of 200 ± 55 nm (ESI, Fig. S4†). Although the elongated morphology is not completely clear, directional growth has been previously observed in micrometer size particles and emulsified particles in an intermediate range of Janus balance.23,41
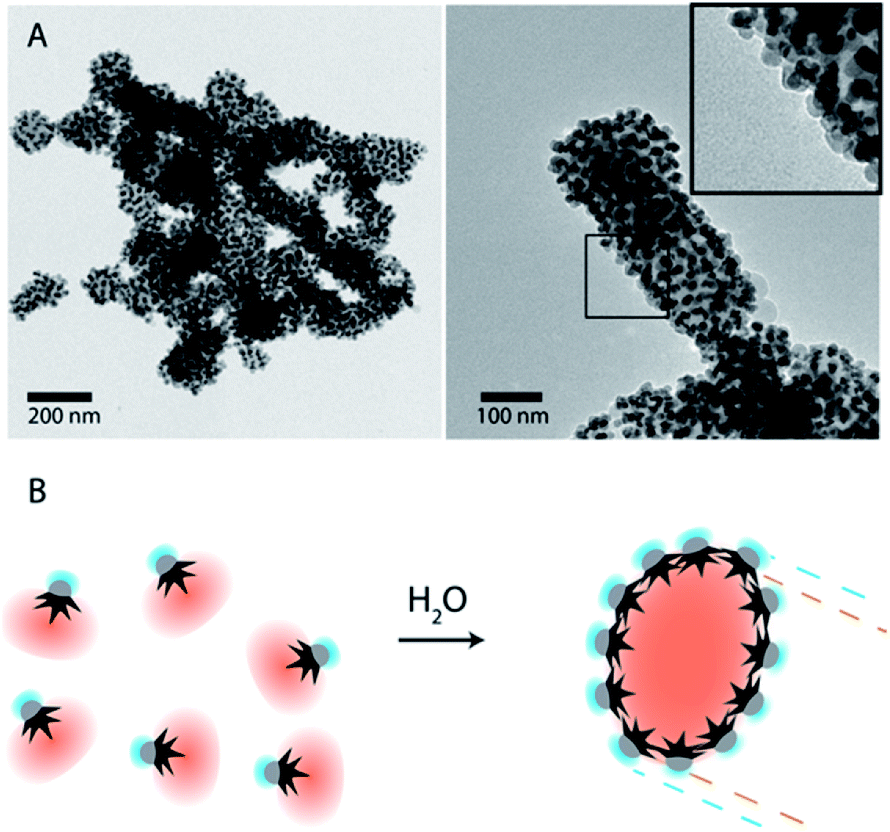 | ||
| Fig. 3 (A) TEM images of nanoparticle-amphiphile assemblies (Fig. 1B1) with long thiol-terminated PS (MW: 20 kg mol−1). Inset shows the Janus nanoparticles with the Fe3O4 facing the exterior of the assemblies. (B) Schematic representation of a self-assembly process by solvent exchange and the formation of assemblies where the Au nanostars are facing the hydrophobic core and the hydrophilic Fe3O4 is facing the exterior. | ||
This previous system can be reversible, and therefore switchable, upon changing the solvent back to DMF or THF; however, this process is either slow or give rise to high dilution. To expand the range of stimuli that can trigger the self-assembly (switchability) and do it in a simpler manner, we substituted the hydrophobic polymer by an ELR. In our case, the ELR used was based on a pentapeptide (VPGVG)n with a tri-cysteine terminal sequence that provided the necessary thiol groups for the Au functionalization, and with an LCST of around ∼30 °C (ESI, Fig. S6†). The Fe3O4 was functionalized with catechol-PEG in the same way as previous nanoparticles. The role of this hydrophilic polymer was, like in the previous cases, to keep the stability of the formed structures upon nanoparticle assembly, offering a hydrophilic PEG layer that surrounds them.
Upon increasing the temperature above the LCST, and despite the relatively high molecular weight of the polymer (∼36 kg mol−1), minimum changes were observed. Only a slight shift in the plasmonic band in the UV-Vis spectra together with a small decrease in the hydrodynamic diameter was observed (ESI, Fig. S7A and B†). These effects can be explained by the contraction of the ELR shell on the nanoparticle Au surface and an increase in its refractive index. Interestingly, this is produced only after a good cleaning procedure. Mildly cleaned nanoparticles (with only one centrifugation cycle after functionalization) produce a distinct increase in the absorbance spectrum and in the hydrodynamic diameter, indicative of nanoparticle interaction. This finding highlights the importance of the free polymer in the solution that might be the real cause of aggregation in many previous reports of thermo-responsive polymer-coated nanoparticles.
Based on this observation, we artificially added free ELR to the smart nanoparticle amphiphiles. By varying the amount of added polymer to the solution, the final size of the assembly could be easily controlled (Fig. 4B). TEM images (Fig. 4C) showed that those assemblies were mainly formed by an ELR hydrogel and surrounded by a sparse layer of nanoparticles as depicted schematically in Fig. 4A. The assemblies observed in TEM images showed a polydisperse size distribution and a clear increase of size with the [free polymer]/[nanoparticle] ratio as observed by DLS. The bigger sizes observed in the TEM images, in comparison with DLS, could be due to the adsorption on the TEM grid causing the spreading of the nanostructure on the amorphous carbon which finally collapses during the drying of the sample. Additional cryo-TEM (ESI Fig. S9†) of the assemblies also confirmed the formation of these self-assembled structures depicted schematically in Fig. 4A. In this case, the sizes of the observed assemblies were limited by the thickness of the glassy ice layer formed in the plunged-freezing preparation.
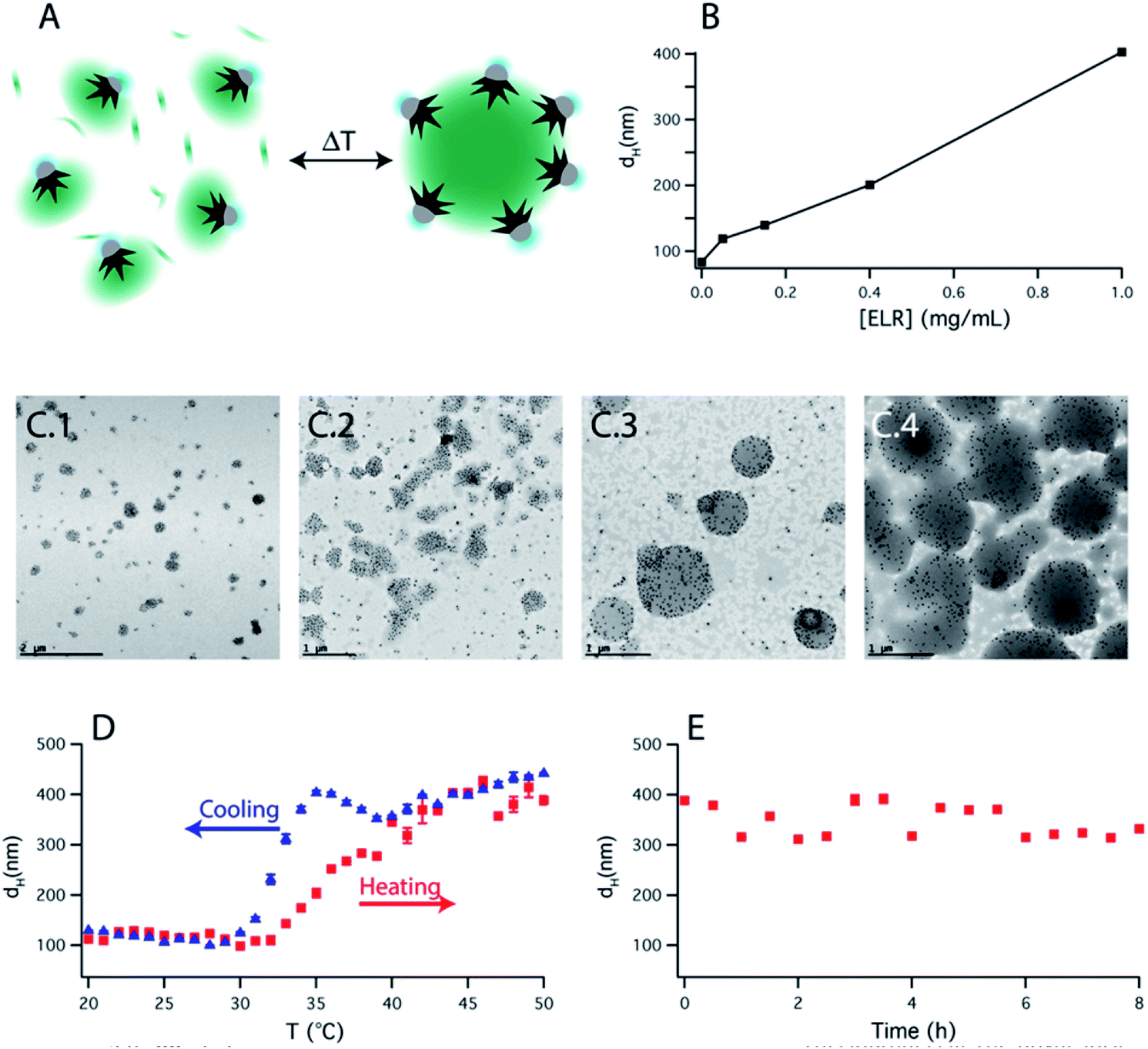 | ||
| Fig. 4 (A) Schematic representation of the temperature-triggered self-assembly of a solution containing ELR functionalized Janus nanoparticles together with free ELR. (B) Hydrodynamic diameter (dH) upon increasing the temperature above the LCST of the ELR as a function of the ELR concentration and at constant nanoparticle concentration. (C) TEM images of the assemblies measured in (B), at ELR concentrations of 0.05, 0.15, 0.4 and 1 mg mL−1. (D) dH in a heating–cooling cycle showing the reversibility of the system. (E) dH of the assemblies measured for several hours at 50 °C. | ||
The self-assembly process was reversible with 2–3 °C of hysteresis in the heating–cooling cycle (Fig. 4D) and once the assemblies were formed, they were shown to be stable over several hours (Fig. 4E). On the other hand, when nanoparticles were not present in solution, upon increasing the temperature, the polypeptide aggregated in micrometer-size macroparticles (Fig. S8C†). Therefore, the nanoparticles act as nano-surfactants inhibiting the growth of the polymer aggregates and stabilizing the assemblies. Assuming that the number of nanoparticles is proportional to the surface of the assemblies and the amount of ELR to the volume of the assemblies, this would produce a linear variation of the diameter of these assemblies with the [free polymer]/[nanoparticle] ratio. This linear variation is observed in Fig. 4B and is similar to the ones observed in emulsified systems.41
This self-assembly can easily be extended to other stimuli by the design of ELRs with different sensitivities.34 Furthermore, thanks to the nanoparticle core properties, the self-assembly process can also be triggered by a hyperthermia transduction mechanism, either with an alternating magnetic field or with light irradiation at plasmonic band wavelengths.33 We tested this last case, where the temperature was raised by irradiating at the NIR. In this case, the temperature increment is highly dependent on the nanoparticle concentration and irradiance. We found out that an Au concentration of only ∼0.33 mM and a relatively low irradiance of 1 W cm−2 (at 808 nm) raised the temperature of the whole sample by more than 20 °C, enough to increase the temperature from room temperature to above the LCST and trigger the self-assembly (see Fig. S10†).
Conclusions
Janus nanoparticles are remarkable building blocks to obtain complex self-assembled suprastructures. We have shown that complex Janus nanostructures, such as Au–Fe3O4 star-sphere nanoparticles, possess unique assembly features. This is highly dependent on the geometry of nanoparticles and the molecular weight of the polymeric shell. For small hydrophobic layers, nanoparticles self-assemble into small micellar clusters, their organization being affected by the nanostar size and shape. A larger polymer shell gives rise to bigger elongated assemblies with oriented nanoparticle nanoamphiphiles. We have also shown that the use of thermo-responsive polymers does not trigger the self-assembly itself. However, the addition of a free polymer into solution allows the formation of nanoparticle stabilized assemblies with tunable size. This self-assembly is reversible and can be equally triggered by the increase in temperature or by the irradiation at the NIR. The work presented here highly contributes to improving our understanding of Janus nanoparticle self-assembly and shows the formation of novel suprastructures with dual magnetic and plasmonic properties that convert the assemblies in very versatile tools for future applications and can be extended to other stimuli-triggered systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. R. acknowledges the financial support of Basque Country Elkartek-KK-2019/00101. T. F. and J. C. R-C acknowledge the funding from the European Commission (NMP-2014-646075), the Spanish Government (PCIN-2015-010 (FunBioPlas), MAT2016-78903-R), Junta de Castilla y León (VA317P18) and Centro en Red de Medicina Regenerativa y Terapia Celular de Castilla y León.References
- F. Tu, B. J. Park and D. Lee, Langmuir, 2013, 29, 12679–12687 CrossRef CAS PubMed.
- M. A. Fernandez-Rodriguez, M. A. Rodriguez-Valverde, M. A. Cabrerizo-Vilchez and R. Hidalgo-Alvarez, Adv. Colloid Interface Sci., 2016, 233, 240–254 CrossRef CAS PubMed.
- H. Fan and A. Striolo, Soft Matter, 2012, 8, 9533–9538 RSC.
- D. Wu, B. P. Binks and A. Honciuc, Langmuir, 2018, 34, 1225–1233 CrossRef CAS PubMed.
- L. L. Schramm, Emulsions, Foams, Suspensions, and Aerosols, Weinheim, Germany, 2014 Search PubMed.
- Y. Liu, J. Hu, X. Yu, X. Xu, Y. Gao, H. Li and F. Liang, J. Colloid Interface Sci., 2017, 490, 357–364 CrossRef CAS PubMed.
- D. J. Cole-Hamilton, Science, 2010, 327, 41–42 CrossRef CAS PubMed.
- G. Agrawal and R. Agrawal, ACS Appl. Nano Mater., 2019, 2, 1738–1757 CrossRef CAS.
- P. Yánez-Sedeño, S. Campuzano and J. M. Pingarrón, Appl. Mater. Today, 2017, 9, 276–288 CrossRef.
- C. Wang, W. Sun, Y. Ye, H. N. Bomba and Z. Gu, Theranostics, 2017, 17, 3504–3516 CrossRef PubMed.
- Q. Xia, H.-M. Ding and Y.-Q. Ma, Nanoscale, 2017, 9, 8982–8989 RSC.
- K. Lee, L. Zhang, Y. Yi, X. Wang and Y. Yu, ACS Nano, 2018, 12, 3646–3657 CrossRef CAS PubMed.
- T. Patiño, X. Arqué, R. Mestre, L. Palacios and S. Sánchez, Acc. Chem. Res., 2018, 51, 2662–2671 CrossRef PubMed.
- J. Reguera, H. Kim and F. Stellacci, Chimia, 2013, 67, 811–818 CrossRef CAS PubMed.
- A. Kirillova, C. Marschelke and A. Synytska, ACS Appl. Mater. Interfaces, 2019, 11, 9643–9671 CrossRef CAS PubMed.
- A. Walther and A. H. E. Müller, Chem. Rev., 2013, 113, 5194–5261 CrossRef CAS PubMed.
- J. Zhang, B. A. Grzybowski and S. Granick, Langmuir, 2017, 33, 6964–6977 CrossRef CAS PubMed.
- V. N. Manoharan, Science, 2015, 349, 1253751 CrossRef PubMed.
- S. Glotzer and M. J. Solomon, Nat. Mater., 2007, 6, 557–562 CrossRef PubMed.
- R. Erhardt, M. Zhang, A. Böker, H. Zettl, C. Abetz, P. Frederik, G. Krausch, V. Abetz and A. H. E. Müller, J. Am. Chem. Soc., 2003, 125, 3260–3267 CrossRef CAS PubMed.
- F. Sciortino, A. Giacometti and G. Pastore, Phys. Rev. Lett., 2009, 103, 237801 CrossRef PubMed.
- A. Walther, X. André, M. Drechsler, V. Abetz and A. H. E. Müller, J. Am. Chem. Soc., 2007, 129, 6187–6198 CrossRef CAS PubMed.
- Q. Chen, J. K. Whitmer, S. Jiang, S. C. Bae, E. Luijten and S. Granick, Science, 2011, 331, 199–202 CrossRef CAS PubMed.
- S. Berger, A. Synytska, L. Ionov, K.-J. Eichhorn and M. Stamm, Macromolecules, 2008, 41, 9669–9676 CrossRef CAS.
- C. Kang and A. Honciuc, ACS Nano, 2018, 12, 3741–3750 CrossRef CAS PubMed.
- C. Kang and A. Honciuc, J. Phys. Chem. Lett., 2018, 9, 1415–1421 CrossRef CAS PubMed.
- H. Hu, F. Ji, Y. Xu, J. Yu, Q. Liu, L. Chen, Q. Chen, P. Wen, Y. Lifshitz, Y. Wang, Q. Zhang and S.-T. Lee, ACS Nano, 2016, 10, 7323–7330 CrossRef CAS PubMed.
- J. Song, B. Wu, Z. Zhou, G. Zhu, Y. Liu, Z. Yang, L. Lin, G. Yu, F. Zhang, G. Zhang, H. Duan, G. D. Stucky and X. Chen, Angew. Chem., 2017, 129, 8222–8226 CrossRef.
- F. Liu, S. Goyal, M. Forrester, T. Ma, K. Miller, Y. Mansoorieh, J. Henjum, L. Zhou, E. Cochran and S. Jiang, Nano Lett., 2019, 19, 1587–1594 CrossRef CAS PubMed.
- R. F. Service, Science, 2005, 309, 95 CrossRef CAS PubMed.
- J. Reguera, D. Jiménez De Aberasturi, N. Winckelmans, J. Langer, S. Bals and L. M. Liz-Marzán, Faraday Discuss., 2016, 191, 47–59 RSC.
- J. Reguera, D. de Aberasturi, M. Henriksen-Lacey, J. Langer, A. Espinosa, B. Szczupak, C. Wilhelm and L. M. Liz-Marzán, Nanoscale, 2017, 9, 9467–9480 RSC.
- A. Espinosa, J. Reguera, A. Curcio, Á. Muñoz-noval, C. Kuttner, A. Van De Walle, L. M. Liz-marzán and C. Wilhelm, Small, 2020, 16, 1904960 CrossRef CAS PubMed.
- J. C. Rodriguez-Cabello, J. Reguera, A. Girotti, M. Alonso and A. M. M. Testera, Prog. Polym. Sci., 2005, 30, 1119–1145 CrossRef.
- J. C. Rodriguez-Cabello, S. Prieto, F. J. J. Arias, J. Reguera, A. Ribeiro, J. Carlos Rodríguez-Cabello, S. Prieto, F. J. J. Arias, J. Reguera and A. Ribeiro, Nanomedicine, 2006, 1, 267–280 CrossRef CAS PubMed.
- A. Girotti, J. Reguera, F. J. F. J. Arias, M. Alonso, A. M. A. M. Testera and J. C. Rodríguez-Cabello, Macromolecules, 2004, 37, 3396–3400 CrossRef CAS.
- J. Reguera, D. W. Urry, T. M. Parker, D. T. McPherson and J. C. Rodriguez-Cabello, Biomacromolecules, 2007, 8, 354–358 CrossRef CAS PubMed.
- A. Girotti, J. Reguera, J. C. J. C. Rodríguez-Cabello, F. J. F. J. Arias, M. Alonso and A. M. A. M. Testera, J. Mater. Sci.: Mater. Med., 2004, 15, 479–484 CrossRef CAS PubMed.
- S. Acosta, L. Quintanilla-Sierra, L. Mbundi, V. Reboto and J. C. Rodríguez-Cabello, Adv. Funct. Mater., 2020, 1909050 CrossRef.
- A. Klinkova, H. Therien-Aubin, R. M. Choueiri, M. Rubinstein and E. Kumacheva, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 18775–18779 CrossRef CAS PubMed.
- T. G. Noguchi, Y. Iwashita and Y. Kimura, Langmuir, 2017, 33, 1030–1036 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: TEM images of nanoparticles and assemblies, UV-Vis spectra of functionalization and self-assembly, MALDI-TOFF and DSC of ELR, Cryo-TEM of thermo-responsive assemblies, photothermal heating profile and UV-Vis. See DOI: 10.1039/d0na00102c |
| This journal is © The Royal Society of Chemistry 2020 |