
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceApplications of Friedel–Crafts reactions in total synthesis of natural products
Majid M. Heravi
 *,
Vahideh Zadsirjan
*,
Pegah Saedi
and
Tayebeh Momeni
*,
Vahideh Zadsirjan
*,
Pegah Saedi
and
Tayebeh Momeni
Department of Chemistry, School of Science, Alzahra University, Vanak, Tehran, Iran. E-mail: mmh1331@yahoo.com; mmheravi@alzahra.ac.ir; z_zadsirjan@yahoo.com; Fax: +98 2188041344; Tel: +98 9121329147
First published on 3rd December 2018
Abstract
Over the years, Friedel–Crafts (FC) reactions have been acknowledged as the most useful and powerful synthetic tools for the construction of a special kind of carbon–carbon bond involving an aromatic moiety. Its stoichiometric and, more recently, its catalytic procedures have extensively been studied. This reaction in recent years has frequently been used as a key step (steps) in the total synthesis of natural products and targeted complex bioactive molecules. In this review, we try to underscore the applications of intermolecular and intramolecular FC reactions in the total syntheses of natural products and complex molecules, exhibiting diverse biological properties.
 Majid M. Heravi | Majid M Heravi was born in 1952 in Mashhad, Iran. He received his BSc degree from the National University of Iran in 1975 and his MSc and PhD degrees from Salford University, England in 1977 and 1980, respectively. He completed his doctoral thesis under the supervision of the late Jim Clarck in Salford University, England. He started his career as a research fellow in Daroupakhsh (a pharmaceutical company) in 1981 in Tehran, Iran and joined Ferdowsi University of Mashhad, Iran as an assistant professor in 1983, and was promoted to associate professor in 1993 and full professor in 1997 in the aforementioned university. In 1999 he moved to Alzahra University in Tehran, Iran as professor of chemistry, where he still works. He has previously been a visiting professor at UC Riverside, California, USA and Hamburg University, Hamburg, Germany. His research interests focus on heterocyclic chemistry, catalysis, organic methodology and green synthetic organic chemistry. |
 Vahideh Zadsirjan | Vahideh Zadsirjan was born in Shiraz, Iran in 1979. She received her BSc in Pure Chemistry from Kharazmi University in 2002, her MSc degree in 2007, and PhD degree in organic chemistry in 2016 under the supervision of Professor Majid M. Heravi from Alzahra University Tehran, Iran. Her research is focused on heterocyclic chemistry, catalysis, organic methodology and green synthetic organic chemistry. |
 Pegah Saedi | Pegah Saedi was born in Tonekabon, Iran in 1992. She received her BSc degree in Pure Chemistry from Kharazmi University in 2015, and her MSc degree in Organic Chemistry in 2018 under the supervision of Professor Majid M. Heravi from Alzahra University, Tehran, Iran. Her research is focused on heterocyclic chemistry, catalysis, organic methodology and green synthetic organic chemistry. |
 Tayebeh Momeni | Tayebeh Momeni was born in 1988 in Karaj, Iran. She received her BSc degree from Imam Khomeini International University in 2010 and her MSc degree from Tehran University under the direction of Professor Mehdi Ghandi in 2012 before moving to Alzahra University, where she became a PhD candidate in Professor Majid. M. Heravi's research group in 2015. |
1. Introduction
Historically, the FC reactions are the well-established set of reactions initially discovered by Charles Friedel and James Crafts in 1877. In these reactions, certain substituents are attached on to a suitable aromatic ring.1 Basically, Friedel–Crafts reactions imply two main sets of reactions. They are alkylation and acylation reactions. In fact, both types of such reaction proceed via the same mechanism, which is the typical reaction of aromatic compounds, electrophilic aromatic substitution.2–5 FC alkylation comprises the alkylation of an appropriate aromatic ring using an alkyl halide, conventionally in the presence of a strong Lewis acid as the catalyst.6 Commonly, anhydrous ferric chloride is used as a catalyst, in which the alkyl group initially attaches itself to the former site of the chloride ion.7 Although this reaction enjoys the advantages already known for electrophilic aromatic substitution, it also suffers from the fact that the final product is more nucleophilic than the reactant, thus undesired over-alkylation may take place, giving unwanted side products. In addition, the reaction is limited to using tertiary carbon and secondary alkylating agents, or else the emerging carbocation (R+) may be subjected to a known carbocation rearrangement.7 Indeed, alkylations via FC reaction are not limited to using just alkyl halides. FC reactions can be successfully performed via the generation of any other appropriate intermediate, such as those carbocationic intermediates that are derived from alkenes in the presence of a protic acid, Lewis acid, enones, and epoxides. During the previous years of FC reactions, several other Lewis acids, such as BF3, BeCl2, TiCl4, SbCl5 or SnCl4, have been proven to act as effective catalysts. Besides, strong Brønsted acids such as H2SO4 and HF, or super acids, such as HF·SbF5 and HSO3 F·SbF5, have also been used successfully and proven to accelerate the FC alkylation reaction. In spite of the great significance of FC alkylation in organic transformation, some major drawbacks and problems still exist, which must be circumvented. For example, the FC alkylation reaction requires stoichiometric or super stoichiometric amounts of a Lewis acid or Brønsted acid. Furthermore, since it needs toxic alkyl halides as a reagent, the formation of large quantities of salts as side products is proven to be inevitable. Thus, development of more eco-friendly strategies and economically feasible chemical processes for such an important reaction is still in great demand. The replacement of the alkyl chlorides by less harmful alkylating agents, for instance alcohols, has undoubtedly been a major development since in this case water is formed as a non-toxic by-product. More importantly, the utilization of activated double bonds and compounds such as styrenes have been found even more advantageous and operative, since no by products are generated at all.The FC acylation reaction is actually the acylation of certain aromatic compounds. For the FC acylation reaction, acyl chlorides are used as common acylating agents. In this version of the FC reaction, frequently Lewis acid catalysts such as AlCl3 can be used along with acid anhydrides as a suitable reagent. The reaction conditions for the FC acylation reaction are exactly as same as those for FC alkylation. It is worthwhile to know that the FC acylation reaction shows several advantages over the alkylation variant. Because of the electron-withdrawing nature of the carbonyl motif, the product, which is actually a ketone, is expectedly less reactive than the substrate, thus undesired multiple acylations do not take place. In addition, since the intermediate is not a carbocation, no rearrangement occurs as the generated carbonium ion is stabilized via a resonance structure in which the positive charge is located on the electronegative oxygen.
By definition, a natural product is a chemical compound or substance that originates from nature and is produced by a living organism.8 In the more extensive sense, a naturally occurring compound implies any substance produced by a living organism.9 Significantly, the total synthesis of natural products, including semi synthesis, is the state of art in synthetic organic chemistry and nowadays plays a key role in the development of organic chemistry as a whole by providing stimulating and useful and especially biologically active natural products as the target. Generally, the total synthesis of natural products is a non-commercial research activity, aimed at deeper understanding of the synthesis of a desired natural product scaffold and, more importantly, the development of more desirable new synthetic approaches. Natural products are also limited to the purified organic compounds isolated from natural sources that are originated by the pathways of primary or secondary metabolism. Many secondary metabolites are cytotoxic and have been chosen and modified through evolution for use as “chemical warfare” agents toward prey, predators, and competing organisms. The terminology of natural product has also been stretched for profitable purposes and thus refers to cosmetics, dietary supplements, and foods obtained from natural sources with no artificial ingredients as additives.10–13 Natural products frequently show therapeutic benefit and many of them have been used as folk medicines for treating diseases for centuries and thus have given some knowledge about lead compounds for drug discovery.14 Natural products can be categorized in accordance with their biological function, biosynthetic pathway, or source.
The applications of FC reactions (alkylation and acylation) in organic synthesis have been extensively studied and reviewed previously.6,15–19 In continuation of our interest in applications of name reactions20–35 in the total synthesis of natural products, herein we try to underline the applications of both FC reactions in the total synthesis of naturally occurring compounds, showing diverse biological properties.
2. Applications of Friedel–Crafts in total synthesis of natural products
2.1. Intramolecular alkylation
Dactylol, 8, an irregular isoprenoid alcohol, was isolated from the Caribbean sea hare Aplysia dactylomela and its structure was identified in 1978 by Schmitz and co-workers.36,37 The total synthesis of the marine sesquiterpenes africanol 1 and dactylol 8 was achieved from bicyclo[5.1.0]octane precursors. In this route, firstly, compound 2 gave carboxylic acid 3, which was transformed into compound 7 via FC cyclization of its acid chloride and acid-mediated dithioketalization (after several steps). Compound 7 was converted into dactylol 8 after several steps. The configuration of the side chain Me substituent is detected to be proper in relation to dactylol 8 but reverse to that existing in africanol 1. Subsequently, africanol 1 was provided from compound 9 upon several steps (Scheme 1).38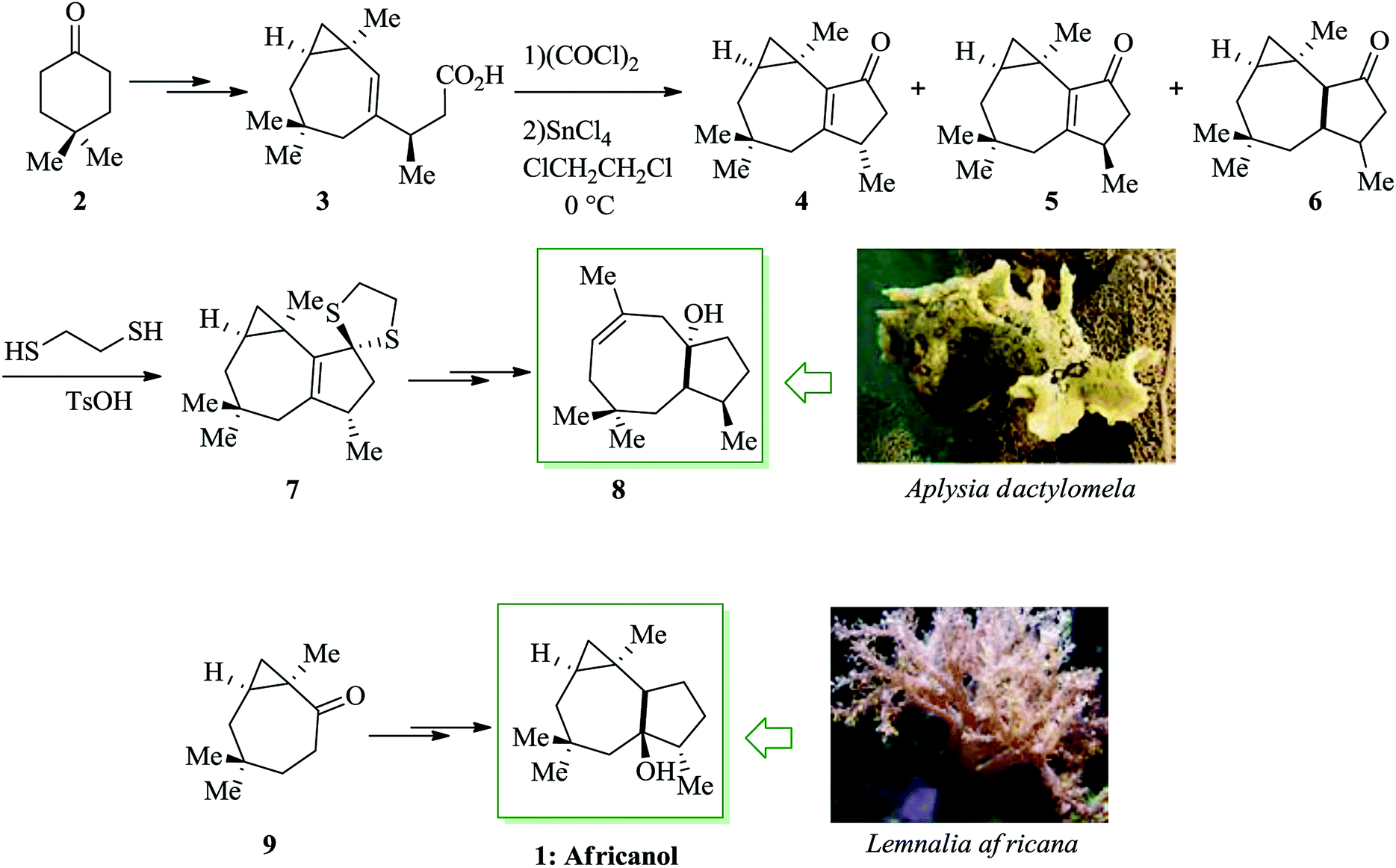 | ||
| Scheme 1 Total synthesis of africanol 1. | ||
Most lactonic aryltetralin lignans, for example peltatins and podophyllotoxin, contain a similar common structure. α-Conidendrin was extracted from both wood39 and waste sulfite liquor.40 Boissin and co-workers developed the total synthesis of natural (−)-conidendrin 10. Conidendrin 10 was provided in eight steps starting from the easily accessible and optically active hemisuccinic ester (R)-(+)-11. The latter gave the monobenzoxy intermediate 12 after several steps, including treatment with BF3·Et2O in dichloromethane at ambient temperature. The benzoxy group of 12 was actually found to be an excellent leaving group during intramolecular FC alkylation. As a result, (−)-α-conidendrin 10 was provided as the desired natural product from 12 in satisfactory yield (Scheme 2).41
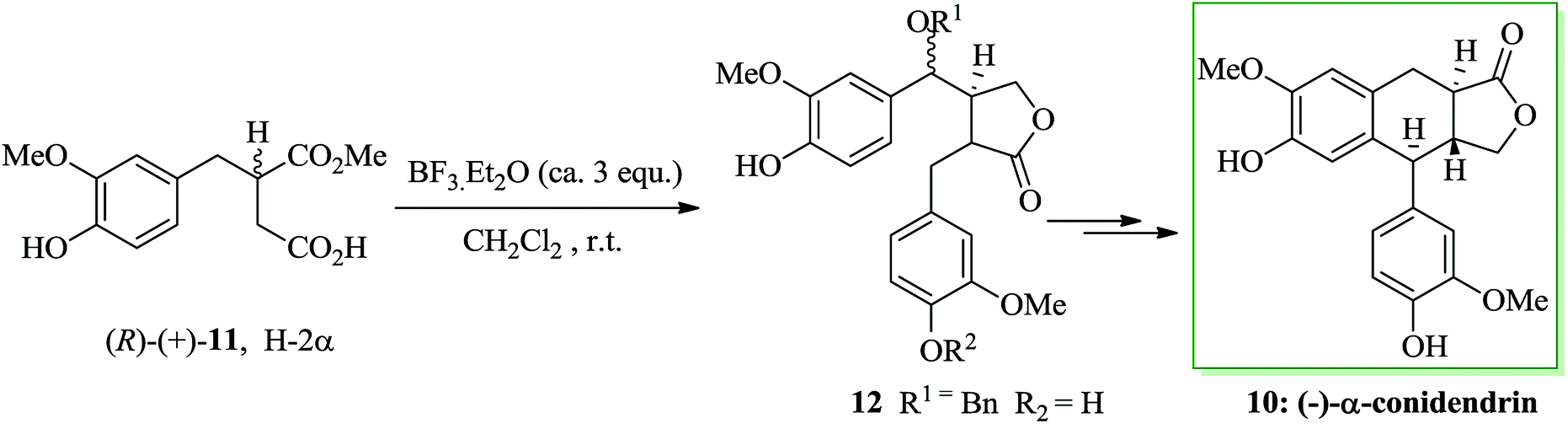 | ||
| Scheme 2 Total synthesis of (−)-α-conidendrin 10. | ||
The Aristotelia alkaloids (−)-serratoline 13, (+)-aristotelone 14 and (−)-alloaristoteline 15 were extracted from Aristotelia chilensis. Serratoline was formerly extracted from Aristotelia serrata, a species native to New Zealand. Heathcock and co-workers reported the total synthesis of (−)-serratoline 13, (+)-aristotelone 14 and (−)-alloaristoteline 15. This approach starts from the reaction between (lS)-(−)-β-pinene 16 and 3-indolylacetonitrile 17 to afford (+)-makomakine 18. Next, an intramolecular FC reaction provided (+)-aristotelone 19, which upon two steps afforded alkaloid 13. Subsequently, base-mediated skeletal rearrangement of 13 provided (+)-aristotelone 14. Finally, alkaloid 14 gave (−)-alloaristoteline 15 upon several steps (Scheme 3).42
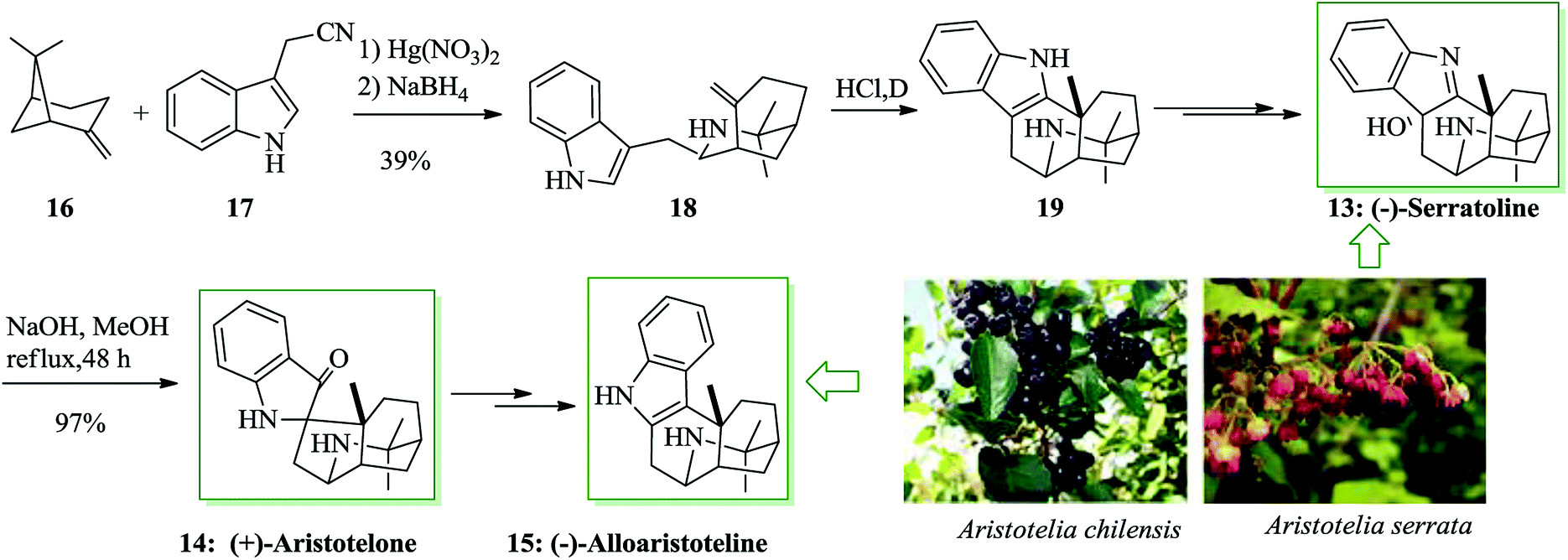 | ||
| Scheme 3 Total synthesis of natural products (−)-serratoline 13, (+)-aristotelone 14 and (−)-alloaristoteline 15. | ||
Phomazarin 20 is the most abundant and extensively studied aza anthraquinone that was extracted from cultures of Phoma terrestris Hansen (Pyrenochaeta terrestris Hansen) in 1940.43 Boger and co-workers reported the total synthesis of phomazarin 20. Initially 2,3-dihydroxybenzaldehyde 21 was converted into 22 after several steps. Compound 22 in the presence of TFAA cleanly led to FC closure of the B-ring and simultaneous MOM deprotection, affording a variable and inconsequential mixture of 23 and 24 with no detection of the intermediate bisphenol. Conditions were planned such that only the single mono-(trifluoroacetate) 23 (88%; TFAA) was obtained. Finally, compound 23 afforded phomazarin 20 upon several steps (Scheme 4).44
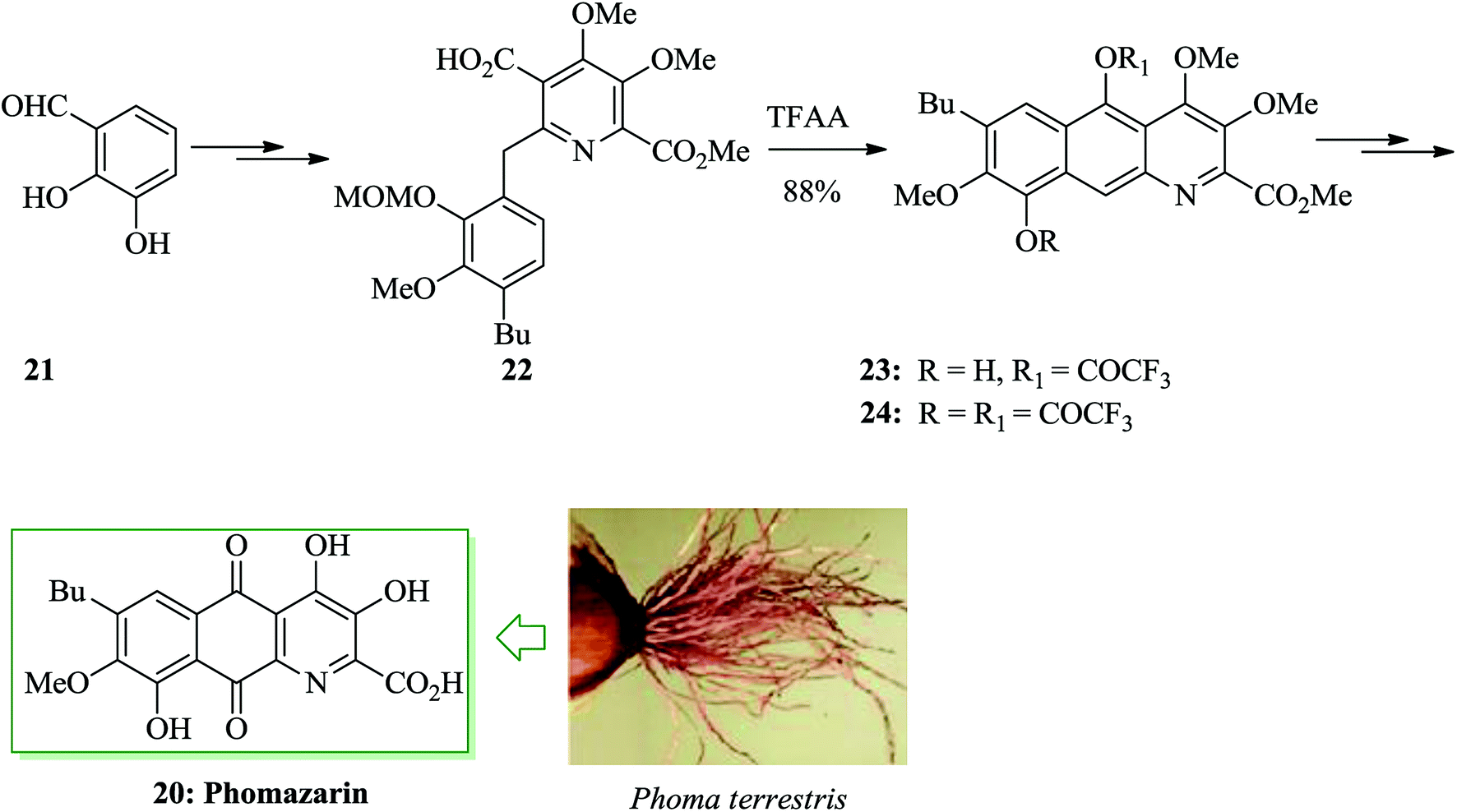 | ||
| Scheme 4 Total synthesis of phomazarin 20. | ||
Alliacol A 25, a sesquiterpene, was extracted in Europe from the culture broth of the fungus Marasmius alliaceus.45 This molecule exhibits antimicrobial properties and prevents DNA synthesis in the ascetic form of Ehrlich carcinoma.46,47 A tandem anodic coupling-FC alkylation methodology was applied to quickly complete the enantioselective synthesis of alliacol A. In this route, firstly, the reaction between compounds 26 and 27 afforded compound 28 after several steps. Then, it was tried to form the tricyclic unit of the desired naturally occurring compound. For this purpose, the alcohol in 28 was transformed into the relevant iodide, which was applied for either the FC alkylation or radical cyclization method. The FC reaction between iodide and AgNO3 in MeOH gave a 71% yield of the corresponding tricyclic product 30 (Scheme 5).48
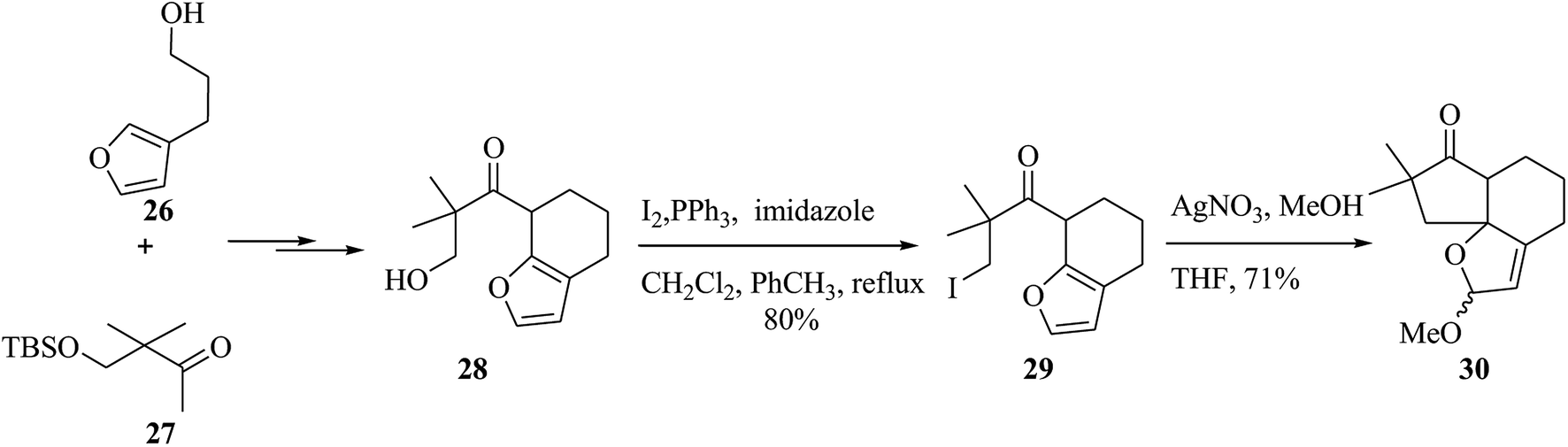 | ||
| Scheme 5 The formation of the tricyclic product 30. | ||
Subsequently, it was attempted to provide substrate 31 containing the B-ring Me group of alliacol A in place. Compounds 26 and 27 gave substrate 31 upon several steps. This compound provided the bicyclic product 32 upon two steps. Using the bicyclic product, the alcohol 32 was transformed into an iodide and the FC cyclization was finished. A 92% extracted yield of the tricyclic product 33 was provided from the FC reaction. Next, compound 33 was converted into (−)-alliacol A 25 upon several steps (Scheme 6).48
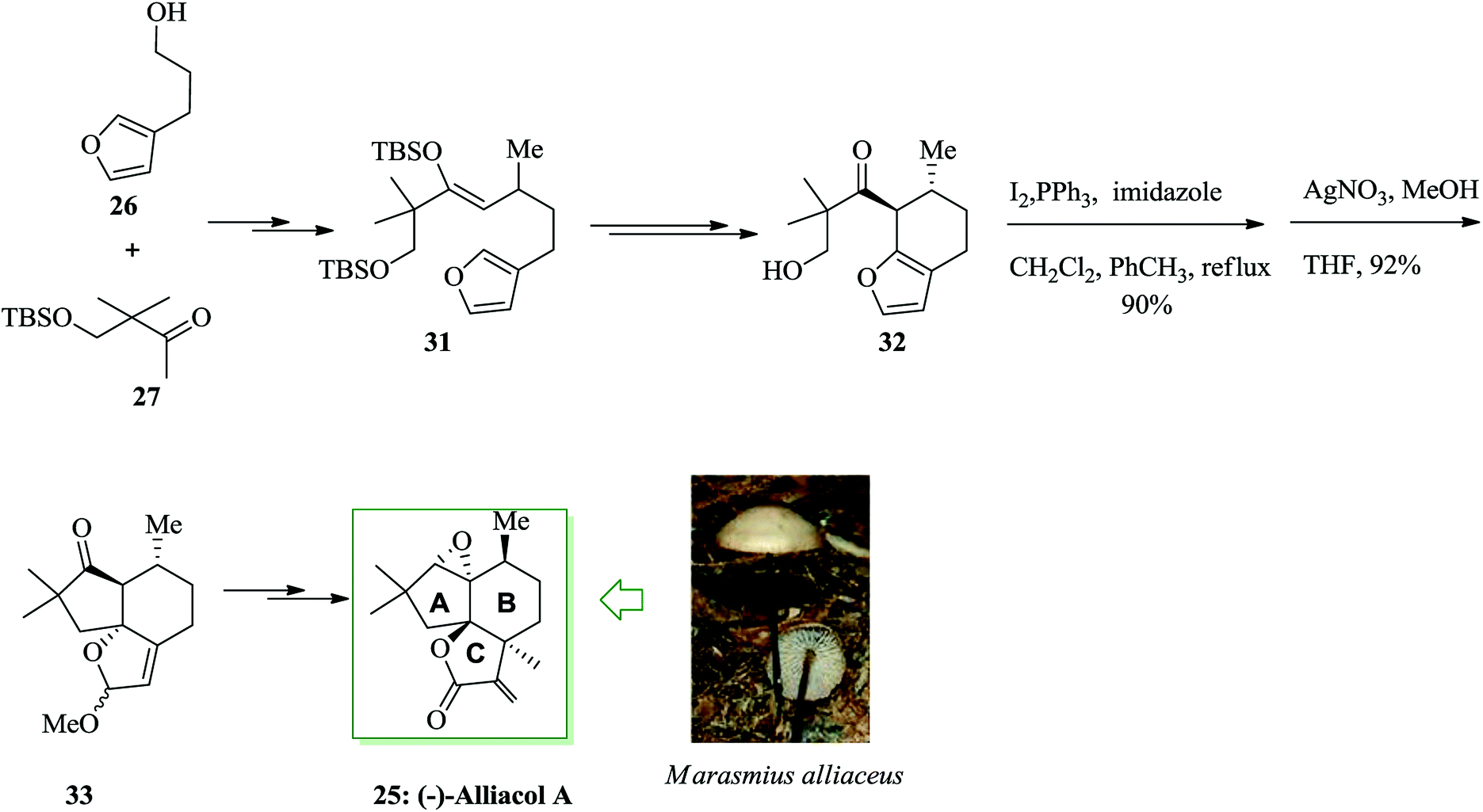 | ||
| Scheme 6 Total synthesis of (−)-alliacol A 25. | ||
In 1990, Meng and co-workers for the first time reported the isolation of pygmaeocin C 34,49 a rearranged abietane diterpenoid, from the roots of Pygmaeopremna herbacea Moldenke. The initial total synthesis of pygmaeocin C 34, in racemic form, was accomplished in 2005 by Liu and co-workers. Total synthesis of pygmaeocin C 34 was accomplished, starting from dimedone 35 in 14 steps with an overall yield of 4.6%. In this route, an intramolecular FC reaction was considered as a key step. Notably, keto aldehyde 37 was synthesized from dimedone 35 and also compound 38 was synthesized from veratrole 36 via several steps. In the following, the reaction of aldehyde 37 and bromide 38 gave the key intermediate 39, upon several steps. It should be mentioned that compound 39 was subjected to a simple intramolecular FC alkylation to give the tricyclic system in the target product. Upon extensive study, it was found that the required cyclization could be obtained from olefin 41 with an extraordinary degree of efficiency. Upon treatment with trifluoroacetic acid (TFA), the latter was submitted to intramolecular FC reaction to afford an 88% yield of the corresponding cyclized product 42 as a result of double bond isomerization with subsequent ring closure. Finally, the tricyclic keto ester 42 was converted into pygmaeocin C 34 upon several steps (Scheme 7).50
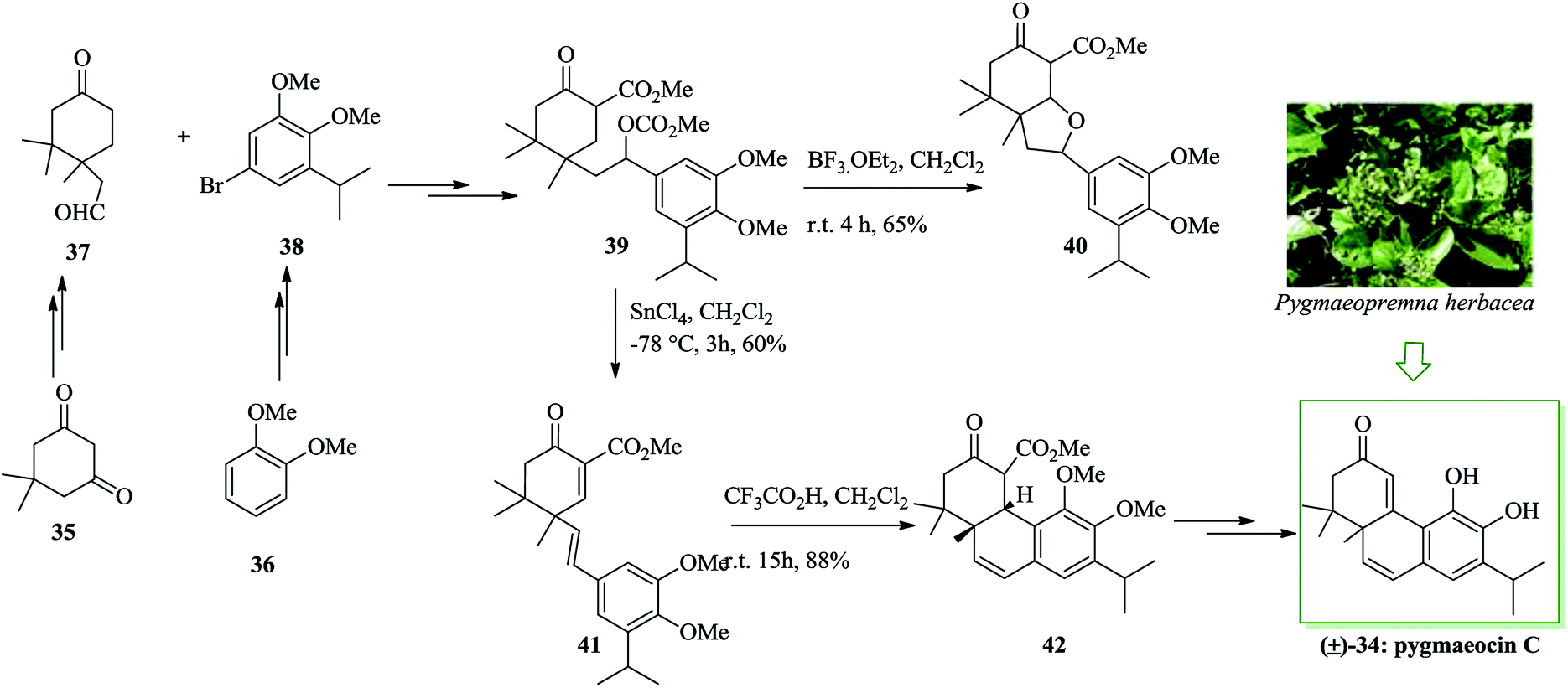 | ||
| Scheme 7 Total synthesis of pygmaeocin C 34. | ||
The myrmicarins, a group of structurally intriguing alkaloids, were initially extracted from the poison gland secretions of the African ant species Myrmicaria opaciventris.51,52 The pyrroloindolizine unit of myrmicarins 43a, 43b, and 43c is a common structural scaffold found in various alkaloids. An asymmetric synthesis of a key dihydroindolizine intermediate 47 for the construction of myrmicarin alkaloids was developed in 2005 by Movassaghi and co-workers. Key conversions in this method contain a stereospecific Pd-mediated N-vinylation, a Cu-mediated asymmetric conjugate reduction, and a regioselective FC reaction. The synthesis of optically potent and isomerically pure samples of (4aR)-myrmicarins 43a, 43b, and 43c and also their respective C4a-epimers was achieved using the above-mentioned protocol. Firstly, the lithium enolate 44 and methyl 4-(dimethoxy)-butyrate 45 were reacted to afford compound 46 in several steps. Next, the dihydroindolizine 47 was provided, which is a key intermediate for the construction of the myrmicarin alkaloids. A regioselective FC reaction of the pyrrole ring upon Brønsted acid activation of the dimethoxyacetal 46 and removal of MeOH was achieved to provide the bicyclic vinyl pyrrole 47. The optimized conditions (acetone–acetic acid–H2O, 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1, 40 °C) were recognized for the quantitative transformation of the β-pyrrolyl ester 46 to the bicyclic vinyl pyrrole to give the corresponding C7a-cyclization product 47. Finally, compound 47 afforded isomerically pure tricyclic myrmicarins 43a–c after several steps (Scheme 8).53
:1:1, 40 °C) were recognized for the quantitative transformation of the β-pyrrolyl ester 46 to the bicyclic vinyl pyrrole to give the corresponding C7a-cyclization product 47. Finally, compound 47 afforded isomerically pure tricyclic myrmicarins 43a–c after several steps (Scheme 8).53
 | ||
| Scheme 8 Total synthesis of tricyclic myrmicarins 43a–c. | ||
Ahmad and co-workers extracted and identified a remarkable polycyclic triterpene from Salvia bucharica, salvadione A 48.54 The tricyclic 6–7–6 unit structure of the triterpene salvadione 48 was provided using an effective method starting from the aryl bromide 50 (provided from cyclohexenone 51) and the alkyl iodide 52 (provided from phenol 49) containing a methylenecyclohexane group at the terminus. Notably, alkylation of 50 with the iodide 52 afforded the tethered system (allylic bromide) 53. An intramolecular FC alkylation reaction of allyl bromide 53 provided compound 54 via cyclization in the presence of zinc chloride and copper(I) chloride in tetrahydrofuran. The synthesis of tricyclic compound 54 demonstrates a formal total synthesis of salvadione 48. Finally, the tricycle 54 afforded salvadione 48 upon several steps (Scheme 9).55
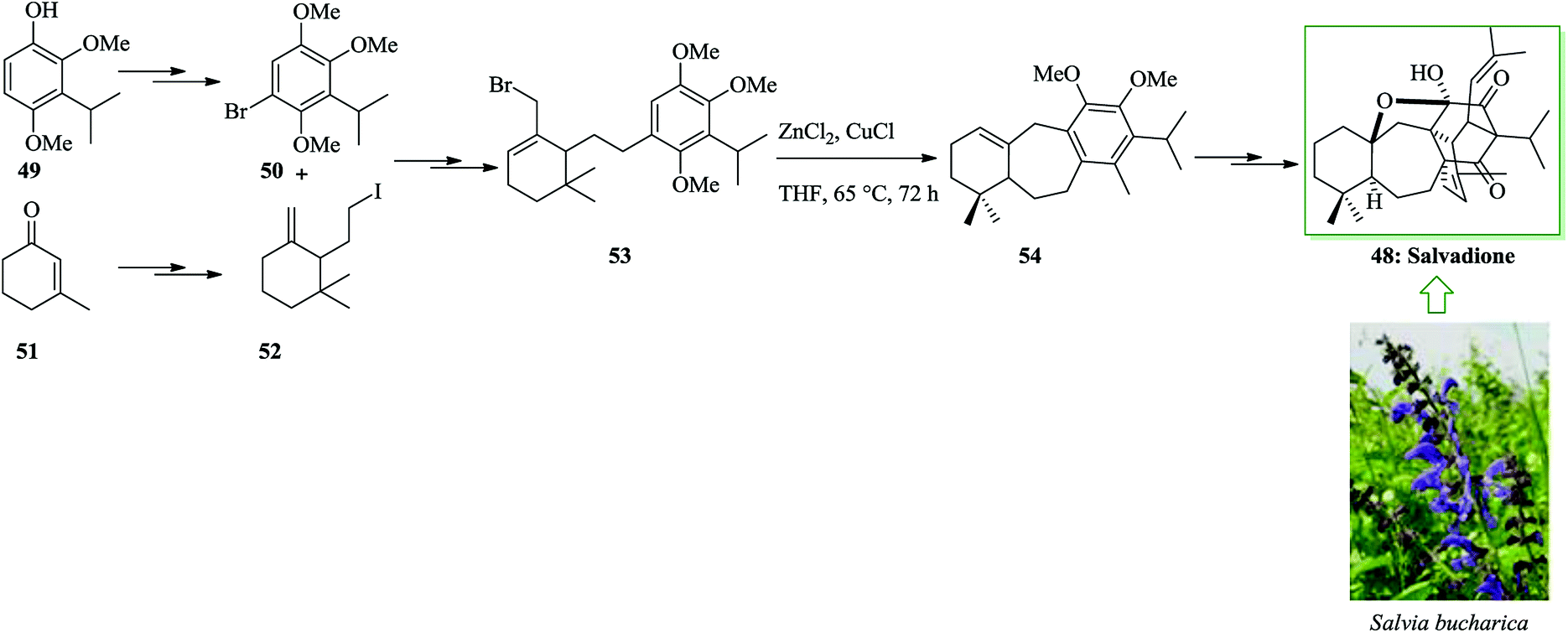 | ||
| Scheme 9 Total synthesis of salvadione 48. | ||
Various aromatic diterpenes with a serrulatane or amphilectane framework have been recognized as bioactive metabolites from marine soft corals, particularly Pseudopterogorgia elisabethae.56 Notable representatives of these compounds are the anti-inflammatory pseudopterosins,57,58 the cytotoxic and antiviral helioporins,59 and the pseudopteroxazoles.60,61 Schmalz et al. in 2007 developed an effective and extremely enantioselective synthetic approach to calamenene 55, a trans-1,4-difunctionalized tetralin derivative, in 6 linear steps and 57% overall yield initiating from 56.62 The reaction was initiated from compound 56, which was converted into the corresponding allylic acetate 57 in moderate overall yield. The 1,4-trans-difunctionalized tetralin framework was selectively provided via a FC-type cationic cyclization reaction using Me2AlCl as a “proton-scavenging” Lewis acid.63 The envisioned trans-dehydrocalamenene 55 was provided in almost quantitative yield and with very moderate de (up to 10:1) (Scheme 10).62
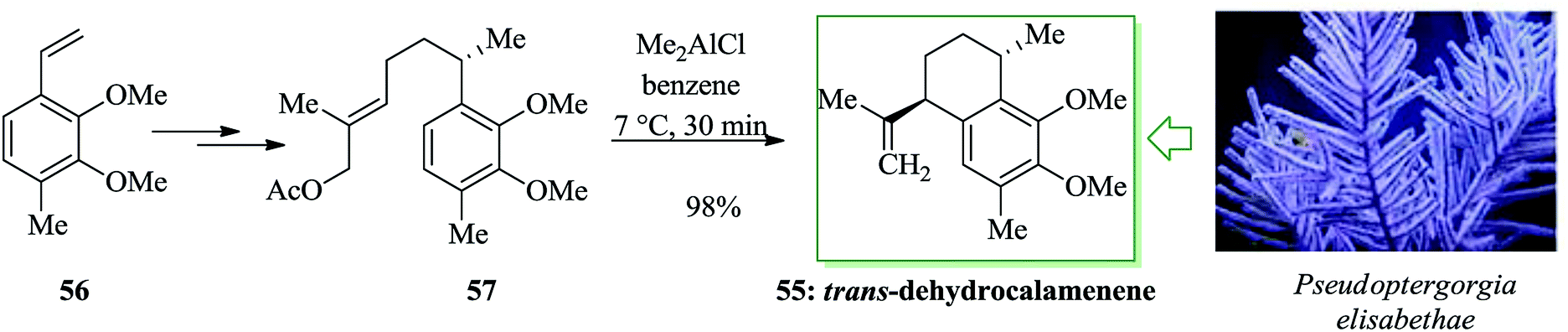 | ||
| Scheme 10 Total synthesis of trans-dehydrocalamenene 55. | ||
Erogorgiaene, extracted together with other diterpenes from the West Indian sea whip Pseudopterogorgia elisabethae, exhibits favorable anti-mycobacterial properties. A total synthesis of erogorgiaene 58 was developed in 16 steps with an overall yield of 8.2%. The synthesis was based on an extremely diastereoselective intramolecular FC reaction of an oxetane obtained through an enantioselective syn aldol coupling. In this route, firstly, easily accessible acid 59 was converted into oxetane 60 in high yield upon several steps. The essential intramolecular FC reaction of oxetane 60 was performed.64 As expected, the reaction progressed easily, at room temperature, providing a single diastereomer 61. Finally, alcohol 61 afforded erogorgiaene 58 upon several steps (Scheme 11).65
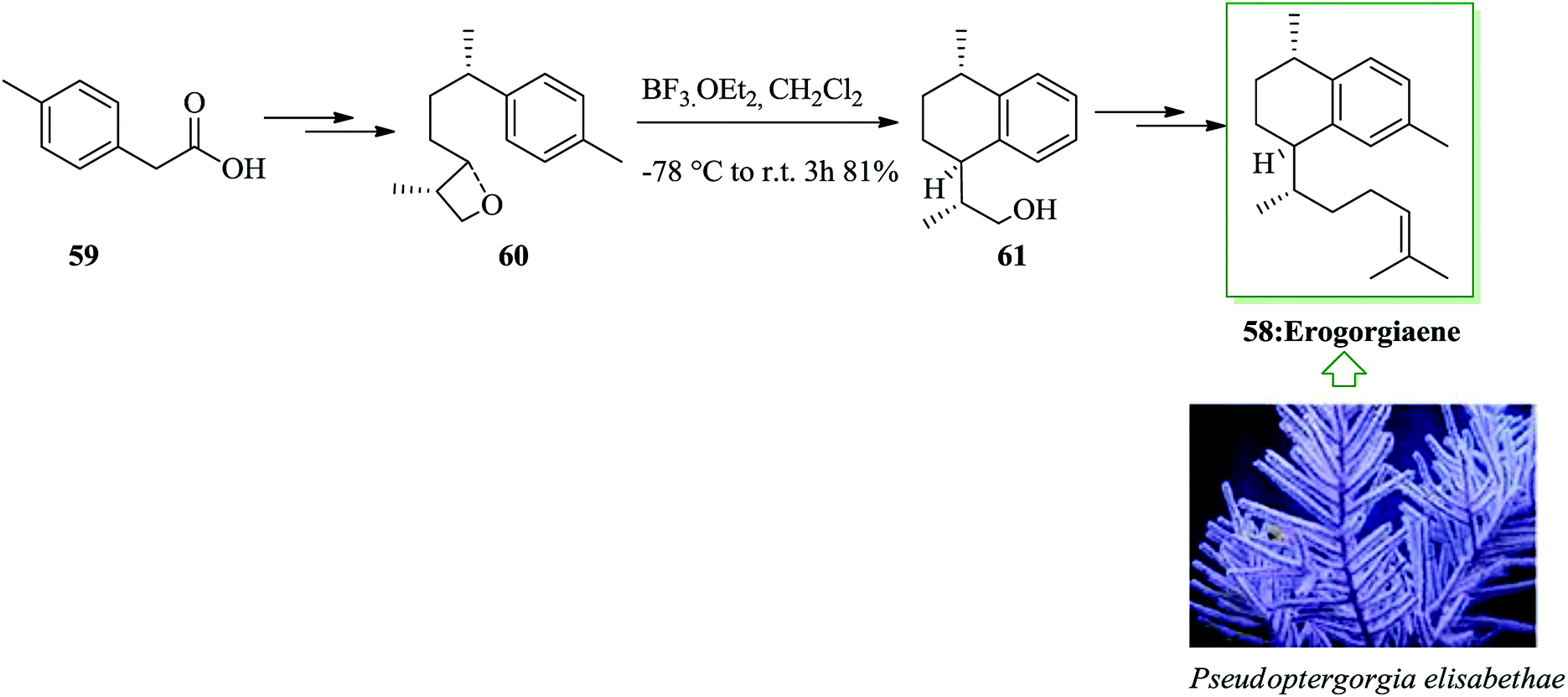 | ||
| Scheme 11 Total synthesis of erogorgiaene 58. | ||
In 2005, Sattler and co-workers extracted and revealed a group of naturally occurring aromatic β-C-glycoside compounds named bruguierols A–C from the stem of the Bruguiera gymmorrhiza mangrove tree.66 In 2007, Jennings and co-workers demonstrated the initial total synthesis of bruguierol C 62 in 7 linear steps from compound 63.67 The main step was an intramolecular FC alkylation that eventually supplied the final natural product. For the synthesis of bruguierol C 62, compound 63 was converted into lactol 64 after several steps. Lastly, an intramolecular trap of the incipient (emerging) oxocarbenium cation using a Marson-type FC alkylation68 permitted the construction of the masked bruguierol C. The two-step reaction sequence including oxocarbenium construction to provide 65 and also intramolecular FC alkylation afforded the corresponding β-C-glycoside product 66. Finally, compound 66 afforded the naturally occurring compound 62 in 85% yield (Scheme 12).67
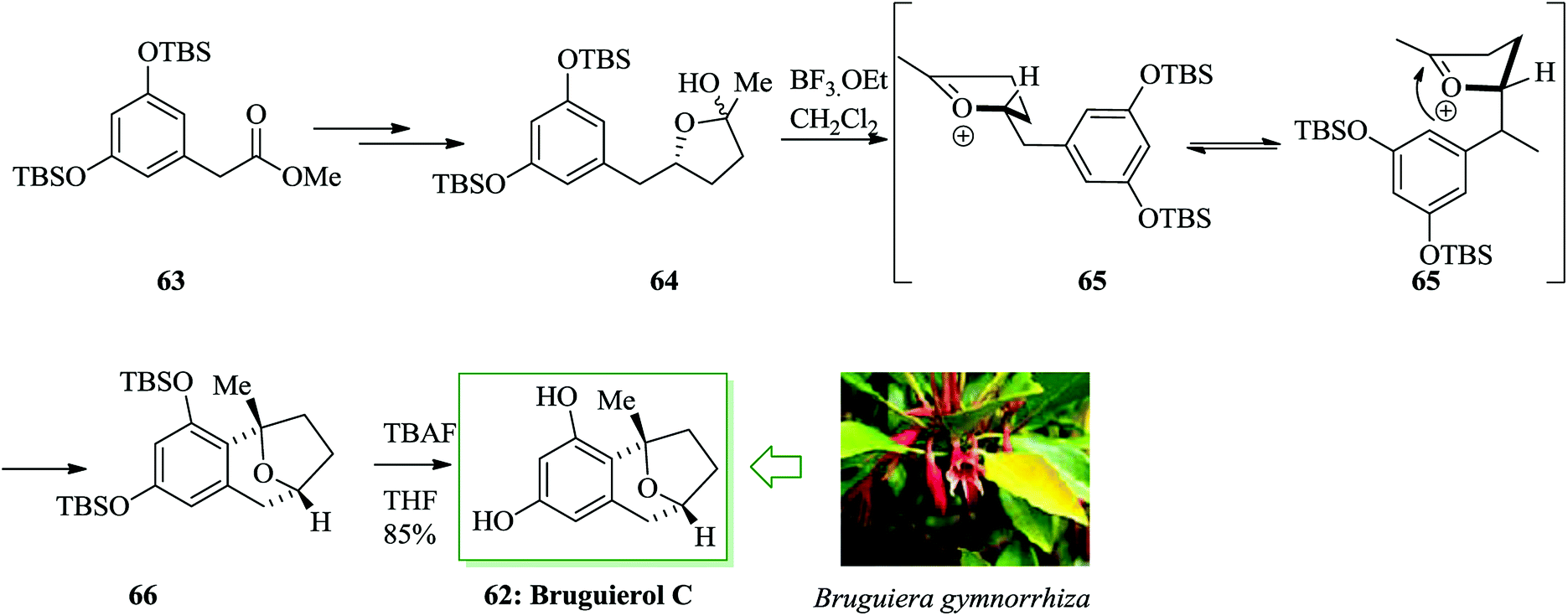 | ||
| Scheme 12 Total synthesis of bruguierol C 62. | ||
Murrayazoline 67, found as mahanimbidine and curryangin, is a carbazole alkaloid extracted as a racemic or an optically active compound from the genus Murraya. Murrayazoline and its corresponding carbazole alkaloids were demonstrated to be have antiplatelet aggregation activity.69 A structural explanation investigation demonstrated that murrayazoline is a hexa-heterocyclic alkaloid composed of N-functionalized carbazole, cyclohexane and dihydropyran constituents.70,71 The total synthesis of (±)-murrayazoline 67 was developed in 2008 by Chida and co-workers.72 The characteristic hexa-heterocyclic structure of 67 was generated using a combination of the intramolecular FC-type Michael addition reaction and palladium-mediated carbon–oxygen coupling reactions. Firstly, the two segments 69 and 71 were provided from 5-amino-2-methylphenol 68 and 1,5-dithiaspiro-[5,5]undecane-9-one 70,73 respectively, in several steps. Next they afforded the corresponding N-functionalized carbazole 72. The reaction of 72 with scandium(III)triflate in water and dichloroethane induced the deprotection of the ethylene ketal substituent, the intramolecular FC-type Michael addition and the deprotection of the O-MOM group, affording pentacyclic ketone 74 in 73% yield.74,75 Finally, compound 74 afforded (±)-murrayazoline 67 after several steps (Scheme 13).72
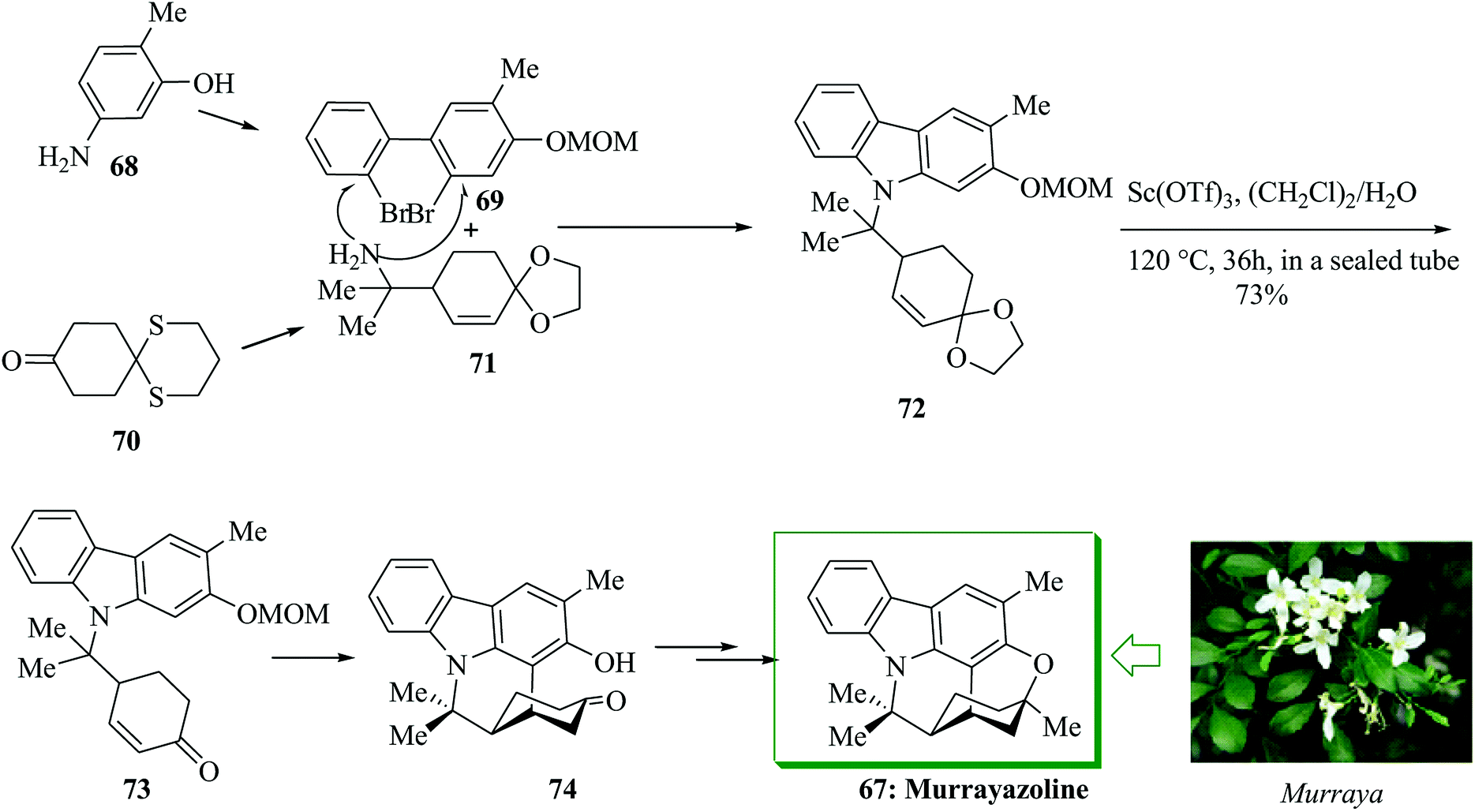 | ||
| Scheme 13 Total synthesis of murrayazoline 67. | ||
Cacalol 75, a sesquiterpene naturally occurring compound, was extracted from the roots of the shrub Psacalium decompositum in northern Mexico,76,77 that has antihyperglycemic,78 anti-inflammatory,79 antimicrobial,79 and antioxidant80 properties. A facile synthesis of cacalol 75 was established from 4-methylanisole 76 in seven steps and 21–25% overall yield. An intramolecular FC alkylation, Baeyer–Villiger oxidation and alkylation are the key reactions in this approach. The synthesis of cacalol 75 was started with ortho-lithiation81 of 4-methylanisole 76 using n-butyllithium in tetrahydrofuran. This anion was alkylated with 5-iodo-1-pentene82 to afford the alkene 77. Subsequently, this molecule was cyclized via an intramolecular FC alkylation using AlCl3 in dichloromethane to afford the tetralin 78. Finally, compound 78 afforded cacalol 75 upon several steps (Scheme 14).83
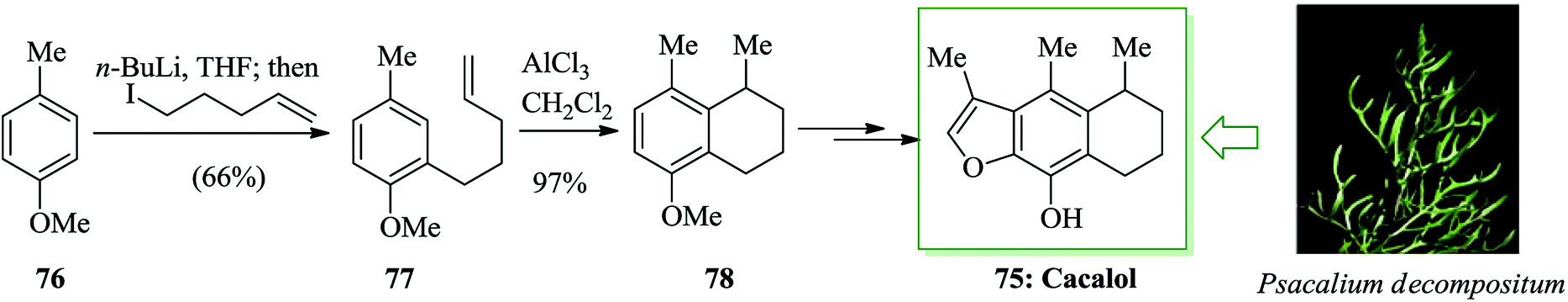 | ||
| Scheme 14 Total synthesis of cacalol 75. | ||
Taiwaniaquinoids, a group of tricyclic diterpenoids containing a 4a-methyltetra- (and hexa-)hydrofluorene framework have been extracted from various East Asian conifers, for example, Taiwania cryptomerioides,84,85 Salvia dichroantha,86 and Thuja standishii.87 They contain taiwaniaquinone H 8085 and dichroanone 79.86 A very significant pathway toward taiwaniaquinoids was reported in 2009 by Alvarez-Manzaneda and co-workers. Key steps are the intramolecular FC alkylation reaction of an aryldiene and the degradative oxidation reaction of a methylenedioxy substituent. Using this strategy, (±)-dichroanone 79 (in three steps, 77% overall yield) and (±)-taiwaniaquinone H 80 (in four steps, 70% overall yield) were provided from α-84a or β-cyclocitral 84b. This group designed a probable synthesis of taiwaniaquinoids. The key intermediate would be the aryldiene 81, which resulted in the arylallyl cation 82 under acidic conditions, which, through a quick intramolecular FC alkylation, afforded the 4a-methyltetrahydrofluorene 83 framework (Scheme 15).88
 | ||
| Scheme 15 Synthesis of 4a-methyltetrahydrofluorene 83 framework. | ||
β-Cyclocitral 84a and α-cyclocitral 84b afforded dichroanone 79 upon several steps. Then, the transformation of this quinone into taiwaniaquinone H 80 was achieved (Scheme 16).88
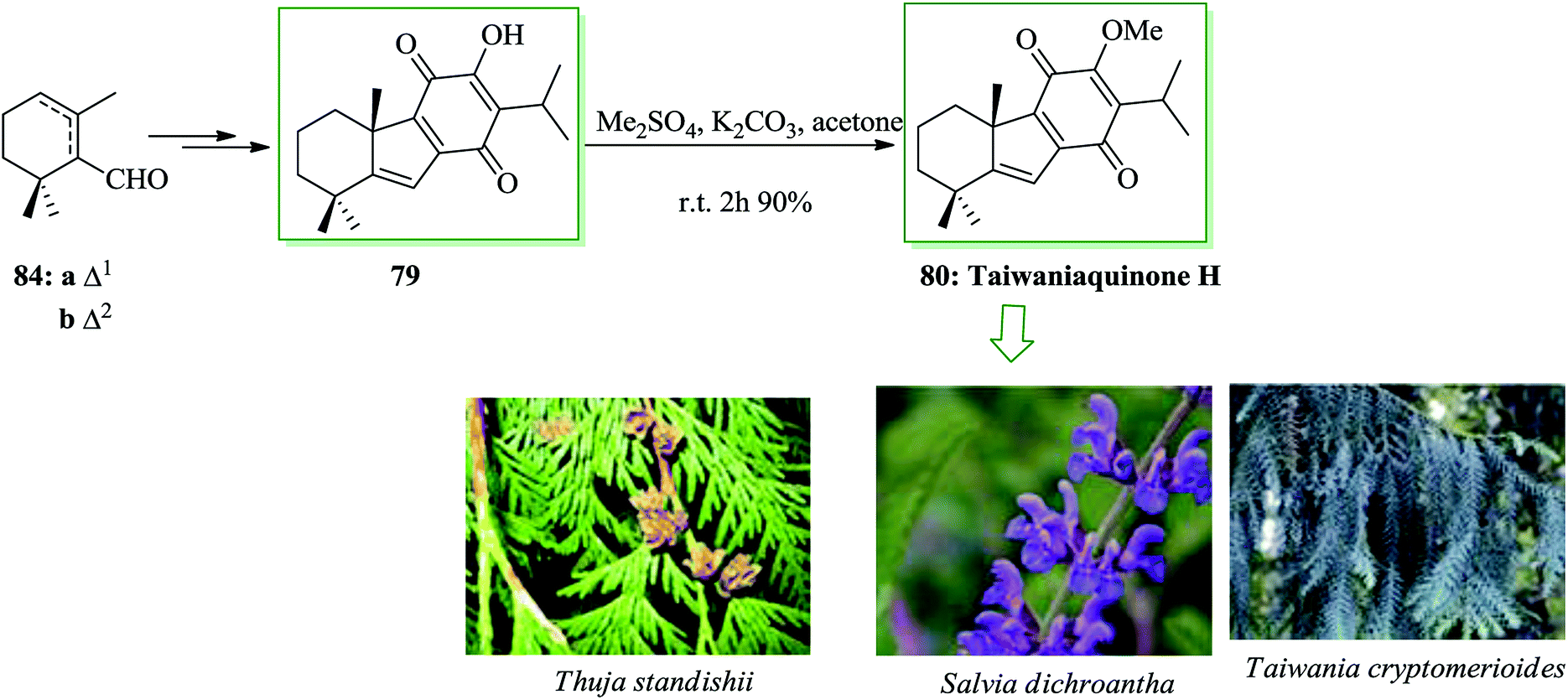 | ||
| Scheme 16 Total synthesis of taiwaniaquinone H 80. | ||
Plicatic acid was identified as the causative agent of occupational asthma.89–91 Plicatic acid was extracted from western red cedar (Thuja plicata) in 1959 by Maclean and co-workers.92 The initial enantioselective total synthesis of (−)-plicatic acid was achieved from eugenol in 12 steps and 14% overall yield. In this approach, a theoretically novel methodology containing an enantioselective epoxidation-intramolecular epoxy-ring-opening FC reaction sequence was performed for the asymmetric formation of a structurally complex 2,7′-cyclolignane framework. The synthesis was initiated from eugenol 86, which was converted into epoxide 87 upon several steps. The key intermediate, α-hydroxy ketone 88, was provided through an intramolecular FC reaction to open the epoxide ring in 86. Noticeably, triflic acid (TfOH) efficiently improved the FC reaction. Finally, compound 88 provided (−)-plicatic acid 85 after several steps (Scheme 17).93
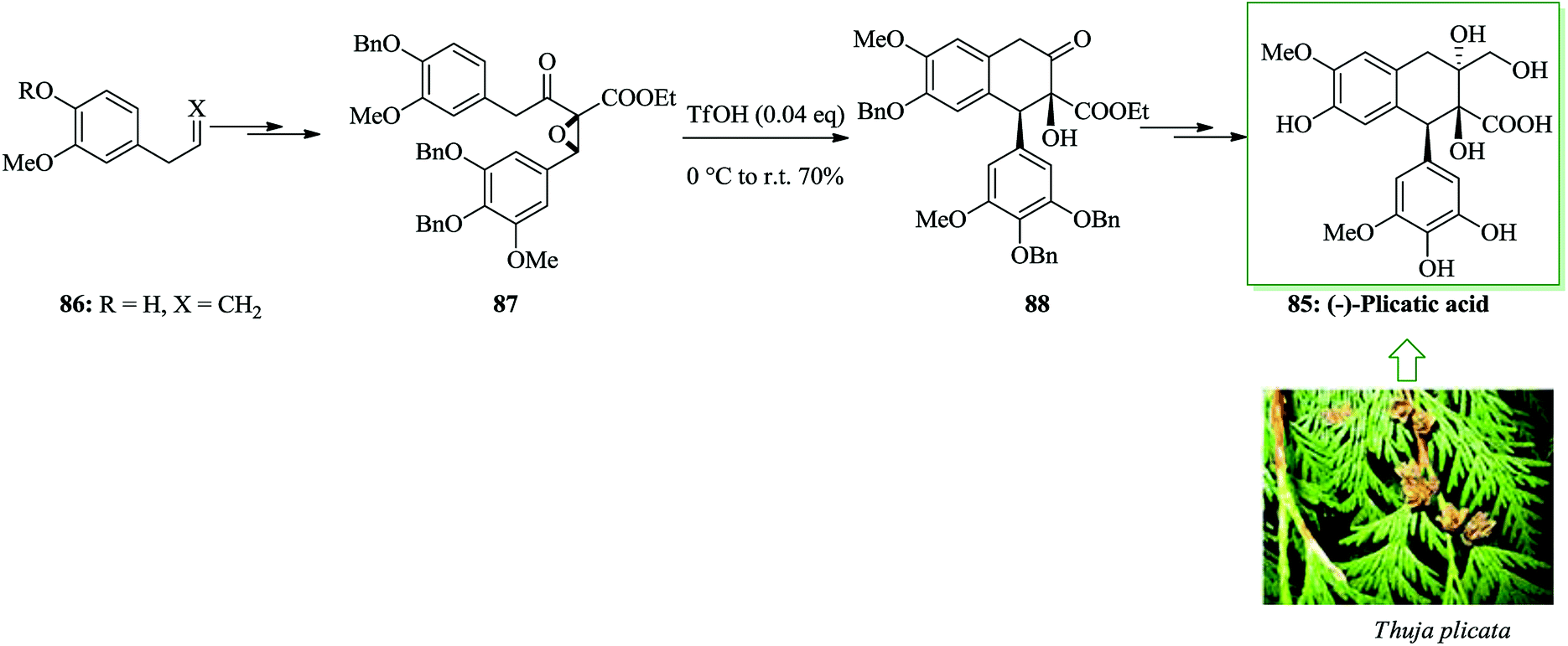 | ||
| Scheme 17 Total synthesis of (−)-plicatic acid 85. | ||
Clavilactone D 89, a tyrosine kinase inhibitor,94 was extracted from cultures of the fungus Clitocybe clavipes.95 A method for the synthesis of clavilactone D was reported in 2009 by Yoshimitsu and co-workers.96 This pathway uses sequential cyclization and FC cyclization to provide a polycyclic lactone fused with an aromatic ring. Total synthesis of clavilactone D 89 was initiated from aldehyde 90. In this route, substrate 92 was synthesized via the aldol reaction of chiral lactone 9197 with aldehyde 90.98 Next, iodo etherification followed via FC-type cyclization of alkenyl alcohol 92 with iodine gave the fused polycyclic compound 94. Finally compound 94 provided clavilactone D 89 upon several steps (Scheme 18).96
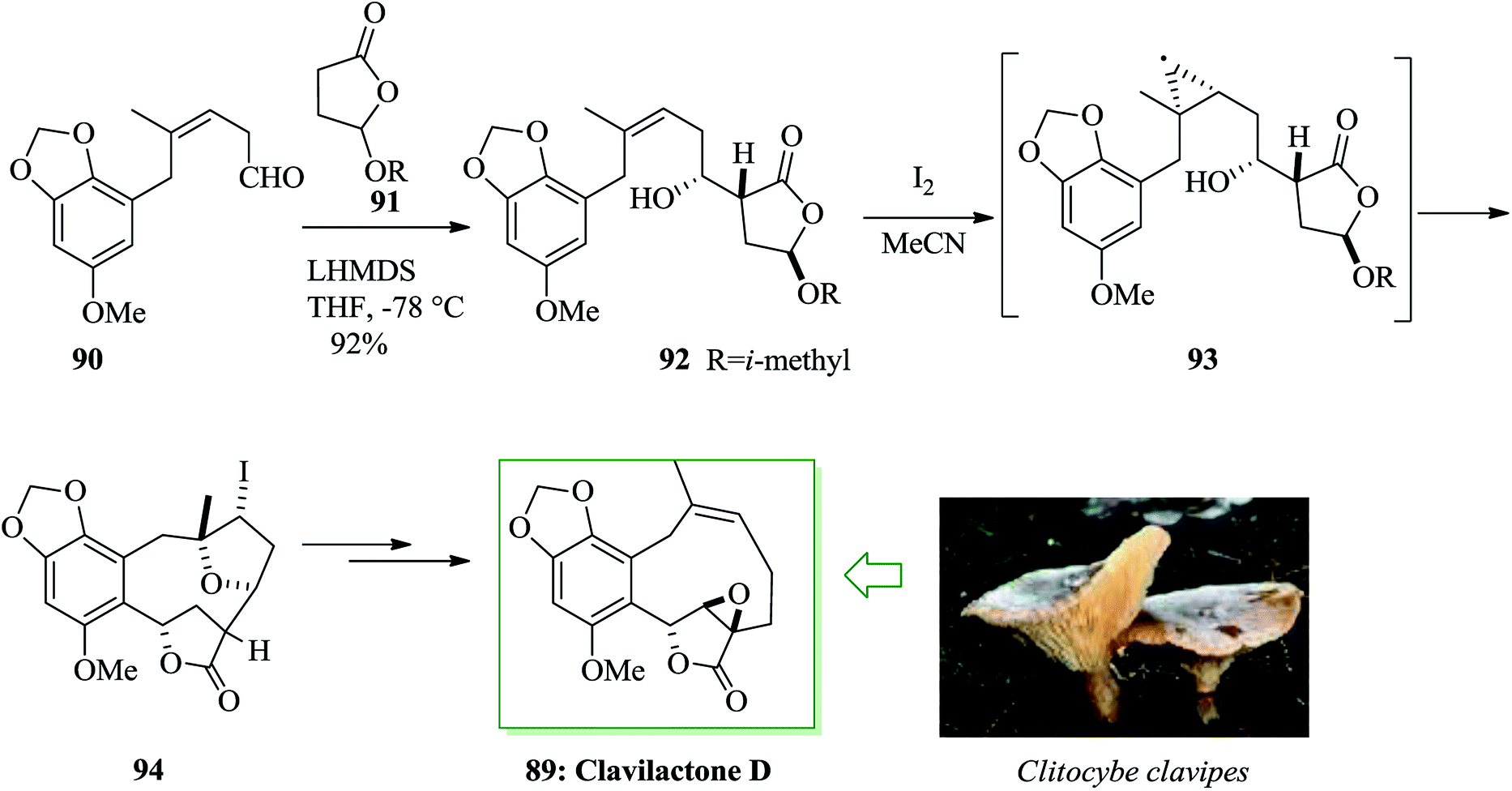 | ||
| Scheme 18 Total synthesis of clavilactone D 89. | ||
Hainanensine 95, racemic, is a structurally distinctive minor alkaloidal constituent identified by Liang and Sun in 1981 from the antileukemia plants Cephalotaxus hainanensis and C. fortunei.99 Hainanensine 95 contains marginal antitumor properties,99 and also Yin et al. demonstrated that saturated derivatives and other structural analogues of 95 have demonstrated a series of remarkable biological properties.100 A simple total synthesis of hainanensine 95 through an efficient acid-catalyzed rearrangement/FC annulation cascade, was developed. Relying on this method, initially, enone 97 was synthesized from 3,4-dihydroisoquinoline 96 after several steps. Upon refluxing enone 97 in formic acid, compound 98 was provided. Seemingly, the construction of rearranged annulation product 98 resulted from more FC-type cyclization-dehydration of the initial ring-expansion intermediates I and II. Subsequently, compound 98 provided hainanensine 95 upon several steps (Scheme 19).101
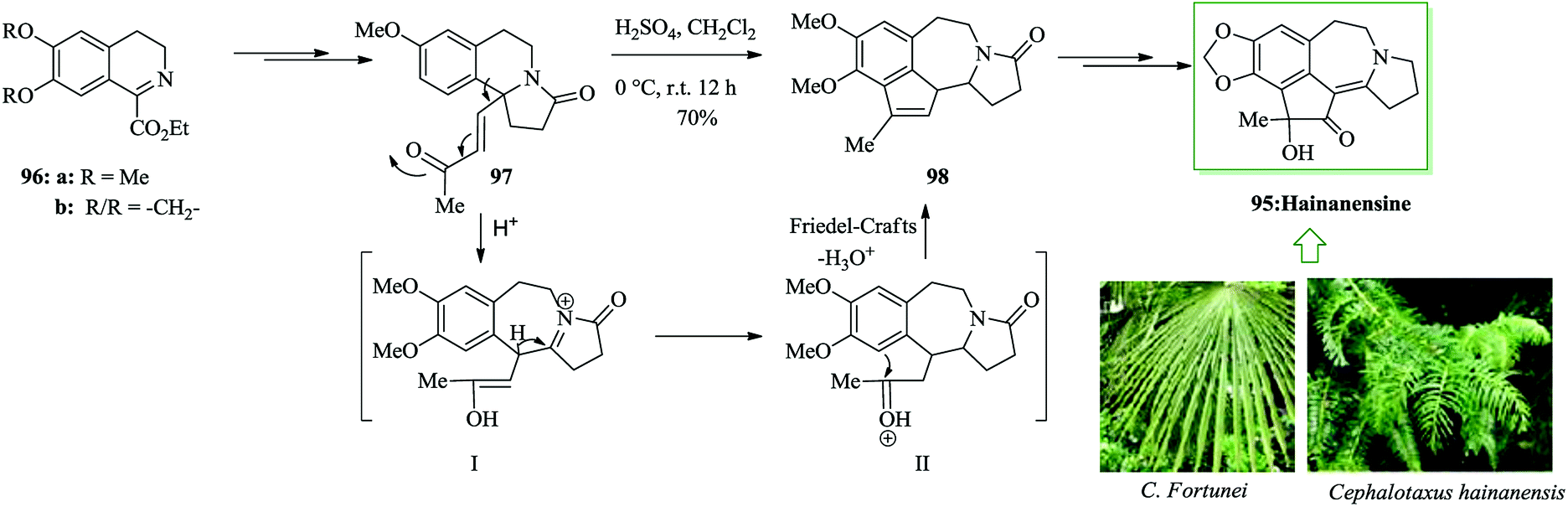 | ||
| Scheme 19 Total synthesis of hainanensine 95. | ||
A rearranged abietane including the cyclohepta[de]naphthalene nucleus, microstegiol 99,102–107 was extracted from roots of a wide range of plants of the genus Salvia. Microstegiol itself was exhibited to contain antileukemic and modest antibacterial properties.102,105–107 For the total synthesis of microstegiol 99, firstly 2,7-dimethoxynaphthalene 100 was converted into compound 101 upon several steps. Exposing tertiary alcohol 101 to sulfuric acid in dichloromethane induced its transformation to 7,7-dimethyltetra-hydrocyclohepta[de]naphthalene 102 (70% yield).108 Next, compound 102 afforded compound 103 upon several steps. Subsequently, 103 using polyphosphoric acid (PPA) in dichloromethane under reflux conditions was transformed to 104 with 80% yield. Afterwards, compound 104 led to the construction of (±)-microstegiol 99 in 64% yield upon several steps (Scheme 20).108
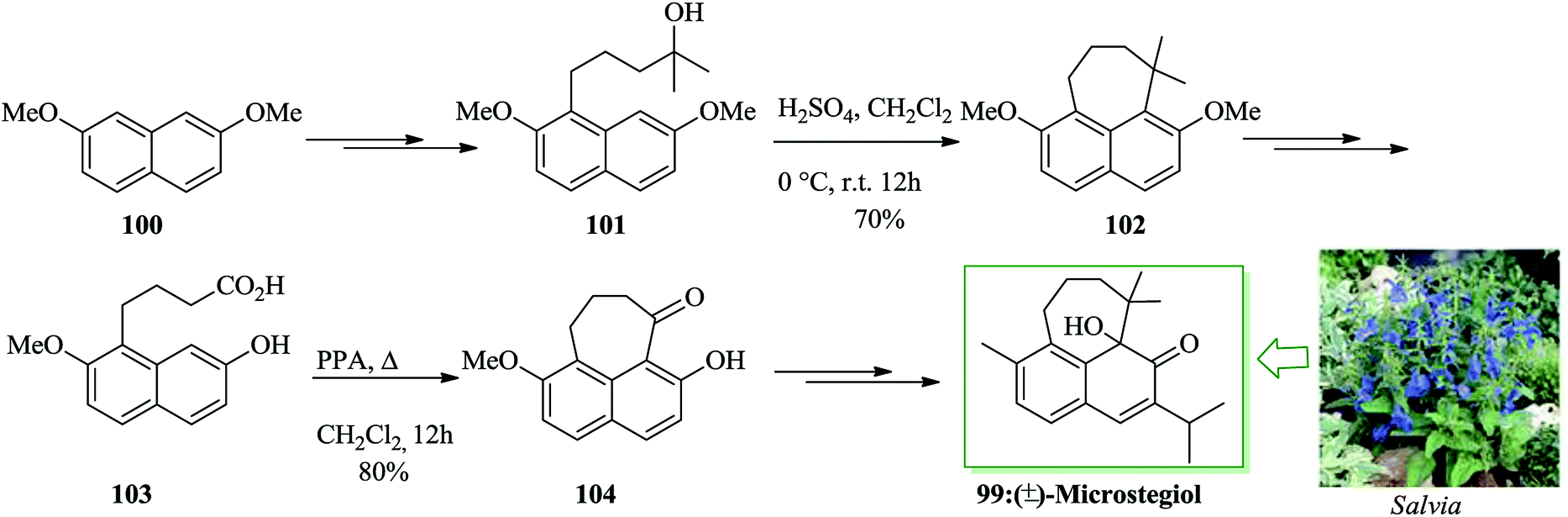 | ||
| Scheme 20 Total synthesis of (±)-microstegiol 99. | ||
The tricyclic alkaloid aphanorphine was extracted from the blue-green alga Aphanizomenon flos-aquae by Clardy and Shimizu in 1988.109 Aphanorphine shares common structural features with other bioactive benzomorphan alkaloids, for example eptazocine, pentazocine and morphine.110–114 A short enantioselective synthesis of (+)-aphanorphine 105 was accomplished in 10 steps and 13% overall yield. A racemic γ-aminoalkene derivative was converted into a 1:1 mixture of enantiomerically enriched diastereomers employing an enantioselective palladium-mediated carboamination. Next, this mixture was transformed into an enantiomerically enriched masked aphanorphine derivative via a FC reaction to make a quaternary all-carbon stereocenter. The catalyst-controlled enantioselective palladium-mediated carboamination reaction of racemic substrate 106 was applied to set the C2 stereocenter of pyrrolidines 107a,b, thus affording a mixture of enantiomerically enriched diastereomers. Subsequently, this mixture of diastereomers was transformed to 109 in a FC alkylation that proceeded through intermediate carbocation 108, and formed the all-carbon quaternary stereocenter in an enantio-convergent carbon–carbon bond-forming stage. A two-step sequence of reduction and demethylation was used to transform 109 to aphanorphine (Scheme 21).115
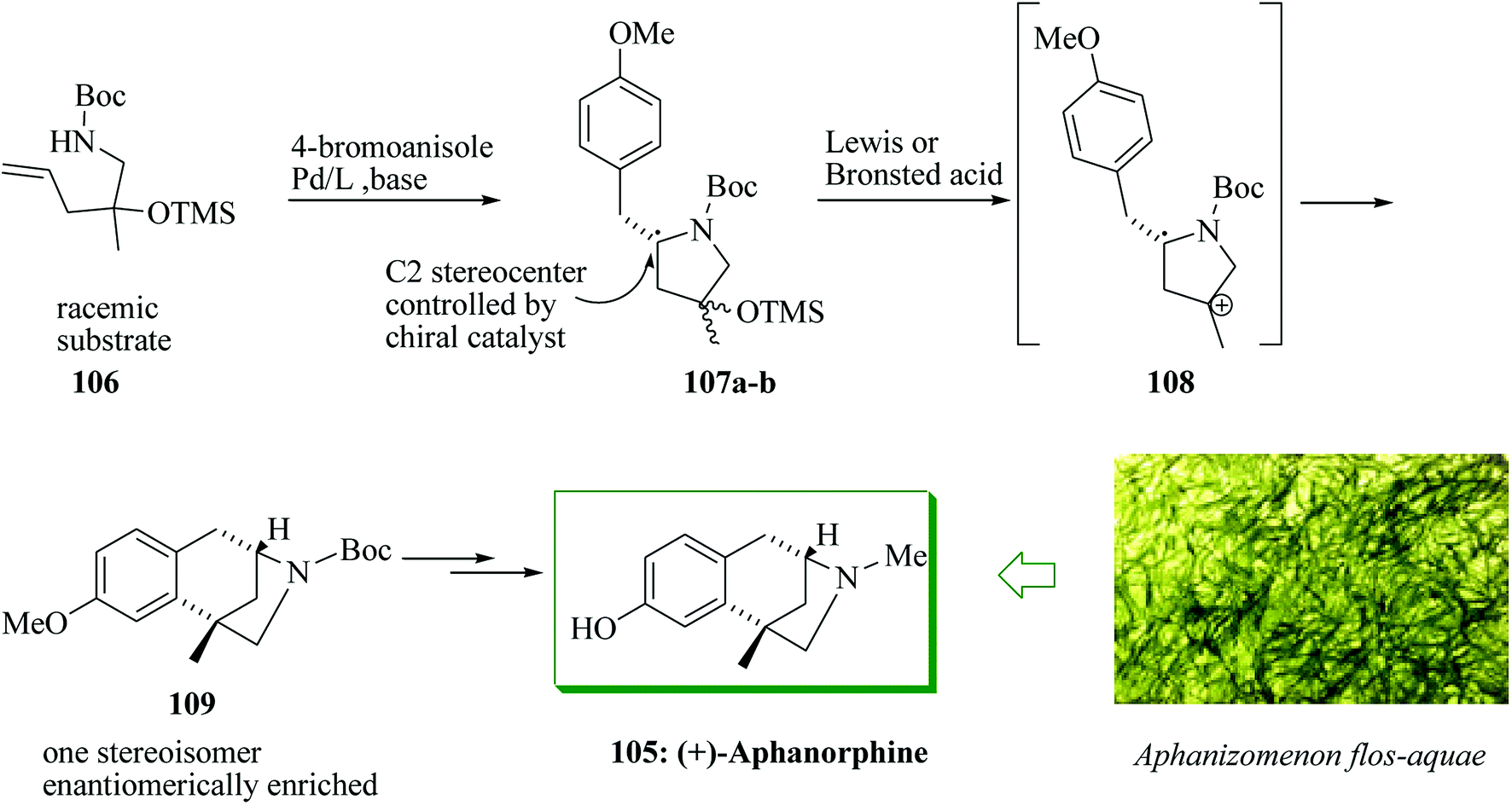 | ||
| Scheme 21 Enantioselective total synthesis of (+)-aphanorphine 105. | ||
The coupling reaction of 106 and 4-bromoanisole was explored for asymmetric alkene carboamination reactions. Based on the optimal conditions, this transformation gave a 1:1 mixture of diastereomers 107a and 107b, which afforded 110a,b. The enantio-convergent intramolecular FC alkylation reaction of the mixture of diastereomers 110a,b afforded 111 in 63% yield.114 Meanwhile, the transformation of 107a,b to 109 failed. The N-tosylated derivative 111 formerly acted as an intermediate in Zhai's syntheses of aphanorphine.114,116 Afterwards, compound 111 was converted into (+)-aphanorphine 105 upon several steps (Scheme 22).115
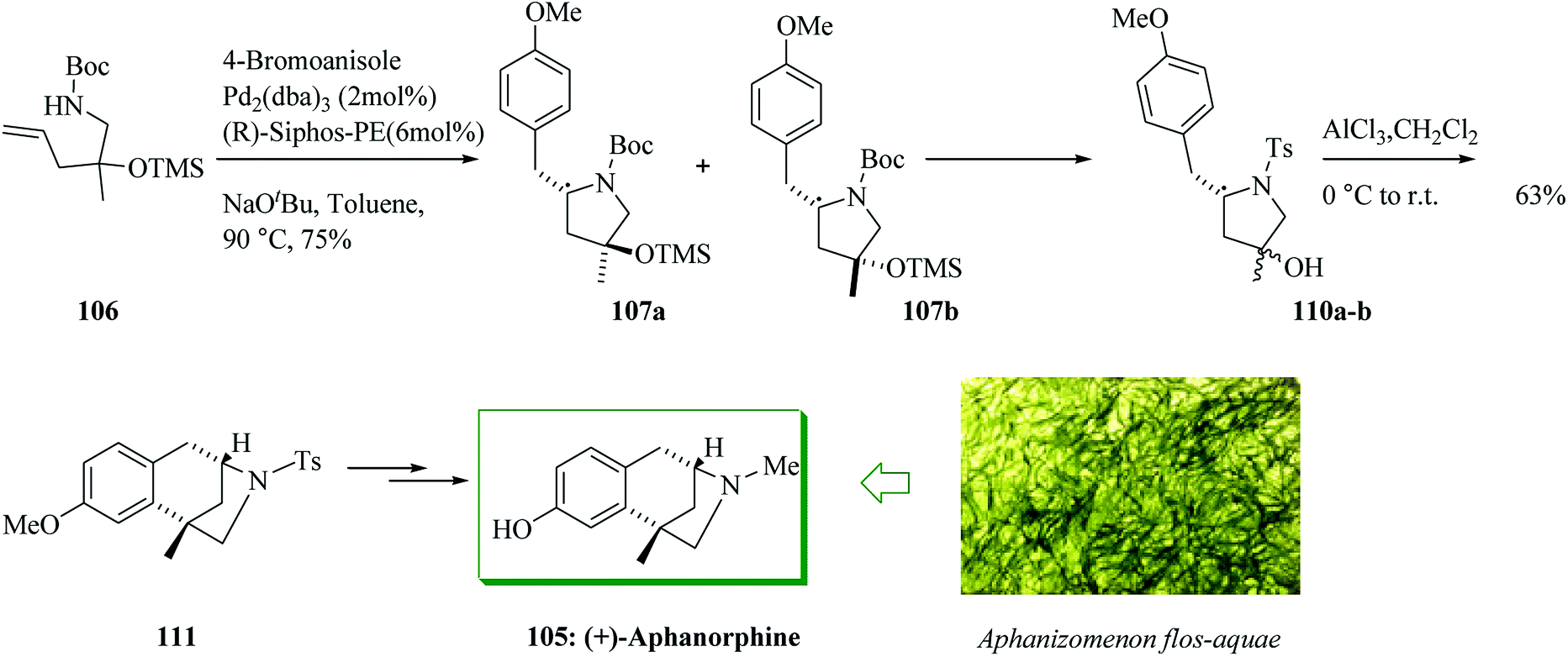 | ||
| Scheme 22 Total synthesis of (+)-aphanorphine 105. | ||
Elisapterosin B 112 was extracted from the gorgonian coral Pseudopterogorgia elisabethae and exhibited antitubercular properties, preventing the growth of Mycobacterium tuberculosis H37Rv.117 As part of a method toward the formation of the antitubercular agent elisapterosin B, it was synthesized from two chiral, non-racemic olefinic substrates and their diastereoselective ring closure, achieved in the presence of Hg salts. In this pathway, the synthesis was initiated from aldehyde 113,118 which was converted into 114 upon several steps. The asymmetric construction of 115 was achieved through an intramolecular FC alkylation reaction of 114. Harmata and co-workers synthesized elisapterosin B 112 by employing benzothiazine chemistry along with Hg(OTf)2-mediated diastereoselective intramolecular FC alkylation (Scheme 23).119
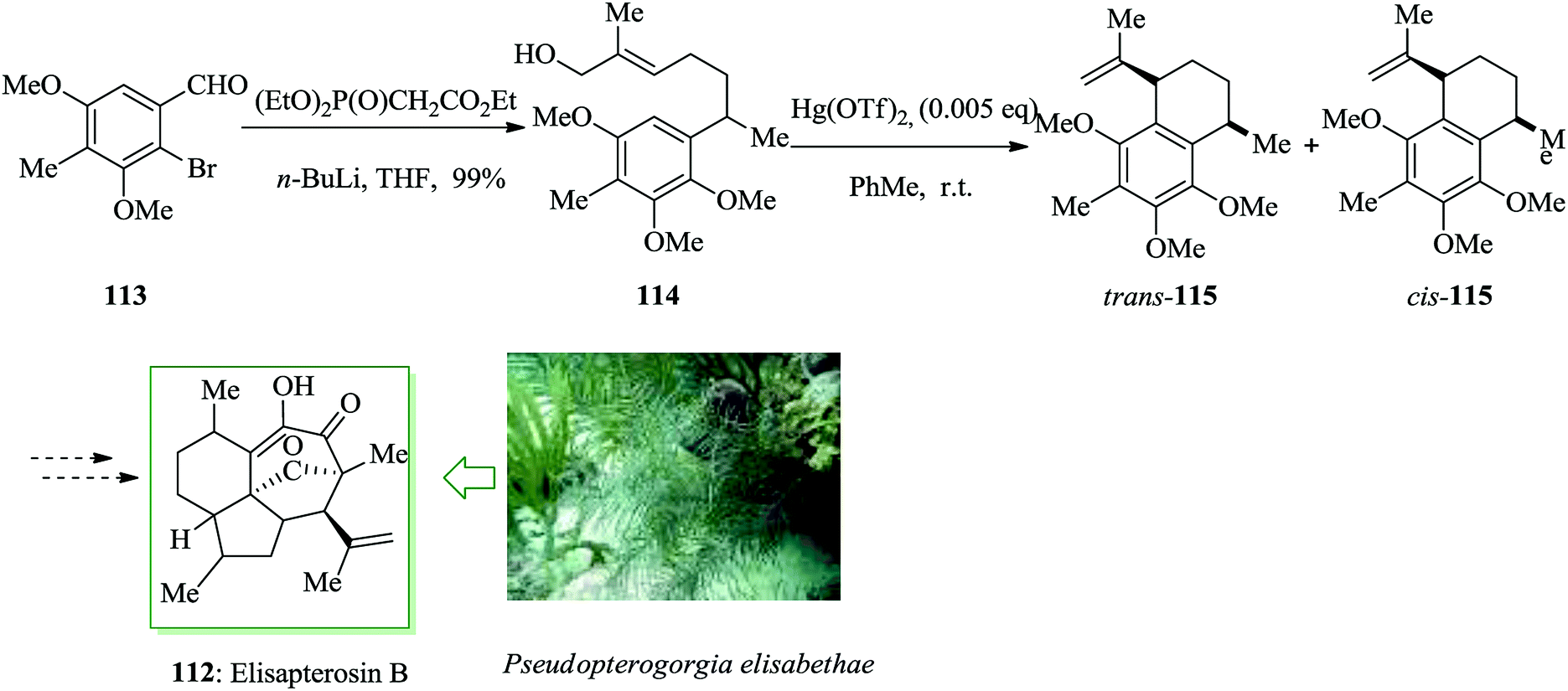 | ||
| Scheme 23 Total synthesis of elisapterosin B 112. | ||
Erogorgiaene 116, together with other structurally relevant diterpenes, has been extracted120 from the West Indian gorgonian octocoral Pseudopterogorgia elisabethae. The formal total synthesis of the antitubercular agent erogorgiaene has been accomplished in 12 steps with an overall yield of 14.2%, initiating from (S)-(−)-citronellal 117. This synthetic method includes an enamine-catalyzed 1,4-addition, an aldol condensation, dehydrogenation, a Wittig olefination reaction, intramolecular FC cyclization, TEMPO-BAIB-catalyzed oxidation, and Evans auxiliary-induced diastereoselective methylation. In this route, total synthesis of erogorgiaene 116 was started from (S)-(−)-citronellal 117, which was converted into allylic alcohol 118 upon several steps. Next, compound 118 was exposed to intramolecular FC cyclization reaction62,119 with BF3·OEt2 in dry CH2Cl2 to provide bicyclic compound 119 in 79% yield with moderate de (8:1). Afterwards, erogorgiaene 116 was provided from compound 119 upon several steps (Scheme 24).121
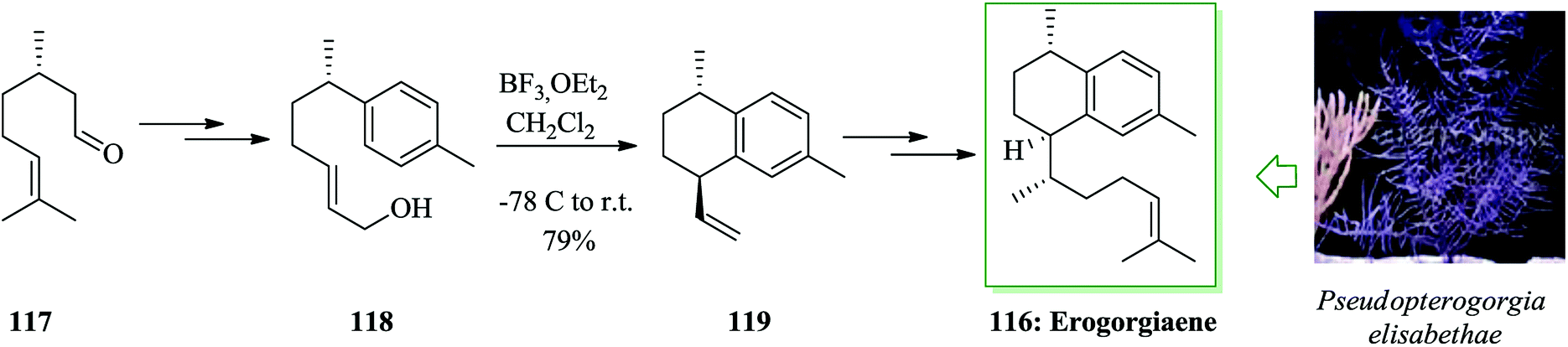 | ||
| Scheme 24 Total synthesis of erogorgiaene 116. | ||
The Cephalotaxus genus is a member of the Cephalotaxaceae group of conifers. They are a fruitful source of several naturally occurring compounds, especially terpenoids (abietanes, troponoids) and alkaloids (cephalotaxine esters), which often exhibit medicinal activities, particularly in the anticancer area.122 An uncommon rearrangement-annulation cascade has been demonstrated by Li and co-workers for the total synthesis of hainanensine 95a and its dimethoxy analogue 95b.101 Initially, isoquinolines 96a/96b provided the intermediate I followed by FC cyclization on the residual ketone II toward tetracyclic intermediates 98a and b. Five more steps were necessary to attain hainanensine 95a and its dimethoxy analogues 95b (Scheme 25).123
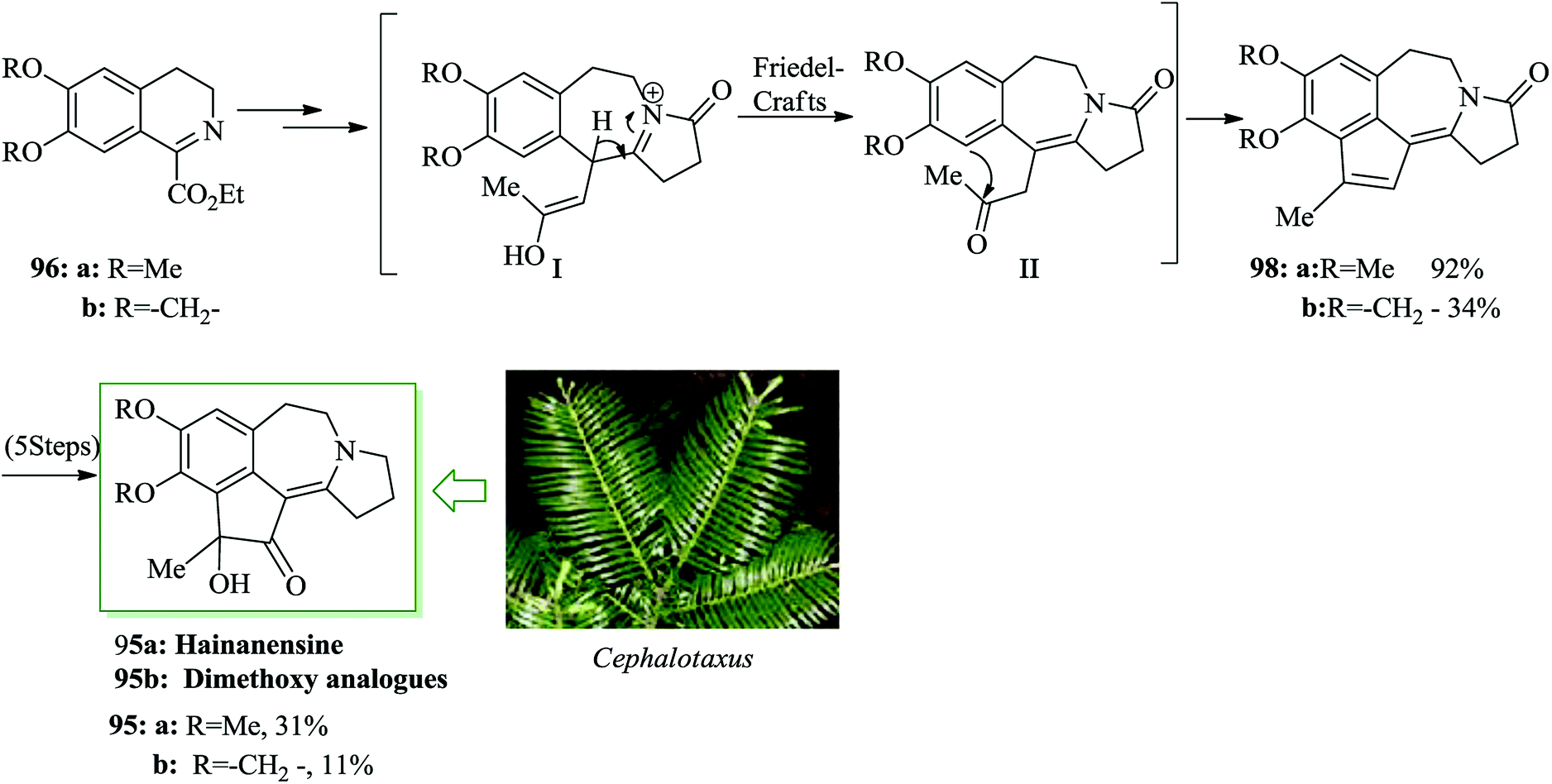 | ||
| Scheme 25 Total synthesis of hainanensine 95a and its dimethoxy analogue 95b. | ||
Laetevirenol A 120, together with laetevirenol B–E, was extracted from the stems and roots of Parthenocissus laetevirens in 2008 by Pan and co-workers.124 Laetevirenol A exhibits potent antioxidant properties, probably because of the existence of a phenanthrene scaffold serving as a free radical scavenger.125–133 The one-pot dehydration/FC alkylation of 121 with p-toluenesulfonic acid in toluene provided the corresponding trans-cyclized product 125 as a single isomer. Finally, global demethylation of 125 with boron tribromide in dichloromethane afforded laetevirenol A 120 in quantitative yield (Scheme 26).134
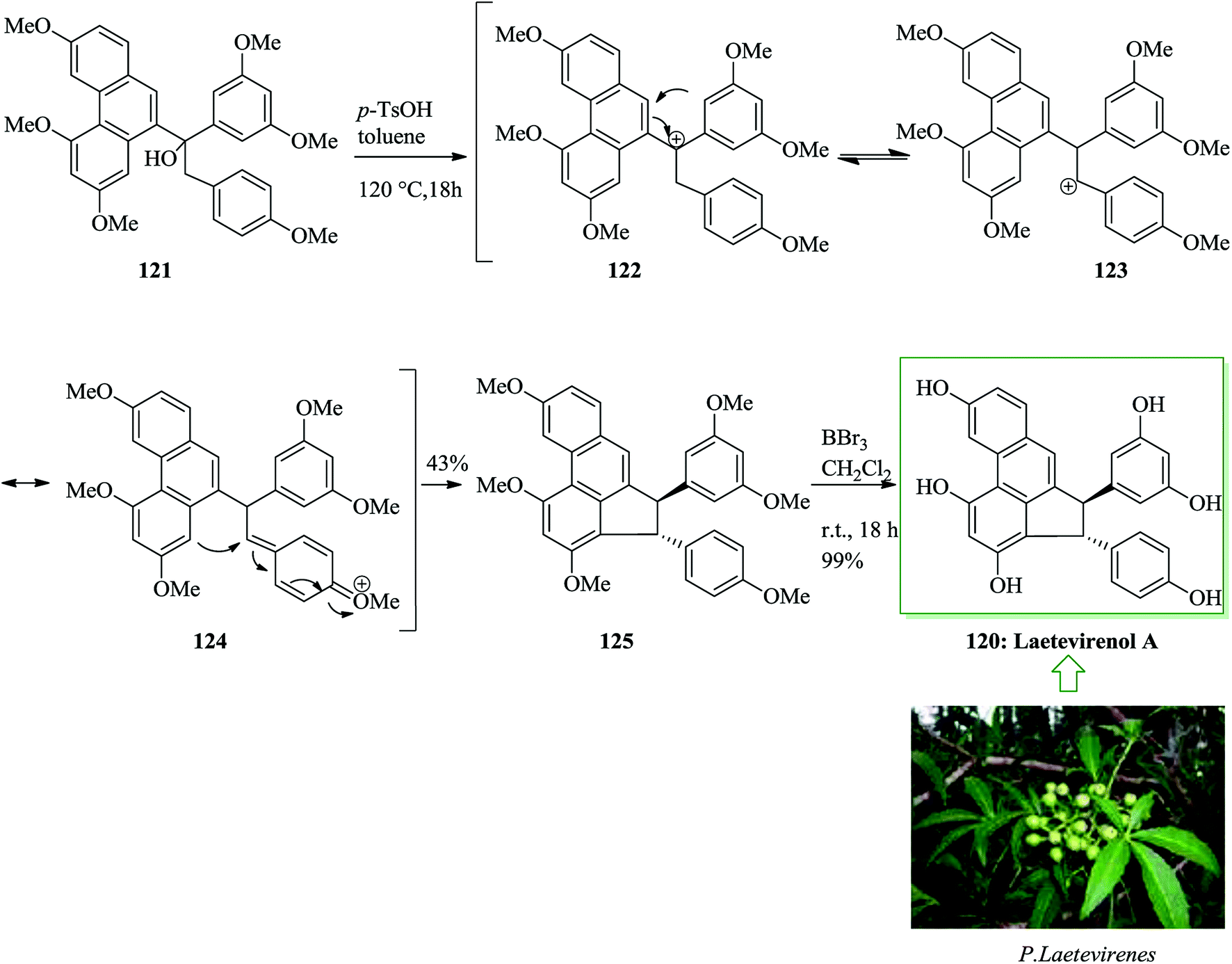 | ||
| Scheme 26 Total synthesis of laetevirenol A 120. | ||
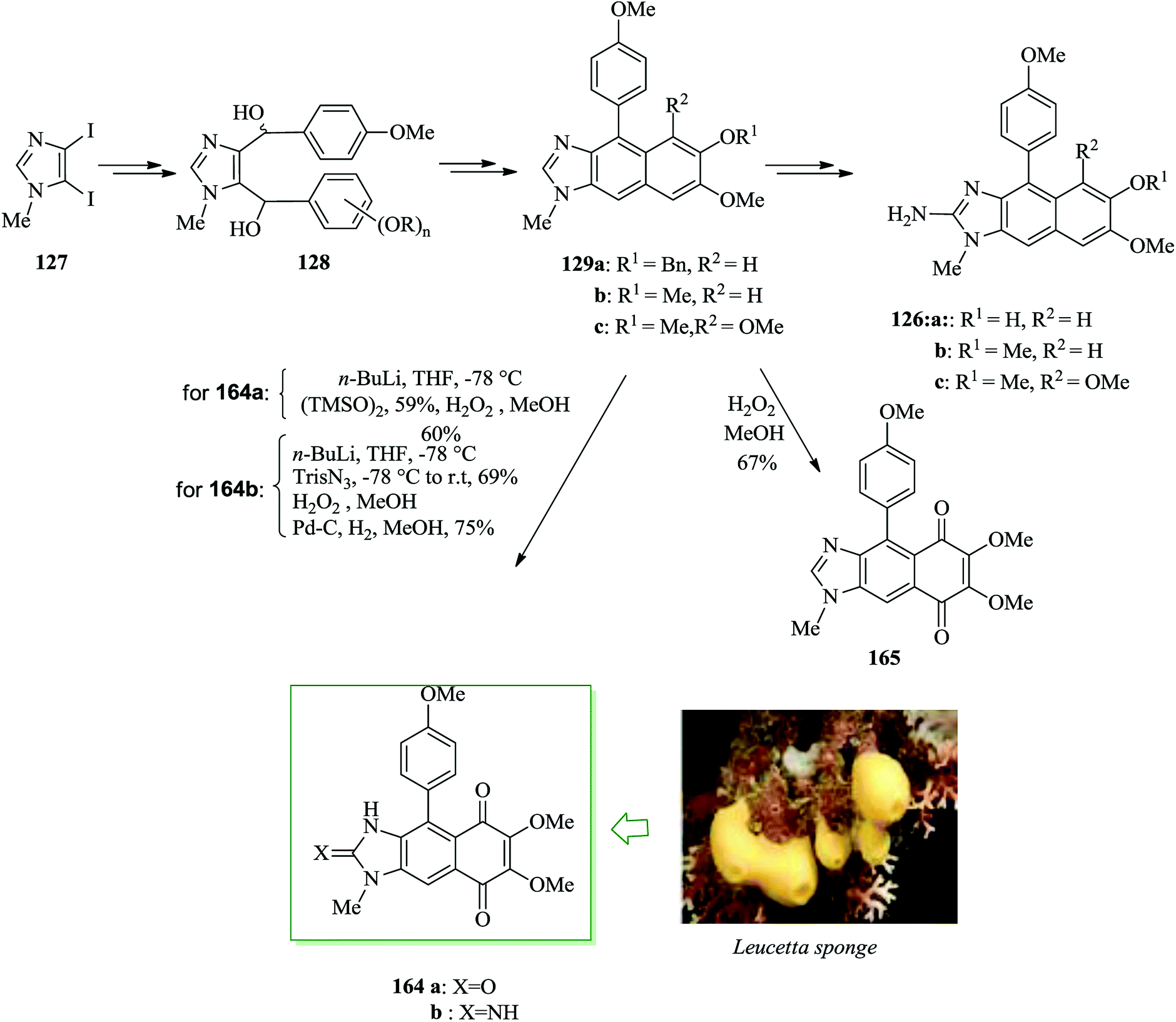
The Leucetta alkaloids, a class of 2-aminoimidazole alkaloids,135 were extracted from sponges of the Clathrina and Leucetta groups.136–138 Structurally, these alkaloids include at least one oxygenated benzyl group. Kealiinines A–C 126a–c139 were extracted by the Proksch group in 2004, together with naamidine H and naamine G. It should be mentioned that no biological activity has been stated by Proksch for 126b and 126c,139 but kealiinine A 126a was found to be potent in the brine shrimp toxicity test.139 Concise total syntheses of the Leucetta-obtained alkaloids, kealiinines A–C, were achieved through an intramolecular FC dehydration sequence of a bis benzylic diol. For the synthesis of kealiinine C 126c, the diol 128c was synthesized from 1-methyl-4,5-diiodoimidazole 127 upon several steps. Pleasantly, the reaction between unpurified 128c and hydrochloric acid led to an intramolecular FC cyclization-dehydration sequence to give the naphthimidazole 129c. Subsequently, compound 129c provided kealiinine C 126c upon several steps. Next, it was tried to make the other two group members, kealiinine A 126a and B 126b. Their syntheses follow largely the similar methodology, simply substituting the suitable benzaldehyde for 126a and 126b, affording the target products after seven steps with 11% overall yield and six steps with 21% overall yield, respectively, starting from compound 127 (Scheme 27).140
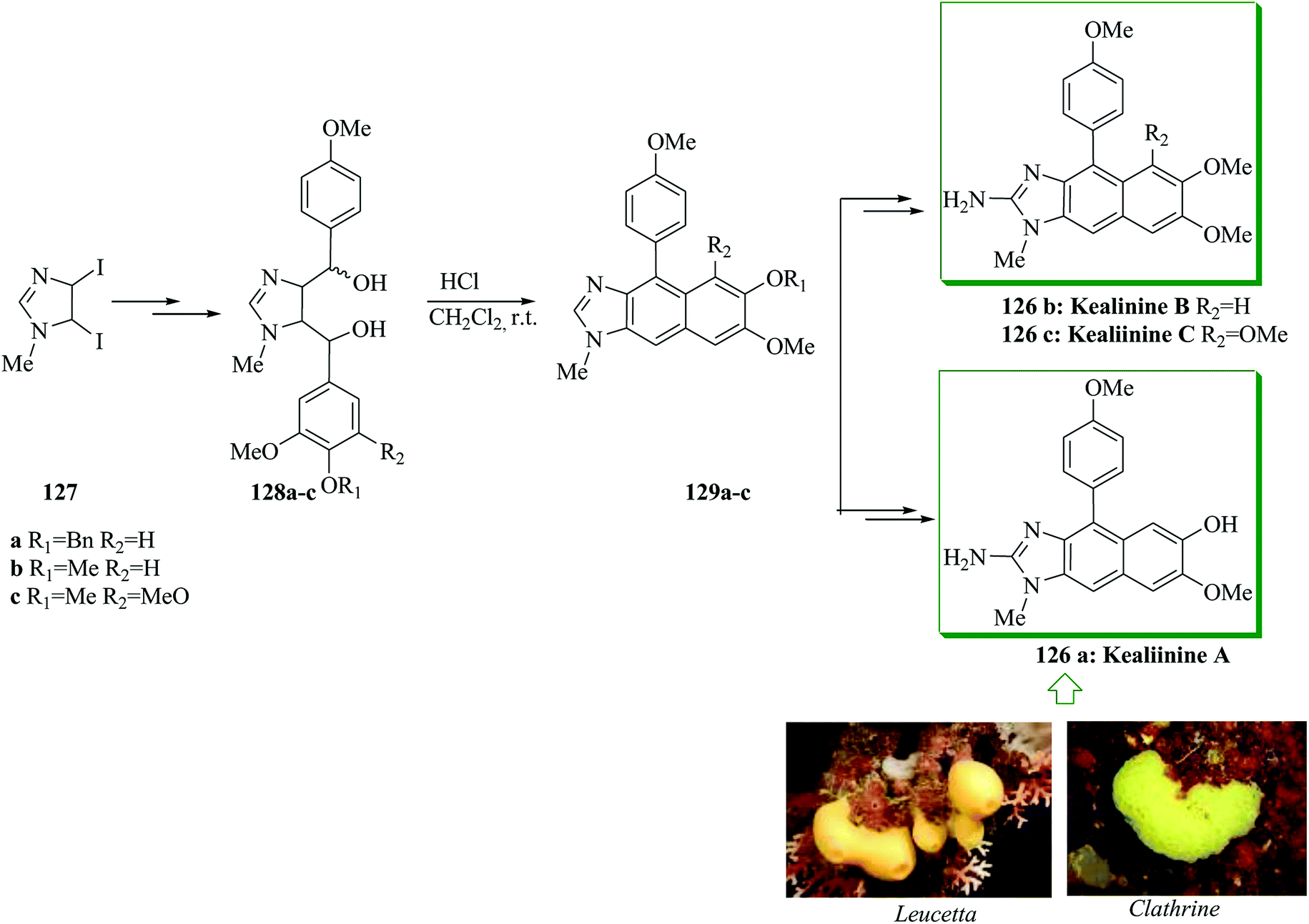 | ||
| Scheme 27 Total synthesis of kealiinines A–C 126a–c. | ||
The dibenzo[a,d]cycloheptene scaffold is a kind of polycyclic framework that occurs broadly in various naturally occurring compounds.141,142 One of the most direct approaches for the formation of this scaffold is the intramolecular FC acylation reaction.143,144 Another common synthetic pathway is the equivalent acid-mediated FC alkylation reaction of olefins.145 The natural diptoindonesin D 130 and pauciflorial F 131 have been extracted from the acetone extract of the tree bark of Hopea dryobalanoides146 and the stem barks of Vatica pauciflora147 respectively. Yang and co-workers established an efficient method for the construction of several dibenzo[a,d] cycloheptenes through FC alkylation reaction. Furthermore, utilizing this approach as the main stage, this group demonstrated short and valuable pathways to form diptoindonesin D 130 and pauciflorial F 131. The formation of diptoindonesin D 130 and pauciflorial F 131 was started with the construction of the biaryl alcohol 134 from the lithiated form of the Me-masked resveratrol bromide 132 and 3,5-dimethoxy benzaldehyde 133.148 With this key intermediate as the substrate, directly, the polyfunctionalized indene 135 was obtained in the presence of trifluoromethane sulfonic anhydride (Tf2O) as a catalyst via FC alkylation reaction. Next, oxidation reaction of compound 135 followed via deprotection afforded paucifloral F. On the other hand, compound 134 provided ketone 136 with 90% yield.149 Then, the carbocyclic unit structure of diptoindonesin D 130 was constructed through FC alkylation reaction, providing 137 in 76% yield. Upon oxidation and deprotection of 137, the target natural diptoindonesin D 130 was provided (Scheme 28).150
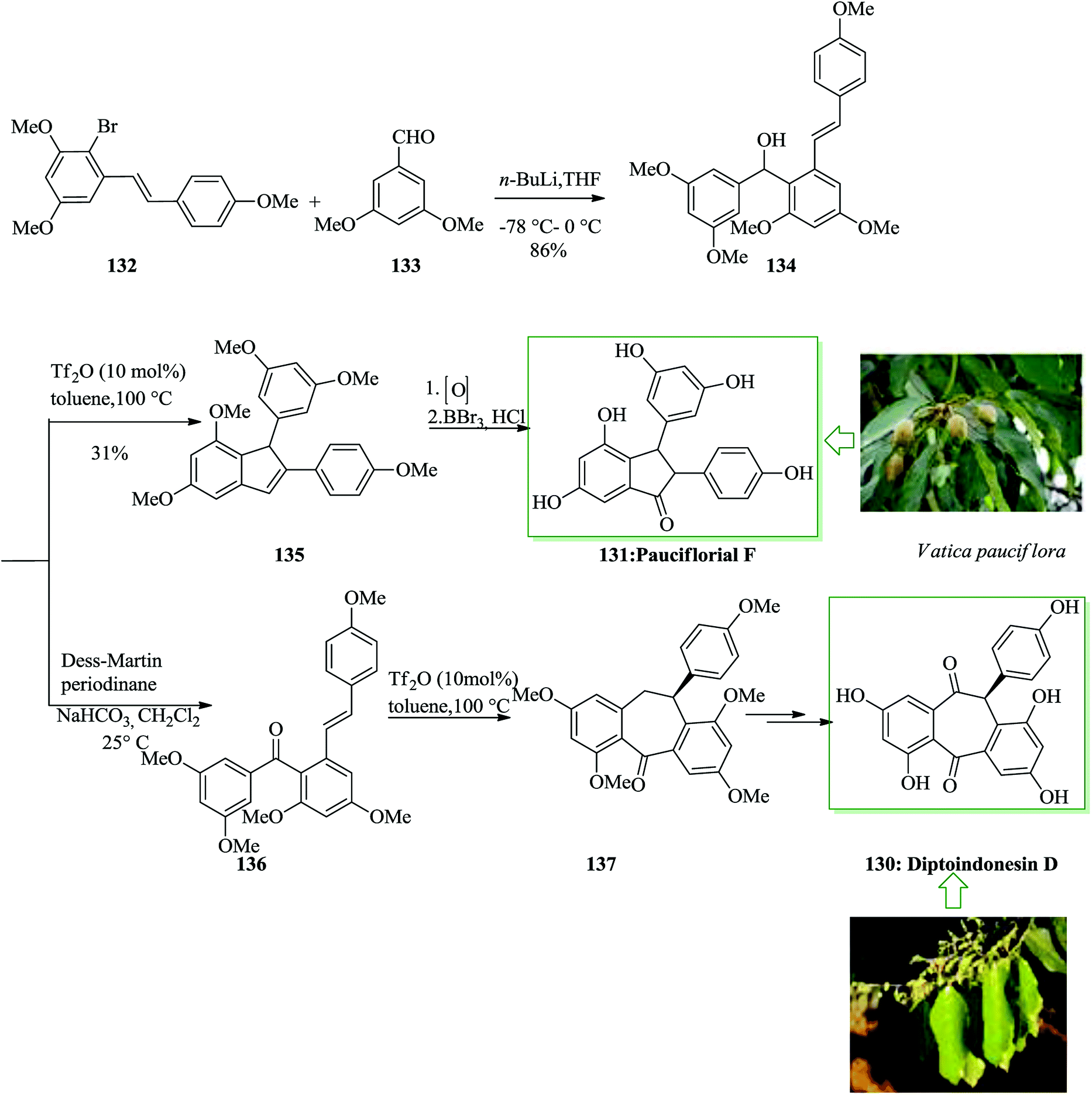 | ||
| Scheme 28 Total synthesis of diptoindonesin D 130 and pauciflorial F 131. | ||
Delle Monache et al. described the extraction of a dihydrophenanthrapyrane natural product from the root of Clusia paralycola, paralycolin A, that shows cytotoxicity against P388 cells and KB.151 The structurally relevant natural product cedrelin A 138 was extracted from the bark of Cedrelinga catenaeformis Duke by Kakisawa et al. in 1991.152 Paralycolin B has been extracted merely as a methylated derivative 139 upon reaction of a resultant mixture of paralycolin A and paralycolin B with diazomethane. Furthermore, these naturally occurring compounds usually contain an isopropenyl substituent at the 10-position of the 9,10-dihydrophenanthrene framework. Hamada and co-workers in 2013 demonstrated the initial asymmetric total syntheses of cedrelin A and methylated paralycolin B, employing mediated enantioselective intramolecular FC allylic alkylation reaction of phenols as the main step.153 Total synthesis of cedrelin A 138 was initiated from aldehyde 140,154 which was transformed into allyl carbonate derivative 141 upon several steps. Next, the mediated enantioselective intramolecular FC allylic alkylation of 141 was examined. With 1.5 equiv. of potassium acetate, the reaction progressed easily to give compound 142 in 98% yield (94% extracted yield) with 66% enantioselectivity. Finally, intermediate 142 has been converted into cedrelin A 138 in 74% yield (12 steps from 140 and 16.5% overall yield) (Scheme 29).153
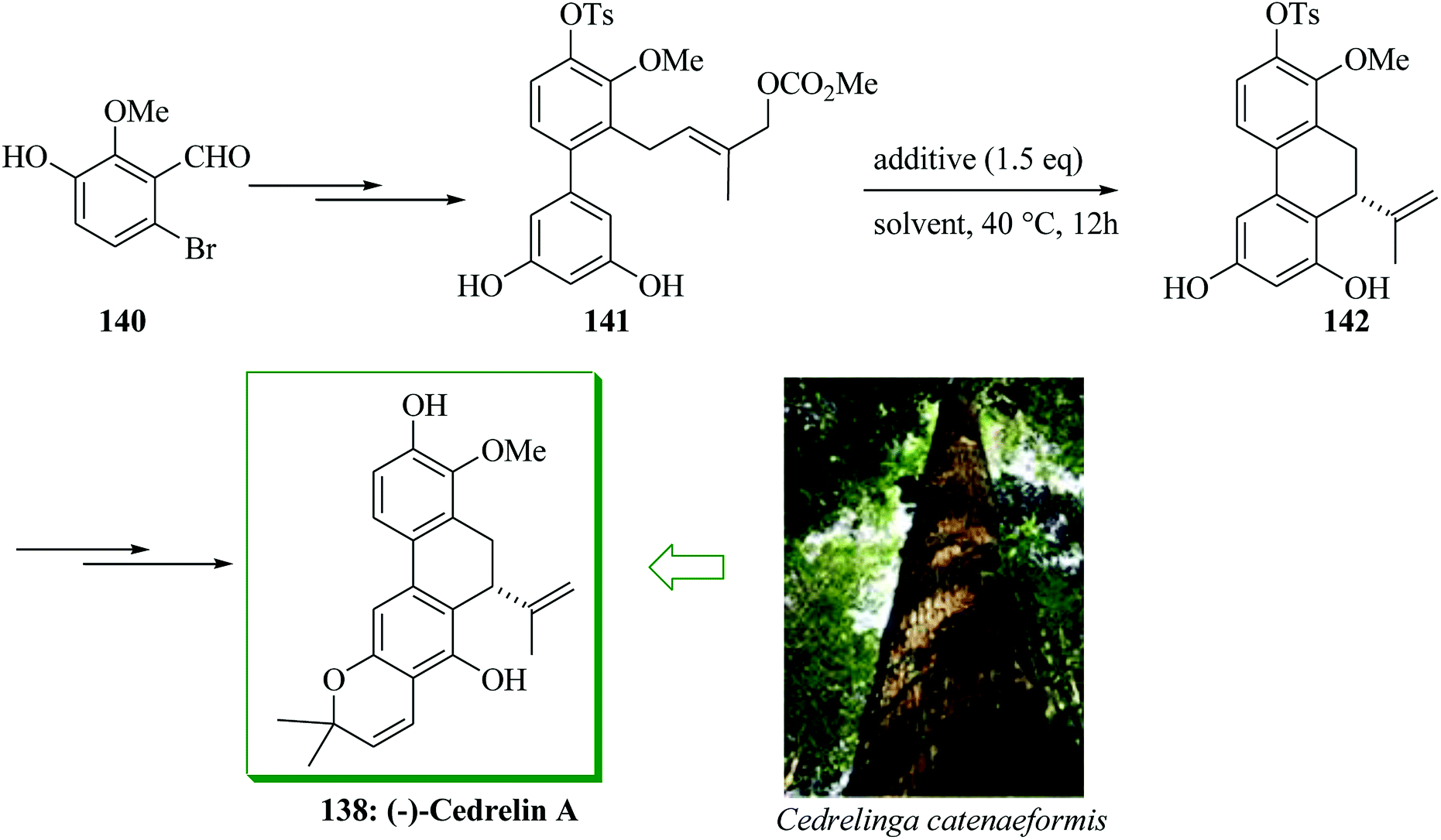 | ||
| Scheme 29 Total synthesis of cedrelin A 138. | ||
Subsequently, the asymmetric total synthesis of methylated paralycolin B 139 was accomplished. First, compound 144 was synthesized from 6-bromoveratraldehyde 143 upon several steps. Enantioselective intramolecular FC allylic alkylation reaction of 144 using Pd(dba)2 and (R,R)-chiral ligand in dichloromethane/methanol mixed solvent afforded 145 in 98% yield with 92% enantioselectivity. Finally, compound 145 provided methylated paralycolin B 139 in 92% yield (10 steps from 143 and 35.2% overall yield) (Scheme 30).154
 | ||
| Scheme 30 Total synthesis of methylated paralycolin B 139. | ||
Members of the genera Ligularia, Euryops, Senecio, and Psacalium of the Asteraceae group are generally dispersed plants rich in furanoeremophilane compounds.155–157 This group of sesquiterpenes includes a linearly fused C6–C6–C4O tricyclic (C12) scaffold having three methyl groups linked to the 4, 5, and 11 positions. An asymmetric total synthesis of the furanoeremophilane sesquiterpene (+)-9-oxoeuryopsin 146 was reported from 2-methyl-2-cyclohexen-1-one 147 in 7% overall yield. This method was accomplished in seven facile synthetic reactions involving conjugate addition-enolate trapping, imidazoylthiocarbonylation reaction, bisdethiocarbonylation, masked cyanohydrin construction-hydrolysis, dehydration, alkaline hydrolysis, and also FC cyclization. The total synthesis of 146 was initiated from 2-methyl-2-cyclohexen-1-one, which was converted into acid 148 upon several steps. Next, the crude acid 148 was transformed into (+)-9-oxoeuyopsin 146 in 59% overall yield via an intramolecular FC cyclization using tin(IV) chloride as the Lewis acid of the spontaneously provided acid chloride (PCl5 in benzene at room temperature) (Scheme 31).158
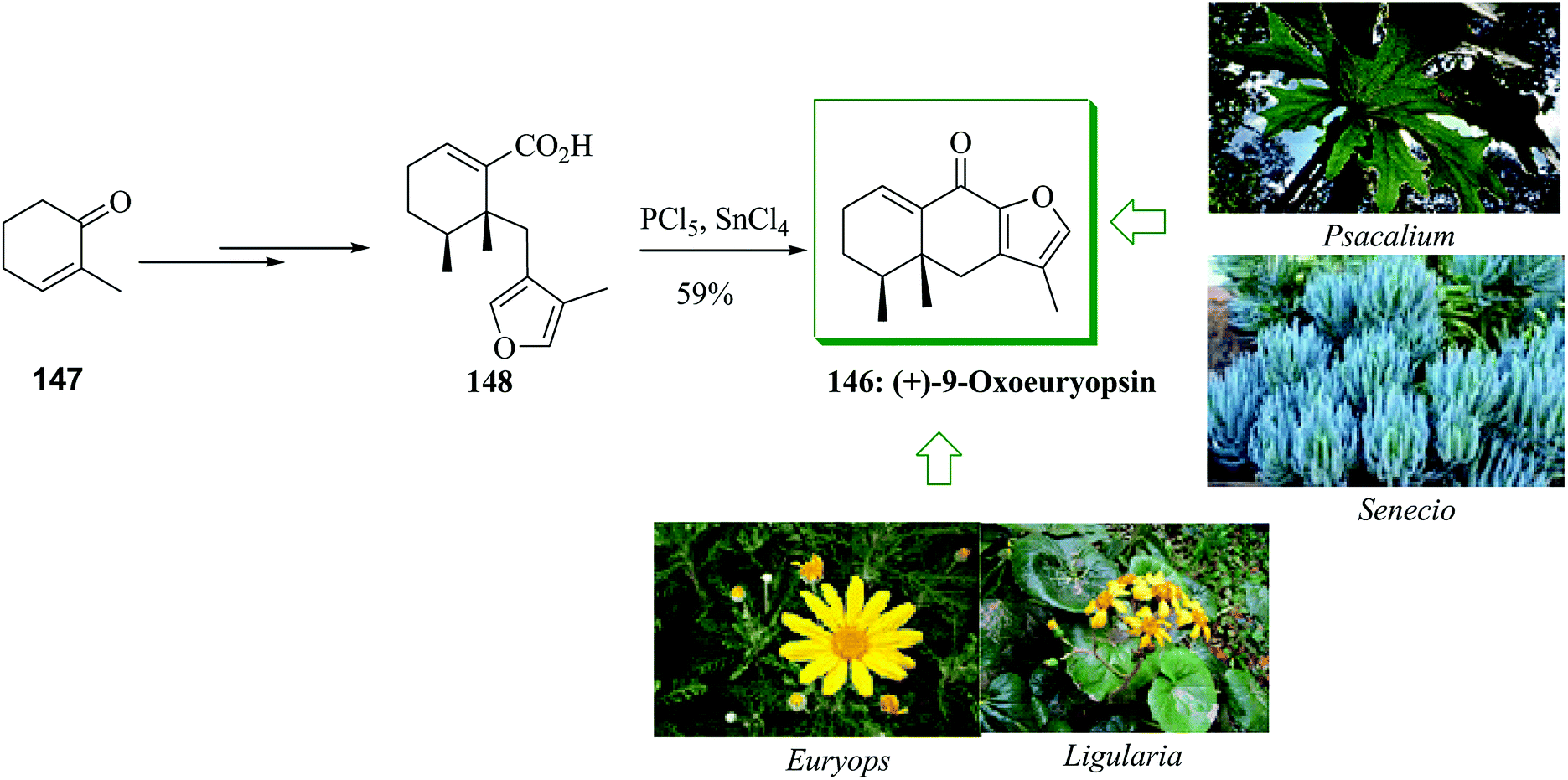 | ||
| Scheme 31 Total synthesis of (+)-9-oxoeuyopsin 146. | ||
Compound 149, an unnamed quinone methide diterpenoid, was initially extracted from the root bark of Bobgunnia madagascariensis.159 A convergent pathway was established to form an antifungal tricyclic o-hydroxy-p-quinone methide diterpenoid 149 and analogues. In 2013, Yang and co-workers finished the total synthesis of the antifungal tricyclic o-hydroxy-p-quinone methide diterpenoid 149. A Stille reaction was applied to introduce the allyl substituent as a protected 2-hydroxyethyl side chain. This group established a BBr3-catalyzed one-pot bis-demethylation/intramolecular FC alkylation reaction to make the tricyclic molecular framework and then completing the total synthesis of 149.160 Compounds 149 and 155 exhibited active cytotoxicity against several strains of pathogenic yeasts tested, but etherification of the 2-hydroxyethyl group resulted in significantly attenuated activity. This total synthesis began from commercially available bromobenzene, which in four steps is converted into 3,4-dimethyoxytoluene 150.161 The latter after several steps gave the primary alcohol 151. The application of the BBr3-catalyzed one-pot reaction resulted in the completion of the synthesis of 149. Therefore, reaction of 150 with BBr3 resulted in bis-demethylation and cyclization in a one-pot manner to generate a tricyclic catechol that, upon oxidation with Ag2O, afforded 149. Selective monoetherification of 149 with dimethoxymethane afforded the analogue 152 (Scheme 32).160
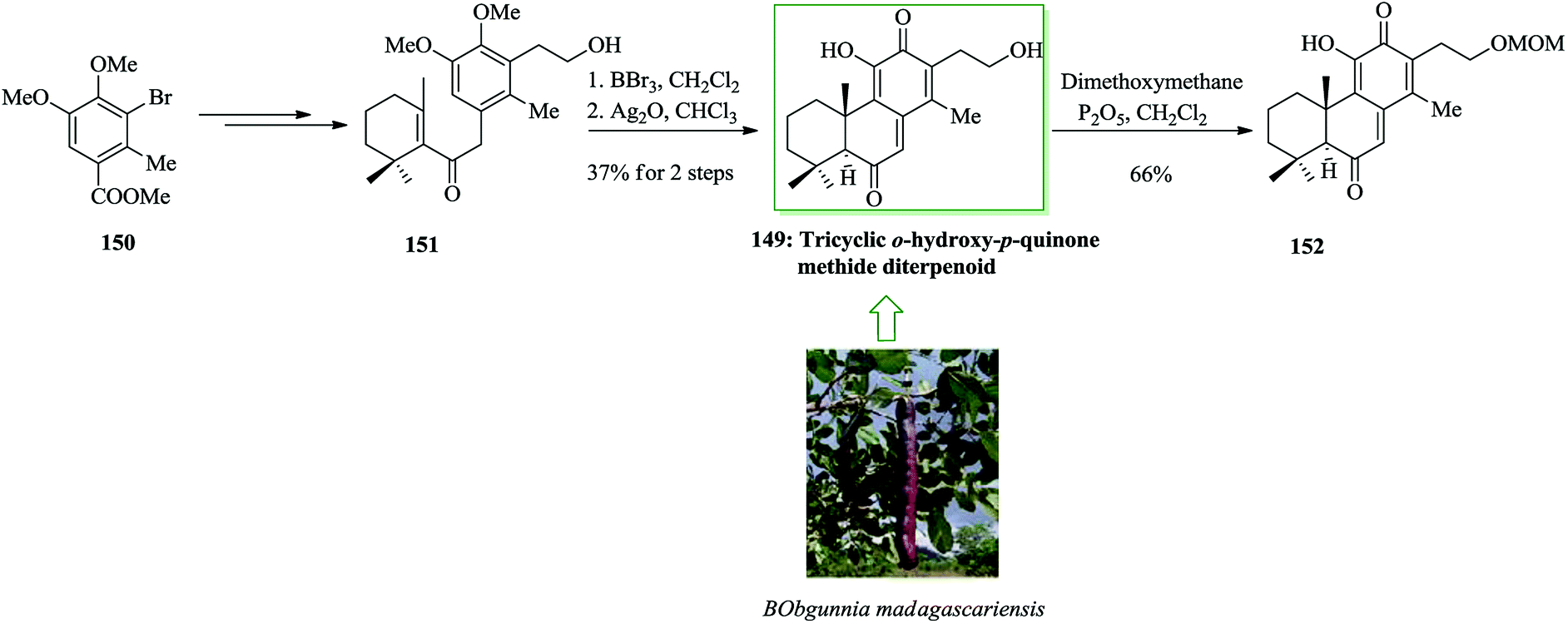 | ||
| Scheme 32 Total synthesis of tricyclic o-hydroxy-p-quinone methide diterpenoid 149 and analogues. | ||
The same group synthesized analogue 155 from carboxylic acid 153.160 Compound 153 afforded enone 154 upon several steps. Compound 155 was provided via BBr3-catalyzed bis-demethylation and intramolecular FC alkylation followed by oxidation of the resulting catechol with Ag2O in one-pot fashion. Compounds 149 and 155 were found to be particularly effective against all yeast strains, including Candida krusei, Candida glabrata, and Candida parapsilosis strains (Scheme 33).161
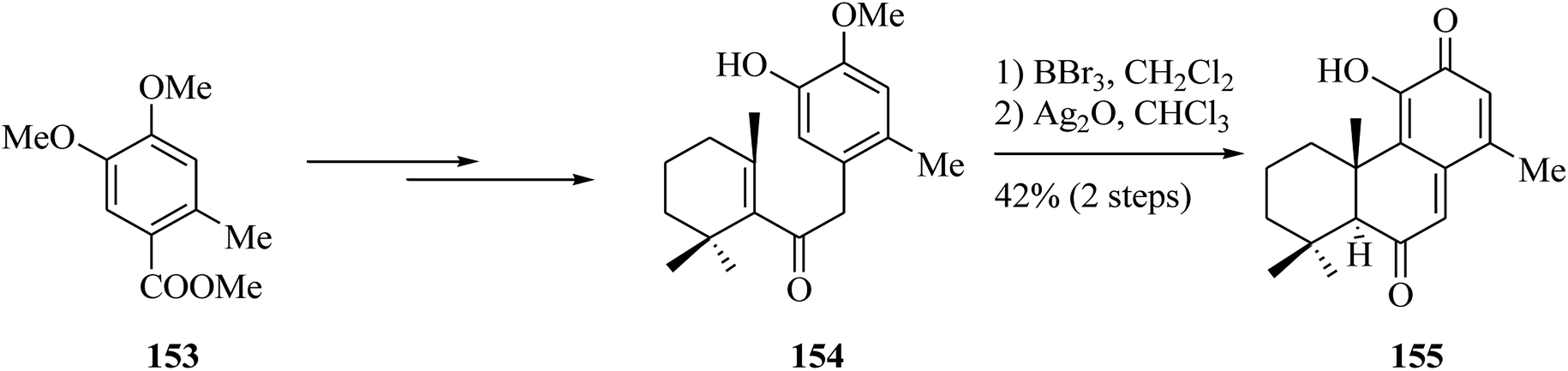 | ||
| Scheme 33 Total synthesis of product 155. | ||
Dimeric epipolythiodiketopiperazine alkaloids are an intriguing group of fungal metabolites notable for their complex molecular scaffolds and biological properties.162,163 (+)-Bionectins A 156 and C 157164 were extracted in 2006 by Zheng and co-workers from fungi of the Bionectra byssicola species. (+)-Bionectins A (+)-156 showed important bacteriostatic activity against methicillin-resistant and quinolone-resistant Staphylococcus aureus Gram-positive eubacteria. The short and effective total synthesis of (+)-bionectins A and C was developed by Movassaghi and co-workers in 2013. This route leading to the total synthesis of these naturally occurring compounds features a new and scalable approach for the synthesis of erythro-β-hydroxytryptophan amino acid. It involves an intramolecular FC reaction of a silyl-tethered indole and a new mercaptan reagent for epipolythiodiketopiperazine (ETP) synthesis that can be unraveled under very mild conditions. Based on this approach, firstly indole-3-carboxaldehyde 158 was converted into the corresponding silyl-tethered indole adduct 159 in 74% yield in several steps. Gratifyingly, a silver-catalyzed intramolecular FC reaction progressed easily in nitroethane to give the C3-(3′-indolyl)-silacyclic product 160a in 68% yield. The structure of a diethyl silyl variant 160b, provided during optimization studies, was confirmed via X-ray analysis. Compound 160a gave the target natural product (+)-bionectin A 156 in 81% yield after several steps. Reductive methylation of (+)-bionectin A 156 with NaBH4 and methyl iodide in pyridine and MeOH gave (+)-bionectin C 157 in 97% yield (Scheme 34).165
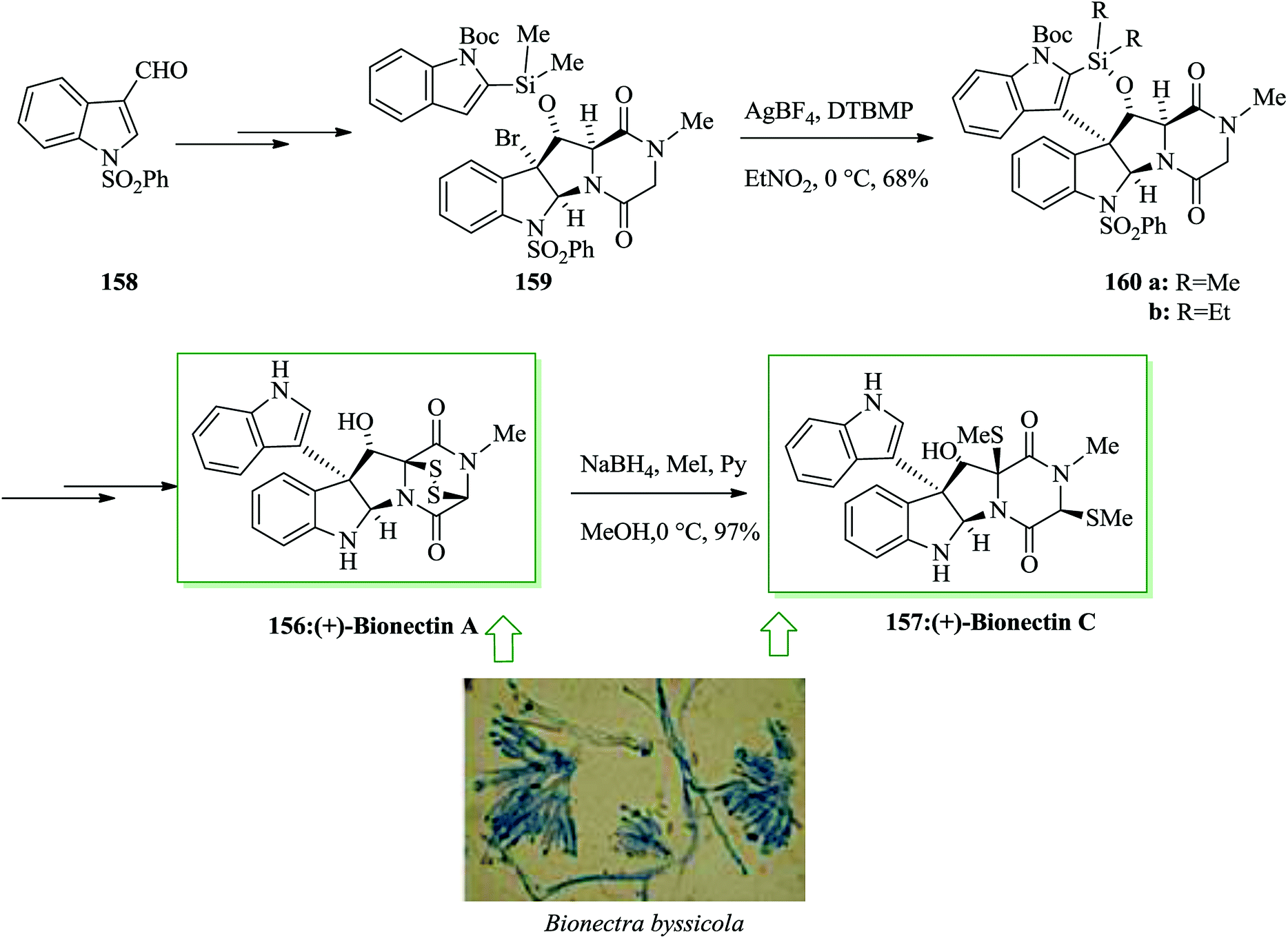 | ||
| Scheme 34 Total synthesis of (+)-bionectins A 156 and C 157. | ||
Ammosamides A–C are marine alkaloids that were initially extracted from the marine Streptomyces strain CNR-698 by Fenical and co-workers in 2009.166 Except for ammosamide D, these natural products have a characteristic pyrroloquinoline unit framework, in which the tricyclic structure contains a pyrrole scaffold bridging the C4 and C5 positions of a quinoline ring system.167,168 Ammosamides A and B exhibit important cytotoxicity against human colon adenocarcinoma HCT-116 cells.166 A total synthesis of ammosamide B 161, a member of the pyrroloquinoline alkaloid group extracted from marine Streptomyces, was developed. The characteristic unit tricyclic structure of 161 was generated through a unique, tandem FC reaction sequence to transform the symmetric tetra-amino functionalized benzene derivative 162 into the tricyclic pyrroloquinoline product 163. The reaction was initiated with an acid-promoted tandem FC reaction of the diamino and dinitro functionalized benzene derivative 162 to form the tricyclic pyrroloquinoline 163. The required substrate 162 was easily synthesized from m-dichlorobenzene in a two-step sequence.
Subsequently, a number of acids were applied to examine the tandem FC reaction and the results demonstrated that the reaction improved in the presence of trifluoroacetic acid (TFA) but does not provide either of the anticipated FC products, only an unidentified oligomeric material. Reactions using the Lewis acids aluminium chloride and titanium tetrachloride also resulted in the construction of insoluble oligomers and no reaction (162 was recovered quantitatively) happened once the less reactive Lewis acids zinc chloride, tin tetrachloride and Yb(OTf)3 were used. The remarkable observation that an indole (side-product) was provided in 52% yield when BF3·Et2O in dichloromethane was used provided the motivation for a broader investigation of solvent effects on this method. When CH3CN or THF was utilized in the presence of BF3·Et2O as the solvent for the improvement of the reaction, insoluble oligomers were provided. In contrast, tricyclic pyrroloquinoline 163, via tandem FC product, produced the expected product in 35% isolated yield once toluene was applied as the solvent. While this reaction was low yielding, it was performed efficiently and no significant side products were detected by TLC analysis. Hence, the poor yield of 163 is probably owing to its insolubility during the work-up and purification processes. Consequently, Nagasawa decided to apply the optimal reaction conditions (BF3·Et2O in toluene) performing the conversion of 162 to 163 in the synthetic pathway to form ammosamide B 161 and skipped the purification stage. A tandem FC reaction of 162 promoted by BF3·Et2O in toluene led to the formation of compound 163. Next, ammosamide B 161 was provided after several steps (Scheme 35).169
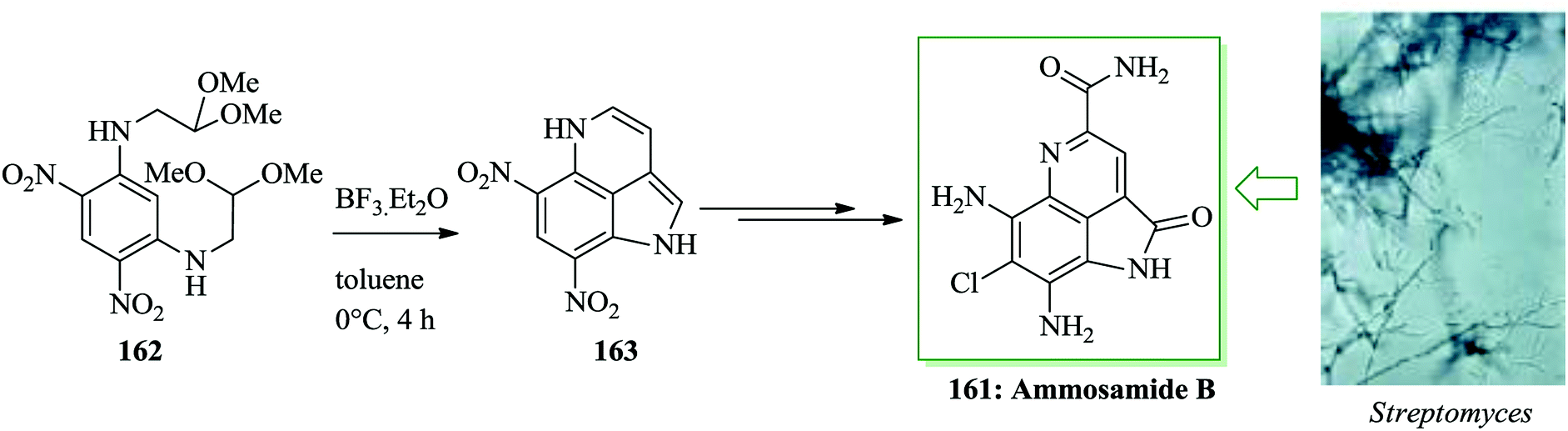 | ||
| Scheme 35 Total synthesis of ammosamide B 161. | ||
Marine sponges provide a wide range of structurally unique secondary metabolites with remarkable biological properties.170,171 Clardy and Scheuer initially extracted kealiiquinone 164a from a Leucetta-obtained sponge containing a unique imidazobenzoquinone scaffold.172 Compound 164a exhibited cytotoxicity, with potentially a unique mechanism of action.173 Schmitz reported the isolation of 2-deoxy-2-aminokealiiquinone 164b, but no bioactivity has been reported for this molecule.174 A brief synthesis for kealiiquinone 164a and 2-deoxy-2-aminokealiiquinone 164b was reported in 2013 by Lovely and co-workers. Advanced intermediates including the full naphthimidazole scaffold are generated via FC reaction followed by oxidation. Total synthesis of 164a and 164b was started from diiodoimidazole 127, which was converted into 128 in several steps the latter undergoes sequential FC reaction/dehydration to give compound 129.140 Subsequently, compound 129 provided kealiiquinone A 164a and the 2-amino congener 164b (Scheme 36).175
 | ||
| Scheme 36 Total synthesis of kealiiquinone A 164a and the 2-amino congener 164b. | ||
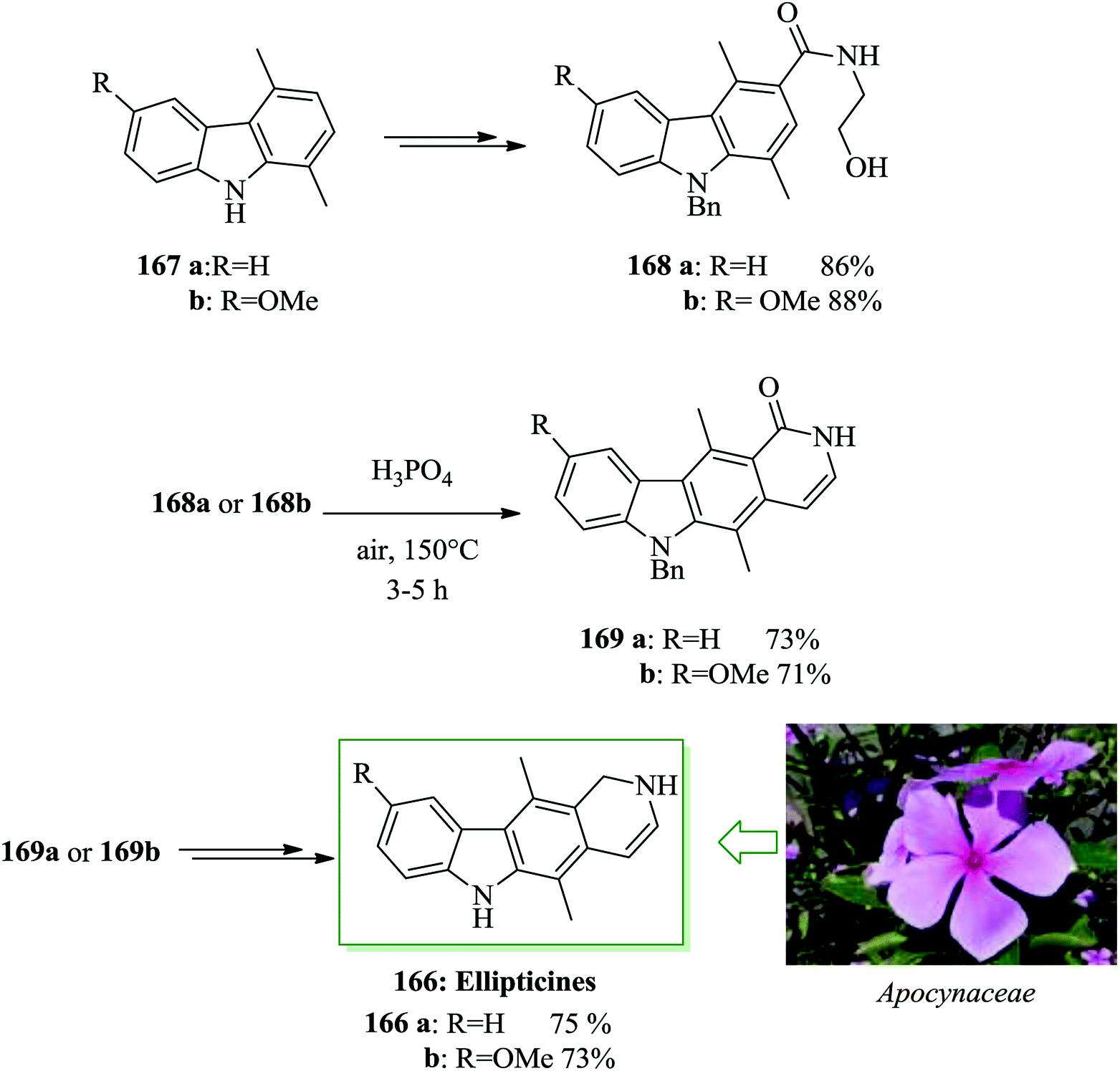
Natural products belonging to the pyrido[4,3-b]carbazole group of alkaloids are well represented in the chemical literature.176–185 Since the first extraction of ellipticine 166a and 9-methoxyellipticine 166b in 1959 by Goodwin and co-workers,177 several other compounds in this group have been extracted from Apocynaceae plants.178 The antineoplastic activity of ellipticine has been developed largely because of the importance of DNA intercalation and inhibition of topoisomerase II.181 Nagarajan and co-workers in 2014 achieved an convenient synthesis of ellipticine 166a and 9-methoxyellipticine 166b in 7 steps with 23% and 25% overall yields, respectively.186 This synthetic method uses a key phosphoric acid-catalyzed FC cyclodehydration for the generation of the pyridine unit. Total synthesis began from 1,4-dimethylcarbazoles (167a and 167b). In this approach, phosphoric acid-catalyzed FC cyclodehydration was employed as a key step for the construction of pyrido[4,3-b]carbazole alkaloids. This synthesis was started from 1,4-dimethylcarbazoles 167a and 167b and afforded the corresponding amides 168a and 168b after several steps. Amides 168a and 168b, were subjected to the critical FC cyclodehydration with subsequent pyridine ring construction. Compounds 168a and 168b in the presence of phosphoric acid provided dihydropyridocarbazolones 169a and 169b in 73% and 71% yields, respectively. Finally, compounds 169a and 169b were converted into ellipticines 166a and 166b after several steps (Scheme 37).186
 | ||
| Scheme 37 Total synthesis of ellipticines 166a and 166b. | ||
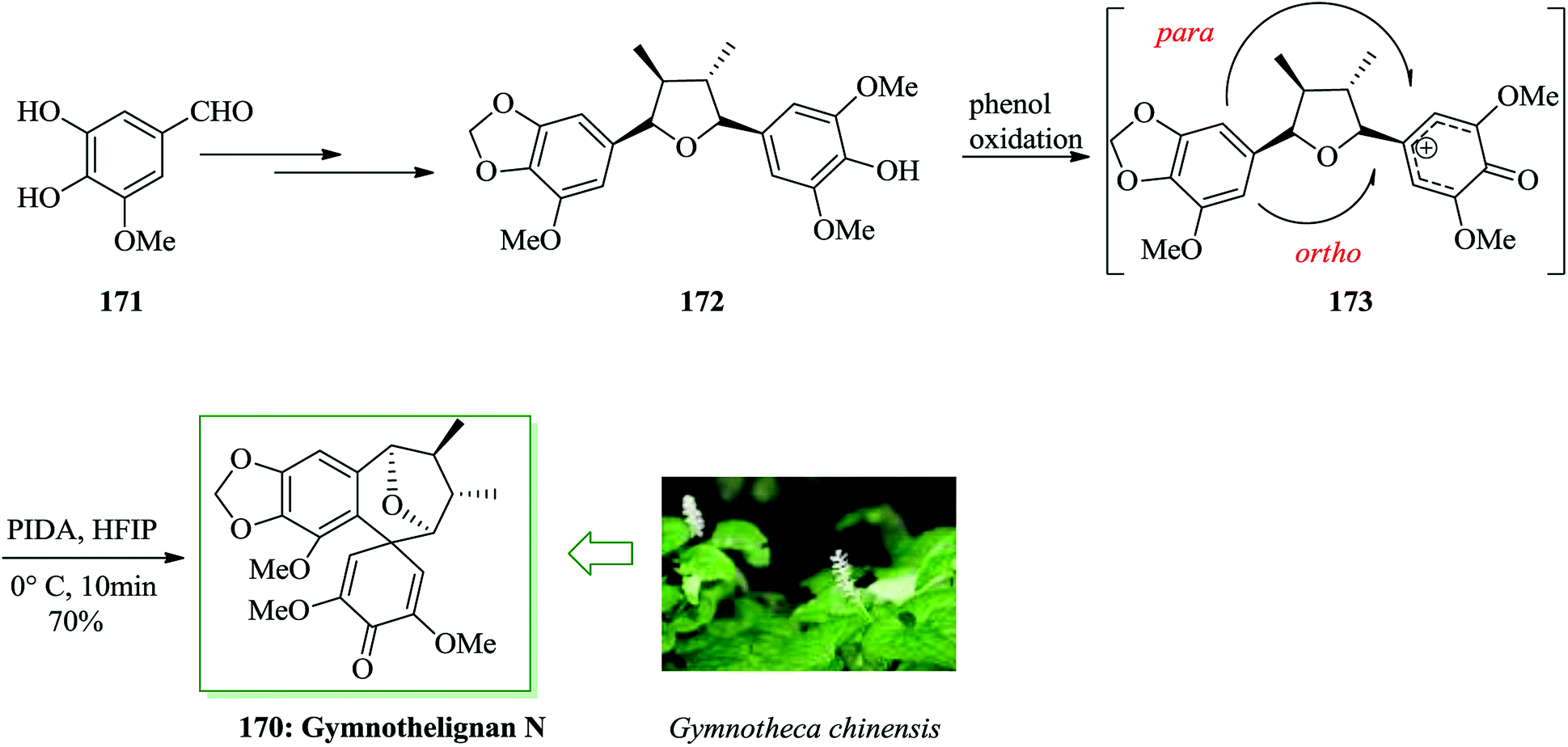
In 2012, Xu and co-workers extracted various tetrahydrofuran-type lignans, gymnothelignans A–O, from Gymnotheca chinensis Decne, a broadly applied perennial Chinese herb of Saururacese.187 Among them, gymnothelignan N attracted the interest of the authors owing to its unique 10,11-benzospiro[5.6]dodec-13,15-dien-14-one scaffold across a tetrahydrofuran ring, which is extraordinary in lignans. Total synthesis of gymnothelignan N 170 was achieved in 13 steps from 5-hydroxyvanilin 171 in 6.7% overall yield. In this approach, a syn Evans aldol reaction, an intramolecular hydrogenative dehydration reaction, and a phenol oxidative dearomatization/FC reaction were considered as the key steps. Total synthesis was started from 5-hydroxyvanilin 171, which was converted into compound 172 via several steps. Using the key intermediate 172, the step was performed to generate the critical bioinspired oxidative FC reaction. However, there are many cases of phenol oxidative dearomatization/FC reaction used in natural product synthesis;188–191 in this substrate, it is considered to be challenging for two aspects. The first one is the desired intramolecular FC reaction, which happens across the tetrahydrofuran ring to generate the seven-membered ring with an all-carbon quaternary center as linkage, while the presumed cation intermediate 173 is constructed in situ. The second one is the site selectivity of the FC reaction. The para and ortho positions of the methoxyl substituent are in competition in this reaction.190,191 Hence, the usual phenol oxidation hypervalent iodine(III) reagents, for example iodobenzene diacetate (PIDA), were considered and PIDA in hexafluoroisopropyl alcohol (HFIP) resulted in the FC product in 70% yield. The selectivity of the ortho/para ratio was found to be 2.6/1. The ortho FC product, so-called gymnothelignan N 170, could be extracted using careful flash chromatography employing dichloromethane/methanol as the eluent system. Thus, the total synthesis of gymnothelignan N 170 was achieved upon several steps from 5-hydroxyvanilin 171 in 6.7% overall yield (Scheme 38).192
 | ||
| Scheme 38 Total synthesis of gymnothelignan N 170. | ||
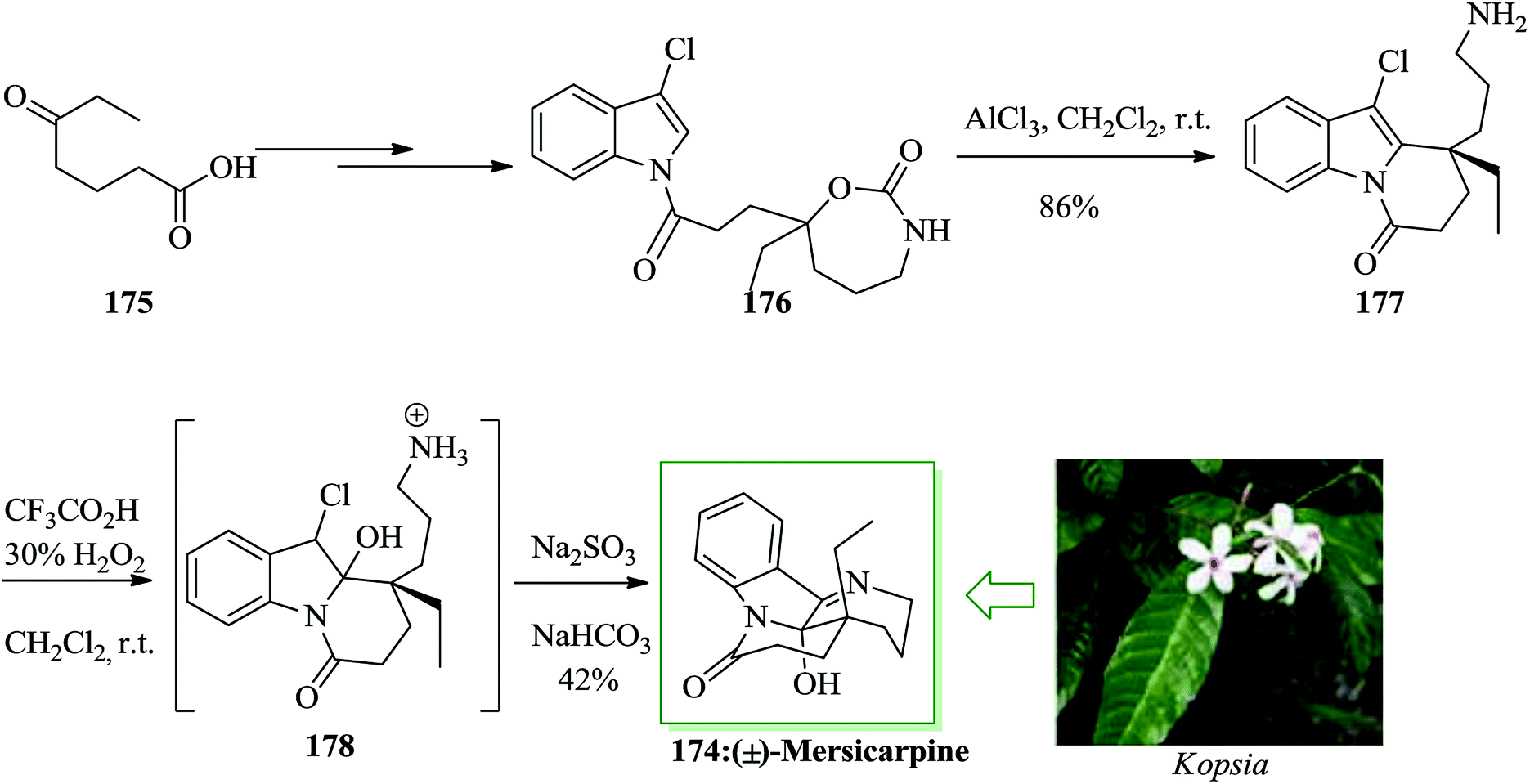
Mersicarpine 174, a structurally fascinating monoterpene indole alkaloid, was extracted from the Kopsia species of plants by Kam and co-workers in 2004.193 This uncommon tetracyclic natural product contains a typical seven-membered cyclic imine and a δ-lactam around a completely functionalized hemiaminal stereogenic center. A short total synthesis of mersicarpine 174 was accomplished using a cyclic carbamate for the construction of a tertiary carbocation. The main step includes intramolecular FC alkylation with this carbocation for the formation of a quaternary carbon center and a subsequent oxidation and cyclization cascade for the generation of a seven-membered cyclic imine. This approach permitted for a rapid synthesis of mersicarpine from a simple intermediate employing straightforward chemical operations in a one-pot fashion. The total synthesis of mersicarpine 174 was initiated from compound 175, which afforded compound 176 upon several steps. The key FC alkylation reaction was employed in the total synthesis of mersicarpine. In this route, compound 176 was reacted with AlCl3, a mixture of trifluoroacetic acid and H2O2 via FC reaction to provide the product 177 in 86% yield. Upon oxidation of 177 and acidity optimization with Na2SO3/NaHCO3, mersicarpine 174 was provided in 42% yield. As a result, the natural product was obtained in 25% overall yield (Scheme 39).194
 | ||
| Scheme 39 Total synthesis of mersicarpine 174. | ||
Naturally occurring diarylheptanoids, a group of secondary plant metabolites,195,196 exhibit active biological properties, for example anticancer, antioxidant, antibacterial, antiosteoporosis, antifungal and antihepatotoxicity activity.195,196 Among them, musellarins A–C represent an uncommon structural group owing the presence of the rare bicyclic tetrahydropyran scaffold. Musellarin A was extracted in 2002 by Kinghorn197 and co-workers from hybrid plant fruits of Musa x paradisiaca in Peru.197,198 In 2014, Tong and co-workers reported the first total syntheses of musellarins A–C in 15–16 steps.199 The key synthetic features are an Achmatowicz rearrangement, a Kishi reduction, and FC cyclization to make the tricyclic scaffold and Heck coupling reaction of aryldiazonium salts to introduce the aryl substituent into the dihydropyran in a 2,6-trans method in the last step of the synthesis. Total syntheses of musellarins A–C were started from isovanillin 180, which was converted into dihydropyranone hemiacetal 181. Kishi reduction of 181 with a combination of trifluoroacetic acid and triethylsilane afforded a 2:1 mixture of dihydropyranone 182 and a surprising FC cyclization adduct 183 in 78% combined yield. In this step the intramolecular FC cyclization reaction of dihydropyranone 182 happened in the presence of BF3·Et2O to give the tricyclic compound 183. Compound 183, after several steps, afforded the key intermediate enol ether 184. Finally, compound 184 was converted into musellarins A–C in 88%, 70% and 80% yields upon several steps (Scheme 40).199
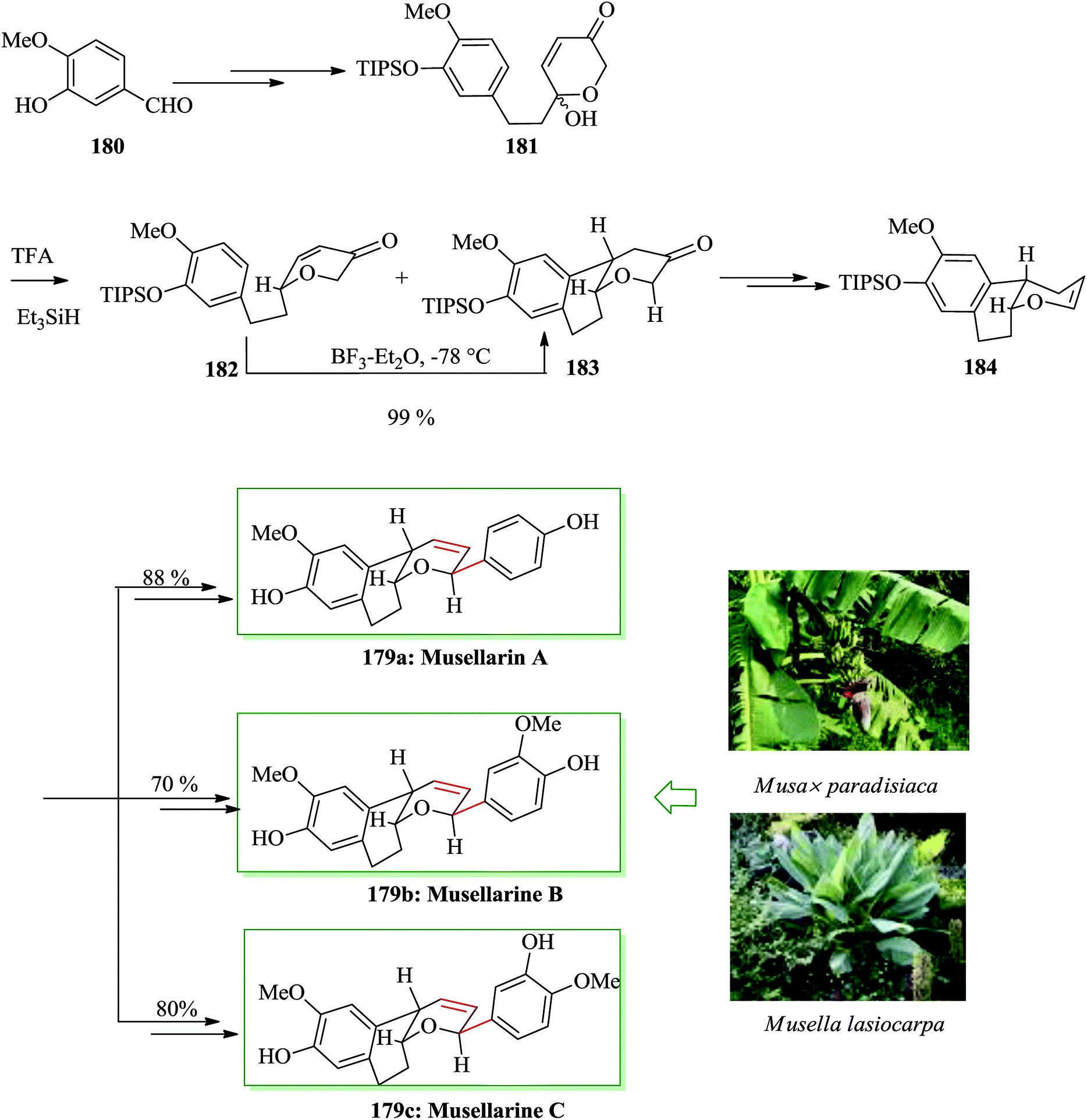 | ||
| Scheme 40 Total synthesis of musellarins A–C 179a–c. | ||
Polycyclic pyrrole-imidazole alkaloids (PIAs) are oroidin-obtained marine naturally occurring compounds200,201 with various and complex frameworks. Most of these alkaloids exhibit multiple biological properties, including immunosuppressive, antitumor and adrenoceptoragonistic properties.202,203 Nagasawa and co-workers in 2014 developed a total synthesis of (+)-cylindradine A through intramolecular FC-type cyclization reaction of pyrrole-aldehyde and oxidative cyclization of tricyclic pyrrolopyrrolidine–guanidine with hypervalent iodine to make the cyclic guanidine structure involving the N,N-aminal scaffold.204 The total synthesis of cylindradine A was initiated from oroidin 186, which was converted into 187 in several steps. Compound 187 containing aldehyde and 4-carbamoylpyrrole functional substituents gave compound 188 via an intramolecular FC-type reaction. Next, compound 188 was converted into 189 after several steps. The FC adduct 190 was provided in 82% yield as a diastereomeric mixture in a ratio of ca. 7:1 (190a:190b) via reaction of 189 with camphorsulfonic acid (CSA). Subsequently, (+)-cylindradine A 185 was provided from compound 190a in 58% yield upon several steps (Scheme 41).204
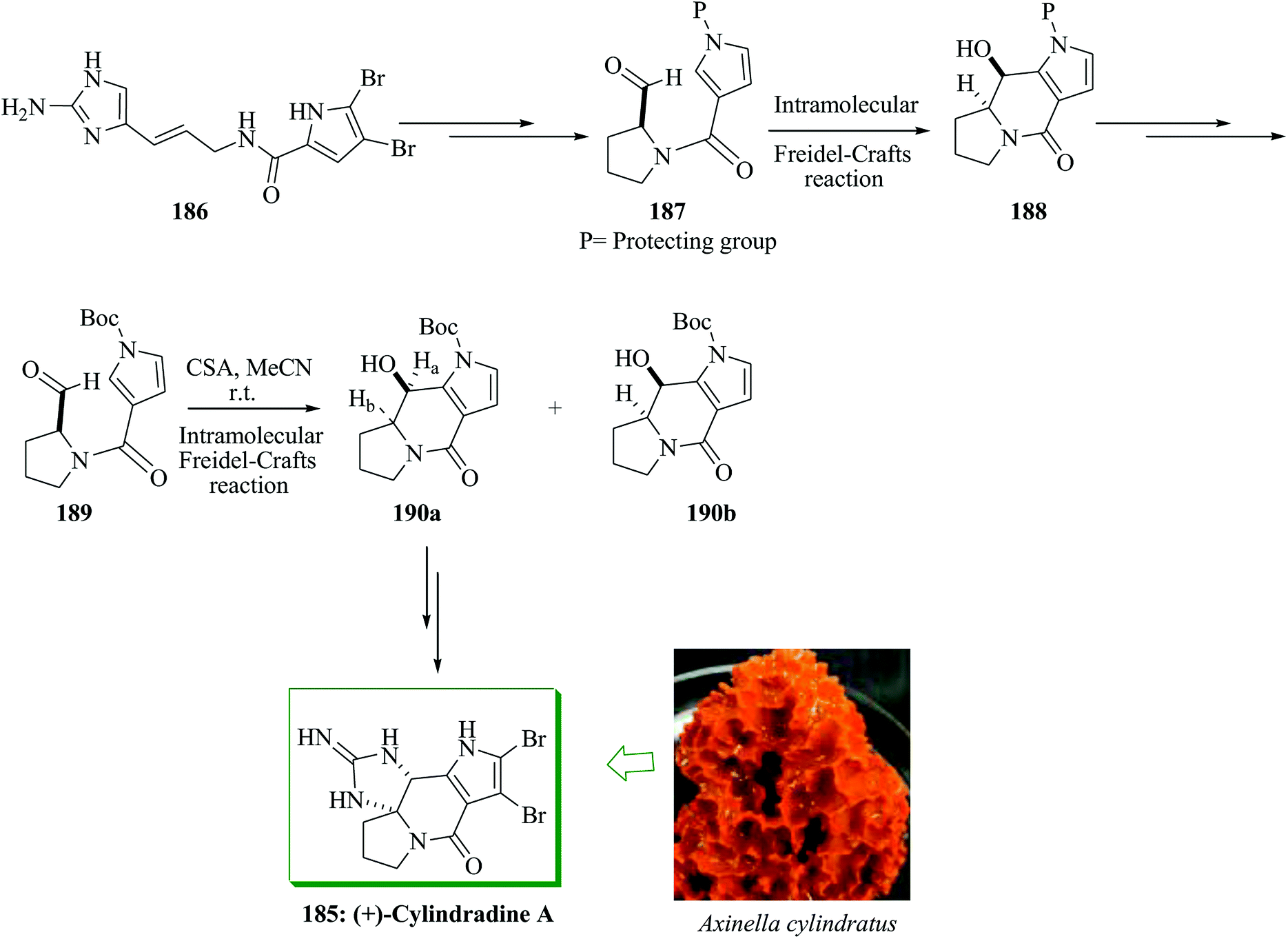 | ||
| Scheme 41 Total synthesis of (+)-cylindradine A 185. | ||
Haouamines were extracted from a tunicate, Aplidium haouarianum, on the southern coast of Spain by Zubía and co-workers in 2003.205 Structurally, haouamines have unique features, for example the cis-fused indeno-tetrahydropyridine and 3-aza-[7]-paracyclophane including a bent aromatic ring. The enantiocontrolled total synthesis of (−)-haouamine B pentaacetate 192 was achieved from 3,4-dimethoxyphenol 193 in 40 steps in 0.055% overall yield with an average 83% yield for every stage. Ellman's diastereoselective Mannich reaction and the scandium(III) triflate-catalyzed intramolecular FC alkylation were applied to make the uncommon indane-fused β-lactam 196. In this route, an intramolecular McMurry coupling reaction was considered as the key step. Tokuyama and co-workers initiated the synthesis of (−)-haouamine B from 3,4-dimethoxyphenol 193, which after several steps was transformed into substrate 194. To avoid the unanticipated cyclization, the benzyl group was changed to the more bulky TIPS substituent with the intention of reducing the nucleophilicity of the oxygen atom and a range of Brønsted/Lewis acids were explored. Upon hydrogenolysis of the benzyl ether, introduction of the TIPS substituent followed by mesylation of the tertiary alcohol gave substrate 195 for the FC alkylation. Compound 195 gave indeno-β-lactam 196. The reaction was accomplished using 2,6-di-tert-butylpyridine (DTBPy). The corresponding compound 196 was provided in 80% yield with the labile TIPS substituent remaining intact. Finally, compound 196 afforded (−)-haouamine B 191 and haouamine B pentaacetate 192 after several steps (Scheme 42).206
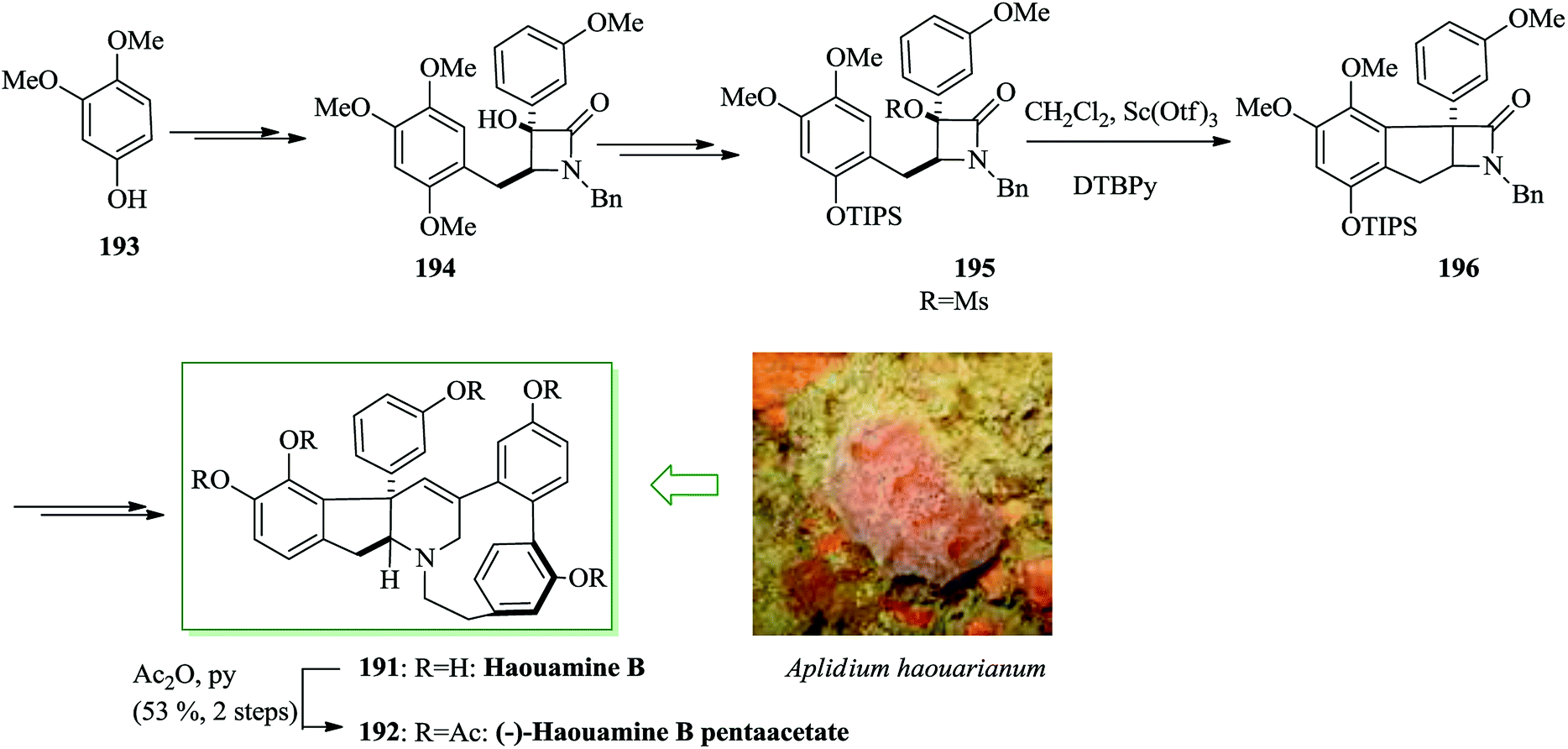 | ||
| Scheme 42 Total synthesis of (−)-haouamine B 191 and haouamine B pentaacetate 192. | ||
Gibberellin207 and ent-kaurane derivatives,208 are naturally occurring diterpene compounds,209 having a complex tetracyclic molecular framework presenting a bicyclo[3.2.1]octane system.210,211 Both gibberellins and ent-kauranes contain remarkable biological properties. Notably, the gibberellins were found to be plant hormones and growth regulators.212–214 Gibberellins were initially extracted from the fungus Gibberella fujikuroi and ent-kaurane diterpenoids were also extracted from the whole plant Sideritis congesta P. H. Davis & Hub.-Mor.215
Malachowski and co-workers in 2015 developed an approach for the formation of tricarbocyclic scaffold via an extension of the asymmetric Birch–Cope sequence along with intramolecular FC alkylation reaction.216 An asymmetric method for the synthesis of the tetracarbocyclic framework of the gibberellins 197a or the ent-kauranes 197b was demonstrated by the same group.216 There is a tetracarbocyclic ring system in the unit of both the gibberellins and the ent-kauranes 197b. The asymmetric formation of tetracyclic diterpene frameworks began from the 5-iodo-salicylate derivative 198. Upon several steps, compounds 199a and 199b were produced. Conversion of these products, 199a and 199b, into the tetracarbocyclic scaffold of the gibberellins 197a or ent-kauranes 197b was achieved via FC alkylation in the presence of BF3·Et2O, which was clearly one of the most charming conjugate additions of an aromatic nucleophile to a cyclohexenone electrophile.217,218 The cyclization happened to generate the cis isomers 200a and 200b, but most of the products were isolated as C-2 epimers, usually as a 1:1 mixture. Finally, the tetracarbocyclic framework of the gibberellins 197a or ent-kauranes 197b was obtained from 200a and 200b upon several steps (Scheme 43).216
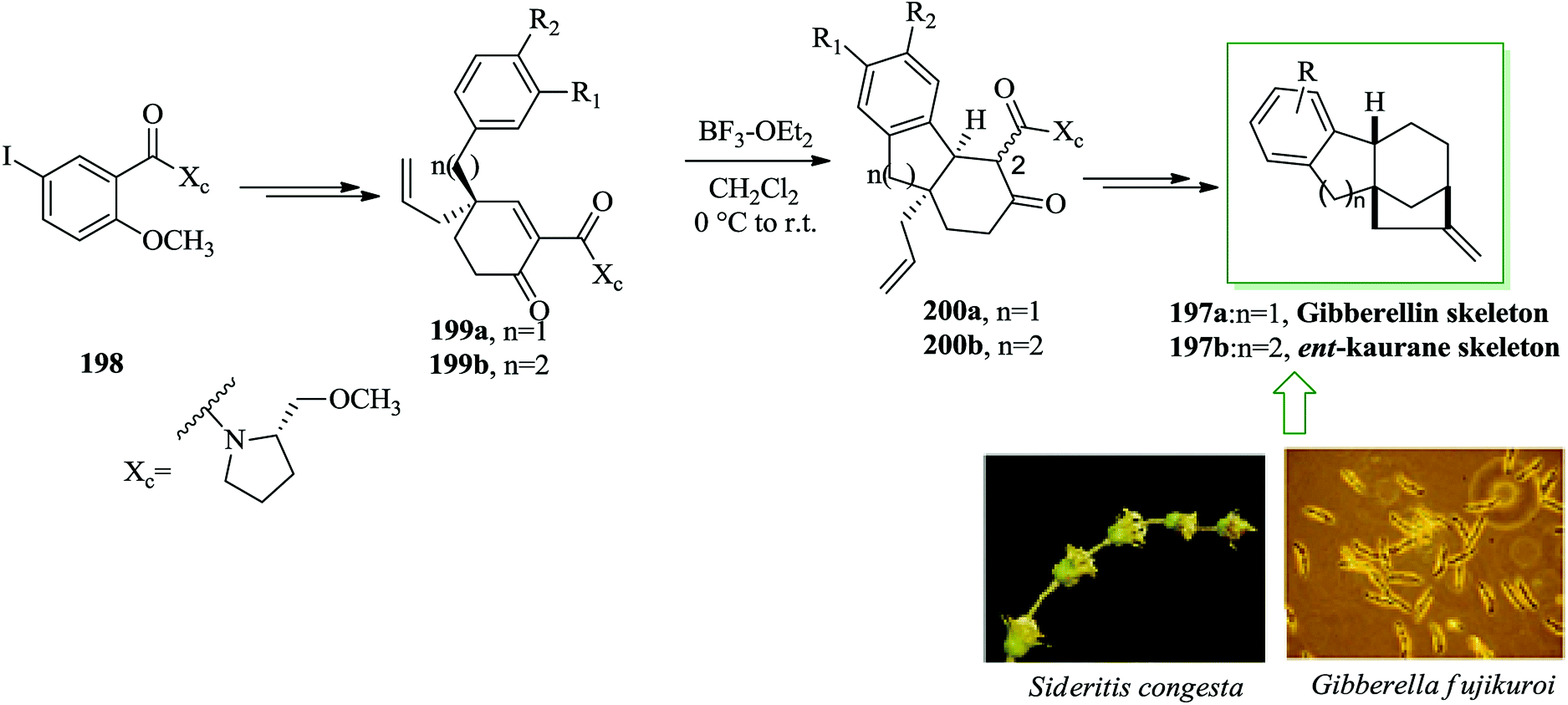 | ||
| Scheme 43 Total synthesis of gibberellins 197a or ent-kauranes 197b. | ||
The alstoscholarisines219 are some fascinating monoterpenoid indole alkaloids having a complex pentacyclic structure extracted from Alstonia scholaris by Luo and co-workers. In 2016, Liang and co-workers reported an asymmetric total synthesis of (−)-alstoscholarisine A 201 in 10 steps.220 This group demonstrated a short and highly asymmetric total synthesis of (−)-alstoscholarisine A 201, an extracted monoterpenoid indole alkaloid having important bioactivity in promoting adult neuronal stem cells proliferation. A highly asymmetric (99% ee), intramolecular iridium-mediated FC alkylation reaction between indole 204 and a secondary allylic alcohol was used to create the first stereogenic center upon which the other three contiguous chiral centers were easily set through an extremely enantioselective tandem 1,4-addition and aldol reaction. Firstly, an acylation reaction between 3-methylindole 202 and 4-vinylbutyrolactone 203 was achieved using trimethylaluminium,221 providing secondary allylic alcohol 204 in 75% yield. Catalytic enantioselective FC alkylation reaction of 204 with the combination of [{Ir(cod)Cl}2], (R)-carreira ligand222–224 and Lewis acid scandium(III) triflate effectively afforded tricycle 205 in 75% yield with 99% ee. The latter was converted into (−)-alstoscholarisine A 201 in several steps (Scheme 44).220
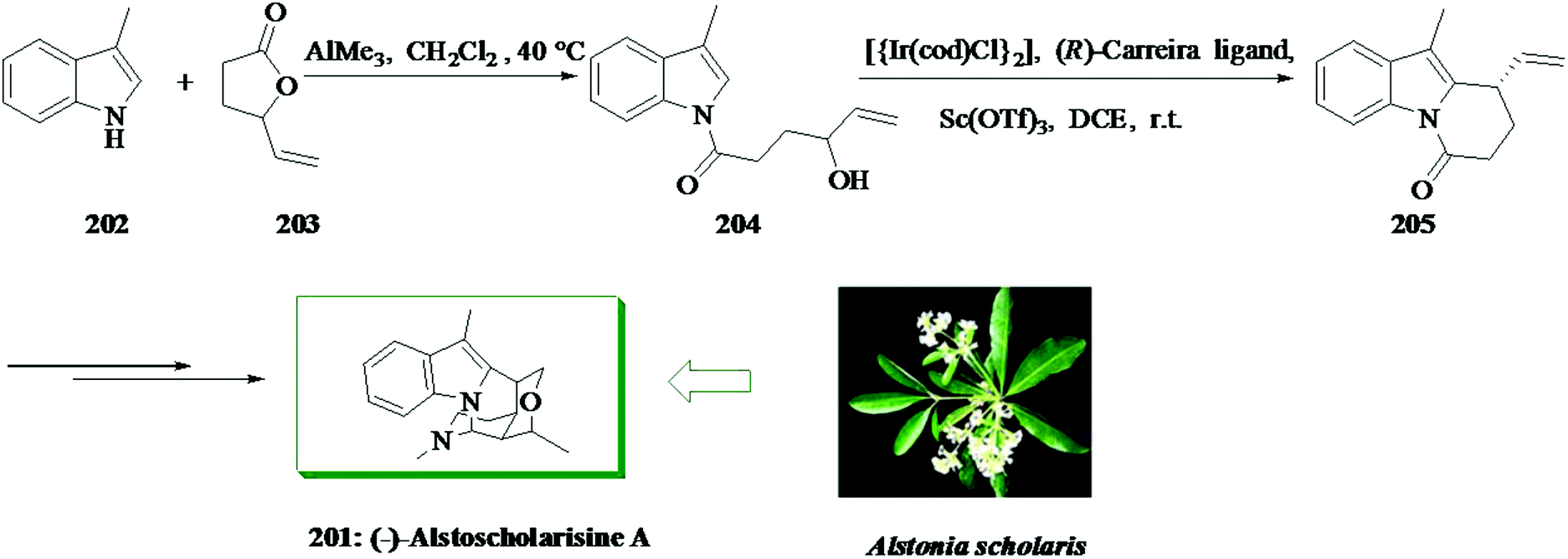 | ||
| Scheme 44 Total synthesis of (−)-alstoscholarisine A 201. | ||
Terpenoids are structurally intriguing molecules with fascinating biological properties.225 For instance, mulinane diterpenoids, initially extracted in 1990 from the Chilean shrub Mulinum crassifolium, have been applied as traditional folk medicine for the treatment of illnesses, for example, diabetes and bronchial and intestinal disorders.226–232 Enantioselective total synthesis of mulinane diterpenoids has been accomplished. In this strategy, an intramolecular FC reaction and Birch reduction were considered as the key steps. Based on this pathway, Xie and co-workers in 2017 reported the divergent asymmetric total synthesis of seven mulinane diterpenoids, namely, 13-epi-mulinolic acid (210, 12 steps, 18% overall yield), mulin-11,13-dien-20-oic acid (211, 13 steps, 17% yield), mulinic acid (214, 14 steps, 11% yield), isomulinic acid (217, 15 steps, 8.6% yield), 16-hydroxy mulin-11,13-dien-20-oic acid (213, 14 steps, 15% yield), mulin-11,13-dien-20-ol (215, 13 steps, 15% yield), and 20-hydroxy mulin-11,13-dienyl acetate (216, 14 steps, 14% yield), along with two analogues, namely, 13-epi-mulinol (212, 12 steps, 17% yield) and 13-epi-mulinolic acid ethyl ester (209, 11 steps, 20% yield), from easily accessible cyclopentenone 206.236
Total synthesis began from 3-isopropyl-2-(ethoxycarbonyl)cyclopentenone 206. Compound 206 gave C-alkylation product 207a in 65% yield, together with a 20% yield of the unanticipated O-alkylation product 207b in several steps. Reaction of 207a with methanesulfonic acid led to an intramolecular FC reaction that afforded tricyclic compound 208 in 91% yields. Subsequently, compound 208 gave 13-epi-mulinolic acid ethyl ester 209 upon several steps. Using compound 209, the same group accomplished the enantioselective total synthesis of mulinane diterpenoids through late-stage functional modification or functionalization. Therefore, the synthesis of seven mulinane diterpenoids from the “biogenic precursor” 209 was initially achieved in several steps. The latter was then converted to various natural products, 13-epi-mulinolic acid 210,233 mulin-11,13-dien-20-oic acid 211,234 mulinic acid 214226 isomulinic acid 217,226 16-hydroxymulin-11,13-dien-20-oic acid 213,235 13-epi-mulinol 212, mulin-11,13-dien-20-ol 215229 and 20-hydroxy mulin-11,13-dienyl acetate 216 (Scheme 45).236
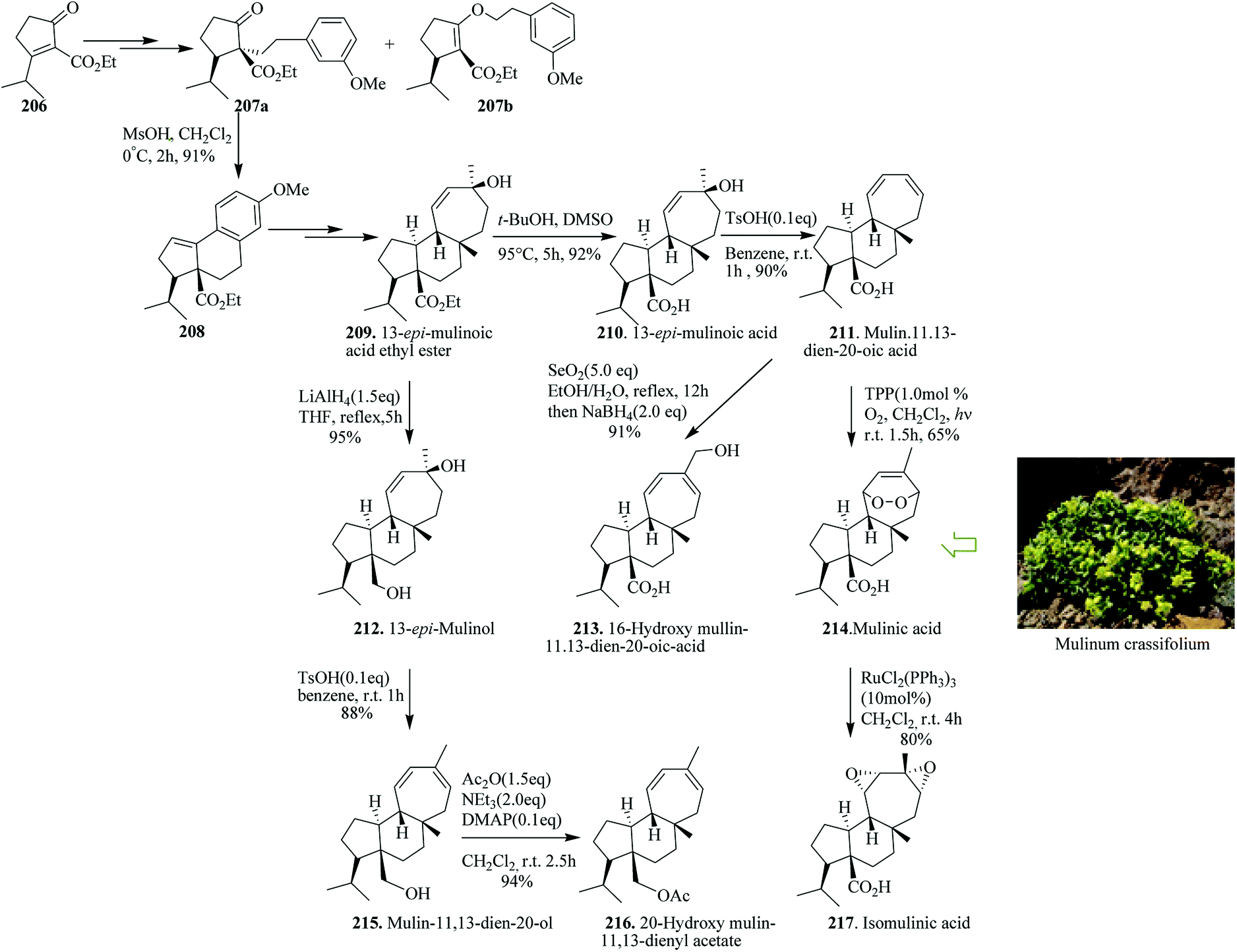 | ||
| Scheme 45 Total synthesis of 13-epi-mulinolic acid ethyl ester 209, 13-epi-mulinolic acid 210, mulin-11,13-dien-20-oic acid 211, 13-epi-mulinol 212, 16-hydroxymulin-11,13-dien-20-oic acid 213, mulinic acid 214 mulin-11,13-dien-20-ol 215, 20-hydroxy mulin-11,13-dienyl acetate 216 and isomulinic acid 217. | ||
Similisines A [(+)-218a] and B [(−)-218a], extracted from Laurencia similis in 2013, are the first example of polybrominated spiro-trisindole alkaloids fused to a five-member ring.237 Similisines A and B are trisindole alkaloids that feature a unique polybrominated [5,5] spirooxindole scaffold fused to a five-membered ring. The first total synthesis of similisines A and B, a pair of unprecedented polybrominated spiro-trisindole enantiomers and all their stereoisomers was achieved in 6 steps from 5,6-dibromoindole 219. In this method, an intramolecular FC cyclization was considered as the key step. Total synthesis of similisines A, B and their stereoisomers started with the construction of trisindole 220, which was obtained from 5,6-dibromoindole 219 upon several steps.238 The five-membered spirocyclic framework of 221 was generated from trisindole 220 via intramolecular FC cyclization. Treatment of 220 with HCl in methanol under microwave irradiation (MWI) provided two racemic isomers (±)-221a (8% yield) and (±)-221b (8% yield), along with the unexpected trisindole 222 in 10% yield. Although the other two corresponding racemic isomers (±)-221c and (±)-221d were detected only in trace amounts, when trisindole 220 was reacted with p-TsOH in DCE under reflux, racemic isomers (±)-221c and (±)-221d were provided in 7% and 16% yields, respectively, along with trace amounts of racemic (±)-221a and (±)-221b. In this case, the unanticipated trisindole 222 was also provided in 10% yield. Therefore, both the racemic stereoisomers (±)-221a–(±)-221d could be obtained via two above-mentioned different conditions. Using the key (±)-221a–(±)-221d, the synthesis of similisines A, B and their stereoisomers was accomplished. Bromination of (±)-221a–(±)-221d with pyridinium tribromide239,240 afforded the corresponding racemic compounds of (±)-218a (85%), (±)-218b (81%), (±)-218c (85%), and (±)-218d (83%), respectively. On the other hand, (±)-218a was identified to be a racemic mixture of natural similisines A and B (Scheme 46).237
 | ||
| Scheme 46 Total synthesis of racemic compounds of (±)-218a, (±)-218b, (±)-218c, and (±)-218d. | ||
2.2. Intermolecular alkylations
Brasiliquinones A–C, unique cytotoxic benz(a)anthraquinones usually found as angucyclines, were initially extracted from pathogenic species of Nocardia. Most of the angucycline antibiotics contain a Me substituent at C-3 while brasiliquinones A–C contain an Et substituent at C-3 providing a unique class of angucyclines. Brasiliquinones B and C are more effective than brasiliquinone A against L I210 tumor cells.241 Brasiliquinone B 223 has been obtained from 7-methoxy-L-tetralone in several steps making use of an FC alkylation reaction as a main stage. Initially, 7-methoxy-L-tetralone 224 gave the relevant tetralin derivative 226 after several steps. FC alkylation reaction of 226 with 3-bromo-4-methoxyphthalide 225 using SnCl4 gave the lactone 227 regiospecifically. Finally, compound 227 provided (+)-brasiliquinone B 223 upon several steps (Scheme 47).242 Deshpande and co-workers in 2002 also used this method for the synthesis of (±)-brasiliquinone B 223.243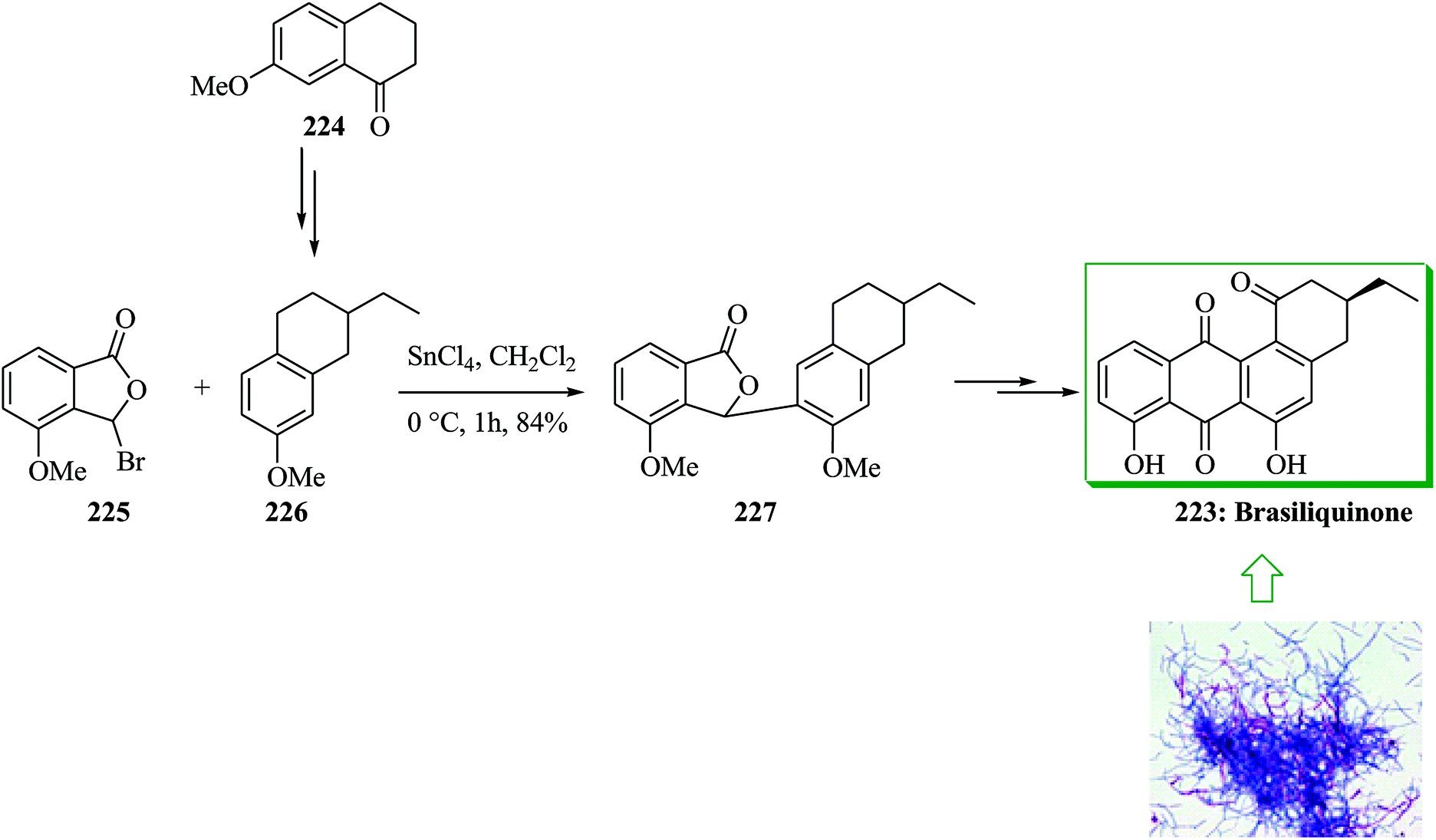 | ||
| Scheme 47 Total synthesis of brasiliquinone B 223. | ||
Methyl 3-(2,4,5-trimethoxyphenyl)propionate 228 has been extracted from the root bark of Cordia alliodora, demonstrating excellent larvicidal and antifungal properties in biological tests.244 Tamariz et al. in 2004 developed a one-step synthesis of the antifungal and larvicidal natural product methyl 3-(2,4,5-trimethoxyphenyl)propionate 228 by the reaction between 1,2,4-trimethoxybenzene with 230 under MWI.245 Even though methyl acrylate 230 failed to afford the desired product 228 under these conditions, the use of sym-tetrach loroethane246 as the solvent at 80 °C for 1 week afforded 228 in 37% yield. The reaction time was shortened to 8 h and the yield improved using MWI (200 W) with the same mixture in a Teflon screw-capped glass tube at 80 °C, affording 228 in 66% yield. The construction of 228, at the present time, is the shortest synthesis of this biologically potent natural product, since the previously reported synthesis, beginning from β-asarone or asaraldehyde, provided 228 in three steps (Scheme 48).247
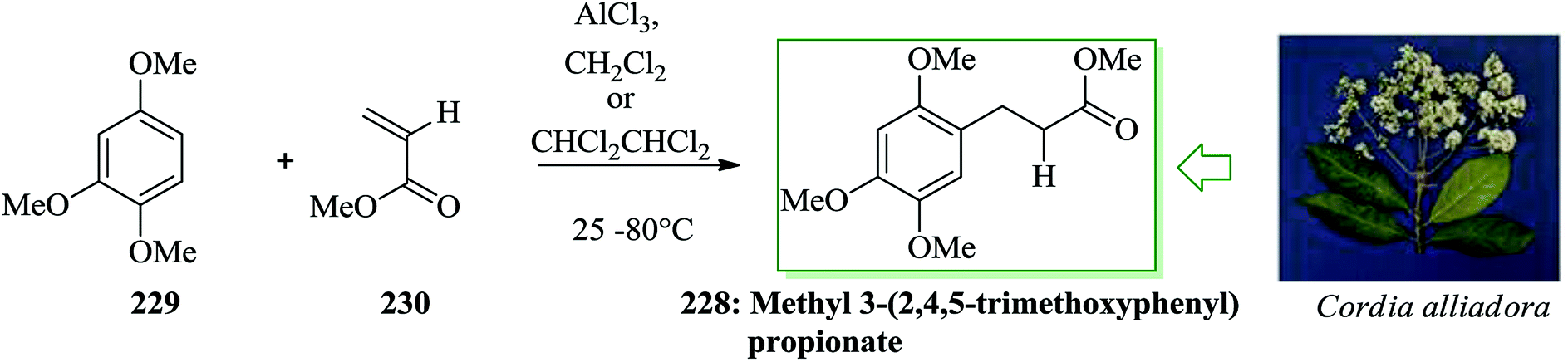 | ||
| Scheme 48 Total synthesis of methyl 3-(2,4,5-trimethoxyphenyl)propionate 228. | ||
Podophyllotoxin 231 and its derivatives are significant members of the lignan group of naturally occurring compounds.248 The biological properties of podophyllotoxin have generated strong attention in the development of synthetic pathways to this natural product.249,250 Bach and co-workers in 2008 reported a six-step total synthesis of enantiomerically pure (−)-podophyllotoxin starting from the Taniguchi lactone 203 in 35% overall yield, using iron(III)-mediated FC alkylation as a key step.251 The synthesis was started from the Taniguchi lactone 203, which is accessible in enantiomerically pure form from 2-butyne-1,4-diol in two steps with a subsequent conventional resolution252 or in six steps via an asymmetric iridium-mediated allylation.253 An aldol reaction with aldehyde 232 gave 233 with high stereoselectivity with respect to the stereogenic center at the α position of the lactone. Compound 233 was reacted with 234 (X = OH) under the optimized reaction conditions (FeCl3 in dichloromethane) to afford the FC alkylation reaction product 235 in 99% yield. Finally, alcohol 233 afforded (−)-podophyllotoxin 231 upon several steps. This synthesis demonstrated that a stereogenic center in the β position to an ester or lactone scaffold can be generated diastereoselectively via a Lewis acid-mediated SN1 reaction if a stereogenic center is already present in the α position (Scheme 49).251
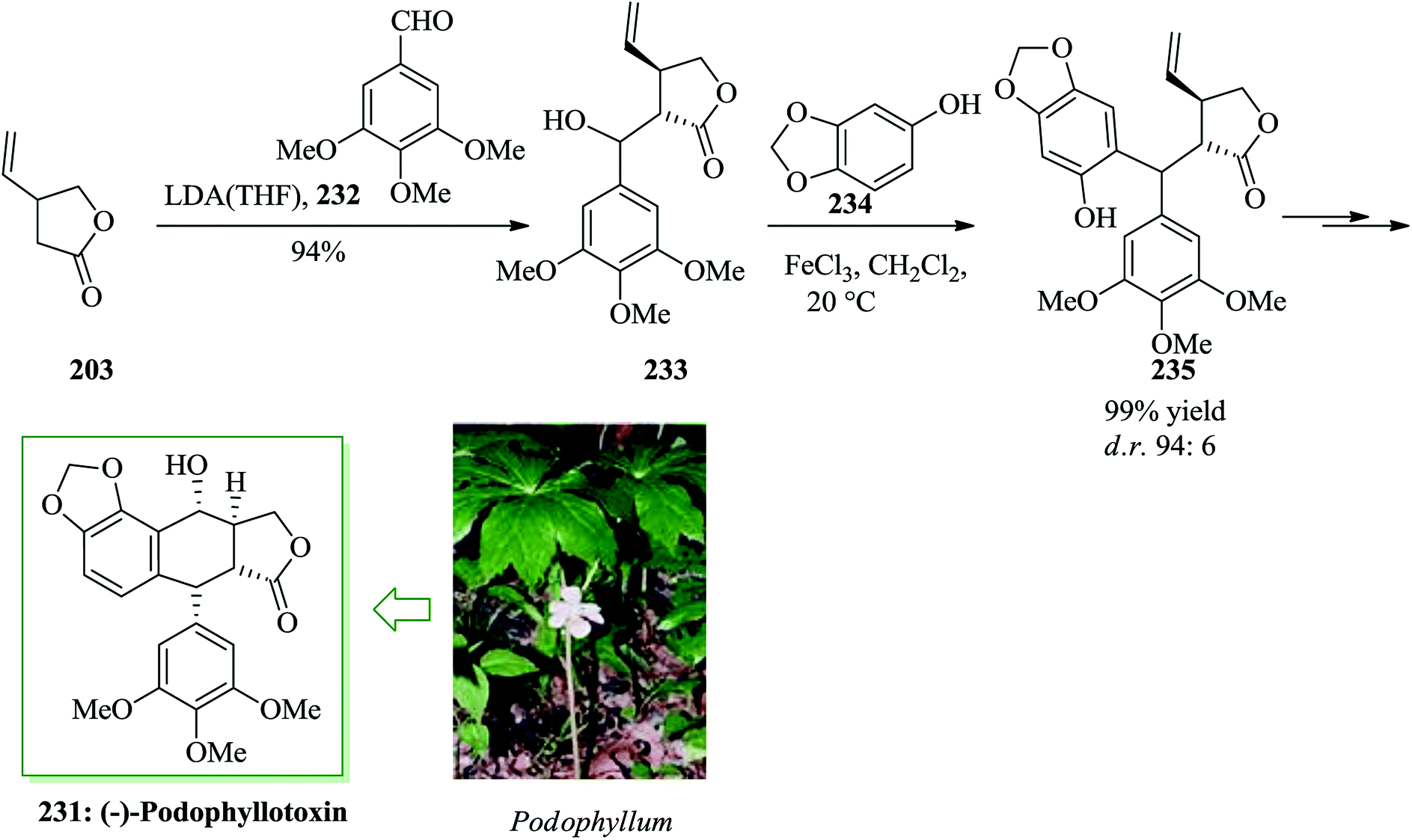 | ||
| Scheme 49 Total synthesis of (−)-podophyllotoxin 231. | ||
Haplophytine 236 is the major indole alkaloid known in the dried leaves of the Mexican plant Haplophyton cimicidum and was first extracted by Snyder et al. in 1952.254–256 The right-half segment is a hexacyclic aspidosperma alkaloid group, so-called aspidophytine, that is provided by the acidic degradation of (+)-haplophytine.257 Fukuyama and co-workers in 2009 reported the initial total synthesis of (+)-haplophytine 236 demonstrating the simple assembly of tricyclic ketone 238 via the intramolecular Mannich reaction. FC alkylation reaction and Fischer indole synthesis were considered as the key steps. For the synthesis of (+)-haplophytine 236, initially, tricyclic ketone 238 was synthesized from 237 after several steps. Using the key tricyclic ketone 237, this group attempted the synthesis of the left-hand segment. 7-Benzyloxyindole 239 was converted into iodoindolenine 240 upon several steps. Next, the FC alkylation reaction progressed easily and provided the corresponding coupling product 242 as the major isomer in nearly a 2.4:1–2:1 diastereoselectivity. In the following, compound 242 afforded 243 after several steps. Finally, hydrazine 243 and ketone 239 provided (+)-haplophytine 236 upon several more steps (Scheme 50).258
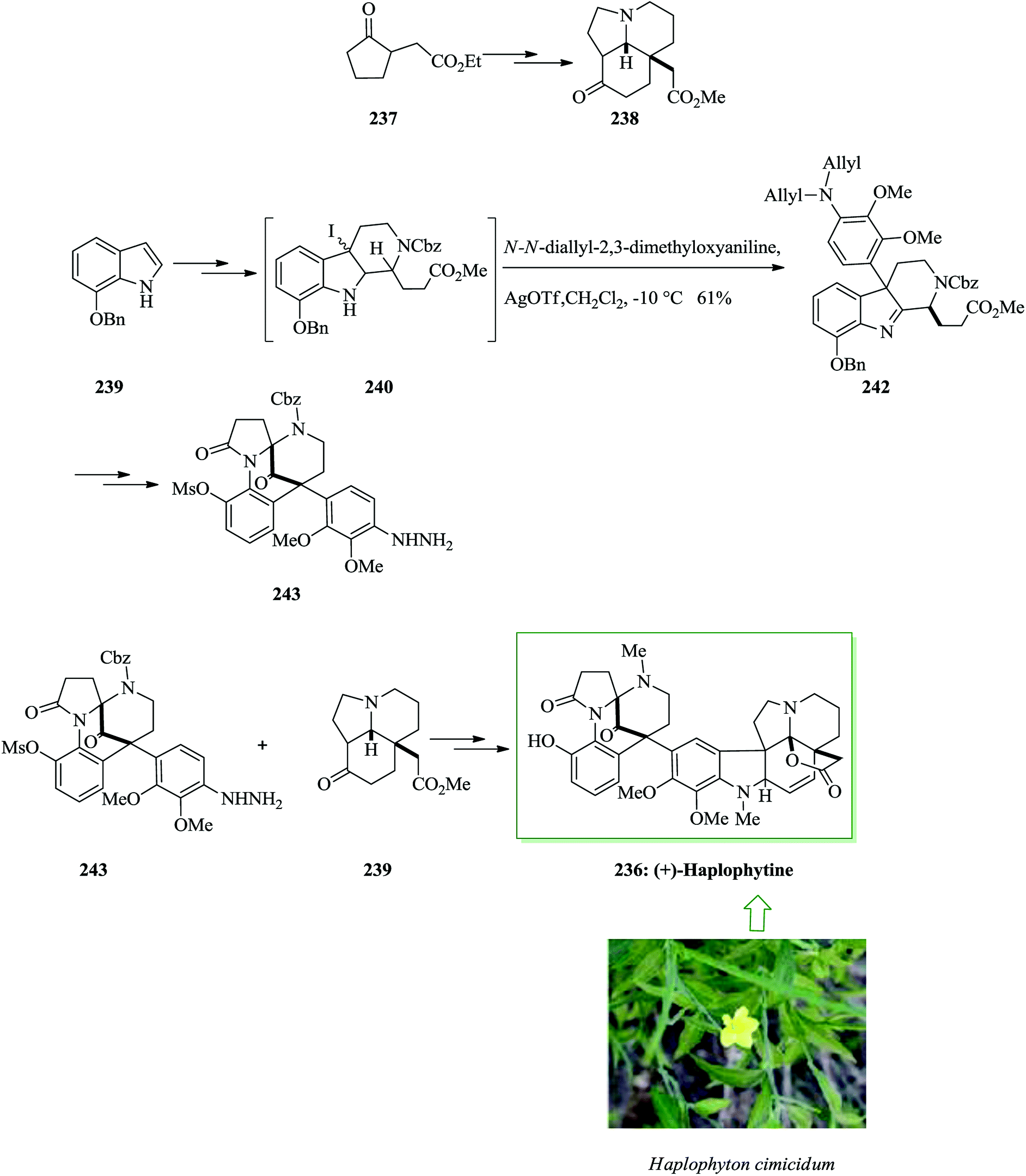 | ||
| Scheme 50 Total synthesis of (+)-haplophytine 236. | ||
Polyphenolic natural products represent a large and growing group of structurally varied compounds exhibiting an extensive series of biological properties.259,260 In 2006, Tan and co-workers revealed the structure and cytotoxic activities against a panel of selected cancer cell lines of hopeanol 245, a polyphenol secondary metabolite extracted from the bark of Hopea exalata.261 A subsequent investigation of H. hainanensis led to the isolation of the structurally related hopeahainol A 244. The total synthesis and biological evaluation of the resveratrol-obtained naturally occurring compounds products hopeanol 244 and hopeahainol A 245 in their racemic and antipodal forms were carried out. In this method, an intramolecular FC reaction and a Grob-type fragmentation were considered as the key steps. It was examined for building the quaternary center of hopeahainol A 244 and hopeanol 245 with appropriate appendages. In the first incursion, the synthesis of the simple triaryl methyl ester 250 via an intermolecular FC-type reaction containing tertiary alcohol 247 as the main substrate and phenol 249 as the external nucleophile was attempted. Therefore, dimethyl oxalate 246 provided the tertiary alcohol methyl ester 247 in 66% overall yield. Satisfyingly, treatment of a solution of tertiary alcohol 247 and phenol 249 in dichloromethane in the presence of excess p-TsOH·H2O resulted in the construction of the desired product 250 in high yield (97%), probably via the intermediacy of carbocation 248 (Scheme 51a). Then, the feasibility of synthesizing the more relevant and advanced model system 257 was attempted. In this case, the corresponding tertiary alcohol methyl ester substrate 253 was synthesized from methyl glyoxalate derivative 251 by addition of Grignard reagent 252 (81% yield). The latter underwent a smooth FC-type reaction with resorcinol 255 providing the desired tetracyclic model system 257 (90% yield), probably via transient intermediate carbocation 254 and diphenolic methyl ester 256. The latter apparently undergoes spontaneous lactonization under the optimized reaction conditions (Scheme 51b).141
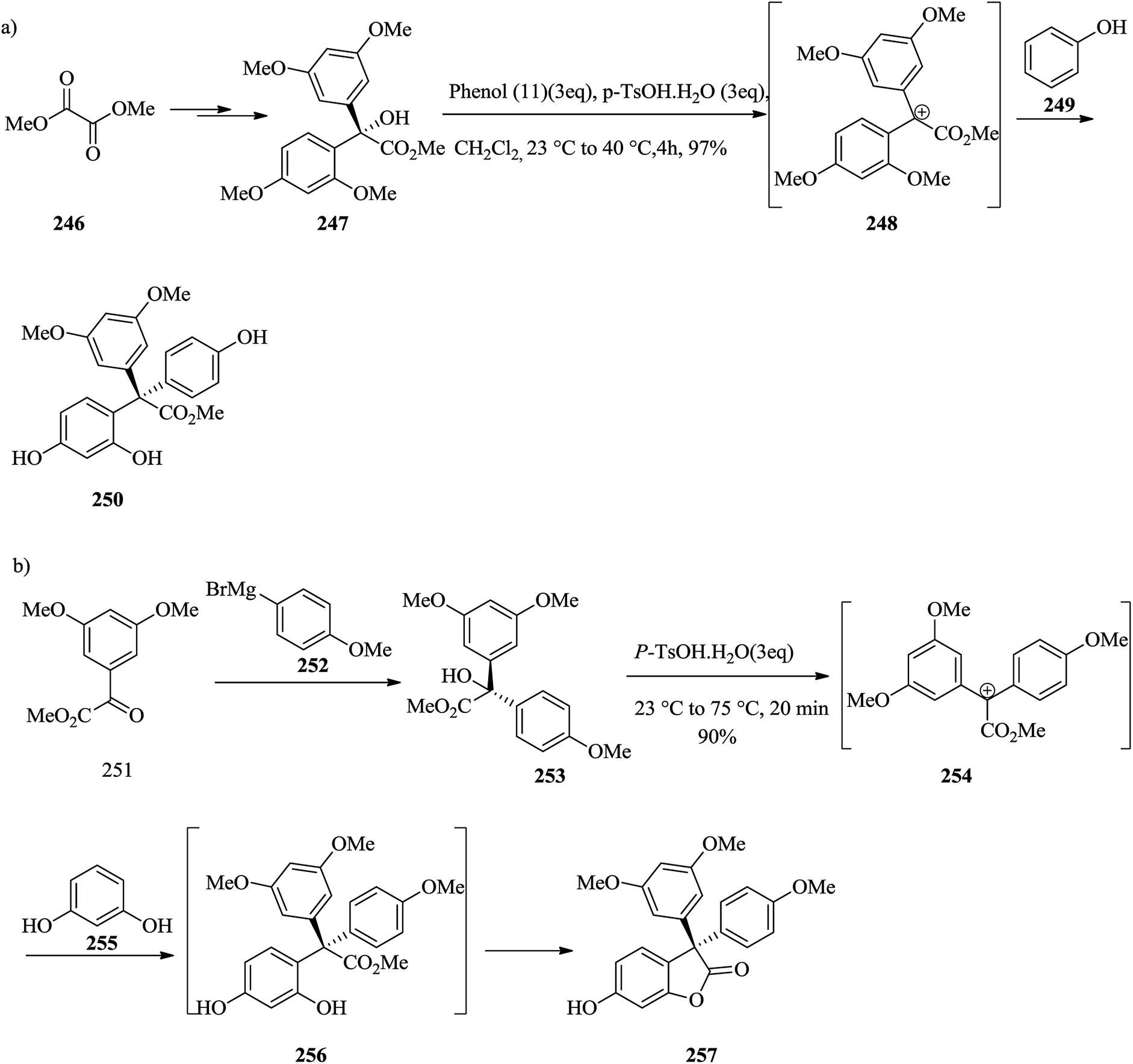 | ||
| Scheme 51 Total synthesis of triaryl methyl ester 250 and tetracyclic model system 257. | ||
The successful formation of model system 257 (comprising four of the six ring systems of hopeahainol A) provided motivation for the next logical step, which was to attempt to use an established approach to assemble the entire carbon framework of hopeahainol A 244. Using protected nucleophilic partner 258 in the FC reaction with tertiary alcohol methyl ester 253 gave regioisomeric product 260 in 80% yield (Scheme 52).141
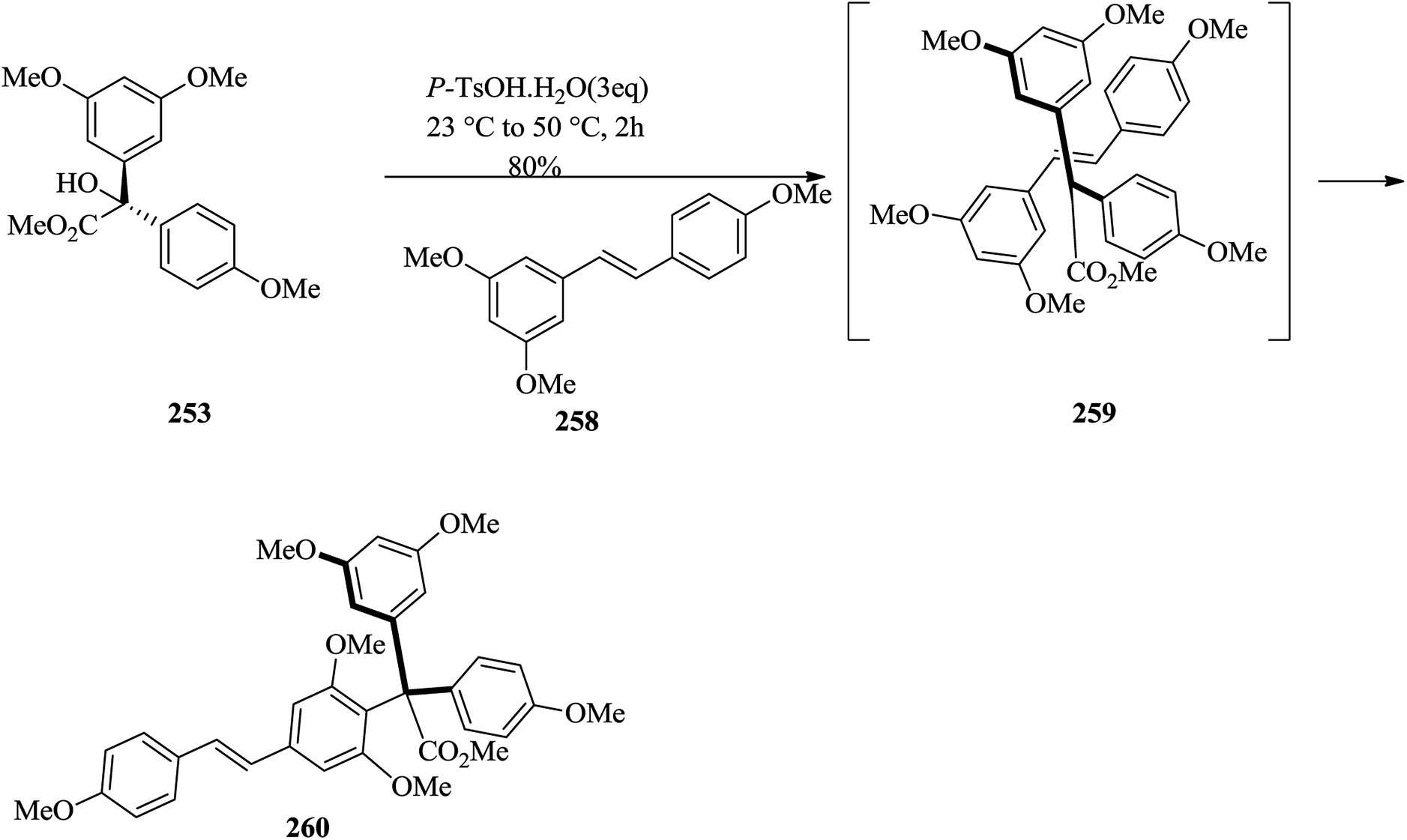 | ||
| Scheme 52 The formation of compound 260. | ||
Next, a simple model study directed toward the formation of model system 263 was designed. Thus, methyl glyoxalate 251 provided tertiary alcohol 261 upon several steps. Treatment of the latter with BF3·Et2O in dichloromethane led to the construction of the corresponding FC reaction product 263 in 86% yield. It is suggested that compound 263 probably formed via generation of intermediate 262 (Scheme 53).141
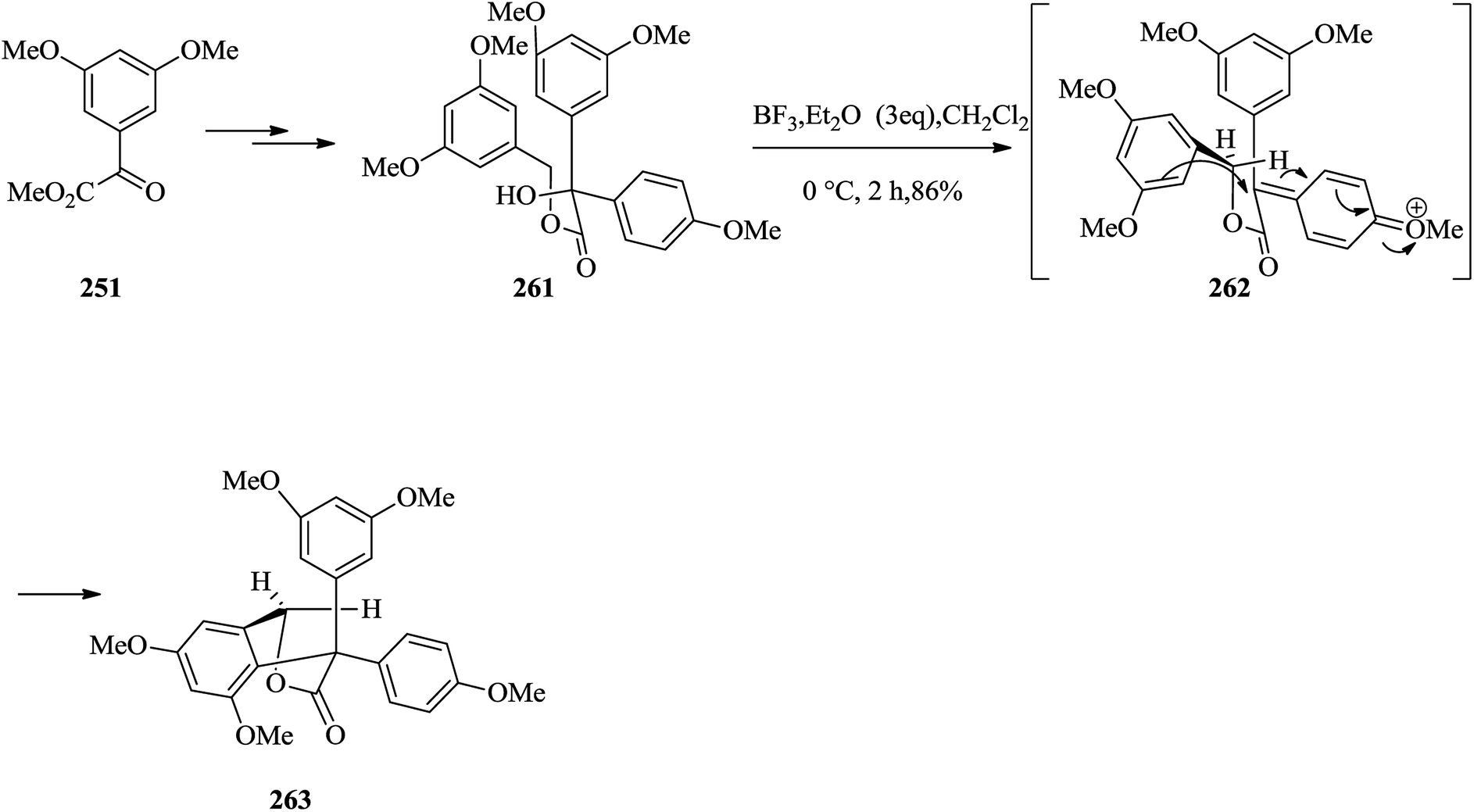 | ||
| Scheme 53 The formation of product 263. | ||
Then, the next phase of the campaign was provided by the initial formation of the diphenolic γ-lactone 268 and/or 269 via hydroxyester 265. The aldehyde 264 provided diphenolic hydroxyester 265 upon several steps. Treatment of 265 (∼1:1 dr) with BF3·Et2O in dichloromethane at room temperature provided diastereomeric products 268 and 269 (86% yield, ∼1:1.3 dr) via FC reaction. Compounds 268 and 269 were probably formed via diastereomeric transition states 266 and 267, respectively (Scheme 54).141
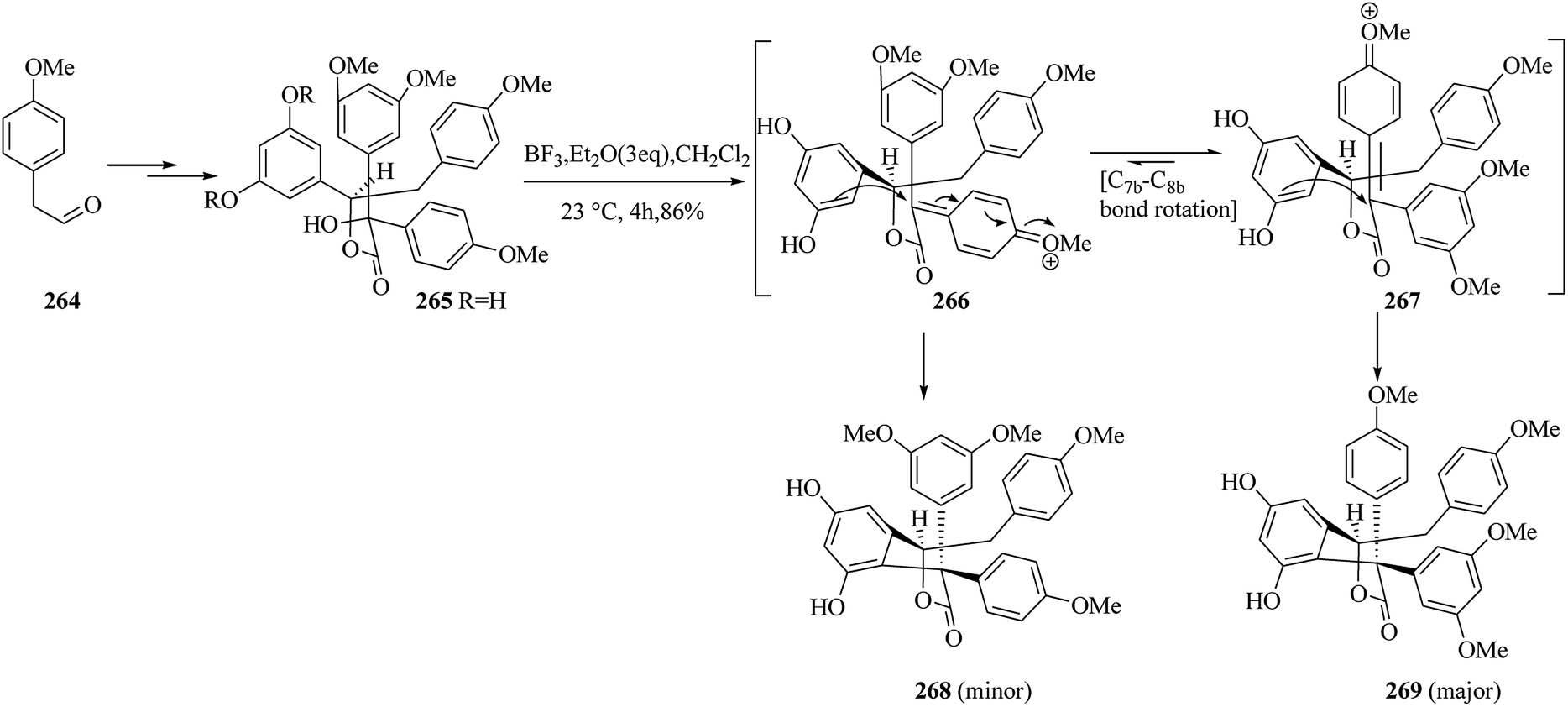 | ||
| Scheme 54 The formation of products 268 and 269. | ||
Compounds 268 and 269 provided epoxide 270 in several steps. Intramolecular FC reaction of epoxide 270 (∼1:1 dr) resulted in the formation of two diastereomeric cyclized products 274 (major) and 273 (minor) in ∼2:1 ratio in the presence of tin(IV) chloride in dichloromethane in 62% yield. Finally, compounds 272 and 273 provided (−)-hopeahainol A 244 in 84% overall yield after several steps. Subsequently, (−)-hopeahainol A 244 was transformed into (−)-hopeanol (−)-245 via several steps (Scheme 55).141
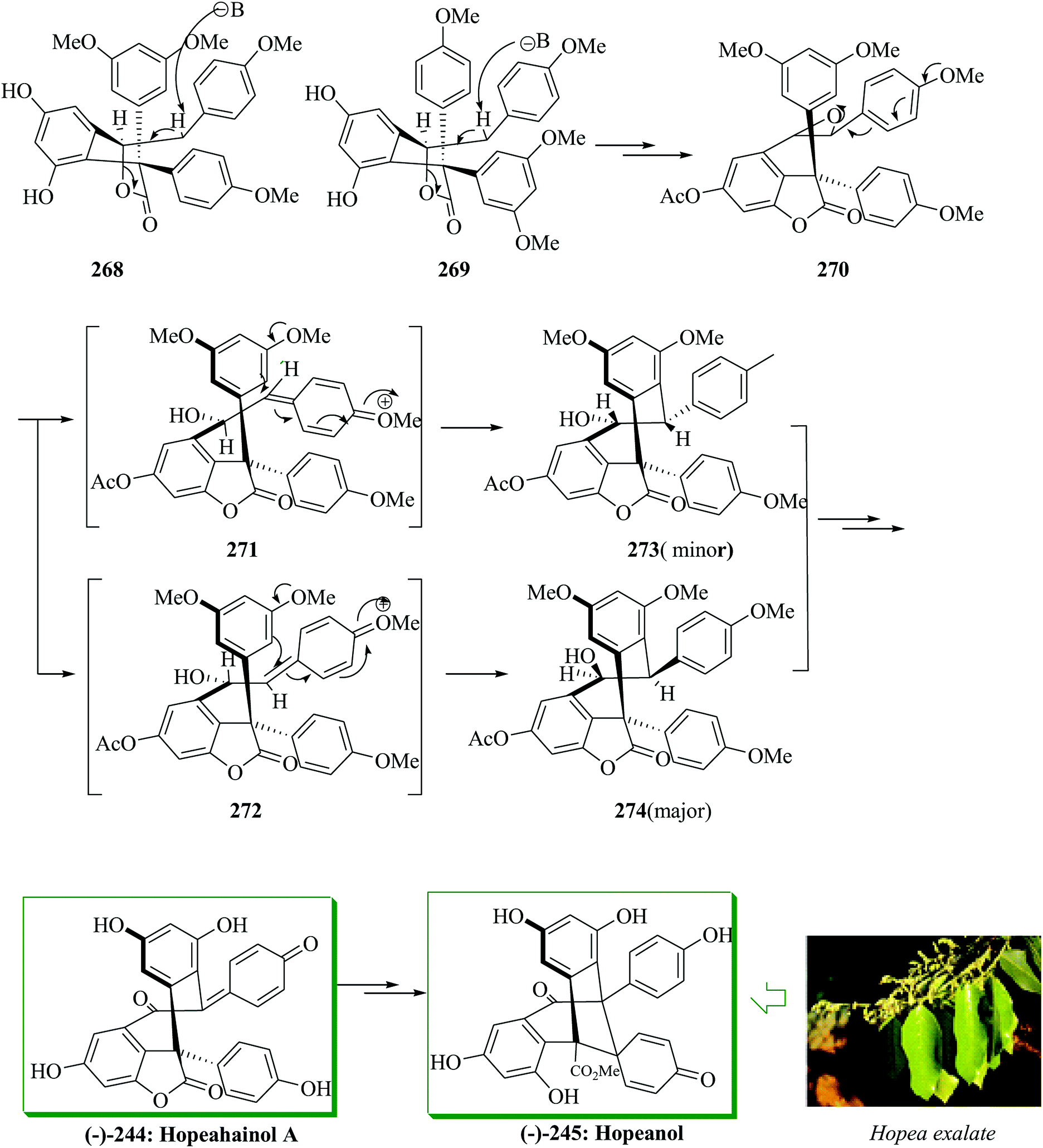 | ||
| Scheme 55 Total synthesis of (−)-hopeahainol A 244 and (−)-hopeanol 245. | ||
Dictyodendrins were initially extracted in 2003 by Fusetani and Matsunaga from the marine sponge Dictyodendrilla verongiformis collected from Nagashima Island in Southern Japan.262 These compounds contain the highly functionalized pyrrolo[2,3-c]carbazole moiety. Tokuyama and co-workers in 2010 reported the first total synthesis of dictyodendrin A 275 and B 276 using an unprecedented one-pot benzyne-catalyzed indoline construction/cross-coupling sequence, employing transmetalation to Cu species.263 In this route, for the synthesis of 275, initially, para-nitrophenol 277 afforded 278 after several steps. The latter initially underwent FC alkylation reaction with 279 in the presence of AgOTf. Then upon pinacolborylation at the bromo group of 278, the azidephenyl group was introduced via Suzuki–Miyaura coupling to afford 281. Finally, azide 281 was converted into dictyodendrin A 275 in 8.2% overall yield over 21 steps, starting from para-nitrophenol 277 (Scheme 56).263
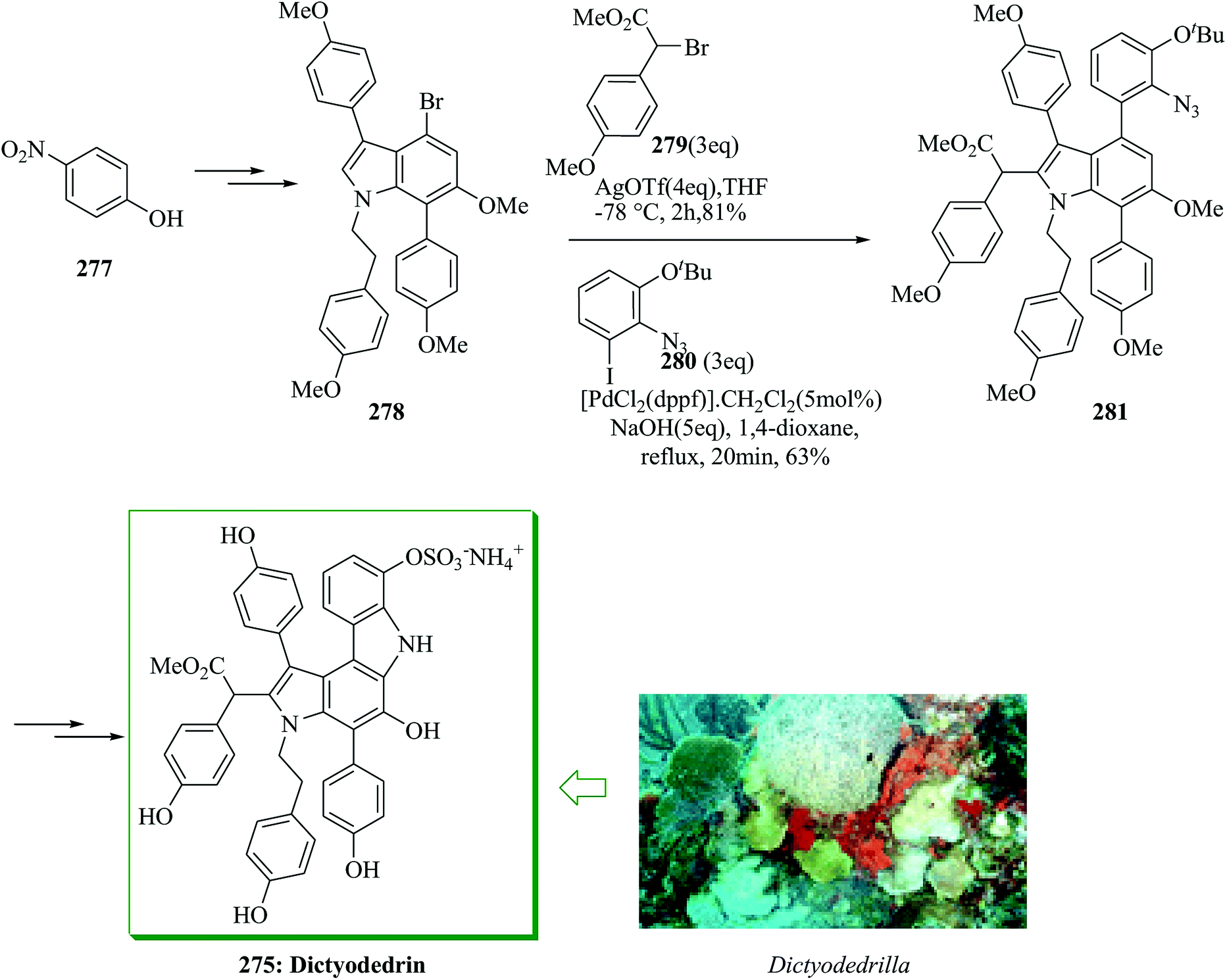 | ||
| Scheme 56 Total synthesis of dictyodendrin A 275. | ||
Subsequently, total synthesis of dictyodendrin B 276 was successfully achieved in 12% overall yield starting from para-nitrophenol 277. By using bromoindole 278, regioselective FC acylation occurred in the presence of zinc chloride264 to afford 283, which was converted into dictyodendrin B 276 in seven steps (Scheme 57).263
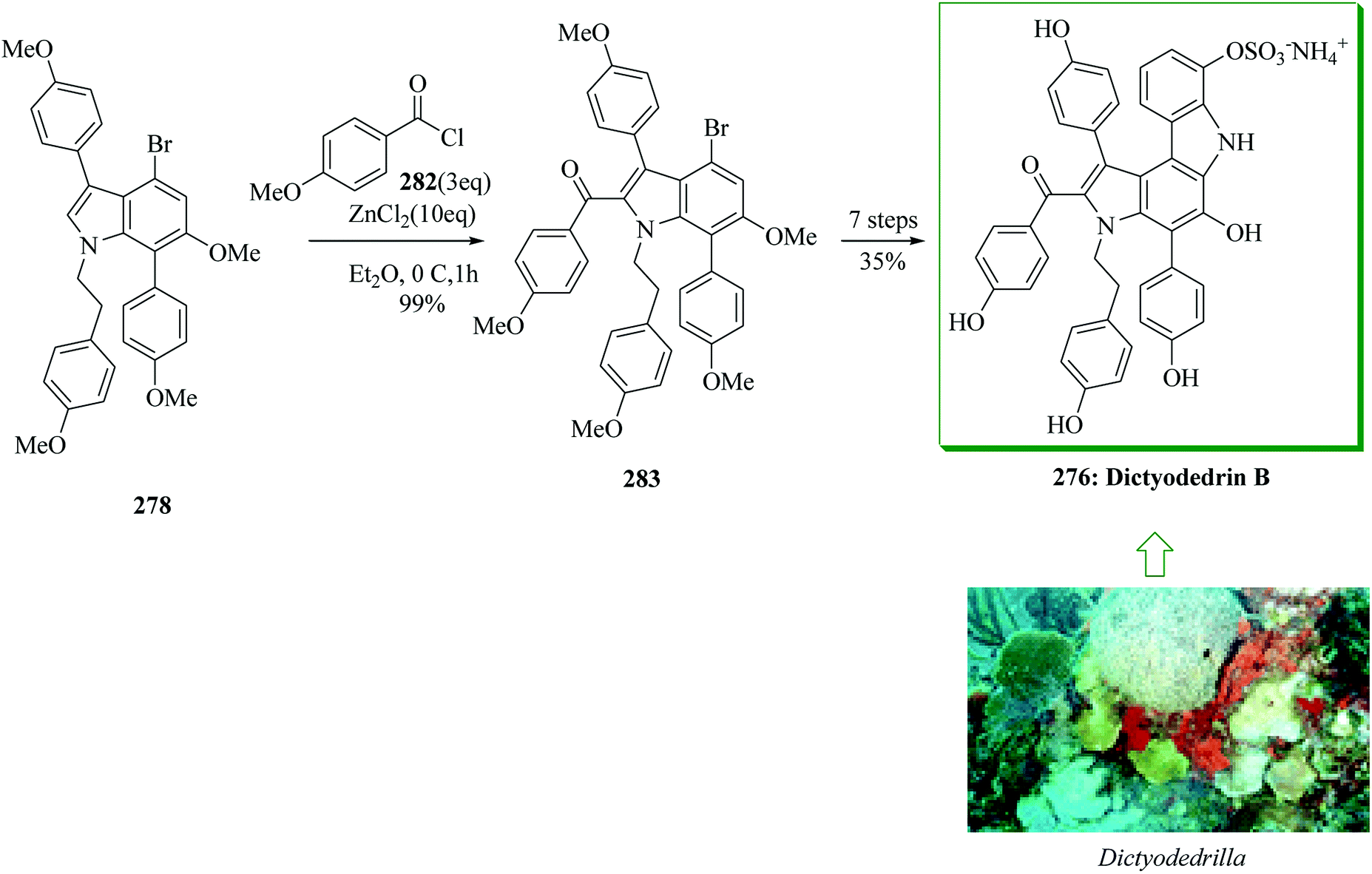 | ||
| Scheme 57 Total synthesis of dictyodendrin B 276. | ||
(−)-Ardeemin 284a and (−)-acetylardeemin 284b were extracted in 1993 from the fungus Aspergillus fischerii.265 (−)-Acetylardeemin 284b is one of the most active preventers of multidrug resistance.266,267 Total synthesis of multidrug-resistant inhibitors (−)-ardeemin 284a, acetylardeemin 284b, and (−)-formylardeemin 285 was accomplished, starting from bromopyrroloinoline 286 in 10 steps by Song and co-workers in 2012. The key step includes direct alkylation of 286 with prenyl tributylstannane 287 to provide 288 through a silver-promoted enantioselective FC reaction. Significant installation of the isoprenyl substituent provided a good overall yield for this FC reaction. Firstly, direct isoprenylation of bromopyrroloindoline 286 with 287 gave 288 through the FC reaction in the presence of silver perchlorate, 287, and caesium carbonate. Based on optimal reaction conditions, the main intermediate 288 was generated from 286 in 91% yield in a single step, leading to the total synthesis of 284 and 285. Next, compound 288 gave (−)-formylardeemin 285 upon several steps. Deformylation of 285 with aqueous sodium hydroxide in methanol afforded (−)-ardeemin 284a in 85% yield. (−)-Acetylardeemin 284b was obtained from 284a employing an already reported method (Scheme 58).266
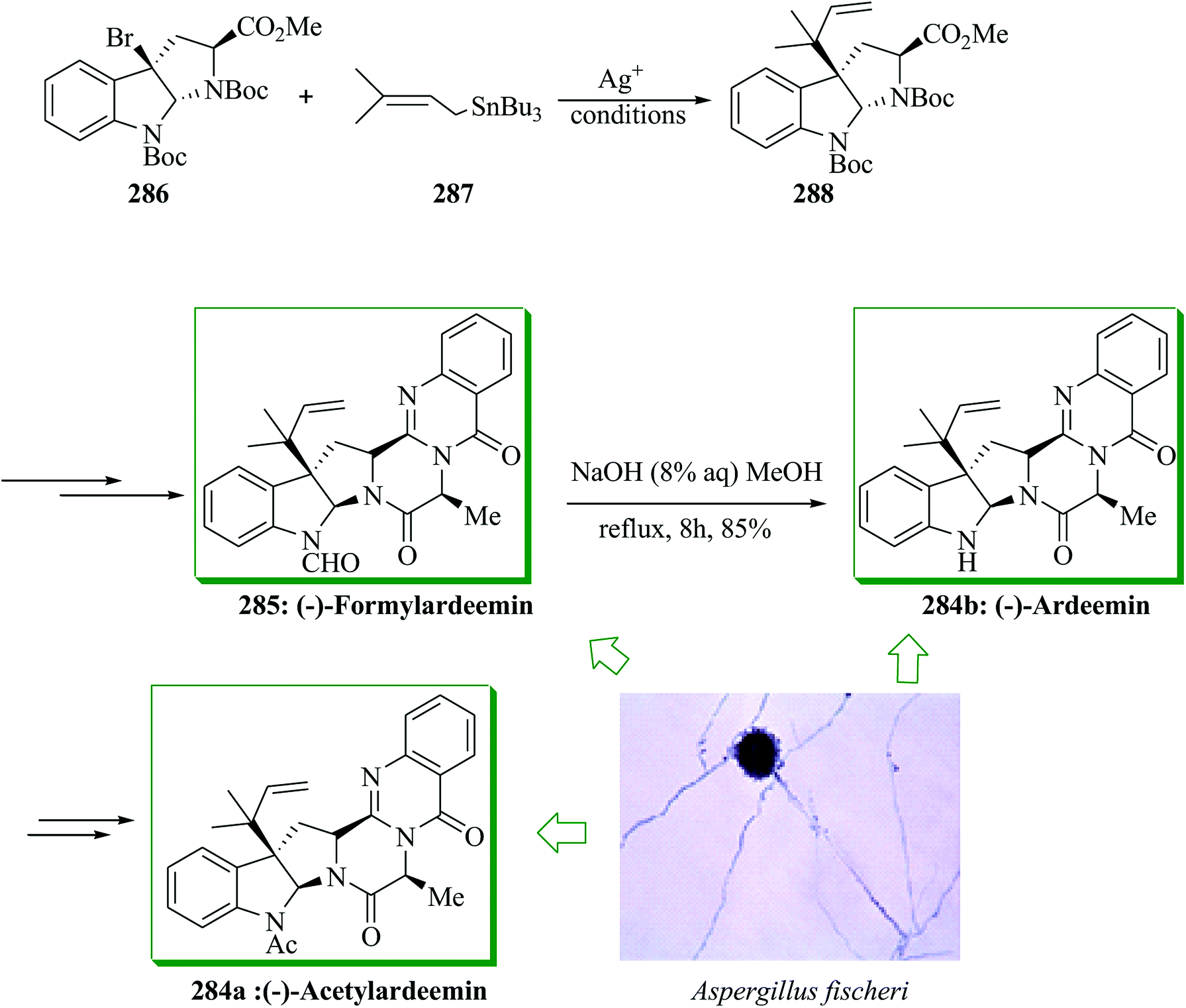 | ||
| Scheme 58 Total synthesis of (−)-ardeemin 284a, (−)-acetylardeemin 284b and (−)-formylardeemin 285. | ||
Epipolythiodiketopiperazine alkaloids, a structurally varied group of secondary fungal metabolites, exhibit a broad range of biological properties, including antiviral, antifungal, antibiotic, and cytotoxic activities.268,269 It is noteworthy that these mycotoxins were characterized by identification of a bridged polysulfide linkage across the cyclic dipeptide substructure.270–276 (+)-Gliocladin B 289,277,278 a new epidithiodiketopiperazine, and (+)-gliocladin C 290, an atypical non-thiolated triketopiperazine, were extracted by Usami and co-workers in 2004 from a strain of Gliocladium roseum OUPS-N132. (+)-Gliocladins show remarkable cytotoxicity against the murine P388 lymphocytic leukemia cellline.277 A short and asymmetric total synthesis of (+)-gliocladin B and C was developed in 2012 by Movassaghi and co-workers. The unified synthesis of (+)-gliocladins B 289 and C 290 was started from the bromocyclization of diketopiperazine (−)-291276,279 in just three steps starting from N-Boc-L-tryptophan and sarcosine methyl ester. Compound 291 reacted with bromine to give endo-tetracyclic bromide (+)-292 in 75% yield (endo-diastereomer). Significantly, coupling of bromide (+)-292 with indole 293 promoted by AgBF4 in nitroethane proceeded smoothly to give the corresponding C3-(3′-indolyl)hexahydropyrroloindole (+)-294 in 83% yield. 5-Bromo-1-triisopropylsilylindole 293 was shown to be a significant nucleophile for the corresponding regio- and stereo-selective FC-type reaction. Finally, (+)-294 provided (+)-gliocladin B 289 and C 290 after several steps (Scheme 59).280
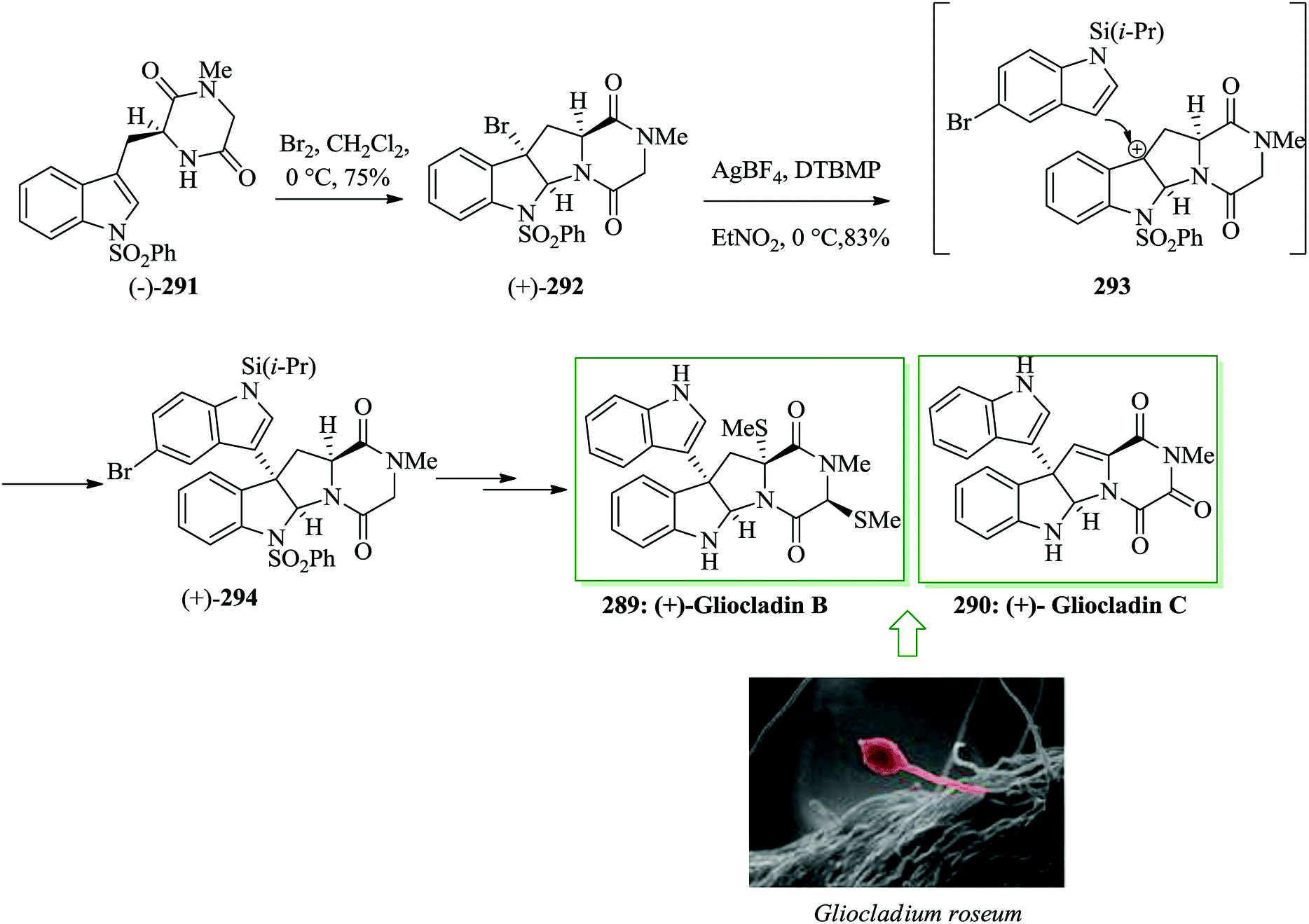 | ||
| Scheme 59 Total synthesis of (+)-gliocladin B 289 and C 290. | ||
Licorice is a popular food additive applied as a sweetener in chewing gums, tobaccos, and candies, and is also one of the most broadly applied medicinal herbs having anticancer281,282 antiparasitic,283 antibacterial,284 superoxide-scavenging285 and antioxidant properties.286 Seven chalcones, licochalcones A–E and G and the closely related compound echinatin, have been extracted and identified from the root of Glycyrrhiza inflata (licorice),287,288 except for the artificial synthetic licochalcone F.289 Among these reported chalcones, licochalcone C shows higher cytotoxicity compared to the analogous licochalcone B.281 In addition, licochalcone C exhibits remarkable inhibitory activity against the PTP1B enzyme.290 Wang and co-workers reported a concise, four-step synthesis of licochalcone C 295a and its regioisomer, tentatively called licochalcone H 295b, via acid-catalyzed Claisen–Schmidt condensation reaction as a main step. Initially, a key intermediate, 2,4-dihydroxy-3-(3-methyl-but-2-en-1-yl)benzaldehyde 298a, was synthesized from the reaction between 2,4-dihydroxybenzaldehyde 296 and 3-hydroxy-3-methyl-1-butene 297. Lewis acid-promoted FC alkylation of 297 in dioxane using BF3·OEt2 as a catalyst gave 298. In this approach, the electron-rich aromatic ring attacked the electrophile in the presence of the Lewis acid catalyst, BF3·OEt2, and 3-hydroxy-3-methyl-1-butene 297, followed by aromatization of the benzene ring to provide the desired compound 298a in 18% yield based on recovery of 2,4-dihydroxybenzaldehyde (70%). The other regioisomer 298b was provided as a major product in 38% yield, probably because of the desirable alkylation of the sterically less demanding C-5 in the FC reaction. Finally, compounds 298a and 298b gave 295a and 295b in 52 and 67% yield, respectively (Scheme 60).291
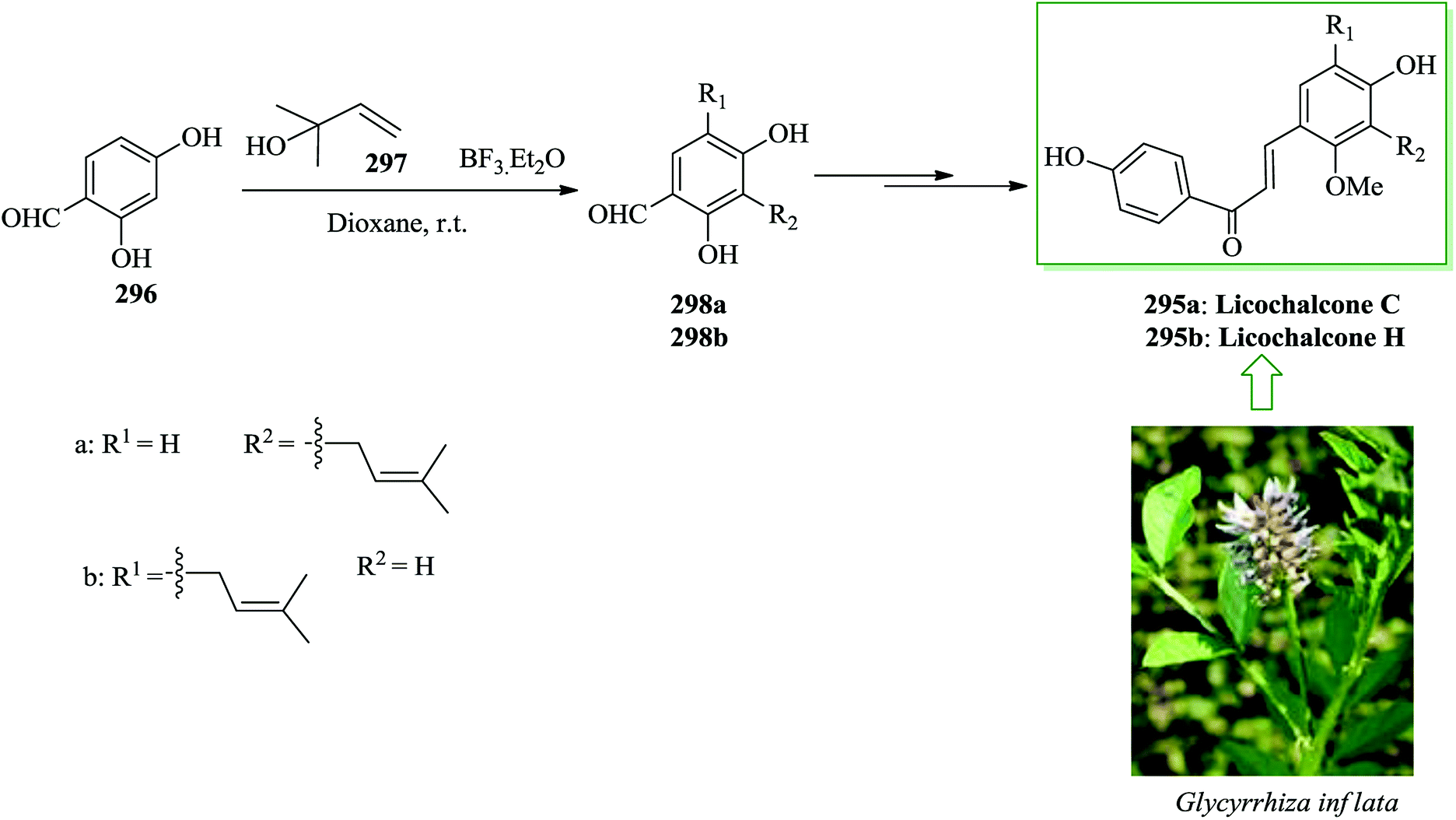 | ||
| Scheme 60 Total synthesis of licochalcone C 295a and its regioisomer licochalcone H 295b. | ||
(±)-Yuehchukene 299, a unique dimeric indole alkaloid, was initially extracted as a racemate from the roots of Murraya paniculata (L.) Jack and other related Murraya species.292,293 Yuehchukene exhibits remarkable anti-implantation property in hamsters and rats. A concise total synthesis of yuehchukene was successfully accomplished through organocatalytic FC alkylation reaction of indole to a sterically encumbered α-alkyl enal with excellent ees (up to 96%) as the vital step. It should be mentioned that a racemization method during the subsequent cyclization steps of the conjugate adduct to yuehchukene has been detected. In this route, at the beginning of exploring feasibility, a FC conjugate addition of dienecarbaldehyde 300 and indole 301 was successfully performed to provide the conjugate addition adduct 304. The best result was obtained in the reaction between two equivalents of 300 and indole 301 in the presence of trifluoroacetic acid in dichloromethane at room temperature, which gave 303 in 22% yield. Various acid additives have been examined with the catalyst 302294 in this reaction, and the best result was obtained under the optimized reaction conditions, affording a 26% yield of the desired product 304 (trans/cis = 73:27) with 60% ee of the trans adduct. The absolute configuration of this FC alkylation adduct has been identified to be (1S, 2R). Next, yuehchukene 299 was provided through alkylation-cyclization of adduct 304 (93% ee) using indole 301 in 36% yield (Scheme 61).295
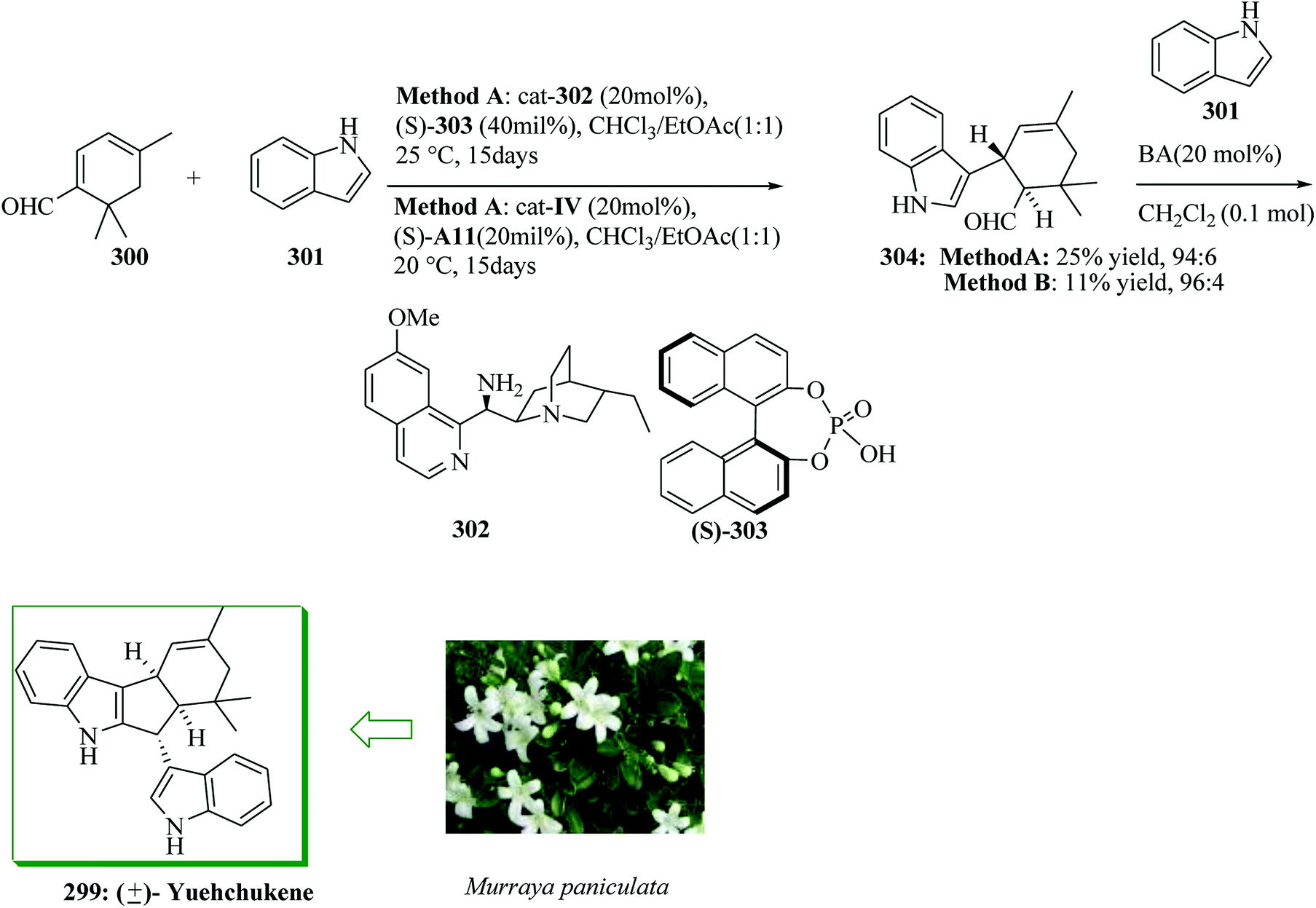 | ||
| Scheme 61 Total synthesis of yuehchukene 299. | ||
Several natural and synthetic heterocyclic quinones have shown significant biological properties, including antiprotozoan, antitumoral, and antibiotic properties.296,297 Many of these compounds contain antineoplastic chemotherapeutic activities.298 Quinones, a group of molecules known in various drugs, were applied in the clinical therapy of solid tumors.299 Ellipticine was extracted from the leaves of Ochrosia elliptica Labill.300 Ellipticine quinone 305301,302 is an essential synthetic intermediate in the Gribble synthesis of ellipticines, which display antitumor activity.303 A straightforward pathway for the synthesis of biologically potent ellipticine quinones was developed in 2014 by Nagarajan and co-workers. The key steps included are FC hydroxyalkylation with subsequent oxidation followed by directed ortho-lithiation of indole-2-carboxylate esters with functionalized quinoline and pyridine carboxaldehydes. For the synthesis of ellipticine quinone 305, firstly, compound 306 and pyridine-3-carboxaldehyde 307 were reacted to afford 308 as acid under FC hydroxyalkylation reaction. The acid 308 was directly transformed into ellipticine quinone 305 using LiTMP in 43% yield (Scheme 62).304
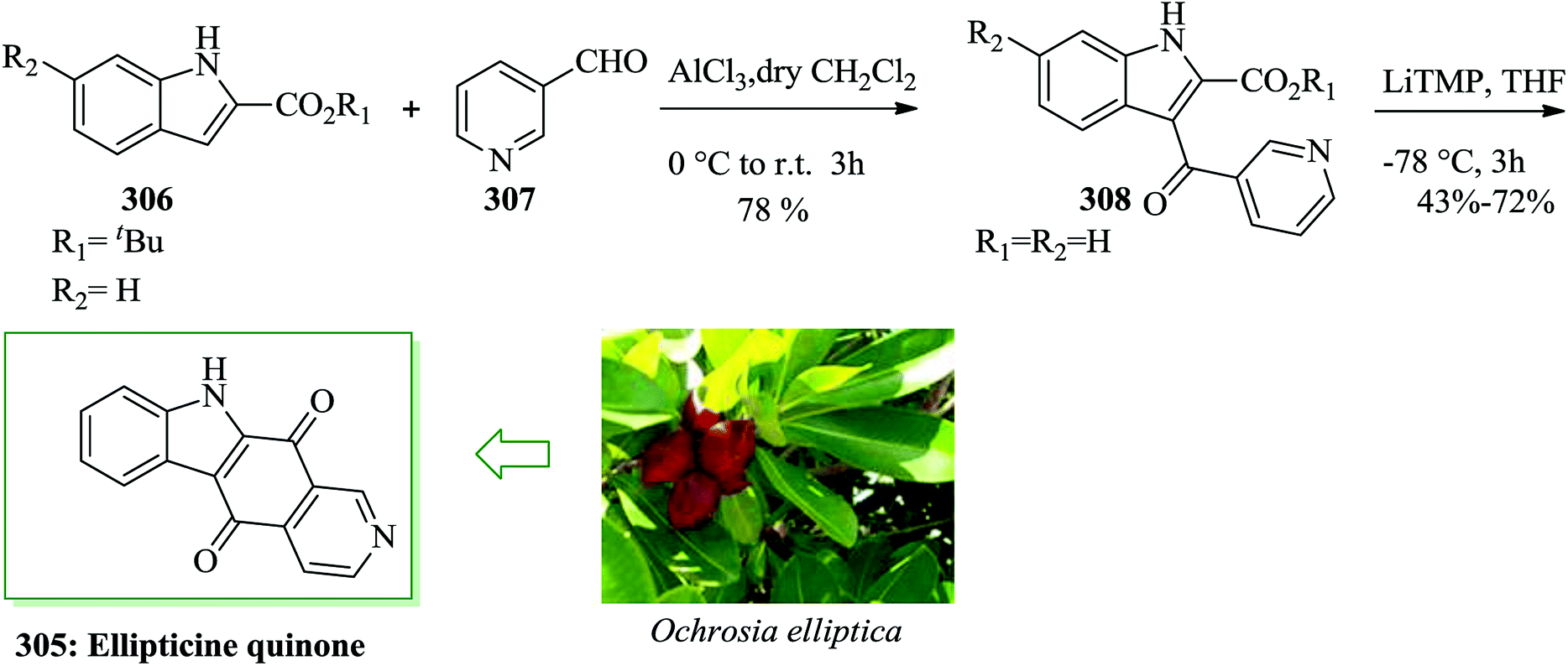 | ||
| Scheme 62 Total synthesis of ellipticine quinone 305. | ||
Studying the extract of leaves of P. adunctum by Orjala and co-workers305 resulted in the extraction of natural adunctin E.305 A wide range of naturally occurring compounds, namely methyllinderatin 310 and linderol A 312, were extracted from the fresh bark of Lindera umbellata by Ichino306 and Mimaki and co-workers,307 respectively. In 2007, Portet and co-workers308 extracted hostmanin A 309 together with methylinderatin 310, adunctin E 311 and relevant naturally occurring compounds from the leaves of Piper hostmannianum. Methyllinderatin 310 exhibited antibacterial effects toward M. luteus and also exhibited potent antiplasmodial activity against chloroquine sensitive and resistant strains of Plasmodium falciparum. (−)-Linderol A 312 inhibited melanin biosynthesis of cultured B-16 melanoma cells without providing any cytotoxicity in the cultured cells.307 Adunctin E 311 and linderol A 312 have four contiguous stereocenters. Dethe et al. in 2015 reported309 protecting group free,310, 311 short and asymmetric total synthesis of (+)-hostmanin A 309, (+)-methyllinderatin 310, (+)-linderol A 312 and adunctin E 311. A one-step method was established for the asymmetric synthesis of hexahydrodibenzofuran derivatives employing a modified FC reaction. The established approach was used for the synthesis of a wide range of naturally occurring compounds involving (+)-hostmanin A, (+)-methyllinderatin, and (−)-linderol A. For the synthesis of these natural products, the diastereoselective modified FC reaction was used,312, 313 which included two needed constituents, dihydrochalcone derivative 314 and alcohol 315. The desired chalcone derivative 314 was provided from acetophenone derivative 313 upon several steps. Using the required segments, the stage was set to examine the key FC coupling reaction of a mixture of 314 and 315 using BF3·OEt2, which resulted in rapid construction of the extremely regio- and diastereo-selective coupling product methyllinderatin 310 in 91% yield with >20:1 diastereomeric ratio (only major diastereomer is depicted in Scheme 63). Moreover, one-step synthesis of (+)-hostmanin A 309 has been accomplished. Therefore, the reaction of dihydrochalcone derivative 314 and alcohol 315 using p-toluenesulfonic acid under reflux in toluene provided (+)-hostmanin A 309 as a single diastereomer with 79% yield, whose spectral data and optical rotation were identical to those reported for the natural product.308 Then, the total synthesis of (−)-linderol A 312 and adunctin E 311 was explored, which were finally obtained from methyllinderatin 310 upon several steps through various pathways.309
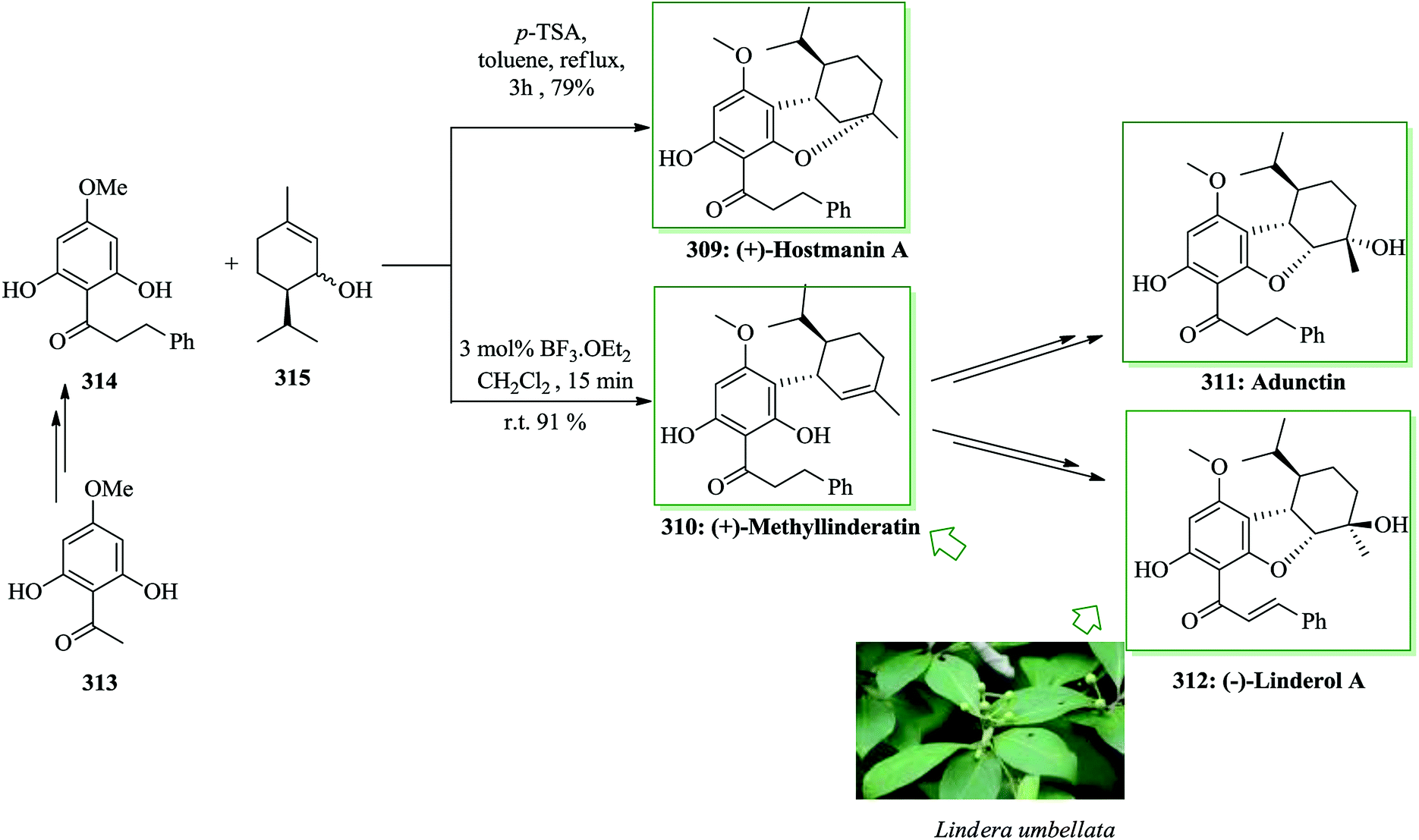 | ||
| Scheme 63 Total synthesis of (+)-hostmanin A 309, (+)-methyllinderatin 310, (+)-linderol A 312 and adunctin E 311. | ||
2.3. Intramolecular acylation
The benzo[c]phenanthridine alkaloid nitidine was first extracted from the root bark and root wood of Zanthoxylum nitidum, a woody climber that grows in most areas of Hong Kong.314 A unique method for the total synthesis of benzophenanthridine alkaloids is presented in the context of the construction of the antileukemic agent nitidine chloride 316. 4,5-Dimethoxyhomophthalic anhydride 317 was reacted with a solution of 3,4-methylenedioxybenzylidenemethylamine 318, which after other steps gave compound 319. Intramolecular FC acylation reaction of 319 with poly(phosphoric acid) afforded the tetracyclic ketone 320. Next, compound 320 gave nitidine, separated as the chloride 316 in 47% yield, along with a 24% yield of the hydrogenolysis product 321 (Scheme 64).315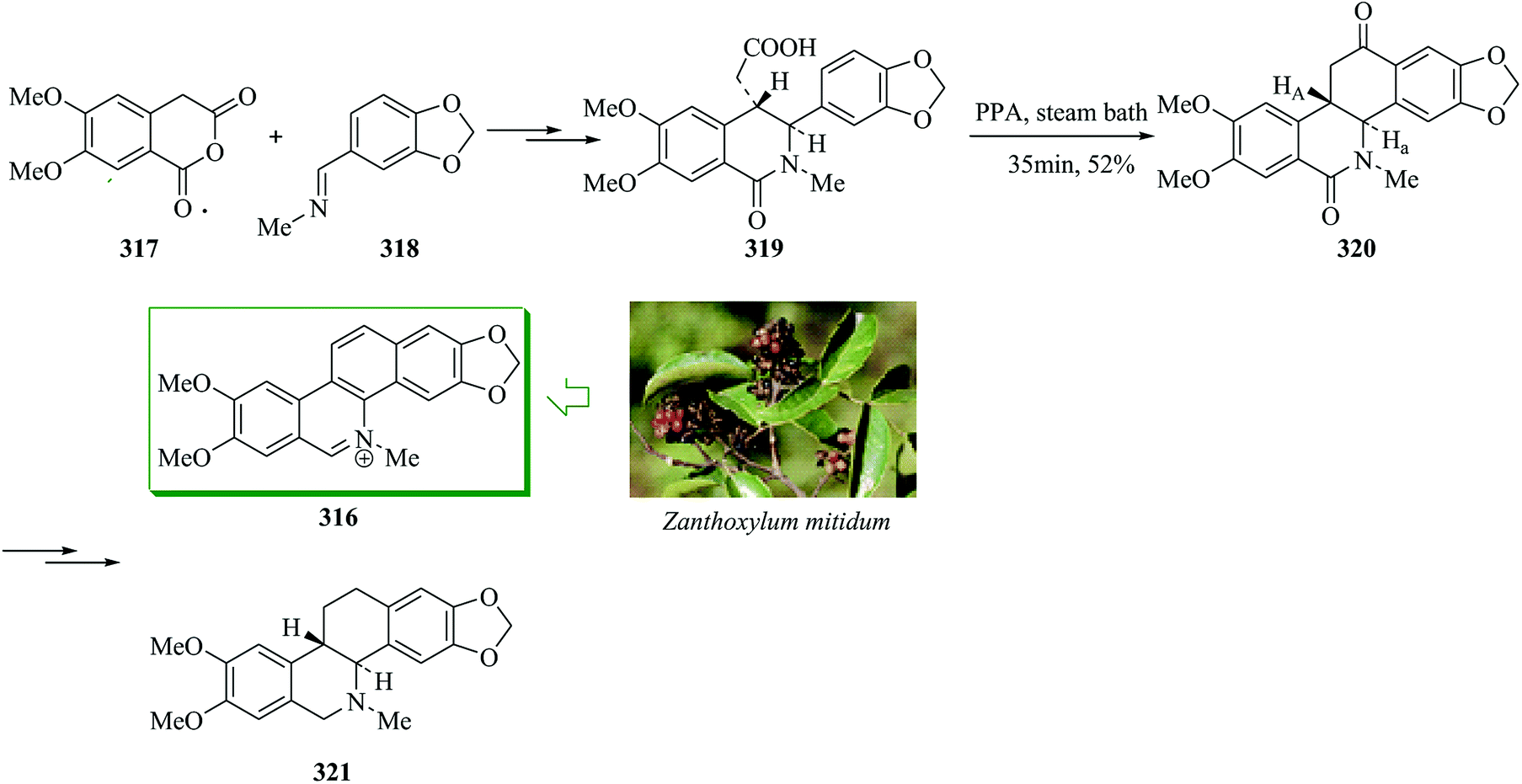 | ||
| Scheme 64 The formation of compounds 316 and 321. | ||
Anthraquinones as naturally occurring compounds extracted from both plants316 and insects317 have stirred up the interest of synthetic organic chemists as significant synthetic targets. Erythroglaucin 322a has been extracted from species of Cortinarius while catenarin 322b was first extracted from cultures of Helminthosporium catenarium. The regiospecific, concise, and effective synthesis of the natural anthraquinones erythroglaucin 322a and catenarin 322b was successfully achieved. The synthesis of erythroglaucin 322a and catenarin 322b was achieved through generation of ortho-metalated N,N-diethylbenzamide as an intermediate. Firstly, 3,5-dimethoxy-N,N-diethylbenzamide 323 afforded a high yield of the o-benzyl-benzoic acid 324 upon several steps, which under mild FC cyclization in the presence of trifluoroacetic anhydride318,319 afforded the anthracenol 325. NMR and IR spectral analysis disclosed evidence for a concentration-dependent anthracenol 325a and anthracenone 325b equilibrium. Finally, compound 325 gave catenarin 322a and erythroglaucin 322b in 22% and 29% overall yields, respectively (Scheme 65).320
 | ||
| Scheme 65 Total synthesis of catenarin 322a and erythroglaucin 322b. | ||
Interleukin-8 (IL-8), a chemoattractant for neutrophils, is provided by macrophages and endothelial cells.321 IL-8 is implicated in a broad series of acute and chronic inflammatory disorders.322 Frondosins A–D were recently extracted from the sponge Dysidea frondosa. In these compounds, the unifying architectural theme is the existence of bicyclo[5.4.0]undecane ring systems in the context of permuted linkages to several hydroquinone-based scaffolds.323 Frondosin B 326324 includes a benzofuran ring system fused to a nor-sesquiterpenoid (14-carbon) scaffold. A concise synthesis of (±)-frondosin B 326, an interleukin-8 receptor antagonist, was accomplished from 5-methoxysalicylaldehyde 327. The seven-membered ring in ketone 329, the common intermediate for both syntheses, was made via FC reaction. This method was initiated from 5-methoxysalicylaldehyde 327, which provided carboxylic acid 328 upon several steps. Fortunately, the critical FC reaction could be performed. Therefore, reaction of 328 with oxalyl chloride and reaction of the resultant acid chloride with SnCl4 afforded the corresponding ketone 329 in moderate yield.325 Finally, compound 329 was converted into (±)-frondosin B 326 after several steps (Scheme 66).326
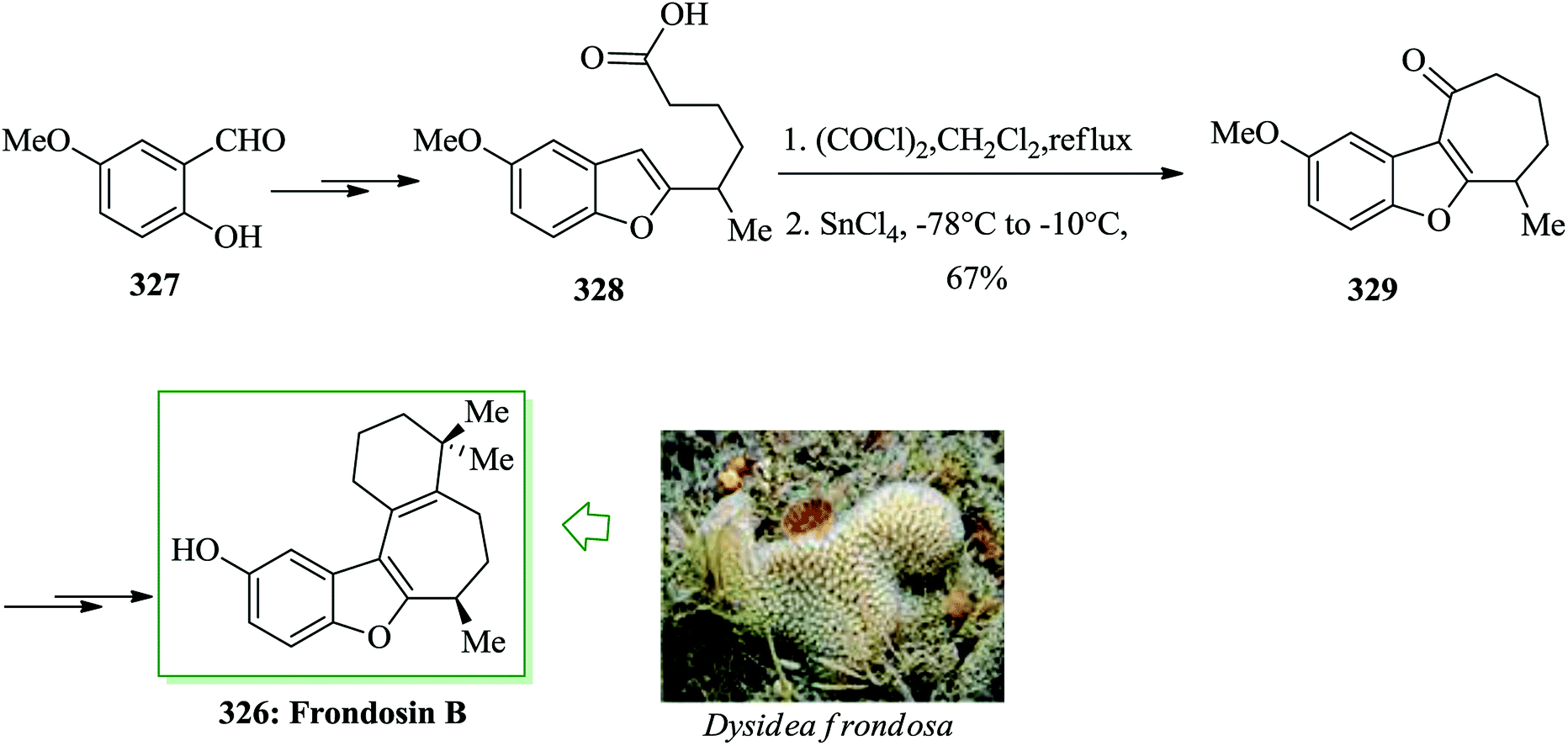 | ||
| Scheme 66 Total synthesis of (±)-frondosin B 326. | ||
Various sesquiterpene metabolites have been identified from species of gorgonians.327 These compounds show remarkable biological properties, including antifungal, cytotoxicity and immunostimulatory activity.328 Echinofuran 330,329 a type of furanosesquiterpenoid tetrahydrolinderazulene, was extracted from the gorgonian Echinogorgia praelonga in 1992.330 The initial total synthesis of (±)-echinofuran 330 was achieved in 12 steps with overall yield of 0.55%. The synthesis of 330 was accomplished using 3-methyl-4-(trimethylsilyl)furan 331 as a precursor. A Suzuki coupling reaction and a Lewis acid-catalyzed FC cyclization reaction were the main steps in the formation of the desired ring system 333 (Scheme 67).
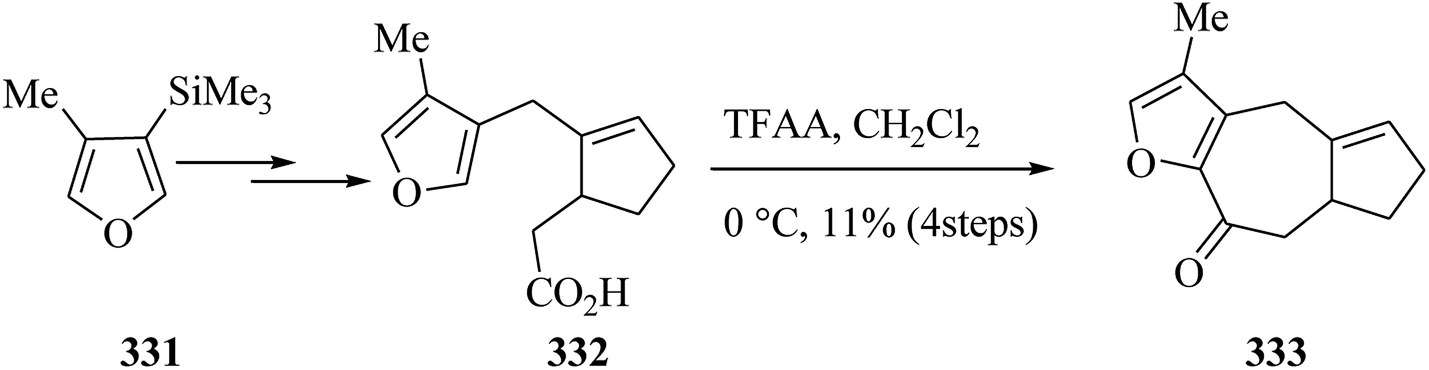 | ||
| Scheme 67 The formation of compound 333. | ||
For the synthesis of (±)-echinofuran 330, next, benzyl ether 335 was obtained starting from 1,3-cyclopentanedione 334, after several steps. Saponification of 335 with sodium hydroxide was followed instantaneously, without more purification, by a FC cyclization reaction, providing 336. Finally, compound 336 was converted into (±)-echinofuran 330 upon several steps (Scheme 68).331
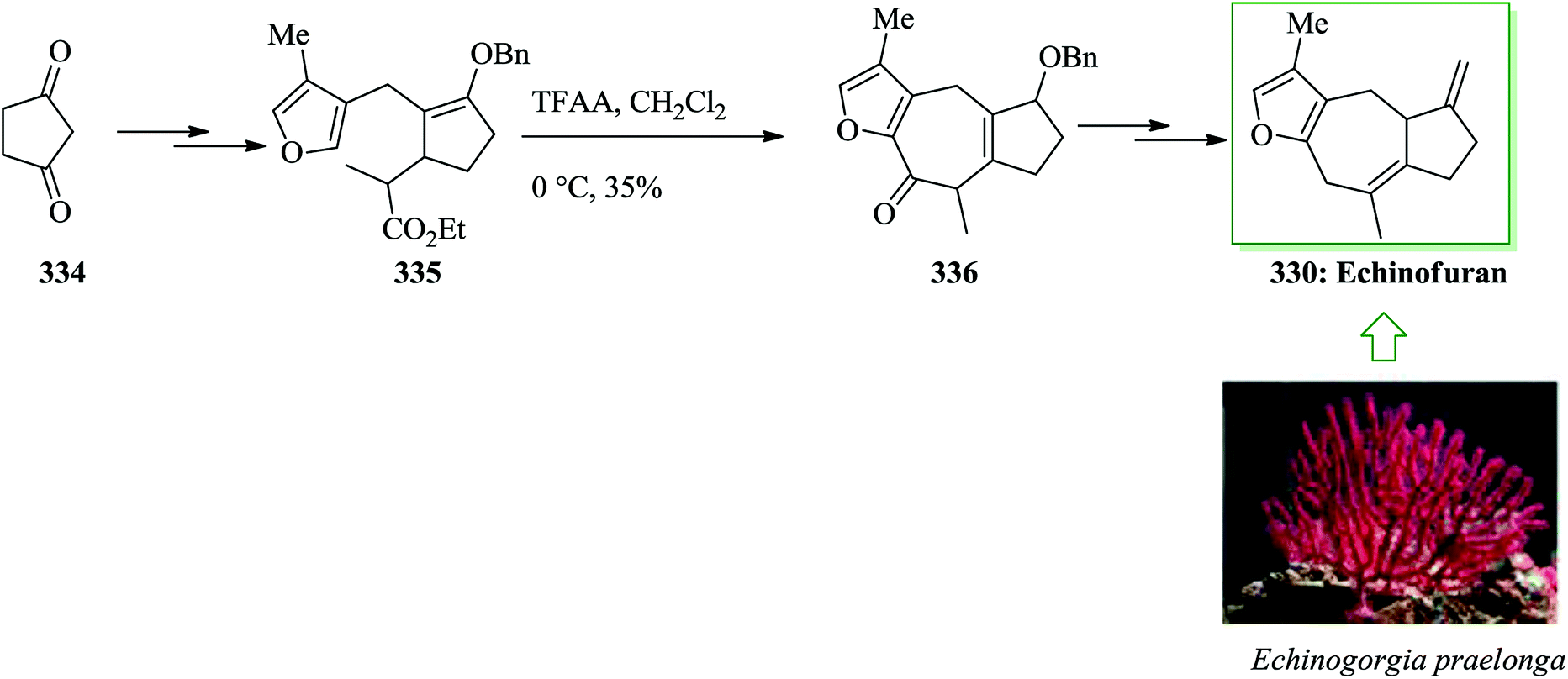 | ||
| Scheme 68 Total synthesis of (±)-echinofuran 330. | ||
Preussomerins A–F were the first members of this group to be reported332 and preussomerins G–L are the most recent additions.333 Structurally, all the preussomerins contain two naphthalene nucleus linked by three oxygen atoms, providing a bis-spiroacetal system. This significant head-to-tail trioxabicyclo[3.3.1]nonane core is a unique natural product unit. The synthesis of newly extracted members of the preussomerin group, preussomerins K and L (4% overall yields in both cases), was demonstrated by Taylor et al.334 The main stages are the functionalization of a 2-arylacetal anion, one-pot FC cyclization-deprotection and reductive opening of epoxides. The total synthesis was commenced from methoxyphenol 338, which afforded the diacid 339 in several steps. The next step of the synthesis was FC cyclization reaction of the diacid 339. Upon activating of the latter as the acid chloride by a homogenous solution of aluminium chloride in nitromethane, the corresponding FC adduct 340 was obtained. However, if the reaction was permitted to stand for longer time in the presence of excess aluminium chloride, two new compounds, identified as 341a and 341b, started forming, demonstrating that upon a quick cyclization, the remaining Lewis acid gradually mediated the removal of the phenolic methoxy groups. Lastly, compound 341b gave preussomerin L 336 and preussomerin K 337 through a different multi-step synthesis (Scheme 69).334
 | ||
| Scheme 69 Total synthesis of preussomerin L 336 and preussomerin K 337. | ||
Preussomerin F was extracted from the coprophilous fungus Preussia isomera by Gloer and co-workers in 1990.335 Preussomerin K was also extracted independently by Isaka et al. from a lichenicolous fungus Microsphaeropsis sp. BCC 3050.336 All of the preussomerins exhibit pronounced antifungal activity as well as preventing Gram-positive bacteria. All the members of the preussomerin group are characterized by the same key structural aspect, containing the naphthalene units linked by three oxygen atoms, providing a bis-spiroacetal functionality. Taylor and co-workers in 2004 developed and reported the first total synthesis of the racemic natural products preussomerin F (3% overall yield, 13 steps) and preussomerins K and L (4% overall yields in both cases, 11 and 10 steps, respectively) from cheap and simply accessible initiating precursors via the usage of a straightforward, non-biomimetic methodology including the functionalization of 2-arylacetal anions, simultaneous one-pot FC cyclization-deprotection and substrate-regiocontrolled hydrogenation as the key steps. The total synthesis of the racemic naturally occurring compounds preussomerins F, K and L started from 4-methoxyphenol, which was transformed into the diacid 339 in several steps. Then the reaction of diacid 339 with a homogenous solution of aluminium chloride in nitromethane at ambient temperature provided the corresponding FC adduct 340 in satisfactory yield (75%). However, if the reaction was permitted to stand for a longer time in the presence of an excess of aluminium chloride, two new compounds, characterized as 341a and 341b, started to appear, demonstrating that after a fast cyclization, the lasting Lewis acid gradually removed the phenolic methoxy substituents. Finally, compound 341b gave preussomerins F, K and L upon several steps (Scheme 70).337
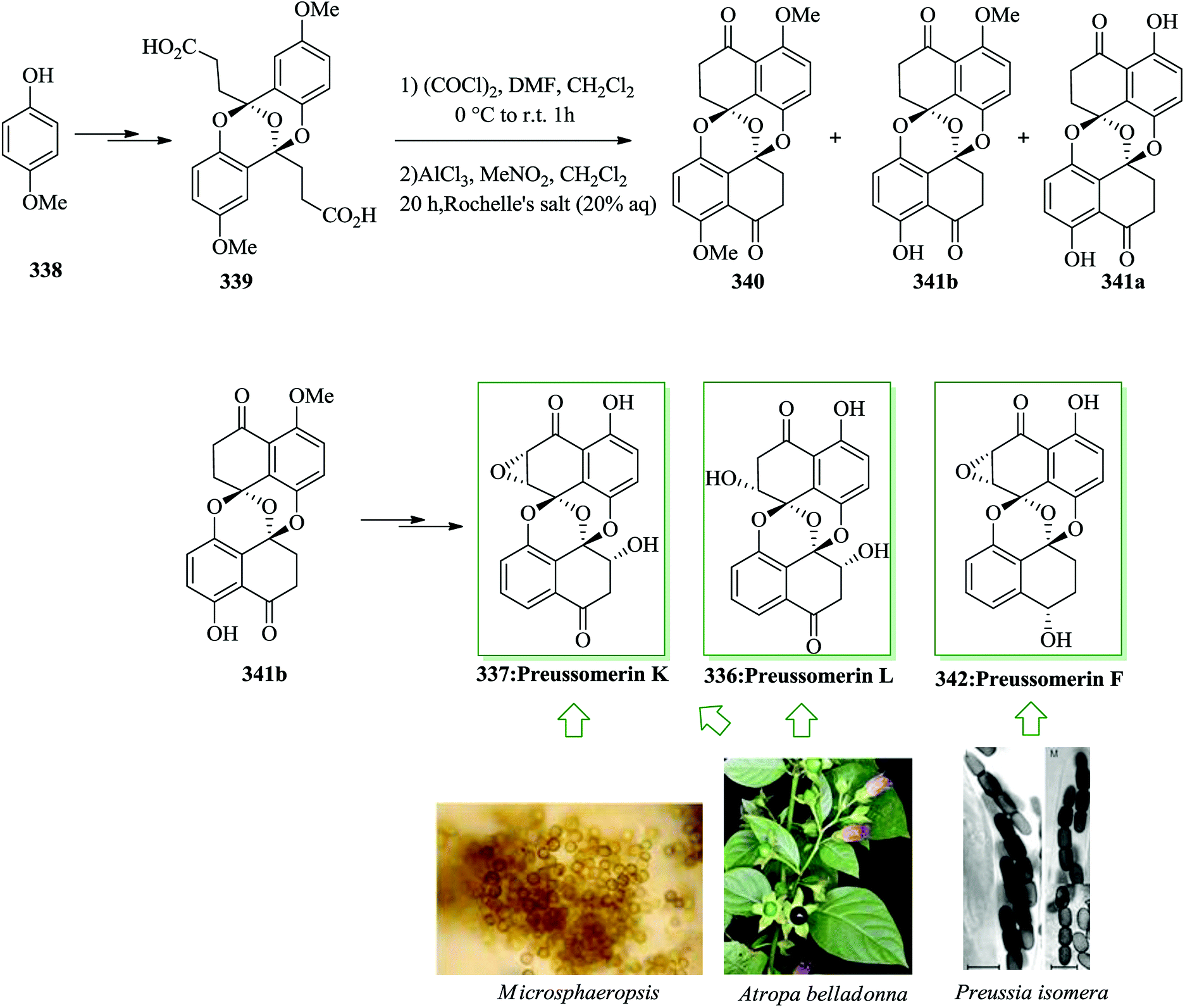 | ||
| Scheme 70 Total synthesis of preussomerins L 336, K 337 and F 342. | ||
The search for phytoalexins in Musa over the years led to the isolation and structural identification of 9- and 4-phenylphenalenones and related compounds, for example dimeric phenylphenalenones, phenylnaphthalic anhydrides, perinaphthenones, and an oxabenzochrysenone.338 4-Methoxy-1H-phenalen-1-one (4-methoxyperinaphthenone) 343, a subunit known in some Musa phytoalexins and relevant naturally occurring compounds from Haemodoraceae, was obtained via a Heck–Fujiwara coupling reaction and a FC acylation as the carbon–carbon bond-forming reactions. It was actually synthesized from 2-methoxynaphthalene in five steps with an overall yield of 36%. The formation of 4-methoxy-1H-phenalen-1-one (4-methoxyperinaphthenone, 343) and other perinaphthenones alike was achieved via the cyclization of b-1-naphthylpropanoic acids provided by the malonic ester synthesis, employing 4-methoxynaphthalenes 344 as the starting material.339 Based on this method, 4-methoxynaphthalene gave 345 upon several steps. Compound 345 was cyclized employing FC reaction conditions and gave 4-methoxy-1H-phenalen-1-one (4-methoxyperinaphthenone) 343 as the sole product in 48% yield (Scheme 71).340
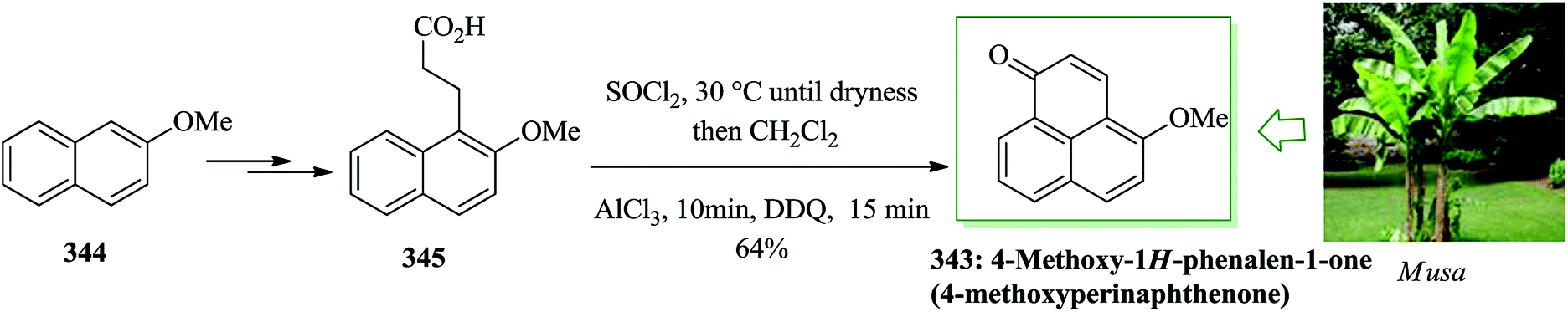 | ||
| Scheme 71 Total synthesis of 4-methoxy-1H-phenalen-1-one (4-methoxyperinaphthenone) 343. | ||
Xestodecalactone C 346 was extracted from the fungus Penicillium cf. montanense, which in turn was extracted from Xestospongia exigua. This molecule is structurally related to a wide range of compounds extracted from terrestrial fungi, involving sporostatin 347. Sporostatin (M5032, 347), extracted from the fungus Sporormiella sp., is an inhibitor of cyclic adenosine 3′,5′-monophosphate phosphodiesterase.341 Total synthesis of xestodecalactone C and epi-sporostatin was developed in 10 steps using Prins cyclizations, Mitsunobu reaction and intramolecular FC acylation reaction. This method is convergent and extremely enantioselective. The synthesis of xestodecalactone 346 and epi-sporostatin 347 was started with chiral homoallyl alcohol 349. The starting material 349 was synthesized in two steps from S-(−)-benzyl glycidyl ether 348. Next, compound 349 was converted into acid 350 after several steps. The corresponding macrolide 351 was provided in 41% yield via intramolecular FC reaction of the carboxylic acid 350 in the presence of a mixture of trifluoroacetic acid and trifluoroacetic acid anhydride.342,343 Demethylation reaction of 351 with freshly synthesized AlI3 afforded the target molecule 346344,345 in 96% yield, whereas the same reaction at ambient temperature provided 347 in 94% yield.342,343 Finally, compound 351 afforded 346 and 347 in 96% and 94% yield, respectively (Scheme 72).346
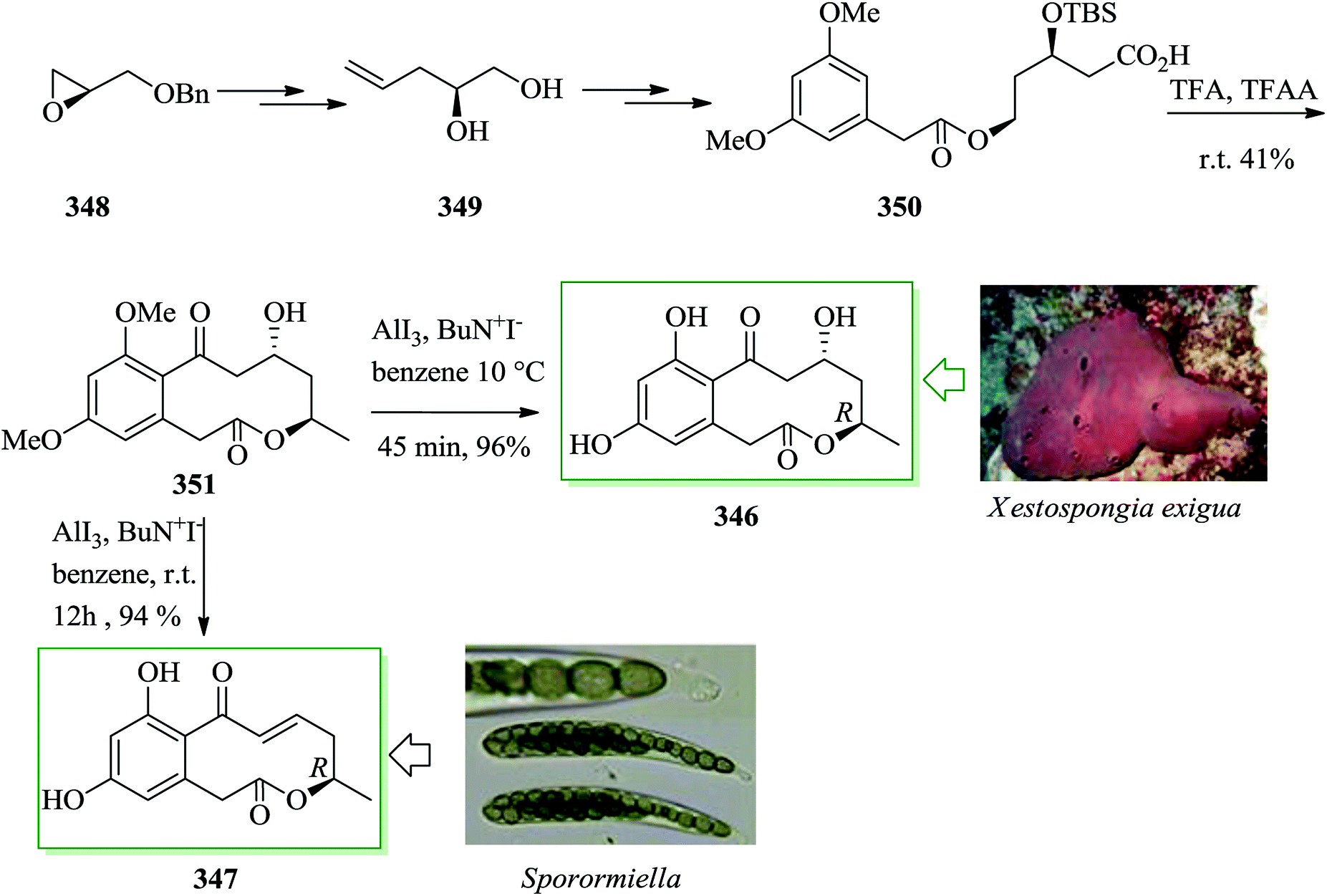 | ||
| Scheme 72 Total synthesis of sporostatin 347. | ||
The aaptamines, first found in nature by Nakamura and co-workers in 1982,347 are marine alkaloids that include a benzo[de][1,6]naphthyridine scaffold.348,349 The aaptamine group involves aaptamine 352, first extracted from an Okinawan specimen of Aaptos aaptos. The aaptamines exhibit various remarkable biological properties. Owing to their antagonistic effects on β-adrenergic receptors, a cardiac property was found for 352.350,351 A formal total synthesis of the marine alkaloid aaptamine 352 has fruitfully been accomplished via the development of a new methodology to provide 2,3,3a,4,5,6-hexahydroaaptamine 356. This alternative access to 356 has been achieved in eight steps and in 11% overall yield, with quinolin-4-one 355 as a key intermediate. The synthesis was started from market-accessible 2,3-dimethoxybenzoic acid 353, which after several steps afforded acid 354. Then, acid 354 was exposed to FC-type acylation reaction with excess PPE in toluene to obtain 355 in 95% yield. Finally, after several steps, intermediate 355 afforded 2,3,3a,4,5,6-hexahydroaaptamine 356, which is an intermediate for the synthesis of aaptamine 352 (Scheme 73).352
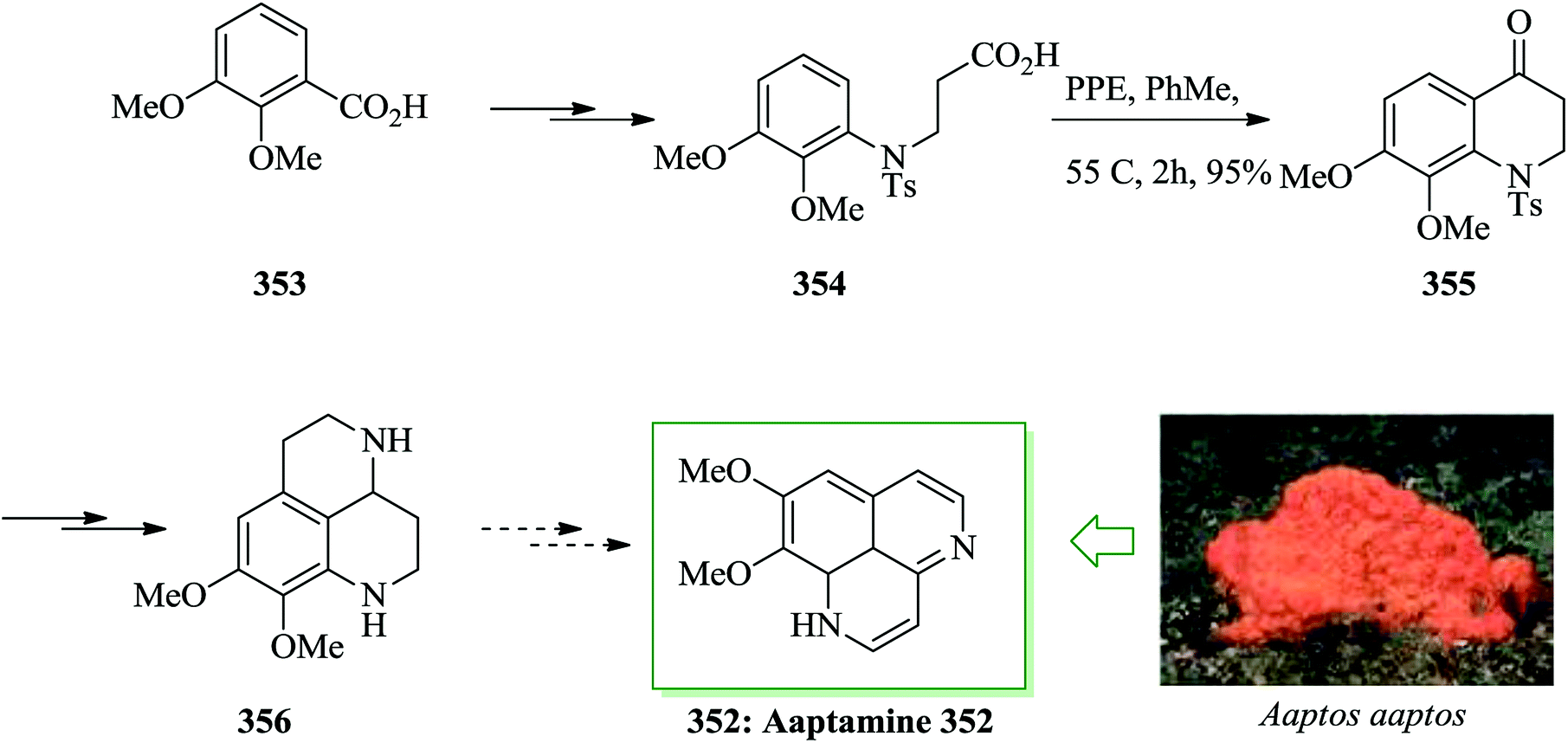 | ||
| Scheme 73 Total synthesis of aaptamine 352. | ||
Sporostatin (M5032, 347), extracted from the fungus of Sporormiella sp., is an inhibitor of cyclic adenosine 3′,5′-monophosphate phosphodiesterase (cAMP-PDE),353 composed of a ten-membered macrolide derivative with a 1,3-di-hydroxybenzene ring. A simple and effective total synthesis of sporostatin was achieved in five steps beginning from (S)-propylene oxide 357. In this method, esterification, cross-metathesis, and intramolecular FC reaction were known as the key steps. Total synthesis of sporostatin (M5032, 347) was started from (S)-propylene oxide 357, which afforded the relevant α,β-unsaturated carboxylic acid 358 after several steps. Then, the macrolide 359 was obtained in 41% yield when the carboxylic acid 358 was exposed to an intramolecular FC reaction with a mixture of trifluoroacetic acid and trifluoroacetic anhydride.344,354–356 Deprotection of the MeO substituents of 359 employing freshly synthesized AlI3 (ref. 357–359) provided the final product 347 in 94% yield (Scheme 74).360
 | ||
| Scheme 74 Total synthesis of sporostatin 347. | ||
In 2002, 10-membered macrolides fused to the 1,3-dihydroxybenzene ring, for example xestodecalactones A, B, and C 346, were extracted from the fungus Penicillium cf. mantanense obtained from the marine sponge Xestospongia exigua.358 Xestodecalactones A–C exhibited antifungal and antibacterial properties.361 A simple and extremely effective enantioselective total synthesis of xestodecalactone C 346, a polyketide natural product, was accomplished using Keck's asymmetric allylation reaction, an iodine-induced electrophilic cyclization, and an intramolecular FC acylation as key steps. The synthesis of xestodecalactone C 346 was initiated from propane-1,3-diol, which was protected with p-methoxybenzyl (PMB) bromide to provide the relevant propan-1-ol 360. Next, compound 360 gave the acid 361 in several steps. The corresponding macrolide 362 was obtained in 40% yield via an intramolecular FC acylation reaction of the carboxylic acid 361 using CF3COOH/(CF3CO)2O346 Finally, compound 362 was transformed into the desired natural product 346 after several steps (Scheme 75).362
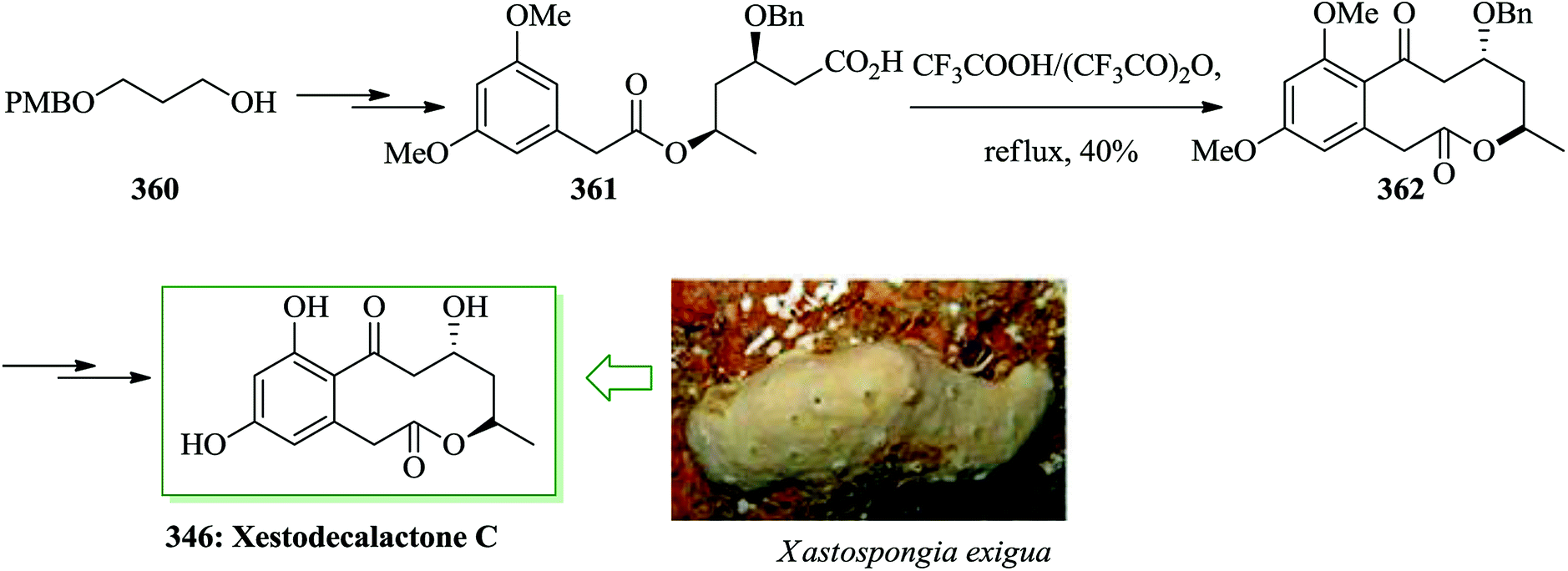 | ||
| Scheme 75 Total synthesis of xestodecalactone C 346. | ||
The curvularins are octaketides composed of a 12-membered macrolide framework fused to a 1,3-dihydroxybenzene scaffold. (11β)-11-Methoxycurvularin 363 is a member of the curvularin group extracted from the mycelium of the hybrid strain ME 005 obtained from Penicillium citreoviride 4692 and 6200 (ref. 363) that demonstrated significant cytotoxicity toward a panel of four human-cancer cell lines.364 A simple and extremely effective enantioselective total synthesis of (11β)-11-methoxycurvularin 363 was accomplished in 2010 by Venkateswarlu and co-workers.365 The synthesis was started with a Cu-catalyzed regioselective ring opening of (2S)-2-methyloxirane 357, a Keck enantioselective allylation and intramolecular FC acylation as key steps. In this strategy, oxirane 357 was used as the starting material, which was converted into compound 364 upon several steps. The corresponding macrolide 365 was obtained in 41% yield via intramolecular FC reaction of the carboxylic acid 364 in the presence of CF3COOH/(CF3CO)2O. Next, demethylation of 365 using freshly synthesized AlI3 afforded the natural product 363 in 96% yield (Scheme 76).365
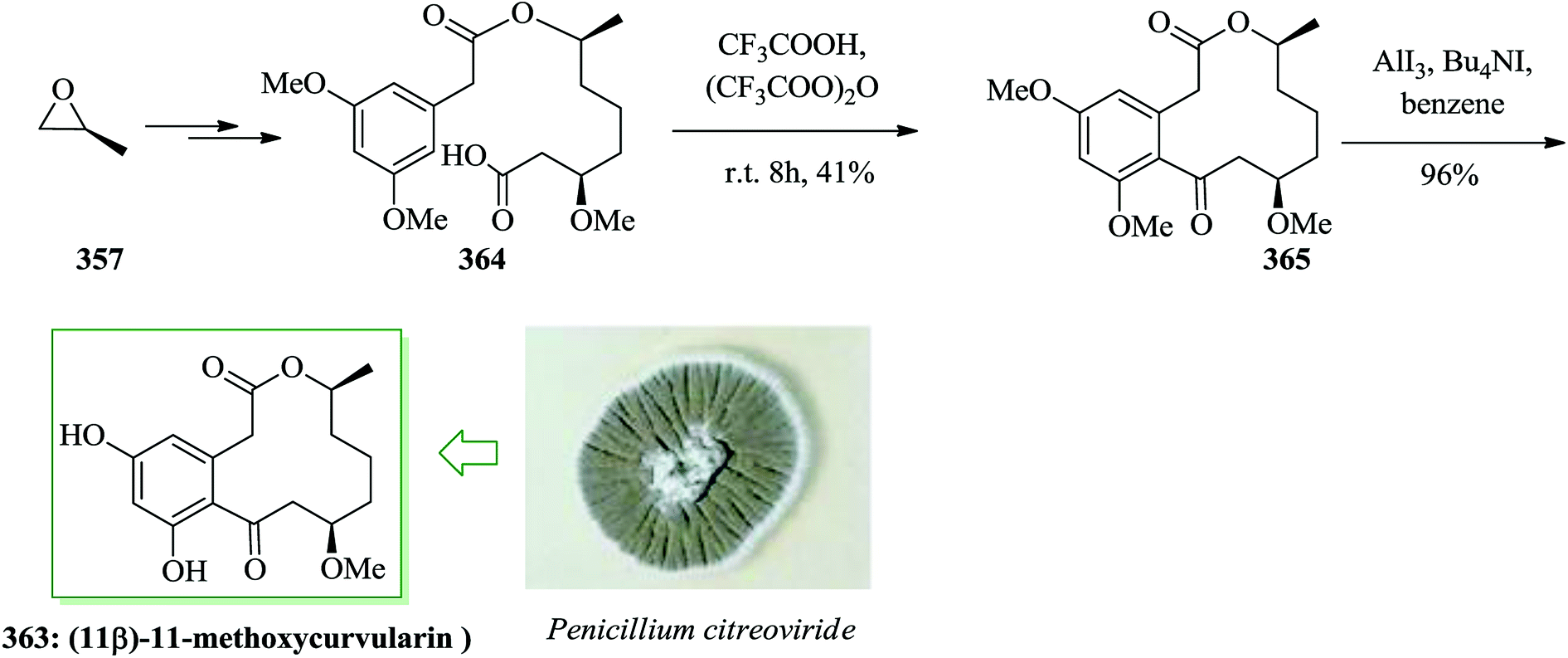 | ||
| Scheme 76 Total synthesis of (11β)-11-methoxycurvularin 363. | ||
Dipterocarpaceae is a rich source of a wide range of biologically potent oligostilbenoids.366,367 Not surprisingly, many remarkable pharmacological functions of this group were also identified, including antifungal, anti-inflammatory, antibacterial, and anticancer properties.368–371 Isolation of a novel oligostilbenoid containing strong cytotoxicity, diptoindonesin G, from the tree bark of Hopea mengarawan was demonstrated by Syah et al.372 This natural product exhibits active immunosuppressive properties.373 A very significant total synthesis of diptoindonesin G 366 was developed using a domino dehydrative cyclization/intramolecular FC acylation/regioselective demethylation reaction of aryloxyketone 371 in the presence of boron trichloride, in which the tetracyclic 6H-anthra[1,9-bc]furan-6-one framework was generated through the 3-arylbenzofuran in a one-pot fashion. Accordingly, a coupling reaction of 367 and 368 took place, which was followed by dehydrative cyclization and subsequent direct arylation at the C2 position of the benzofuran scaffold to afford the key intermediate 369. The latter was then transformed into compound 370 upon treatment with NaOH and then TFAA by intramolecular FC acylation reaction. Upon treatment with BBr3 the latter was converted into G 366 in 93% yield (Scheme 77).374
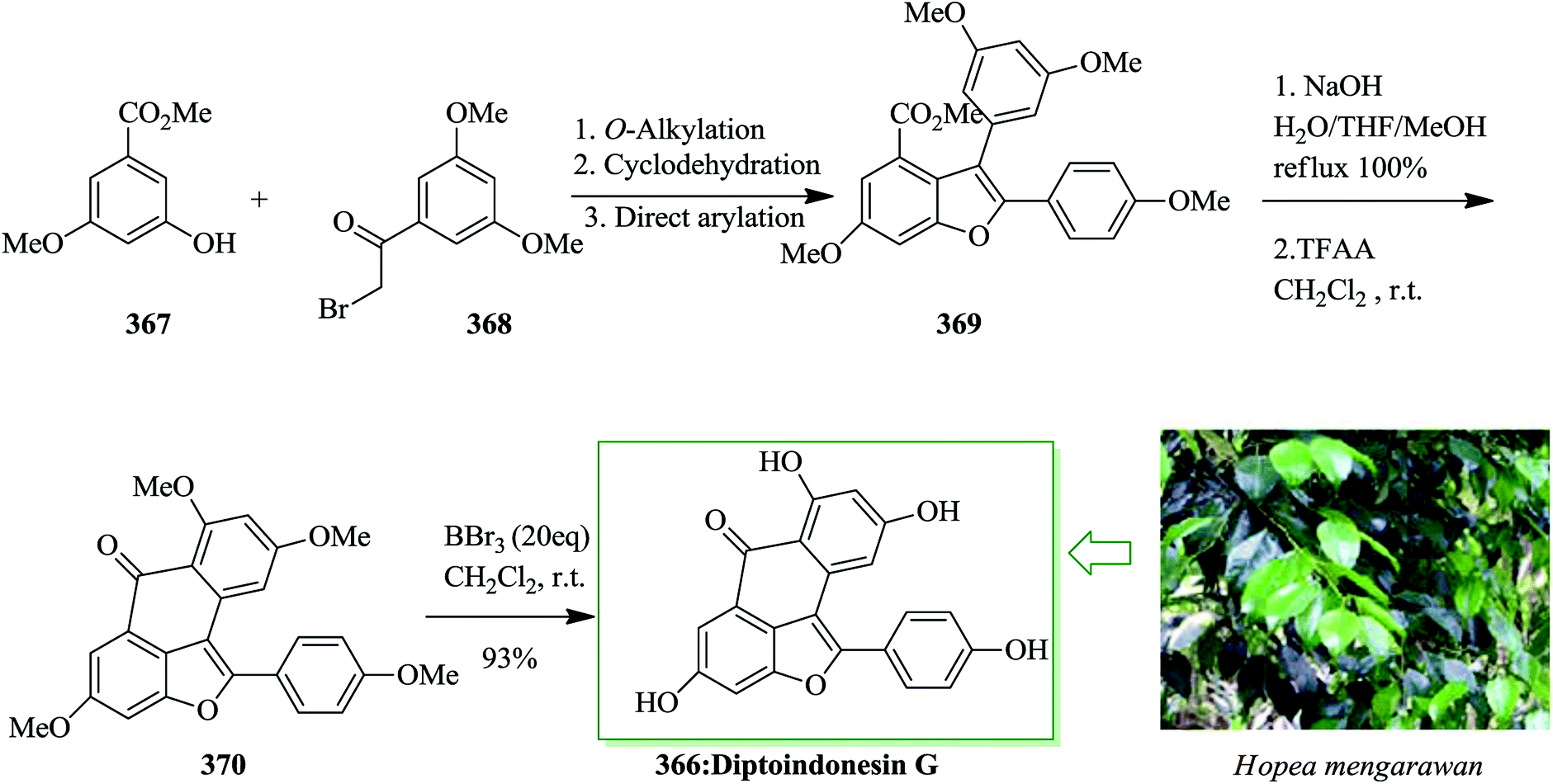 | ||
| Scheme 77 Total synthesis of diptoindonesin G 366. | ||
The same strategy was applied for the synthesis of diptoindonesin G 366 using compound 371 instead of 370 as the precursor. Compound 371 was treated with BCl3 in CH2Cl2 to give the demethylated compound 372 in 95% yield.375–378 Finally, the latter was transformed into the desired natural product diptoindonesin G 366 upon several steps (Scheme 78).374
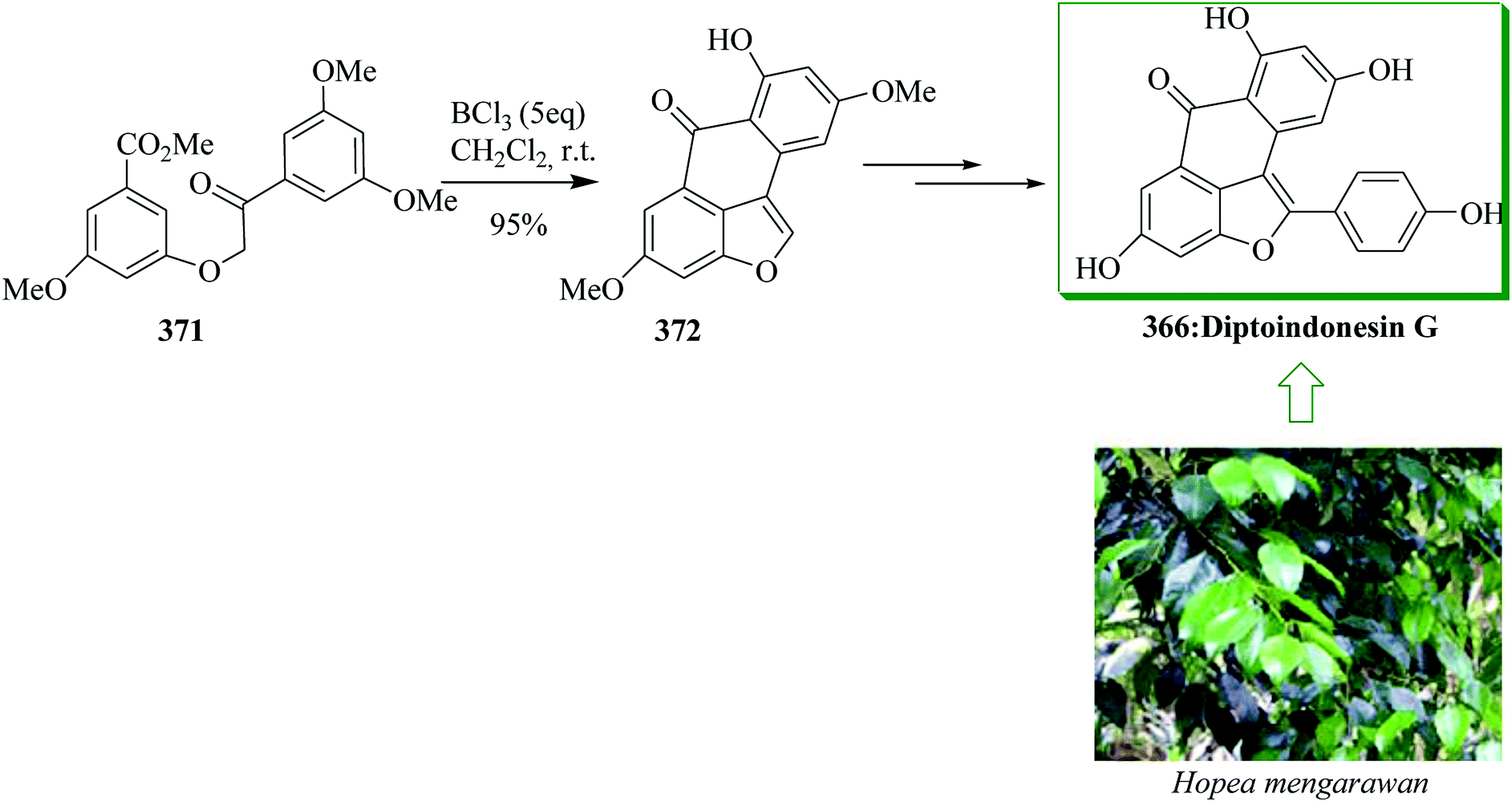 | ||
| Scheme 78 Total synthesis of diptoindonesin G 366. | ||
Kinamycin antibiotics379,380 have been extracted from of Streptomyces murayamaensis culture broth381,382 with antitumor and antiviral properties.382 The structure was at first identified as N-cyanobenzo[b]carbazoloquinone. The structure of so-called ‘prekinamycin’ extracted by Gould was revised to, temporarily, diazobenzo[b]fluorene 373.383 This compound with the structure 373 was extracted as a metabolite provided by S. murayamensis mutant MC2 (ref. 384) and finally named as prekinamycin.385 Total synthesis of prekinamycin 373 was accomplished through Suzuki coupling reaction of naphthaleneboronic acid and a bromobenzene derivative, intramolecular FC reaction of 2-(naphthalen-2-yl)benzoic acid, and diazotization in ten steps starting from 3,5-dimethylphenol 374, in 7% yield. Initially, 3,5-dimethylphenol 374 was converted into carboxylic acid 375 upon several steps. The intramolecular FC reaction of 375 with (CF3CO)2O386 or POCl3/K2CO3 (ref. 387) in a one-pot fashion resulted in a complex mixture. Transformation of acid 375 to acid chloride 376, followed by intramolecular FC reaction in the presence of tin(IV) chloride, afforded the key MOM-deprotected benzo[b]fluorenone 377 in merely 4% yield. Finally, compound 377 was transformed into the desired natural product prekinamycin 373 in 47% yield (Scheme 79).388
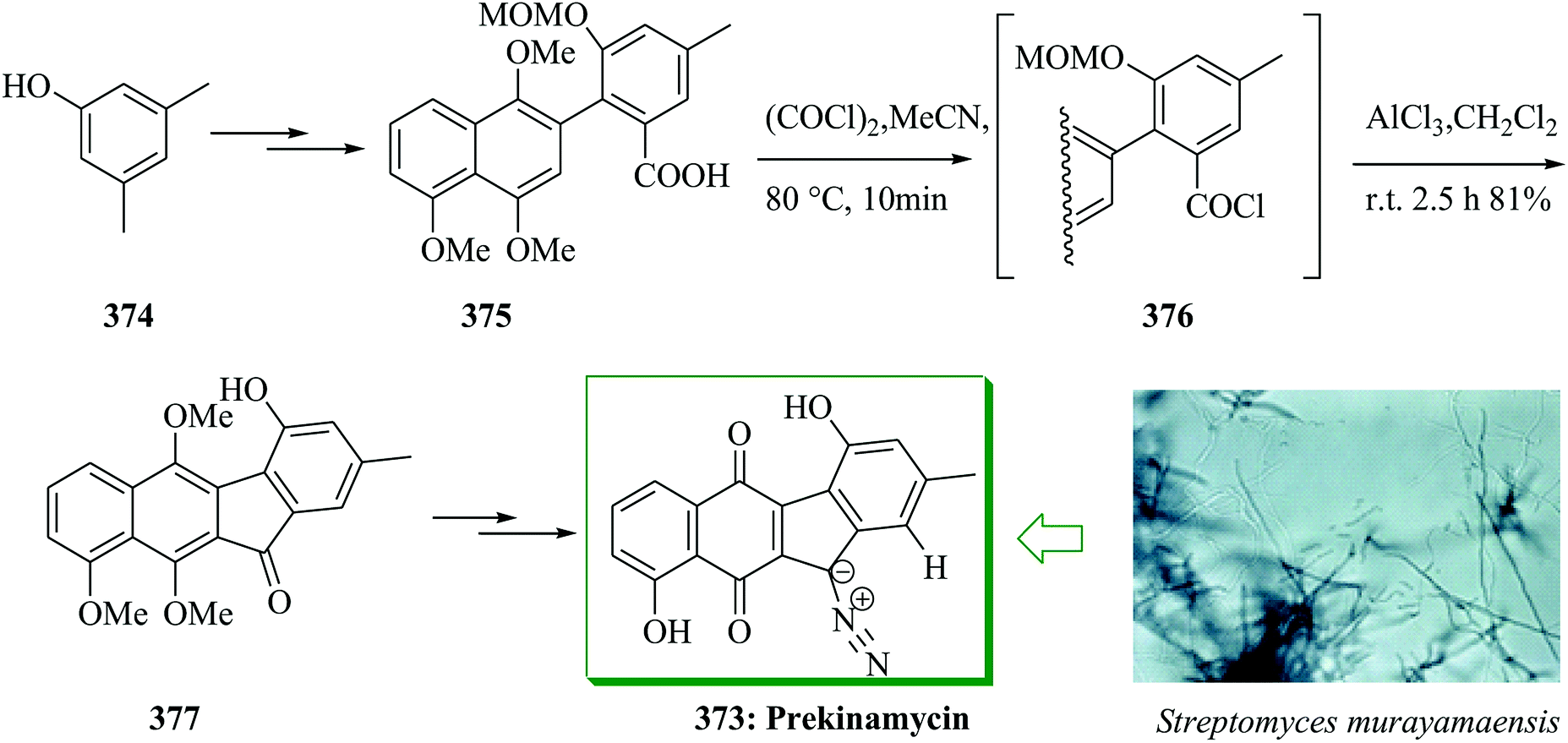 | ||
| Scheme 79 Total synthesis of prekinamycin 373. | ||
Diazobenzofluorene is a naturally occurring compound, classified as a group of structurally complex molecules bearing a tetracyclic (ABCD) scaffold along with a diazo group. Some epoxykinamycins contain FL-120B and the closely related FL-120B′ 378.389 Monomeric diazobenzofluorenes were found to exhibit antitumor activity. Enantioselective synthesis of FL-120B′ was accomplished employing Sharpless asymmetric epoxidation, Stille cross-coupling, and intramolecular FC reactions as key steps. The synthesis of FL-120B′ represents the first total synthesis of an epoxide-comprising, diazobenzofluorene natural product. Firstly, quinone 379 gave carboxylic acid 380 upon several steps. Then, carboxylic acid 380 in the presence of trifluoroacetic anhydride (TFAA) underwent an intramolecular FC acylation, affording the corresponding ketone 381. Finally, ketone 381 was transformed into FL-120B′ 378 after several steps (Scheme 80).390
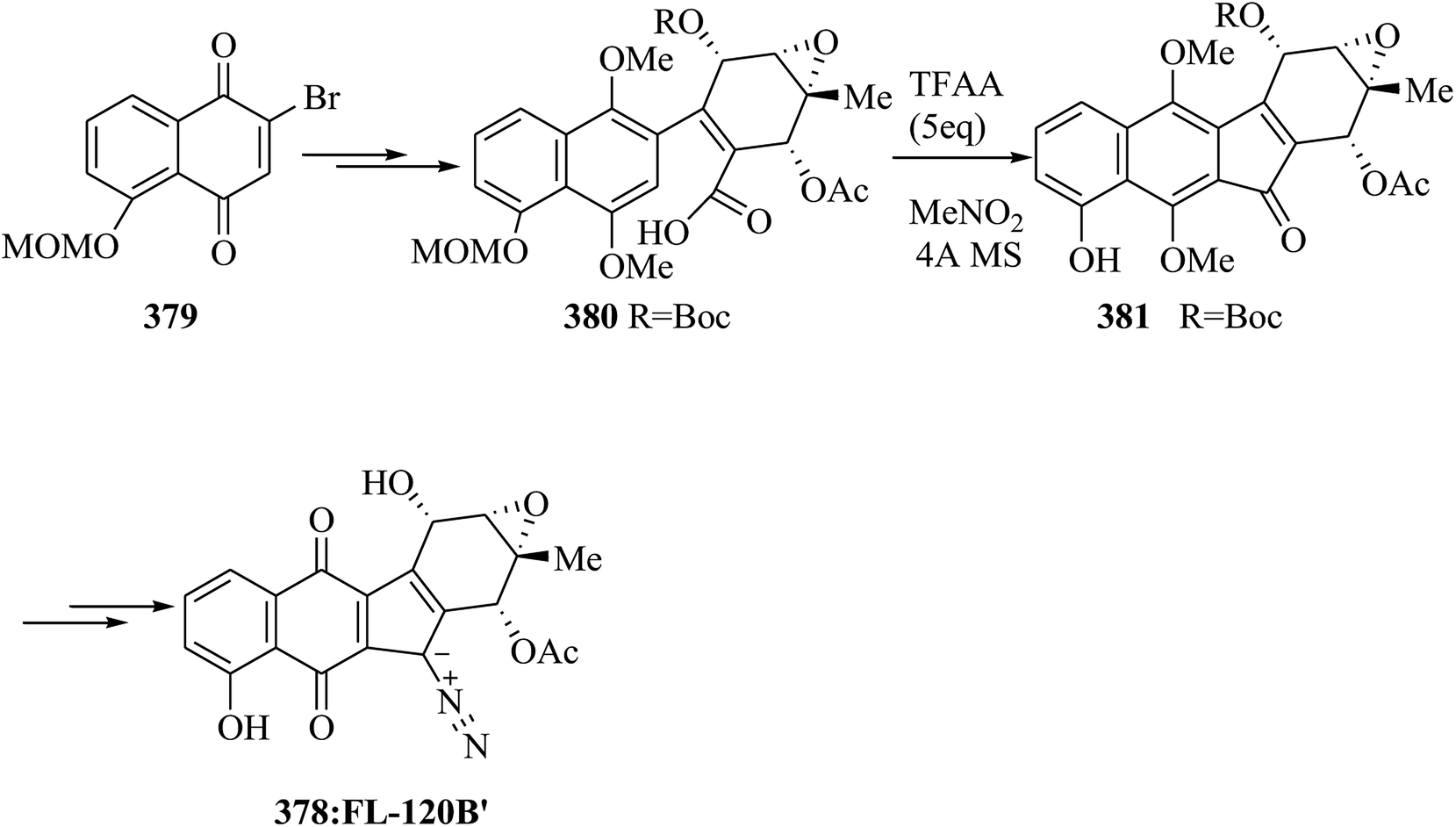 | ||
| Scheme 80 Total synthesis of FL-120B′ 378. | ||
The trikentrins 382 and 384 were extracted from the marine sponge Trikentrion flabelliforme by Capon391 and exhibited antibacterial properties. In a concise total synthesis of (+)-cis-trikentrin A, initially 4-ethyl-7-vinylindole underwent nickel(II)-mediated enantioselective hydrovinylation to afford the desired 2-but-3-enyl derivative 384a in excellent yield (99%) and high ee (96%). Compound 384a was converted into the corresponding carboxylic acid 385a in several steps. The latter was subjected to an intramolecular FC reaction and a Wittig reaction affording the exo-methylene compound 387a in about 70% yield from the acid 385a. Subsequently, compound 387a was converted into the natural product (+)-cis-trikentrin 382 after several steps. Noticeably, synthesis of a more complex natural product, (+)-cis-trikentrin B, demonstrates the application of the hydrovinylation product, 384b, and a late-step functionalization of a 6,8-dimethylcyclopent[g]indole nucleus. Employing the aforementioned protocol, 384b was transformed into (+)-cis-trikentrin-B 383 after several steps (Scheme 81).392
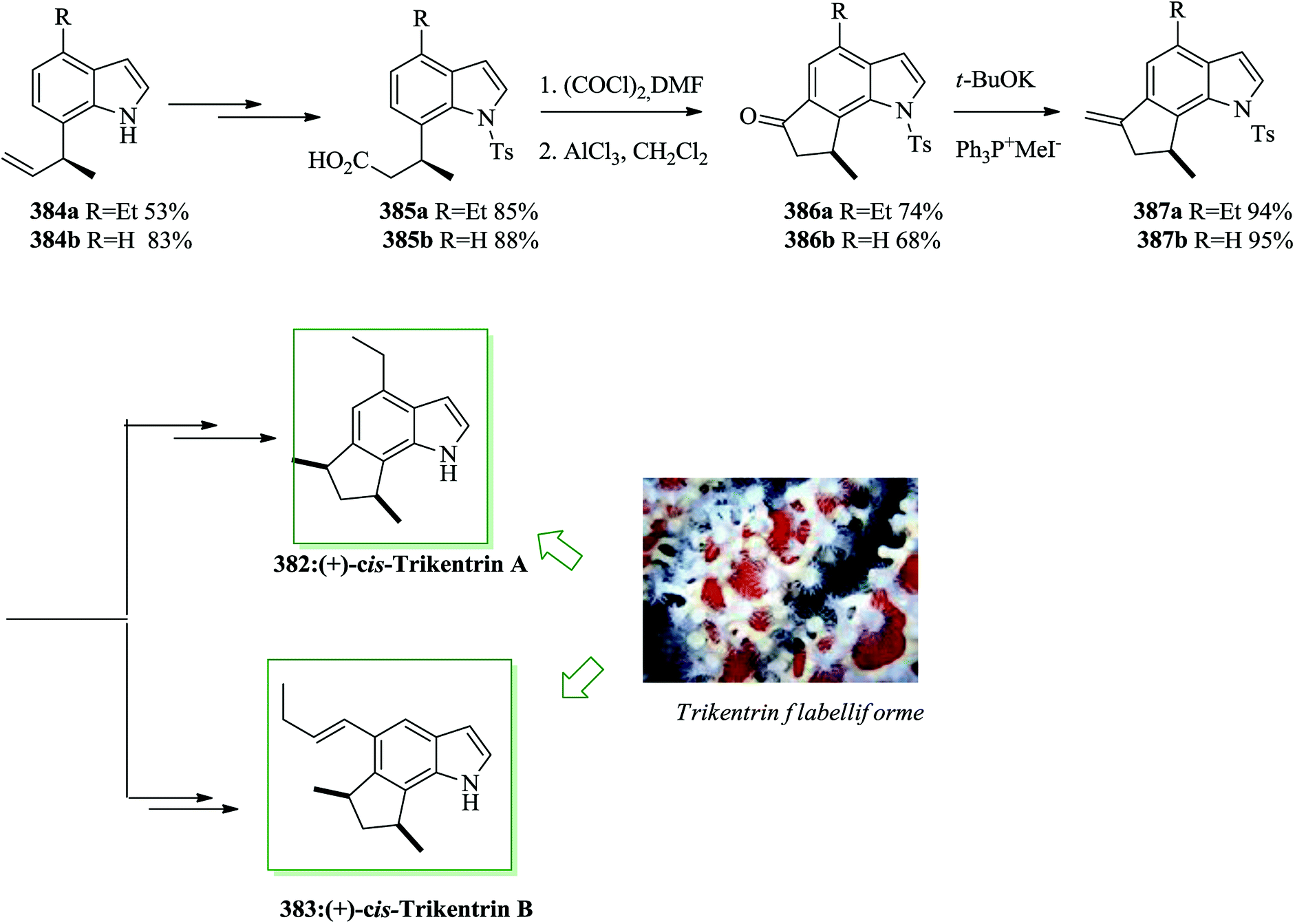 | ||
| Scheme 81 Total synthesis of (+)-cis-trikentrin 382 and (+)-cis-trikentrin-B 383. | ||
Amaryllidaceae isocarbostyril alkaloids have been attractive synthetic targets for organic synthetic chemists during the last two decades.393,394 The isolation of trans-dihydronarciclasine 388 from the Chinese medical plant Zephyranthes candida was demonstrated in 1990.395 An extremely enantioselective and effective total synthesis of trans-dihydronarciclasine 388 in an entirely substrate-controlled strategy from easily accessible chiral starting materials was developed in which the desired target 388 was obtained in 16% overall yield. The total synthesis of trans-dihydronarciclasine 388 was commenced by preparing triacetate 390 from the enantiomerically enriched allylic alcohol 389 via several steps. Using 1.1 equivalents of Tf2O and 1.5 equivalents of 2-chloropyridine, and heating at 35 °C, the product 392 was obtained with very high regioselectivity (12.5:1) and in 76% yield. After two steps, the desired target (+)-388 was obtained (Scheme 82).396
 | ||
| Scheme 82 Total synthesis of trans-dihydronarciclasine 388. | ||
Colchicine,397,398 extracted from Colchicum autumnale, is a cytostatic drug that strongly binds to tubulin, the main constitutive protein of microtubules.399 Colchicine and its structural analogues, for example allocolchicine400 and combretastatin A-4,401 symbolize promising lead structures for the development of new anticancer agents addressing the colchicine binding site of tubulin.402,403 and 5,6,7-trimethoxy-4-(1-methyl-1H-indol-6-yl)-2H-chromen-2-one,404 respectively that were known to prevent microtubule assembly in vitro at nanomolar concentrations. Schmalz and co-workers in 2012 provided a unique group of indole-comprising allocolchicine analogues. Using 3,4,5-trimethoxy-phenylpropionic acid 394, the target compounds rac-393a and rac-393b were obtained in 11 steps with overall yields of 14% and 3%, respectively, through a Suzuki cross-coupling reaction and a FC cyclization reaction as the key carbon–carbon bond-forming reactions. Firstly, 3,4,5-trimethoxyphenylpropionic acid 394 afforded a clear solution including acid chloride 395 upon several steps. In a first attempt to induce the projected FC cyclization, ZnCl2 was added to a crude solution of 395 in dichloromethane at ambient temperature. The corresponding ketones 396a and 396b were obtained in merely 5% yield (as a 4:1 mixture). Finally, compounds 396a and 396b provided the indole-obtained allocolchicinoids rac-393a and rac-393b, respectively, after several steps (Scheme 83).405
 | ||
| Scheme 83 Total synthesis of allocolchicinoids rac-393a and rac-393b. | ||
The kibdelones are a unique group of bioactive heterocyclic polyketides provided by a rare soil actinomycete, Kibdelosporangium sp. (MST-108465). Significantly, kibdelones A–C were found to be active at low nanomolar concentrations against tumor cell lines.406 The total synthesis of kibdelone A 397 was achieved through In(III)-mediated arylation of a heterocyclic quinone monoketal and iodine-catalyzed oxidative photochemical electrocyclization. A one-pot oxa-Michael/FC method was tried using monomethoxy phloroglucinol 398407 to produce the first simplified analogues of kibdelones. Under the optimal conditions, using the extremely activated HFIP ester 399 resulted in the finding that tetrahydroxanthone construction based on basic conditions was possible in a one-pot method, avoiding extraction of the acid- and heat-sensitive vinylogous carbonate 400. Furthermore, the oxa-Michael reaction progressed in a site-selective manner at ambient temperature; however, the FC cyclization reaction needed heating to 60 °C using K3PO4 to provide tetrahydroxanthone 401 in 41% yield. The usage of base was needed for the FC cyclization reaction by the isolation of vinylogous carbonate 400 and its subsequent thermolysis in dimethylformamide (60 °C), which was shown to be unsuccessful. Final deprotection of 401 gave the simplified kibdelone tetrahydroxanthone analogue 402 (Scheme 84).408
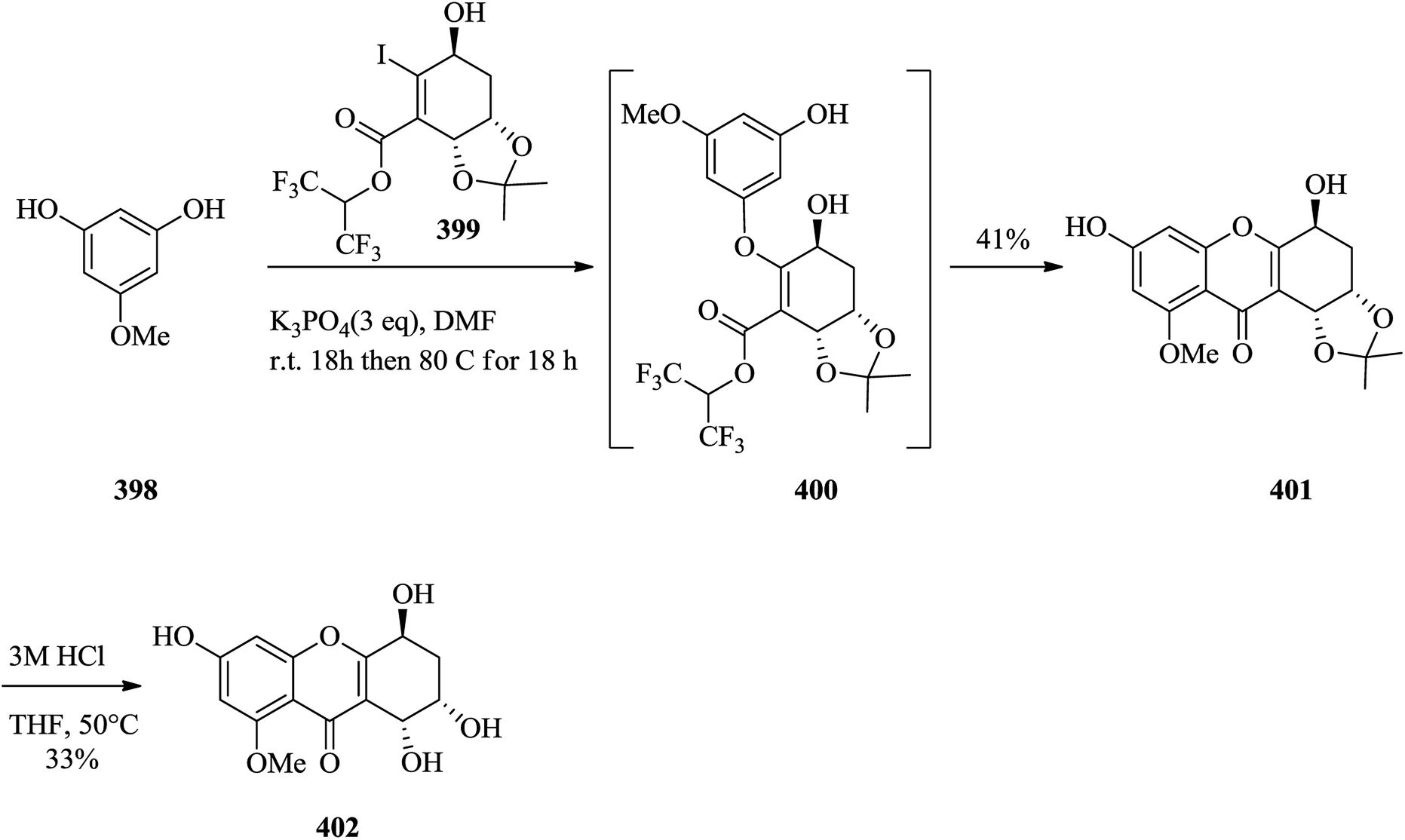 | ||
| Scheme 84 The formation of the simplified kibdelone tetrahydroxanthone analogue 402. | ||
Then, total synthesis of the triol unit of kibdelone A 397 was examined. The reaction of 403 and 404a–c gave the corresponding products 405a–c. Additional FC cyclization was explored employing a one- or two-pot method with the naturally occurring compounds 405a–c based on thermal, Lewis acid-mediated, and N-methylimidazole-improved conditions; however, in these cases merely initiating material, aromatized F-ring, or retro-Michael products were detected. Cyclization reaction was accomplished employing a two-step sequence through saponification/cyanuric chloride activation of vinylogous carbonates 405b and 405c in 52% and 42% yield over two steps, respectively. Lastly, deprotection of the acetonide scaffold of the F-ring and oxidation reaction of the B-ring employing ceric ammonium nitrate (CAN) in H2O/acetonitrile gave kibdelone A 397 in moderate yield (Scheme 85).408
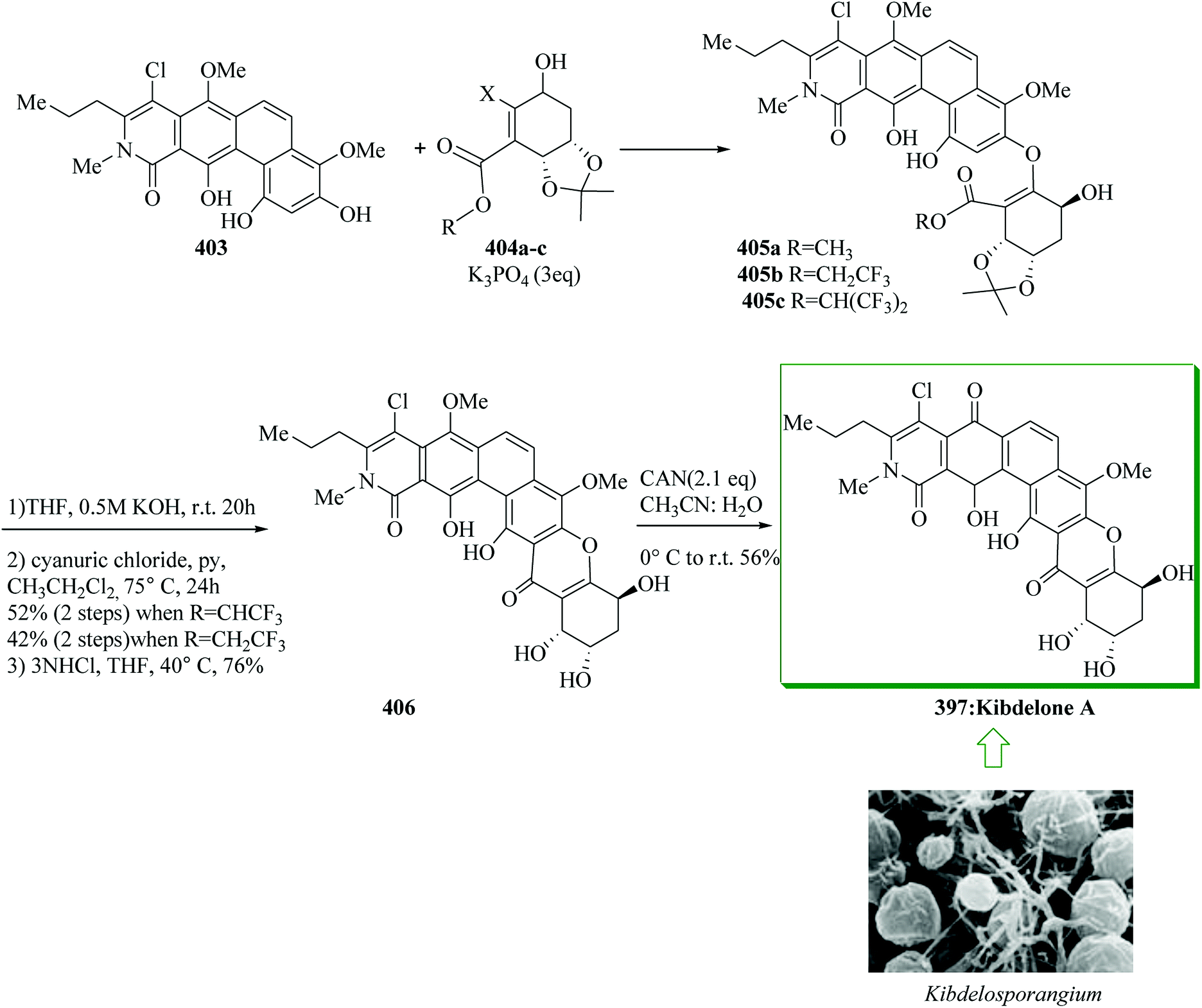 | ||
| Scheme 85 Total synthesis of kibdelone A 397. | ||
The Caribbean octocoral Pseudopterogorgia elisabethae is a chemically prolific species that has attracted the attention of scientists.409 Sandresolide B 407410–412 was first reported in 1999.413 The total synthesis of the diterpenoid sandresolide B from a common furan framework was achieved and reported in 2013 by Trauner and co-workers. Key stages contain Pd-catalyzed carbonylation, lanthanide mediated ring closure, Myers alkylation reaction, intramolecular FC acylation, photooxygenation, and a Kornblum–DeLaMare rearrangement.414 Total synthesis of 407 was started from ketone 408, which was converted into compound 409 upon several steps. Next, compound 409 afforded acid 410 upon several steps. Using the key acid 410, the seven-membered ring of the sandersolides was provided through an intramolecular FC acylation reaction. Activation of the acid with trifluoroacetic anhydride followed by gentle heating with zinc chloride afforded the corresponding product 411. It should be mentioned that short reaction times and stoichiometric ZnCl2 were key to this ring closure. Finally, sandresolide B, 407, was obtained from 411 in 51% yield over two seps (Scheme 86).414
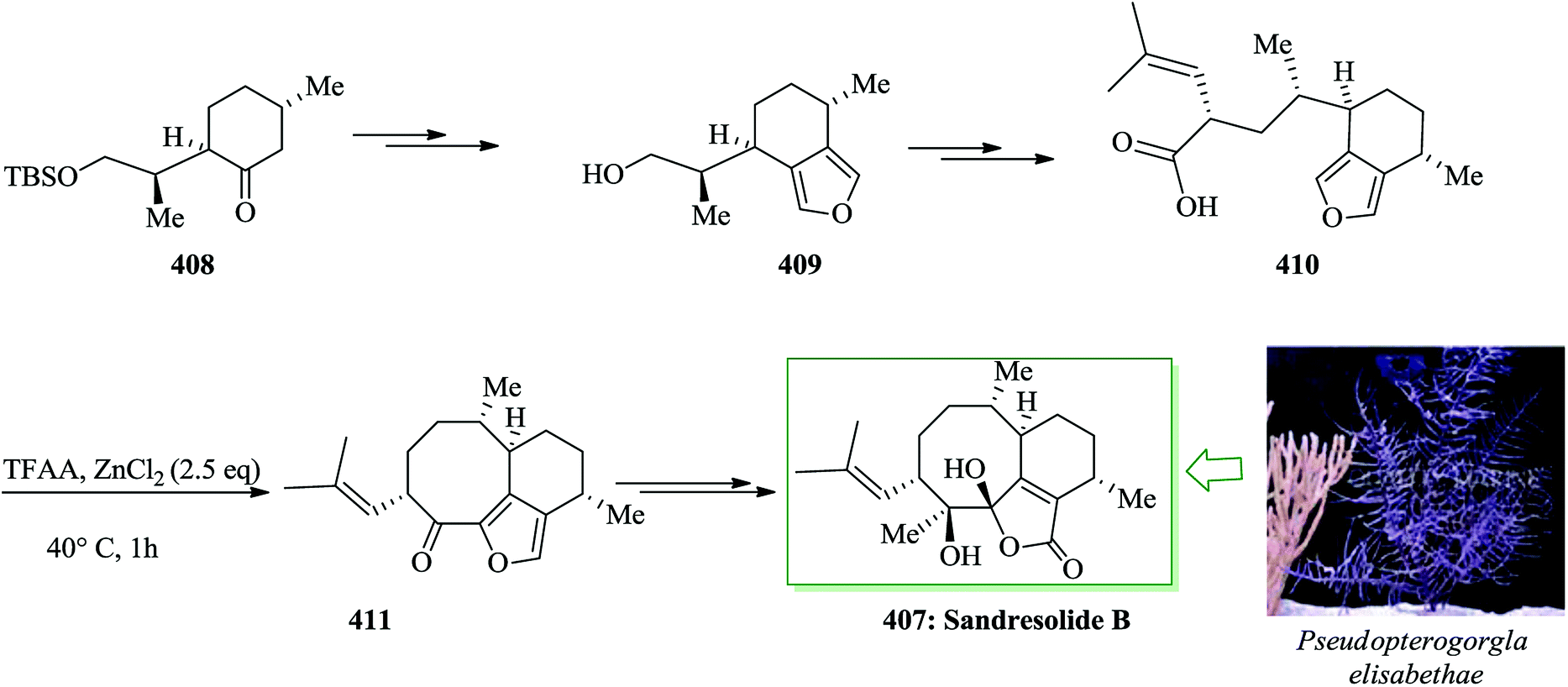 | ||
| Scheme 86 Total synthesis of sandresolide B 407. | ||
Daphenylline 412, extracted from the fruit of Daphniphyllum longeracemosum by Hao and co-workers in 2009, is the first member of the Daphniphyllum alkaloids that includes a benzene ring in the unit structure.415 Daphenylline contains a rearranged 22-norcalyciphylline A type framework,416 the highly fused hexacyclic system of which has generated significant interest in the chemical community. Total synthesis of (−)-daphenylline, a hexacyclic Daphniphyllum alkaloid, was accomplished in 2016 by Fukuyama and co-workers.417 In this approach, an asymmetric Negishi coupling reaction, an intramolecular FC reaction, Sonogashira coupling and Claisen rearrangement are considered as key steps. Total synthesis of (−)-daphenylline 412 was initiated from 6-methoxyindan-1-one 413, which was converted into carboxylic acid 414 after several steps. Next, an intramolecular FC reaction of 414 was achieved by reaction with trifluoroacetic anhydride (TFAA) and trifluoroacetic acid (TFA) to provide cyclic ketone 415. Finally, daphenylline 412 was obtained after several steps (Scheme 87).417
 | ||
| Scheme 87 Total synthesis of daphenylline 412. | ||
In 2010, sparstolonin B (SsnB) 416 was first extracted from the Chinese herb Spaganium stoloniferum by Liang et al.418 In 2015, Sun and co-workers reported that sparstolonin B can prevent high-fat-diet-induced obesity in rats, and also prevents LPS-induced production of MCP-1, interleukin-6, and tumor necrosis factor-α in 3T3-L1 adipocytes.419 Shibata and co-workers in 2017 developed a unique method for the syntheses of sparstolonin B (SsnB) 416 with an overall yield of 38% over 10 steps from the affordable, market-accessible initiating precursor methyl 5-nitro-2-furoate. In this method, Diels–Alder reaction and intramolecular FC reaction were found as the key steps. Total synthesis of 416 was started with aromatic substitution of methyl 5-nitro-2-furoate with sodium 4-methoxybenzenolate as a nucleophile to afford the cyclic anhydride 419 in 95% yield. The intramolecular FC reaction of anhydride 419 in benzene afforded a mixture of the corresponding ester 420 and a side-product 420b, provided via elimination of an O-methyl substituent.420 Without more purification, reaction of the product mixture with iodomethane afforded intermediate 420 in 83% yield over two steps. Lastly, compound 420 gave sparstolonin B (SsnB) 416 upon several steps (Scheme 88).421
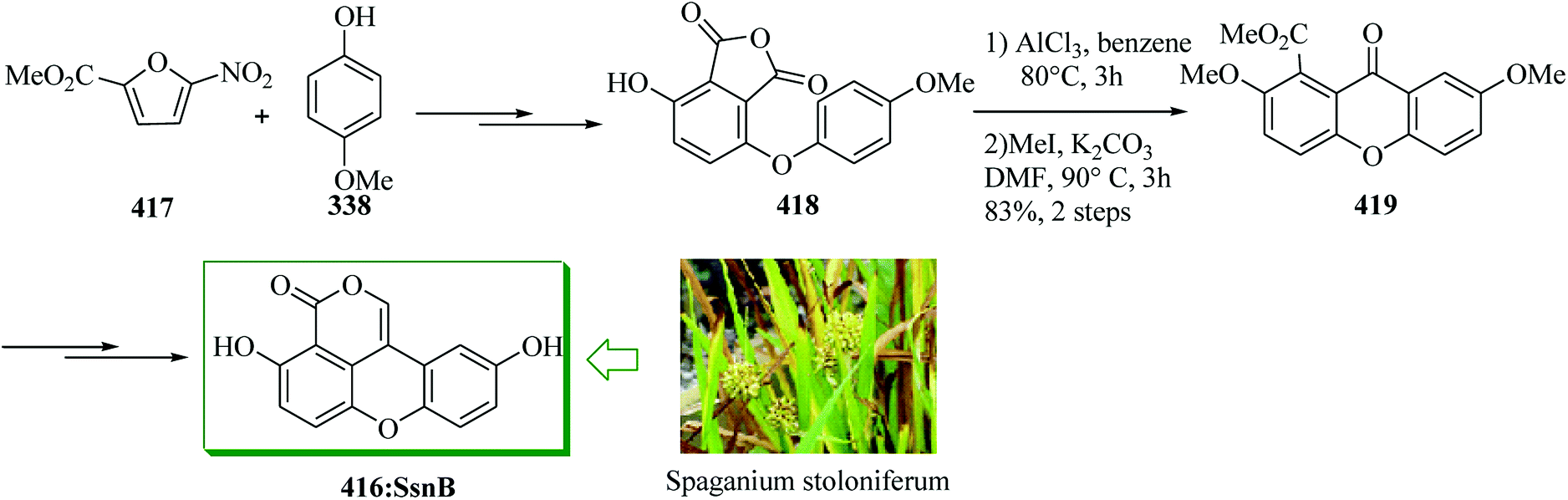 | ||
| Scheme 88 Total synthesis of sparstolonin B (SsnB) 416. | ||
2.4. Intermolecular acylation
The lignan lactone picropodophyllin 420 has been obtained from various species of Podophyllum.422,423 Gensler and co-workers demonstrated total synthesis of picropodopyllin. Initially, 3, 4-methylenedioxy-3′,4′,5′-trimethoxybenzophenone 422 was synthesized from the FC reaction between methylenedioxybenzene 421 and trimethoxybenzoyl chloride 422. Subsequently, compound 423 provided picropodophyllin 420 after several steps (Scheme 89).424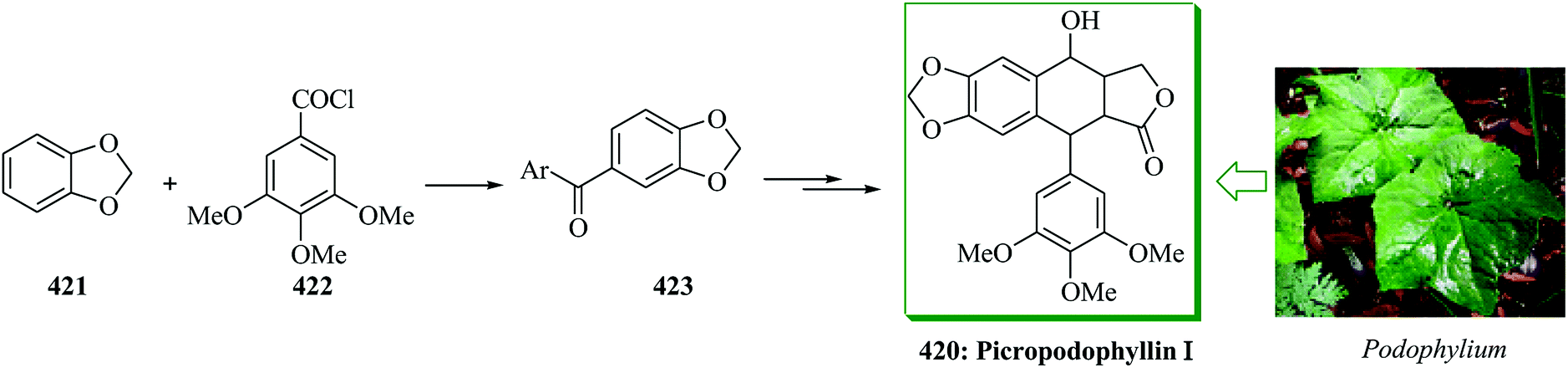 | ||
| Scheme 89 Total synthesis of picropodophyllin 420. | ||
Isoflavones425 are the most abundant subset of the flavonoid group of compounds, which also includes pterocarpans, rotenoids,426 and coumestans.427 Structurally, isoflavones are highly functionalized and oxygenated derivatives of 3-phenylchromans. Jamaicin 424a has been extracted from Piscidia erythrina bark root.428 Calopogonium isoflavone-B 424b was extracted from ether extracts of Calopogonium mucunoides seeds. The FC acylation reaction has been examined on various acid-sensitive substrates. Based on the proper conditions of changing Lewis acids, solvents, and reaction temperatures, the acylation indeed occurred, therefore obviating the necessity for functional group protection-deprotection sequences. By the use of these methods, the naturally occurring isoflavones jamaicin 424a and calopogonium isoflavone-B 424b have been obtained. Jamaicin 424a and calopogonium isoflavone-B 424b have been obtained from the deoxybenzoin systems 427b and 427a, respectively, by any of a wide range of excellent formylation methodology.429,430 These crucial and sensitive deoxybenzoins were synthesized directly via the FC acylation reaction between 2,2-dimethyl-5-hydroxychrom-3-ene 425 and a homologated piperonal-426a or sesamol-426b obtained acid halide (Scheme 90).431
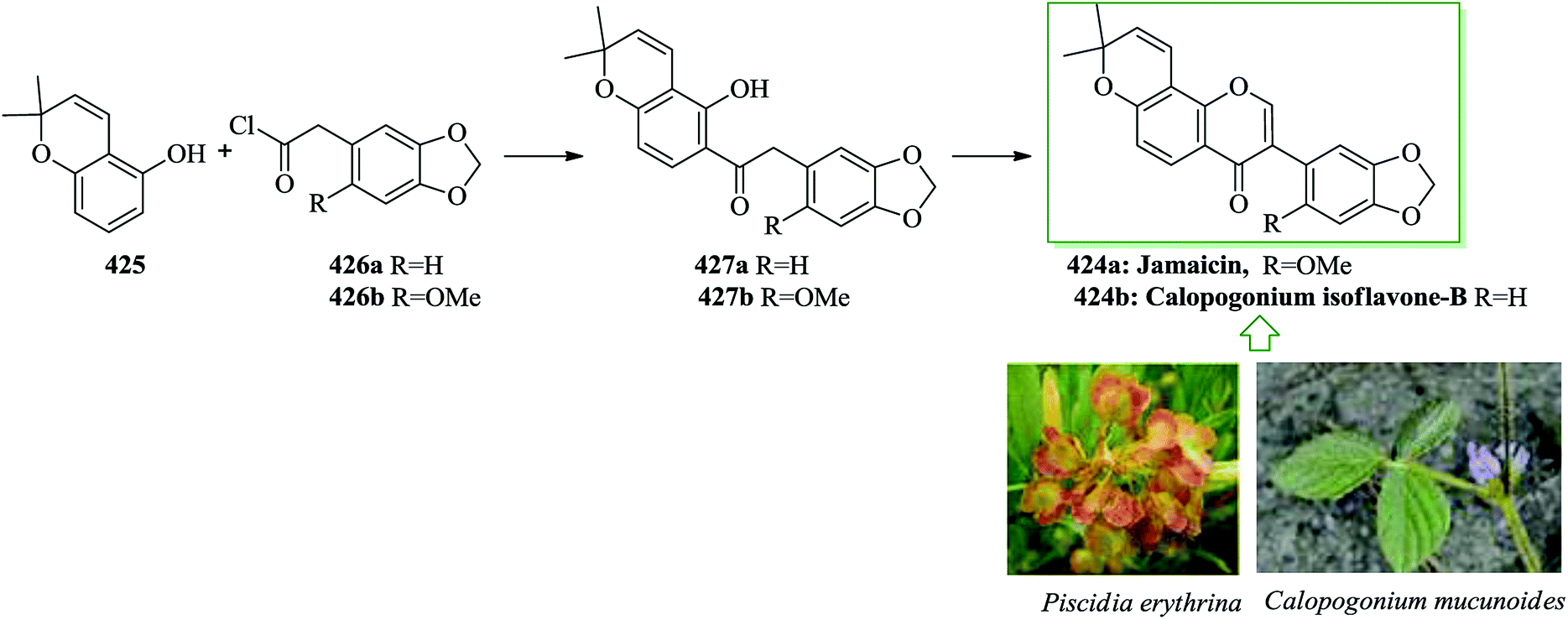 | ||
| Scheme 90 Total synthesis of jamaicin 424a and calopogonium isoflavone-B 424b. | ||
The tricyclic diterpenes sempervirol and totarol incorporate a trans-fused octahydrophenanthrene core as the basic carbocyclic scaffold. Totarol includes a modified abietane framework and has been extracted as a major constituent of the heartwood of Eodocarpus totara.432 The diterpene sempervirol also contains a rearranged abietane framework and has been extracted433 from the resin from Cupressus sempervirens. Mukherjee and co-workers developed stereocontrolled total synthesis of (±)-totaryl methyl ether 428 and (+)-semperviryl methyl ether 429.434 Firstly, 2-acetyl-6-methoxynaphthalene 430 was converted into compound 431 via several steps. The relevant methyl ester 431 was reacted with AcCl in the presence of anhydrous aluminium chloride to provide the 1-acetyl compound 432 in 84% yield. FC acylation reaction of 2-methoxynaphthalene afforded435 a mixture of 6-acyl and 1-acyl derivatives. Compound 432 provided the trans-fused ketones 434 and 433 via several steps. Huang–Minlon reduction436 of 433 gave (+)-totaryl methyl ether 428 in 84% yield. An analogous reduction of the ketone 434 provided octahydrophenanthrene 435a. FC acylation reaction between 435a and AcCl using tin(IV) chloride gave the methyl ketone 435b as the only product in 80% yield. Reaction of 435b with MeMgI and catalytic hydrogenolysis of the resulting carbinol in acetic acid with a few drops of perchloric acid gave (+)-semperviryl methyl ether 429 in 80% yield (Scheme 91).434
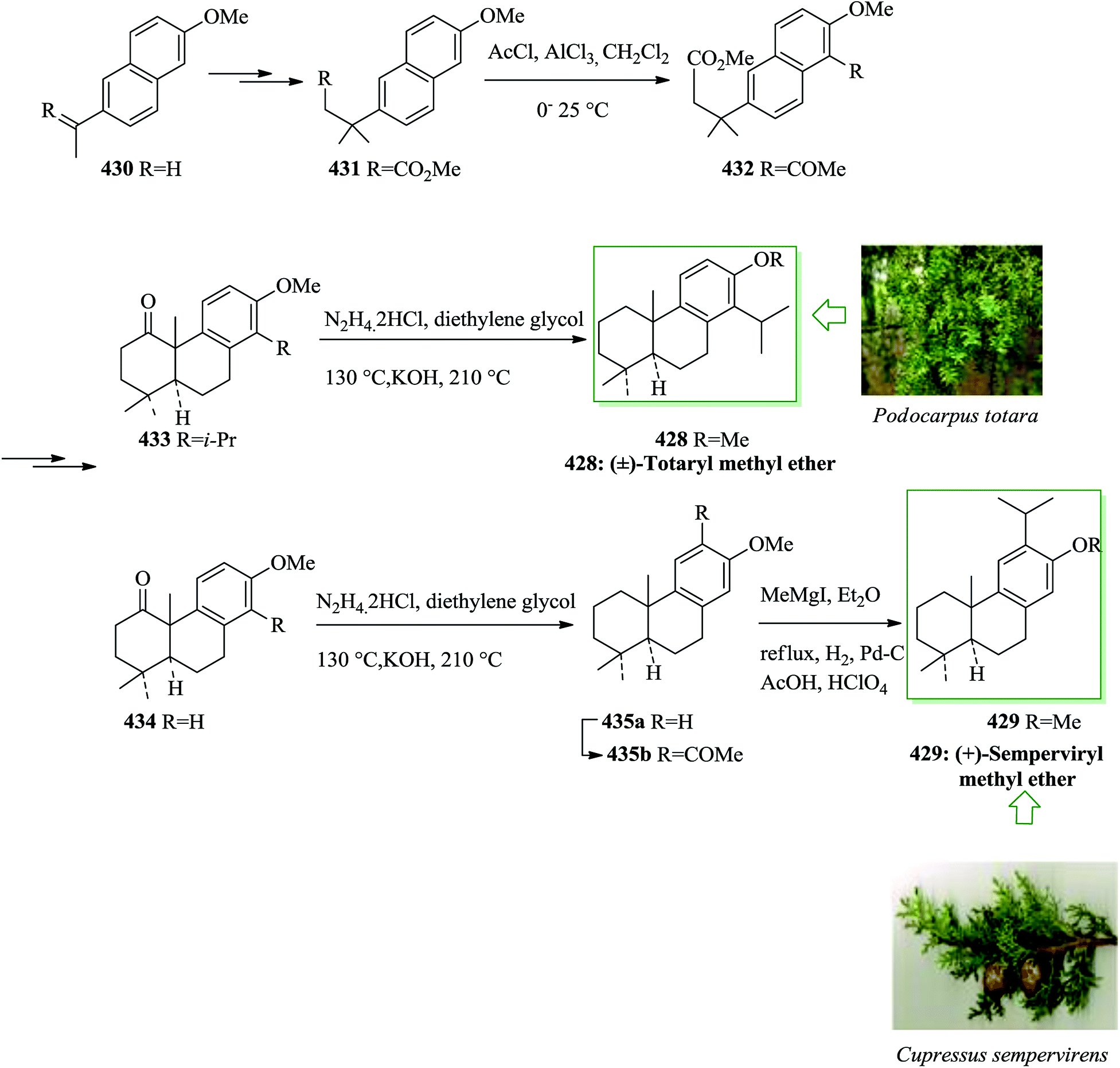 | ||
| Scheme 91 Total synthesis of (±)-totaryl methyl ether 428 and (+)-semperviryl methyl ether 429. | ||
Duocannycin A 436 437–439 extracted from Streptomyces sp. was found to be an enormously active antitumor antibiotic that is effective against several strains of experimental cancer cell lines. Total synthesis of 436 was accomplished using methoxycarbonylation of the C4-position of the 5-aminoindoline by way of the isatin and subsequent Dieckmann cyclization to the methyl 2-methylindoxyl-2-carboxylate as key steps. Total synthesis of 436 was started from 6-methoxy-3-methylaniline 437. Next, one carbon unit was introduced effectively using isatin derivative 439 obtainable via the FC reaction of N-alkyl derivative 438 with oxalyl chloride. Finally, compound 439 was converted into dl-436 after several steps (Scheme 92).440
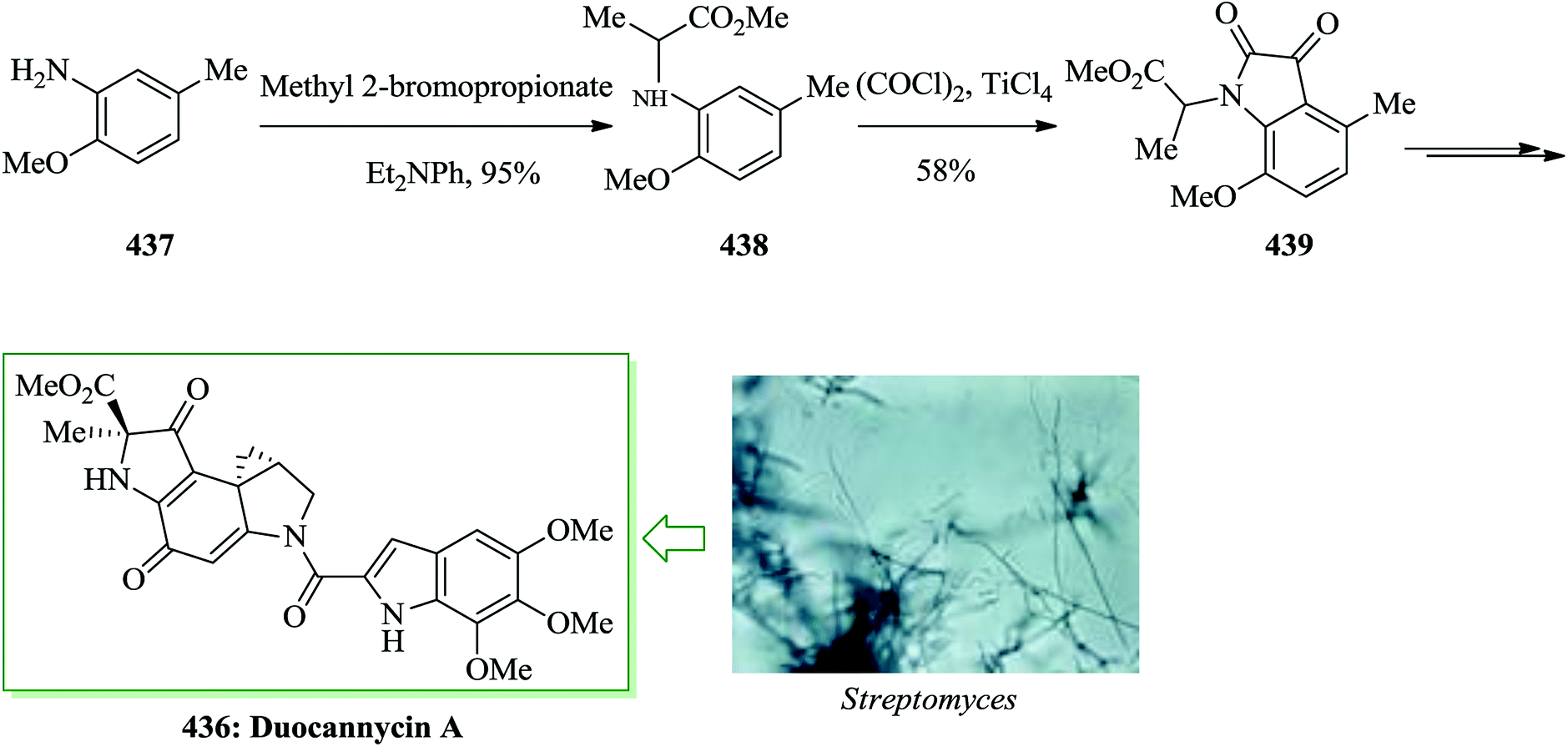 | ||
| Scheme 92 Total synthesis of duocannycin A 436. | ||
Ikimine A has been extracted from Niphates sp.441 The cytostatic ikimine A 440 was synthesized via acylation of 2-methyl-3-(2-thiophene)-L-propylacetate 441, reductive de-sulfurization, and functional group transformation to methyl branched racemic and nonracemic chiral 3-alkylpyridines. SnCl4 mediated the acylation reaction between 2-methyl-3-(2-thiophene)-L-propylacetate 441 and the acid chloride 442 to afford the ketone 443. Reaction with TiCl4 or H3PO4 as FC catalysts or acylation reaction with in situ made acyl trifluoroacetates using phosphoric acid442 afford lower yields. Lastly, compound 443 afforded ikimine A (α-methyl-3-pyridinedodecanal-O-methyloxime) 440 via several steps (Scheme 93).443
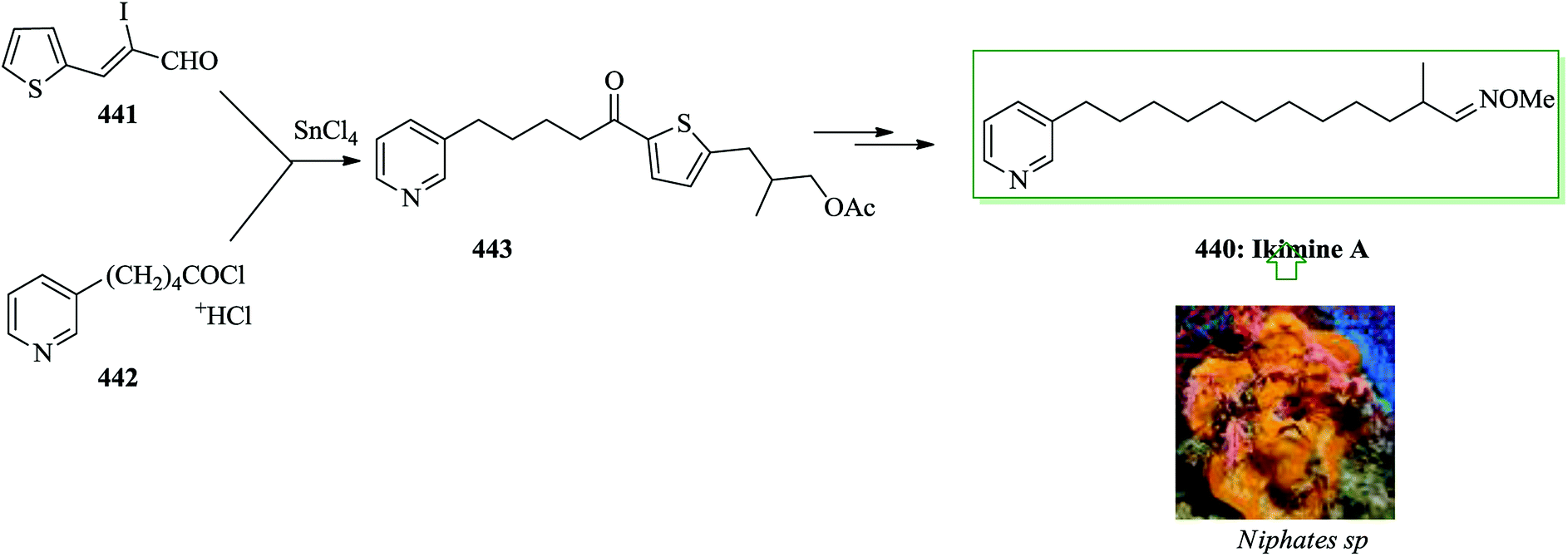 | ||
| Scheme 93 Ikimine A (α-methyl-3-pyridinedodecanal-O-methyloxime) 440. | ||
Tolypocladin 444 has been extracted from Tolypocladium inflatum DSM 915 and identified as 3-methyl-5,6(or 7),8-trihydroxy-2-aza-anthraquinone-(9,10).444 Total synthesis of the microbial metabolite tolypocladin 444 and isomeric isotolypocladin 445 was developed through FC acylation reaction via the condensation of 2-aza-anthraquinone-(9,10) ring. The total synthesis was commenced by the reaction of l,2,4-trimethoxy-benzene 229 and 2-methyl-pyridine 4,5-dicarboxylic acid anhydride 446 based on FC condition reactions. The resultant key products 447 and 448 can be separated though recrystallization. Therefore, cyclization and demethylation of the key products 447 and 448 using sulfuric acid afforded 444 (Scheme 94).445
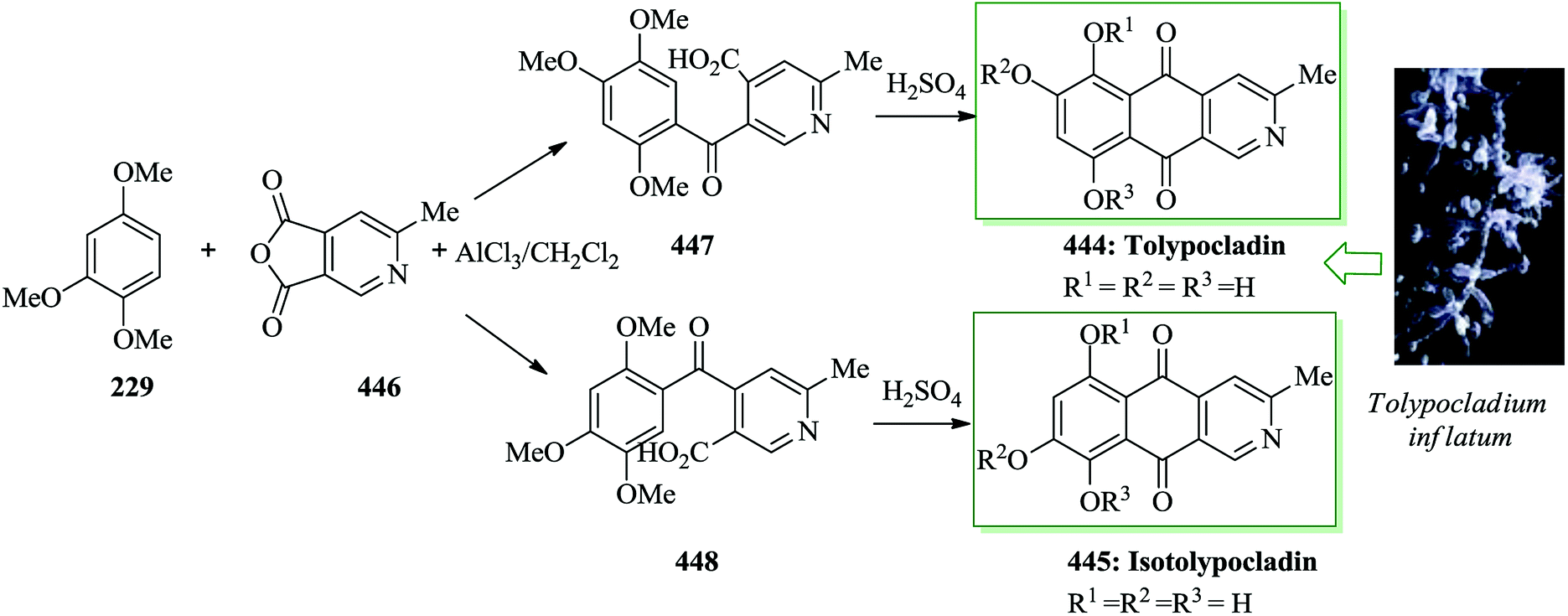 | ||
| Scheme 94 Total synthesis of tolypocladin 444 and isomeric isotolypocladin 445. | ||
(+)-Calanolide A,446 initially extracted from the dried fruits and twigs of Calophyllum lanigerum var. Austrocoriaceum, is a non-nucleoside reverse transcriptase inhibitor currently in clinical trials as an anti-HIV agent. Zhang and co-workers in 2000 used an investigation method to perform the FC acylation reaction that was applied in the total synthesis of (+)-calanolide A 449. A FC acylation reaction was applied to form 5,7-dihydroxy-4-propyl-8-propionylcoumarin 452 as a key intermediate for the formation of (+)-calanolide A. This reaction was performed by reacting 5,7-dihydroxycoumarin 450 with 1 equiv. of propionic anhydride aluminium chloride under reflux in dichloroethane (DCE). The optimal reaction conditions used 7 equiv. of aluminium chloride in 1.0 mL of nitromethane, with a predicted excellent yield (97%) (Scheme 95).447
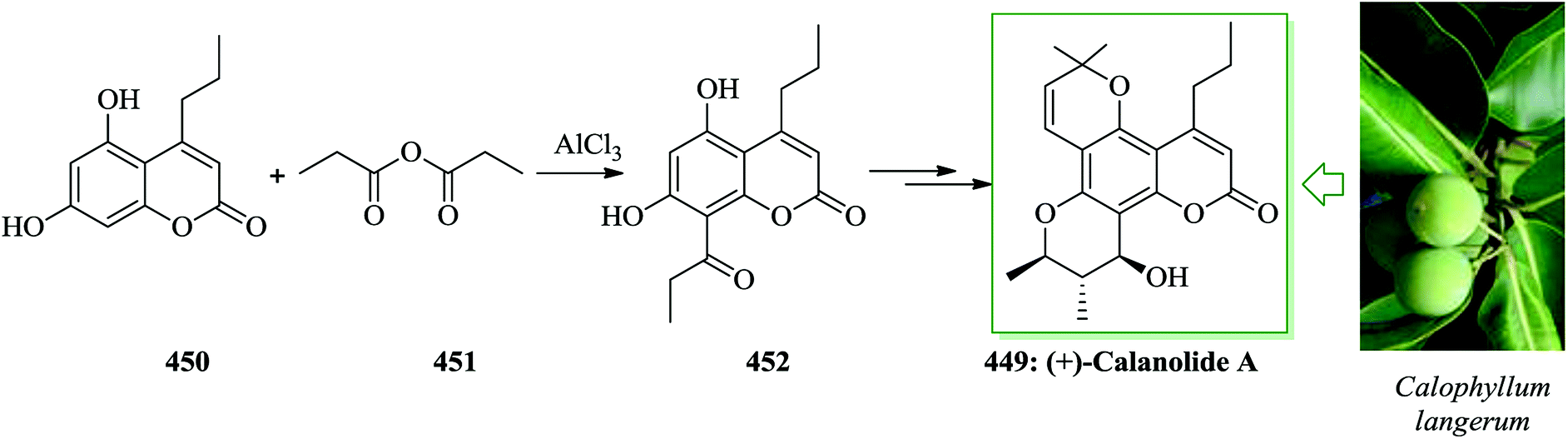 | ||
| Scheme 95 Total synthesis of (+)-calanolide A 449. | ||
Kaempferol 453448,449 and other flavonoids have been found to be potential antitumor agents and human immunodeficiency virus (HIV) type 1 integrase inhibitors.450 Kaempferol and its 3-O-glycoside derivatives were identified in several plants, vegetables, fruits, and beverages, for example Taiwan Bauhinia,451 Chamaecyparis formosensis,452 Inula britannica,453 French beans,454 asparagus,455 blueberries,456 grape fruit juice,457 teas,458 and honey.459 Kaempferol 453 was obtained in seven steps with 47% overall yield from 1,3,5-trimethoxybenzene 454. Firstly, the FC reaction of trimethoxy benzene 454 and 4-methoxycinnamic acid chloride 455 with AlBr3 as a catalyst provided o-hydroxy-chalcone 456 in 35% yield after several steps. Finally, compound 456 provided the corresponding product 453 upon several steps (Scheme 96).460
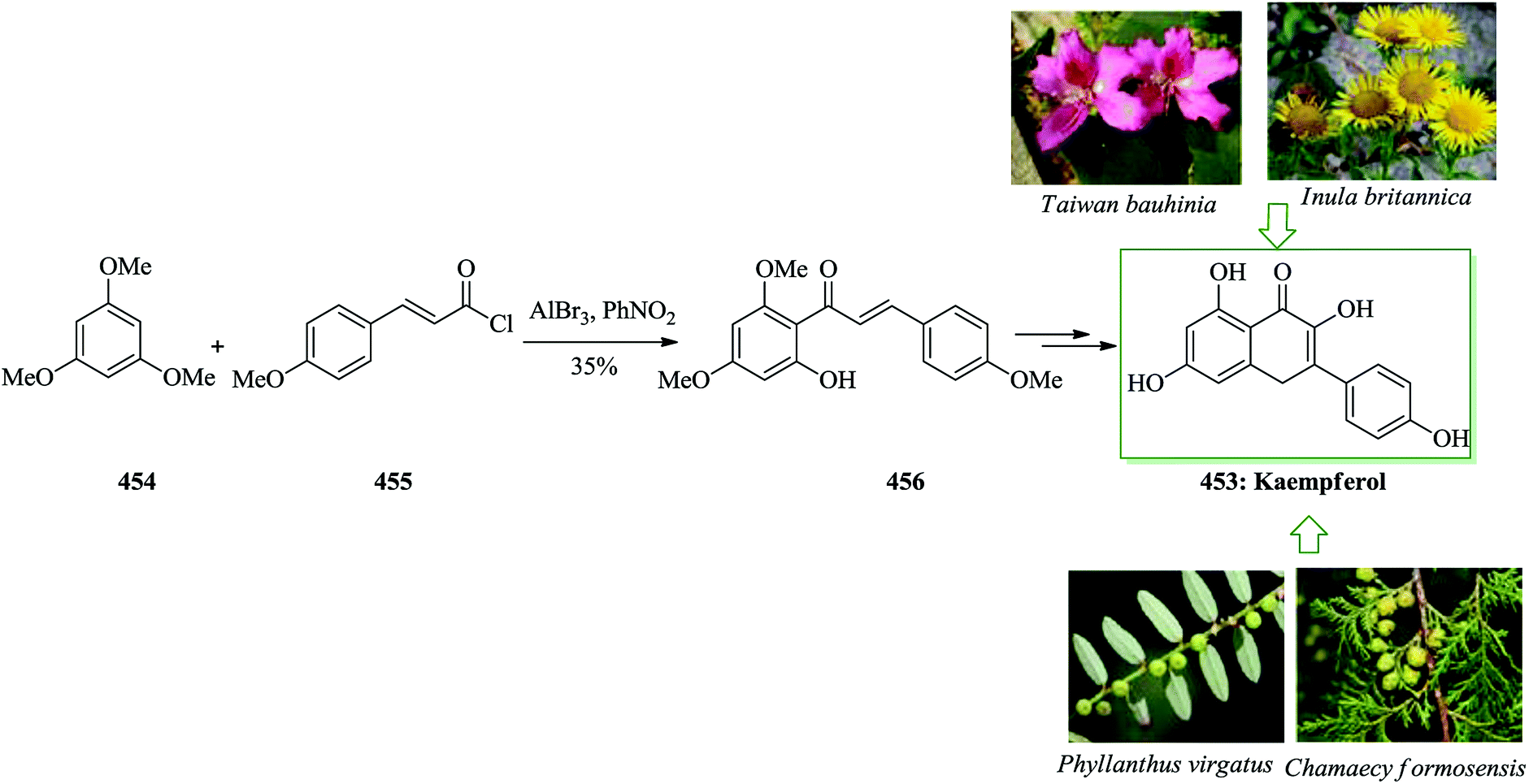 | ||
| Scheme 96 Total synthesis of kaempferol 453. | ||
Several naturally occurring phenylanthraquinones, namely knipholone 457a, knipholone anthrone 457b, 6′-O-methylknipholone 457c and 4′-O-demethylknipholone 457d, demonstrated moderate to high antiplasmodial activity in vitro against Plasmodium falciparum,461 the carrier of the most lethal malaria tropica. M-Knipholone 457a has been extracted from Kniphofia foliosa.462 Bringmann et al. in 2002 demonstrated the initial significant synthetic method toward natural knipholone-type phenylanthraquinones. Firstly, lactone 458 afforded the racemic biaryl 459.463 With this substrate, the introduction of the C-acetyl substituent succeeded both directly, via FC acetylation reaction, and in a two-step method, via an ortho-selective Fries reaction464 on ester 461, and afforded the C-acetyl compound 460 in moderate yields, without any removal of the methyl ether. Therefore, compound 460 afforded 6′-O-methylknipholone 457c, the first natural phenylanthraquinone. Selective mono-O-demethylation of 457c at C-6′ employing aluminium bromide afforded the target molecule knipholone 457a, reduction of which afforded its anthrone 457b. The fully O-demethylated derivative 457d was obtained by employing aluminium bromide in excess (Scheme 97).463
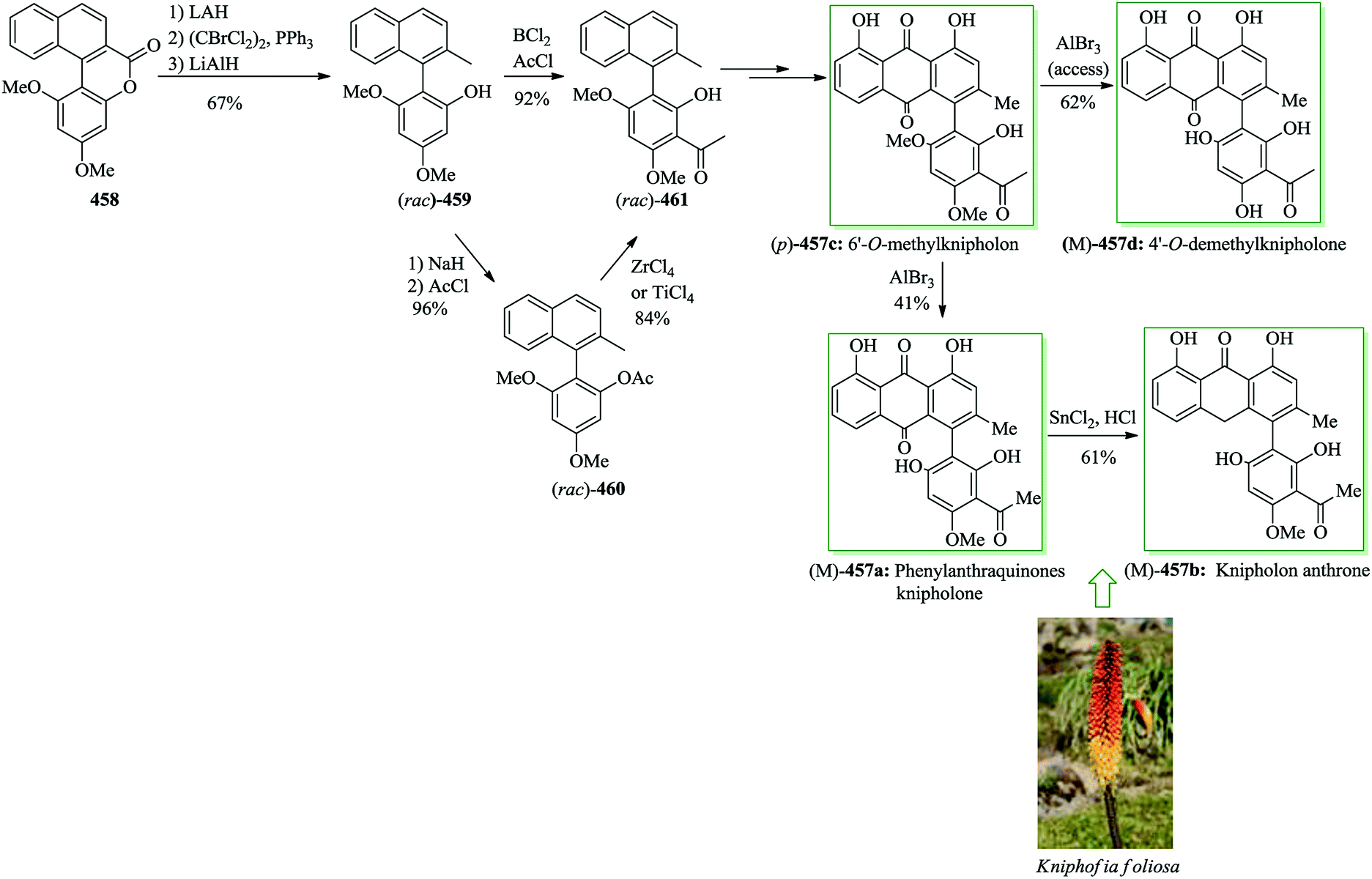 | ||
| Scheme 97 Total synthesis of phenylanthraquinones knipholone 457a, knipholone anthrone 457b, 6′-O-methylknipholone 457c and 4′-O-demethylknipholone 457d. | ||
The polycitones A 462 and B 463, polybrominated pyrrole alkaloids, were extracted by Kashman et al.465 from marine ascidians of the genus Polycitor. Polycitone A is an active inhibitor of retroviral reverse transcriptases and cellular DNA polymerases.466 Polycitone B 463 was obtained in four steps from pyrrole dicarboxylic acid 464, involving FC reaction of the desired acid chloride with anisole. The transformation of 462 into polycitone A 463 was accomplished in two steps through Mitsunobu alkylation of the pyrrolic NH group. Polycitone A was obtained in 18% overall yield and gave the probability of changing the groups on the pyrrole ring. This synthetic method was started from dicarboxylic acid 464, which reacted with oxalyl chloride followed by elimination of the solvent and rigorous drying, which afforded the crude acid chloride 465. Compound 465 was treated with anisole under FC reaction conditions to provide the diketone 466 in 66% yield. In the following, compound 466 afforded polycitone B 462 after several steps and then polycitone B 462 was transformed into polycitone A 463 after several steps (Scheme 98).356
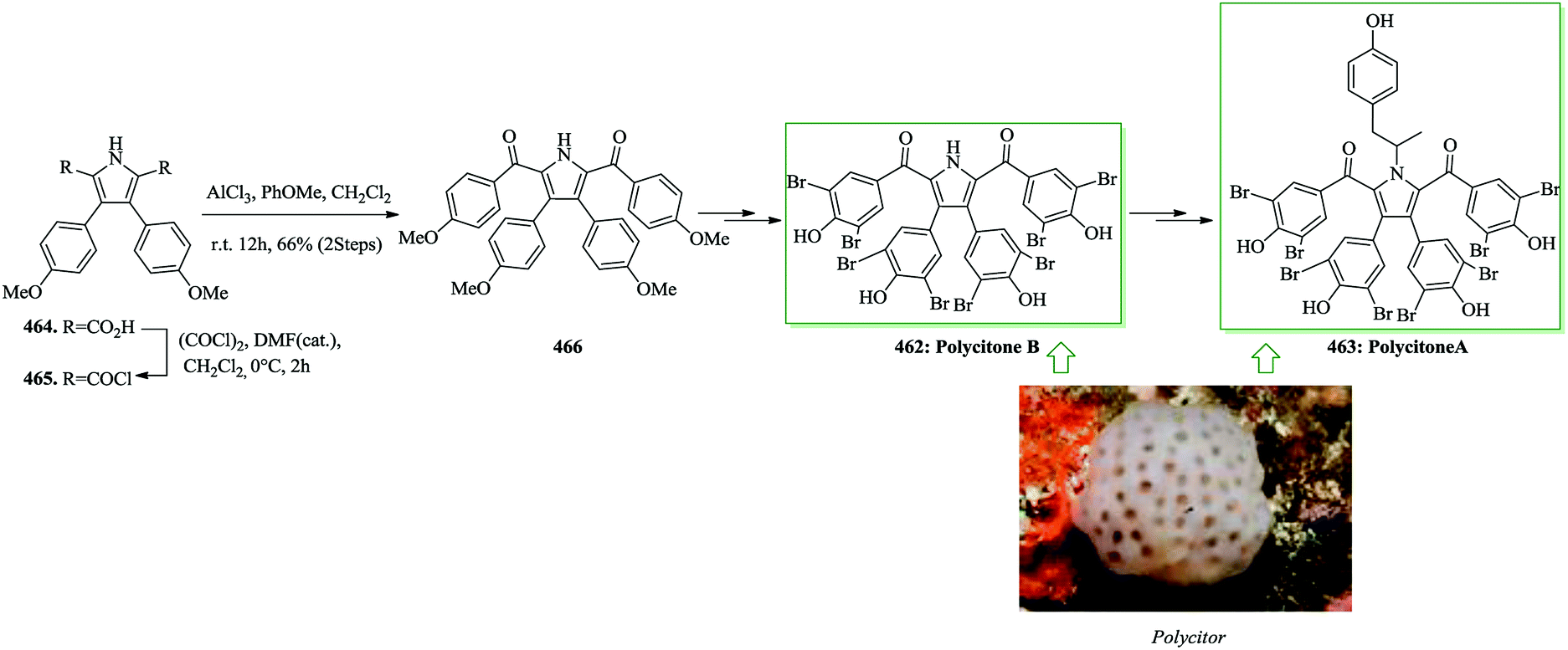 | ||
| Scheme 98 Total synthesis of polycitone B 462 and polycitone A 463. | ||
The unique inhibitors of 3α-hydroxysteroid dehydrogenase, 0231A and 0231B, have been extracted from a fermentation broth of Streptomyces sp. HKI0231.467 These compounds are promising as lead structures for anti-inflammatory agents. 0231A and 0231B have a unique benz[c,d]indol-3(1H)-one scaffold in their molecules. Nakatsuka and co-workers in 2003 reported the synthesis of 0231B 467 in 10 steps (8.1% overall yield) from 6-methylindole 468.468 Upon masking the nitrogen with a pivaloyl group, the introduction of a trans-p-methylcinnamoyl group was achieved at the 3-position via FC acylation,469 and subsequent de-protection of the pivaloyl group gave the trans-p-methylcinnamoyl derivative 469 in an 87% overall yield from 468. Finally, compound 469 was converted into 0231B 467 upon several steps (Scheme 99).468
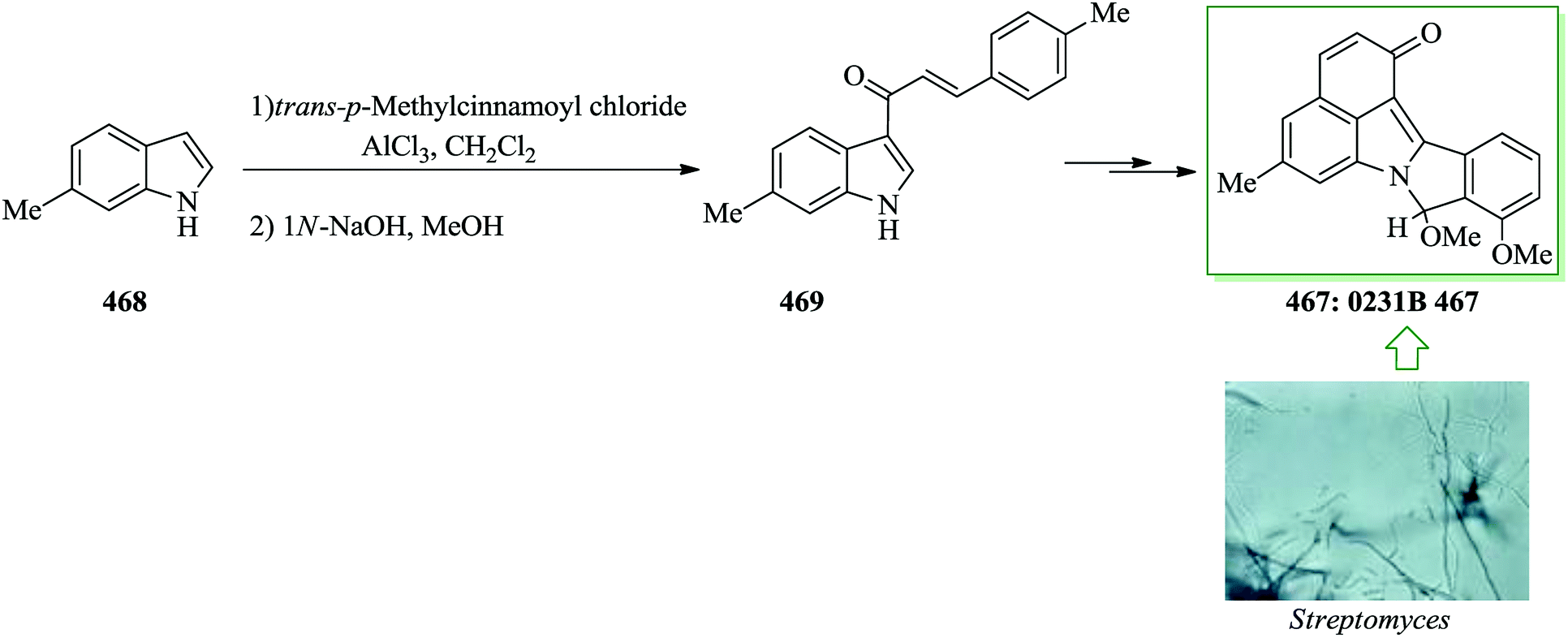 | ||
| Scheme 99 Total synthesis of 0231B 467. | ||
Various diterpenoids have the 4a-methyltetra- (and hexa-)hydrofluorene framework, for example, taiwaniaquinols A, B, and D and taiwaniaquinone D and H, as well as diverse structurally relevant diterpenoids. Dichroanone, dichroanal B, and standishinal were extracted from Taiwania cryptomerioides, Salvia dichroantha and Thuja standishii.84,86,470 A significant acid-promoted domino FC acylation/alkylation reaction was known as the key step. Significantly, the formal total syntheses of diterpenoids (±)-taiwaniaquinol B 470 and (±)-dichroanone 471 were performed. Total synthesis of (±)-taiwaniaquinol B 470 was accomplished in 2008 by She and co-workers.471 Total synthesis of 470 was accomplished from the reaction of 1,3-dimethoxy-2-isopro-pylbenzene 472 and cyclogeranic acid 473 through the domino FC acylation/alkylation reaction to give the corresponding tricyclic 474 as a single diastereomer in 70% yield. Next, compound 474 was converted into 470 after several steps (Scheme 100).
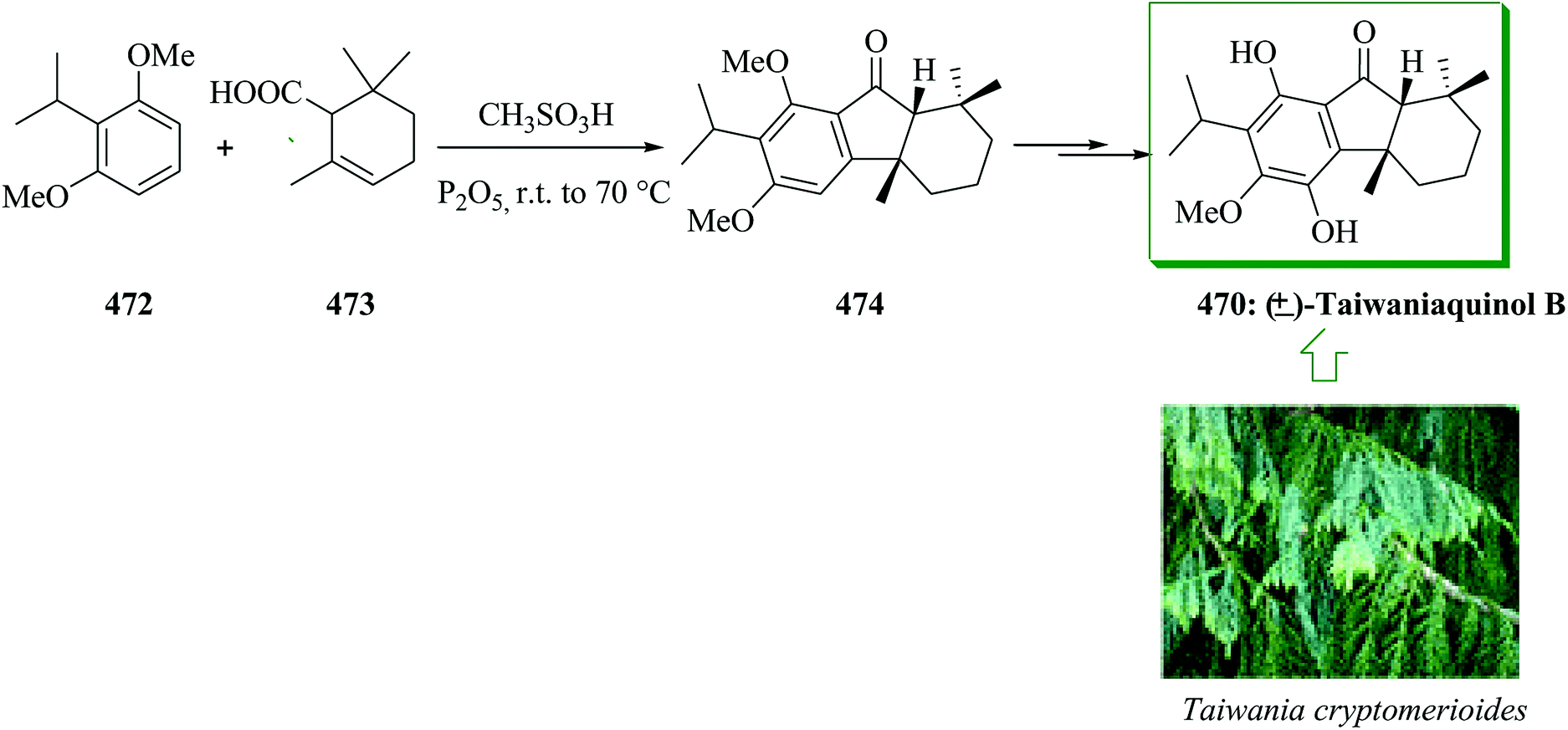 | ||
| Scheme 100 Total synthesis of (±)-taiwaniaquinol B 470. | ||
Subsequently, the total synthesis of (±)-dichroanone 471 was accomplished and reported. Accordingly, the reaction between isopropylanisole 475 and geranic acid 477 or cyclogeranic acid 476 in the presence of P2O5 in neat CH3SO3H provided a mixture of regioisomers in 70% and 72% overall yield, respectively. The major isomer 478 was obtained by employing the domino FC acylation/alkylation reaction of compound 475 with geranic acid 477, giving a 64% yield after purification by column chromatography. Finally, compound 478 afforded (±)-dichroanone 471 in several steps and its spectra were found to be identical with those formerly reported (Scheme 101).86,471,472
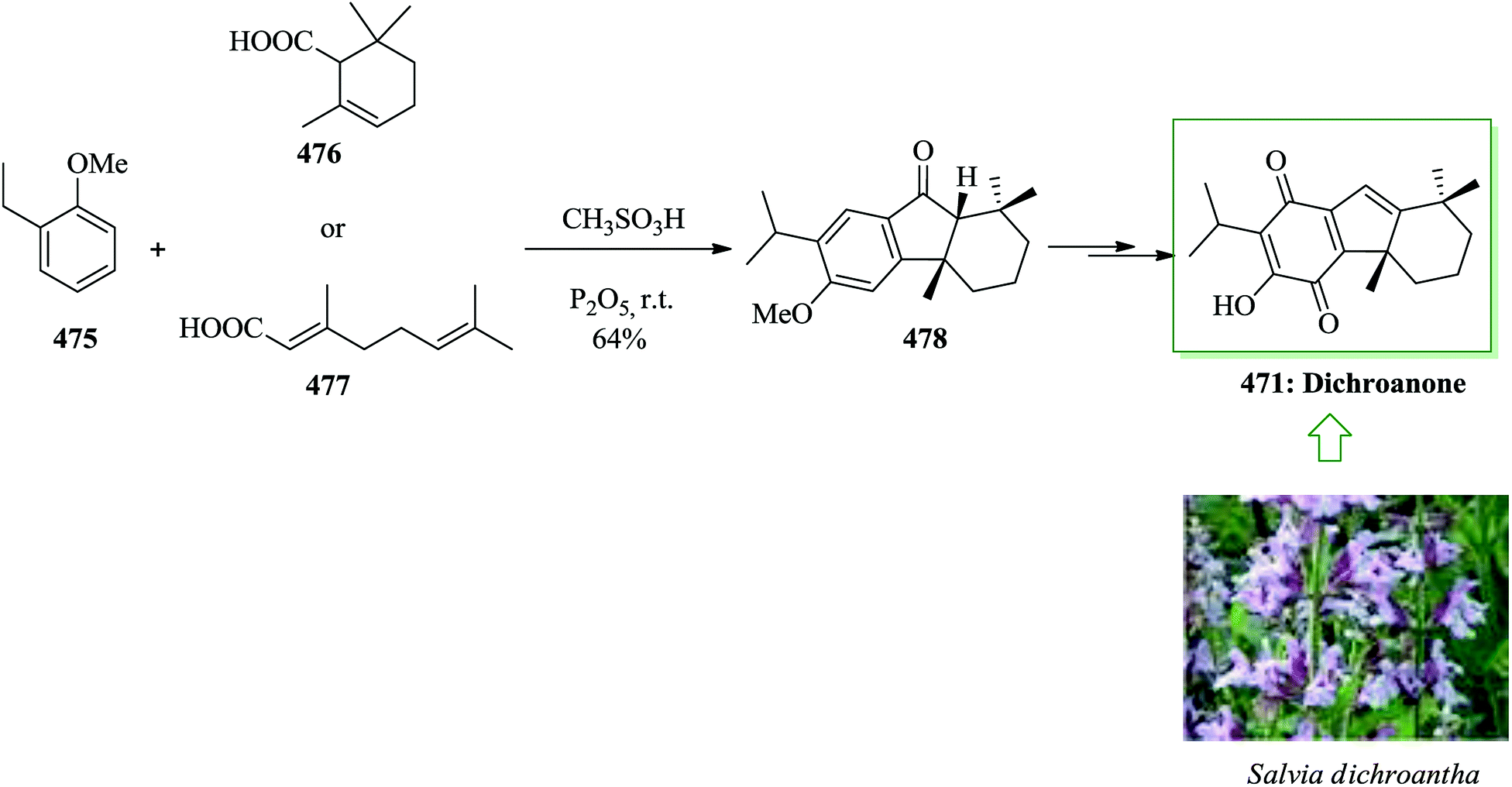 | ||
| Scheme 101 Total synthesis of (±)-dichroanone 471. | ||
Protein tyrosine phosphatase 1B (PTP1B) has aroused intensive research interest because of its involvement in the insulin signaling cascade as a major negative regulator.473 Bioassay-guided separation of the ethanol extract using a wide range of chromatographic techniques resulted in a series of bromophenols. One of them, named bis-(2,3-dibromo-4,5-dihydroxyphenyl)-methane, exhibited surprising inhibitory activity against PTP1B. Bis-(2,3-dibromo-4,5-dihydroxyphenyl)-methane 479, which was extracted from red algae Rhodomela confervoides, was demonstrated as a natural bromophenol with important inhibition against PTP1B. Total synthesis of compound 479 was achieved with an overall yield of 24%. Initially, 3,4-dimethoxybenzoic acid 481 on reaction with 1,2-dimethoxybenzene 36 using polyphosphoric acid at 80 °C for one hour easily gave 481 in 85% yield.474 Finally, compound 481 afforded compound 479 after several steps in 24% overall yield (Scheme 102).475
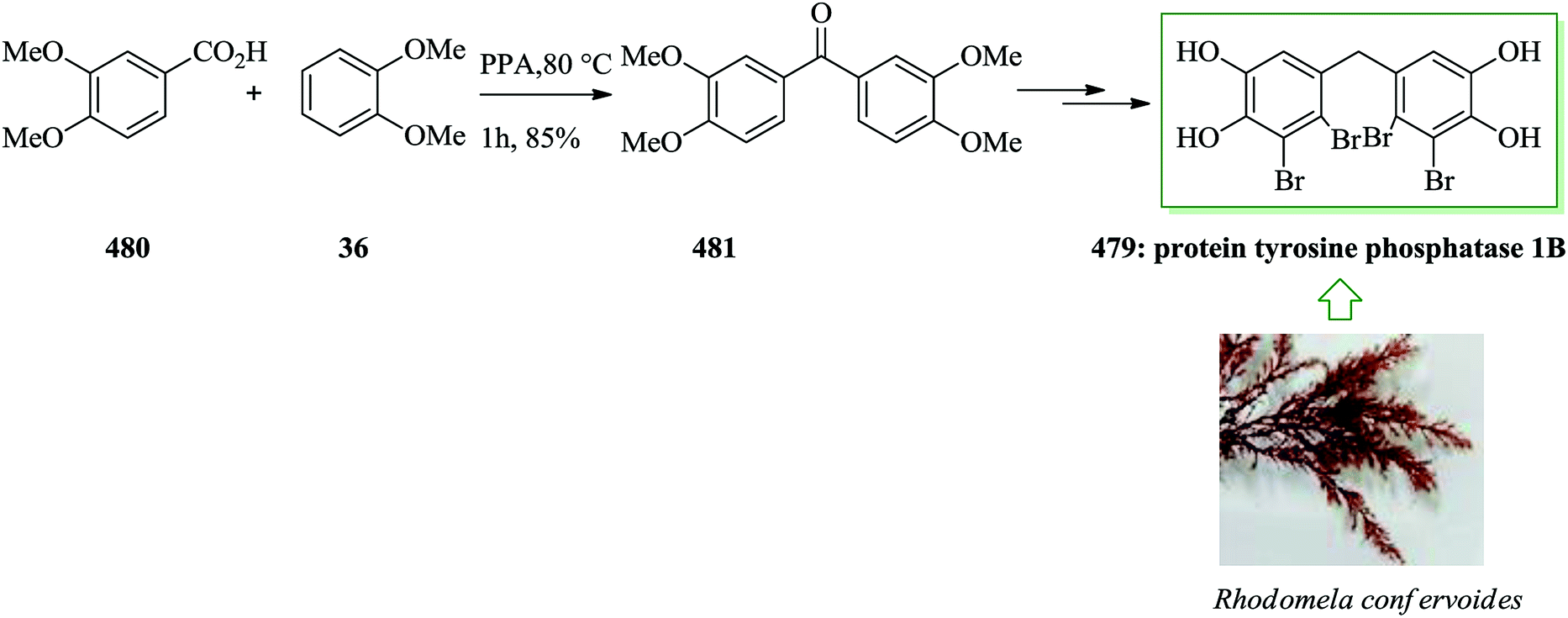 | ||
| Scheme 102 Total synthesis of protein tyrosine phosphatase 1B (PTP1B). | ||
The topopyrones, a group of planar anthraquinone polyphenols, were identified by the orientation of the pyrone ring appended on one side as well as the presence or lack of a chlorine at C7. Topopyrones A 482a and B 482b contain a chloro group at C7, i.e., that part of the molecule distant from the pyrone ring, while topopyrones C 482c and D 482d are unfunctionalized at C7. The topopyrones were extracted from the culture broths of two fungi, Phoma sp. BAUA2861 and Penicillium sp. BAUA4206. Particularly, topopyrones A, B, C, and D selectively inhibited the growth of yeast expressing human topoisomerase I. The topopyrones exhibit a unique group of extremely cytotoxic topoisomerase I poisons. Efficient total synthesis of all four naturally occurring members of this group was achieved in 2008 by Hecht et al.476 Main steps are Diels–Alder reaction and a titanium-catalyzed ortho-directed FC acylation. For the synthesis of topopyrone A 482a, B 482b, C 482c and D 482d, initially quinone 483477 was converted into phenol 484 in several steps. At this point TiCl4 was selected to perform regiocontrolled FC acylations. Actually, it was known that reaction of 484 in 1,2-dichloroethane with titanium tetrachloride and AcCl under reflux introduced the needed acyl side chain selectively at the ortho position giving 485 in 78% yield. Finally, compound 485 gave topopyrone A 482a, B 482b, C 482c and D 482d after several steps (via different routes) (Scheme 103).476
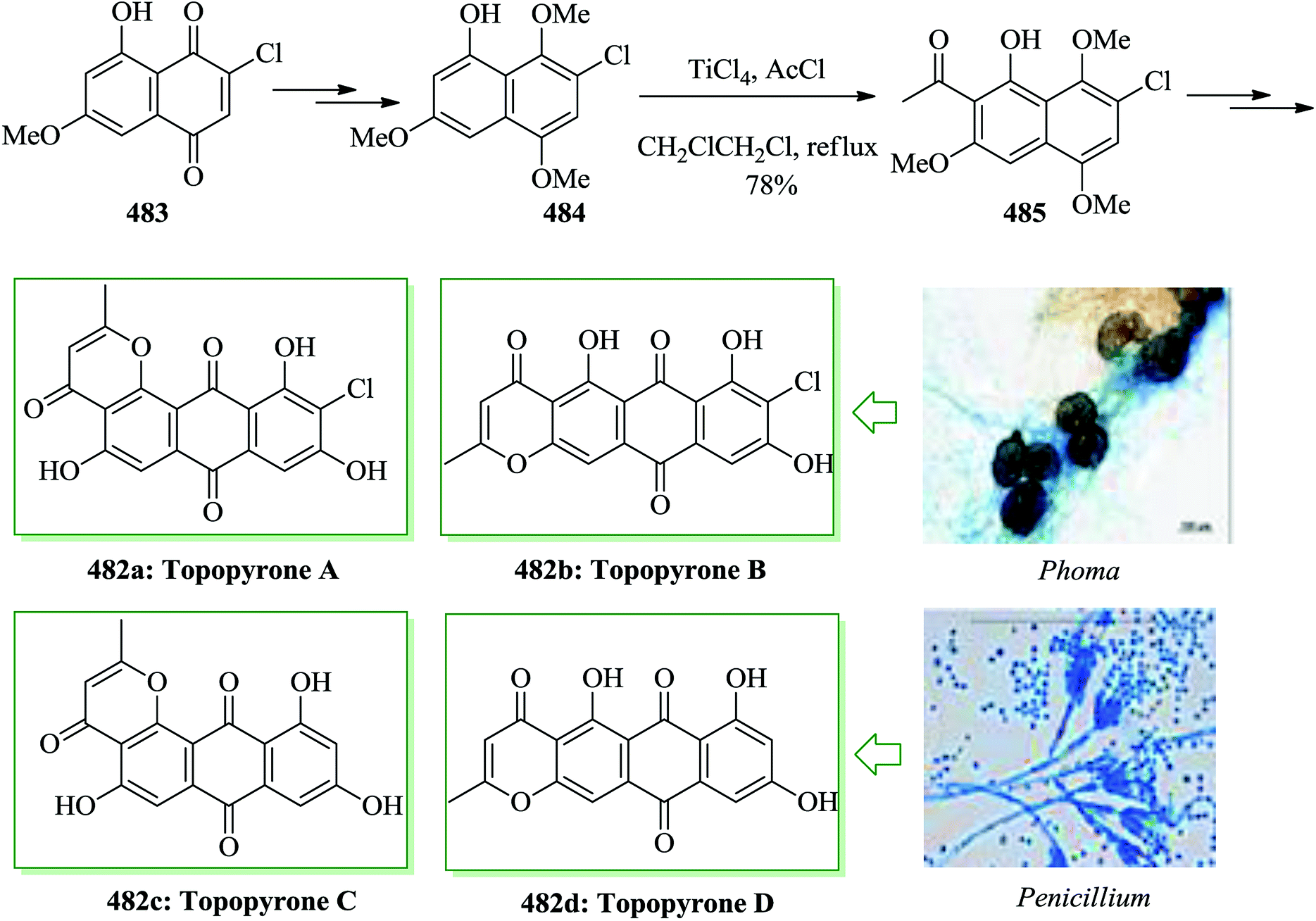 | ||
| Scheme 103 Total synthesis of topopyrone A 482a, B 482b, C 482c and D 482d. | ||
Phenanthroindolizidine alkaloids, extracted from Pergularia, Cynanchum, Tylophora, and some genera of the Asclepiadaceae group, show significant biological, pharmacological,478,479 and antitumor properties.480–483 (−)-Antofine 486c, extracted from Cynanchum komarovii, exhibited high antiviral activity against the tobacco mosaic virus (TMV).484,485 Wang et al. in 2008 reported the total synthesis of tylophorine 486a, deoxytylophorinine 486b, and antofine 486c starting from pyrrole in 48%, 44%, and 46% overall yields, respectively.486 In this route for the synthesis of phenanthroindolizidine alkaloids 486a, 486b, and 486c, the reaction of benzeneacetic acid 487a–c and aromatic aldehyde 488a–c afforded phenanthrene-9-carboxylic acid 489a–c via several steps. Then, the intermolecular FC acylation of the corresponding acyl chloride (synthesized by chlorination of acid 489a with oxalyl chloride) and pyrrole was catalyzed by tin(IV) chloride to form pyrrolidine ring system 490a under mild conditions in 79% yield. After three steps, the racemic tylophorine 486a was provided. Thus, the shortest synthetic pathway to a large-scale construction of tylophorine 486a was accomplished starting from pyrrole under mild conditions without any protecting group in 48% overall yield. This group demonstrated the construction of deoxytylophorinine 486b and antofine 486c. Deoxytylophorinine 486b and antofine 486c were obtained in 44% and 46% overall yields, respectively, in six steps including a Perkin condensation reaction, intramolecular oxidative coupling reaction of acids, chlorination of acids 489b,c, and subsequent intermolecular FC reactions with pyrrole in one pot, deketonization of 2-acylpyrroles 490b,c, catalytic hydrogenation of 2-alkylpyrroles, and Pictet–Spengler cyclomethylenation. The versatility and flexibility of this approach were exhibited by the large-scale construction of three representative phenanthroindolizidine alkaloids, tylophorine 486a, deoxytylophorinine 486b, and antofine 486c (Scheme 104).486
 | ||
| Scheme 104 Total synthesis of tylophorine 486a, deoxytylophorinine 486b, and antofine 486c. | ||
Lamellarin G, a marine natural product, includes a 5-oxa-6b-aza dibenzo[a,i]fluoren-6-one framework. Lamellarins were extracted from the prosobranch mollusk Lamellaria sp. and the ascidians.487 Some of these lamellarins488,489 show active biological properties,490 for example cytotoxicity to a series of cancer cell lines, cell division inhibition, and immunomodulation. A modular synthesis of the lamellarin G trimethyl ether was established using various reactions in sequence, including FC acylation, esterification, haloarylation, and oxidative cyclization. This method provided the complete structure of lamellarin G trimethyl ether in four steps with 44% overall yield. Total synthesis of lamellarin G trimethyl ether was accomplished in 2009 by Yadav et al.491 In this approach, 4-(3,4-dimethoxy-phenyl)-4-oxo-but-2-enoic acid 494 was obtained from veratrole 492 and maleic anhydride via FC reaction.492 Subsequently, compound 494 afforded the desired target product lamellarin G trimethyl ether 491 upon several steps (Scheme 105).491
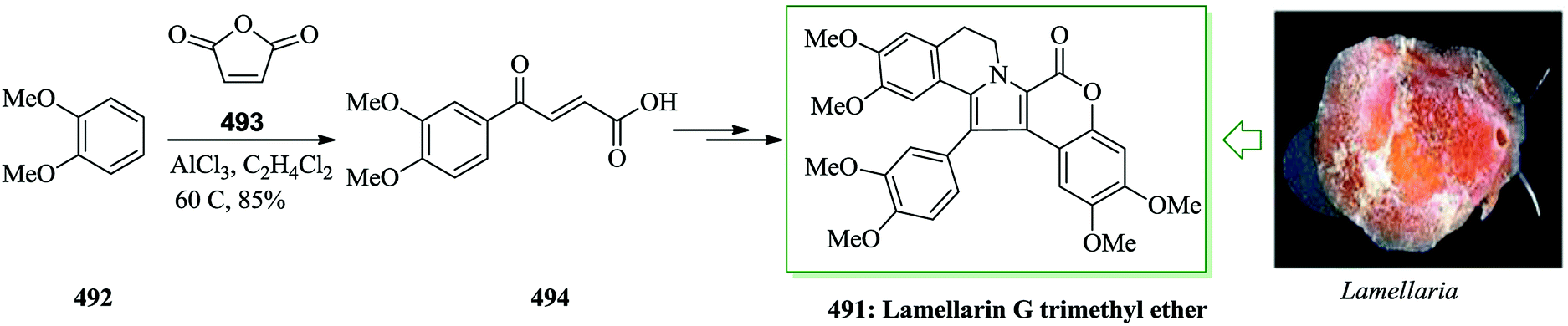 | ||
| Scheme 105 Total synthesis of lamellarin G trimethyl ether 491. | ||
The epimeric ialibinones A and B 495, uncommon types of PPAP, were extracted from the leaves of Hypericum papuanum by Sticher et al. in 2000.493 They show antibacterial activity against cytotoxic against KB cell lines.493,494 The tricyclic natural products ialibinone A and ialibinone B were synthesized as a 41:59 mixture in four steps starting from phloroglucinol. The synthetic sequence included acylation reaction of phloroglucinol through FC reaction, double prenylation, dearomatizing methylation, and oxidative free radical cyclization. Initially, the bis-prenylated acylphloroglucinol 497 was synthesized in two steps from phloroglucinol 496 via FC acylation reaction. Next, compound 497 provided the target natural products, ialibinone A 495a and ialibinone B 495b, as an inseparable mixture in 35% overall yield (Scheme 106).495
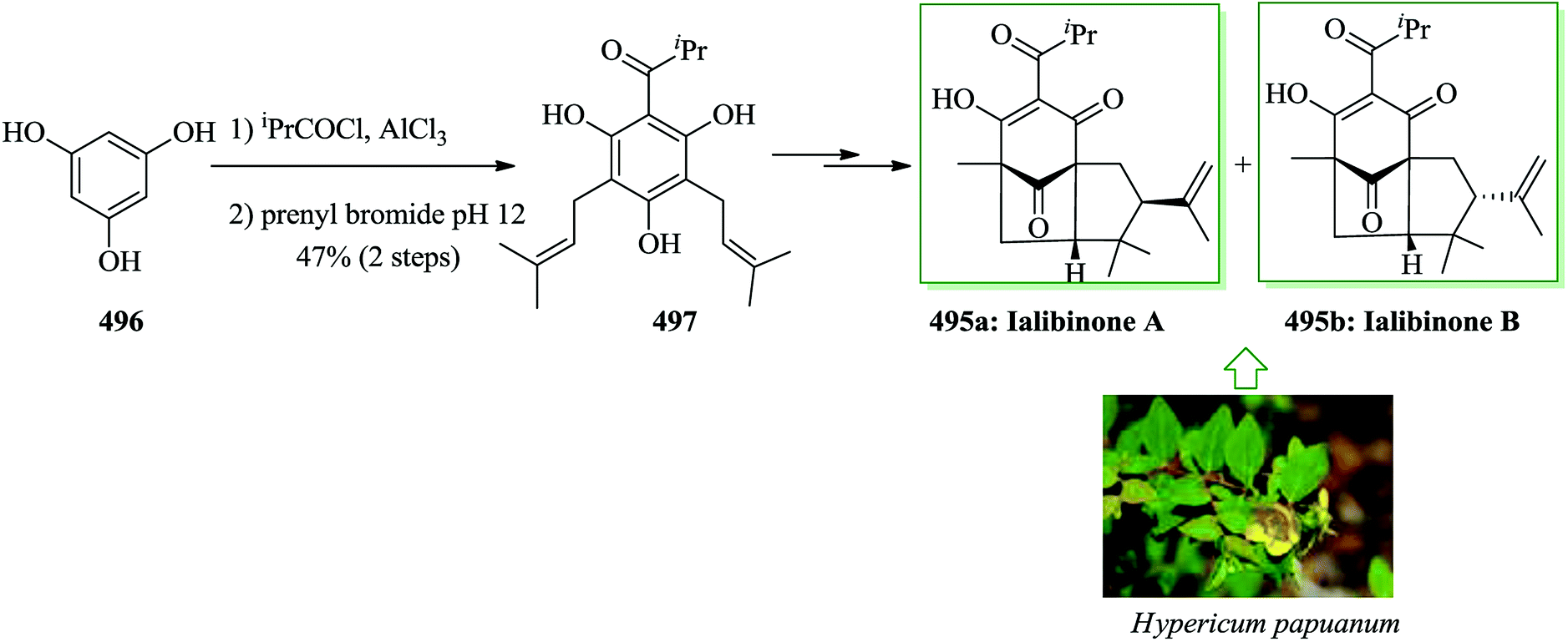 | ||
| Scheme 106 Total synthesis of ialibinone A 495a and ialibinone B 495b. | ||
Lactonamycin 498, extracted from a culture broth of Streptomyces rishiriensis MJ773-88K4, exhibited active antimicrobial properties against Gram-positive bacteria and vancomycin-resistant Enterococcus (VRE) as well as antitumor properties.496,497 Structurally, compound 498 contains a hexacyclic system and a glycosidic bond at the t-alcohol.497 The first total synthesis of lactonamycin 498 was accomplished in 2010 by Tatsuta et al.498 The synthesis includes sequential intramolecular conjugate addition reaction of alcohols to the acetylenic ester, asymmetric glycosylation of the tertiary alcohol, and Michael–Dieckmann type cyclization with the thioester. The synthesis of lactonamycin 498 was started from 4-bromo-3,5-dimethylphenol 499, that after two steps gave ester 500.499 Then, FC-type formylation reaction500 of 500, gave aldehyde 501. Upon several steps, compound 501 gave lactonamycin 498 (Scheme 107).498
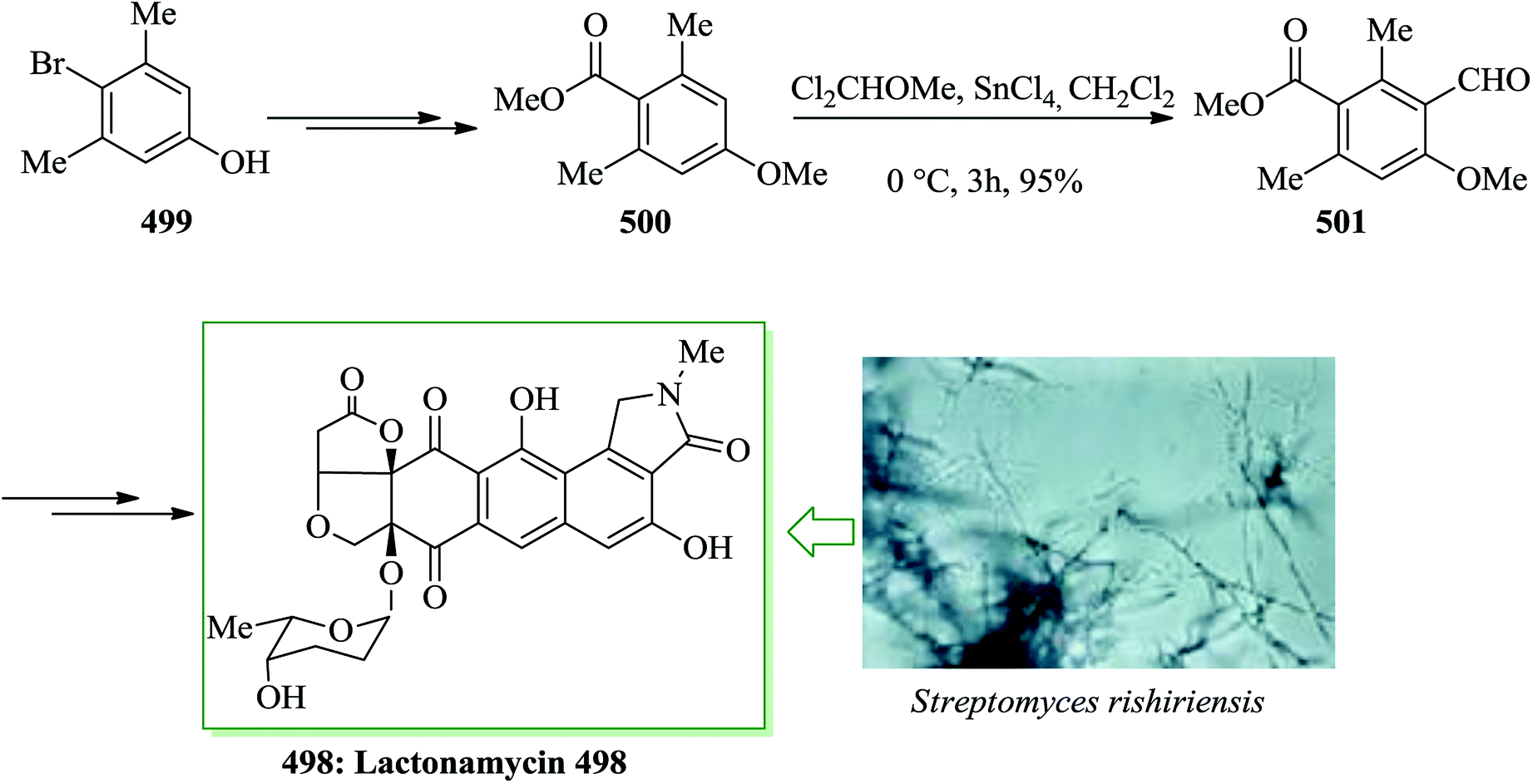 | ||
| Scheme 107 Total synthesis of lactonamycin 498. | ||
Myrtucommulone A 502 was developed in 1974 by Kashman et al. as a substance known in the common myrtle Myrtus communis L.501 Jauch et al. in 2010 reported synthesis of myrtucommulone A 502 from isobutyryl phloroglucinol 504, isobutyraldehyde, and syncarpic acid in one step. Isobutyryl phloroglucinol 504 is easily accessible via FC acylation of phloroglucinol 496 in 70–80% yield.502,503 Compound 496 after three steps gave syncarpic acid.504,505 Compound 503 is also accessible from 496 upon several steps. In this part, two acid-catalyzed FC alkylation reactions occurred consecutively in a one-pot reaction. Running the FC alkylation under basic conditions needed removal of the acid and extraction of 505 as a crude product prior to reaction with 504, which had been deprotonated with two equivalents of sodium hydride in tetrahydrofuran. Finally, the synthesis of 502 was completed in three hours at ambient temperature in quantitative yield (Scheme 108).506
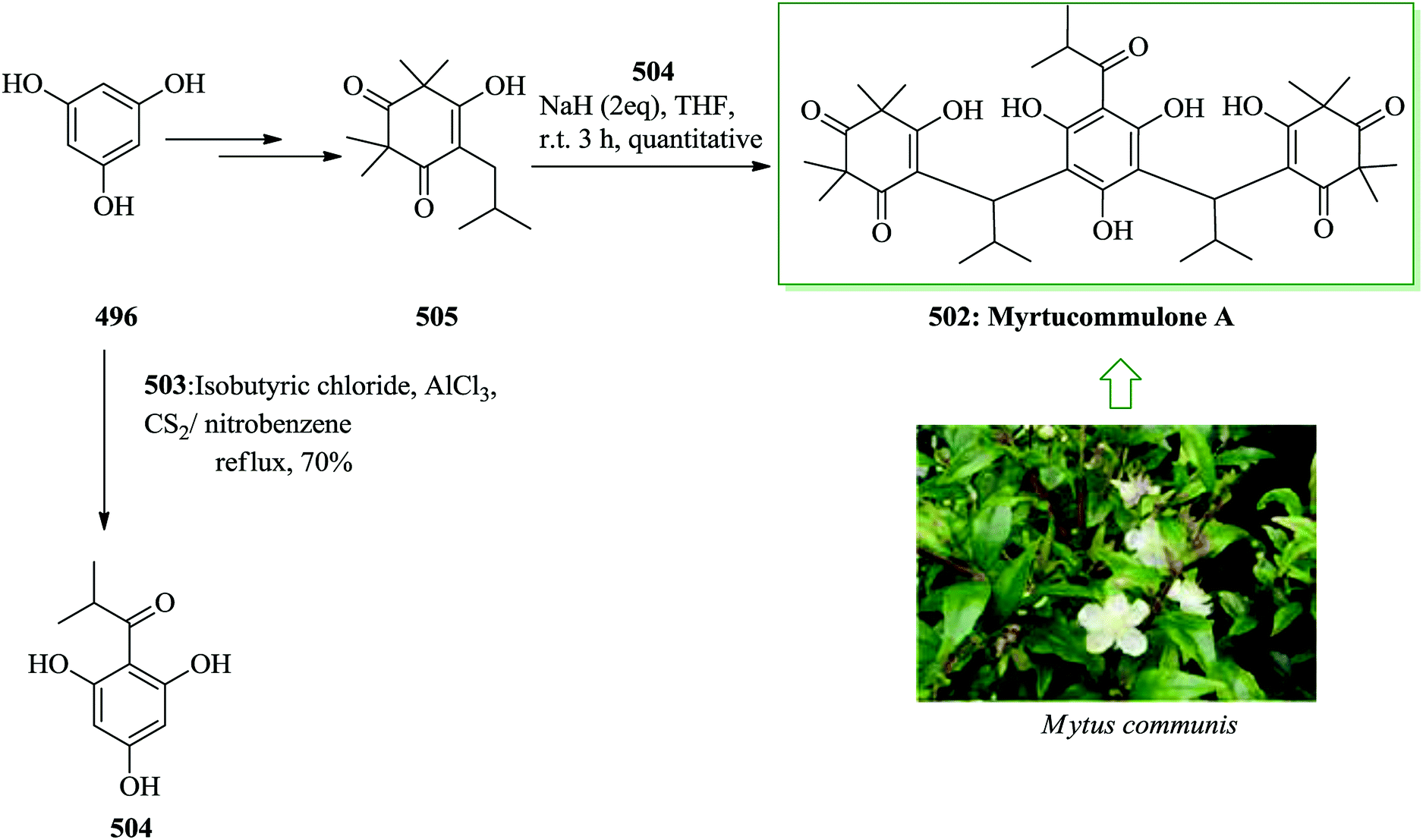 | ||
| Scheme 108 Total synthesis of myrtucommulone A 502. | ||
The tylophora alkaloids have obtained synthetic and medicinal attention owing to their panoply of biological properties, such as antibacterial, anticancer, antifungal, antiviral, and anti-inflammatory properties.507 Antofine and 13aα-secoantofine were extracted from the root of Cynanchum paniculatum Kitagawa (Asclepiadaceae)508 and from aerial parts of Cynanchum vincetoxicum,509 respectively.510 A fast synthetic method to the tylophora alkaloids antofine and 13aα-secoantofine was achieved. The key step in this synthetic methodology was the multicatalytic aminochlorocarbonylation/FC acylation reaction of a protected 4-pentenyl amine to give a β-pyrrolidinyl ketone intermediate. Thus, after exposing the easily accessible nosyl protected alkenyl amine 508 and veratrole to aminochlorocarbonylation, it was gratifying to find that the β-pyrrolidinyl ketone 509 could be isolated in high yield. Finally, compound 509 afforded 13aα-secoantofine and antofine in 19% and 48% overall yield, respectively (Scheme 109).511
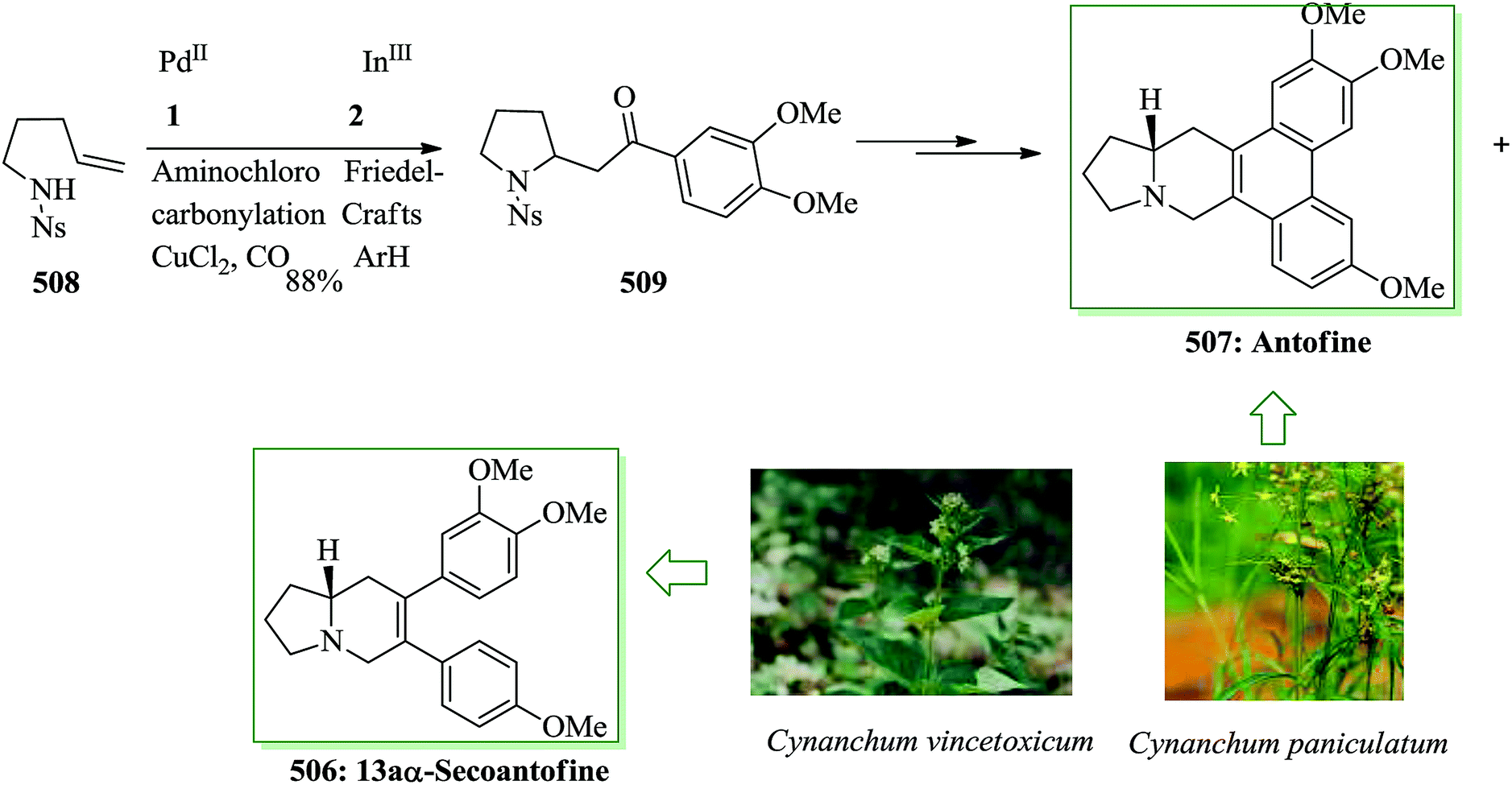 | ||
| Scheme 109 Total synthesis of 13aα-secoantofine 506 and antofine 507. | ||
Cytosporone B 510, extracted from Cytospora sp. CR200 and Dothiorella sp. HTF3,512 exhibits antibacterial activity.512–515 The total synthesis of cytosporone B was achieved from 1-bromo-3,5-dimethoxybenzene. The key steps are sequential Grignard reaction and Lemieux-van Rudloff oxidation followed by a deprotection of the methyl aromatic ether to phenol and subsequent FC acylation. In this route, total synthesis of cytosporone B was started from 1-bromo-3,5-dimethoxybenzene 511. Next, it was converted into the key intermediate 512 via three steps. Then, with the key intermediate 512 obtained by oxidation and esterification, the sequential FC acylation and deprotection were accomplished subsequently. The FC reaction progressed with 86% yield of the purified product. The ether in 512 was deprotected to phenol 514 using BBr3, followed by etherification with benzyl bromide to provide the intermediate 515 (56% yield, two steps). The hydroxyl protection step was followed by an attempt at acylation of this protected phenol with octanoyl chloride catalyzed by aluminium chloride in 83% yield. Finally, the debenzylation of ethyl 2-(3,5-bis(benzyloxy)-2-octanoylphenyl)acetate 516 under H2 progressed very easily (95% yield) and very pure cytosporone B 510 was obtained (Scheme 110).512
 | ||
| Scheme 110 Total synthesis of cytosporone B 510. | ||
Bruguierol A 517 was extracted and identified by Sattler et al. from the stem of Bruguiera gymnorrhiza tree in 2005.66 This natural product has a unique structure characterized by a 2,3-benzofused 8-oxabicyclo[3.2.1]octane core. Total synthesis of (±)-bruguierol A 517 was achieved in 10 steps and with an overall 16.8% yield. The embedded unique 8-oxabicyclo[3.2.1]octane unit framework in this natural product was generated through a novel Sc(OTf)3-catalyzed intramolecular [3 + 2] cycloaddition of cyclopropane. The synthesis of bruguierol A 517 was achieved via FC acylation reaction of 3-bromoanisole 518 to give compound 519 in 72% yield.516,517 Finally, (±)-bruguierol A 517 was synthesized after several steps from compound 519 (Scheme 111).
 | ||
| Scheme 111 Total synthesis of bruguierol A 517. | ||
The malyngamides, usual metabolites of Lyngbya majuscula, are N-functionalized amides of long chain fatty acids. A subgroup of malyngamides contains a communal and remarkable terminal vinyl chloride functionality, for example malyngamides A, B, M, and isomalyngamides A, B, M. Malyngamide M 521 was extracted from the Hawaiian red alga Gracilaria coronopifolia.518 It was the first example of a natural aromatized malyngamide that possesses a special terminal vinyl chloride substructure. The first concise and significant asymmetric synthesis of malyngamide M 521 was achieved in nine steps from o-cresol in 12% overall yield. The key steps included the Wittig reaction, FC acetylation, amidation, and isomerization reaction. The isomalyngamide M 520 was also obtained in 2010 by Cao et al.519 In this pathway, total synthesis of malyngamide M 521 was started from o-cresol 522. FC acetylation reaction of 522 with chloroacetonitrile using boron trichloride and aluminium trichloride afforded phenol 523 in 78% yield.520 Next, after several steps, compound 523 gave isomalyngamide 520 in 83% yield. Finally, compound 520 was transformed into a mixture of 521/Z-521 (2.5:1) when exposed to UV light using benzophenone in dichloromethane for 8 h at room temperature.521 Fortunately, the two isomers could be simply separated by flash chromatography over silica gel, and pure 521 was provided in 67% yield (Scheme 112).519
 | ||
| Scheme 112 Total synthesis of isomalyngamide 520 and malyngamide M 521. | ||
Marinopyrrole A 524 is an alkaloid endowed with promising antibiotic properties against methicillin-resistant Staphylococcus aureus (MRSA)523 that was extracted from an obligate marine Streptomyces. This structurally uncommon molecule exists at room temperature as enantiopure M-(−)-atropisomers. Nicolaou et al. developed total synthesis of the antibiotic marinopyrrole A 524 in five steps and 16% overall yield from aminopyrrole 525.524 The synthesis of marinopyrrole (−)-524 (natural) and (+)-524 (unnatural) was started from aminopyrrole 525, which was converted into tricycle 526 upon several steps. FC arylation reaction of 526 and acid chloride 527, catalyzed by aluminium chloride in dichloromethane, resulted in the marinopyrrole unit structure 528 in 64% yield. Finally, compound 528 afforded racemic marinopyrrole A [(±)-524] via several steps (Scheme 113).524
 | ||
| Scheme 113 Total synthesis of marinopyrrole A (±)-524. | ||
In a similar method, mono-arylated marinopyrrole 530 was synthesized from bis-pyrrole 529 (obtained from compound 525), via a three-step sequence containing a FC C-arylation with acid chloride 527, tetra-chlorination with NCS in acetonitrile, and demethylation with BBr3 in dichloromethane to provide the target product 530 in 39% overall yield (Scheme 114).525
 | ||
| Scheme 114 Total synthesis of mono-arylated marinopyrrole 530. | ||
Biaryl compounds, containing a 6,7,6-ring system, include a wide range of naturally occurring compounds and synthetic pharmaceuticals, for example alkaloids as well as cyclolignans. Several of them showed activity against various cancer cell lines. Lin et al. extracted a unique sesquiterpenoid, tenuifolin 531, from the stems of Cinnamomum tenuifolium.526 In the structure of 531 exists a similar moiety to allocolchicine and a preliminary bioassay indicated that 531 only exhibited weak anti-proliferative activity against tumor cell line DU145.526 The first total synthesis of a sesquiterpenoid, tenuifolin 531, was accomplished in seven linear steps from market-accessible benzodioxole 421. A FC acylation reaction of 421 using ethyl 2-chloro-2-oxoacetate 532 gave compound 533 in 92% yield. Next, compound 533 was converted into bromoester 534 via several steps. Lastly, bromoester 534 and aldehyde 536 afforded tenuifolin 531 in excellent yield (91%) (Scheme 115).527
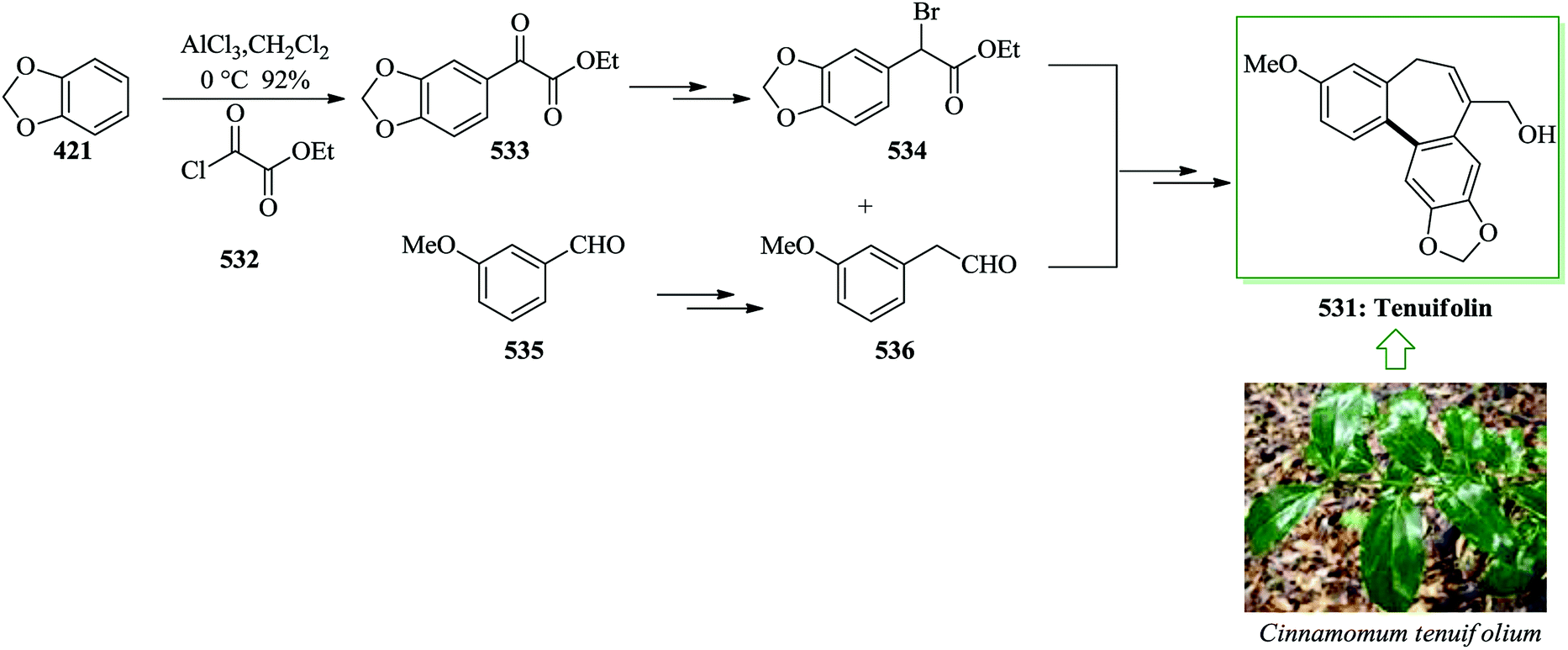 | ||
| Scheme 115 Total synthesis of tenuifolin 531. | ||
Dictyodendrins were extracted in 2003 by Fusetani and Matsunaga from the extracts of a Japanese marine invertebrate, the marine sponge Dictyodendrilla verongiformis, which was from Nagashima Island in Kagoshima, Japan. These compounds were the first marine alkaloids that contain inhibitory activity against telomerase.528 A highly effective total synthesis of dictyodendrins A–E was accomplished. The synthesis demonstrates a novel benzyne-catalyzed one-pot indoline construction/cross-coupling sequence. Firstly, 2,6-dibromo-phenylethylamine 539 was transformed into the pivotal indole 278 via removal of the Boc group, DDQ oxidation to the indole,529 and attachment of apara anisylethyl group onto the nitrogen atom. Then, the stage was set for the installation of subunits on the indole 2-position for the synthesis of dictyodendrin A 275, B 276, and E 538. 2-Acylindole derivative 283 was provided in almost quantitative yield through FC acylation reaction264 with para-methoxybenzoyl chloride 282 and zinc chloride as a promoter (Table 1, entry 1). In contrast, installation of the para-anisyl-acetate group was needed for synthesis of dictyo-dendrin A 275 (Table 1, entry 2). It was found that silver triflate530 was effective and the reaction progressed to afford the corresponding product 541 in high yield (Table 1, entry 3). To provide the intermediate to dictyodendrin E 538, it was necessary to introduce the para-methoxybenzyl group onto the indole 2-position. Unfortunately, reaction with ZnCl2 or silver triflate did not afford the corresponding product at all. It was assumed that the difficulty with para-methoxybenzylation was because of the high reactivity of para-methoxybenzyl chloride 540 and the presence of electron-rich aromatic rings in the substrate, which induce nonselective benzylation. Moreover, compound 278 afforded intermediates 541 and 542. Finally, compound 541 provided the dictyodendrins upon several steps (Scheme 116).531
| Entry | RX | Lewis acid | Product | Yield [%] |
|---|---|---|---|---|
| a Reaction condition: ZnCl2 (10 equiv.), RX (2 equiv.), Et2O, 0 °C.b Reaction condition: AgOTf (4 equiv.), RX (3 equiv.), THF, −78 °C.c Complex mixture.d 540 two equivalents. Reaction temperature: −78 to 0 °C. | ||||
| 1a | 282 | ZnCl2 | 283 | 99 |
| 2a | 279 | ZnCl2 | 541 | — |
| 3b | 279 | AgOTf | 541 | 81 |
| 4a | 540 | ZnCl2 | 542 | Tracec |
| 5b | 540d | AgOTf | 542 | — |
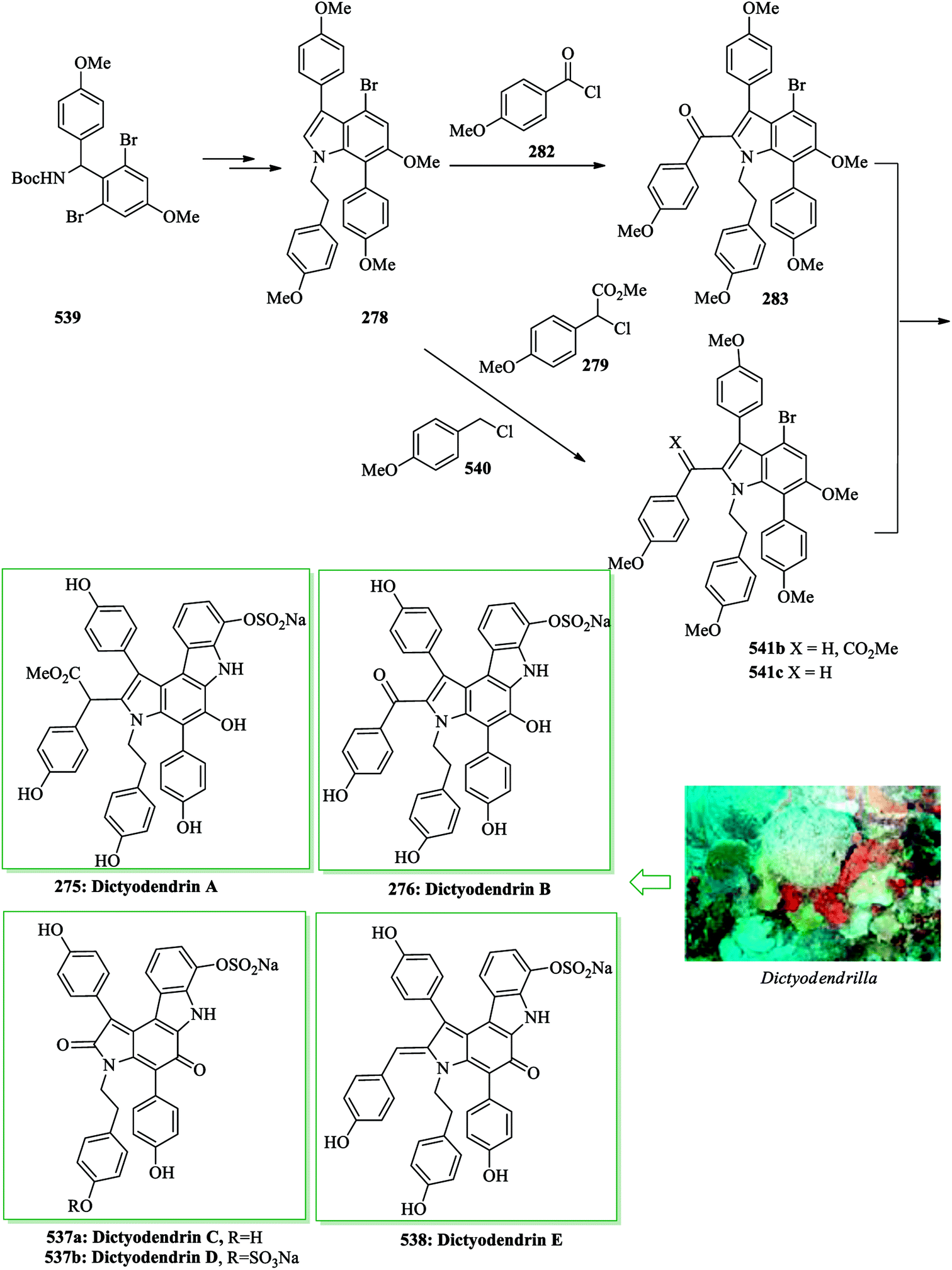 | ||
| Scheme 116 Total synthesis of dictyodendrins A–E. | ||
Miles et al. extracted cadinane sesquiterpene lactones heritol 544532 and heritonin 543533 from the sap of the mangrove plant Heritiera littoralis in the Philippines, which were demonstrated to possess ichthyotoxicity in parts per million quantities to Tilapia nilotica fingerlings. Chavan et al. reported the highly diastereoselective total synthesis of racemic heritonin 543 and heritol 544 via intramolecular cyclization on a preprovided sensitive butenolide functionality from market-purchasable initiating precursors in eight and nine purification operation in 43% and 33% overall yield, respectively. Synthesis of heritol 544 and heritonin 543 was started with FC acylation reaction of o-cresol methyl ether 545 and allylacetyl chloride in CH2Cl2 at ambient temperature to provide the desired allylic keto compound 546 in 95% yield. Next, compound 546 was converted into acid 547 via several steps. Then, compound 547 provided the key intermediate tetralone 548 through intramolecular FC acylation in 95% yield (over two steps). Compound 548 afforded compound 549 via several steps. The latter was converted into heritonin 543 in 96% yield and lastly compound 543 gave heritol 544 in 80% yield (Scheme 117).534
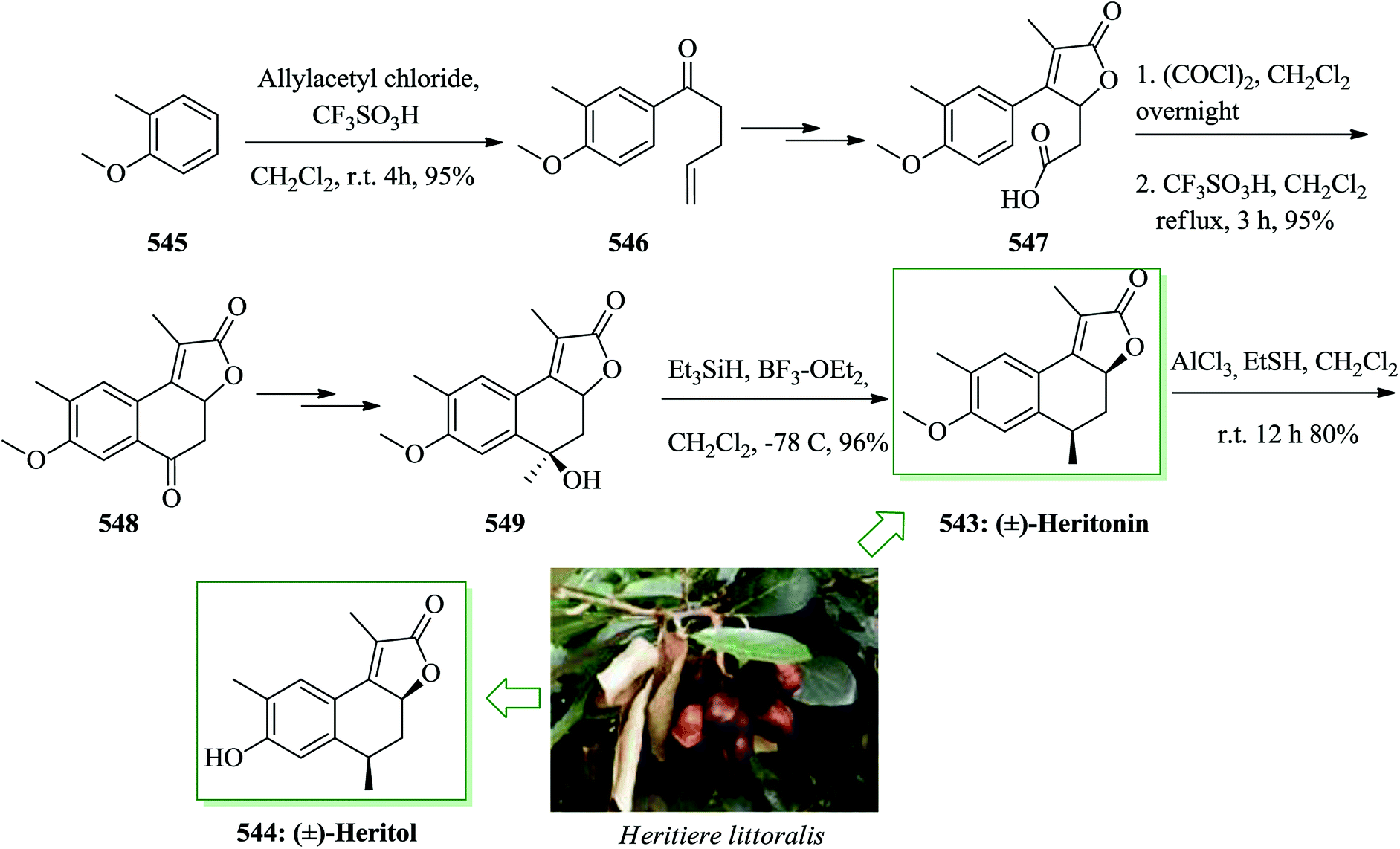 | ||
| Scheme 117 Total synthesis of (±)-heritonin 543 and (±)-heritol 544. | ||
Lysidicins A–C were extracted from Lisidicie rhodostegia Hance (Fabaceae) by Yu et al. in 2006535 and the isolation of lysidicins D–H was reported by the same group in 2007536 and 2010.537 Meanwhile, among the lysidicin group, lysidicin A 553 has the most unique and complicated structure in which two acetals form a spiro[furan-furofuran] ring system. Watanabe et al. in 2012 reported the initial total synthesis of (±)-lysidicin A significantly through single and cascade Claisen rearrangements and FC acylation reaction with AgOTf.538 In this method, the overall yield was 3.5% in 15 steps from diol 551. The total synthesis was started from diol 551, which was converted into benzyl ether 552 after several steps. Next, FC acylation reaction of 552 gave 553a and 553b in 27% and 35% yield (in two steps), respectively. Both 550a and 550b are regioisomeric compounds of lysidicin A; they were separately exposed to acid-mediated isomerization of the acetalic spiro[furan-furofuran] ring system and finally total synthesis of (±)-lysidicin A 550 was achieved (Scheme 118).538
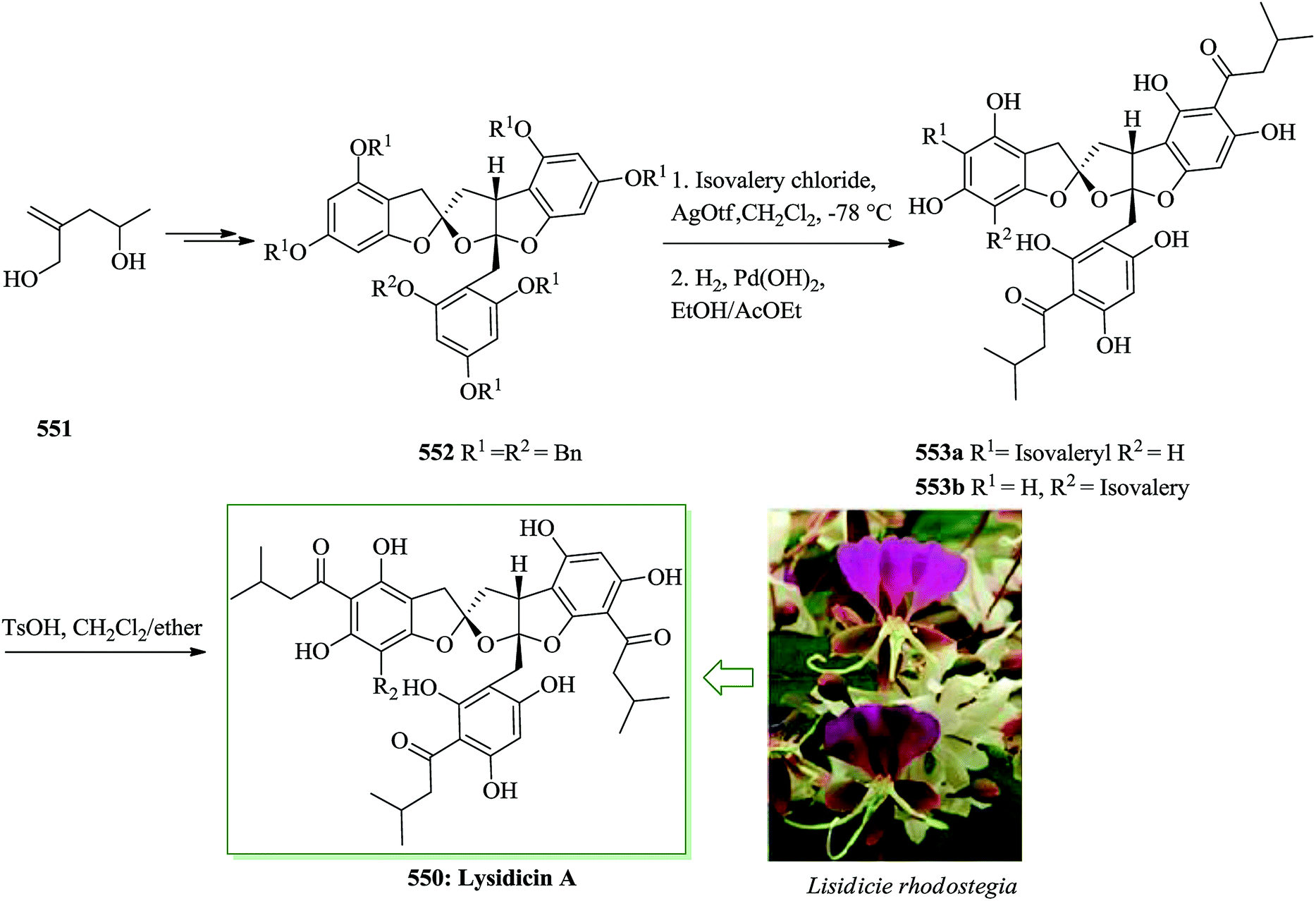 | ||
| Scheme 118 Total synthesis of lysidicin A 550. | ||
The cultivation of Streptomyces armeniacus under specific conditions resulted in the discovery of the armeniaspiroles, a unique group of compounds biosynthetically related to streptopyrroles.539 Armeniaspiroles, a unique group of natural products extracted from Streptomyces armeniacus, are identified by a novel spiro[4.4]non-8-ene moiety. Several derivatives of armeniaspiroles were obtained via halogenation, alkylation, addition/removal or reduction. A total synthesis of the 5-chloro analogue of armeniaspirole A was achieved in a linear six-step sequence. 5-Chloro-armeniaspirole A shows moderate activity against a series of multidrug-resistant, Gram-positive bacterial pathogens. A biomimetic synthesis of the armeniaspirole unit was envisaged starting from 556. The pyrrolo-phenone precursor 556 was easily accessible in two steps from N-methyl pyrrole and 2-methoxy-benzoyl chloride under modified FC conditions, followed by Lewis acid removal of the methyl ether. Subsequently, compound 556 was converted into a mixture of mono-, di- and tri-brominated products 557, 558 and 559 with the corresponding spiro framework, the main product being the dibromo derivative (Scheme 119).540
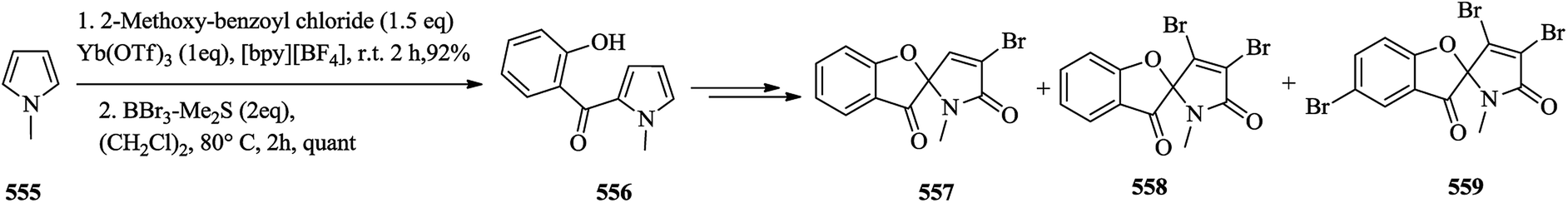 | ||
| Scheme 119 Formation of a mixture of mono-, di- and tri-brominated products 557, 558 and 559. | ||
A refined method to armeniaspiroles with the alkyl chain substitutions is shown in Scheme 120. 2,6-Dimethoxybromobenzene gave compound 561 in 70% yield. FC reaction of the latter with pyrrole-2-carbonyl chloride 563 afforded compound 564 in a satisfactory 60% yield. Compound 564 gave compound 554 in 72% yields after several steps. The two enantiomers of racemic 5-chloro-armeniaspirole A 554 were finally separated by chiral chromatography to afford enantiopure 5-chloro-armeniaspirole A 565 (R-configuration) and 566 (S-configuration).540
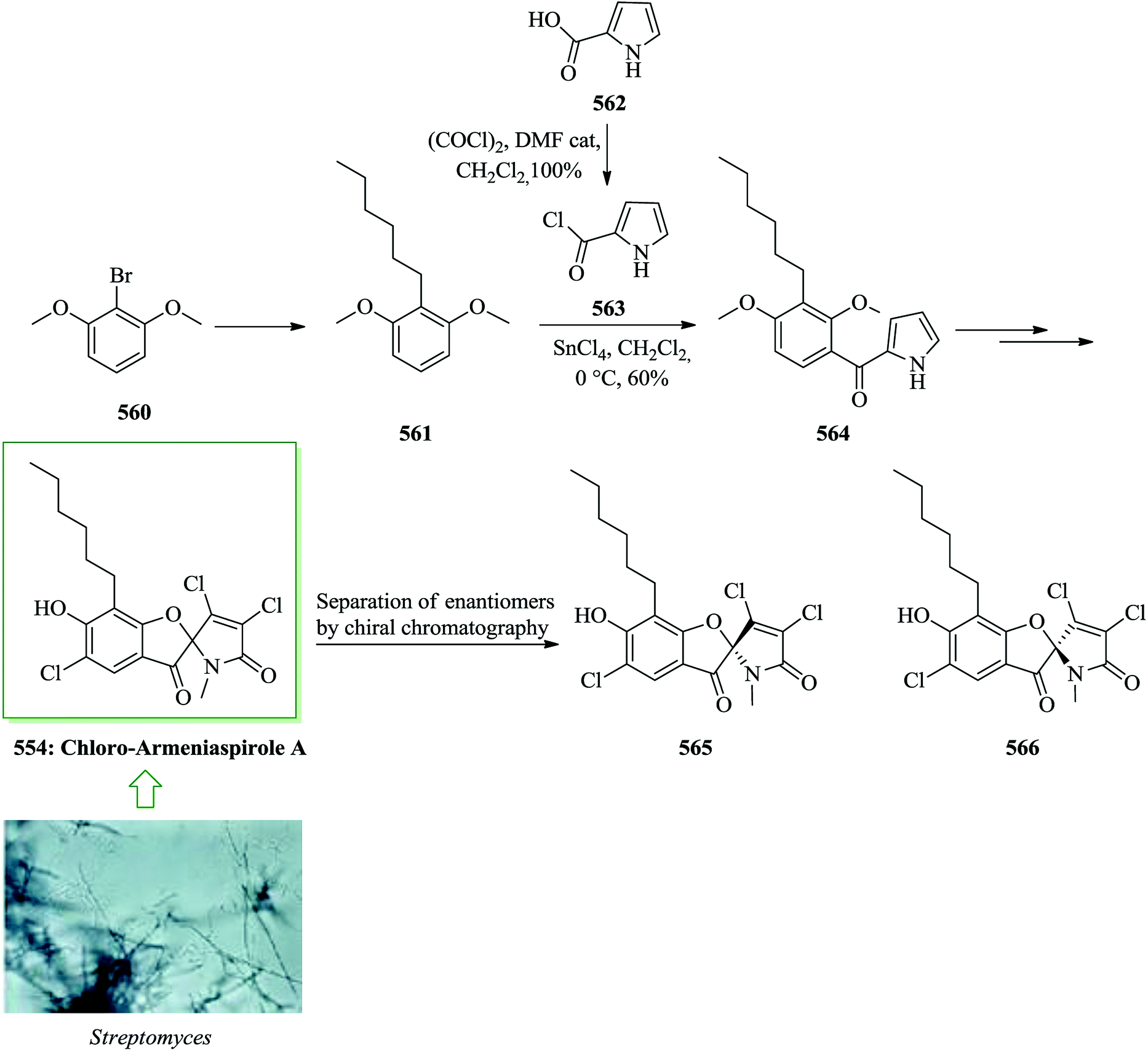 | ||
| Scheme 120 Total synthesis of 5-chloro-armeniaspirol 554. | ||
Norartocarpin 567 and artocarpin 568 are natural isoprenylated flavonoids isolated from the genus Artocarpus.541–543 They have a wide range of remarkable biological properties, including inhibitor effects on melanin biosynthesis and 5α-reductase, as well as antibacterial and cytotoxic activity.544 The total syntheses of norartocarpin and artocarpin, two biologically remarkable natural flavonoids with two regioisomeric isoprenyl side chains, were accomplished for the first time through a linear reaction sequence of 9 and 12 steps with overall yields of 14% and 3.5%, respectively, starting from 1,3,5-trimethoxybenzene. Starting from 1,3,5-trimethoxybenzene 454, twice sequential FC acylation of 454 in CH2Cl2 with aluminium chloride using acetyl chloride and 3-methylbutanoyl chloride as the acylated reagents, respectively, gave the intermediate 570 in moderate yields (75% for two steps). In the course of FC acylation, the corresponding demethylation simultaneously happened, which may be related to the effect of two carbonyl groups at ortho positions. On the other hand, if the two FC reactions were accomplished in a reversed sequence, 2,4,6-trimethoxyacetylbenzene 569 rather than the desired target molecule 570 was obtained as the major product, i.e., the isopentanoyl group was removed through reverse FC reaction during the second step. Finally, compounds 567 and 568 were obtained from 570 via several steps (Scheme 121).545
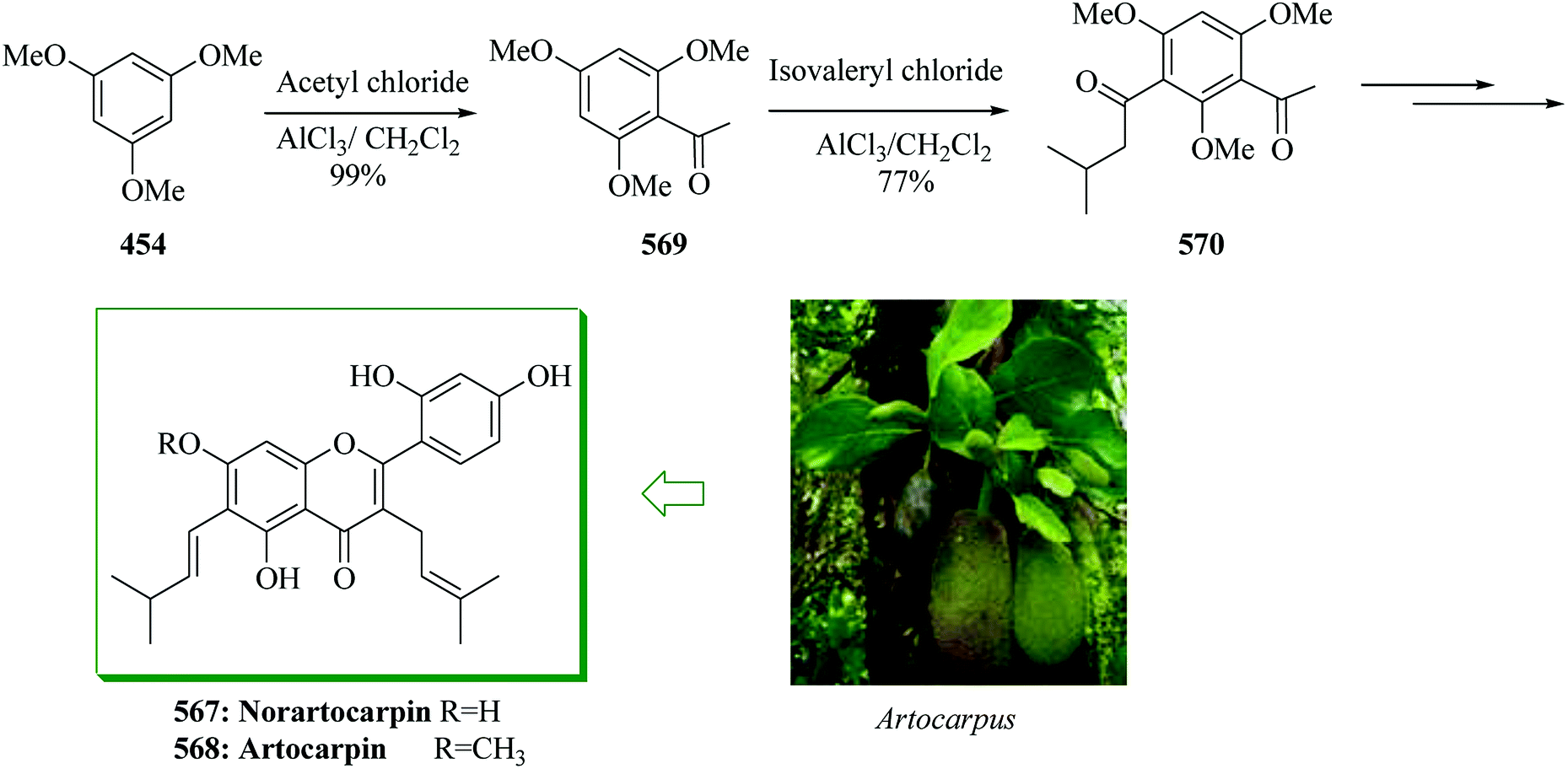 | ||
| Scheme 121 Synthesis of norartocarpin 567 and artocarpin 568. | ||
(+)-R-Mimosifoliol 571, a neoflavonoid isolated from the rootwood of Aeschynomene mimosifolia, demonstrated weak activity in a DNA-strand scission assay.546 Racemic (±)-mimosifoliol was obtained in 5 steps through an o-quinone methide intermediate and subsequently (+)-mimosifoliol was obtained through cycloaddition of the same intermediate with a chiral enol ether.547 A two-step method was developed to vinylate the diarylmethanes at the bridging CH2 with lateral lithiation and formylation followed by a Wittig reaction. This strategy was used in the racemic synthesis of the natural product mimosifoliol. Completion of the total synthesis of (±)-mimosifoliol needed a protected hydroxyl substituent. For this purpose, compound 572 was protected with a tert-butyldimethylsilyl group to afford 573. The latter was acylated to provide ketone 574. Careful control of the temperature of the FC reaction was needed to prevent accidental deprotection. Finally, compound 574 afforded (±)-mimosifoliol 571 after several steps (Scheme 122).548
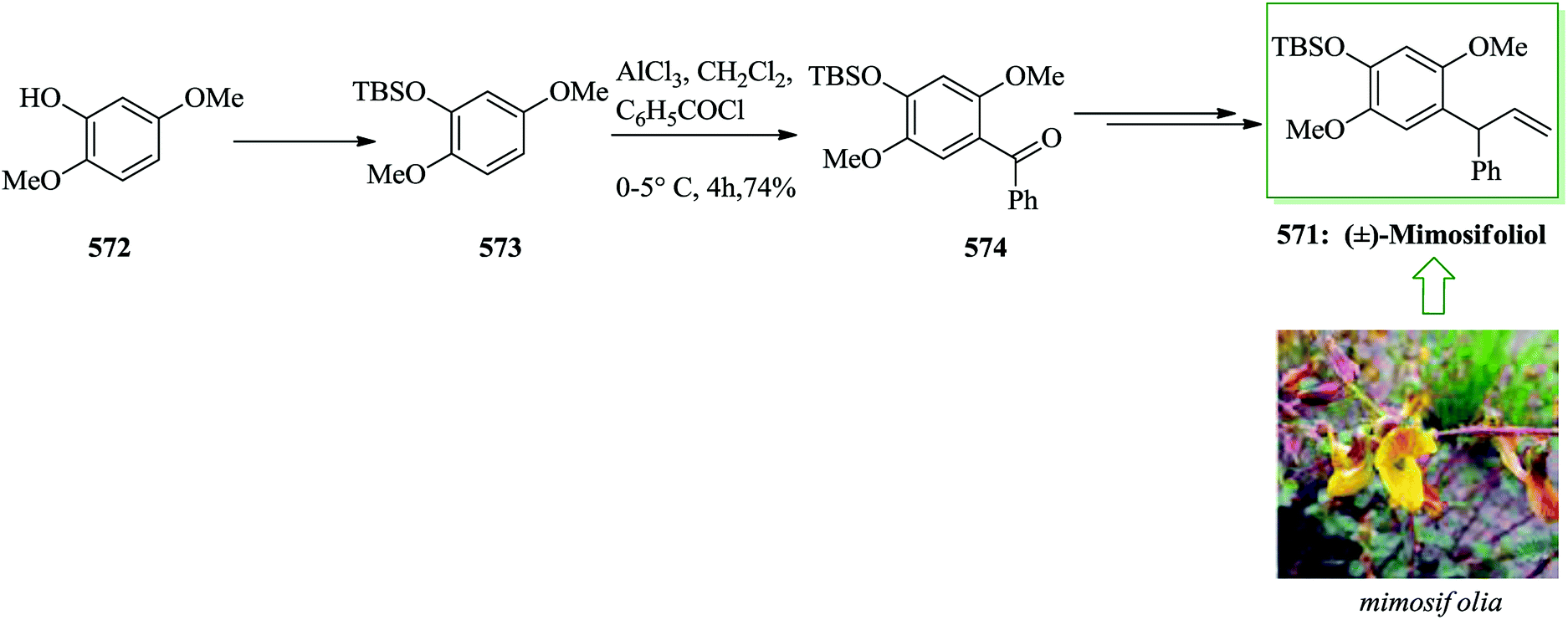 | ||
| Scheme 122 Total synthesis of (±)-mimosifoliol 571. | ||
The flowering plant Rhodomyrtus tomentosa (Aiton) Hassk. of the group Myrtaceae is applied in Thailand to treat a wide range of ailments. Rhodomyrtone 576 was isolated from the ethanol extract. The same compound was extracted from the bark of small twigs of Eucalyptus globulus Labill, which is a member of the Myrtaceae group.549,550 This acylphloroglucinol exhibits significant antibacterial activity. Additional acylphloroglucinols could be isolated from the leaves of R. tomentosa, which contain the isomer rhodomyrtosone B 575.551 In 2013, Maier et al. developed a concise synthesis of the isomeric acylphloroglucinols rhodomyrtosone B 575 and rhodomyrtone 576.552 In this pathway, for the synthesis of 575, phloroglucinol 496 was converted into the cross-conjugated enedione 577 via several steps. Since enone 577 is prone to isomerization,553 it was instantaneously treated with the enolate made via the reaction of acylphlorglucinol 504 with sodium hydride. In this way, a reasonable yield of the key intermediate 578 could be obtained. Refluxing a benzene solution of hydroxy ketone 578 in the presence of catalytic quantities of p-toluenesulfonic acid resulted in rhodomyrtosone B 575 in 69% yield (Scheme 123).552
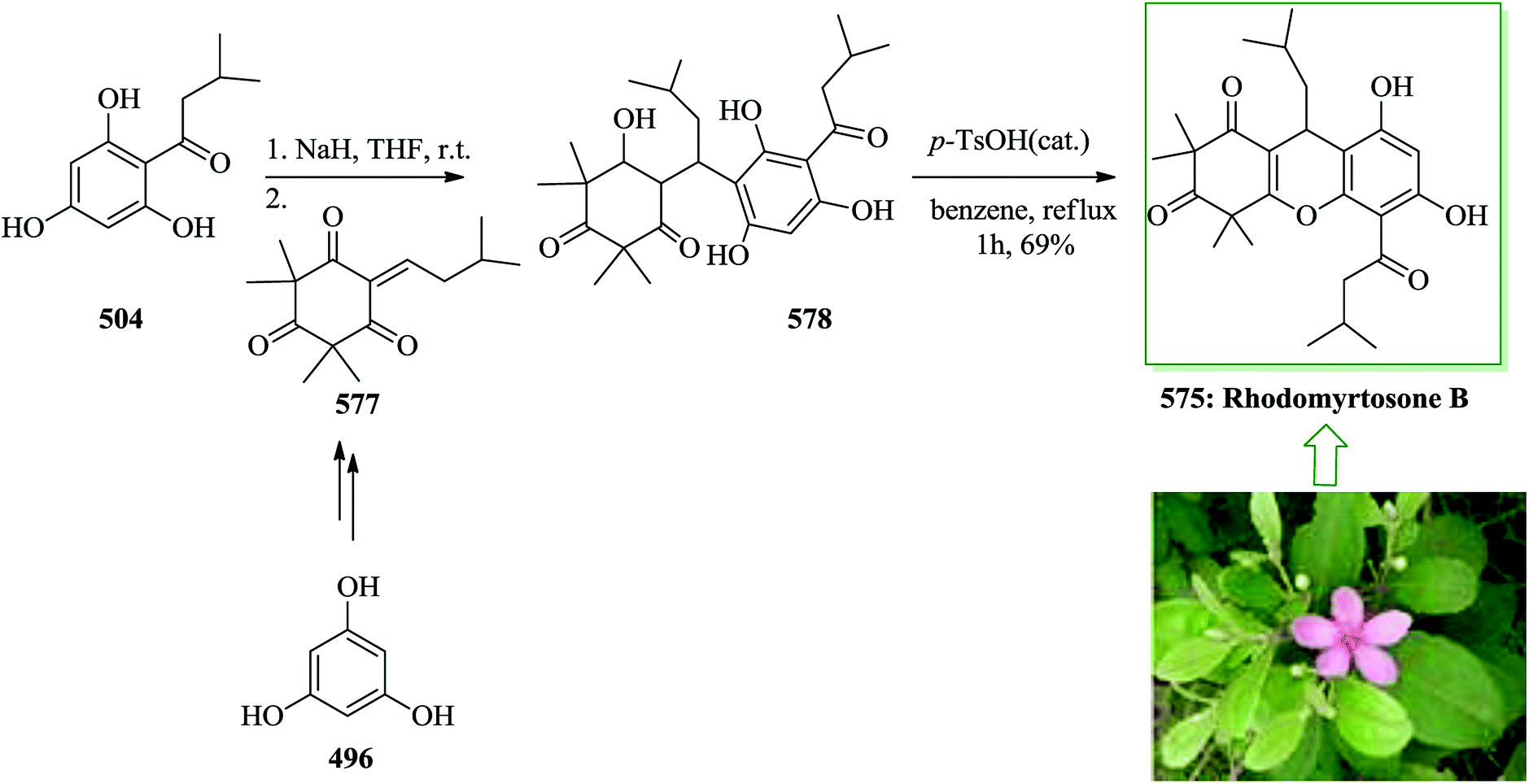 | ||
| Scheme 123 Total synthesis of rhodomyrtosone B 575. | ||
The other isomer, the antibiotic rhodomyrtone 576, was obtained from 578 through a sequence of acid-catalyzed cyclization, retro FC reaction, and reacylation (Scheme 124).552
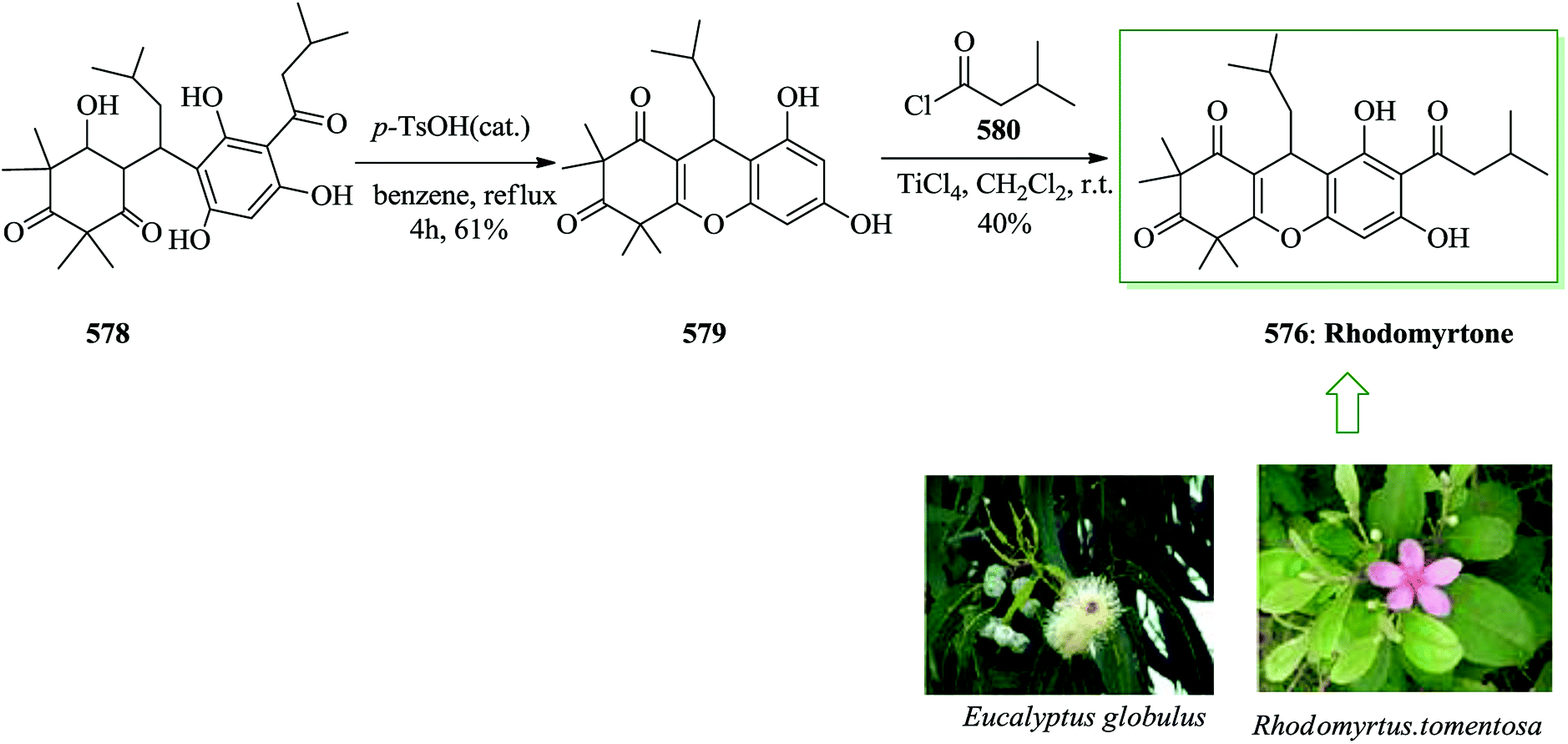 | ||
| Scheme 124 Synthesis of rhodomyrtone 576. | ||
8,5′-Neolignans having an 8-aryl-2,3-dihydrobenzofuran framework are the most plentiful natural products known in various groups of plants. These dihydrobenzofuran neolignans exhibit a wide range of biological activities, including cytotoxic, antiviral, and antifungal.554,555 (+)-Conocarpan 581, isolated from the wood of Conocarpus erectus by Hayashi and Thomson in 1975,556 showed a range of biological activities, including insecticidal, antifungal, anti-inflammatory, and antitrypanosomal.557–559 The enantioselective synthesis of natural (+)-conocarpan was reported in 2013 by Chen et al.560 The highlights of the synthesis are the asymmetric hydrogenation of prochiral ketones and intramolecular ring closure. Total synthesis of (+)-conocarpan 581 was started from 2′-fluoro-5′-bromophenyl acetic acid 582. The acid was transformed into acid chloride 583 followed by FC reaction with a slight excess of anisole to provide ketone 584 in 92% yields over two steps. Subsequently, compound 584 provided (+)-conocarpan 581 in 84% yield (Scheme 125).560
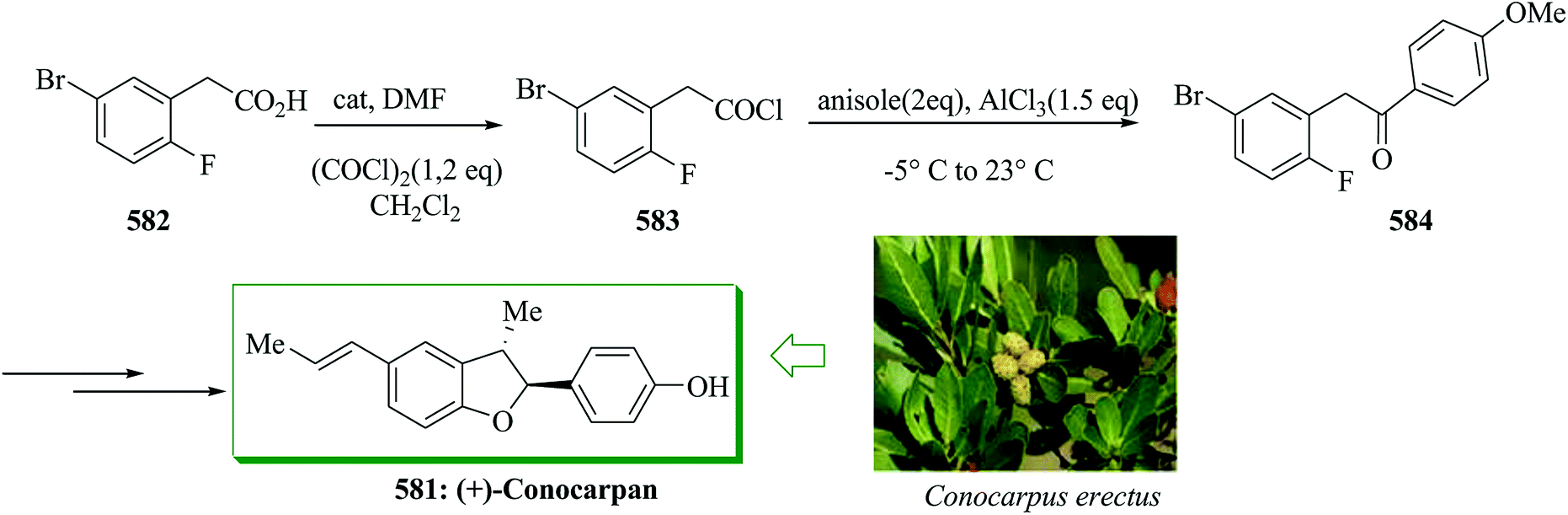 | ||
| Scheme 125 Enantioselective synthesis of (+)-conocarpan 581. | ||
Marmycin A, a unique angucycline analogue, was isolated by Fenical et al. in 2007 from the culture broth of a marine sediment-obtained actinomycete related to the genus Streptomyces. FC acylation and Dess–Martin oxidation were considered as the key steps in the synthesis of marmycin A 585. In this route, firstly anthracen-1-amine 589 and glycal 588 gave the C1′ N-glycosidation product 590. Next, N-trifluoroacetylation of 590 using TFAA/triethylamine followed by O-deprotection with ammonium resulted in a FC acylation product 591b in 70% overall yield, other than the desired N-Boc product 591a. Finally, compound 591b provided compound 585 in several steps (Scheme 126).561
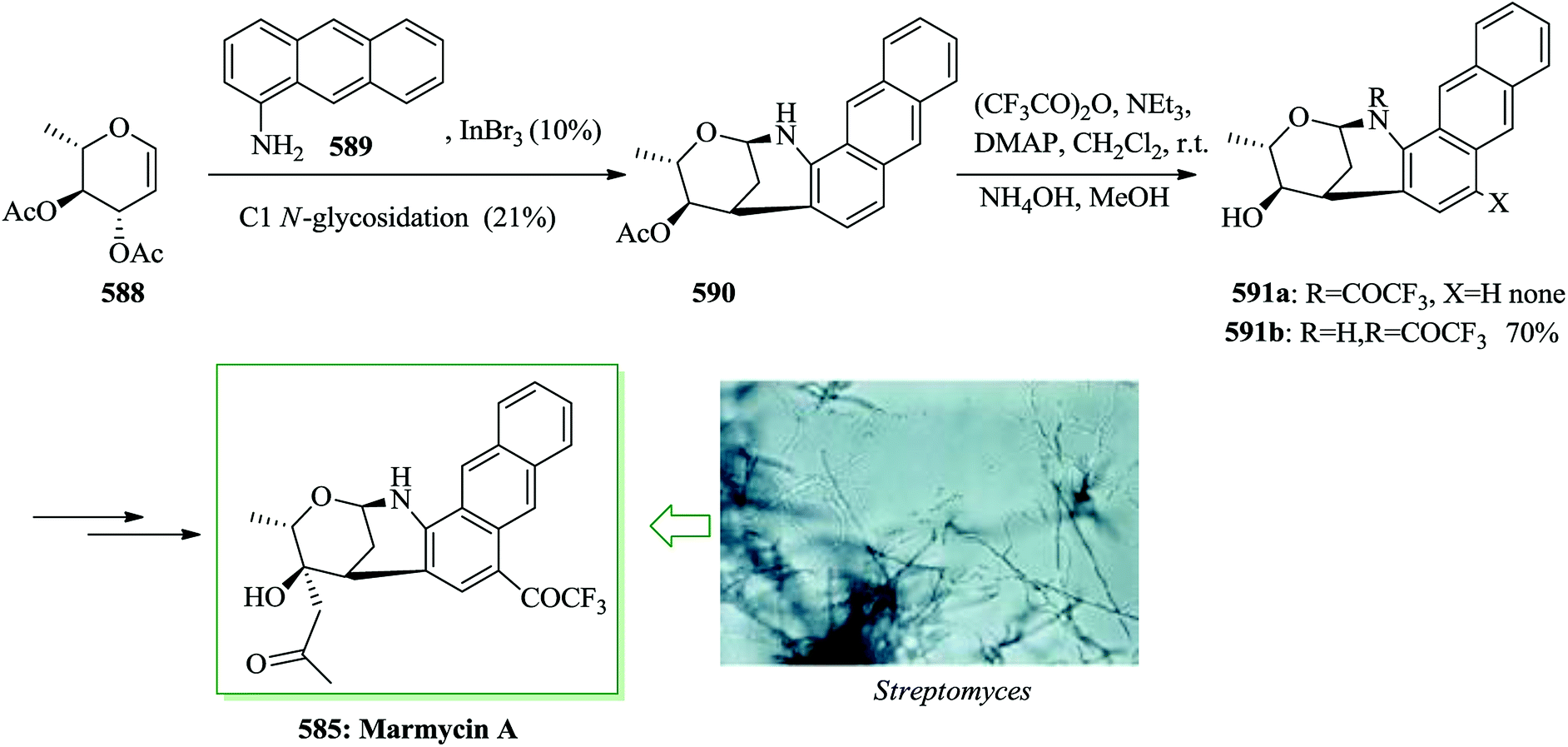 | ||
| Scheme 126 Total synthesis of marmycin A 585. | ||
To confirm the FC acylation using (CF3CO)2O and the nucleophilic addition occurring on the carbonyl group, diastereomeric N-glycosidation products 592 and 593 were reacted with (CF3CO)2O/triethylamine/DMAP and then ammonium to afford FC acylation products 594 and 595, respectively, in 70% overall yield. Quaternary alcohols 586 and 587 were obtained from 594 and 595, respectively, in 55% overall yield after several steps (Scheme 127).561
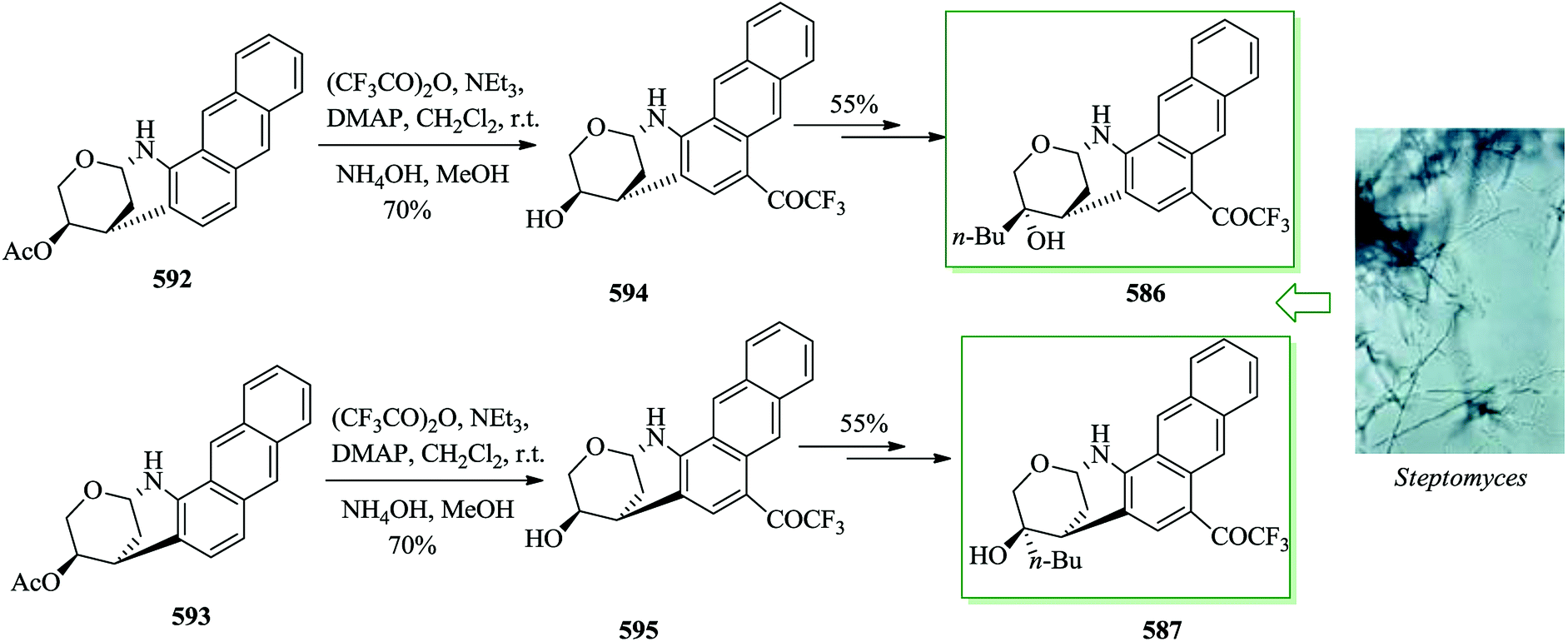 | ||
| Scheme 127 The formation of quaternary alcohols 586 and 587. | ||
Marinamide 596 and its methyl ester 602 are two isoquinolinone alkaloids that were isolated from the metabolite of mixed fermentation of two mangrove endophytic fungi (strains no. 1924 and 3893) from the South China Sea. These two compounds were identified as 4-(2-pyrrolyl)-1-isoquinolone-3-carboxylic acid 596 and methyl 4-(2-pyrrolyl)-1-isoquinolone-3-carboxylate 602, respectively. Compounds 596 and 602 exhibited significant antibacterial and562 antitumor properties. The first total synthesis of isoquinolinone alkaloid marinamide 596 and its methyl ester 602 was developed in 2013 by Ji et al.563 The key steps included a regioselective FC reaction of 1-benzyl-1H-pyrrole to generate the intermediate 601. The synthesis of intermediate 601 is the key step for the formation of marinamide and its methyl ester, which included a regioselective FC acylation of 1-benzyl-1H-pyrrole to generate a 2-acylpyrrole derivative. Phthalic anhydride 597 afforded the acyl chloride 598 after several steps. Compound 598 reacted with 599, which was obtained by reacting pyrrole 600 with benzyl bromide in anhydrous dimethyl sulfoxide, via the catalysis of zinc powder in toluene at ambient temperature to effectively give the key intermediate 601 in 68% yield.564 Compound 601 gave the methyl ester of marinamide 602.565 Finally, hydrolysis of 602 with hydrochloric acid gave marinamide 596 (Scheme 128).563
 | ||
| Scheme 128 Total synthesis of marinamide 596. | ||
The naphtho[2,3-c]furandiones (isofuranonaphthoquinones), a relatively small group of secondary metabolites with various biological properties, have been isolated from fungal, botanical, bacterial and insect sources. Monosporascone 603 was isolated from the fungus Gelasinospora pseudoreticulata.566 Monosporascone is the only known isofuranonaphthoquinone with oxygenation at the 5 and 7 positions. The first total synthesis of the natural isofuranonaphthoquinone monosporascone 603 was exhibited in 2014 by Piggott et al.567 The five-step synthesis established involves a silver acetylide–acid chloride coupling, domino Diels–Alder-retro-Diels–Alder reaction, and an intramolecular FC acylation reaction. Total synthesis of the natural product monosporascone 603 was accomplished in 57% yield overall. The synthesis of monosporascone 603 was started from the reaction of silver acetylide 605568 and acid chloride 604,569 which after four steps gave acid chloride 606. Reaction of acid chloride 606 with five equivalents of aluminium chloride,570 with an extended reaction period to permit selective demethylation of the perimethoxy group, gave monosporascone 603 in satisfactory yield (Scheme 129).571
 | ||
| Scheme 129 Total synthesis of monosporascone 603. | ||
In 2006, Merck researchers reported the discovery of a unique and active antibiotic, (−)-platensimycin 607, isolated from Streptomyces platensis strain MA7327, which originated from South Africa.572 Lear et al. developed its total synthesis via a four-step construction of the aromatic amine segment and an improved stereocontrolled assembly of the ketolide segment, (−)-platensic acid. Key synthetic advances contain a modified Lieben haloform reaction, a sterically controlled chemo- and diastereo-selective organocatalytic conjugate reduction and a bismuth(III)-mediated FC cyclization. The longest linear sequence is 21 steps with an overall yield of 3.8% from eugenol. In this approach, firstly, compound 608 was converted into lactol 609 via several steps. A practical limitation of this method is the excessive requirement for very toxic and ecologically harmful tin(IV)chloride to drive the FC arylation. Hence, Bi(OTf)3 was found to be the most superior in reactivity and gave 610 in 94% yield. Subsequently, compound 610 was converted into the tetracyclic dienone 613 after several steps. Then, the Lewis acid-catalyzed cyclization of the cis-tosyl lactol 611, which could be accessed directly from 608, was explored. The reaction of 611 with tin(IV) chloride needed a greater excess (8–10 equiv.) to achieve a high-yielding FC transformation. Inspired by Bartoli, Sambri, and co-workers,573 they eventually turned to lithium perchlorate as a cocatalyst to Bi(OTf)3 to help drive the FC cyclization of 611. The best catalytic combination of 5 mol% Bi(OTf)3 with 3 equivalents of lithium perchlorate eventually provided 612 in 94% yield. Subsequently, compound 612 was converted into 613 via several steps and then compound 613 was transformed into 614 via several more steps (Scheme 130).574
 | ||
| Scheme 130 Total synthesis of compound 614. | ||
A unique synthesis of the aromatic section of platensimycin 607 was accomplished. This synthesis was started with FC acylation of 615 with acetic acid in hot PPA to provide the acetophenone 616. Next, compound 616 was converted into compound 617 via several steps (Scheme 131).574
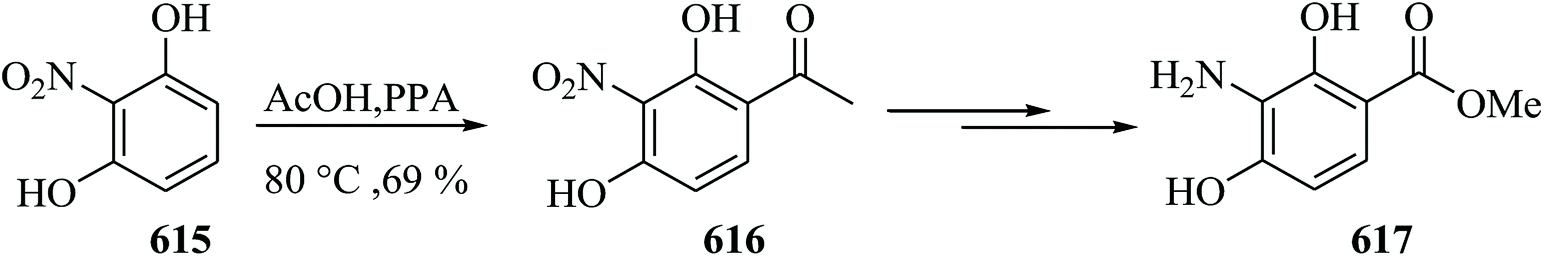 | ||
| Scheme 131 The formation of compound 617. | ||
Finally, platensic acid 614 and the aniline core 617 afforded (−)-platensimycin 607 in 60% yield upon several steps (Scheme 132).574
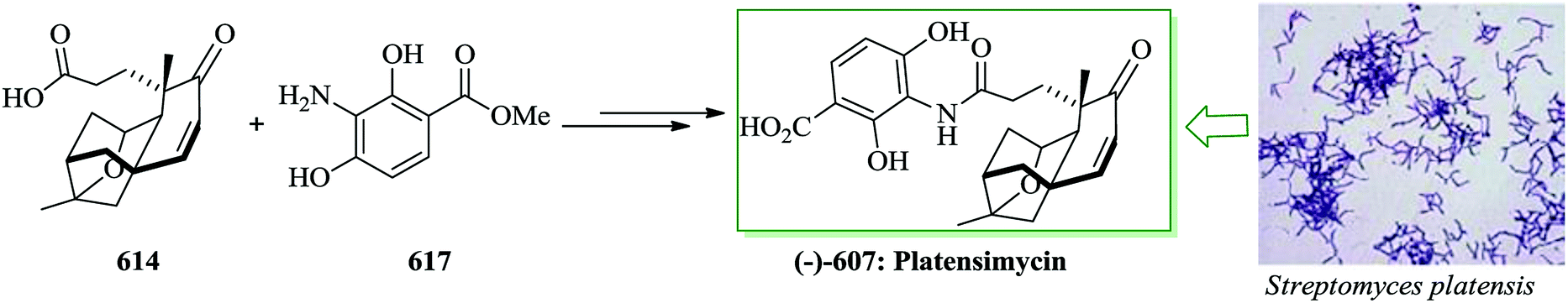 | ||
| Scheme 132 Total synthesis of (−)-platensimycin 607. | ||
The natural product 2,4,6-trihydroxy-3-geranyl-acetophenone (tHGA) isolated from the medicinal plant Melicope ptelefolia was demonstrated to show lipoxygenase (LOX) inhibitory activity. It is known that LOX plays a significant role in inflammatory response as it catalyzes the oxidation of unsaturated fatty acids.575,576 Shaari et al. in 2014 reported synthesis of tHGA analogues through simple FC acylation and alkylation reactions.577 In this method, they reported the formation of 3-geranyl-1-(2′-methylpropanoyl)phloroglucinol 618, a natural product known in Hypericum empetrifolium. Direct FC acylation of phloroglucinol 496 was achieved with isobutyryl chloride as the acylating agent using anhydrous AlCl3 to provide compound 619.578 Subsequently, the geranyl scaffold was introduced through electrophilic substitution of geranyl bromide with anhydrous K2CO3 as the base in dry MeOH under reflux to afford the FC alkylation product 618 in satisfactory yield (19.5%) (Scheme 133).577
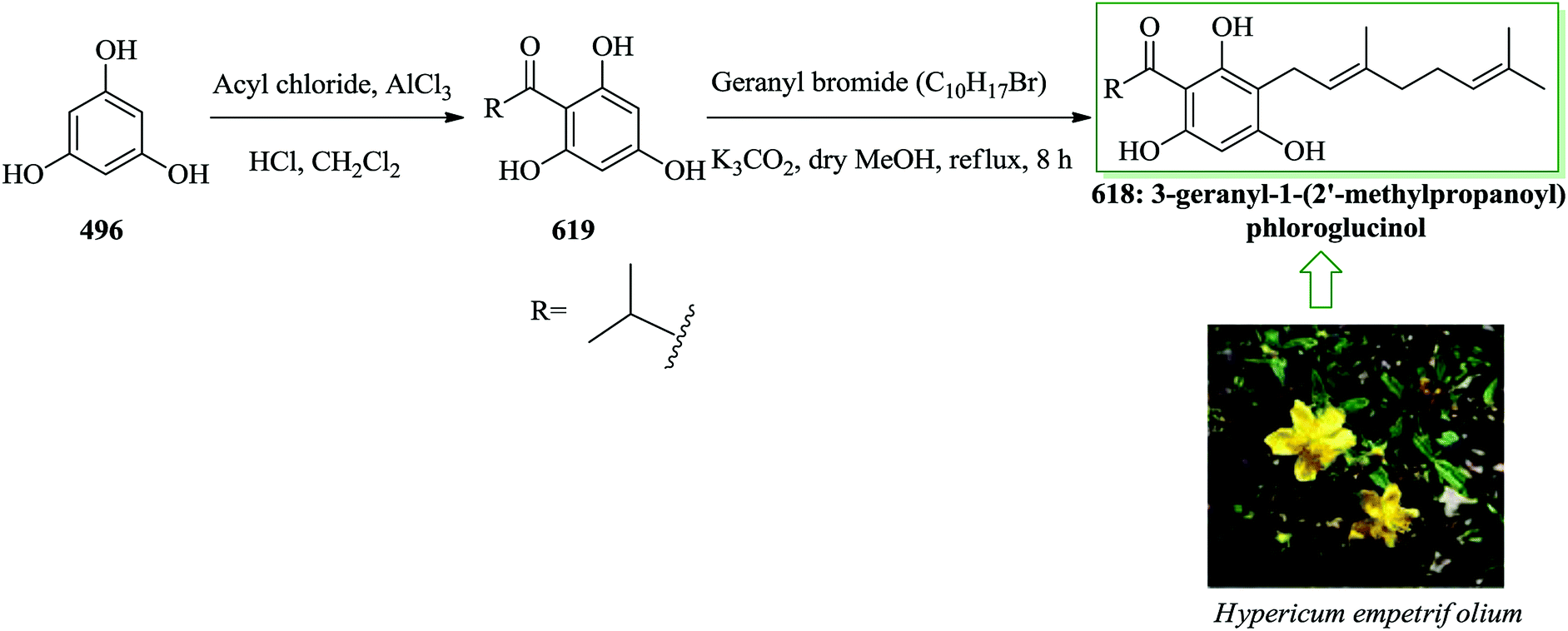 | ||
| Scheme 133 Total synthesis of 3-geranyl-1-(2′-methylpropanoyl)phloroglucinol 618. | ||
The merochlorins, a structurally uncommon group of four chlorinated meroterpenoids, have potent antimicrobial activity and were isolated from a marine strain of Streptomyces bacteria.579 Merochlorin A 620 contains four contiguous stereocenters embedded in a bicyclo[3.2.1]octanone scaffold. The complex, polycyclic structures of merochlorins A, obtained from a unique terpene side chain, have aroused the interest of chemists.580,581
Total synthesis of merochlorin A was exhibited alongside the biosynthetic studies of George et al. in 2015.582 Synthesis of merochlorin A 620 was started with a FC reaction of methyl 3,5-dimethoxyphenylacetate 621 and chloroacetyl chloride to afford ketone 623 in 56% yield. Finally, merochlorin A 620 was obtained from compound 623 in 42% yield via several steps (Scheme 134).582
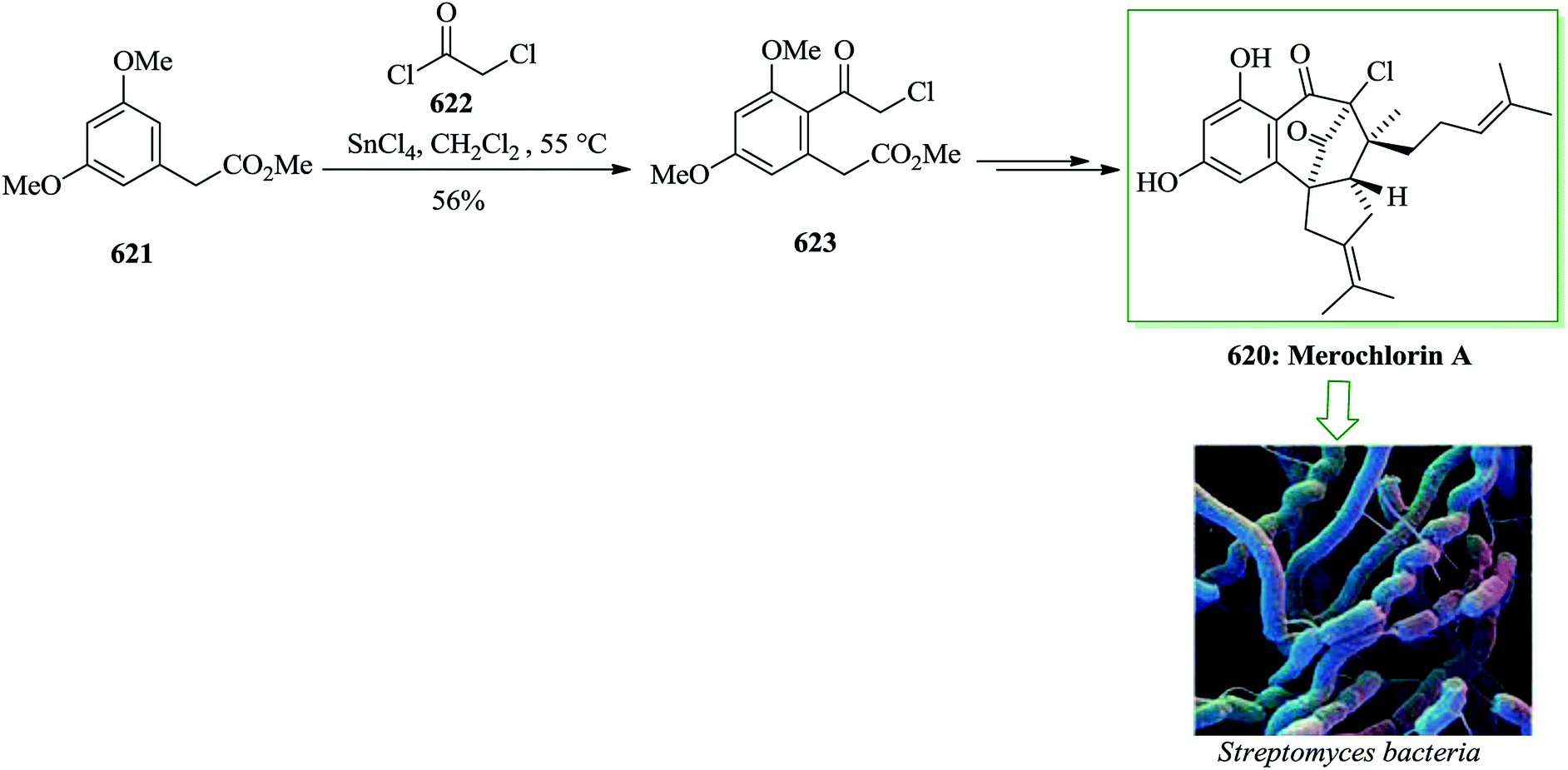 | ||
| Scheme 134 Total synthesis of merochlorin A 620. | ||
Curvulone B 624, a cis-di functionalized tetrahydropyran natural product, is one of a wide range of relevant compounds isolated from the marine fungus Curvularia sp., which was isolated from the marine alga Gracilaria folifera.583 A total synthesis of curvulone B was achieved using a FC reaction and a highly cis-selective intramolecular oxa-Michael addition. This synthetic method was accomplished in ten steps and 39% overall yield. Total synthesis of Curvulone B 624 was started from 3,5-dihydroxybenzoic acid 625. Next, compound 625 afforded ester 626 upon several steps. The FC acylation reaction of ester 626 was performed with whole regioselectivity with a 2-chlorobenzyl protecting group to afford ketone 628 in 89% yields. Finally, ketone 628 afforded the target natural product 624 via several steps (Scheme 135).584
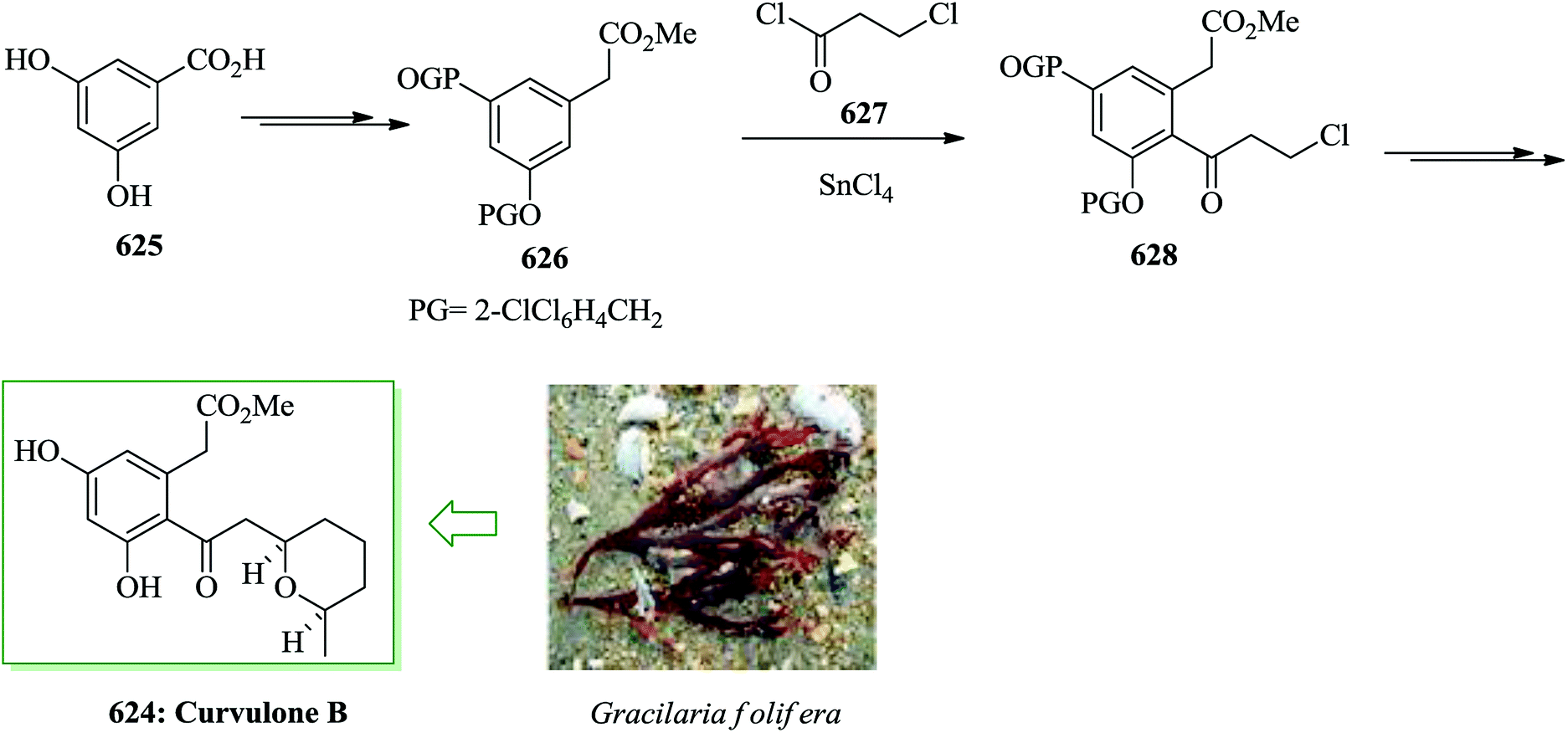 | ||
| Scheme 135 Total synthesis of curvulone B 624. | ||
Triumphalone 629 and isotriumphalone 630, highly oxidized monomeric phloroglucinols585,586 with two stereogenic centers, were extracted from Melaleuca triumphalis by Brophy et al.587 The first total synthesis of (±)-triumphalone was accomplished in 8 steps from phloroglucinol. Synthetic triumphalone was converted into (±)-isotriumphalone in one step. In this route, firstly phloroglucinol 496 was converted into 632 through the FC acylation reaction in 82% yield.588 Subsequently, (±)-triumphalone 629 was provided from compound 632 via several steps and also compound 629 was transformed to (±)-isotriumphalone 630 in one step. An spontaneous change of 629 to 630 was detected. The structure change gradually occurred to afford a 3:7 mixture of 629 and 630 upon standing for 24 months (Scheme 136).589
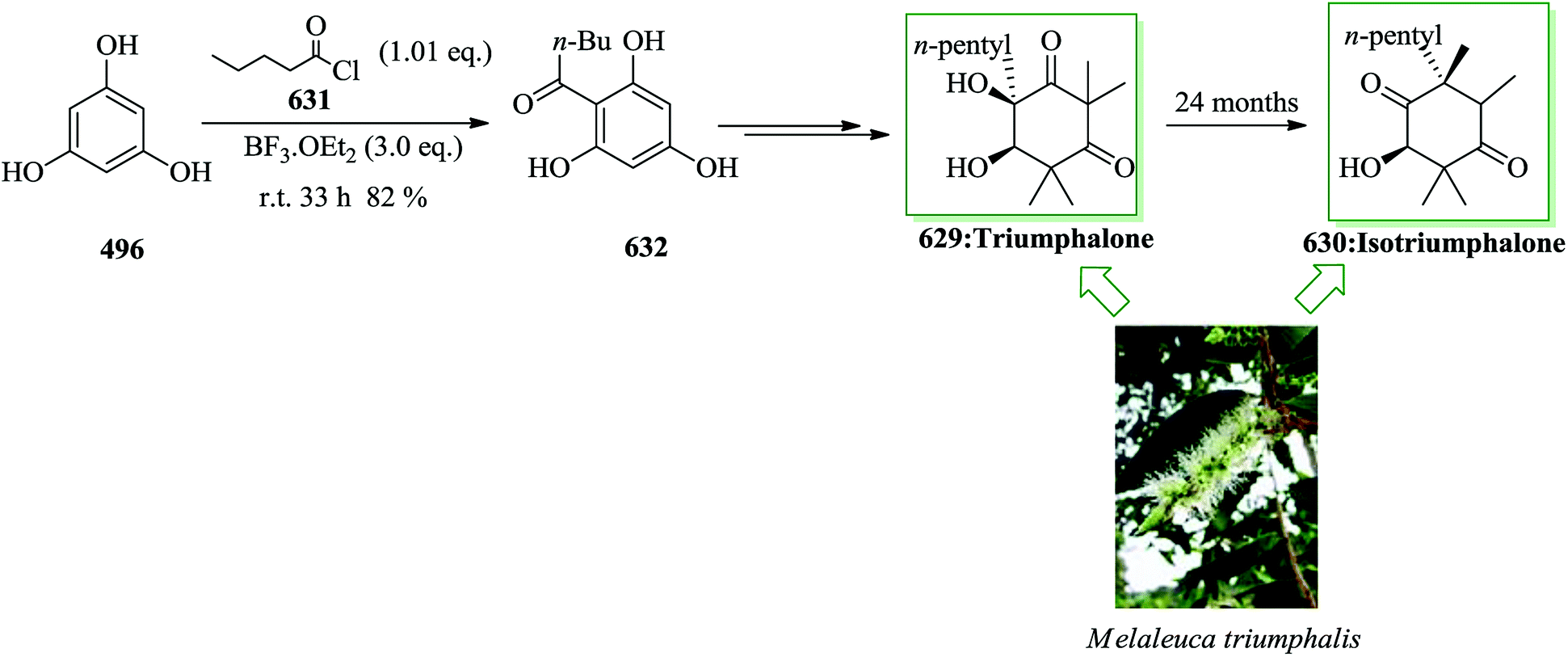 | ||
| Scheme 136 Total synthesis of triumphalone 629 and isotriumphalone 630. | ||
Flavonoids, a group of polyphenolic compounds that are abundant in plants,590 include dietary components of potential importance to health.591 In addition, flavanoids are emerging as a potentially significant unique group of pharmaceutical lead substrates.590 Among the several classes of flavonoids, dihydrochalcones exhibit a series of biologically remarkable activities, including antioxidant,592,593 antiinflammatory,594 antileishmanial,595 antidiabetic,596 anticancer597 and molluscidal598 properties.599 Peng et al. described the isolation and identification of a significant dihydrochalcone, named taccabulin E 633, from extracts of the plant species Tacca chantrieri and Tacca integrifolia.600 Spring et al. in 2015 reported the concise and divergent total synthesis of dihydrochalcone 633,601 which was started from catechol 634. Next, FC acylation reaction of 634 permitted access to 635 in moderate yield.602,603 Finally, compound 635 was converted into taccabulin E 633 in 94% yield upon several steps (Scheme 137).601
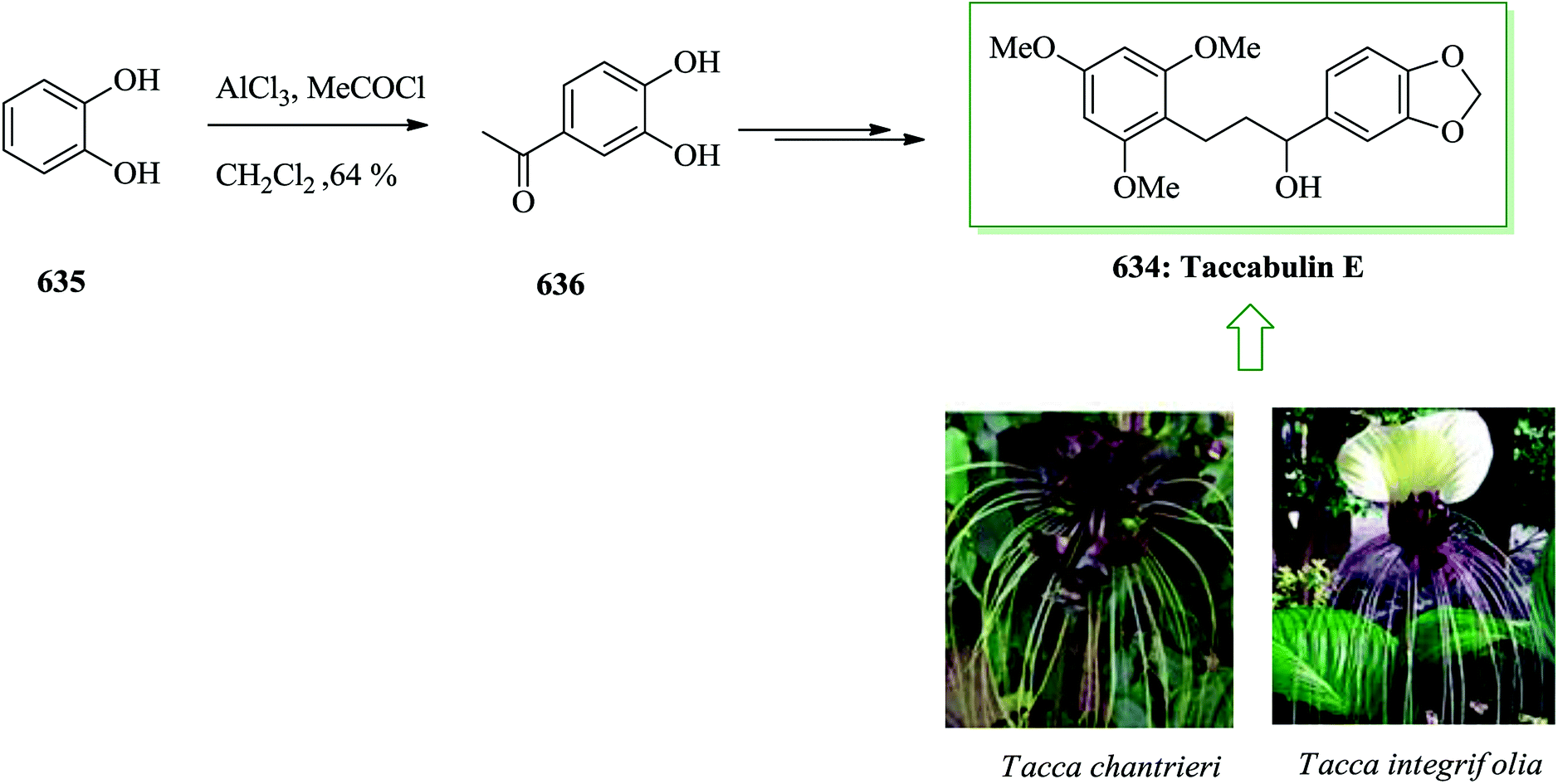 | ||
| Scheme 137 Total synthesis of taccabulin E 633. | ||
Diarylheptanoids, with two aryl groups at C(1) and C(7) of a C7 chain, are a significant group of natural products isolated from nature.604–608 Generally, diarylheptanoids are categorized into three groups comprising linear diarylheptanoids, macrocyclic biarylheptanoids, and macrocyclic diaryl ether heptanoids. Sun et al.609 isolated two furan-cyclized diarylheptanoids, 637 and 638, from rhizomes of Alpinia officinarum Hance (Zingiberaceae) as minor constituents and they were demonstrated to have satisfactory cytotoxicity against the IMR-32 human neuroblastoma cell line. Seçen et al. in 2015 reported a synthetic strategy for the formation of two natural diarylheptanoids, 2-benzyl-5-(2-phenylethyl)furan 637 and 2-methoxy-4-{[5-(2-phenyl-ethyl)furan-2-yl]methyl}phenol 638.610 Total synthesis of the natural product 637 was started from benzyl bromide 639, which afforded 2-(2-phenylethyl)furan 640 via several steps. FC acylation of compound 640 with benzoyl chloride using aluminium chloride afforded ketone 641 in 73% yield. Finally, the reduction of 641 afforded 637 in 81% yield (Scheme 138).610
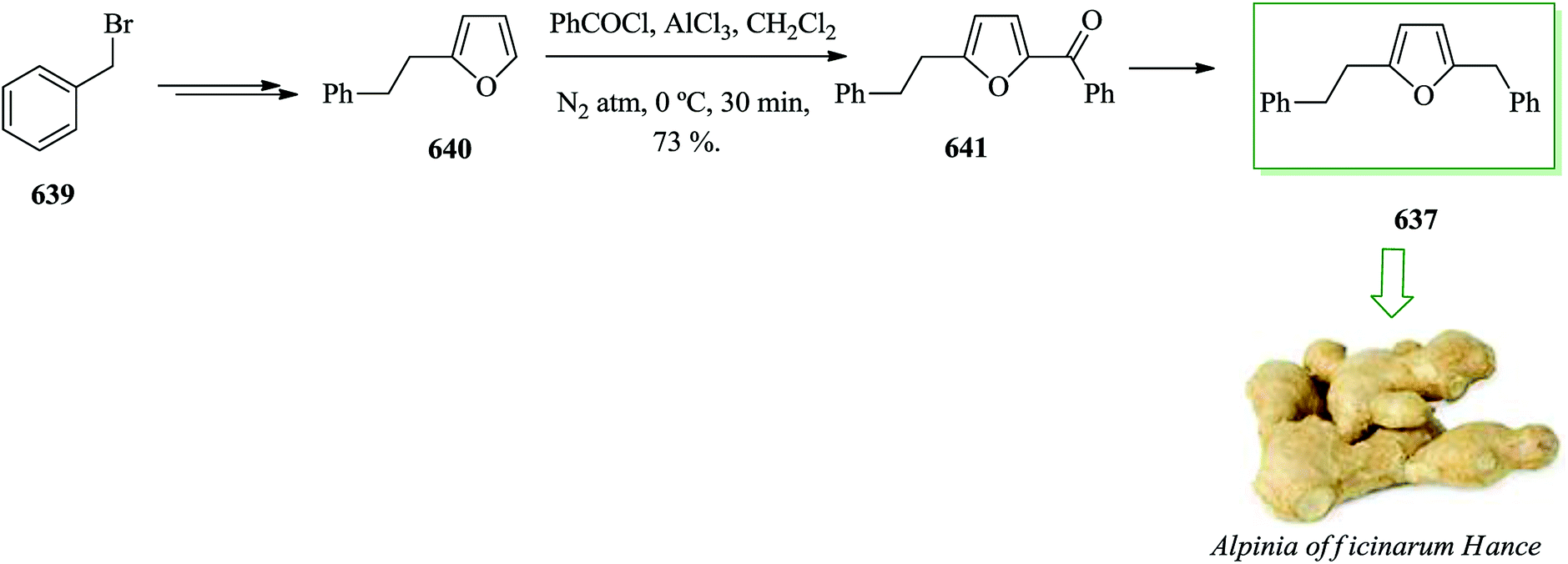 | ||
| Scheme 138 Total synthesis of furan-cyclized diarylheptanoid 637. | ||
Then, this group used 2-(2-phenylethyl)furan 640 as the initiating precursor to form natural product 638. In this route, compound 640 was reacted with 4-(acetyloxy)-3-methoxybenzoyl chloride 642 via FC reaction to afford ketone 643 in a satisfactory yield (59%). Subsequently, natural product 642 was synthesized from compound 643 upon several steps (83%) (Scheme 139).610
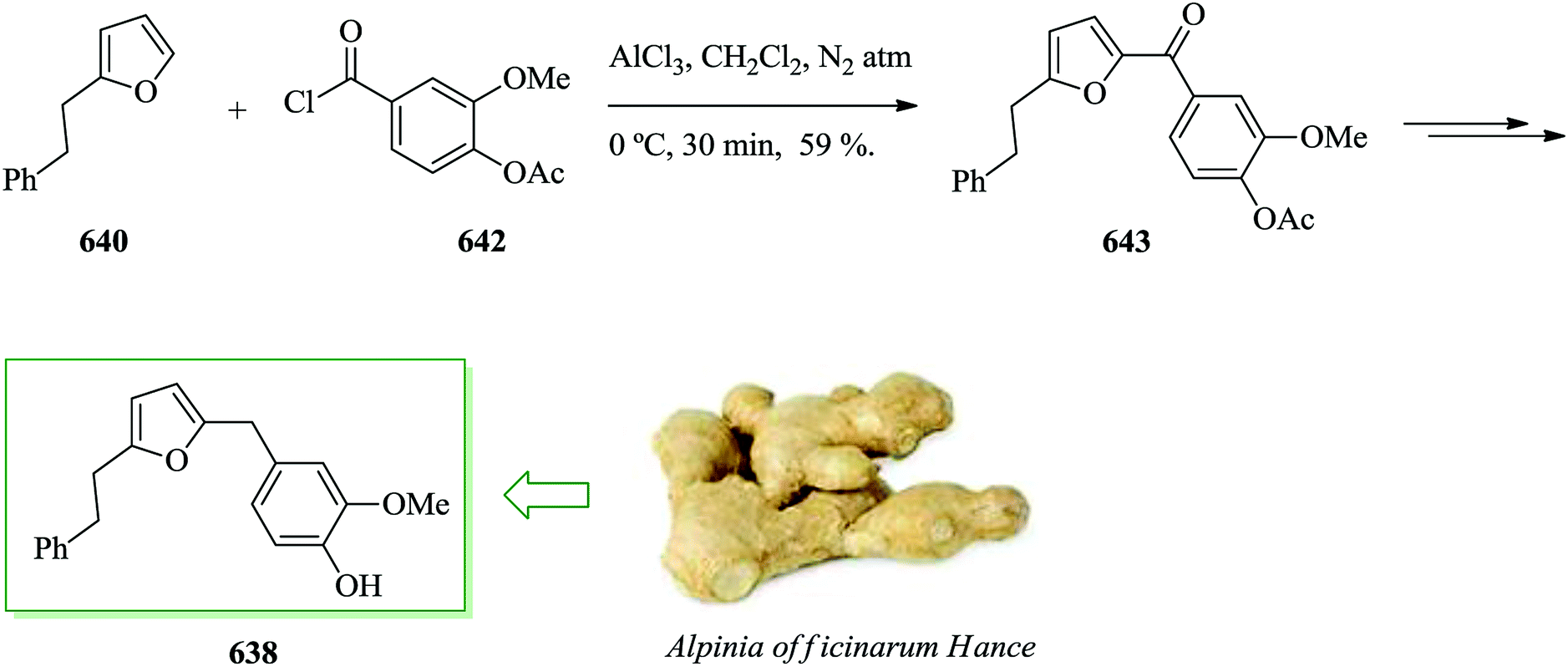 | ||
| Scheme 139 Total synthesis of furan-cyclized diarylheptanoid 638. | ||
Stigmatellin A and B, two unique antibiotics, were isolated from the gliding bacterium Stigmatella aurantiaca.611 They are powerful inhibitors of electron transport in chloroplasts and mitochondria. The absolute configuration of Stigmatellin A was confirmed as (S,S,S,S) by chemical correlation.612 A significant and enantioselective method for the formal total synthesis of stigmatellin A was developed in 2017 by Yadav et al.613 The key steps included in this synthesis are desymmetrization, FC acylation reaction, regioselective demethylation, Baker–Venkataraman rearrangement and Grubbs cross-metathesis. Total synthesis of stigmatellin A was started from tetramethoxy benzene 645. The FC acylation reaction of tetramethoxy benzene 645 with propanoyl chloride using aluminium chloride gave the corresponding ketone 646.614 Finally, compound 646 gave stigmatellin A 644 after several steps (Scheme 140).613
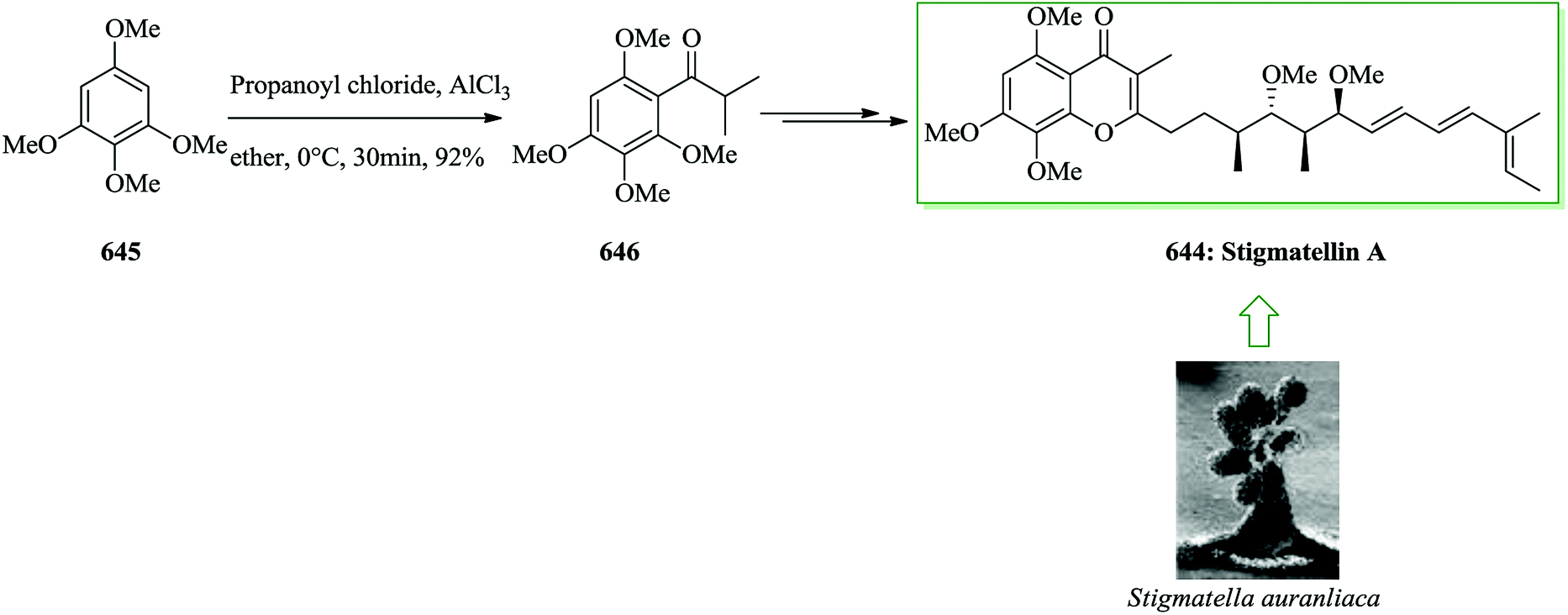 | ||
| Scheme 140 Total synthesis of stigmatellin A 644. | ||
γ-Lycorane is an alkaloid isolated from the plants of the Amaryllidacae group.615 Various methods for the synthesis of γ-lycorane 647 have been reported, despite, as several authors have pointed out, its apparent lack of useful pharmacological activities.616,617 A total synthesis of γ-lycorane was accomplished using N-tosylpyrrole as a key framework. The synthesis uses both an intermolecular and an intramolecular FC reaction, and also an entirely diastereoselective hydrogenation of a late-step pyrrole intermediate. For the synthesis of γ-lycorane 647, N-tosyl pyrrole 648 was subjected to an FC acylation reaction with succinic anhydride. The resulting acid 650 afforded the corresponding alcohol 651 via several steps. The reaction between alcohol 651 and amberlyst-15 easily afforded the corresponding FC product 652 in 82% yield by construction of the novel C–C bond at the α-position. Finally, compound 652 gave γ-lycorane 647 via several steps (Scheme 141).618
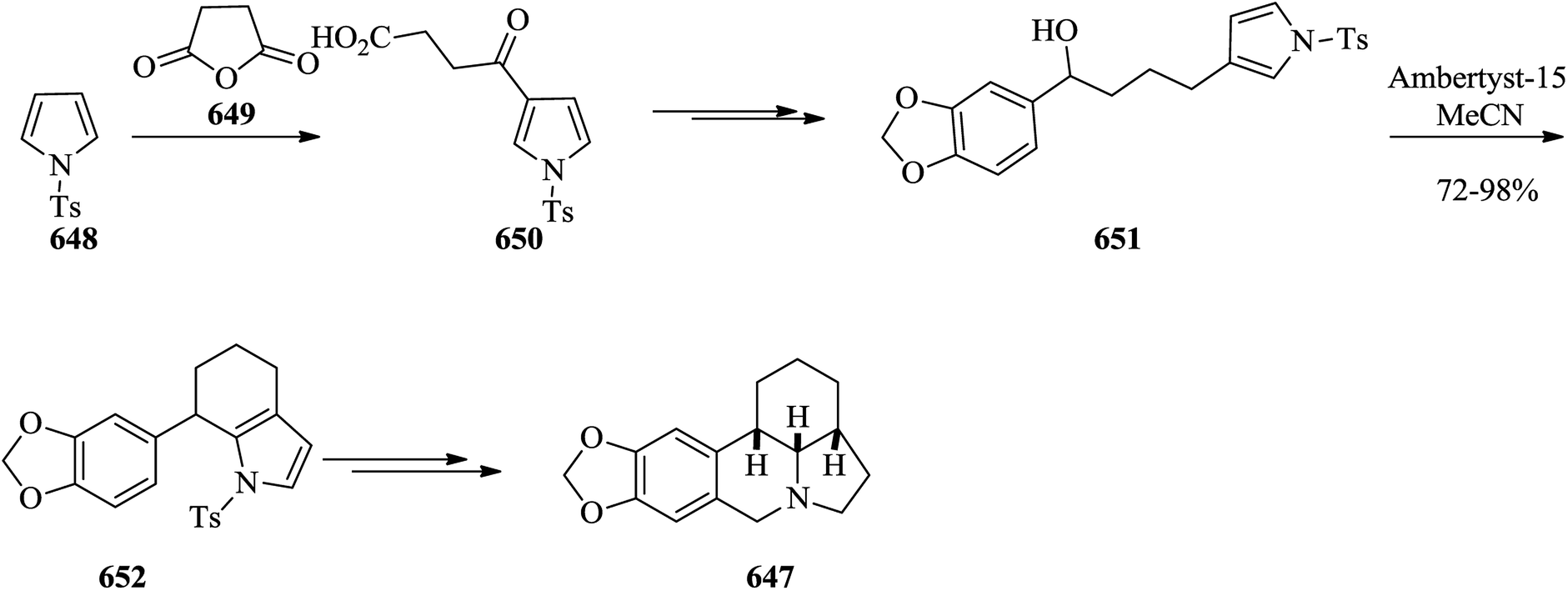 | ||
| Scheme 141 Total synthesis of γ-lycorane 647. | ||
Calothrixin A and B are unique indolo[3,2-j]phenanthridine alkaloids that were isolated from Calothrix cyanobacteria in 1999.619 These natural products show significant biological activities, for example antimalarial and anticancer, and they inhibit bacterial RNA polymerase.620 The total synthesis of oxacalothrixin B, an isostere of the biologically significant carbazoloquinone alkaloid calothrixin B, was accomplished from 2-acetyl-3-methylbenzofuran. In this route, initially FC acylation reaction of 2-methylbenzofuran 654621 using CH3COCl with tin(IV) chloride in dry dichloromethane at 0 °C gave 3-acetyl-2-methylbenzo[b]furan 655 in moderate yield. Next, compound 655 afforded oxacalothrixin B 653a–f in moderate yields after several steps (Scheme 142).622
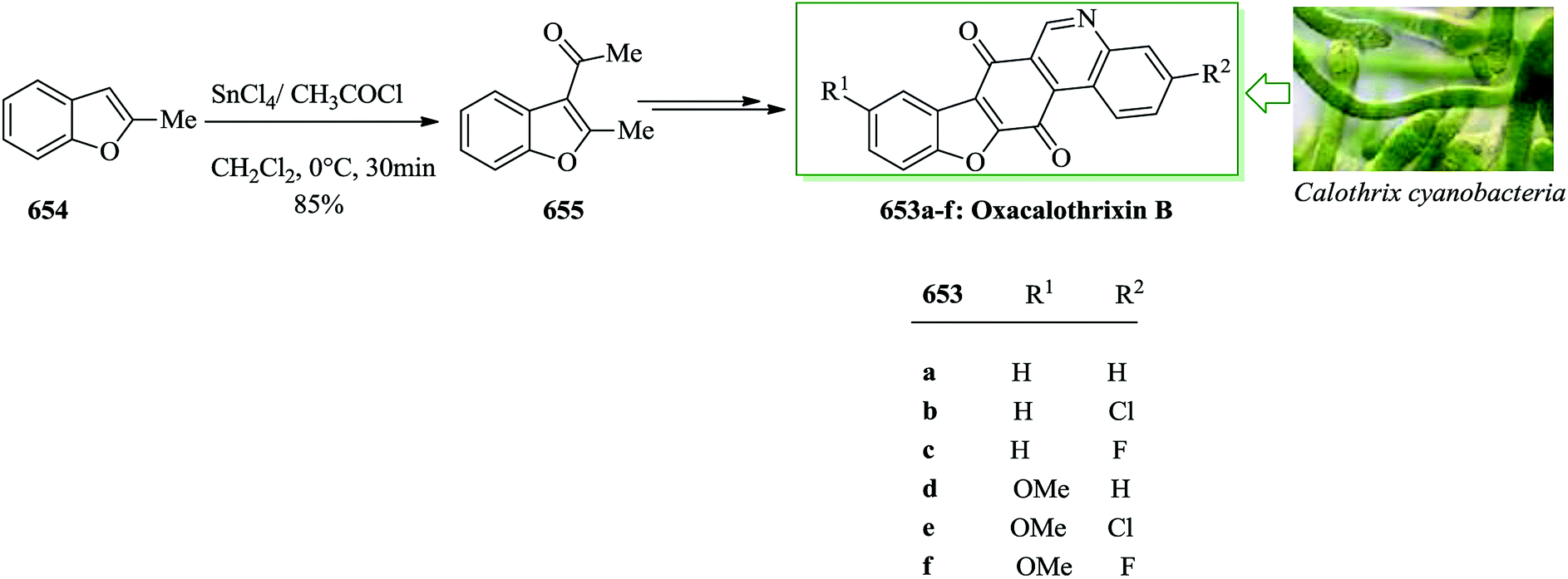 | ||
| Scheme 142 Total synthesis of oxacalothrixin B 653a–f. | ||
Coumarins constitute a significant group of heterocyclic compounds that are known as benzo-α-pyrones, wherein a pyran ring is fused with a benzene ring. Two unique geranylated coumarins, mammeasins C and D (656 and 657), were isolated together with 20 other coumarins.623 Compounds 656 and 657 are rare coumarins, wherein a dioxaphenalene type scaffold is generated by attaching a pyran ring to the coumarin unit. Both these compounds exhibit active aromatase inhibitory activity comparable to that of aminoglutethimide, which was applied as a reference standard.623 The first total synthesis of the geranylated pyranocoumarins, mameasins C 656 and D 657, aromatase inhibitors isolated from the flowers of Mammea siamensis, was achieved in five steps, starting from phloroglucinol 496. Based on this method, total synthesis of 656 and 657 was started from 496, in which 496 was exposed to FC acylation with crotonyl chloride. This reaction gave the corresponding chromanone 5,7-dihydroxy-2-methylchroman-4-one 659, albeit in a low yield of 31%. Next compound 659 provided pyranocoumarin 660 in 69% yield. FC acylation reaction of 660 with butyryl chloride or isobutyryl chloride was applied for the acylation of phenolic compounds,506 and two regioisomers (663/664) in 4:1 and 5:1 ratio were provided. Finally, compounds 663a and 663b afforded the corresponding 656 and 657 in 23% and 15% yield after 2 steps, respectively. In addition, examining of the intermediates obtained in the synthetic route to 656 and 657 revealed that de-geranylated pyranocoumarins 663 and 664 exhibit superior aromatase inhibitory activity as compared to the natural products 656 and 657 (Scheme 143).624
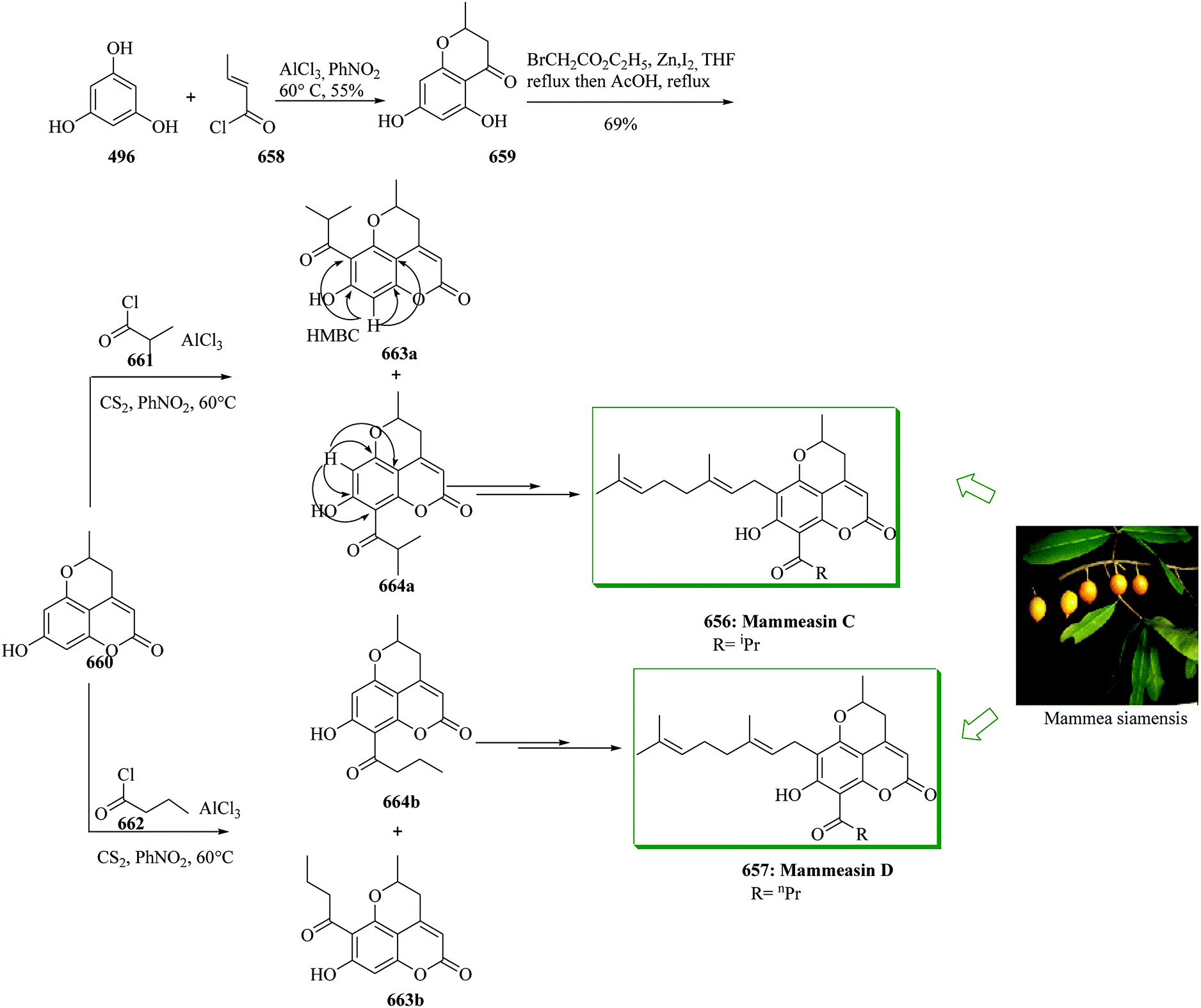 | ||
| Scheme 143 Total synthesis of mammeasins C 656 and 657. | ||
Isoflavone C-glycosides, in which the sugar scaffold is attached by a carbon–carbon bond directly to the isoflavone ring, are not simply hydrolyzed in acidic gastric juices, unlike O-glycosides and aglycone. These glycosides exhibit several biological properties, including radioprotective,625 anti-myocardial ischemic,626 mitogenic and colony-stimulating,627 and antidiabetic628 activities. Among these compounds, puerarin, which is known mainly in Pueraria radix, exhibits a strong anti-myocardial ischemic influence.629 Two isoflavone C-glycosides (6-tert-butyl puerarin and 6-tert-butyl-4′-methoxypuerarin) were obtained using FC acetylation reaction and Vilsmeier–Haack cyclization in five steps with overall yields of 14.6% and 14.2%, respectively. Initially, C-glucosyl acetophenone 666 was converted into 2 C-β-D-glucopyranoside 667 after several steps. The key intermediate deoxybenzoin 669 was obtained from FC acetylation reaction of 667 using anhydrous AlCl3 with a 63.6% yield.630,631 Lastly, compound 669 gave 6-tert-butyl-4′-methoxypuerarin 665a and 6-tert-butyl puerarin 665b with 95.5% and 98.6% yields,632,633 respectively (Scheme 144).634
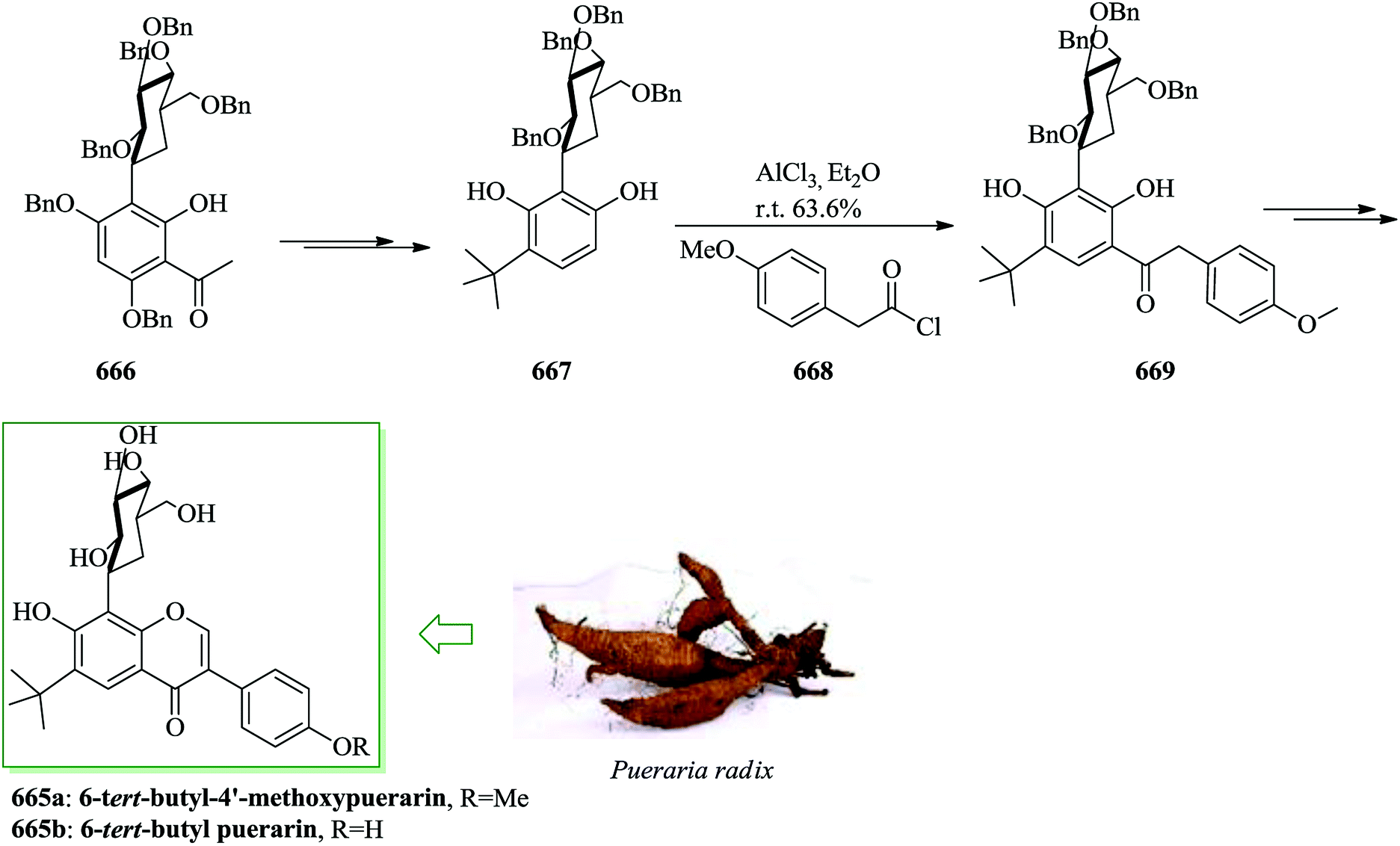 | ||
| Scheme 144 Total synthesis of 6-tert-butyl-4′-methoxypuerarin 665a and 6-tert-butyl puerarin 665b. | ||
2.5. Miscellaneous
With the increase in the advanced age population, neurodegenerative disorders, for example Alzheimer's and Parkinson's disease, are emerging as a major social issue, therefore resulting in great demand for new therapeutic drugs to prevent these diseases.635 Significant efforts at searching for small-molecule-based natural products with neurotrophic activities resulted in the isolation of (−)-talaumidin 670 from Brazilian Aristolochia arcuata Masters.636 Talaumidin and its analogues show important neurite outgrowth-promoting and neuroprotective activities in the primary cultured rat corticaland additionally in the hippocampal neurons.636 The initial asymmetric total synthesis of neurotrophic (−)-talaumidin 670 was developed in 16 steps from 4-benzyloxy-3-methoxybenzaldehyde in ca. 10.7% overall yield. In this method, the key steps are Evans asymmetric anti-aldol reaction, hydroboration/oxidation and epimerization and FC arylation reaction. Total synthesis of (−)-talaumidin 670 was started from the reaction between 4-benzyloxy-3-meth-oxybenzaldehyde 671 and (S)-4-benzyl-3-propionyl-2-oxazolidinone 672 to provide five-membered acetal 673 via several steps. Then, FC-type arylation reaction of 673 with 1,2-methylenedioxybenzene 421 using tin(IV) chloride in dichloromethane afforded only the corresponding (5S)-675 in 89% yield along with 2% of talaumidin 670. Finally, debenzylation of 675 with Pd(OH)2 in ethanol provided (−)-(2S,3S,4S,5S)-670 in 77% yield (Scheme 145).637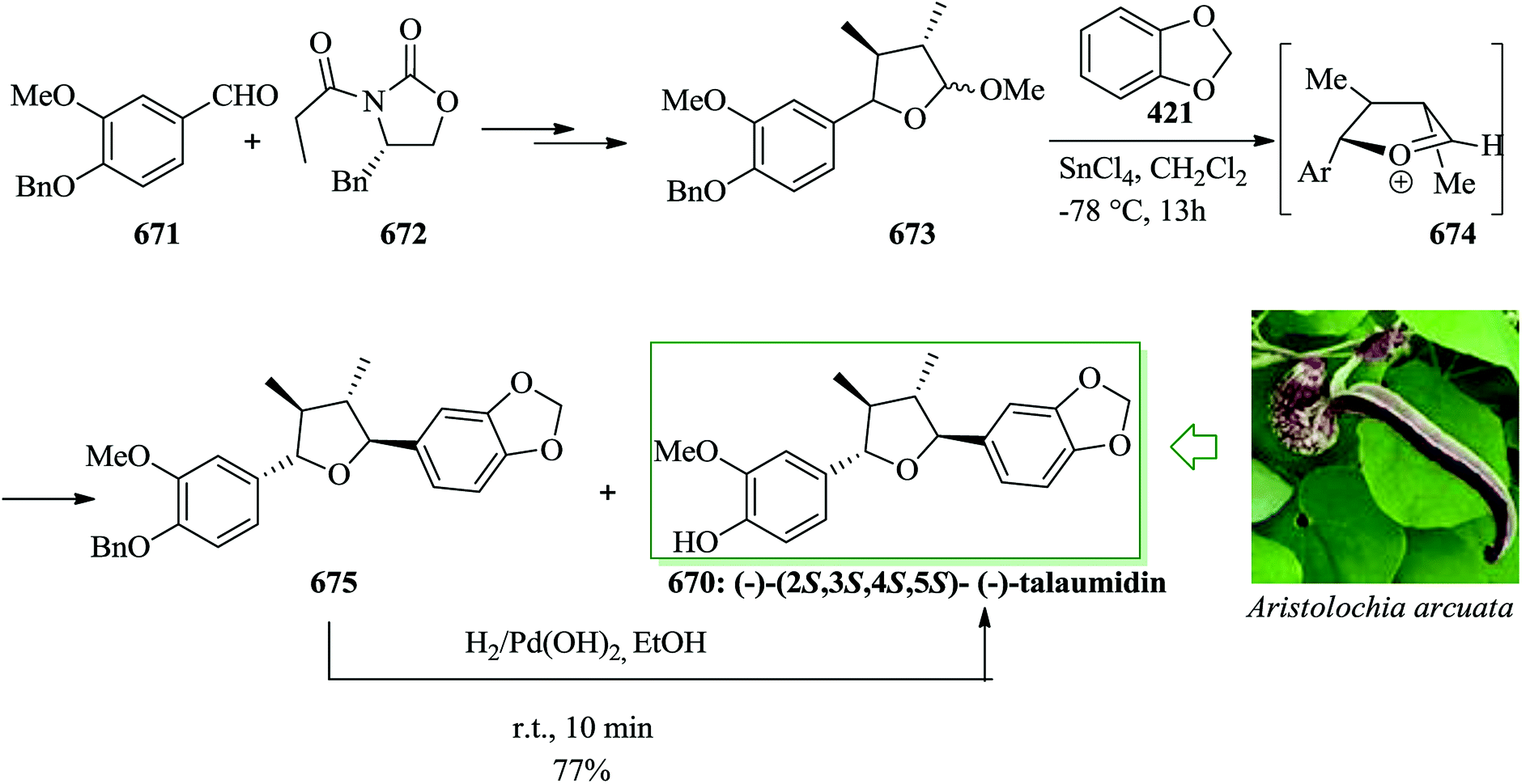 | ||
| Scheme 145 Total synthesis of (−)-talaumidin 670. | ||
An oxidative FC reaction containing various aromatic compounds catalyzed by a hypervalent iodine reagent was accomplished by employing polyfunctionalized phenols. This reaction provided rapid access to extremely functionalized compounds, including a dienone, a quaternary carbon center, and an aromatic ring. The product's framework is found in various naturally occurring compounds. Based on this method, total synthesis of compounds belonging to the Amaryllidaceae alkaloids group, for example O-methyljoubertiamine, mesembrine, and its natural derivative the dihydro-O-methylsceletenone, was accomplished in eight/nine steps. The synthetic pathway to these molecules features a significant transformation on the basis of a Fukuyama and Michael-retro-Michael tandem method. The synthesis of 679 was started from 2,4,6-trimethylphenol 676, which afforded compound 677 via several steps. Subsequently, reaction of compound 678a or 678b with TBAF leads in satisfactory yield to the bicyclic compound 679 with complete regioselectivity regarding the alkene, including the silyl substituent, the latter being subsequently removed under the reaction conditions. This oxidative FC reaction permits quick access to substituted synthons, including a quaternary carbon center, a dienone, and an aromatic scaffold, and various functionalities can be present on the side chain. Such an intermediate could provide some opportunity for total synthesis of different naturally occurring compounds. The most well-known belong to the group of Amaryllidaceae alkaloids (Scheme 146).638
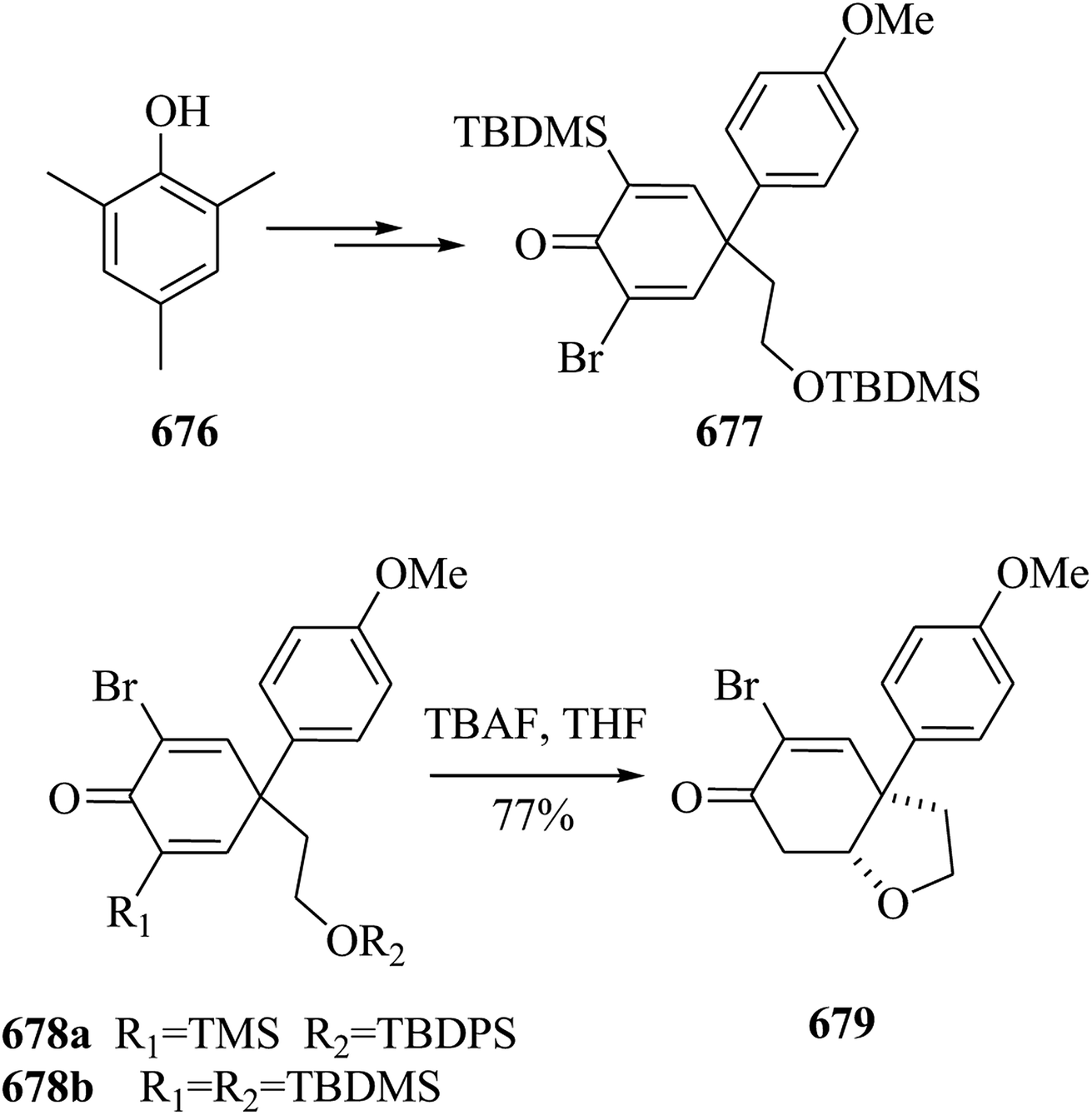 | ||
| Scheme 146 The formation of the bicyclic compound 679. | ||
Based on this method, two alkaloids of this group were synthesized. Mesembrine 680a and O-methyljoubertiamine 681 are alkaloids present in Sceletium tortuosum. They were demonstrated to be very active serotonin reuptake inhibitors, potent at very low doses. The 4,5-dihydro-4′-O-methylsceletenone 680b is a simpler natural derivative of mesembrine extracted from Aptenia cordifolia. Starting from 2-(4-hydroxyphenyl)ethanol 682, total synthesis of mesembrine and 4,5-dihydro-4′-O-methylsceletenone was achieved via several steps in 86% yield. Furthermore, the transformation of 4,5-dihydro-4′-O-methylsceletenone, 680b, into O-methyljoubertiamine 681 was accomplished by reaction with iodomethane (Scheme 147).638
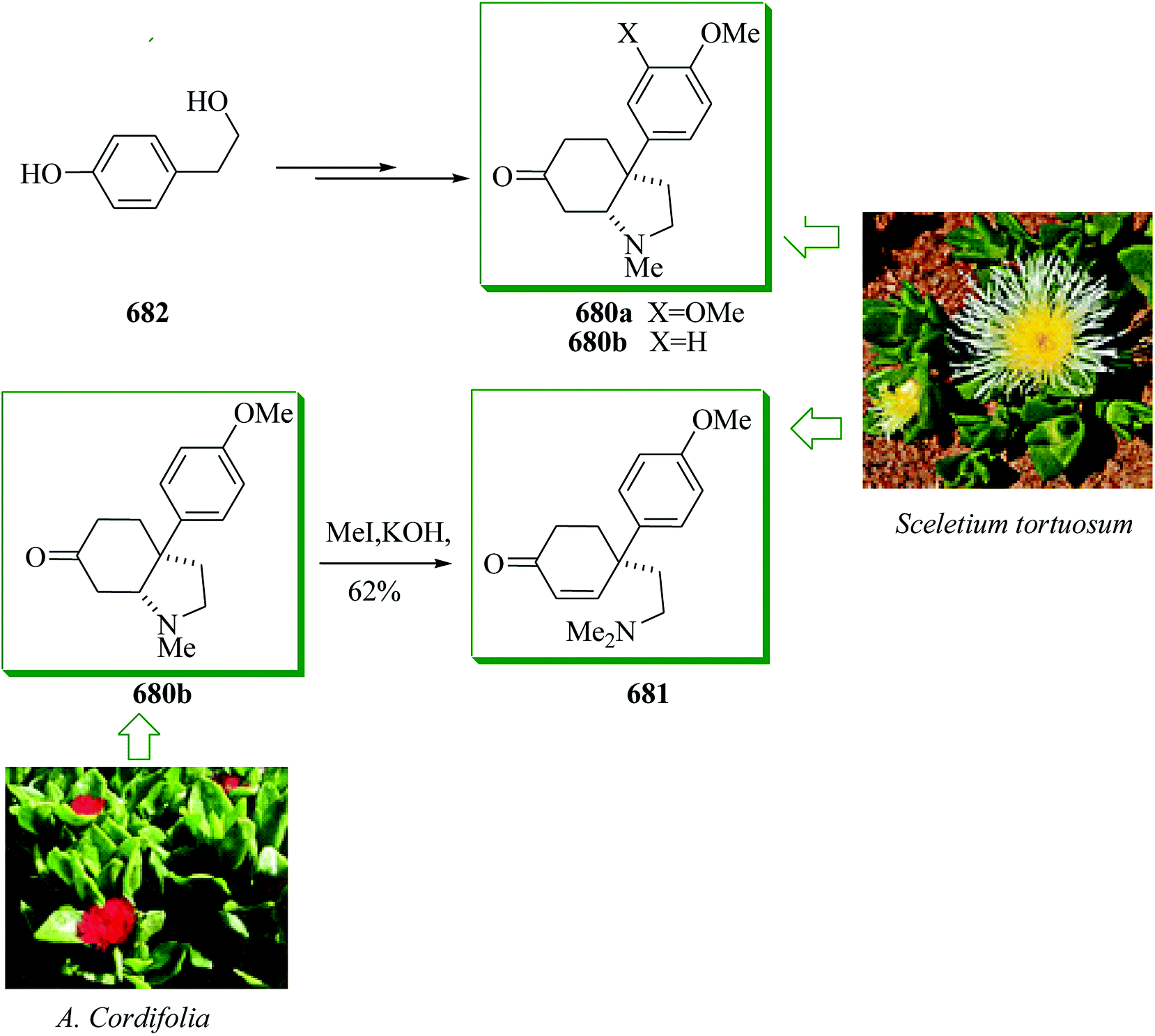 | ||
| Scheme 147 The formation of 4,5-dihydro-4′-O-methylsceletenone 680b into O-methyljoubertiamine 681. | ||
(2)-Talaumidin 683 and (2)-galbelgin 684, naturally occurring lignans, were isolated from Aristolochia arcuata636 and Piper futokadsura.639 They are 2,5-diaryl-3,4-dimethyl-tetrahydrofuran lignans.640 Among them, (2)-talaumidin 683 demonstrated important neurotrophic activity in the primary culture of rat cortical neurons and could serve as a promising lead compound for the treatment of neurodegenerative disorders, for example Alzheimer's and Parkinson's disease,641 whereas (2)-galbelgin 684 exhibits anti-HBV activity.642 (2)-Talaumidin 683 and (2)-galbelgin 684 were obtained starting from 4-pentenoic acid with an overall yield of about 17.8 and 16.9%, respectively. The key steps contain an Evans asymmetrical anti-aldol reaction, TBS protection, hydroboration, oxidation, and FC arylation. Firstly, 4-pentenoic was converted into 686 via several steps. Next, the methyl acetal 686 was transformed into 2,5-diaryl-3,4-dimethyltetrahydrofuran via FC-type arylation reaction using tin(IV) chloride in dichloromethane.637,643 The FC-type arylation reaction was performed with the epimerization at the C2 position of 686 to give 2,3-trans-3,4-trans-4,5-trans-tetrahydrofuran 687a and 687b as a single diastereomer. Lastly, removal of the Bn protecting group in 687a afforded (2)-talaumidin 683 (90%). Furthermore, the total synthesis of (2)-galbelgin 684 was achieved in two steps from the intermediate 687b (Scheme 148).644
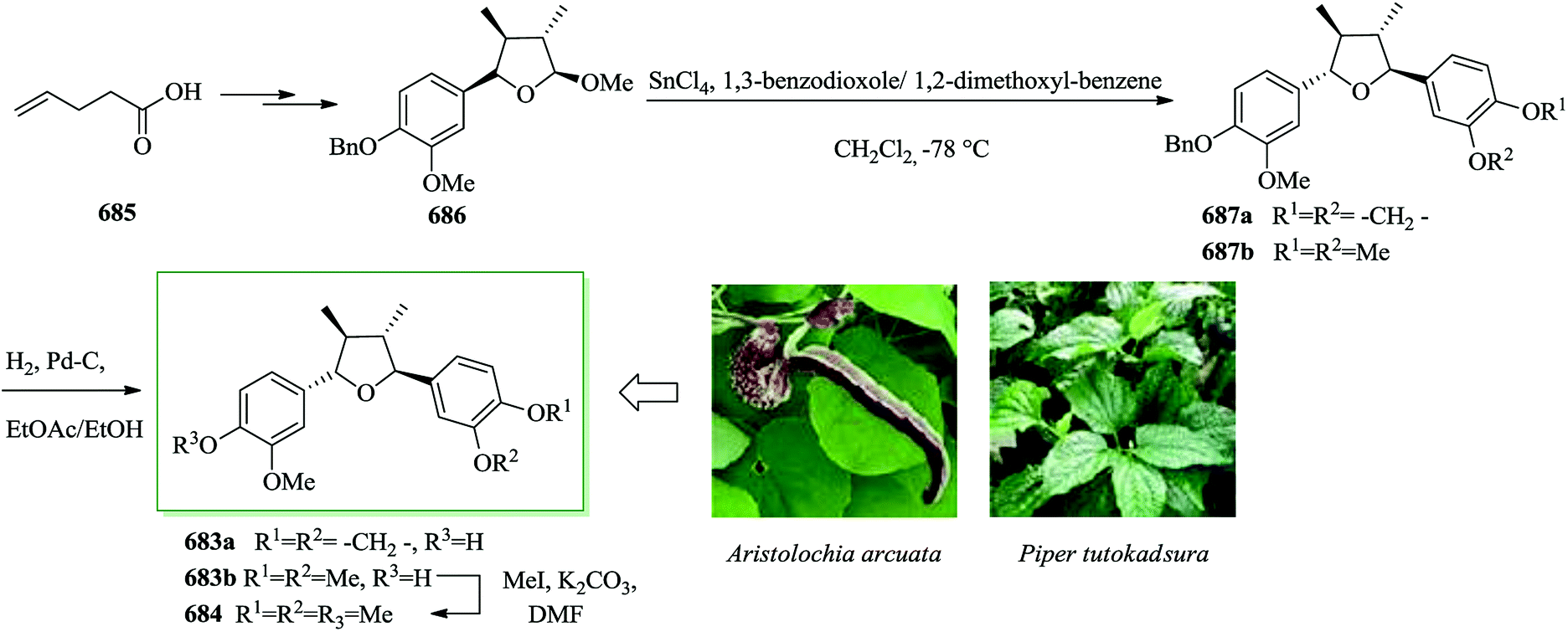 | ||
| Scheme 148 Total synthesis of (2)-talaumidin 683 and (2)-galbelgin 684. | ||
Extracts from the plants Pycnanthus angolensis and Holostylis reniformis were applied to treat malaria throughout Africa and Brazil, respectively.645 Extracted lignans from these and other plants,250 such as (−)-8′-epi-aristoligone 689, have demonstrated promising antiplasmodial activity against a chloroquine-resistant strain of Plasmodium falciparum.250 The development of a unique one-pot oxidative [3,3] rearrangement/FC arylation reaction permitted the fast and stereocontrolled synthesis of several tetralone- and naphthyl-type lignan natural products with antimalarial activity. For the synthesis of 689 and 688, initially hydrazine 691 serves as a linchpin for the reaction of aryl aldehyde 690 with arene 693 via initial construction of hydrazone 692, which is followed by a hypervalent-iodide oxidative [3,3] rearrangement/FC arylation in one-pot fashion to provide benzhydryl derivative 694. Next, via several steps, the one-pot oxidative [3,3] rearrangement/FC arylation of 695 with 1,2-dimethoxybenzene 36 progressed with complete chirality transfer to provide benzhydryl 696 in 77% yield. Subsequently, compound 696 was transformed into (−)-8′-epi-aristoligone 689 in 28% yield and (−)-cyclogalgravin 688 in 73% yield via several steps (Scheme 149).646
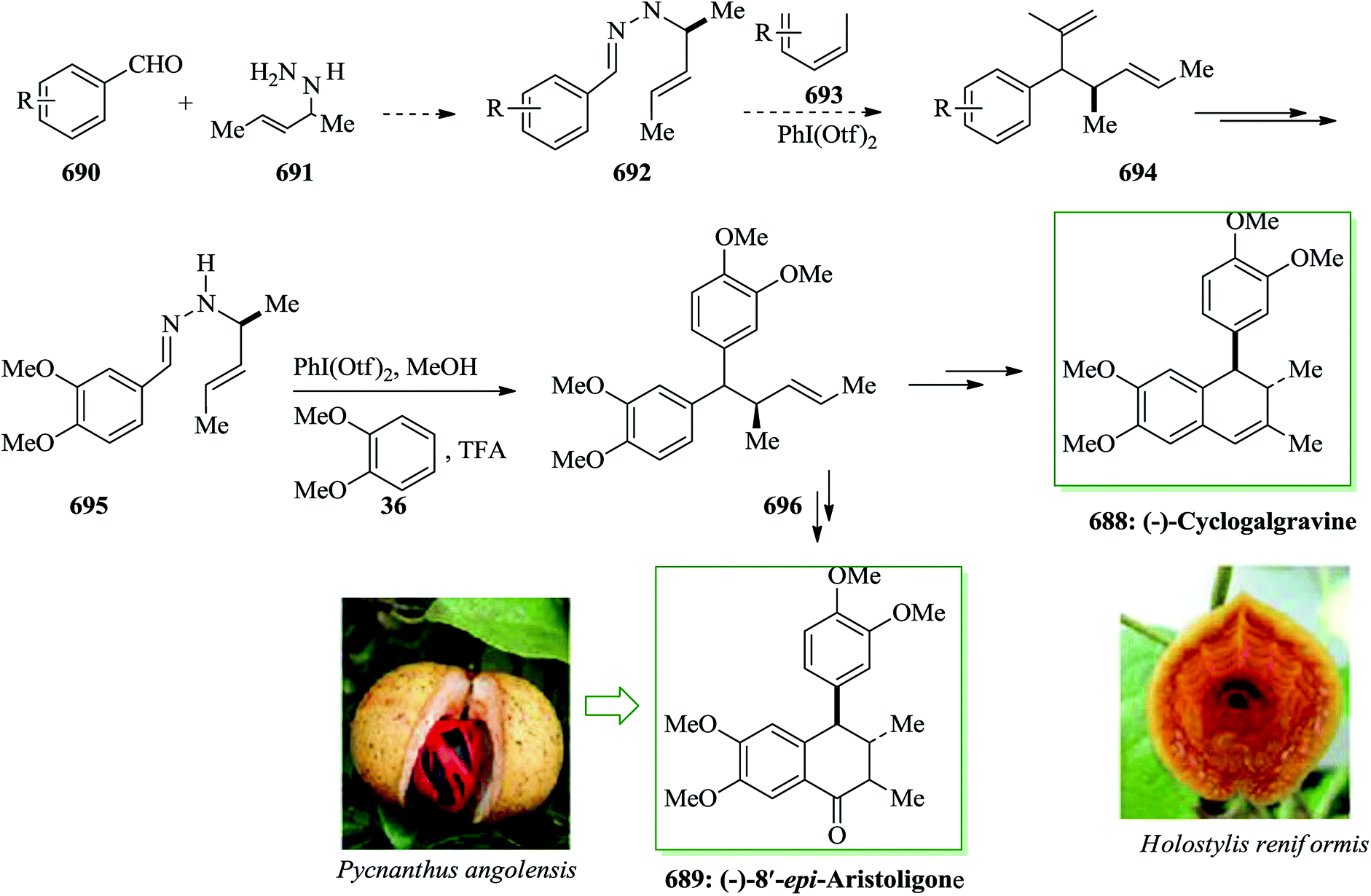 | ||
| Scheme 149 Total synthesis of (−)-cyclogalgravin 688 and (−)-8′-epi-aristoligone 689. | ||
Based on this method, the total synthesis of 697 and 698 was achieved as follows. Construction of hydrazone 699 from piperonal followed via oxidative [3,3] rearrangement and FC arylation reaction with 1,2-dimethoxybenzene 36 resulted in the formation of diastereomeric benzhydryl 700 (80% yield, 5:1 d.r.). Next, the final two natural products, (−)-8′-epi-aristotetralone 698 and (−)-pycnanthulignene B 697, were synthesized in 43% and 70% yield, respectively, from compound 700 via several steps (Scheme 150).646
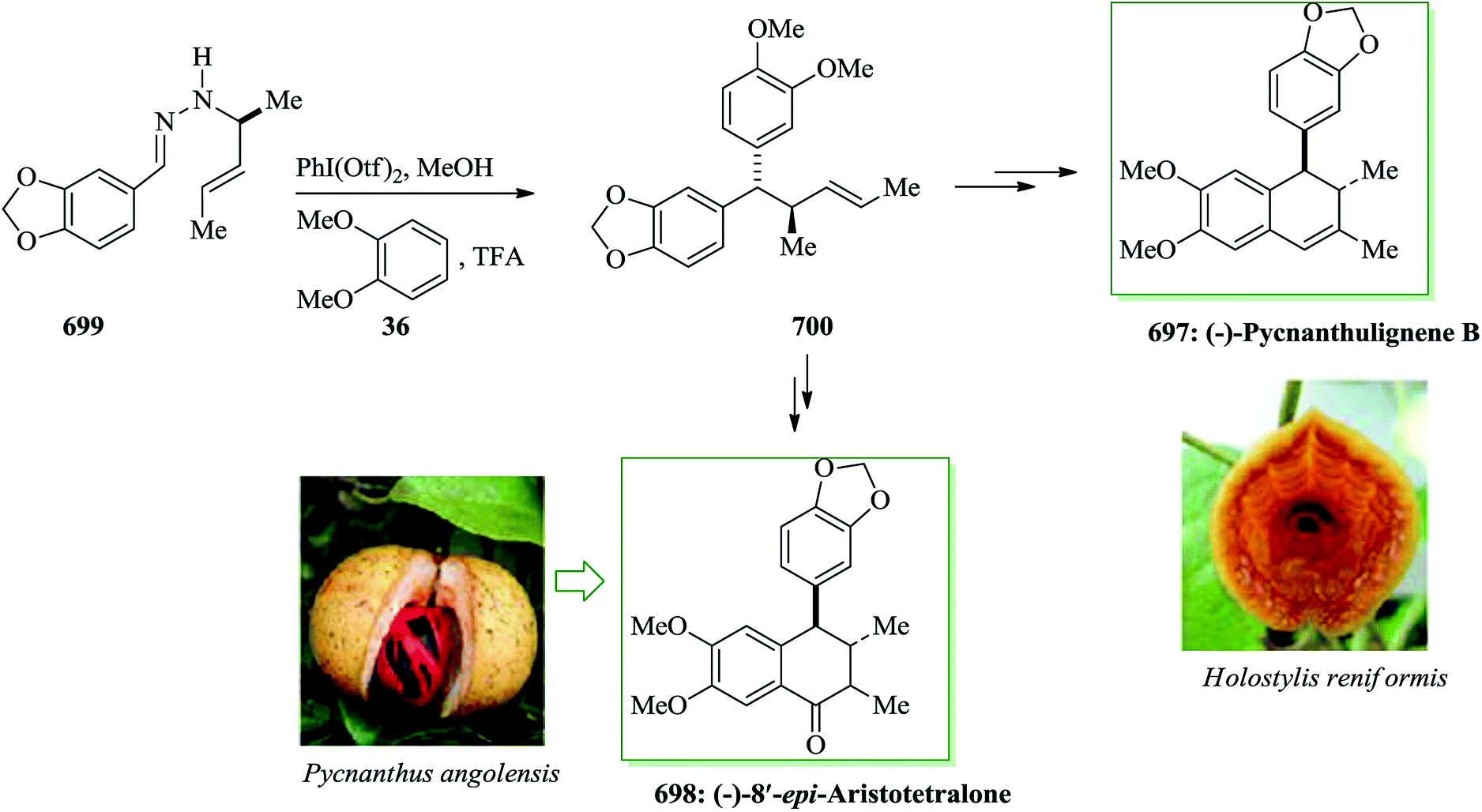 | ||
| Scheme 150 Total synthesis of (−)-pycnanthulignene B 697 and (−)-8′-epi-aristotetralone 698. | ||
Santalin Y occurs in “red sandalwood”, the hardwood of Pterocarpus santalinus. The yellow pigment santalin Y was isolated by Nohara et al. in 1995 as a minor component.647 A biomimetic total synthesis of santalin Y, a structurally complex but racemic natural product, was developed in 2015 by Strych et al. Santalin Y was obtained via seven steps (longest linear sequence) and in 8% overall yield. The key steps were a (3 + 2) cycloaddition reaction and an intramolecular FC reaction. The total synthesis of santalin Y was started from isoflavylium ion 702, which was converted into a tetrahydrofuran intermediate 703 via several steps. Next, a subsequent intramolecular FC cyclization provided santalin Y (Scheme 151).648
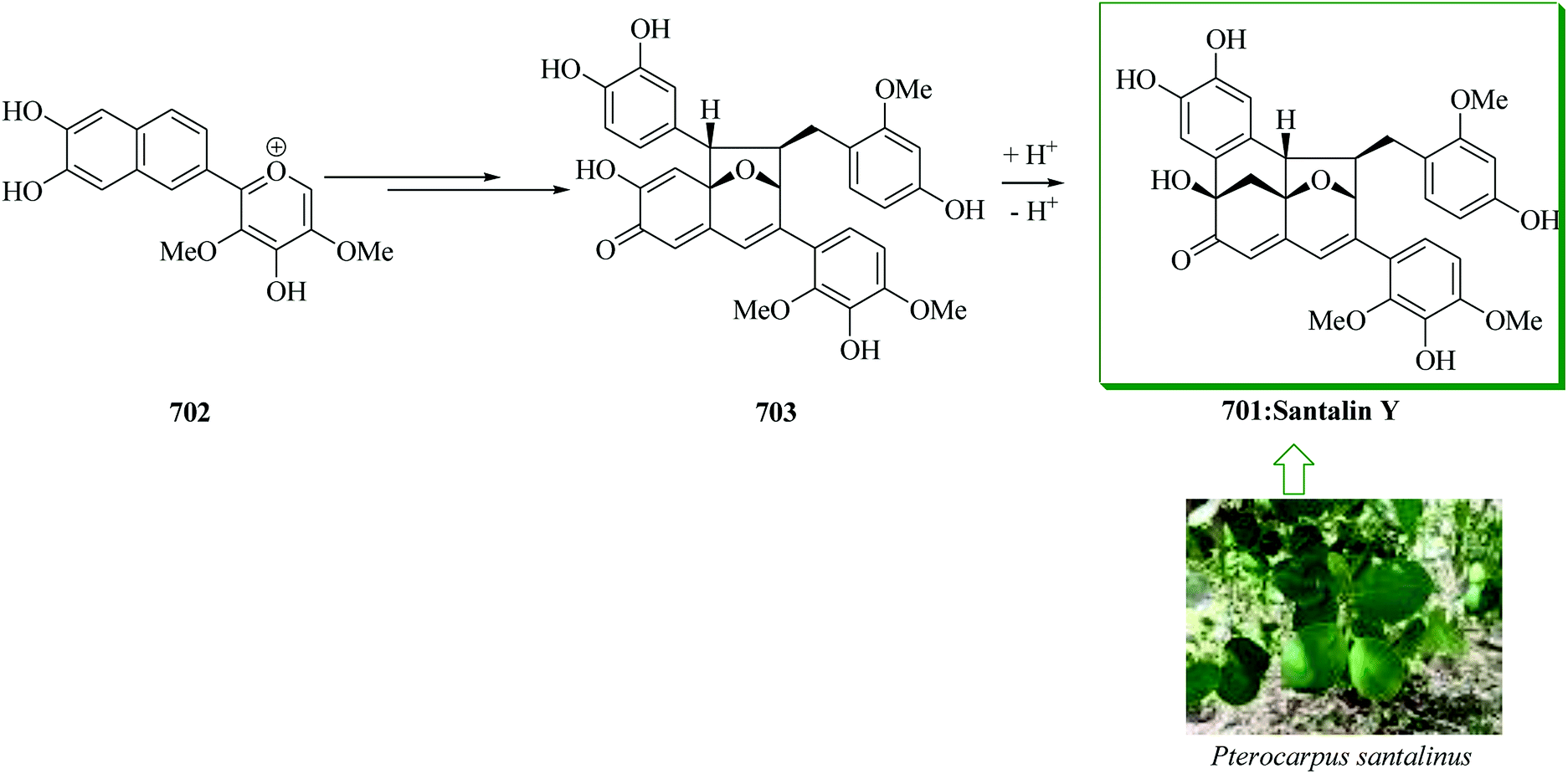 | ||
| Scheme 151 Total synthesis of santalin Y 701. | ||
Epipolythiodiketopiperazine alkaloids represent a structurally complex and biologically potent group of secondary fungal metabolites.162,268,649 Several members of this group of natural products share a cyclo-tryptophan unit and an eponymous epipolythiodiketopiperazine (ETP) substructure. Among them, (+)-luteoalbusin A 704 and (+)-luteoalbusin B 705 were isolated from the marine fungi Acrostalagmus luteoalbus SCSIO F457. The first total synthesis of (+)-luteoalbusins A and B was developed in 2015 by Movassaghi et al.650 The total synthesis of alkaloids (+)-704 and (+)-705 was started with the Ag-catalyzed FC arylation of diketopiperazine (+)-707. Reaction of a solution of C3-bromo diketopiperazine (+)-707 in CH2Cl2 with indoles 706 and silver(I) hexafluoroantimonate using 2,6-di-tert-butyl-4-methylpyridine (DTBMP) gave the corresponding C3-indolylhexacycle (+)-708 in 77% yield. Lastly, compound 708 produced (+)-luteoalbusins A 704 and B 705 via several steps (Scheme 152).650
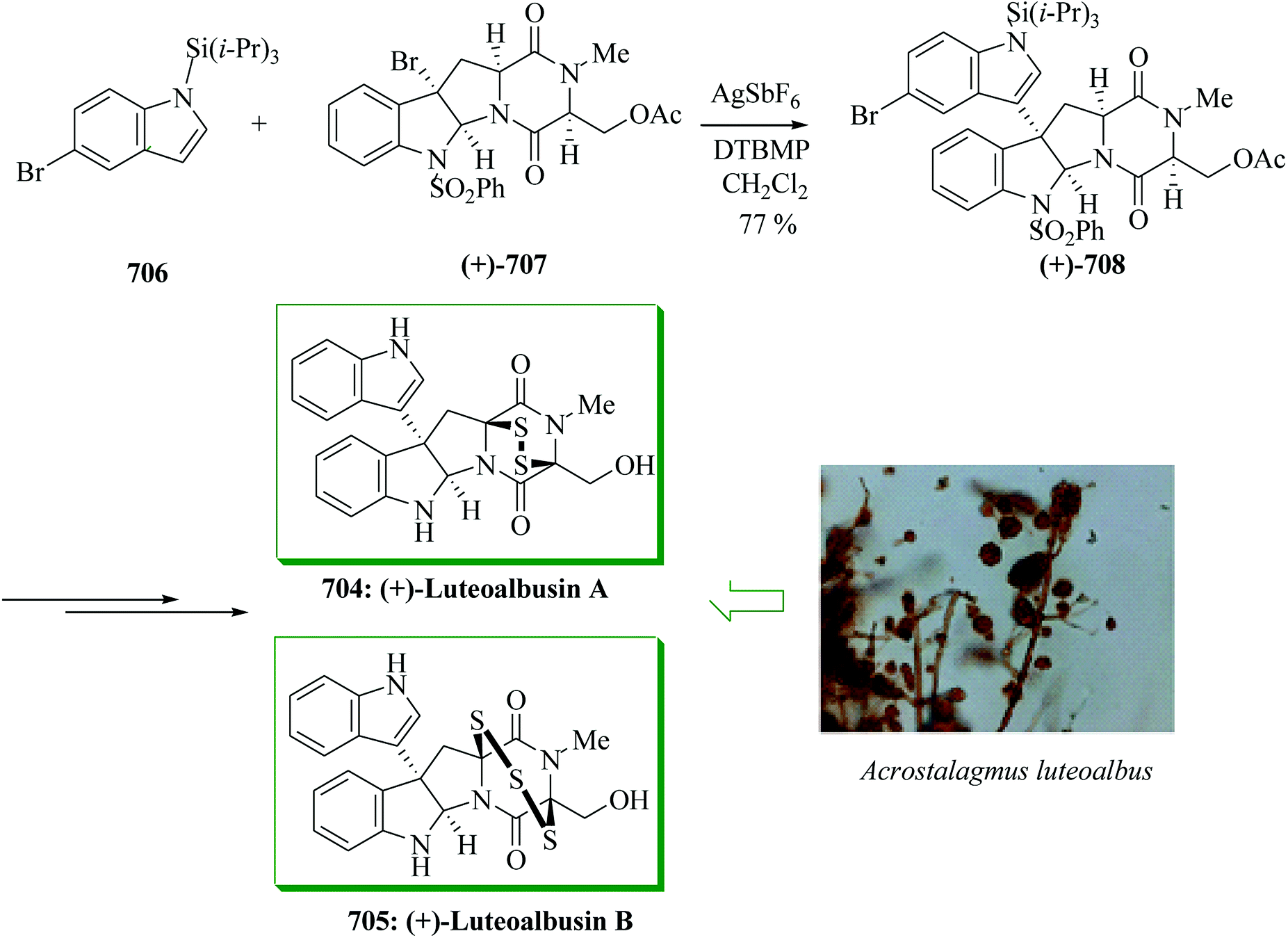 | ||
| Scheme 152 Total synthesis of (+)-luteoalbusin A 704 and (+)-luteoalbusin B 705. | ||
In 2011, Ding et al. identified sespenine 709, which is a structurally uncommon polycyclic molecule isolated from Streptomyces sp. HKI0595.651 Li et al. in 2016 reported a ten-step (the longest linear sequence) synthesis of this molecule from market-accessible precursors.652 Sharpless enantioselective epoxidation, Stille–Miyata coupling reaction and Prins cyclization/FC/retro FC were considered as the key steps. Total synthesis of sespenine 709 was started from methyl indole-2-carboxylate 710. Initially, the feasibility of FC-type allylation at the C3 position of 710 was investigated.653,654 In this pathway, stannane 711 was synthesized from 710 through a two-step sequence described by Routier et al.655 Iodination based on basic conditions gave the desired C3 iodide, which was transformed to 711 via a methodology-mediated stannylation [Pd(PPh3)4, Me3SnSnMe3]. Afterwards, sespenine 709 was obtained after several steps (Scheme 153).652
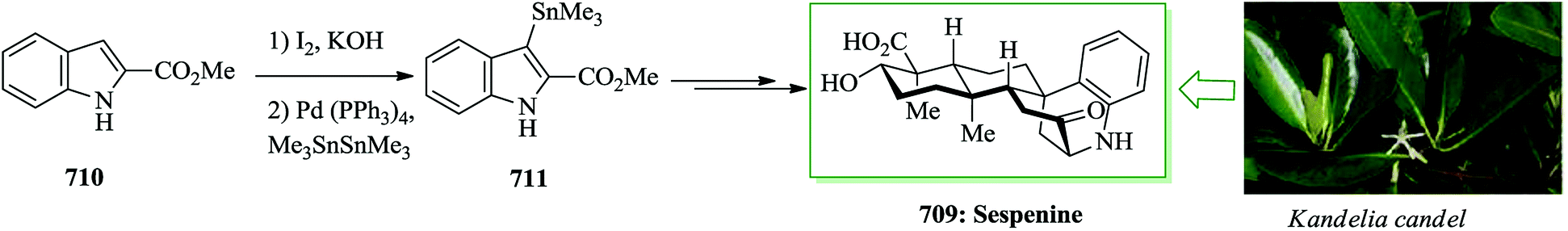 | ||
| Scheme 153 Total synthesis of sespenine 709. | ||
3. Conclusion
In conclusion, in this review, we tried to underscore the significance and importance of the FC reaction as an old reaction with a new and interesting perspective owing to its applications in the important and new field of total synthesis of naturally occurring compounds. We showed how this old reaction can play an important and key role in the total synthesis of natural products proven to exhibit diverse biological activities. Indeed, nowadays, the FC reaction is considered as one of the most significant basic reaction classifications in the total synthesis of the most important class of natural products. The important compounds from biological points of views are totally synthesized via a pathway including at least one step involving this old but useful FC reaction as a key step. As a result, the FC reaction as an old but extremely useful reaction is frequently applied in the total synthesis of natural products, particularly those with various biological activities.Conflicts of interest
There are no conflicts to declare.References
- C. Friedel and J. M. Crafts, Compt. Rend., 1877, 84, 1392–1395 Search PubMed.
- C. C. Price, Org. React., 1962, 3, 1 Search PubMed.
- J. Groves, Chem. Soc. Rev., 1972, 1, 73–97 RSC.
- S. C. Eyley and F. Ince, Synlett, 1991, 4, 721–723 Search PubMed.
- H. Heaney, Compr. Org. Synth., 1991, 2, 733–752 Search PubMed.
- M. Rueping and B. J. Nachtsheim, Beilstein J. Org. Chem., 2010, 6, 1–31 Search PubMed.
- M. B. Smith and J. March, March's advanced organic chemistry: reactions, mechanisms, and structure, John Wiley & Sons, 2007 Search PubMed.
- J. Gershenzon and N. Dudareva, Nat. Chem. Biol., 2007, 3, 408–414 CrossRef CAS PubMed.
- G. Samuelson, Drugs of natural origin: a textbook of pharmacognosy, Taylor & Francis Group, 1999 Search PubMed.
- J. R. Hanson, Natural products: the secondary metabolites, Royal Society of Chemistry, 2003 Search PubMed.
- D. Williams and T. Lemke, Foye's Principles of Medicinal Chemistry, Lippincott Williams Wilkins, Philadelphia, 5th edn, 2002, vol. 1, p. 25, ISBN 0-683-30737 Search PubMed.
- R. A. Maplestone, M. J. Stone and D. H. Williams, Gene, 1992, 115, 151–157 CrossRef CAS PubMed.
- P. Hunter, EMBO Rep., 2008, 9, 838–840 CrossRef CAS PubMed.
- J. W.-H. Li and J. C. Vederas, Science, 2009, 325, 161–165 CrossRef PubMed.
- N. Calloway, Chem. Rev., 1935, 17, 327–392 CrossRef CAS.
- G. A. Olah and G. S. Prakash, Across Conventional Lines: Selected Papers of George A. Olah, World scientific, 2003 Search PubMed.
- R. M. Roberts and A. A. Khalaf, Friedel–Crafts alkylation chemistry: a century of discovery, Marcel Dekker Inc, 1984 Search PubMed.
- T. B. Poulsen and K. A. Jørgensen, Chem. Rev., 2008, 108, 2903–2915 CrossRef CAS PubMed.
- N. A. Paras and D. W. MacMillan, J. Am. Chem. Soc., 2001, 123, 4370–4371 CrossRef CAS PubMed.
- M. M. Heravi and E. Hashemi, Tetrahedron, 2012, 45, 9145–9178 CrossRef.
- M. M. Heravi, E. Hashemi and F. Azimian, Tetrahedron, 2014, 1, 7–21 CrossRef.
- M. M. Heravi and V. Zadsirjan, Tetrahedron: Asymmetry, 2013, 24, 1149–1188 CrossRef CAS.
- M. M. Heravi, E. Hashemi and N. Nazari, Mol. Diversity, 2014, 18, 441–472 CrossRef CAS PubMed.
- M. M. Heravi and V. F. Vavsari, RSC Adv., 2015, 5, 50890–50912 RSC.
- M. M. Heravi, E. Hashemi and N. Ghobadi, Curr. Org. Chem., 2013, 17, 2192–2224 CrossRef CAS.
- M. M. Heravi, T. B. Lashaki and N. Poorahmad, Tetrahedron: Asymmetry, 2015, 26, 405–495 CrossRef CAS.
- M. M. Heravi, V. Zadsirjan and Z. Bozorgpour Savadjani, Curr. Org. Chem., 2014, 18, 2857–2891 CrossRef CAS.
- M. M. Heravi and N. Nazari, Curr. Org. Chem., 2015, 19, 2358–2408 CrossRef CAS.
- M. M. Heravi, V. Zadsirjan and B. Farajpour, RSC Adv., 2016, 6, 30498–30551 RSC.
- M. M. Heravi, T. Ahmadi, M. Ghavidel, B. Heidari and H. Hamidi, RSC Adv., 2015, 5, 101999–102075 RSC.
- M. M. Heravi, V. Zadsirjan, H. Hamidi and P. H. T. Amiri, RSC Adv., 2017, 7, 24470–24521 RSC.
- M. M. Heravi, V. Zadsirjan, M. Esfandyari and T. B. Lashaki, Tetrahedron: Asymmetry, 2017, 28, 987–1043 CrossRef CAS.
- M. M. Heravi, T. B. Lashaki, B. Fattahi and V. Zadsirjan, RSC Adv., 2018, 8, 6634–6659 RSC.
- M. M. Heravi, S. Rohani, V. Zadsirjan and N. Zahedi, RSC Adv., 2017, 7, 52852–52887 RSC.
- A. T. K. Koshvandi, M. M. Heravi and T. Momeni, Appl. Organomet. Chem., 2018, 32 Search PubMed.
- R. C. Gadwood, R. M. Lett and J. E. Wissinger, J. Am. Chem. Soc., 1986, 108, 6343–6350 CrossRef CAS.
- F. J. Schmitz, K. H. Hollenbrek and D. J. Vanderah, Tetrahedron, 1978, 34, 2719–2722 CrossRef CAS.
- L. A. Paquette and W. H. Ham, J. Am. Chem. Soc., 1987, 109, 3025–3036 CrossRef CAS.
- F. Brauns, J. Org. Chem., 1945, 10, 216–218 CrossRef CAS.
- I. A. Pearl, J. Org. Chem., 1945, 10, 219–221 CrossRef CAS.
- P. Boissin, R. Dhal and E. Brown, Tetrahedron Lett., 1989, 30, 4371–4374 CrossRef CAS.
- D. Stoermer and C. H. Heathcock, J. Org. Chem., 1993, 58, 564–568 CrossRef CAS.
- F. Kögl, G. Van Wessem and O. Elsbach, Recl. Trav. Chim. Pays-Bas, 1945, 64, 23–29 CrossRef.
- D. L. Boger, J. Hong, M. Hikota and M. Ishida, J. Am. Chem. Soc., 1999, 121, 2471–2477 CrossRef CAS.
- T. J. King, I. W. Farrell, T. G. Halsall and V. Thaller, J. Chem. Soc., Chem. Commun., 1977, 727–728 RSC.
- T. Anke, W. Watson, B. Giannetti and W. Steglich, J. Antibiot., 1981, 34, 1271–1277 CrossRef CAS PubMed.
- H. Hoffmann and J. Rabe, Angew. Chem., Int. Ed. Engl., 1985, 24, 94–110 CrossRef.
- J. Mihelcic and K. D. Moeller, J. Am. Chem. Soc., 2004, 126, 9106–9111 CrossRef CAS PubMed.
- Q. Meng and M. Hesse, Helv. Chim. Acta, 1990, 73, 455–459 CrossRef CAS.
- C.-L. Chin, D. D.-P. Tran, K.-S. Shia and H.-J. Liu, Synlett, Georg Thieme Verlag, 2005 Search PubMed.
- F. Schröder, S. Franke, W. Francke, H. Baumann, M. Kaib, J. M. Pasteels and D. Daloze, Tetrahedron, 1996, 52, 13539–13546 CrossRef.
- F. Schröder, V. Sinnwell, H. Baumann, M. Kaib and W. Francke, Angew. Chem., Int. Ed., 1997, 36, 77–80 CrossRef.
- M. Movassaghi and A. E. Ondrus, Org. Lett., 2005, 7, 4423–4426 CrossRef CAS PubMed.
- V. U. Ahmad, M. Zahid, M. S. Ali, Z. Ali, A. Jassbi, M. Abbas, J. Clardy, E. Lobkovsky, R. Tareen and M. Z. Iqbal, J. Org. Chem., 1999, 64, 8465–8467 CrossRef CAS.
- M. E. Maier and A. Bayer, Eur. J. Org. Chem., 2006, 2006, 4034–4043 CrossRef.
- T. J. Heckrodt and J. Mulzer, in Natural products synthesis II, Springer, 2005, pp. 1–41 Search PubMed.
- A. Majdalani and H.-G. Schmalz, Synlett, 1997, 1997, 1303–1305 CrossRef.
- P. J. Kocienski, A. Pontiroli and L. Qun, J. Chem. Soc., Perkin Trans. 1, 2001, 2356–2366 RSC.
- S. E. Lazerwith, T. W. Johnson and E. Corey, Org. Lett., 2000, 2, 2389–2392 CrossRef CAS PubMed.
- M. Harmata and X. Hong, Org. Lett., 2005, 7, 3581–3583 CrossRef CAS PubMed.
- R. G. Kerr, A. C. Kohl and T. A. Ferns, J. Ind. Microbiol. Biotechnol., 2006, 33, 532–538 CrossRef CAS PubMed.
- S. Werle, T. Fey, J. M. Neudörfl and H.-G. Schmalz, Org. Lett., 2007, 9, 3555–3558 CrossRef CAS PubMed.
- B. B. Snider, D. J. Rodini, M. Karras, T. C. Kirk, E. A. Deutsch, R. Cordova and R. T. Price, Tetrahedron, 1981, 37, 3927–3934 CrossRef CAS.
- F. Muehlthau and T. Bach, Synthesis, 2005, 2005, 3428–3436 Search PubMed.
- J. Yadav, A. Basak and P. Srihari, Tetrahedron Lett., 2007, 48, 2841–2843 CrossRef CAS.
- L. Han, X. Huang, I. Sattler, U. Moellmann, H. Fu, W. Lin and S. Grabley, Planta Med., 2005, 71, 160–164 CrossRef CAS PubMed.
- D. M. Solorio and M. P. Jennings, J. Org. Chem., 2007, 72, 6621–6623 CrossRef CAS PubMed.
- C. M. Marson, J. Campbell, M. B. Hursthouse and K. A. Malik, Angew. Chem., Int. Ed., 1998, 37, 1122–1124 CrossRef CAS PubMed.
- T. S. Wu, Y. Y. Chan, M. J. Liou, F. W. Lin, L. S. Shi and K. T. Chen, Phytother. Res., 1998, 12, S80–S82 CrossRef CAS.
- T.-S. Wu, M.-L. Wang and P.-L. Wu, Phytochemistry, 1996, 43, 785–789 CrossRef CAS.
- H. Furukawa, T. Wu, T. Ohta and C. Kuoh, Chem. Pharm. Bull., 1985, 33, 4132–4138 CrossRef CAS.
- A. Ueno, T. Kitawaki and N. Chida, Org. Lett., 2008, 10, 1999–2002 CrossRef CAS PubMed.
- H. Rupp, W. Schwarz and H. Musso, Eur. J. Inorg. Chem., 1983, 116, 2554–2563 CAS.
- W. Zhuang, T. Hansen and K. A. Jørgensen, Chem. Commun., 2001, 347–348 RSC.
- S. Yamazaki and Y. Iwata, J. Org. Chem., 2006, 71, 739–743 CrossRef CAS PubMed.
- J. Romo and P. Joseph-Nathan, Tetrahedron, 1964, 20, 2331–2337 CrossRef.
- H. Kakisawa, Y. Inouye and J. Romo, Tetrahedron Lett., 1969, 10, 1929–1932 CrossRef.
- W. D. Inman, J. Luo, S. D. Jolad, S. R. King and R. Cooper, J. Nat. Prod., 1999, 62, 1088–1092 CrossRef CAS PubMed.
- M. Jimenez-Estrada, R. R. Chilpa, T. R. Apan, F. Lledias, W. Hansberg, D. Arrieta and F. A. Aguilar, J. Ethnopharmacol., 2006, 105, 34–38 CrossRef CAS PubMed.
- K. Shindo, M. Kimura and M. Iga, Biosci., Biotechnol., Biochem., 2004, 68, 1393–1394 CrossRef CAS PubMed.
- V. Snieckus, Chem. Rev., 1990, 90, 879–933 CrossRef CAS.
- A. Padwa and N. Kamigata, J. Am. Chem. Soc., 1977, 99, 1871–1880 CrossRef CAS.
- B. L. Kedrowski and R. W. Hoppe, J. Org. Chem., 2008, 73, 5177–5179 CrossRef CAS PubMed.
- W.-H. Lin, J.-M. Fang and Y.-S. Cheng, Phytochemistry, 1995, 40, 871–873 CrossRef CAS.
- C.-I. Chang, J.-Y. Chang, C.-C. Kuo, W.-Y. Pan and Y.-H. Kuo, Planta Med., 2005, 71, 72–76 CrossRef CAS PubMed.
- K. Kawazoe, M. Yamamoto, Y. Takaishi, G. Honda, T. Fujita, E. Sezik and E. Yesilada, Phytochemistry, 1999, 50, 493–497 CrossRef CAS.
- H. Ohtsu, M. Iwamoto, H. Ohishi, S. Matsunaga and R. Tanaka, Tetrahedron Lett., 1999, 40, 6419–6422 CrossRef CAS.
- E. Alvarez-Manzaneda, R. Chahboun, E. Cabrera, E. Alvarez, R. Alvarez-Manzaneda, R. Meneses, H. Es-Samti and A. Fernandez, J. Org. Chem., 2009, 74, 3384–3388 CrossRef CAS PubMed.
- A. Cartier, H. Chan, J.-L. Malo, L. Pineau, K. Tse and M. Chan-Yeung, J. Allergy Clin. Immunol., 1986, 77, 639–645 CrossRef CAS PubMed.
- D. N. Weissman and D. M. Lewis, Occup. Med., 2000, 15, 385–398 CAS.
- M. Chan-Yeung, H. Chan, K. Tse, H. Salari and S. Lam, J. Allergy Clin. Immunol., 1989, 84, 762–768 CrossRef CAS PubMed.
- J. Gardner, G. Barton and H. Maclean, Can. J. Chem., 1959, 37, 1703–1709 CrossRef CAS.
- B.-F. Sun, R. Hong, Y.-B. Kang and L. Deng, J. Am. Chem. Soc., 2009, 131, 10384–10385 CrossRef CAS PubMed.
- G. Cassinelli, C. Lanzi, T. Pensa, R. A. Gambetta, G. Nasini, G. Cuccuru, M. Cassinis, G. Pratesi, D. Polizzi and M. Tortoreto, Biochem. Pharmacol., 2000, 59, 1539–1547 CrossRef CAS PubMed.
- L. Merlini, G. Nasini, L. Scaglioni, G. Cassinelli and C. Lanzi, Phytochemistry, 2000, 53, 1039–1041 CrossRef CAS PubMed.
- T. Yoshimitsu, S. Nojima, M. Hashimoto, K. Tsukamoto and T. Tanaka, Synthesis, 2009, 2009, 2963–2969 CrossRef.
- O. M. Moradei and L. A. Paquette, Org. Synth., 2003, 66–74 CAS.
- M. E. Layton, C. A. Morales and M. D. Shair, J. Am. Chem. Soc., 2002, 124, 773–775 CrossRef CAS PubMed.
- N. Sun and X. Liang, Acta Chim. Sin., 1981, 16, 24 CAS.
- D. Yin, C. Xu, Y. Gao, D. Liu, S. Wen and J. Guo, Acta Pharm. Sin., 1992, 27, 824–829 CAS.
- Z.-W. Zhang and W.-D. Z. Li, Org. Lett., 2010, 12, 1649–1651 CrossRef CAS PubMed.
- A. Ulubelen, G. Topcu, N. Tan, L.-J. Lin and G. A. Cordell, Phytochemistry, 1992, 31, 2419–2421 CrossRef CAS.
- A. Ulubelen, U. Sönmez and G. Topcu, Phytochemistry, 1997, 44, 1297–1299 CrossRef CAS.
- A. Ulubelen, G. Topçu, H.-B. Chai and J. M. Pezzuto, Pharm. Biol, 1999, 37, 148–151 CrossRef CAS.
- A. Ulubelen, S. Öksüz, U. Kolak, C. Bozok-Johansson, C. Çelik and W. Voelter, Planta Med., 2000, 66, 458–462 CrossRef CAS PubMed.
- G. Topcu, E. N. Altiner, S. Gozcu, B. Halfon, Z. Aydogmus, J. Pezzuto, B.-N. Zhou and D. G. Kingston, Planta Med., 2003, 69, 464–467 CrossRef CAS PubMed.
- A. Kabouche, N. Boutaghane, Z. Kabouche, E. Seguin, F. Tillequin and K. Benlabed, Fitoterapia, 2005, 76, 450–452 CrossRef CAS PubMed.
- R. A. Taj and J. R. Green, J. Org. Chem., 2010, 75, 8258–8270 CrossRef CAS PubMed.
- N. Gulavita, A. Hori, Y. Shimizu, P. Laszlo and J. Clardy, Tetrahedron Lett., 1988, 29, 4381–4384 CrossRef CAS.
- M. Medjahdi, J. C. González-Gómez, F. Foubelo and M. Yus, Eur. J. Org. Chem., 2011, 2011, 2230–2234 CrossRef.
- M. Li, P. Zhou and H. F. Roth, Synthesis, 2007, 2007, 55–60 CrossRef.
- J. F. Bower, P. Szeto and T. Gallagher, Chem. Commun., 2005, 5793–5795 RSC.
- Z. Ma and H. Zhai, Synlett, 2007, 2007, 0161–0163 CrossRef.
- Z. Ma, H. Hu, W. Xiong and H. Zhai, Tetrahedron, 2007, 63, 7523–7531 CrossRef CAS.
- D. N. Mai, B. R. Rosen and J. P. Wolfe, Org. Lett., 2011, 13, 2932–2935 CrossRef CAS PubMed.
- P. A. Donets, J. L. Goeman, J. Van der Eycken, K. Robeyns, L. Van Meervelt and E. V. Van der Eycken, Eur. J. Org. Chem., 2009, 2009, 793–796 CrossRef.
- A. D. Rodríguez, C. Ramírez, I. I. Rodríguez and C. L. Barnes, J. Org. Chem., 2000, 65, 1390–1398 CrossRef.
- M. Harmata, W. Ying and C. L. Barnes, Tetrahedron Lett., 2009, 50, 2326–2328 CrossRef CAS.
- W. Ying, C. L. Barnes and M. Harmata, Tetrahedron Lett., 2011, 52, 177–180 CrossRef CAS PubMed.
- A. D. Rodríguez and C. Ramírez, J. Nat. Prod., 2001, 64, 100–102 CrossRef.
- J. S. Yadav, B. Thirupathaiah and A. Al Khazim Al Ghamdi, Eur. J. Org. Chem., 2012, 2012, 2072–2076 CrossRef CAS.
- L. Fu, N. Li and R. Mill, Flora of China, 1999, vol. 4, pp. 85–88 Search PubMed.
- H. Abdelkafi and B. Nay, Nat. Prod. Rep., 2012, 29, 845–869 RSC.
- S. He, B. Wu, Y. Pan and L. Jiang, J. Org. Chem., 2008, 73, 5233–5241 CrossRef CAS PubMed.
- L. Li, G. E. Henry and N. P. Seeram, J. Agric. Food Chem., 2009, 57, 7282–7287 CrossRef CAS PubMed.
- T. Ito, Y. Akao, H. Yi, K. Ohguchi, K. Matsumoto, T. Tanaka, M. Iinuma and Y. Nozawa, Carcinogenesis, 2003, 24, 1489–1497 CrossRef CAS PubMed.
- K.-S. Huang, M. Lin, L.-N. Yu and M. Kong, Tetrahedron, 2000, 56, 1321–1329 CrossRef CAS.
- H. J. Kim, E. J. Chang, S. H. Cho, S. K. Chung, H. D. Park and S. W. Choi, Biosci., Biotechnol., Biochem., 2002, 66, 1990–1993 CrossRef CAS PubMed.
- J.-R. Dai, Y. F. Hallock, J. H. Cardellina and M. R. Boyd, J. Nat. Prod., 1998, 61, 351–353 CrossRef CAS PubMed.
- A. E. Bala, A. Kollmann, P. H. Ducrot, A. Majira, L. Kerhoas, R. Delorme and J. Einhorn, Pestic. Sci., 1999, 55, 206–208 CrossRef CAS.
- S. Sotheeswaran and V. Pasupathy, Phytochemistry, 1993, 32, 1083–1092 CrossRef CAS.
- C. W. Choi, Y. H. Choi, M.-R. Cha, Y. S. Kim, G. H. Yon, K. S. Hong, W.-K. Park and S. Y. Ryu, Planta Med., 2011, 77, 374–376 CrossRef CAS PubMed.
- M. H. Jang, X. L. Piao, J. M. Kim, S. W. Kwon and J. H. Park, Phytother. Res., 2008, 22, 544–549 CrossRef CAS PubMed.
- Y. L. Choi, B. T. Kim and J.-N. Heo, J. Org. Chem., 2012, 77, 8762–8767 CrossRef CAS PubMed.
- Z. Jin, Nat. Prod. Rep., 2011, 28, 1143–1191 RSC.
- P. B. Koswatta and C. J. Lovely, Nat. Prod. Rep., 2011, 28, 511–528 RSC.
- J. Sullivan, R. Giles and R. Looper, Curr. Bioact. Compd., 2009, 5, 39–78 CrossRef CAS.
- M. Roué, E. Quévrain, I. Domart-Coulon and M.-L. Bourguet-Kondracki, Nat. Prod. Rep., 2012, 29, 739–751 RSC.
- W. Hassan, R. Edrada, R. Ebel, V. Wray, A. Berg, R. van Soest, S. Wiryowidagdo and P. Proksch, J. Nat. Prod., 2004, 67, 817–822 CrossRef CAS PubMed.
- J. Das, P. B. Koswatta, J. D. Jones, M. Yousufuddin and C. J. Lovely, Org. Lett., 2012, 14, 6210–6213 CrossRef CAS PubMed.
- K. Nicolaou, Q. Kang, T. R. Wu, C. S. Lim and D. Y.-K. Chen, J. Am. Chem. Soc., 2010, 132, 7540–7548 CrossRef CAS PubMed.
- K. e. C. Nicolaou, T. R. Wu, Q. Kang and D. Y. K. Chen, Angew. Chem., 2009, 121, 3492–3495 CrossRef.
- A. Dorn, V. Schattel and S. Laufer, Bioorg. Med. Chem. Lett., 2010, 20, 3074–3077 CrossRef CAS PubMed.
- S. A. Laufer, G. M. Ahrens, S. C. Karcher, J. S. Hering and R. Niess, J. Med. Chem., 2006, 49, 7912–7915 CrossRef CAS PubMed.
- Y.-P. Xue and W.-D. Z. Li, J. Org. Chem., 2011, 76, 57–64 CrossRef CAS PubMed.
- E. H. Hakim, L. D. Juliawaty, Y. M. Syah, L. bin Din, E. L. Ghisalberti, J. Latip, I. M. Said and S. A. Achmad, Z. Naturforsch., C: J. Biosci., 2005, 60, 723–727 Search PubMed.
- T. Ito, T. Tanaka, M. Iinuma, K.-i. Nakaya, Y. Takahashi, R. Sawa, J. Murata and D. Darnaedi, J. Nat. Prod., 2004, 67, 932–937 CrossRef CAS PubMed.
- L. Botella and C. Nájera, Tetrahedron, 2004, 60, 5563–5570 CrossRef CAS.
- P. S. Baran and N. Z. Burns, J. Am. Chem. Soc., 2006, 128, 3908–3909 CrossRef CAS PubMed.
- G.-W. Wang, H.-L. Wang, D. A. Capretto, Q. Han, R.-B. Hu and S.-D. Yang, Tetrahedron, 2012, 68, 5216–5222 CrossRef CAS.
- F. Delle Monache, G. Delle Monache, J. F. Cavalcanti and R. M. Pinheiro, Tetrahedron Lett., 1987, 28, 563–566 CrossRef CAS.
- K. Ezaki, M. Satake, T. Kusumi and H. Kakisawa, Tetrahedron Lett., 1991, 32, 2793–2796 CrossRef CAS.
- Y. Suzuki, N. Matsuo, T. Nemoto and Y. Hamada, Tetrahedron, 2013, 69, 5913–5919 CrossRef CAS.
- R. Bandichhor, A. N. Lowell and M. C. Kozlowski, J. Org. Chem., 2011, 76, 6475–6487 CrossRef CAS PubMed.
- A. Pinder, in Fortschritte der Chemie Organischer Naturstoffe/Progress in the Chemistry of Organic Natural Products, Springer, 1977, pp. 81–186 Search PubMed.
- A. Romo de Vivar, A.-L. Pérez-Castorena, A. Arciniegas and J. L. Villaseñor, J. Mex. Chem. Soc., 2007, 51, 160–172 CAS.
- Y. Saito, M. Hattori, Y. Iwamoto, Y. Takashima, K. Mihara, Y. Sasaki, M. Fujiwara, M. Sakaoku, A. Shimizu and X. Chao, Tetrahedron, 2011, 67, 2220–2231 CrossRef CAS.
- A. L. Silva, R. A. Toscano and L. A. Maldonado, J. Org. Chem., 2013, 78, 5282–5292 CrossRef CAS PubMed.
- F. Schaller, L. Rahalison, N. Islam, O. Potterat, K. Hostettmann, H. Stoeckli-Evans and S. Mavi, Helv. Chim. Acta, 2000, 83, 407–413 CrossRef CAS.
- J. Huang, D. Foyle, X. Lin and J. Yang, J. Org. Chem., 2013, 78, 9166–9173 CrossRef CAS PubMed.
- J. K. Kim, Y. H. Kim, H. T. Nam, B. T. Kim and J.-N. Heo, Org. Lett., 2008, 10, 3543–3546 CrossRef CAS PubMed.
- D. M. Gardiner, P. Waring and B. J. Howlett, Microbiology, 2005, 151, 1021–1032 CrossRef CAS PubMed.
- N. Boyer, K. C. Morrison, J. Kim, P. J. Hergenrother and M. Movassaghi, Chem. Sci., 2013, 4, 1646–1657 RSC.
- C.-J. Zheng, C.-J. Kim, K. S. Bae, Y.-H. Kim and W.-G. Kim, J. Nat. Prod., 2006, 69, 1816–1819 CrossRef CAS PubMed.
- A. Coste, J. Kim, T. C. Adams and M. Movassaghi, Chem. Sci., 2013, 4, 3191–3197 RSC.
- C. C. Hughes, J. B. MacMillan, S. P. Gaudêncio, P. R. Jensen and W. Fenical, Angew. Chem., Int. Ed., 2009, 48, 725–727 CrossRef CAS PubMed.
- A. Miyanaga, J. E. Janso, L. McDonald, M. He, H. Liu, L. Barbieri, A. S. Eustáquio, E. N. Fielding, G. T. Carter and P. R. Jensen, J. Am. Chem. Soc., 2011, 133, 13311–13313 CrossRef CAS PubMed.
- Y. Aotani, H. Nagata and M. Yoshida, J. Antibiot., 1997, 50, 543–545 CrossRef CAS PubMed.
- Y. Takayama, T. Yamada, S. Tatekabe and K. Nagasawa, Chem. Commun., 2013, 49, 6519–6521 RSC.
- J. W. Blunt, B. R. Copp, R. A. Keyzers, M. H. G. Munro and M. R. Prinsep, Nat. Prod. Rep., 2012, 29, 144–222 RSC.
- J. C. Morris and A. J. Phillips, Nat. Prod. Rep., 2011, 28, 269–289 RSC.
- R. K. Akee, T. R. Carroll, W. Y. Yoshida, P. J. Scheuer, T. J. Stout and J. Clardy, J. Org. Chem., 1990, 55, 1944–1946 CrossRef CAS.
- S. Nakamura, N. Tsuno, M. Yamashita, I. Kawasaki, S. Ohta and Y. Ohishi, J. Chem. Soc., Perkin Trans. 1, 2001, 1, 429–436 RSC.
- X. Fu, J. R. Barnes, T. Do and F. J. Schmitz, J. Nat. Prod., 1997, 60, 497–498 CrossRef CAS.
- J. Das, A. Bhan, S. S. Mandal and C. J. Lovely, Bioorg. Med. Chem. Lett., 2013, 23, 6183–6187 CrossRef CAS PubMed.
- M. Alvarez and J. A. Joule, Tetrahedron Lett., 2001, 42, 15–317 Search PubMed.
- S. Goodwin, A. Smith and E. Horning, J. Am. Chem. Soc., 1959, 81, 1903–1908 CrossRef CAS.
- R. Woodward, G. Iacobucci and I. Hochstein, J. Am. Chem. Soc., 1959, 81, 4434–4435 CrossRef CAS.
- P. Juret, A. Tanguy, A. Girard, J. Le Talaer, J. Abbatucci, N. Dat-Xuong, J. Le Pecq and C. Paolett, Eur. J. Cancer, 1978, 14, 205–206 CrossRef CAS PubMed.
- H.-J. Knölker and K. R. Reddy, Chem. Rev., 2002, 102, 4303–4428 CrossRef.
- M. Stiborová, C. A. Bieler, M. Wiessler and E. Frei, Biochem. Pharmacol., 2001, 62, 1675–1684 CrossRef.
- C. M. Miller and F. O. McCarthy, RSC Adv., 2012, 2, 8883–8918 RSC.
- D. Mal, B. K. Senapati and P. Pahari, Tetrahedron, 2007, 63, 3768–3781 CrossRef CAS.
- H. Y. Lee, G. S. Chen, C. S. Chen and J. W. Chern, J. Heterocycl. Chem., 2010, 47, 454–458 CAS.
- M. Dračínský, J. Sejbal, B. Rygerová and M. Stiborová, Tetrahedron Lett., 2007, 48, 6893–6895 CrossRef.
- N. Ramkumar, M. S. Raghavendra and R. Nagarajan, Synlett, 2014, 25, 2791–2793 CrossRef CAS.
- D. He, L. Ding, H. Xu, X. Lei, H. Xiao and Y. Zhou, J. Org. Chem., 2012, 77, 8435–8443 CrossRef CAS PubMed.
- L. H. Mejorado and T. R. Pettus, J. Am. Chem. Soc., 2006, 128, 15625–15631 CrossRef CAS PubMed.
- K. e. C. Nicolaou, D. J. Edmonds, A. Li and G. S. Tria, Angew. Chem., 2007, 119, 4016–4019 CrossRef.
- Y. Kita, M. Arisawa, M. Gyoten, M. Nakajima, R. Hamada, H. Tohma and T. Takada, J. Org. Chem., 1998, 63, 6625–6633 CrossRef CAS.
- H. Tohma, H. Morioka, S. Takizawa, M. Arisawa and Y. Kita, Tetrahedron, 2001, 57, 345–352 CrossRef CAS.
- H. Li, Y. Zhang, X. Xie, H. Ma, C. Zhao, G. Zhao and X. She, Org. Lett., 2014, 16, 4440–4443 CrossRef CAS PubMed.
- T.-S. Kam, G. Subramaniam, K.-H. Lim and Y.-M. Choo, Tetrahedron Lett., 2004, 45, 5995–5998 CrossRef CAS.
- Z. Lv, Z. Li and G. Liang, Org. Lett., 2014, 16, 1653–1655 CrossRef PubMed.
- A. l. Biechy and S. Z. Zard, Org. Lett., 2009, 11, 2800–2803 CrossRef CAS PubMed.
- J. Zhu, G. Islas-Gonzalez and M. Bois-Choussy, Org. Prep. Proced. Int., 2000, 32, 505–546 CrossRef CAS.
- D. S. Jang, E. J. Park, M. E. Hawthorne, J. S. Vigo, J. G. Graham, F. Cabieses, B. D. Santarsiero, A. D. Mesecar, H. H. Fong and R. G. Mehta, J. M. Pezzuto, and A. D. Kinghorn, J. Agric. Food Chem., 2002, 50, 6330–6334 CrossRef CAS.
- L.-B. Dong, J. He, X.-Y. Li, X.-D. Wu, X. Deng, G. Xu, L.-Y. Peng, Y. Zhao, Y. Li and X. Gong, Nat. Prod. Bioprospect., 2011, 1, 41–47 CrossRef CAS.
- Z. Li, T.-F. Leung and R. Tong, Chem. Commun., 2014, 50, 10990–10993 RSC.
- A. Al Mourabit and P. Potier, Eur. J. Org. Chem., 2001, 2001, 237–243 CrossRef.
- H. Hoffmann and T. Lindel, Synthesis, 2003, 2003, 1753–1783 Search PubMed.
- T. Imaoka, M. Iwata, T. Akimoto and K. Nagasawa, Nat. Prod. Bioprospect., 2013, 8, 961–964 CAS.
- N. M. Hewlett and J. J. Tepe, Org. Lett., 2011, 13, 4550–4553 CrossRef CAS PubMed.
- M. Iwata, K. Kanoh, T. Imaoka and K. Nagasawa, Chem. Commun., 2014, 50, 6991–6994 RSC.
- L. Garrido, E. Zubía, M. J. Ortega and J. Salvá, J. Org. Chem., 2003, 68, 293–299 CrossRef CAS PubMed.
- Y. Momoi, K. i. Okuyama, H. Toya, K. Sugimoto, K. Okano and H. Tokuyama, Angew. Chem., 2014, 126, 13431–13435 CrossRef.
- L. N. Mander, Nat. Prod. Rep., 2003, 20, 49–69 RSC.
- P. A. García, A. B. De Oliveira and R. Batista, Molecules, 2007, 12, 455–483 CrossRef.
- J. R. Hanson, Nat. Prod. Rep., 2009, 26, 1156–1171 RSC.
- M. Presset, Y. Coquerel and J. Rodriguez, Chem. Rev., 2012, 113, 525–595 CrossRef PubMed.
- L. Zhu, S.-H. Huang, J. Yu and R. Hong, Tetrahedron Lett., 2015, 56, 23–31 CrossRef CAS.
- E. H. Colebrook, S. G. Thomas, A. L. Phillips and P. Hedden, J. Exp. Biol., 2014, 217, 67–75 CrossRef CAS PubMed.
- J.-M. Davière and P. Achard, Development, 2013, 140, 1147–1151 CrossRef PubMed.
- P. J. Davies, The plant hormones: their nature, occurrence, and functions, in Plant hormones, Springer, Dordrecht, 2010, pp. 1–15 Search PubMed.
- G. Topçu, A. Ertaş, M. Öztürk, D. Dinçel, T. Kılıç and B. Halfon, Phytochem. Lett., 2011, 4, 436–439 CrossRef.
- S. J. Burke, W. P. Malachowski, S. K. Mehta and R. Appenteng, Org. Biomol. Chem., 2015, 13, 2726–2744 RSC.
- D. F. Taber and R. B. Sheth, J. Org. Chem., 2008, 73, 8030–8032 CrossRef CAS PubMed.
- H.-J. Liu and D. D.-P. Tran, Tetrahedron Lett., 1999, 40, 3827–3830 CrossRef CAS.
- X.-W. Yang, C.-P. Yang, L.-P. Jiang, X.-J. Qin, Y.-P. Liu, Q.-S. Shen, Y.-B. Chen and X.-D. Luo, Org. Lett., 2014, 16, 5808–5811 CrossRef CAS PubMed.
- X. Liang, S.-Z. Jiang, K. Wei and Y.-R. Yang, J. Am. Chem. Soc., 2016, 138, 2560–2562 CrossRef CAS PubMed.
- C. Beemelmanns, V. Blot, S. Gross, D. Lentz and H. U. Reissig, Eur. J. Org. Chem., 2010, 2010, 2716–2732 CrossRef.
- M. A. Schafroth, D. Sarlah, S. Krautwald and E. M. Carreira, J. Am. Chem. Soc., 2012, 134, 20276–20278 CrossRef CAS PubMed.
- J. Y. Hamilton, D. Sarlah and E. M. Carreira, J. Am. Chem. Soc., 2013, 135, 994–997 CrossRef CAS PubMed.
- O. F. Jeker, A. G. Kravina and E. M. Carreira, Angew. Chem., 2013, 125, 12388–12391 CrossRef.
- T. J. Maimone and P. S. Baran, Nat. Chem. Biol., 2007, 3, 396–407 CrossRef CAS PubMed.
- L. A. Loyola, G. Morales, B. Rodriguez, J. Jiménez-Barbero, M. C. d. l. Torre, A. Perales and M. R. Torres, Tetrahedron, 1990, 46, 5413–5420 CrossRef CAS.
- L. A. Loyola, G. Morales, M. C. De La Torre, S. Pedreros and B. Rodríguez, Phytochemistry, 1990, 29, 3950–3951 CrossRef CAS.
- C. Areche, B. Sepulveda, A. S. Martin, O. Garcia-Beltran, M. Simirgiotis and A. Canete, Org. Biomol. Chem., 2014, 12, 6406–6413 RSC.
- C. Areche, F. Rojas-Alvarez, C. Campos-Briones, C. Lima, E. G. Pérez and B. Sepúlveda, J. Pharm. Pharmacol., 2013, 65, 1231–1238 CrossRef CAS PubMed.
- G. A. Wächter, S. G. Franzblau, G. Montenegro, E. Suarez, R. H. Fortunato, E. Saavedra and B. N. Timmermann, J. Nat. Prod., 1998, 61, 965–968 CrossRef PubMed.
- L. A. Loyola, J. Bórquez, G. Morales, A. San-Martín, J. Darias, N. Flores and A. Giménez, Phytochemistry, 2004, 65, 1931–1935 CrossRef CAS PubMed.
- G. A. Wächter, G. Matooq, J. J. Hoffmann, W. M. Maiese, M. P. Singh, G. Montenegro and B. N. Timmermann, J. Nat. Prod., 1999, 62, 1319–1321 CrossRef.
- L. A. Loyola, J. Bórquez, G. Morales and A. San-Martín, Phytochemistry, 2000, 53, 961–963 CrossRef CAS PubMed.
- L. A. Loyola, J. Bórquez, G. Morales and A. S. Martin, Phytochemistry, 1997, 44, 649–651 CrossRef CAS.
- C. Areche, L. A. Loyola, J. Borquez, J. Rovirosa and A. San-Martin, Magn. Reson. Chem., 2008, 46, 765–768 CrossRef CAS PubMed.
- Y. T. Liu, L. P. Li, J. H. Xie and Q. L. Zhou, Angew. Chem., Int. Ed., 2017, 56, 12708–12711 CrossRef CAS PubMed.
- W.-S. Sun, S. Su, R.-X. Zhu, G.-Z. Tu, W. Cheng, H. Liang, X.-Y. Guo, Y.-Y. Zhao and Q.-Y. Zhang, Tetrahedron Lett., 2013, 54, 3617–3620 CrossRef CAS.
- B. P. Bandgar and K. A. Shaikh, Tetrahedron Lett., 2003, 44, 1959–1961 CrossRef CAS.
- S. Luo, C. A. Zificsak and R. P. Hsung, Org. Lett., 2003, 5, 4709–4712 CrossRef CAS PubMed.
- P. Feng, Y. Fan, F. Xue, W. Liu, S. Li and Y. Shi, Org. Lett., 2011, 13, 5827–5829 CrossRef CAS PubMed.
- M. Tsuda, H. Sato, Y. Tanaka, K. Yazawa, Y. Mikami, T. Sasaki and J. i. Kobayashi, J. Chem. Soc., Perkin Trans. 1, 1996, 1773–1775 RSC.
- M. L. Patil, H. B. Borate, D. E. Ponde, B. M. Bhawal and V. H. Deshpande, Tetrahedron Lett., 1999, 40, 4437–4438 CrossRef CAS.
- M. L. Patil, H. B. Borate, D. E. Ponde and V. H. Deshpande, Tetrahedron, 2002, 58, 6615–6620 CrossRef CAS.
- J.-R. Ioset, A. Marston, M. P. Gupta and K. Hostettmann, J. Nat. Prod., 2000, 63, 424–426 CrossRef CAS.
- R. Aguilar, A. Benavides and J. Tamariz, Synth. Commun., 2004, 34, 2719–2735 CrossRef CAS.
- A. Chatterjee and B. Hazra, Tetrahedron, 1977, 33, 1983–1987 CrossRef CAS.
- A. K. Sinha, R. Dogra and B. P. Joshi, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2002, 41B, 635–638 CAS.
- M. Gordaliza, P. A. García, J. M. Miguel del Corral, M. A. Castro and M. A. Gómez-Zurita, Toxicon, 2004, 44, 441–459 CrossRef CAS PubMed.
- R. S. Ward, Phytochem. Rev., 2003, 2, 391–400 CrossRef CAS.
- R. S. Ward, Nat. Prod. Rep., 1999, 16, 75–96 RSC.
- D. Stadler and T. Bach, Angew. Chem., Int. Ed., 2008, 47, 7557–7559 CrossRef CAS PubMed.
- K. Kondo and F. Mori, Chem. Lett., 1974, 3, 741–742 CrossRef.
- C. Gnamm, S. Foerster, N. Miller, K. Broedner and G. Helmchen, Synlett, 2007, 2007, 0790–0794 CrossRef.
- J. Saxton, in The Alkaloids: Chemistry and Physiology, Elsevier, 1965, vol. 8, pp. 673–678 Search PubMed.
- E. F. Rogers, H. R. Snyder and R. F. Fischer, J. Am. Chem. Soc., 1952, 74, 1987–1989 CrossRef CAS.
- H. R. Snyder, R. F. Fischer, J. F. Walker, H. E. Els and G. A. Nussberger, J. Am. Chem. Soc., 1954, 76, 2819–2825 CrossRef CAS.
- P. Yates, F. N. MacLachlan, I. D. Rae, M. Rosenberger, A. G. Szabo, C. R. Willis, M. P. Cava, M. Behforouz, M. V. Lakshmikantham and W. Zeiger, J. Am. Chem. Soc., 1973, 95, 7842–7850 CrossRef CAS.
- H. Ueda, H. Satoh, K. Matsumoto, K. Sugimoto, T. Fukuyama and H. Tokuyama, Angew. Chem., Int. Ed., 2009, 48, 7600–7603 CrossRef CAS PubMed.
- C. S. Yang, J. D. Lambert and S. Sang, Arch. Toxicol., 2009, 83, 11–21 CrossRef CAS PubMed.
- L. Bonfili, V. Cecarini, M. Amici, M. Cuccioloni, M. Angeletti, J. N. Keller and A. M. Eleuteri, FEBS J., 2008, 275, 5512–5526 CrossRef CAS PubMed.
- H. M. Ge, C. Xu, X. T. Wang, B. Huang and R. X. Tan, Eur. J. Org. Chem., 2006, 2006, 5551–5554 CrossRef.
- K. Warabi, S. Matsunaga, R. W. M. van Soest and N. Fusetani, J. Org. Chem., 2003, 68, 2765–2770 CrossRef CAS PubMed.
- K. Okano, H. Fujiwara, T. Noji, T. Fukuyama and H. Tokuyama, Angew. Chem., 2010, 122, 6061–6065 CrossRef.
- E. Duval and G. D. Cuny, Tetrahedron Lett., 2004, 45, 5411–5413 CrossRef CAS.
- J. P. Karwowski, M. Jackson, R. R. Rasmussen, P. E. Humphrey, J. B. Poddig, W. L. Kohl, M. H. Scherr, S. Kadam and J. B. Mcalpine, J. Antibiot., 1993, 46, 374–379 CrossRef CAS PubMed.
- K. M. Depew, S. P. Marsden, D. Zatorska, A. Zatorski, W. G. Bornmann and S. J. Danishefsky, J. Am. Chem. Soc., 1999, 121, 11953–11963 CrossRef CAS.
- S. Takiguchi, T. Iizuka, Y.-s. Kumakura, K. Murasaki, N. Ban, K. Higuchi and T. Kawasaki, J. Org. Chem., 2010, 75, 1126–1131 CrossRef CAS PubMed.
- U. Anthoni, C. Christophersen and P. H. Nielsen, Alkaloids: Chem. Biol. Perspect., 1999, 13, 163–236 CAS.
- P. Waring, R. D. Eichner and A. Müllbacher, Med. Chem. Res., 1988, 8, 499–524 CAS.
- T. Rezanka, M. Sobotka, J. Spízek and K. Sigler, Anti-Infect. Agents Med. Chem., 2006, 5, 187–224 CrossRef CAS.
- C. L. Chai and P. Waring, Redox Rep., 2000, 5, 257–264 CrossRef CAS PubMed.
- P. Trown, Biochem. Biophys. Res. Commun., 1968, 33, 402–407 CrossRef CAS PubMed.
- T. Hino and T. Sato, Tetrahedron Lett., 1971, 12, 3127–3129 CrossRef.
- Y. Kishi, T. Fukuyama and S. Nakatsuka, J. Am. Chem. Soc., 1973, 95, 6492–6493 CrossRef CAS PubMed.
- Y. Kishi, S. Nakatsuka, T. Fukuyama and M. Havel, J. Am. Chem. Soc., 1973, 95, 6493–6495 CrossRef CAS PubMed.
- J. Kim, J. A. Ashenhurst and M. Movassaghi, Science, 2009, 324, 238–241 CrossRef CAS PubMed.
- Y. Usami, J. Yamaguchi and A. Numata, Heterocycles, 2004, 63, 1123–1129 CrossRef CAS.
- B. V. Bertinetti, M. A. Rodriguez, A. M. Godeas and G. M. Cabrera, J. Antibiot., 2010, 63, 681 CrossRef CAS PubMed.
- M. Movassaghi, M. A. Schmidt and J. A. Ashenhurst, Angew. Chem., 2008, 120, 1507–1509 CrossRef.
- N. Boyer and M. Movassaghi, Chem. Sci., 2012, 3, 1798–1803 RSC.
- E. J. Park, H. R. Park, J. S. Lee and J. Kim, Planta Med., 1998, 64, 464–466 CrossRef CAS PubMed.
- G. Yoon, B. Y. Kang and S. H. Cheon, Arch. Pharmacal Res., 2007, 30, 313–316 CrossRef CAS.
- S. F. Nielsen, M. Chen, T. G. Theander, A. Kharazmi and S. B. Christensen, Bioorg. Med. Chem. Lett., 1995, 5, 449–452 CrossRef.
- H. Haraguchi, K. Tanimoto, Y. Tamura, K. Mizutani and T. Kinoshita, Phytochemistry, 1998, 48, 125–129 CrossRef CAS PubMed.
- H. Haraguchi, H. Ishikawa, K. Mizutani, Y. Tamura and T. Kinoshita, Bioorg. Med. Chem., 1998, 6, 339–347 CrossRef CAS PubMed.
- T. Nomura and T. Fukai, in Fortschritte der Chemie organischer Naturstoffe/Progress in the Chemistry of Organic Natural Products, Springer, 1998, pp. 1–140 Search PubMed.
- T. Saitoh and S. Shibata, Tetrahedron Lett., 1975, 16, 4461–4462 CrossRef.
- G. Yoon, Y. Do Jung and S. H. Cheon, Chem. Pharm. Bull., 2005, 53, 694–695 CrossRef CAS PubMed.
- Y. Na, J.-H. Cha, H.-G. Yoon and Y. Kwon, Chem. Pharm. Bull., 2009, 57, 607–609 CrossRef CAS PubMed.
- G. Yoon, W. Lee, S.-N. Kim and S. H. Cheon, Bioorg. Med. Chem. Lett., 2009, 19, 5155–5157 CrossRef CAS PubMed.
- Z. Wang, Y. Cao, S. Paudel, G. Yoon and S. H. Cheon, Arch. Pharmacal Res., 2013, 36, 1432–1436 CrossRef CAS PubMed.
- Y.-C. Kong, K.-F. Cheng, R. C. Cambie and P. G. Waterman, J. Chem. Soc., Chem. Commun., 1985, 47–48 RSC.
- Y.-C. Kong, K.-F. Cheng, K.-H. Ng, P. P.-H. But, S.-X. Yu, H.-T. Chang, R. C. Cambie, T. Kinoshita, W.-s. Kan and P. G. Waterman, Biochem. Syst. Ecol., 1986, 14, 491–497 CrossRef CAS.
- C. Cassani, R. Martín-Rapún, E. Arceo, F. Bravo and P. Melchiorre, Nat. Protoc., 2013, 8, 325 CrossRef CAS PubMed.
- N. S. Dange, B.-C. Hong and G.-H. Lee, RSC Adv., 2014, 4, 59706–59715 RSC.
- Y. S. Tsizin, Chem. Heterocycl. Compd., 1978, 14, 925–940 CrossRef.
- P. D. Bass, D. A. Gubler, T. C. Judd and R. M. Williams, Chem. Rev., 2013, 113, 6816–6863 CrossRef CAS PubMed.
- I. Kock, D. Heber, M. Weide, U. Wolschendorf and B. Clement, J. Med. Chem., 2005, 48, 2772–2777 CrossRef CAS PubMed.
- L. Z. Benet, D. Kroetz, L. Sheiner, J. Hardman and L. Limbird, Goodman and Gilman's the pharmacological basis of therapeutics, 1996, pp. 3–27 Search PubMed.
- A. Clarysse, A. Brugarolas, P. Siegenthaler, R. Abele, F. Cavalli, R. De Jager, G. Renard, M. Rozencweig, H. H. Hansen and E. E. C. T. Group, Eur. J. Cancer Clin. Oncol., 1984, 20, 243–247 CrossRef CAS PubMed.
- G. W. Gribble, M. G. Saulnier, M. P. Sibi and J. A. Obaza-Nutaitis, J. Org. Chem., 1984, 49, 4518–4523 CrossRef CAS.
- M.-L. Bennasar, T. Roca and F. Ferrando, J. Org. Chem., 2005, 70, 9077–9080 CrossRef CAS PubMed.
- P. H. Bernardo, C. L. Chai, G. A. Heath, P. J. Mahon, G. D. Smith, P. Waring and B. A. Wilkes, J. Med. Chem., 2004, 47, 4958–4963 CrossRef CAS PubMed.
- N. Ramkumar and R. Nagarajan, J. Org. Chem., 2014, 79, 736–741 CrossRef CAS PubMed.
- J. Orjala, A. D. Wright, C. A. Erdelmeier, O. Sticher and T. Rali, Helv. Chim. Acta, 1993, 76, 1481–1488 CrossRef CAS.
- K. Ichino, Phytochemistry, 1989, 28, 955–956 CrossRef CAS.
- Y. Mimaki, A. Kameyama, Y. Sashida, Y. Miyata and A. Fujii, Chem. Pharm. Bull., 1995, 43, 893–895 CrossRef CAS PubMed.
- B. Portet, N. Fabre, V. Roumy, H. Gornitzka, G. Bourdy, S. Chevalley, M. Sauvain, A. Valentin and C. Moulis, Phytochemistry, 2007, 68, 1312–1320 CrossRef CAS PubMed.
- D. H. Dethe and B. D. Dherange, J. Org. Chem., 2015, 80, 4526–4531 CrossRef CAS PubMed.
- I. S. Young and P. S. Baran, Nat. Chem., 2009, 1, 193 CrossRef CAS PubMed.
- E. Roulland, Angew. Chem., Int. Ed., 2011, 50, 1226–1227 CrossRef CAS PubMed.
- C. C. Price and M. Lund, J. Am. Chem. Soc., 1940, 62, 3105–3107 CrossRef CAS.
- F. Mühlthau, D. Stadler, A. Goeppert, G. A. Olah, G. S. Prakash and T. Bach, J. Am. Chem. Soc., 2006, 128, 9668–9675 CrossRef PubMed.
- H. R. Arthur, W. Hui and Y. Ng, J. Chem. Soc., 1959, 1840–1845 RSC.
- M. Cushman and L. Cheng, J. Org. Chem., 1978, 43, 286–288 CrossRef CAS.
- R. H. Thomson, Naturally Occurring Quinones, Academic Press, 2nd edn, 1971 Search PubMed.
- K. Brown, Chem. Soc. Rev., 1975, 4, 263–288 RSC.
- R. Ferrier and J. Tedder, J. Chem. Soc., 1957, 1435–1437 CAS.
- K. Kim, M. Spatz and F. Johnson, Tetrahedron Lett., 1979, 20, 331–334 CrossRef.
- S. De Silva, M. Watanabe and V. Snieckus, J. Org. Chem., 1979, 44, 4802–4808 CrossRef CAS.
- R. C. Hoch, I. U. Schraufstätter and C. G. Cochrane, J. Lab. Clin. Med., 1996, 128, 134–145 CrossRef CAS PubMed.
- M. Seitz, B. Dewald, N. Gerber and M. Baggiolini, J. Clin. Invest., 1991, 87, 463–469 CrossRef CAS PubMed.
- R. J. Capon, in Studies in Natural Products Chemistry, Elsevier, 1995, vol. 15, pp. 289–326 Search PubMed.
- A. D. Patil, A. J. Freyer, L. Killmer, P. Offen, B. Carte, A. J. Jurewicz and R. K. Johnson, Tetrahedron, 1997, 53, 5047–5060 CrossRef CAS.
- G. Bhide, N. Tikotkar and B. Tilak, Tetrahedron, 1960, 10, 223–229 CrossRef CAS.
- M. Inoue, M. W. Carson, A. J. Frontier and S. J. Danishefsky, J. Am. Chem. Soc., 2001, 123, 1878–1889 CrossRef CAS PubMed.
- M. K. Li and P. J. Scheuer, Tetrahedron Lett., 1984, 25, 587–590 CrossRef CAS.
- S. Sakemi and T. Higa, Experientia, 1987, 43, 624–625 CrossRef CAS PubMed.
- H. Fukui, N. Yoshikawa and M. Tabata, Phytochemistry, 1984, 23, 301–305 CrossRef CAS.
- J.-i. Tanaka, H. Miki and T. Higa, J. Nat. Prod., 1992, 55, 1522–1524 CrossRef CAS.
- H.-K. Yim, Y. Liao and H. N. Wong, Tetrahedron, 2003, 59, 1877–1884 CrossRef CAS.
- H. A. Weber and J. B. Gloer, J. Org. Chem., 1991, 56, 4355–4360 CrossRef CAS.
- K. Krohn, U. Flörke, M. John, N. Root, K. Steingröver, H.-J. Aust, S. Draeger, B. Schulz, S. Antus and M. Simonyi, Tetrahedron, 2001, 57, 4343–4348 CrossRef CAS.
- E. Quesada, M. Stockley and R. J. Taylor, Tetrahedron Lett., 2004, 45, 4877–4881 CrossRef CAS.
- H. A. Weber, N. C. Baenziger and J. B. Gloer, J. Am. Chem. Soc., 1990, 112, 6718–6719 CrossRef CAS.
- P. Seephonkai, M. Isaka, P. Kittakoop, P. Palittapongarnpim, S. Kamchonwongpaisan, M. Tanticharoen and Y. Thebtaranonth, Planta Med., 2002, 68, 45–48 CrossRef CAS PubMed.
- E. Quesada, M. Stockley, J. P. Ragot, M. E. Prime, A. C. Whitwood and R. J. Taylor, Org. Biomol. Chem., 2004, 2, 2483–2495 RSC.
- T. Kamo, N. Hirai, K. Iwami, D. Fujioka and H. Ohigashi, Tetrahedron, 2001, 57, 7649–7656 CrossRef CAS.
- R. Cooke, B. Johnson and W. Segal, Aust. J. Chem., 1958, 11, 230–235 CrossRef CAS.
- J. Nanclares, J. Gil, J. Saez, B. Schneider and F. Otálvaro, Tetrahedron Lett., 2008, 49, 3844–3847 CrossRef CAS.
- F. Sponga, L. Cavaletti, A. Lazzarini, A. Borghi, I. Ciciliato, D. Losi and F. Marinelli, J. Biotechnol., 1999, 70, 65–69 CrossRef CAS.
- Q. Liang, J. Zhang, W. Quan, Y. Sun, X. She and X. Pan, J. Org. Chem., 2007, 72, 2694–2697 CrossRef CAS PubMed.
- D. Konwar, R. C. Boruah and J. S. Sandhu, Tetrahedron Lett., 1990, 31, 1063–1064 CrossRef CAS.
- G. Bringmann, G. Lang, M. Michel and M. Heubes, Tetrahedron Lett., 2004, 45, 2829–2831 CrossRef CAS.
- R. Kasar, R. Khan, V. Deshpande and N. Ayyangar, Tetrahedron Lett., 1991, 32, 1599–1600 CrossRef CAS.
- J. Yadav, N. Thrimurtulu, K. U. Gayathri, B. S. Reddy and A. Prasad, Tetrahedron Lett., 2008, 49, 6617–6620 CrossRef CAS.
- H. Nakamura, J. i. Kobayashi, Y. Ohizumi and Y. Hirata, Tetrahedron Lett., 1982, 23, 5555–5558 CrossRef CAS.
- S. Aoki, H. Wei, K. Matsui, R. Rachmat and M. Kobayashi, Bioorg. Med. Chem., 2003, 11, 1969–1973 CrossRef CAS PubMed.
- B. F. Bowden, B. J. McCool and R. H. Willis, J. Org. Chem., 2004, 69, 7791–7793 CrossRef CAS PubMed.
- H. Nakamura, J. i. Kobayashi, Y. Ohizumi and Y. Hirata, J. Chem. Soc., Perkin Trans. 1, 1987, 173–176 RSC.
- Y. Ohizumi, A. Kajiwara, H. Nakamura and J. i. Kobayashi, J. Pharm. Pharmacol., 1984, 36, 785–786 CrossRef CAS PubMed.
- E. L. Larghi, B. V. Obrist and T. S. Kaufman, Tetrahedron, 2008, 64, 5236–5245 CrossRef CAS.
- Y. Murakami, A. Ishii, S. Mizuno, S. Yaginuma and Y. Uehara, Anticancer Res., 1999, 19, 4145–4149 CAS.
- T. Yoshino, F. Ng and S. J. Danishefsky, J. Am. Chem. Soc., 2006, 128, 14185–14191 CrossRef CAS PubMed.
- F. Bracher and B. Schulte, Eur. J. Org. Chem., 1997, 1997, 1979–1982 Search PubMed.
- A. T. Kreipl, C. Reid and W. Steglich, Org. Lett., 2002, 4, 3287–3288 CrossRef CAS PubMed.
- S. Lai, Y. Shizuri, S. Yamamura, K. Kawai, Y. Terada and H. Furukawa, Tetrahedron Lett., 1989, 30, 2241–2244 CrossRef CAS.
- R. A. Edrada, M. Heubes, G. Brauers, V. Wray, A. Berg, U. Gräfe, M. Wohlfarth, J. Mühlbacher, K. Schaumann, G. Bringmann and P Proksch, J. Nat. Prod., 2002, 65, 1598–1604 CrossRef CAS.
- M. V. R. Reddy, A. J. Yucel and P. V. Ramachandran, J. Org. Chem., 2001, 66, 2512–2514 CrossRef CAS PubMed.
- J. Yadav, N. Thrimurtulu, K. U. Gayathri, B. S. Reddy and A. Prasad, Synlett, 2009, 2009, 790–792 CrossRef.
- V. Bernan, M. Greenstein and W. Maiese, in Advances in applied microbiology, Elsevier, 1997, vol. 43, pp. 57–90 Search PubMed.
- K. Rajesh, V. Suresh, J. J. P. Selvam, C. Bhujanga Rao and Y. Venkateswarlu, Helv. Chim. Acta, 2009, 92, 1866–1872 CrossRef CAS.
- S. Lai, Y. Shizuri, S. Yamamura, K. Kawai and H. Furukawa, Bull. Chem. Soc. Jpn., 1991, 64, 1048–1050 CrossRef CAS.
- J. He, E. K. Wijeratne, B. P. Bashyal, J. Zhan, C. J. Seliga, M. X. Liu, E. E. Pierson, L. S. Pierson, H. D. VanEtten and A. L. Gunatilaka, J. Nat. Prod., 2004, 67, 1985–1991 CrossRef CAS PubMed.
- K. Rajesh, V. Suresh, J. J. P. Selvam, D. C. Babu and Y. Venkateswarlu, Helv. Chim. Acta, 2010, 93, 147–152 CrossRef CAS.
- Y. Takaya and M. Niwa, Trends Heterocycl. Chem., 2001, 7, 41–54 Search PubMed.
- T. Shen, X.-N. Wang and H.-X. Lou, Nat. Prod. Rep., 2009, 26, 916–935 RSC.
- U. Samaraweera, S. Sotheeswaran and M. U. S. Sultanbawa, Phytochemistry, 1982, 21, 2585–2587 CrossRef CAS.
- H. Kurihara, J. Kawabata, S. Ichikawa, M. Mishima and J. Mizutani, Phytochemistry, 1991, 30, 649–653 CrossRef CAS.
- M. Ohyama, T. Tanaka, T. Ito, M. Iinuma, K. F. Bastow and K.-H. Lee, Bioorg. Med. Chem. Lett., 1999, 9, 3057–3060 CrossRef CAS PubMed.
- K.-S. Huang, M. Lin and G.-F. Cheng, Phytochemistry, 2001, 58, 357–362 CrossRef CAS PubMed.
- L. D. Juliawaty, E. H. Hakim, S. A. Achmad, Y. M. Syah, J. Latip and I. M. Said, Nat. Prod. Commun., 2009, 4, 947–950 CAS.
- H. M. Ge, W. H. Yang, Y. Shen, N. Jiang, Z. K. Guo, Q. Luo, Q. Xu, J. Ma and R. X. Tan, Chem.–Eur. J., 2010, 16, 6338–6345 CrossRef CAS PubMed.
- K. Kim and I. Kim, Org. Lett., 2010, 12, 5314–5317 CrossRef CAS PubMed.
- T.-L. Ho, Tandem organic reactions, John Wiley & Sons, 1992 Search PubMed.
- L. F. Tietze, G. Brasche and K. Gericke, Domino reactions in organic synthesis, John Wiley & Sons, 2006 Search PubMed.
- L. F. Tietze and U. Beifuss, Angew. Chem., Int. Ed. Engl., 1993, 32, 131–163 CrossRef.
- I. Kim, S. G. Kim, J. Choi and G. H. Lee, Tetrahedron, 2008, 64, 664–671 CrossRef CAS.
- S. J. Gould, Chem. Rev., 1997, 97, 2499–2510 CrossRef CAS PubMed.
- J. Marco-Contelles and M. T. Molina, Curr. Org. Chem., 2003, 7, 1433–1442 CrossRef CAS.
- S. Ito, T. Matsuya, S. Omura, M. Otani, A. Nakagawa, H. Takeshima, Y. Iwai, M. Ohtani and T. Hata, J. Antibiot., 1970, 23, 315–317 CrossRef CAS PubMed.
- T. Hata, S. Omura, Y. Iwai, A. Nakagawa, M. Otani, S. Ito and T. Matsuya, J. Antibiot., 1971, 24, 353–359 CrossRef CAS PubMed.
- S. J. Gould, N. Tamayo, C. R. Melville and M. C. Cone, J. Am. Chem. Soc., 1994, 116, 2207–2208 CrossRef CAS.
- S. J. Gould, J. Chen, M. C. Cone, M. P. Gore, C. R. Melville and N. Tamayo, J. Org. Chem., 1996, 61, 5720–5721 CrossRef CAS.
- P. J. Proteau, Y. Li, J. Chen, R. T. Williamson, S. J. Gould, R. S. Laufer and G. I. Dmitrienko, J. Am. Chem. Soc., 2000, 122, 8325–8326 CrossRef CAS.
- X. Lei and J. A. Porco, J. Am. Chem. Soc., 2006, 128, 14790–14791 CrossRef CAS PubMed.
- H. Ishii, I.-S. Chen, S. Ueki, T. Masuda, K. Morita and T. Ishikawa, J. Chem. Soc., Perkin Trans. 1, 1987, 2415–2420 RSC.
- S. Kimura, S. Kobayashi, T. Kumamoto, A. Akagi, N. Sato and T. Ishikawa, Helv. Chim. Acta, 2011, 94, 578–591 CrossRef CAS.
- H.-C. Lin, S.-C. Chang, N.-L. Wang and L.-R. Ceng, J. Antibiot., 1994, 47, 675–680 CrossRef CAS PubMed.
- S. S. Scully and J. A. Porco Jr, Angew. Chem., Int. Ed., 2011, 50, 9722–9726 CrossRef CAS PubMed.
- R. J. Capon, J. K. Macleod and P. J. Scammells, Tetrahedron, 1986, 42, 6545–6550 CrossRef CAS.
- W. Liu, H. J. Lim and T. RajanBabu, J. Am. Chem. Soc., 2012, 134, 5496–5499 CrossRef CAS PubMed.
- Z. Jin, Nat. Prod. Rep., 2011, 28, 1126–1142 RSC.
- A. Kornienko and A. Evidente, Chem. Rev., 2008, 108, 1982–2014 CrossRef CAS PubMed.
- G. R. Pettit, G. M. Cragg, S. B. Singh, J. A. Duke and D. L. Doubek, J. Nat. Prod., 1990, 53, 176–178 CrossRef CAS.
- S. Hwang, D. Kim and S. Kim, Chem.–Eur. J., 2012, 18, 9977–9982 CrossRef CAS PubMed.
- H. G. Capraro, A. Brossi, A. Tropolonic colchicum alkaloids, in The Alkaloids: Chemistry and Pharmacology, Academic Press, 1984, vol. 23, pp. 1–70 Search PubMed.
- T. Graening and H. G. Schmalz, Angew. Chem., 2004, 116, 3292–3318 CrossRef.
- K. W. Wood, W. D. Cornwell and J. R. Jackson, Curr. Opin. Pharmacol., 2001, 1, 370–377 CrossRef CAS PubMed.
- O. Boyé and A. Brossi, in The Alkaloids: Chemistry and Pharmacology, Elsevier, 1992, vol. 41, pp. 125–176 Search PubMed.
- G. C. Tron, T. Pirali, G. Sorba, F. Pagliai, S. Busacca and A. A. Genazzani, J. Med. Chem., 2006, 49, 3033–3044 CrossRef CAS PubMed.
- C. Rappl, P. Barbier, V. Bourgarel-Rey, C. Grégoire, R. Gilli, M. Carre, S. Combes, J.-P. Finet and V. Peyrot, Biochemistry, 2006, 45, 9210–9218 CrossRef CAS PubMed.
- D. G. I. Kingston, J. Nat. Prod., 2009, 72, 507–515 CrossRef CAS PubMed.
- O. G. Ganina, E. Daras, V. Bourgarel-Rey, V. Peyrot, A. N. Andresyuk, J.-P. Finet, A. Y. Fedorov, I. P. Beletskaya and S. Combes, Bioorg. Med. Chem., 2008, 16, 8806–8812 CrossRef CAS PubMed.
- N. Sitnikov, J. Velder, L. Abodo, N. Cuvelier, J. Neudörfl, A. Prokop, G. Krause, A. Y. Fedorov and H. G. Schmalz, Chem.–Eur. J., 2012, 18, 12096–12102 CrossRef CAS PubMed.
- R. Ratnayake, E. Lacey, S. Tennant, J. H. Gill and R. J. Capon, Chem.–Eur. J., 2007, 13, 1610–1619 CrossRef CAS PubMed.
- K. Oda, N. Nishizono and Y. Tamai, Heterocycles, 2005, 65, 1985–1988 CrossRef CAS.
- D. K. Winter, M. A. Endoma-Arias, T. Hudlicky, J. A. Beutler and J. A. Porco Jr, J. Org. Chem., 2013, 78, 7617–7626 CrossRef CAS PubMed.
- T. Heckrodt and J. Mulzer, Natural Product Synthesis II: Targets, Methods, Concepts, Springer, 2005 Search PubMed.
- C. C. Hughes, A. K. Miller and D. Trauner, Org. Lett., 2005, 7, 3425–3428 CrossRef CAS PubMed.
- A. K. Miller, C. C. Hughes, J. J. Kennedy-Smith, S. N. Gradl and D. Trauner, J. Am. Chem. Soc., 2006, 128, 17057–17062 CrossRef CAS PubMed.
- P. A. Roethle, P. T. Hernandez and D. Trauner, Org. Lett., 2006, 8, 5901–5904 CrossRef CAS PubMed.
- A. D. Rodríguez, C. Ramírez and I. I. Rodríguez, Tetrahedron Lett., 1999, 40, 7627–7631 CrossRef.
- I. T. Chen, I. Baitinger, L. Schreyer and D. Trauner, Org. Lett., 2013, 16, 166–169 CrossRef PubMed.
- Q. Zhang, Y.-T. Di, C.-S. Li, X. Fang, C.-J. Tan, Z. Zhang, Y. Zhang, H.-P. He, S.-L. Li and X.-J. Hao, Org. Lett., 2009, 11, 2357–2359 CrossRef CAS PubMed.
- H. Morita and J. i. Kobayashi, Org. Lett., 2003, 5, 2895–2898 CrossRef CAS PubMed.
- R. Yamada, Y. Adachi, S. Yokoshima and T. Fukuyama, Angew. Chem., 2016, 128, 6171–6174 CrossRef.
- Q. Liang, Q. Wu, J. Jiang, J. a. Duan, C. Wang, M. D. Smith, H. Lu, Q. Wang, P. Nagarkatti and D. Fan, J. Biol. Chem., 2011, 286, 26470–26479 CrossRef CAS PubMed.
- M. Wang, L. Xiu, J. Diao, L. Wei and J. Sun, Eur. J. Pharmacol., 2015, 769, 79–85 CrossRef CAS PubMed.
- A. Oleinik and E. Adamskaya, Chem. Heterocycl. Compd., 1983, 19, 1221–1224 CrossRef.
- S. Nishigaki, Y. Shibata and K. Tanaka, J. Org. Chem., 2017, 82, 11117–11125 CrossRef CAS PubMed.
- A. Stoll, J. Renz and A. Von Wartburg, Helv. Chim. Acta, 1954, 37, 1747–1762 CrossRef CAS.
- A. Von Wartburg, E. Angliker and J. Renz, Helv. Chim. Acta, 1957, 40, 1331–1357 CrossRef.
- W. J. Gensler, C. M. Samour, S. Y. Wang and F. Johnson, J. Am. Chem. Soc., 1960, 82, 1714–1727 CrossRef CAS.
- P. M. Dewick, The Flavonoids: Advances in Research, ed. J. B. Harborne and T. J. Mabry, Chapman and Hall, London, 1982, p. 535 Search PubMed.
- A. Jain, A. Kumar and A. Kohli, J. Sci. Ind. Res., 1978, 37, 606–621 CAS.
- M. Darbarwar, V. Sundaramurthy and N. S. Rao, J. Sci. Ind. Res., 1976, 35, 297–312 CAS.
- O. Stamm, H. Schmid and J. Büchi, Helv. Chim. Acta, 1958, 41, 2006–2021 CrossRef CAS.
- A. Pelter and S. Foot, Synthesis, 1976, 1976, 326 CrossRef.
- R. J. Bass, J. Chem. Soc., Chem. Commun., 1976, 78–79 RSC.
- P. F. Schuda and W. A. Price, J. Org. Chem., 1987, 52, 1972–1979 CrossRef CAS.
- T. Easterfield and J. McDowell, Trans. R. Soc. N. Z., 1915, 48, 518–520 Search PubMed.
- L. Mangoni and R. Caputo, Tetrahedron Lett., 1967, 8, 673–675 CrossRef.
- S. Das, S. Bhattachryya and D. Mukherjee, Tetrahedron, 1992, 48, 9101–9110 CrossRef CAS.
- C. Giordano, M. Villa and R. Annunziata, Synth. Commun., 1990, 20, 383–392 CrossRef CAS.
- H. Minlon, J. Am. Chem. Soc., 1946, 68, 2487–2488 CrossRef.
- M. Ichimura, K. Muroi, K. Asano, I. Kawamoto, F. Tomita, M. Morimoto and H. Nakano, J. Antibiot., 1988, 41, 1285–1288 CrossRef CAS PubMed.
- T. Yasuzawa, T. Iida, K. i. Muroi, M. Ichimura, K. Takahashi and H. Sano, Chem. Pharm. Bull., 1988, 36, 3728–3731 CrossRef CAS PubMed.
- I. Takahashi, K.-I. Takahashi, M. Ichimura, M. Morimoto, K. Asano, I. Kawamoto, F. Tomita and H. Nakano, J. Antibiot., 1988, 41, 1915–1917 CrossRef CAS PubMed.
- Y. Fukuda, Y. Itoh, K. Nakatani and T. Shiro, Tetrahedron, 1994, 50, 2793–2808 CrossRef CAS.
- J. i. Kobayashi, C.-m. Zeng, M. Ishibashi, H. Shigemori, T. Sasaki and Y. Mikami, J. Chem. Soc., Perkin Trans. 1, 1992, 1291–1294 RSC.
- C. Galli, Synthesis, 1979, 1979, 303–304 CrossRef.
- F. Bracher and T. Papke, Nat. Prod. Lett., 1994, 4, 223–226 CrossRef CAS.
- U. Gräfe, W. Ihn, D. Tresselt, N. Miosga, U. Kaden, B. Schlegel, E.-J. Bormann, P. Sedmera and J. Novák, Biol. Met., 1990, 3, 39–44 CrossRef.
- W. Werner, U. Gräfe, W. Ihn, D. Tresselt, S. Winter and E. Paulus, Tetrahedron, 1997, 53, 109–118 CrossRef CAS.
- Y. Kashman, K. R. Gustafson, R. Fuller, J. McMahon, M. Currens, J. R. Buckheit, S. Hughes, G. Cragg and M. Boyd, J. Med. Chem., 1992, 35, 2735–2743 CrossRef CAS PubMed.
- J. Zhang, E. W. Kirchhoff, D. E. Zembower, N. Jimenez, P. Sen, Z.-Q. Xu and M. T. Flavin, Org. Process Res. Dev., 2000, 4, 577–580 CrossRef CAS.
- N. Chikao, E. Nobuyasu, T. Shinkichi, M. Akihisa, K. Koji and F. Masako, Biol. Chem., 1987, 51, 139 Search PubMed.
- M. Joyeux, A. Lobstein, R. Anton and F. Mortier, Planta Med., 1995, 61, 126–129 CrossRef CAS PubMed.
- M. R. Fesen, K. W. Kohn, F. Leteurtre and Y. Pommier, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 2399–2403 CrossRef CAS.
- Y. H. Kuo and M. H. Yeh, J. Chin. Chem. Soc., 1997, 44, 379–383 CrossRef CAS.
- T.-C. Lin, J.-M. Fang and Y.-S. Cheng, Phytochemistry, 1999, 51, 793–801 CrossRef CAS.
- E. J. Park, Y. Kim and J. Kim, J. Nat. Prod., 2000, 63, 34–36 CrossRef CAS.
- J. Hempel and H. Böhm, J. Agric. Food Chem., 1996, 44, 2114–2116 CrossRef CAS.
- T. Kartnig, A. Gruber and J. Stachel, Planta Med., 1985, 51, 288 CrossRef.
- F. Kader, B. Rovel, M. Girardin and M. Metche, Food Chem., 1996, 55, 35–40 CrossRef CAS.
- A. Freiburghaus, H. Ha, J. Chen, P. Leuenberger and F. Follath, Eur. J. Clin. Pharmacol., 1995, 48, 367–371 CrossRef.
- M. G. Hertog, P. C. Hollman and B. Van de Putte, J. Agric. Food Chem., 1993, 41, 1242–1246 CrossRef CAS.
- F. Ferreres, F. A. Tomáas-Barberáan, M. I. Gil and F. Tomáas-Lorente, J. Sci. Food Agric., 1991, 56, 49–56 CrossRef CAS.
- Y. J. Lee and T. D. Wu, J. Chin. Chem. Soc., 2001, 48, 201–206 CrossRef CAS.
- G. Bringmann, D. Menche, M. Bezabih, B. M. Abegaz and R. Kaminsky, Planta Med., 1999, 65, 757–758 CrossRef CAS PubMed.
- E. Dagne and W. Steglich, Phytochemistry, 1984, 23, 1729–1731 CrossRef CAS.
- G. Bringmann, D. Menche, J. Kraus, J. Mühlbacher, K. Peters, E.-M. Peters, R. Brun, M. Bezabih and B. M. Abegaz, J. Org. Chem., 2002, 67, 5595–5610 CrossRef CAS PubMed.
- D. C. Harrowven and R. F. Dainty, Tetrahedron Lett., 1996, 37, 7659–7660 CrossRef CAS.
- A. Rudi, T. Evan, M. Aknin and Y. Kashman, J. Nat. Prod., 2000, 63, 832–833 CrossRef CAS.
- S. Loya, A. Rudi, Y. Kashman and A. Hizi, Biochem. J., 1999, 344, 85 CrossRef CAS PubMed.
- P. Kleinwaechter, B. Schlegel, I. Groth, A. Haertl and U. Graefe, J. Antibiot., 2001, 54, 510–512 CrossRef.
- T. Komoda, Y. Shinoda and S.-i. Nakatsuka, Biosci., Biotechnol., Biochem., 2003, 67, 659–662 CrossRef CAS PubMed.
- S.-i. Nakatsuka, K. Teranishi and T. Goto, Tetrahedron Lett., 1994, 35, 2699–2700 CrossRef CAS.
- W.-H. Lin, J.-M. Fang and Y.-S. Cheng, Phytochemistry, 1996, 42, 1657–1663 CrossRef CAS.
- S. Tang, Y. Xu, J. He, Y. He, J. Zheng, X. Pan and X. She, Org. Lett., 2008, 10, 1855–1858 CrossRef CAS PubMed.
- R. M. McFadden and B. M. Stoltz, J. Am. Chem. Soc., 2006, 128, 7738–7739 CrossRef CAS PubMed.
- A. R. Saltiel and C. R. Kahn, Nature, 2001, 414, 799 CrossRef CAS PubMed.
- M. Harig, B. Neumann, H. G. Stammler and D. Kuck, Eur. J. Org. Chem., 2004, 2004, 2381–2397 CrossRef.
- J. Li, S. J. Guo, H. Su, L. J. Han and D. Y. Shi, Chin. Chem. Lett., 2008, 19, 1290–1292 CrossRef CAS.
- M. A. Elban and S. M. Hecht, J. Org. Chem., 2008, 73, 785–793 CrossRef CAS PubMed.
- J. Savard and P. Brassard, Tetrahedron, 1984, 40, 3455–3464 CrossRef CAS.
- J. P. Michael, Nat. Prod. Rep., 2001, 18, 520–542 RSC.
- E. Gellert, J. Nat. Prod., 1982, 45, 50–73 CrossRef CAS.
- A. G. Damu, P.-C. Kuo, L.-S. Shi, C.-Y. Li, C.-S. Kuoh, P.-L. Wu and T.-S. Wu, J. Nat. Prod., 2005, 68, 1071–1075 CrossRef CAS PubMed.
- P.-L. Wu, K. Rao, C.-H. Su, C.-S. Kuoh and T.-S. Wu, Heterocycles, 2002, 57, 2401–2408 CrossRef CAS.
- R. S. Gupta and L. Siminovitch, Biochemistry, 1977, 16, 3209–3214 CrossRef CAS PubMed.
- E. Gellert and R. Rudzats, J. Med. Chem., 1964, 7, 361–362 CrossRef CAS PubMed.
- T.-y. An, R.-q. Huang, Z. Yang, D.-k. Zhang, G.-r. Li, Y.-C. Yao and J. Gao, Phytochemistry, 2001, 58, 1267–1269 CrossRef CAS PubMed.
- Z.-Q. Huang, Y.-X. Liu, Z.-J. Fan, Q.-M. Wang, G.-R. Li, Y.-C. Yao, X.-S. Yu and R.-Q. Huang, Fine Chem. Intermed., 2007, 37, 20–24 CAS.
- K.-L. Wang, M.-Y. Lü, Q.-M. Wang and R.-Q. Huang, Tetrahedron, 2008, 64, 7504–7510 CrossRef CAS.
- B. S. Davidson, Chem. Rev., 1993, 93, 1771–1791 CrossRef CAS.
- R. J. Andersen, D. J. Faulkner, C. H. He, G. D. Van Duyne and J. Clardy, J. Am. Chem. Soc., 1985, 107, 5492–5495 CrossRef CAS.
- R. A. Davis, A. R. Carroll, G. K. Pierens and R. J. Quinn, J. Nat. Prod., 1999, 62, 419–424 CrossRef CAS PubMed.
- A. Carroll, B. Bowden and J. Coll, Aust. J. Chem., 1993, 46, 489–501 CrossRef CAS.
- J. S. Yadav, K. U. Gayathri, B. V. S. Reddy and A. R. Prasad, Synlett, 2009, 2009, 43–46 CrossRef.
- G. Baddeley, S. Makar and M. Ivinson, J. Chem. Soc., 1953, 3969–3971 RSC.
- K. Winkelmann, J. Heilmann, O. Zerbe, T. Rali and O. Sticher, J. Nat. Prod., 2000, 63, 104–108 CrossRef CAS.
- K. Winkelmann, J. Heilmann, O. Zerbe, T. Rali and O. Sticher, Helv. Chim. Acta, 2001, 84, 3380–3392 CrossRef CAS.
- N. S. Simpkins and M. D. Weller, Tetrahedron Lett., 2010, 51, 4823–4826 CrossRef CAS.
- N. Matsumoto, T. Tsuchida, M. Maruyama, R. Sawa, N. Kinoshita, Y. Homma, Y. Takahashi, H. Iinuma, H. Naganawa and T. Sawa, J. Antibiot., 1996, 49, 953–954 CrossRef CAS PubMed.
- N. Matsumoto, T. Tsuchida, M. Maruyama, N. Kinoshita, Y. Homma, H. Iinuma, T. Sawa, M. Hamada, T. Takeuchi and N. Heida, J. Antibiot., 1999, 52, 269–275 CrossRef CAS PubMed.
- K. Tatsuta, H. Tanaka, H. Tsukagoshi, T. Kashima and S. Hosokawa, Tetrahedron Lett., 2010, 51, 5546–5549 CrossRef CAS.
- M. E. Jung and J. A. Hagenah, J. Org. Chem., 1987, 52, 1889–1902 CrossRef CAS.
- A. D. Patten, N. N. Huy and S. J. Danishefsky, J. Org. Chem., 1988, 53, 1003–1007 CrossRef CAS.
- Y. Kashman, A. Rotstein and A. Lifshitz, Tetrahedron, 1974, 30, 991–997 CrossRef CAS.
- R. Mani and K. Venkataraman, Curr. Sci., 1954, 23, 220–221 CAS.
- N. Gokan, H. Kikuchi, K. Nakamura, Y. Oshima, K. Hosaka and Y. Kubohara, Biochem. Pharmacol., 2005, 70, 676–685 CrossRef CAS PubMed.
- W. Crow, T. Osawa, K. Platz and D. Sutherland, Aust. J. Chem., 1976, 29, 2525–2531 CrossRef CAS.
- M. Bolte, W. Crow and S. Yoshida, Aust. J. Chem., 1982, 35, 1411–1419 CrossRef CAS.
- H. Müller, M. Paul, D. Hartmann, V. Huch, D. Blaesius, A. Koeberle, O. Werz and J. Jauch, Angew. Chem., Int. Ed., 2010, 49, 2045–2049 CrossRef PubMed.
- J. P. Michael, Nat. Prod. Rep., 2005, 22, 603–626 RSC.
- S. K. Lee, K.-A. Nam and Y.-H. Heo, Planta Med., 2003, 69, 21–25 CrossRef CAS PubMed.
- D. Stærk, J. Christensen, E. Lemmich, J. Ø. Duus, C. E. Olsen and J. W. Jaroszewski, J. Nat. Prod., 2000, 63, 1584–1586 CrossRef.
- D. Stærk, A. K. Lykkeberg, J. Christensen, B. A. Budnik, F. Abe and J. W. Jaroszewski, J. Nat. Prod., 2002, 65, 1299–1302 CrossRef.
- L. M. Ambrosini, T. A. Cernak and T. H. Lambert, Tetrahedron, 2010, 66, 4882–4887 CrossRef CAS.
- S. F. Brady, M. M. Wagenaar, M. P. Singh, J. E. Janso and J. Clardy, Org. Lett., 2000, 2, 4043–4046 CrossRef CAS PubMed.
- J. D. Hall, N. W. Duncan-Gould, N. A. Siddiqi, J. N. Kelly, L. A. Hoeferlin, S. J. Morrison and J. K. Wyatt, Bioorg. Med. Chem., 2005, 13, 1409–1413 CrossRef CAS PubMed.
- T. Ohzeki and K. Mori, Biosci., Biotechnol., Biochem., 2003, 67, 2584–2590 CrossRef CAS PubMed.
- Y. Zhan, X. Du, H. Chen, J. Liu, B. Zhao, D. Huang, G. Li, Q. Xu, M. Zhang and B. C. Weimer, Nat. Chem. Biol., 2008, 4, 548 CrossRef CAS PubMed.
- X. H. Cheng and C. J. Fu, Chin. J. Chem., 2007, 25, 1762–1765 CrossRef CAS.
- F. Wang, Y. J. Zhang, H. Wei, J. Zhang and W. Zhang, Tetrahedron Lett., 2007, 48, 4083–4086 CrossRef CAS.
- Y. Kan, T. Fujita, H. Nagai, B. Sakamoto and Y. Hokama, J. Nat. Prod., 1998, 61, 152–155 CrossRef CAS PubMed.
- J. Chen, Z.-F. Shi, L. Zhou, A.-L. Xie and X.-P. Cao, Tetrahedron, 2010, 66, 3499–3507 CrossRef CAS.
- T. Toyoda, K. Sasakura and T. Sugasawa, J. Org. Chem., 1981, 46, 189–191 CrossRef CAS.
- L. Xu, J. Jin, M. Lal, P. Daublain and M. Newcomb, Org. Lett., 2007, 9, 1837–1840 CrossRef CAS PubMed.
- R. Ciochina and R. B. Grossman, Chem. Rev., 2006, 106, 3963–3986 CrossRef CAS PubMed.
- C. C. Hughes, A. Prieto-Davo, P. R. Jensen and W. Fenical, Org. Lett., 2008, 10, 629–631 CrossRef CAS PubMed.
- K. Nicolaou, N. L. Simmons, J. S. Chen, N. M. Haste and V. Nizet, Tetrahedron Lett., 2011, 52, 2041–2043 CrossRef CAS PubMed.
- R. H. Furneaux and P. C. Tyler, J. Org. Chem., 1999, 64, 8411–8412 CrossRef CAS PubMed.
- R.-J. Lin, M.-J. Cheng, J.-C. Huang, W.-L. Lo, Y.-T. Yeh, C.-M. Yen, C.-M. Lu and C.-Y. Chen, J. Nat. Prod., 2009, 72, 1816–1824 CrossRef CAS PubMed.
- C. Tang, Z. Li, Y. Wang, J. Xu, L. Kong, H. Yao and X. Wu, Tetrahedron Lett., 2011, 52, 3275–3278 CrossRef CAS.
- K. Shin-ya, K. Wierzba, K.-i. Matsuo, T. Ohtani, Y. Yamada, K. Furihata, Y. Hayakawa and H. Seto, J. Am. Chem. Soc., 2001, 123, 1262–1263 CrossRef CAS PubMed.
- N. S. Girgis, H. B. Cottam and R. K. Robins, J. Heterocycl. Chem., 1988, 25, 361–366 CrossRef CAS.
- T. Sasaki, A. Nakanishi and M. Ohno, J. Org. Chem., 1982, 47, 3219–3224 CrossRef CAS.
- H. Tokuyama, K. Okano, H. Fujiwara, T. Noji and T. Fukuyama, Chem.–Asian J., 2011, 6, 560–572 CrossRef CAS PubMed.
- D. H. Miles, D. S. Lho, A. A. De la Cruz, E. D. Gomez, J. A. Weeks and J. L. Atwood, J. Org. Chem., 1987, 52, 2930–2932 CrossRef CAS.
- D. H. Miles, A.-M. Ly, V. Chittawong, A. A. de la Cruz and E. D. Gomez, J. Nat. Prod., 1989, 52, 896–898 CrossRef CAS.
- S. P. Chavan, S. Garai and U. R. Kalkote, Tetrahedron, 2012, 68, 8509–8514 CrossRef CAS.
- Y.-C. Hu, X.-F. Wu, S. Gao, S.-S. Yu, Y. Liu, J. Qu, J. Liu and Y.-B. Liu, Org. Lett., 2006, 8, 2269–2272 CrossRef CAS PubMed.
- X.-F. Wu, Y.-C. Hu, S. Gao, S.-S. Yu, Y.-H. Pei, W.-Z. Tang and X.-Z. Huang, J. Asian Nat. Prod. Res., 2007, 9, 471–477 CrossRef CAS PubMed.
- X.-F. Wu, Y.-C. Hu, S.-S. Yu, N. Jiang, J. Ma, R.-X. Tan, Y. Li, H.-N. Lv, J. Liu and S.-G. Ma, Org. Lett., 2010, 12, 2390–2393 CrossRef CAS PubMed.
- Y. Ogura, K. Ishigami and H. Watanabe, Tetrahedron, 2012, 68, 1723–1728 CrossRef CAS.
- C. Dufour, J. Wink, M. Kurz, H. Kogler, H. Olivan, S. Sablé, W. Heyse, M. Gerlitz, L. Toti and A. Nußer, Chem.–Eur. J., 2012, 18, 16123–16128 CrossRef CAS PubMed.
- C. Couturier, A. Bauer, A. Rey, C. Schroif-Dufour and M. Broenstrup, Bioorg. Med. Chem. Lett., 2012, 22, 6292–6296 CrossRef CAS PubMed.
- T. Zhao, G. R. Yan, S. L. Pan, H. Y. Wang and A. J. Hou, Chem. Biodiversity, 2009, 6, 2209–2216 CrossRef CAS PubMed.
- E. T. Arung, K. Shimizu and R. Kondo, Chem. Biodiversity, 2007, 4, 2166–2171 CrossRef CAS PubMed.
- A.-R. Han, Y.-J. Kang, T. Windono, S. K. Lee and E.-K. Seo, J. Nat. Prod., 2006, 69, 719–721 CrossRef CAS PubMed.
- E. T. Arung, K. Shimizu and R. Kondo, Planta Med., 2006, 72, 847–850 CrossRef CAS PubMed.
- W.-J. Zhang, J.-F. Wu, P.-F. Zhou, Y. Wang and A.-J. Hou, Tetrahedron, 2013, 69, 5850–5858 CrossRef CAS.
- F. Fullas, L. J. Kornberg, M. C. Wani, M. E. Wall, N. R. Farnsworth, T. E. Chagwedera and A. D. Kinghorn, J. Nat. Prod., 1996, 59, 190–192 CrossRef CAS PubMed.
- C. Selenski and T. R. Pettus, J. Org. Chem., 2004, 69, 9196–9203 CrossRef CAS PubMed.
- I. Slabu, S. B. Rossington, P. M. Killoran, N. Hirst and J. A. Wilkinson, Tetrahedron Lett., 2013, 54, 1489–1490 CrossRef CAS.
- G. A. Mohamed and S. R. Ibrahim, ARKIVOC, 2007, 15, 281–291 Search PubMed.
- M. I. Choudhary, N. Khan, M. Ahmad, S. Yousuf, H.-K. Fun, S. Soomro, M. Asif, M. A. Mesaik and F. Shaheen, Org. Lett., 2013, 15, 1862–1865 CrossRef CAS PubMed.
- A. Hiranrat and W. Mahabusarakam, Tetrahedron, 2008, 64, 11193–11197 CrossRef CAS.
- M. Morkunas, L. Dube and M. E. Maier, Tetrahedron, 2013, 69, 8559–8563 CrossRef CAS.
- P. Kalsi, J. Singh, W. Crow and B. Chhabra, Phytochemistry, 1987, 26, 3367–3369 CrossRef CAS.
- H. Yeo, J. H. Lee and J. Kim, Arch. Pharmacal Res., 1999, 22, 306 CrossRef CAS.
- Y. Tezuka, M. Terazono, T. Kusumoto, I. Tomoco, Y. Hatanaka, S. Kadota, M. Hattori and O. Namba, Helv. Chim. Acta, 2000, 83, 29 CrossRef.
- T. Hayashi and R. H. Thomson, Phytochemistry, 1975, 14, 1085–1087 CrossRef CAS.
- S. Apers, A. Vlietinck and L. Pieters, Phytochem. Rev., 2003, 2, 201–217 CrossRef CAS.
- P. S. Luize, T. Ueda-Nakamura, B. P. Dias Filho, D. A. G. Cortez and C. V. Nakamura, Biol. Pharm. Bull., 2006, 29, 2126–2130 CrossRef CAS.
- L. Baumgartner, S. Sosa, A. G. Atanasov, A. Bodensieck, N. Fakhrudin, J. Bauer, G. D. Favero, C. Ponti, E. H. Heiss and S. Schwaiger, J. Nat. Prod., 2011, 74, 1779–1786 CrossRef CAS PubMed.
- C.-y. Chen and M. Weisel, Synlett, 2013, 24, 189–192 CrossRef CAS.
- X. Zhang, X. Jiang, C. Ding, Q. Yao and A. Zhang, Org. Biomol. Chem., 2013, 11, 1383–1389 RSC.
- F. Zhu and Y. Lin, Chin. Sci. Bull., 2006, 51, 1426 CAS.
- C.-L. Feng, S.-G. Zhang, J.-Q. Chen, J. Cai and M. Ji, Chin. Chem. Lett., 2013, 24, 767–769 CrossRef CAS.
- J. S. Yadav, B. V. S. Reddy, G. Kondaji, R. Srinivasa Rao and S. Praveen Kumar, Tetrahedron Lett., 2002, 43, 8133–8135 CrossRef CAS.
- A. Merz, R. Schropp and E. Dötterl, Synthesis, 1995, 1995, 795–800 CrossRef.
- R. D. Stipanovic, J. Zhang, B. D. Bruton and M. H. Wheeler, J. Agric. Food Chem., 2004, 52, 4109–4112 CrossRef CAS PubMed.
- K. A. Punch and M. J. Piggott, Org. Biomol. Chem., 2014, 12, 2801–2810 RSC.
- B. J. Albert and K. Koide, J. Org. Chem., 2008, 73, 1093–1098 CrossRef CAS PubMed.
- G. Pickaert, M. Cesario and R. Ziessel, J. Org. Chem., 2004, 69, 5335–5341 CrossRef CAS PubMed.
- M. J. Piggott and D. Wege, Aust. J. Chem., 2003, 56, 691–702 CrossRef CAS.
- H. Fujimoto, H. Okuyama, Y. Motohashi, E. Yoshida and M. Yamazaki, JSM Mycotoxins, 1995, 1995, 61–66 CrossRef.
- J. Wang, S. M. Soisson, K. Young, W. Shoop, S. Kodali, A. Galgoci, R. Painter, G. Parthasarathy, Y. S. Tang and R. Cummings, Nature, 2006, 441, 358 CrossRef CAS PubMed.
- G. Bartoli, M. Locatelli, P. Melchiorre and L. Sambri, Eur. J. Org. Chem., 2007, 2007, 2037–2049 CrossRef.
- S. T. C. Eey and M. J. Lear, Chem.–Eur. J., 2014, 20, 11556–11573 CrossRef CAS PubMed.
- S. Suryati, Phytochemicals and Anti-inflammatory Activity of Melicope ptelefolia Champ Ex, Benth Masters's Thesis, Universiti Putra Malaysia, Malaysia, 2005 Search PubMed.
- K. Shaari, S. Safri, F. Abas, N. H. Lajis and D. Israf, Nat. Prod. Res., 2006, 20, 415–419 CrossRef CAS PubMed.
- C. H. Ng, K. Rullah, M. F. F. M. Aluwi, F. Abas, K. W. Lam, I. S. Ismail, R. Narayanaswamy, F. Jamaludin and K. Shaari, Molecules, 2014, 19, 11645–11659 CrossRef PubMed.
- W. Reininger and A. Hartl, US Pat., 4053517, U.S. Patent and Trademark Office, Washington, DC, 1977.
- L. Kaysser, P. Bernhardt, S.-J. Nam, S. Loesgen, J. G. Ruby, P. Skewes-Cox, P. R. Jensen, W. Fenical and B. S. Moore, J. Am. Chem. Soc., 2012, 134, 11988–11991 CrossRef CAS PubMed.
- H. P. Pepper and J. H. George, Angew. Chem., Int. Ed., 2013, 52, 12170–12173 CrossRef CAS PubMed.
- R. Meier, S. Strych and D. Trauner, Org. Lett., 2014, 16, 2634–2637 CrossRef CAS PubMed.
- H. P. Pepper and J. H. George, Synlett, 2015, 26, 2485–2490 CrossRef CAS.
- J. Dai, K. Krohn, U. Flörke, G. Pescitelli, G. Kerti, T. Papp, K. E. Kövér, A. C. Bényei, S. Draeger and B. Schulz, Eur. J. Org. Chem., 2010, 2010, 6928–6937 CrossRef.
- R. W. Bates, K. Wang, G. Zhou and D. Z. Kang, Synlett, 2015, 26, 751–754 CrossRef CAS.
- I. P. Singh and S. B. Bharate, Nat. Prod. Rep., 2006, 23, 558–591 RSC.
- I. P. Singh, J. Sidana, S. B. Bharate and W. J. Foley, Nat. Prod. Rep., 2010, 27, 393–416 RSC.
- J. J. Brophy, D. C. Craig, R. J. Goldsack, C. J. Fookes, D. N. Leach and P. G. Waterman, Phytochemistry, 2006, 67, 2085–2089 CrossRef CAS PubMed.
- G. Wei and B. Yu, Eur. J. Org. Chem., 2008, 2008, 3156–3163 CrossRef.
- E. Nishimura, Y. Ohfune and T. Shinada, Tetrahedron Lett., 2015, 56, 539–541 CrossRef CAS.
- B. Wu, W. Zhang, Z. Li, L. Gu, X. Wang and P. G. Wang, J. Org. Chem., 2011, 76, 2265–2268 CrossRef CAS PubMed.
- A. Briot, C. Baehr, R. Brouillard, A. Wagner and C. Mioskowski, J. Org. Chem., 2004, 69, 1374–1377 CrossRef CAS PubMed.
- D. H. Silva, Y. Zhang, L. A. Santos, V. S. Bolzani and M. G. Nair, J. Agric. Food Chem., 2007, 55, 2569–2574 CrossRef CAS PubMed.
- P. W. Snijman, E. Joubert, D. Ferreira, X.-C. Li, Y. Ding, I. R. Green and W. C. Gelderblom, J. Agric. Food Chem., 2009, 57, 6678–6684 CrossRef CAS PubMed.
- J. Somsrisa, P. Meepowpan, S. Krachodnok, H. Thaisuchat, S. Punyanitya, N. Nantasaen and W. Pompimon, Molecules, 2013, 18, 6898–6907 CrossRef CAS PubMed.
- A. Hermoso, I. A. Jiménez, Z. A. Mamani, I. L. Bazzocchi, J. E. Piñero, A. G. Ravelo and B. Valladares, Bioorg. Med. Chem., 2003, 11, 3975–3980 CrossRef CAS PubMed.
- D. Ahmed, V. Kumar, M. Sharma and A. Verma, BMC Complementary Altern. Med., 2014, 14, 155 CrossRef PubMed.
- J. Zhang and Y. Qin, 2013 21st International Conference on Geoinformatics, 2013 Search PubMed.
- E. Lemmich, C. O. Adewunmi, P. Furu, A. Kristensen, L. Larsen and C. E. Olsen, Phytochemistry, 1996, 42, 1011–1013 CrossRef CAS.
- E. Szliszka, Z. P. Czuba, B. Mazur, A. Paradysz and W. Krol, Molecules, 2010, 15, 5336–5353 CrossRef CAS PubMed.
- J. Peng, A. L. Risinger, C. Da, G. A. Fest, G. E. Kellogg and S. L. Mooberry, J. Nat. Prod., 2013, 76, 2189–2194 CrossRef CAS PubMed.
- T. H. Sum, T. J. Sum, J. E. Stokes, W. R. Galloway and D. R. Spring, Tetrahedron, 2015, 71, 4557–4564 CrossRef CAS.
- A. T. Tran, N. P. West, W. J. Britton and R. J. Payne, ChemMedChem, 2012, 7, 1031–1043 CrossRef CAS PubMed.
- D. Maes, M. E. Riveiro, C. Shayo, C. Davio, S. Debenedetti and N. De Kimpe, Tetrahedron, 2008, 64, 4438–4443 CrossRef CAS.
- G. Keserü and M. Nógrádi, in Studies in Natural Products Chemistry, Elsevier, 1995, vol. 17, pp. 357–394 Search PubMed.
- P. Ruedi and M. Juch, Curr. Org. Chem., 1999, 3, 623–646 CrossRef CAS.
- C. Per, U. P. Claeson, P. Tuchinda and V. Reutrakul, in Studies in Natural Products Chemistry, Elsevier, 2002, vol. 26, pp. 881–908 Search PubMed.
- H. Lv and G. She, Nat. Prod. Commun., 2010, 5, 1687–1708 CAS.
- H. Lv and G. She, Rec. Nat. Prod., 2012, 6, 321–333 CAS.
- Y. Sun, K. Tabata, H. Matsubara, S. Kitanaka, T. Suzuki and K. Yasukawa, Planta Med., 2008, 74, 427–431 CrossRef CAS PubMed.
- H. Seçinti and H. Seçen, Helv. Chim. Acta, 2015, 98, 938–944 CrossRef.
- G. Höfle, B. Kunze, C. Zorzin and H. Reichenbach, Eur. J. Org. Chem., 1984, 1984, 1883–1904 Search PubMed.
- D. Enders and S. Osborne, J. Chem. Soc., Chem. Commun., 1993, 424–426 RSC.
- J. Yadav, G. Revathi and B. S. Reddy, Tetrahedron Lett., 2017, 58, 3943–3946 CrossRef CAS.
- T. Horie, H. Tominaga, Y. Kawamura, T. Hada, N. Ueda, Y. Amano and S. Yamamoto, J. Med. Chem., 1991, 34, 2169–2176 CrossRef CAS PubMed.
- B. D. Chapsal and I. Ojima, Org. Lett., 2006, 8, 1395–1398 CrossRef CAS PubMed.
- R. J. Huntley and R. L. Funk, Tetrahedron Lett., 2011, 52, 6671–6674 CrossRef CAS PubMed.
- K. Tomooka, M. Suzuki, K. Uehara, M. Shimada and T. Akiyama, Synlett, 2008, 2008, 2518–2522 CrossRef.
- B. N. Do Doan, X. Y. Tan, C. M. Ang and R. W. Bates, Synthesis, 2017, 49, 4711–4716 CrossRef.
- R. W. Rickards, J. M. Rothschild, A. C. Willis, N. M. de Chazal, J. Kirk, K. Kirk, K. J. Saliba and G. D. Smith, Tetrahedron, 1999, 55, 13513–13520 CrossRef CAS.
- N. T. Doan, P. R. Stewart and G. D. Smith, FEMS Microbiol. Lett., 2001, 196, 135–139 CrossRef CAS PubMed.
- R. AI and N. A. Bumagin, J. Organomet. Chem., 1998, 560, 163–167 CrossRef.
- B. M. Ramalingam and A. K. Mohanakrishnan, Tetrahedron Lett., 2017, 58, 2919–2922 CrossRef CAS.
- K. Ninomiya, K. Shibatani, M. Sueyoshi, S. Chaipech, Y. Pongpiriyadacha, T. Hayakawa, O. Muraoka and T. Morikawa, Chem. Pharm. Bull., 2016, 64, 880–885 CrossRef CAS PubMed.
- G. Tanabe, N. Tsutsui, K. Shibatani, S. Marumoto, F. Ishikawa, K. Ninomiya, O. Muraoka and T. Morikawa, Tetrahedron, 2017, 73, 4481–4486 CrossRef CAS.
- H. H. Kinfe, H. S. Long, M. A. Stander and B. E. Van Wyk, S. Afr. J. Bot., 2015, 100, 75–79 CrossRef CAS.
- Z. Dong, J. Guo, X. Xing, X. Zhang, Y. Du and Q. Lu, Biomed. Pharmacother., 2017, 89, 297–304 CrossRef CAS PubMed.
- K. Kawaguchi, S. de Mello Alves, T. Watanabe, S. Kikuchi, M. Satake and Y. Kumazawa, Planta Med., 1998, 64, 653–655 CrossRef CAS PubMed.
- Y. Tao, Y. Jiang, W. Li and B. Cai, Anal. Methods, 2016, 8, 4211–4219 RSC.
- S. Liu, C. Zhang, Q. Shi, G. Li, M. Song, Y. Gao, C. Xu, H. Xu, B. Fan, S. Yu, C. Zheng, Q. Zhu, B. Wu, L. Peng, H. Xiong, Q. Wu and S. Liang, Brain Res. Bull., 2014, 101, 57–63 CrossRef CAS PubMed.
- Y. Zou, S. Zhang, G. Wang, X. Wen, S. Liu, T. Peng, Y. Gao and L. Wang, J. Carbohydr. Chem., 2016, 35, 387–395 CrossRef CAS.
- J. F. W. McOmie and S. A. Saleh, Tetrahedron, 1973, 29, 4003–4005 CrossRef CAS.
- N. Öztaşkın, Y. Çetinkaya, P. Taslimi, S. Göksu and İ. Gülçin, Bioorg. Chem., 2015, 60, 49–57 CrossRef PubMed.
- D. Shi, J. Li, B. Jiang, S. Guo, H. Su and T. Wang, Bioorg. Med. Chem. Lett., 2012, 22, 2827–2832 CrossRef CAS PubMed.
- Y. Zou, S. Zhang, X. Wen, S. Liu, T. Peng, Y. Gao and L. Wang, Tetrahedron Lett., 2017, 58, 2835–2837 CrossRef CAS.
- M. P. Mattson, Nat. Rev. Mol. Cell Biol., 2000, 1, 120 CrossRef CAS PubMed.
- H. Zhai, M. Nakatsukasa, Y. Mitsumoto and Y. Fukuyama, Planta Med., 2004, 70, 598–602 CrossRef CAS PubMed.
- T. Esumi, D. Hojyo, H. Zhai and Y. Fukuyama, Tetrahedron Lett., 2006, 47, 3979–3983 CrossRef CAS.
- K. C. Guerard, C. Sabot, L. Racicot and S. Canesi, J. Org. Chem., 2009, 74, 2039–2045 CrossRef CAS PubMed.
- T. Konishi, T. Konoshima, A. Daikonya and S. Kitanaka, Biol. Pharm. Bull., 2005, 53, 121–124 CAS.
- S. Hanessian and G. J. Reddy, Synlett, 2007, 2007, 0475–0479 CrossRef.
- H. Zhai, T. Inoue, M. Moriyama, T. Esumi, Y. Mitsumoto and Y. Fukuyama, Biol. Pharm. Bull., 2005, 28, 289–293 CrossRef CAS.
- R.-L. Huang, C.-F. Chen, H.-Y. Feng, L.-C. Lin and C.-J. Chou, J. Chin. Clin. Med., 2001, 12, 179–192 Search PubMed.
- S. Aketani, K. Tanaka, K. Yamamoto, A. Ishihama, H. Cao, A. Tengeiji, S. Hiraoka, M. Shiro and M. Shionoya, J. Med. Chem., 2002, 45, 5594–5603 CrossRef CAS PubMed.
- P. Xue, L.-P. Wang, X.-Z. Jiao, Y.-J. Jiang, Q. Xiao, Z.-G. Luo, P. Xie and X.-T. Liang, J. Asian Nat. Prod. Res., 2009, 11, 281–287 CrossRef CAS PubMed.
- V. F. de Andrade-Neto, T. da Silva, L. M. X. Lopes, V. E. do Rosário, F. de Pilla Varotti and A. U. Krettli, Antimicrob. Agents Chemother., 2007, 51, 2346–2350 CrossRef PubMed.
- J. C. Reddel, K. E. Lutz, A. B. Diagne and R. J. Thomson, Angew. Chem., 2014, 126, 1419–1422 CrossRef.
- J. Kinjo, H. Uemura, T. Nohara, M. Yamashita, N. Marubayashi and K. Yoshihira, Tetrahedron Lett., 1995, 36, 5599–5602 CrossRef CAS.
- S. Strych, G. Journot, R. P. Pemberton, S. C. Wang, D. J. Tantillo and D. Trauner, Angew. Chem., Int. Ed., 2015, 54, 5079–5083 CrossRef CAS PubMed.
- N. J. Patron, R. F. Waller, A. J. Cozijnsen, D. C. Straney, D. M. Gardiner, W. C. Nierman and B. J. Howlett, BMC Evol. Biol., 2007, 7, 174 CrossRef PubMed.
- T. C. Adams, J. N. Payette, J. H. Cheah and M. Movassaghi, Org. Lett., 2015, 17, 4268–4271 CrossRef CAS PubMed.
- L. Ding, A. Maier, H.-H. Fiebig, W.-H. Lin and C. Hertweck, Org. Biomol. Chem., 2011, 9, 4029–4031 RSC.
- Y. Sun, Z. Meng, P. Chen, D. Zhang, M. Baunach, C. Hertweck and A. Li, Org. Chem. Front., 2016, 3, 368–374 RSC.
- Z. Liu, L. Liu, Z. Shafiq, D. Wang and Y.-J. Chen, Lett. Org. Chem., 2007, 4, 256–260 CrossRef CAS.
- X. Zhu and A. Ganesan, J. Org. Chem., 2002, 67, 2705–2708 CrossRef CAS PubMed.
- A. Bourderioux, A. Ouach, V. Beneteau, J.-Y. Mérour and S. Routier, Synthesis, 2010, 2010, 783–790 CrossRef.
| This journal is © The Royal Society of Chemistry 2018 |