
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A fluorescence switching sensor based on graphene quantum dots decorated with Hg2+ and hydrolyzed thioacetamide for highly Ag+-sensitive and selective detection
Pimpisa
Kaewanan
a,
Phitchan
Sricharoen
a,
Nunticha
Limchoowong
a,
Thitiya
Sripakdee
b,
Prawit
Nuengmatcha
c and
Saksit
Chanthai
 *a
*a
aMaterials Chemistry Research Center, Department of Chemistry, Center of Excellence for Innovation in Chemistry, Faculty of Science, Khon Kaen University, 123 Mittraphab Road, Khon Kaen 40002, Thailand. E-mail: sakcha2@kku.ac.th; Fax: +66 43202373; Tel: +66 43009700 ext. 42174-5
bChemistry Program, Faculty of Science and Technology, Sakon Nakhon Rajabhat University, Sakon Nakhon 47000, Thailand
cDepartment of Chemistry, Faculty of Science and Technology, Nakhon Si Thammarat Rajabhat University, Nakhon Si Thammarat 80280, Thailand
First published on 13th October 2017
Abstract
A selective fluorescent sensor based on graphene quantum dots (GQDs) was developed for the determination of silver ions (Ag+). The GQDs were prepared by the citric acid pyrolysis method. In the presence of mercury ions (Hg2+), the fluorescence intensity of the GQDs decreased linearly and it was fully recovered by the hydrolysis of thioacetamide (TAA), giving hydrogen sulfide in the reaction system. This research study was aimed at using the fluorescence turn-off sensor for the selective determination of Ag+. Upon the addition of Ag+, the fluorescence intensity of the generated sulfide-(Hg2+ quenched GQDs) decreased as a linear function of the Ag+ concentration. Then, the acquired GQDs showed steady, selective, and highly sensitive detection of Ag+. The experimental parameters affecting the fluorescence turn-on/off sensor were investigated and optimized. The optimum conditions included 4 μM Hg2+ concentration, 70 μM TAA concentration, solution pH of 7 and a 5 min reaction time. Under the optimized conditions, the working linear concentration range, limit of detection and limit of quantification for Ag+ were 0.5–10.0, 0.18 and 0.60 μM, respectively. The proposed method was successfully applied for the selective determination of trace amounts of Ag+ in five real water samples with satisfying levels of recovery (89.31–114.08%).
Introduction
Silver ions (Ag+) are included in the list of heavy metal ions, and their potential toxicity for the environment and human body has drawn people’s attention. Owing to their broad employment in many industries, such as electronics, photography, mirrors, and pharmacy, a large amount of silver is released into the environment annually from industrial waste and emissions, especially into sludge waste and even surface waters.1,2 It has also been found that silver is toxic to humans at concentrations as high as 0.9 μM in drinking water.3 Considering these facts, applying a simple, rapid and accurate method to monitor silver ions at trace levels in various samples is of great importance. Various techniques have been used for the determination of silver in real samples, such as the spectrophotometric method,4 graphite furnace atomic absorption spectroscopy (GFAAS),5 high-resolution continuum source flame atomic absorption spectrometry (HR-CS FAAS),6 inductively coupled plasma optical emission spectrometry (ICP-OES),7 differential pulse anodic stripping voltammetry (DPASV),8 and spectrofluorimetry.9,10 Although these techniques are highly sensitive and selective, they require tedious sample preparation and preconcentration procedures, expensive instruments, and professional personnel.Recently, nanomaterials have been used to build sensitive analytical sensors for the analysis of various inorganic and organic substances including trace amounts of hazardous substances in the environment. Such sensors possess certain advantages, e.g. high sensitivity, short analysis time, low cost, and simple preparation and treatment.11 Graphene quantum dots (GQDs) have generated enormous excitement because of their superiority in a variety of advantageous properties. They were discovered very recently as a class of zero-dimensional graphitic nanomaterials with lateral dimensions less than 100 nm, and they have either a single layer, double layers, or a few layers (3 to <10).12,13 Compared with organic dyes and semiconductive quantum dots (QDs), GQDs are superior in terms of their excellent properties, such as their high photostability against photobleaching and blinking, biocompatibility and low toxicity.15 Moreover, similar to graphene, GQDs have excellent characteristics such as large surface areas, large diameters, fine surface grafting via the π–π conjugated network or surface groups, and other special physical properties.16,17 Furthermore, the carboxyl and hydroxyl groups at their edge enable them to display excellent water solubility and suitability for successive functionalization with various organic, inorganic, polymeric or biological species. For these reasons, GQDs have attracted significant attention worldwide. From the literature, a small number of fluorescent sensors for Ag+ have been reported compared with other metal sensors.18–21 Furthermore, most of the fluorescent sensors for Ag+ showed limitations in their practical application due to their low water solubility, serious toxicity even at relatively low concentrations, and other drawbacks. Hence, it remains essential to develop much simpler, more efficient and robust methods for silver ion probing that can be used for the selective detection of Ag+ in real samples.
In this research, a selective fluorescent sensor for the determination of silver ions (Ag+) was developed based on GQDs. The GQDs were prepared by the citric acid pyrolysis method. In the presence of mercury ions (Hg2+), the fluorescence intensity of the GQDs decreased linearly and it was fully recovered by the hydrolysis of thioacetamide (TAA). This study is aimed at using a fluorescent sensor for the determination of Ag+. Upon the addition of Ag+, the fluorescence intensity of the TAA-(Hg2+ quenched GQDs) decreased as a linear function of the Ag+ concentration. In addition, the optimum conditions including the Hg2+ and TAA concentrations, pH of the solution and reaction time were investigated in detail.
Experimental
Chemicals and reagents
All of the chemicals used were of analytical grade. The citric acid, sodium hydroxide, silver chloride, potassium chloride, and barium chloride were purchased from Ajax Fine Chem Pty. Ltd. (Australia). The mercury nitrate, cobalt nitrate hexahydrate, lead nitrate, zinc nitrate hexahydrate, cadmium nitrate tetrahydrate, copper nitrate trihydrate, potassium cyanide, potassium iodide, thioacetamide (99.0%), and sodium sulfide were purchased from Sigma-Aldrich (Germany).Apparatus
UV-Vis absorption spectra were obtained using an Agilent 8453 spectrophotometer (Agilent, Germany). Emission spectra were recorded using an RF-5301PC spectrofluorophotometer (Shimadzu, Japan) with excitation and emission slit widths of 5 nm. A quartz cuvette (1 cm path length) was used for both the UV-visible absorption and fluorescence measurements. Transmission electron microscopy (TEM) images were obtained using a JEOL1200 (JEOL Ltd., Japan). The functional groups of the GQDs were characterized by an attenuated total reflectance-Fourier transform infrared (ATR-FTIR) spectrometer (TENSOR 27, Bruker, Germany). A pH meter (UB-10 Ultra Basic, Denver, USA) and analytical balance (AC121S, Sartorius, USA) were also used in this study.Synthesis and characterization of the GQDs
The GQDs were prepared from citric acid by a pyrolysis method with a modified procedure.12–14 Briefly, 2.0 g of citric acid was added into a 250 mL beaker. The beaker was heated to 200 °C using a paraffin oil bath for about 5 min. The solid citric acid was slowly liquated and had a yellow color. 100 mL of 0.25 M NaOH was added into the beaker with continuous stirring for 30 min at room temperature. The obtained sample solution was neutralized to pH 7 with HCl, and the GQD stock solution was stored at 4 °C before use.The quenching effect of the GQDs using Hg2+
20 μL of a 2.0 mg mL−1 GQD solution and 1 mL of 1 M phosphate buffer with a pH of 7.4 were mixed in a 10 mL volumetric flask. Then, various concentrations of Hg2+ were added to an aliquot of the GQD solution (10 mL final volume) at room temperature. The Hg2+ quenched fluorescence spectra of each of the GQD solutions were recorded immediately at λex/λem 370/460 nm. Then, such spectral measurements were used to plot the quenching calibration curve for Hg2+.To confirm the selectivity of the Hg2+ quenched GQDs, the following procedure was carried out. An individual stock solution of various metal ions (0.01 mM) was prepared by the dissolution of metal salts in ultrapure water. 10 μL of the GQD solution was mixed with 100 μL of 0.01 mM solutions of each of the metal ions (10 μM final concentration) and adjusted to a final volume of 10 mL in a volumetric flask with DI water.
The selectivity of the Hg2+–GQDs fluorescence turn-on by the hydrolysis of TAA
The fluorescence measurement of the GQDs was performed using a 10 mL volumetric flask containing 20 μL of GQDs (2.0 mg mL−1) mixed with 4 μM Hg2+ in a 0.1 M phosphate buffer solution with a pH of 7. Various concentrations of TAA and Na2S as a reference were added to the solution mixture and adjusted to 10 mL with DI water. The emission spectra of the test solution were recorded with an excitation wavelength of 370 nm. In addition, the effect of the concentration of ammonium acetate solution was investigated in order to find out any common ion effects, i.e. acetate and ammonium ions, except hydrogen sulfide, generated by the hydrolysis of TAA.The selectivity of the Hg2+–GQDs–TAA fluorescence turn-off sensor for Ag+ detection
The fluorescence measurement of the GQDs was performed using 20 μL of GQDs (2.0 mg mL−1) mixed with 4 μM Hg2+ and 70 μM TAA in a 0.1 M phosphate buffer solution with a pH of 7. Various concentrations of Ag+ were added to the mixture solution and adjusted to various pH values with a 10 mL final volume with DI water. The emission spectra of the test solution were also recorded with an excitation wavelength of 370 nm. Spectral measurements were used to plot the quenching calibration curve for Ag+.The optimization of the conditions for the fluorescence turn-off sensor for Ag+
The fluorescence measurement of the GQDs was performed in a phosphate buffer solution at a pH of 7 in a 10 mL volumetric flask. Briefly, 1 mL of the GQDs (200 mg L−1) was mixed with 4 μM Hg2+ and 70 μM TAA in a 0.1 M phosphate buffer solution at a pH of 7. Then, various concentrations of Ag+ were added to the sample solution. Finally, the volume of the solution was adjusted to 10 mL with DI water. The emission spectra of the sample mixtures were recorded with a spectrofluorometer.To obtain the optimum conditions of the proposed sensor, the following parameters were studied using the same procedure as mentioned above. The effect of the pH of the solution was studied by varying the pH of the solution from 3–12 using an acetate buffer (pH 3–5) and phosphate buffer (pH 6–12). The incubation times were studied from 0–1 h. The effect of the concentration of Hg2+ was investigated by varying the concentration from 0 to 10 μM.
The selectivity of the fluorescent sensor
To evaluate the selectivity of the proposed fluorescent sensor, the following procedure was carried out. An individual stock solution of each of the various metal ions (0.01 mM), including Li+, Na+, K+, Ba2+, Ca2+, Cd2+, Co2+, Cu2+, Fe2+, Fe3+, Mg2+, Mn2+, Ni2+, Pb2+, Zn2+, Cr3+ and Al3+ ions, was prepared by the dissolution of their metal salts in DI water. Then 100 μL of their stock solutions were added to 10 mL volumetric flasks each containing 1 mL of the GQDs (200 mg L−1), 4 μM Hg2+, and 70 μM TAA in a 0.1 M phosphate buffer solution with a pH of 7. Finally, the volume of the reaction mixture was adjusted to 10 mL with DI water.Results and discussion
The characterization of the synthesized GQDs
Fig. 1(a) shows the UV-Vis absorption spectrum of the synthesized GQDs, which exhibits two absorption bands at around 230 nm. The broadened absorption band centered at 230 nm, resulting in nearly no fluorescence signal, is attributed to the π–π* electronic transition of the aromatic sp2 domains.22 The other typical absorption peak centered at 365 nm is assigned to the n–π* transition of the graphitic sp2 domains.23 The photoluminescence (PL) spectra of the synthesized GQDs show strong peaks at 460 nm when excited at 370 nm and the full width at half maximum (FWHM) is about 100 nm, which is approximately the same as those of most reported GQDs.24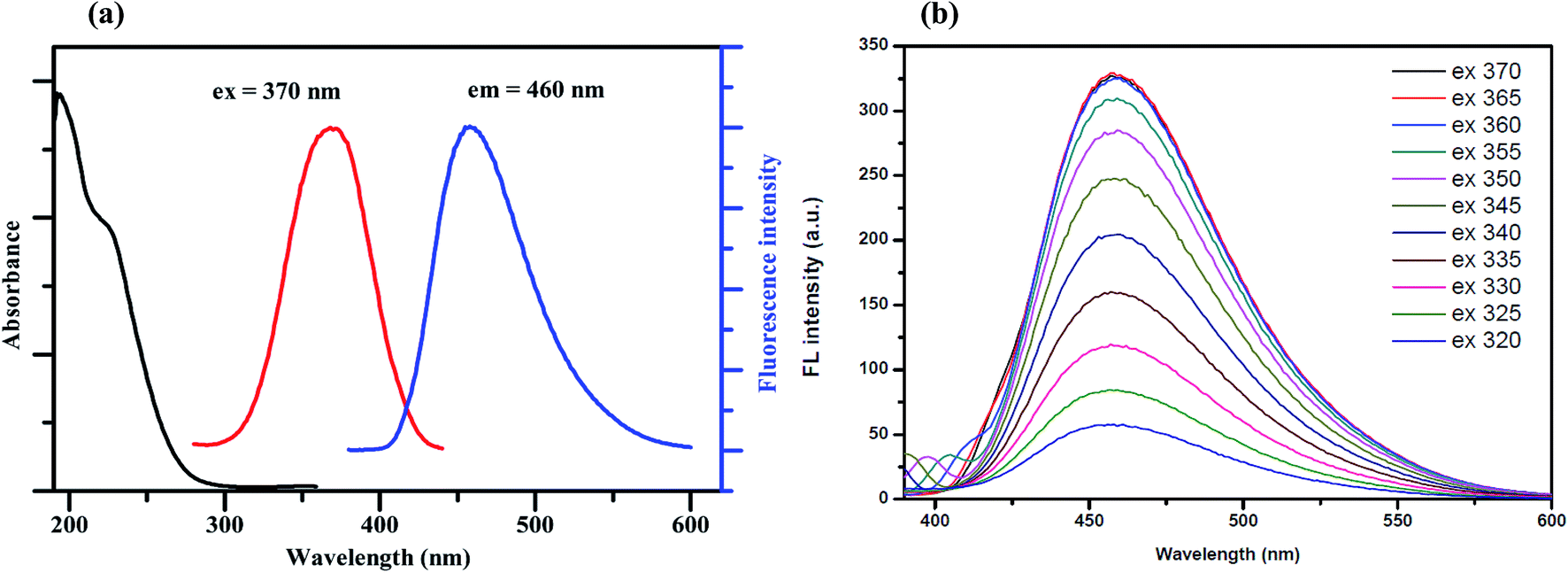 | ||
| Fig. 1 (a) The absorption (black line), fluorescence excitation (red line) and fluorescence emission (blue line) spectra of the GQDs and (b) the fluorescence spectra of the GQDs at different excitation wavelengths. | ||
To further characterize the optical properties of the synthesized GQDs, a detailed PL investigation was carried out with different excitation wavelengths (Fig. 1(b)). When the excited wavelengths change from 320 to 370 nm, the PL intensities increase markedly, but the fluorescence emission peak remains unshifted. This excitation-independent PL behavior is also attributed to a similar trend to the behaviour of previously reported GQDs.25
Fig. 2(a) shows the FT-IR spectrum of the GQDs. The strong bands at around 1556 cm−1 are due to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching mode of polycyclic aromatic hydrocarbons,26 indicating that the GQDs retain their graphene structure. Moreover, the bands at around 1718 cm−1 correspond to the carboxyl groups, and the bands at around 3440 cm−1 are attributed to the stretching vibrations of the hydroxyl groups. The peaks at 1364–1068 cm−1 are attributed to the C–O bond in the COH/COC (epoxy) groups.27
C stretching mode of polycyclic aromatic hydrocarbons,26 indicating that the GQDs retain their graphene structure. Moreover, the bands at around 1718 cm−1 correspond to the carboxyl groups, and the bands at around 3440 cm−1 are attributed to the stretching vibrations of the hydroxyl groups. The peaks at 1364–1068 cm−1 are attributed to the C–O bond in the COH/COC (epoxy) groups.27
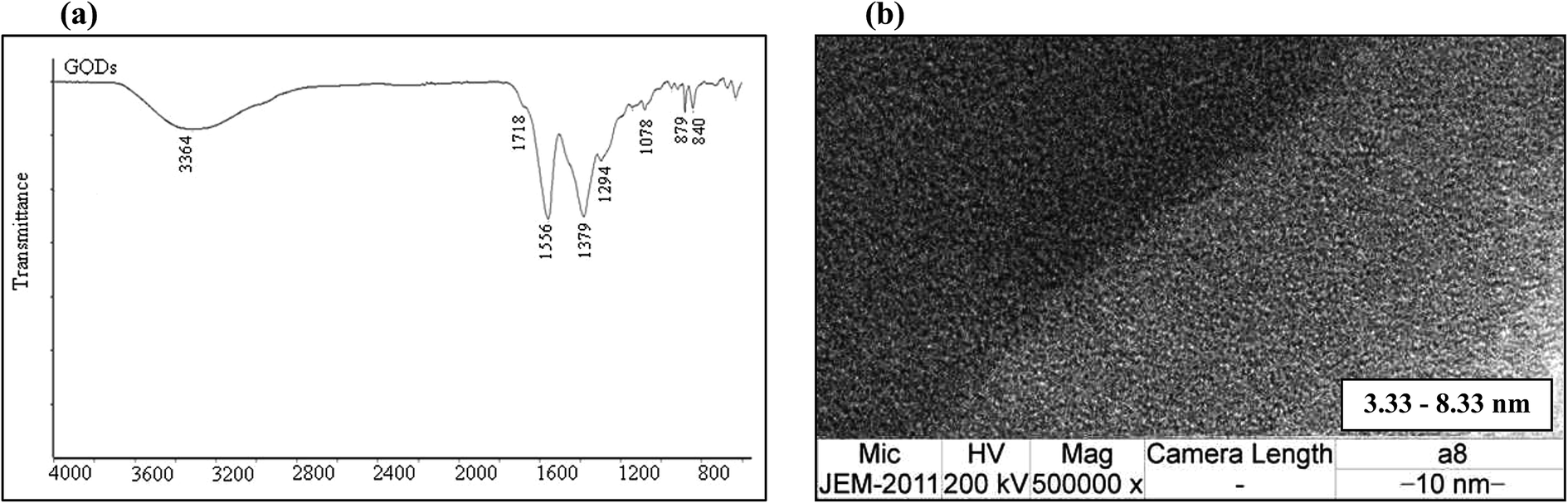 | ||
| Fig. 2 (a) The FT-IR spectrum and (b) TEM image of the GQDs. | ||
To determine the particle size of the obtained GQDs, the sample was characterized by transmission electron microscopy (TEM). Fig. 2(b) shows the image of the GQDs. Using image J software, the GQD particles, which had a diameter in the range of 3.33 nm to 8.33 nm, are shown to be distributed uniformly. The small particle size of the GQDs provides evidence for the fluorescence intensity.
The quenching effect of the metal ions on the fluorescence intensity of the GQDs
To test the effect of the metal ions on the fluorescence intensity of the GQDs, various cations were studied including Ag+, K+, Na+, Ba2+, Ca2+, Cd2+, Co2+, Cu2+, Fe2+, Fe3+, Hg2+, Mg2+, Mn2+, Ni2+, Pb2+, Zn2+, Cr3+, Fe3+, Sb3+, and Al3+ as shown in Fig. 3.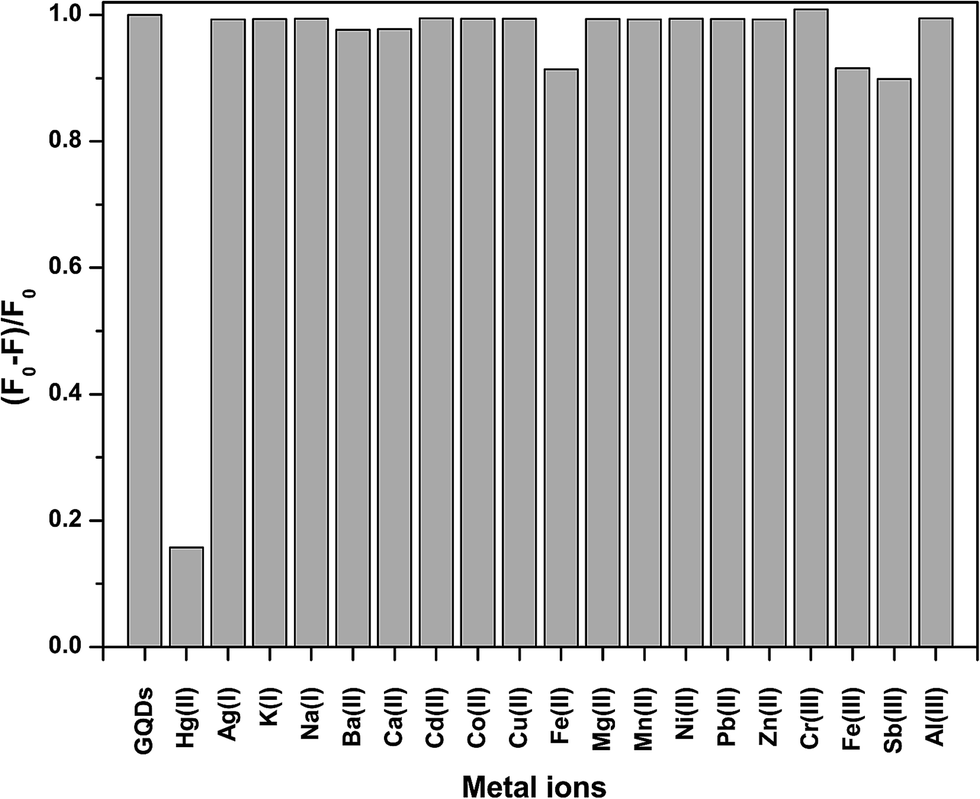 | ||
| Fig. 3 The quenching of the fluorescence intensity of the GQDs by different metal ions. All the cation concentrations were 100 μM. F and F0 are the emission intensities of the GQDs at 460 nm in the presence and absence of the metal ions. | ||
From Fig. 3, it was clearly found that Hg2+ could quench the fluorescence intensity of the GQDs very strongly, while both the Fe2+ and Fe3+ ions had a slight quenching effect. The other ions scarcely quenched the fluorescence intensity of the GQDs. The high selectivity may be attributed to the fact that the Hg2+ ions have a stronger affinity towards the carboxylic groups on the GQD surface than other reported metal ions.28 The quenching mechanism presumably occurs via either electron or energy transfer from the GQDs to the Hg2+ ions.29
The optimum conditions for the quenching effect of the GQDs by mercury ions
The optimum concentration of Hg2+ was studied as shown in Fig. 4(a) and (b). When the Hg2+ concentration increased, the emission intensity of the GQDs gradually decreased. A linear relationship was obtained when the maximum intensity of the GQDs at 460 nm was plotted against the Hg2+ concentration in the range of 1–4 μM. When the concentration of Hg2+ was 4 μM, the fluorescence intensity of the GQDs was efficiently quenched. Therefore, 4 μM Hg2+ was used for the further studies.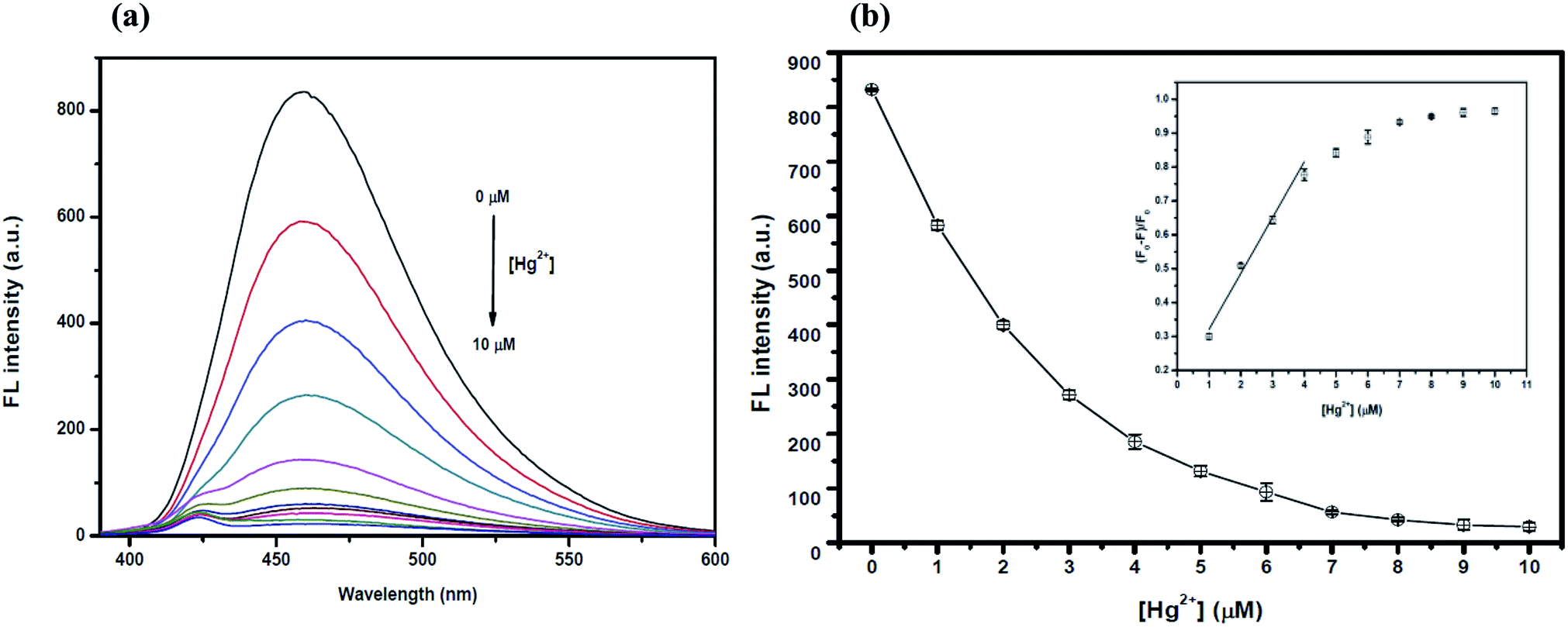 | ||
| Fig. 4 (a) The changes in the emission spectra of the GQDs at different Hg2+ concentrations (0–10 μM) and (b) changes in the emission intensity at 460 nm versus the Hg2+ concentration (inset, the plot of (F0 − F)/F0versus the concentration of Hg2+). | ||
To improve the quenching effect of Hg2+, several experimental parameters were optimized including the reaction time and solution pH value. After the addition of 4 μM Hg2+, the fluorescence (FL) spectrum of the GQDs (pH 7.0 and 20 mg L−1 GQDs), was recorded at every 5 min interval. Thus, the FL intensity of the GQDs at 460 nm decreased as much as 82% within a minute after the addition of Hg2+ (Fig. 5(a)), then the FL intensity gradually decreased until 5 min, and then finally it remained constant over the next 60 min. From the results, it is suggested that the quenching process is almost completed within 5 min.
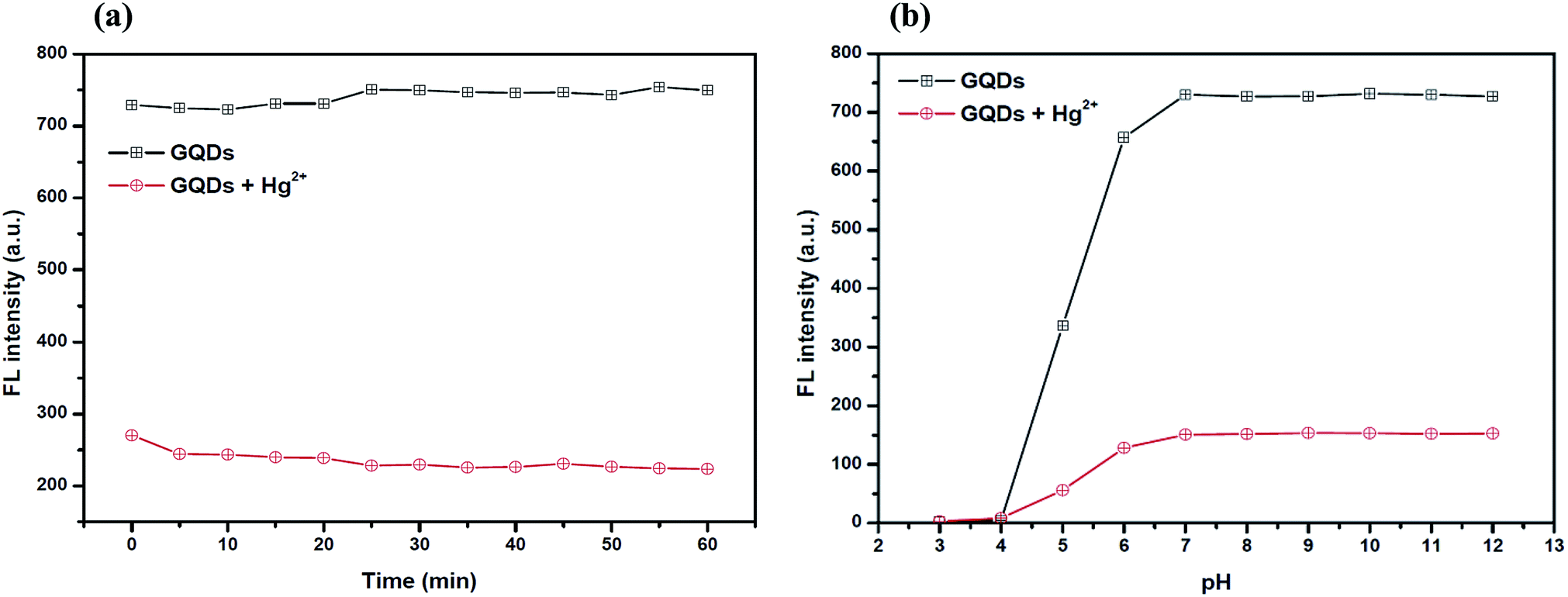 | ||
| Fig. 5 (a) The changes in the fluorescence intensity of the GQDs at different reaction times. Conditions: 4 μM Hg2+ in 0.1 M phosphate buffer at a pH of 7.0 and (b) the changes in the fluorescence intensity of the GQDs at different values of pH. Conditions: 4 μM Hg2+ and 5 min reaction time. | ||
For the effect of the pH, a series of acetate and phosphate buffers at different pH values were prepared, and then the GQD solutions (20 mg L−1 final concentration) together with 4 μM Hg2+ were added into each different buffer solution (pH: 3.0, 4.0, 5.0, 6.0, 7.0, 8.0, 9.0, 10.0, 11.0 and 12.0 in 0.1 M). The FL intensity was measured at 460 nm from each of the two buffered solutions of GQD only and GQDs with Hg2+ (Fig. 5(b)). The FL intensity of the GQDs increased gradually with pH values from 3.0–7.0 and stabilized under alkaline conditions. It was implied that a suitable quenching effect occurred with pH > 7.0. Thus, the working pH for a sufficiently sensitive and stable quenching of Hg2+ was 7.0.
The optimum conditions for the fluorescence turn-on of the Hg2+ quenched GQDs using the hydrolysis of thioacetamide
TAA is an active organosulfur, which undergoes hydrolysis giving hydrogen sulfide in the solution. As shown in reaction (1), the homogeneously generated hydrogen sulfide and sulfide ions (HS− and S2−) can act in similar roles to the thiol group, then they can bind to Hg2+ through Hg–S interactions. This causes the Hg2+–GQD complex to dissociate, resulting in the restoration of the fluorescence intensity of the GQDs.30| CH3CSNH2 + 2H2O → CH3CO2− + NH4+ + H2S | (1) |
To make sure that H2S can have a fluorescence enhancing effect on the Hg2+–GQDs, a common sulfide salt was used. In this regard, a solution of Na2S was added to the Hg2+–GQDs reaction mixture and then the fluorescence intensity of the GQD system was measured compared with that of the Hg2+–GQDs mixture using TAA. From the results (Fig. 6), it was found that both kinds of the two sulfide solutions provided a similar trend, suggesting that the H2S generated from TAA could enhance the fluorescence intensity of the Hg2+–GQDs.
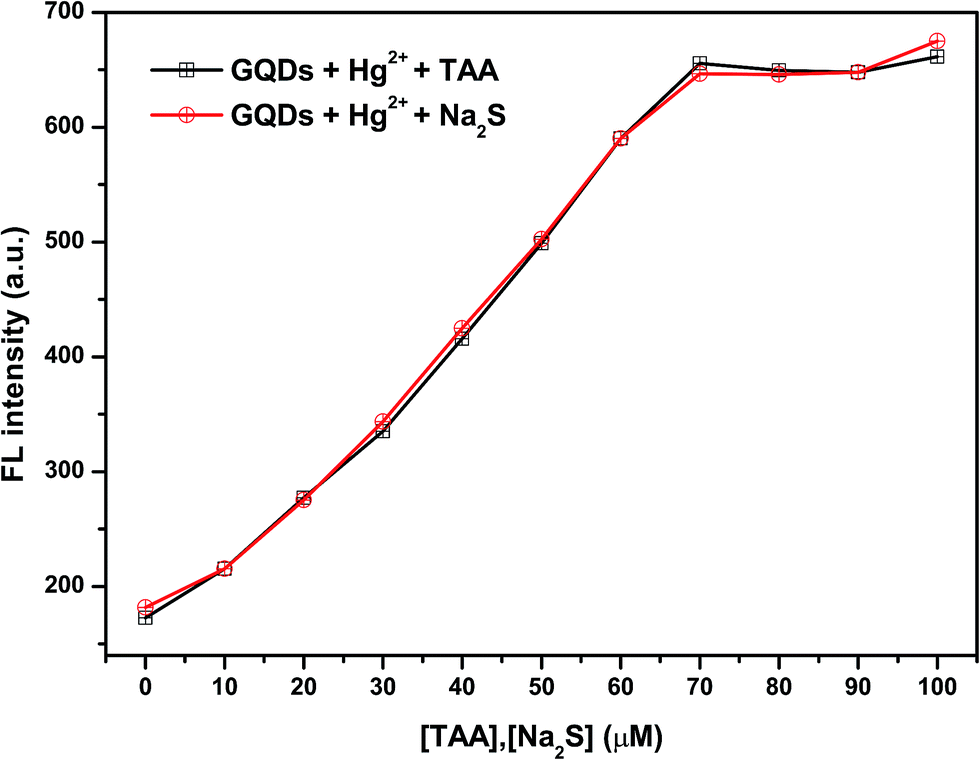 | ||
| Fig. 6 The effect of different concentrations of TAA or Na2S on the fluorescence intensity of the Hg2+–GQDs. | ||
However, the optimum concentration of TAA was also studied as shown in Fig. 7(a) and (b). When the concentration of TAA increased, the FL intensity of the Hg2+–GQDs was gradually restored. A linear relationship was obtained when plotting the maximum intensity of the Hg2+–GQDs at 460 nm against the TAA concentration in the range of 0–70 μM (Fig. 7(b)). At 70 μM TAA, the fluorescence intensity of the Hg2+–GQDs was efficiently restored. Therefore, 70 μM TAA was selected for further studies.
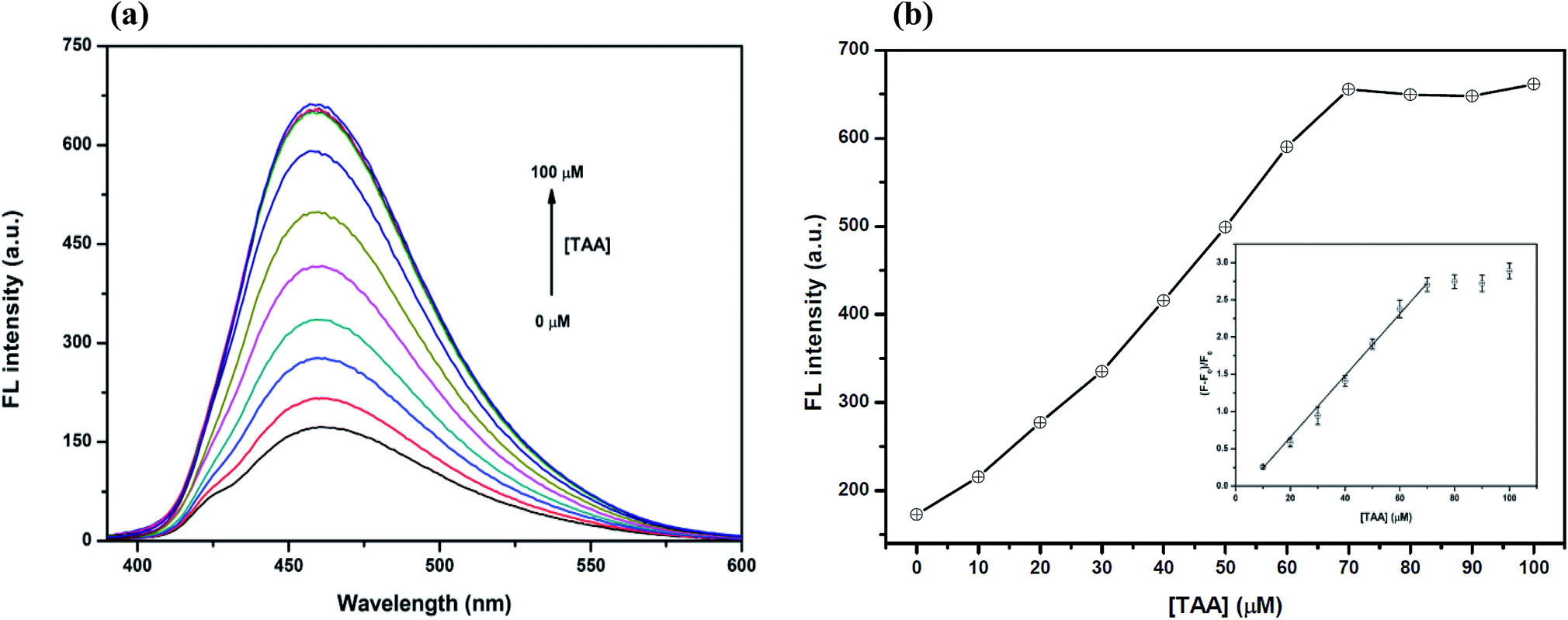 | ||
| Fig. 7 (a) The changes in the FL spectra of the Hg2+–GQDs at different TAA concentrations (0–100 μM). Conditions: 4 μM Hg2+ in a 0.1 M phosphate buffer with a pH of 7.0 and (b) the changes in the FL intensity at 460 nm versus the TAA concentration. Conditions: 4 μM Hg2+ in 0.1 M phosphate buffer with a pH of 7.0 (inset, plot of (F0 − F)/F0versus the concentration of TAA). | ||
Also, to improve the fluorescence restoration efficiency of the Hg2+–GQD complex, the reaction time and pH value were optimized (Fig. 8(a)). After the addition of 70 μM TAA, the FL intensity of the GQD system (pH 7.0) was recorded every 5 min and it increased rapidly. The results suggested that the restoration process is completed within a minute.
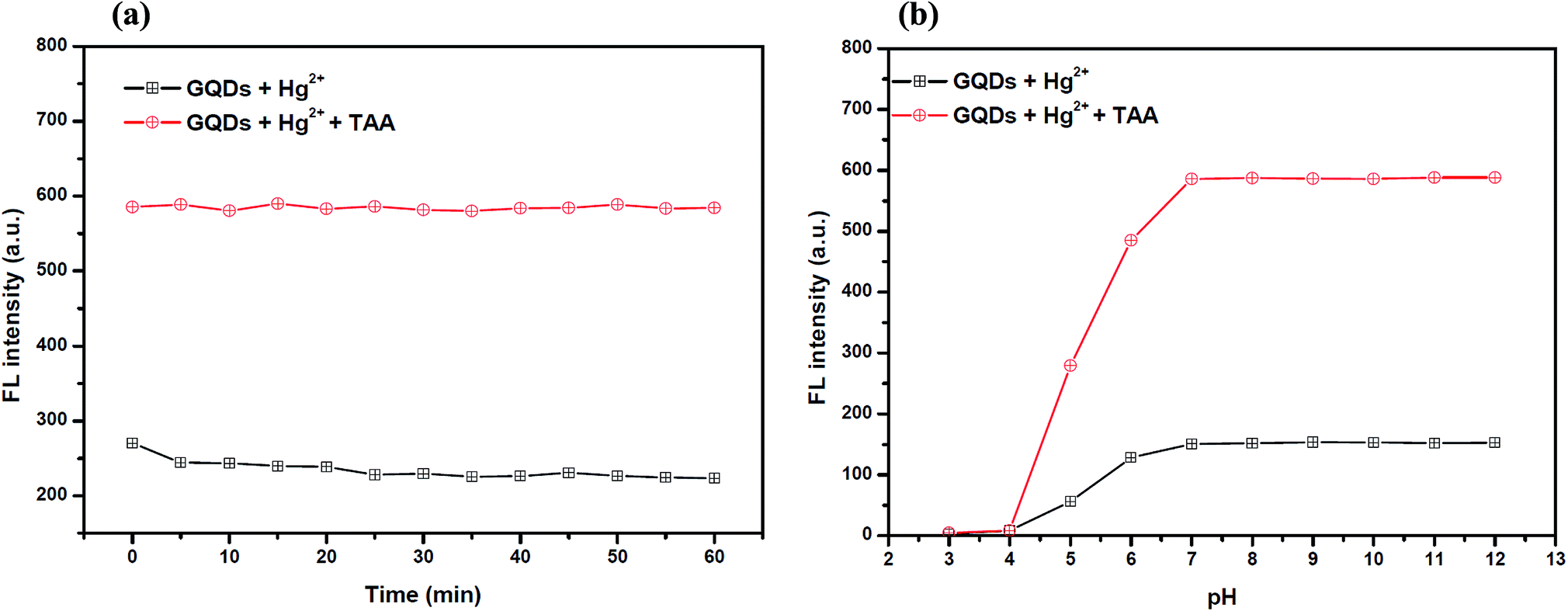 | ||
| Fig. 8 (a) The changes in the FL intensity of the Hg2+–GQDs at different reaction times. Conditions: 0.1 M phosphate buffer pH 7.0, 4 μM Hg2+ and 70 μM TAA and (b) the changes in the FL intensity of the Hg2+–GQDs at different pH values. Conditions: 4 μM Hg2+, 70 μM TAA and 5 min reaction time. | ||
To study the effect of the pH, 70 μM TAA was added to the Hg2+–GQD complex in the buffer solution at different pH values. As the results show in Fig. 8(b), the FL intensity of the GQDs increased gradually with the pH increasing from 3.0–7.0 and stabilized under alkaline conditions. It is implied that a suitable FL enhancing effect occurred with pH > 7.0. Thus, the working pH of the hydrolysis of TAA for a sufficiently sensitive and stable FL enhancing effect was 7.0.
The optimum conditions for the fluorescence turn-off sensor for silver ions
The overall assay strategy for the sensing of Ag+ is shown in Fig. 9. The GQDs showed strong blue fluorescence in an aqueous buffer solution. Upon the addition of Hg2+, it bound to the GQDs resulting in fluorescence quenching of the GQDs. But when TAA was added, the TAA could react with the Hg2+ ion through Hg–S interaction. When the Hg2+–GQD complex dissociated, the fluorescence of the GQDs was restored. Lastly, when Ag+ ions were added to the reaction mixture, the fluorescence intensity of the GQDs decreased. The FL turn-off mechanism possibly occurs, and this means that the Ag+ ion has more affinity to bind with a sulfur atom on the thiol group compared with the mercury ion. Thus, when Ag+ was added to the mixture, it would bind with the sulfur atom of H2S and break the Hg–S bond. Therefore, the free Hg2+ ion would bind to the GQDs again, leading to the fluorescence turn-off sensing of the GQD system.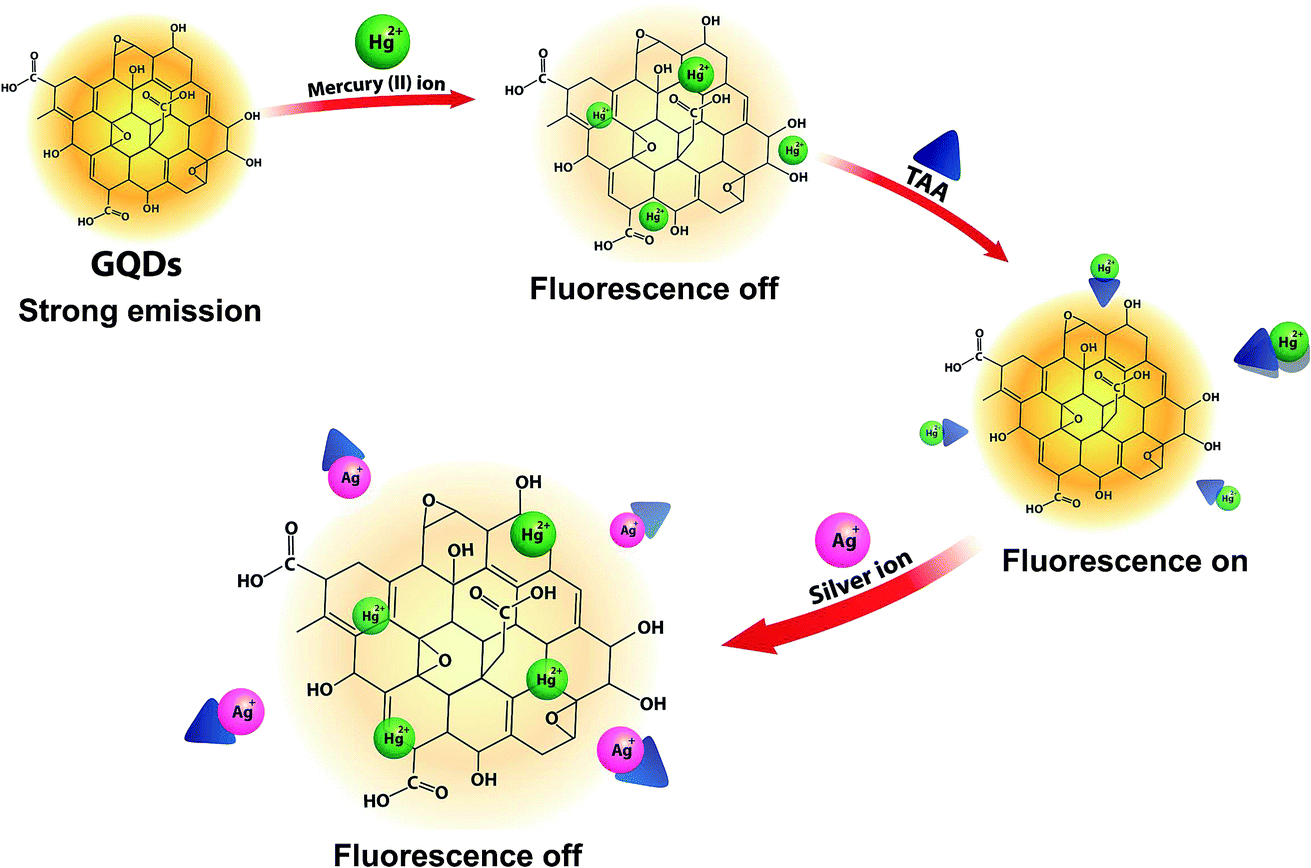 | ||
| Fig. 9 A schematic illustration of the FL turn-off/on/off mechanism of the GQD-based Ag+ sensing. | ||
As shown in Fig. 10(a), the fluorescence intensity of the GQDs gradually decreased when the concentration of Ag+ increased. A linear relationship was obtained when the maximum intensity of the GQDs at 460 nm was plotted against the Ag+ concentration in the range of 0.5–10 μM (Fig. 10(b)) with the Stern–Volmer plot shown in Fig. 10(c).
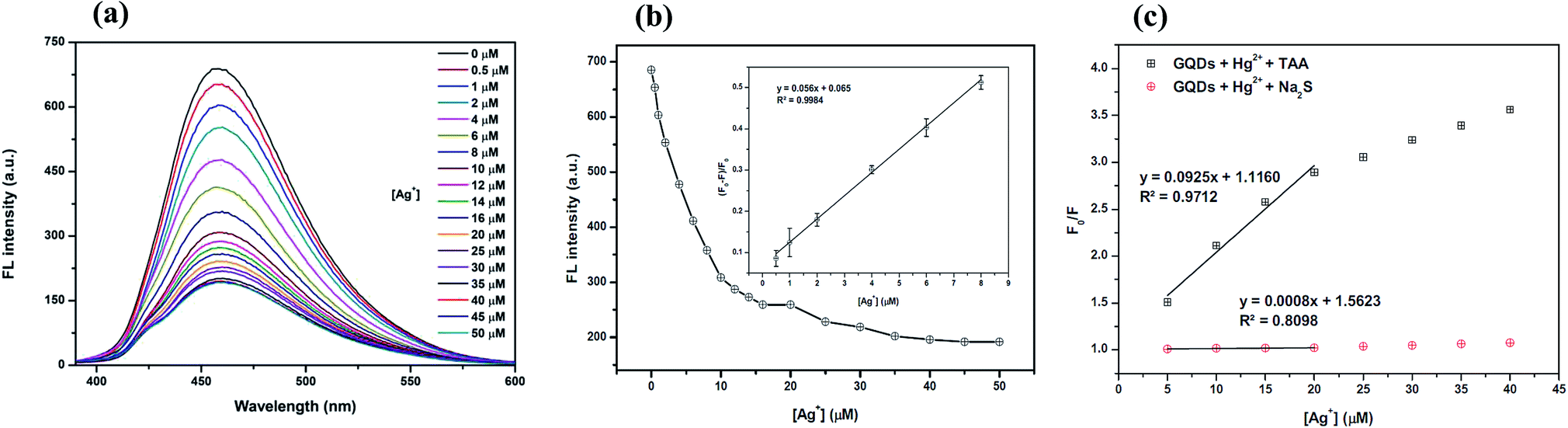 | ||
| Fig. 10 (a) The changes in the FL intensity of the Hg2+–GQDs at different Ag+ concentrations (0–50 μM), (b) the changes in the FL intensity at 460 nm versus the Ag+ concentration (inset, the plot of (F0 − F)/F0versus the concentration of Ag+) and (c) the Stern–Volmer plots of the Ag+ quenching effect for both Hg2+–GQDs in the presence of either TAA or Na2S solution. | ||
To improve the performance of the quenching of Ag+, several experimental conditions, i.e. the reaction time and solution pH value, were optimized. After the addition of 10 μM Ag+, the FL spectrum of the GQDs was recorded at five min intervals. The FL intensity of the quenched GQDs at 460 nm rapidly decreased and remained constant within 5 min (Fig. 11(a)), and the FL intensity remained constant over the next 60 min. The results suggest that the quenching process is completed within 5 min.
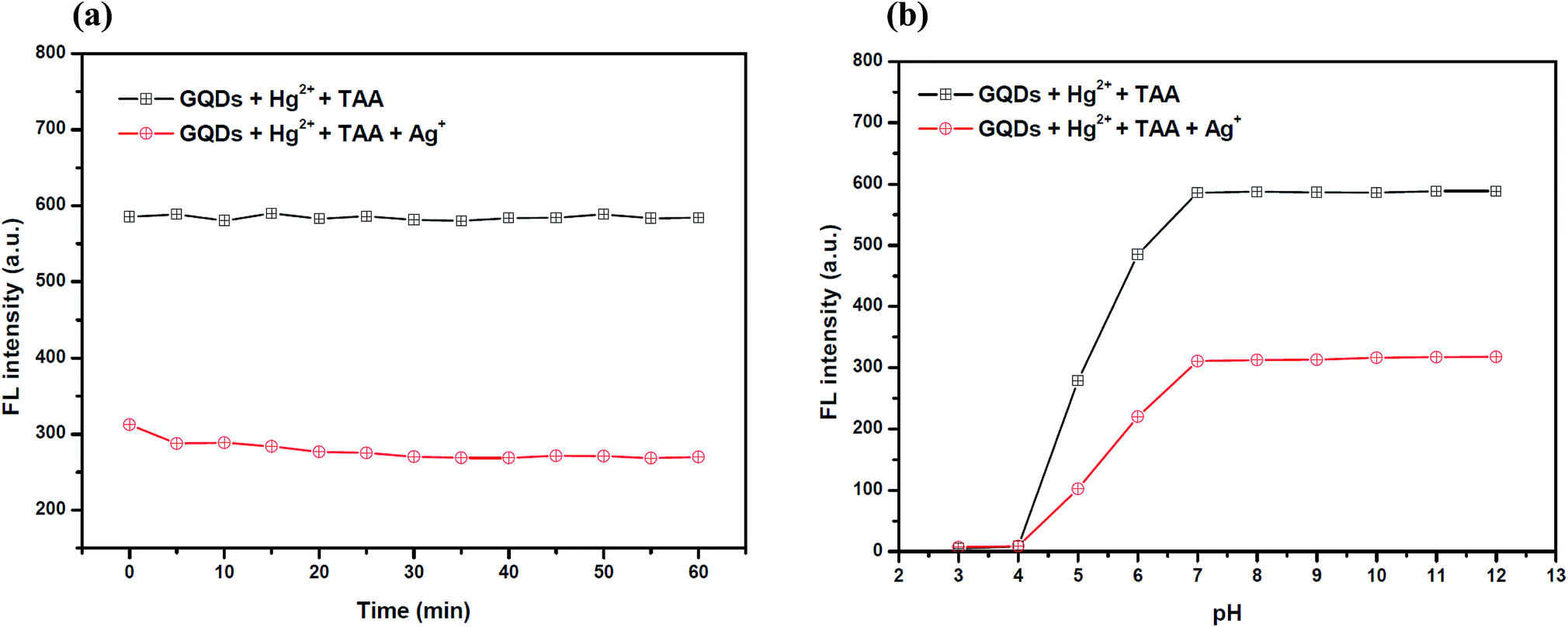 | ||
| Fig. 11 (a) The changes in the FL intensity of the Hg2+–GQDs at different reaction times. Conditions: 0.1 M phosphate buffer with a pH of 7.0, 4 μM Hg2+, 70 μM TAA and 10 μM Ag+ and (b) the changes in the FL intensity of the Hg2+–GQDs at different pH values. Conditions: 4 μM Hg2+, 70 μM TAA, 10 μM Ag+ and a 5 min reaction time. | ||
The effect of the pH on the FL sensing system was studied from pH 3–12 in two selected buffer solutions. 10 μM Ag+ solution was added to the GQD solution at different pH values, as shown in Fig. 11(b). The FL intensity of the GQDs increased gradually from pH 3.0–7.0 and stabilized under the alkaline conditions. It is implied that the most suitable quenching effect occurred with pH > 7.0. Thus, the working solution pH for the Ag+ quenching effect was also found to be 7.0.
The selective determination of the silver ion
To study the selectivity of the developed GQD fluorescent sensor for Ag+ detection, the values of (F − F0)/F0 for the GQD system in the presence of 18 different cations, Ag+, Li+, Na+, K+, Ba2+, Ca2+, Cd2+, Co2+, Cu2+, Fe2+, Fe3+, Mg2+, Mn2+, Ni2+, Pb2+, Zn2+, Cr3+, and Al3+, were plotted using a 10 μM concentration of each metal. Fig. 12 shows that the addition of Ag+ to the reaction mixture of the GQD system resulted in fluorescence quenching, whereas the other remaining cations had no significant effects under the same experimental conditions.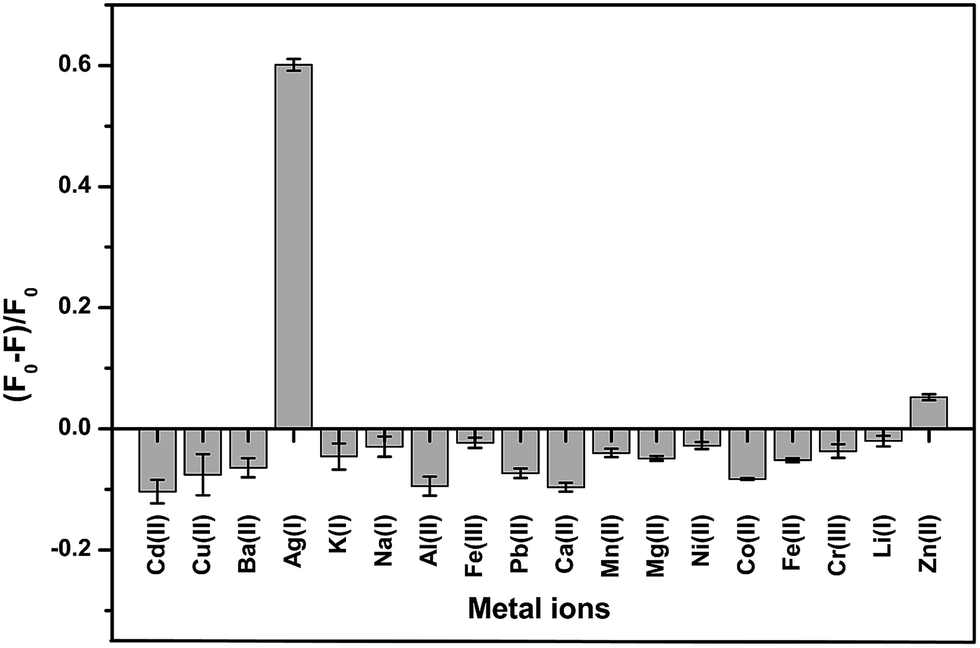 | ||
| Fig. 12 The selectivity of the GQD FL sensor for different cations (10 μM) under the optimum conditions. | ||
Method validation
The analytical characteristics of the developed method were validated under the optimized conditions in terms of the linearity, limit of detection (LOD), limit of quantification (LOQ), precision and reproducibility (expressed as the relative standard deviation (RSD) of the calibration curve slope obtained from both intra-day and inter-day analysis) to estimate the efficiency and feasibility of the method for use with tap and drinking water samples. The linearity values are from 0.5–10.0 μM (R2 = 0.9918). The linear calibration graph is as follows: y = 32.14x + 63.66 (where y is the fluorescence intensity and x is the concentration of Ag+). The LOD, defined as 3SD/m (where SD is the standard deviation of the low concentration of Ag+ and m is the slope of the calibration graph), was 0.18 μM. While the LOQ, defined as 10SD/m, was 0.60 μM. The precision, which was evaluated in terms of the repeatability (data from 3 × 3 independent standard preparations, intra-day RSD), was 0.63% and the reproducibility (work performed during 11 × 3 consecutive days, inter-day RSD) was 2.45%, indicating an acceptable repeatability of the method.The analysis of real samples
To demonstrate the applicability and reliability of the developed method, it was successfully applied to five water samples including tap water, three brands of drinking water (Brand 1, Brand 2, and Brand 3), and one brand of natural drinking water (Brand 4). The amounts of Ag+ in each sample tested were obtained as shown in Table 1. The results showed that Ag+ was not detected in all of these samples. In addition, to evaluate the matrix effect, the accuracy of the method was verified by calculating the recovery study in the real samples. Each sample was spiked with three concentrations (2.0, 4.0, and 6.0 μM) of the standard solution of Ag+. Then, the relative percentage recoveries were calculated as follows:31,32| % recovery = [(Cfound − Creal)/Cadded] × 100 | (2) |
| Sample | Added (μM) | Found (μM) | % recovery ± SD (μM) |
|---|---|---|---|
| a n.d. = not detected. | |||
| Tap water | — | n.d. | — |
| 2 | 2.03 | 101.52 ± 2.99 | |
| 4 | 4.56 | 114.08 ± 2.99 | |
| 6 | 6.53 | 108.98 ± 3.49 | |
| Drinking water (Brand 1) | — | n.d. | — |
| 2 | 1.85 | 92.85 ± 4.17 | |
| 4 | 4.03 | 100.87 ± 1.08 | |
| 6 | 5.90 | 94.34 ± 3.18 | |
| Drinking water (Brand 2) | — | n.d. | — |
| 2 | 2.01 | 100.30 ± 4.15 | |
| 4 | 3.98 | 99.43 ± 4.34 | |
| 6 | 5.75 | 95.81 ± 1.02 | |
| Drinking water (Brand 3) | — | n.d. | — |
| 2 | 1.99 | 99.49 ± 3.13 | |
| 4 | 3.76 | 93.92 ± 1.74 | |
| 6 | 5.36 | 89.31 ± 2.61 | |
| Natural drinking water (Brand 4) | — | n.d. | — |
| 2 | 2.08 | 104.20 ± 4.76 | |
| 4 | 4.11 | 102.89 ± 2.56 | |
| 6 | 5.77 | 96.11 ± 1.97 | |
Conclusion
A highly sensitive and selective fluorescent sensor for the detection of Ag+ based on Hg2+ quenched GQDs in the presence of TAA was obtained. This novel approach based on GQDs for Ag+ determination was carried out at a trace level. The GQDs were simply prepared from citric acid pyrolysis and characterized by molecular absorption and emission spectrophotometry, FT-IR spectroscopy and TEM. The optimum conditions for the fluorescence turn-on/off sensing probe, including the effects of the solution pH, the concentration of the GQDs, Hg2+, Ag+, and TAA, and the reaction time, were investigated. The Stern–Volmer plot for the Ag+ quencher in the presence of homogeneously generated sulfide from TAA showed higher sensitivity compared with that of the common sulfide salt used.The selectivity of the fluorescence quenching for Ag+ compared to various interfering metal ions was demonstrated. The analytical features of merit, including accuracy, precision, linearity, and limits of detection (LOD) and quantification (LOQ), were validated. The application of the developed method for the trace determination of Ag+ in tap water and drinking samples was demonstrated.Conflicts of interest
The authors have declared no conflict of interest.Acknowledgements
The authors thank the Higher Education Research Promotion and National Research University Project of Thailand, the Office of the Higher Education Commission, the Food and Functional Food Research Cluster of Khon Kaen University, the Materials Chemistry Research Center, Department of Chemistry, and the Center of Excellence for Innovation in Chemistry (PERCH-CIC), Thailand for the financial support.References
- R. sedghi, M. Shojaee, M. Behbahani and M. R. Nabid, RSC Adv., 2015, 5, 67418–67426 RSC.
- G. Absalan, M. Akhond, L. Sheikhian and D. M. Goltz, Anal. Methods, 2011, 3, 2354–2359 RSC.
- C. Z. Lai, M. A. Fierke, R. C. Costa, J. A. Gladysz, A. Stein and P. Buhlmann, Anal. Chem., 2010, 82, 7634–7640 CrossRef CAS.
- R. E. Godoy and A. G. Perez, Analyst, 1986, 111, 1297–1299 RSC.
- J. A. Lopez-Lopez, J. A. Jonsson, M. Garcia-Vargas and C. Moreno, Anal. Methods, 2014, 6, 1462–1467 RSC.
- E. Kariptas, C. Er, E. Kiray and H. Ciftci, Anal. Methods, 2016, 8, 4285–4292 RSC.
- L. M. Hallerstig, P. Granath, L. Lindgren and M. Tranberg, Anal. Methods, 2017, 9, 149–153 RSC.
- L. Liu, C. Wang and G. Wang, Anal. Methods, 2013, 5, 5812–5822 RSC.
- S. Zhan, Y. Wu, L. He, F. Wang, X. Zhan, P. Zhou and S. Qiu, Anal. Methods, 2012, 4, 3997–4002 RSC.
- D.-S. Lin, J.-P. Lai, H. Sun, Z. Yang and Y. Zuo, Anal. Methods, 2014, 6, 1517–1522 RSC.
- M. M. Rahman and A. M. Asiri, RSC Adv., 2015, 5, 63252–63263 RSC.
- P. Sricharoen, N. Limchoowong, Y. Areerob, P. Nuengmatcha, S. Techawongstien and S. Chanthai, Ultrason. Sonochem., 2017, 37, 83–93 CrossRef CAS.
- N. Limchoowong, P. Sricharoen, Y. Areerob, P. Nuengmatcha, T. Sripakdee, S. Techawongstien and S. Chanthai, Food Chem., 2017, 230, 388–397 CrossRef CAS.
- Y. Dong, J. Shao, C. Chen, H. Li, R. Wang, Y. Chi, X. Lin and G. Chen, Carbon, 2012, 50, 4738–4743 CrossRef CAS.
- S. Zhu, S. Tang, J. Zhang and B. Yang, Chem. Commun., 2012, 48, 4527–4539 RSC.
- Z. Fan, S. Li, F. Yuan and L. Fan, RSC Adv., 2015, 5, 19773–19789 RSC.
- S. Zhou, H. Xu, W. Gan and Q. Yuan, RSC Adv., 2016, 6, 110775–110788 RSC.
- L. Liu, D. Zhang, G. Zhang, J. Xiang and D. Zhu, Org. Lett., 2008, 10, 2271–2274 CrossRef CAS.
- X. Zhu, S. Fu, W. K. Wong and W. Y. Wong, Tetrahedron Lett., 2008, 49, 1843–1846 CrossRef CAS.
- L. Liu, G. X. Zhang, J. F. Xiang, D. Q. Zhang and D. B. Zhu, Org. Lett., 2010, 10, 4581–4584 CrossRef.
- F. Wang, R. Nandhakumar, J. H. Moon, K. M. Kim and J. Y. Lee, Inorg. Chem., 2011, 50, 2240–2245 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang and S. V. Dubono, Science, 2004, 306, 666–669 CrossRef CAS.
- M. Xie, Y. Su, X. Lu, Y. Zhang, Z. Yang and Y. Zhang, Mater. Lett., 2013, 93, 161–164 CrossRef CAS.
- Y. Li, Y. Hu, Y. Zhao, G. Shi, L. Deng and Y. Hou, Adv. Mater., 2011, 23, 776–780 CrossRef CAS.
- S. K. Cushing, L. Ming, F. Huang and N. Wu, ACS Nano, 2014, 8, 1002–1013 CrossRef CAS.
- X. Wang, X. Sun, J. Lao, H. He, T. Cheng, M. Wang, S. Wang and F. Huang, Colloids Surf., B, 2014, 122, 638–644 CrossRef CAS.
- H. Wang, Q. Hao, X. Yang, L. Lu and X. Wang, Electrochem. Commun., 2009, 11, 1158–1161 CrossRef CAS.
- F. Chai, T. Wang, L. Li, H. Liu, L. Zhang, Z. Su and C. Wang, Nanoscale Res. Lett., 2010, 5, 1856–1860 CrossRef CAS.
- W. B. Lu, X. Y. Qin, S. Liu, G. H. Chang, Y. W. Zhang, Y. L. Luo, A. M. Asiri, A. O. Al-Youbi and X. P. Sun, Anal. Chem., 2012, 84, 5351–5357 CrossRef CAS PubMed.
- Z. Wu, W. Li, J. Chen and C. Yu, Talanta, 2014, 119, 538–543 CrossRef CAS.
- P. Sricharoen, N. Limchoowong, T. Sripakdee, P. Nuengmatcha and S. Chanthai, Anal. Methods, 2017, 9, 3810–3818 RSC.
- P. Sricharoen, N. Limchoowong, S. Techawongstien and S. Chanthai, Food Chem., 2016, 203, 386–393 CrossRef CAS PubMed.
- Z. S. Qian, J. J. Ma, X. Y. Shan, H. Feng, L. X. Shao and J. R. Chen, Chem.–Eur. J., 2014, 20, 2254–2263 CrossRef CAS.
- X. Gao, Y. Lu, R. Zhang, S. He, J. Ju, M. Liu, L. Lia and W. Chen, J. Mater. Chem. C, 2015, 3, 2302–2309 RSC.
- X. L. Li, Y. W. He, J. S. Ryu and S. I. Yang, New J. Chem., 2014, 38, 503–506 RSC.
- A. Suryawanshi, M. Biswal, D. Mhamane, R. Gokhale, S. Patil, D. Guin and S. Ogale, Nanoscale, 2014, 6, 11664–11670 RSC.
| This journal is © The Royal Society of Chemistry 2017 |