
Tacrine-allyl/propargylcysteine–benzothiazole trihybrids as potential anti-Alzheimer's drug candidates†
Asha Hiremathadab,
Karam Chanda,
A. Raquel Estevesc,
Sandra M. Cardosocd,
Rona R. Ramsaye,
Sílvia Chavesa,
Rangappa S. Keri*b and
M. Amélia Santos*a
aCentro de Química Estrutural, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais 1, 1049-001 Lisboa, Portugal. E-mail: masantos@ist.utl.pt; Tel: +351-218419273
bCentre for Nano and Material Sciences, Jain University, Jain Global Campus, Bangalore, Karnataka 562112, India. E-mail: keriphd@gmail.com; Tel: +91-8027577199
cCNBC – Centro de Neurociências e Biologia Celular, Universidade de Coimbra, Coimbra, Portugal
dFaculdade de Medicina, Universidade de Coimbra, 3030 Coimbra, Portugal
eBiomedical Sciences Research Complex, University of St Andrews, Biomolecular Sciences Building, North Haugh, St Andrews KY16 9ST, UK
First published on 25th May 2016
Abstract
On continuing our research into new drug candidates for Alzheimer's disease (AD), we have designed, synthesized and evaluated a series of multifunctional trihybrid agents. The design strategy was based on the incorporation of a benzothiazole (BTA) moiety on a series of very recently reported bihybrids, resulting from the conjugation of a tacrine (TAC) with natural based moieties, namely S-allylcysteine (SAC) (garlic constituent) and S-propargylcysteine (SPRC). Thus, in addition to the acetylcholinesterase inhibition (AChEI) and anti-ROS capacity of the bihybrids (TAC–SAC/SPRC), the new trihybrids (TAC–SAC/SPRC–BTA) appeared endowed with a 5-fold capacity for inhibition of amyloid beta-peptide (Aβ) aggregation. The BTA moiety led also to considerable enhancement of the AChEI on the trihybrids, which molecular modeling suggested as being due to simultaneous binding to the catalytic active site and peripheral anionic site of AChE. The trihybrids were also assessed for MAO inhibition, but resulted in lower activity than expected, ascribed to the low accessibility of the propargyl groups to the enzyme active site. Finally, the effects of the compounds on the viability of neuroblastome cells stressed with Aβ42 and H2O2 showed moderate cell protection. Overall, the performed studies illustrate the importance (and limitations) of enclosing several molecular scaffolds in one molecular entity to allow the modulation of multiple AD targets.
1. Introduction
Alzheimer's disease (AD) is one of the most devastating age-related neurodegenerative disorders with no cure so far, characterized by progressive memory loss and other cognitive impairments. The increase of life expectancy of humans and the exponential rise in the number of cases of AD support the need for developing an effective treatment for this frontline health problem.1 Although its etiology has not yet been well understood, patient brains are characterized by β-amyloid (Aβ) deposits (plaques) around neurons and neurofibrillary tangles (NFT) inside neurons,2 increased oxidative stress and disruption of metal homeostasis.3 Other prominent features of AD are disturbances in neurotransmitters, especially an acetylcholine (cholinergic) deficit due to its hydrolysis by acetylcholinesterase (AChE). Since around two decades ago, AD patients have been mainly treated with inhibitors of AChE (AChEI), but with limited therapeutic success. In fact they can provide amelioration of symptoms (improving patient memory) for a limited time window and for mild situations, but cannot alter the course of the disease.4 The multifactorial nature and the complexity of AD is believed to be the main reason for the absence of effective drugs, and has lead to the emergence of multitargeting drugs. Under this approach, one molecular entity is conveniently polyfunctionalized to enable its simultaneous binding interaction with several of the most important targets, but mostly focused on inhibition of AChE and Aβ-aggregation and/or on the control of the brain oxidative stress.5 Monoamine oxidase (MAO) has also been considered an important target for the treatment of AD,6 since it catalyzes the oxidative deamination of the monoamine based neurotransmitters, such as serotonine and dopamine, with the concomitant production of H2O2 that can promote the Fenton reaction and formation of reactive oxygen species (ROS).7In this context, the development of multitargeting drugs for treatment of AD has been a challenge for many research groups.8–10 Most of the multitarget drug design strategies have been driven by the recognized importance of maintaining the cholinergic functions (cholinergic hypothesis). Thus, they include a main molecular framework based on approved anti-AD drugs (AChE inhibitors), which is modified (hybridized) with other functional units to hit other disease targets. Among the USA FDA anti-AD drugs that are AChEI (e.g. tacrine, donepezil, rivastigmine, and galanthamine),11 tacrine was the first and most potent AChEI (discontinued due to side effects such as hepatotoxicity),12 with superiority in the structural modification, thus making it a privileged structure for incorporation into the overall molecular skeletons of the multi-target-directed drugs for the treatment of AD.13 In fact, we can find tacrine hybrids containing fragments of donepezil or galanthamine, while also a number of hybrids containing a structural fragment derived from natural based products such as alkaloids, flavonoids or others.
An important part of our current research has also been a focus on the development of acetylcholine esterase inhibitors functionalized for the inhibition/modulation of other AD targets, namely Aβ aggregation, oxidative stress and redox-active metal ions.14–16 Very recently, the conjugation of tacrine (TAC) with S-allylcysteine (SAC) and S-propargylcysteine (SPRC), two natural based products with neuroprotective and antioxidant roles (e.g. garlic derivatives),17,18 resulted in a set of bihybrids (TAC–SAC and TAC–SPRC) with important neuroprotecting properties which, besides the expected AChEI and anti-oxidant properties, presented also inhibitory activity for the self- and Cu-induced Aβ aggregation.19,20 Interestingly, N-propargylamine is also an important pharmacophore used in the design of MAO inhibitors.8,21 In this work we report the design, synthesis and biological evaluation of a new set of trihybrids (see Fig. 1) which, in addition to the functional groups presented in the above bihybrids, include also a benzothiazole (BTA) moiety to enhance the inhibition of Aβ aggregation, since BTA is a derivative of thioflavine T (ThT), a well known Aβ dye widely used in the design of AD drugs.14,22 Furthermore this new set of eight hybrids was designed by selection of suitable linkers to enable a dual mode binding, via the tacrine and BTA moieties, with both active binding sites of AChE. Besides the description of the design and synthetic procedures, we include herein the results and discussion of the bioassays, namely the inhibition of AChE, Aβ aggregation, MAOs and anti-oxidant properties, as well as their effect on the neuronal cell viability upon being subjected to model stressors of AD.
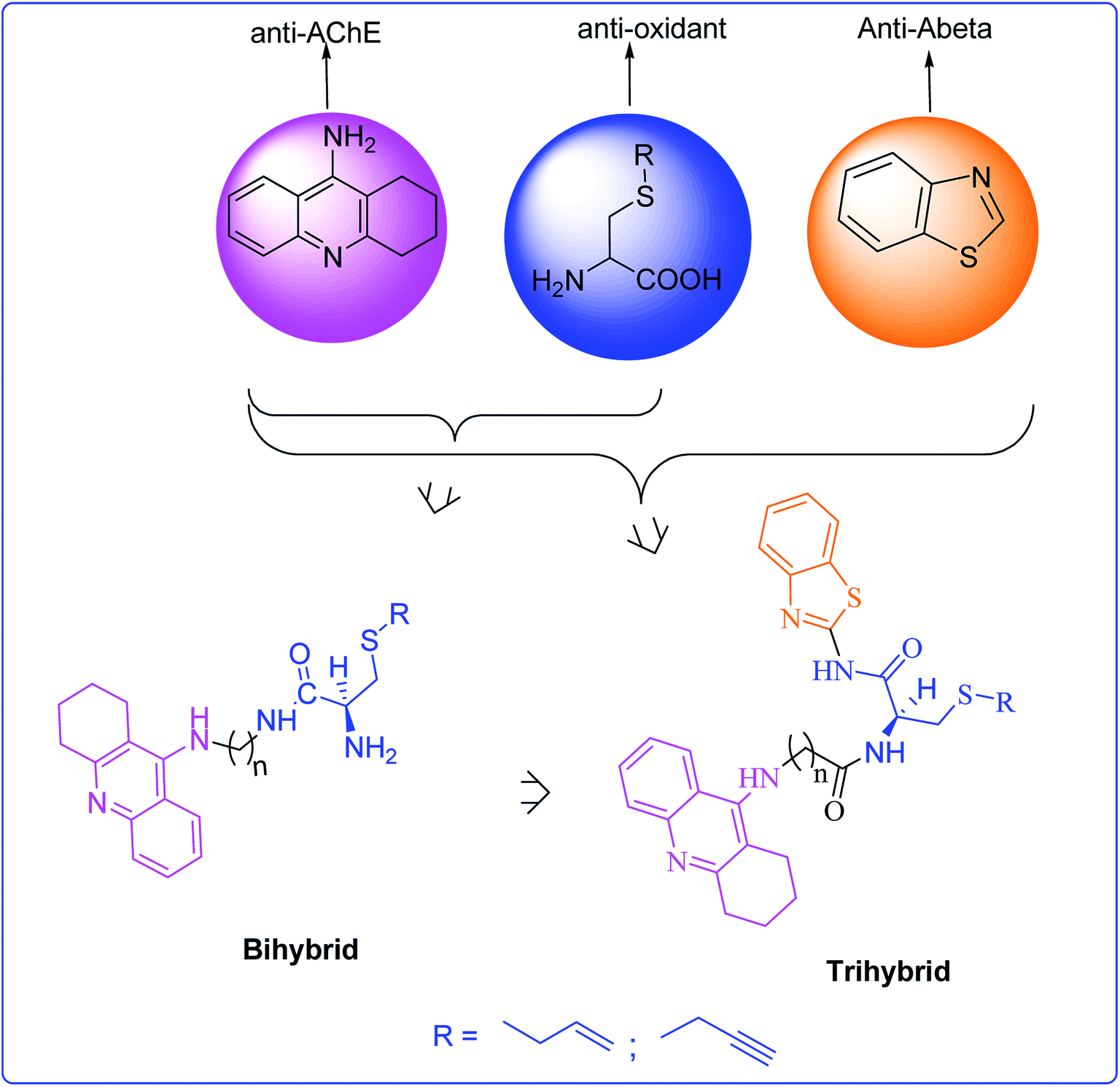 | ||
| Fig. 1 Design strategy for the novel trihybrids TAC–SAC–BTA and TAC–SPRC–BTA. | ||
2. Results and discussions
2.1. Molecular design and modeling
The design of the new trihybrids, to be subsequently prepared and studied (Fig. 2), involved a preliminary selection of the three molecular scaffolds, to address for main biological symptoms associated with AD, and then docking simulations were carried out to set the optimal length of the linker between the main frameworks.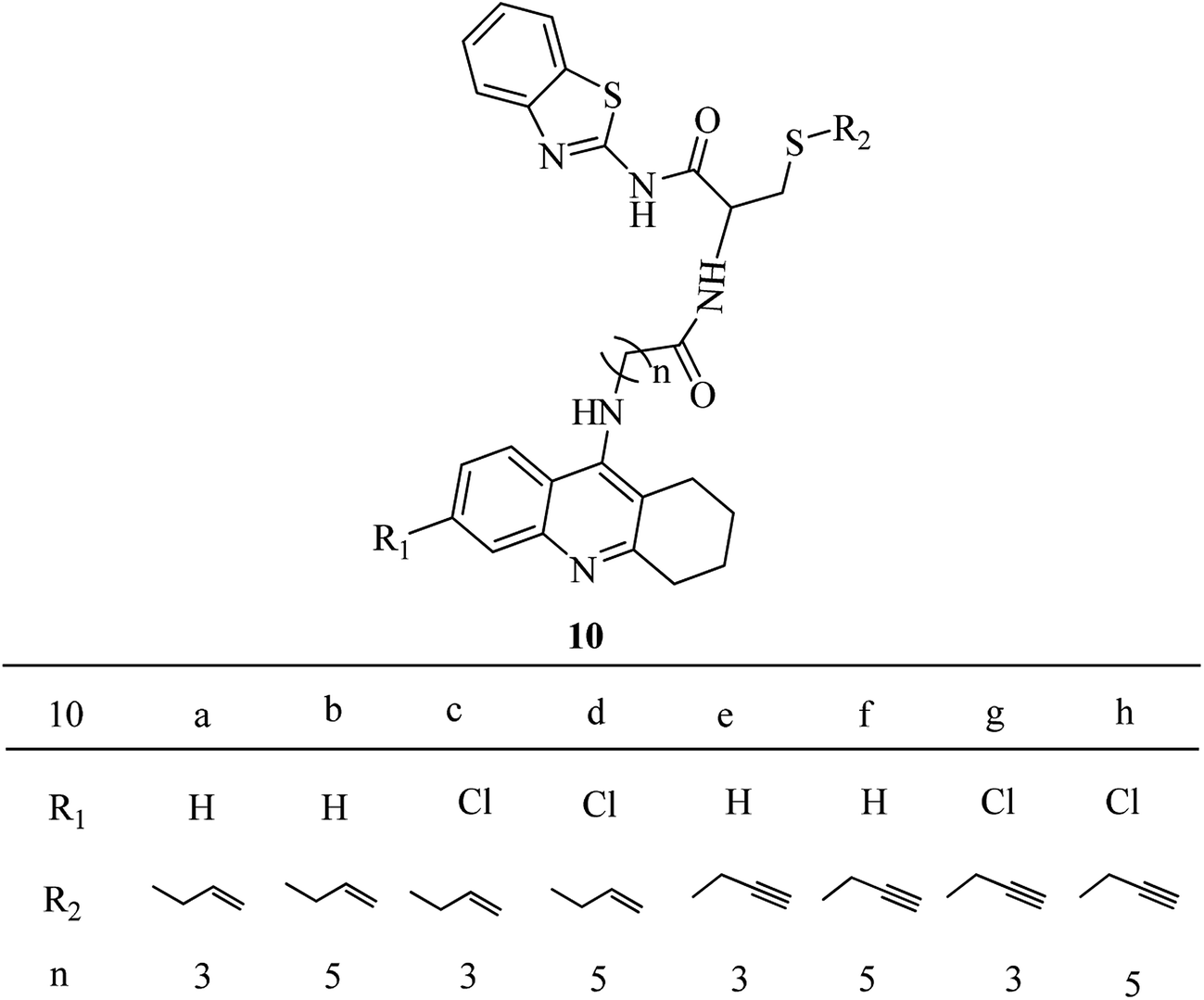 | ||
| Fig. 2 Synthesized compounds: TAC–SAC–BTA (R2 = allyl) and TAC–SPRC–BTA (R2 = propargyl). | ||
The selection of the main molecular moieties was based on our previous works, specifically: tacrine (TAC) and the natural based S-allylcysteine (SAC) or S-propargylcysteine (SPRC) to endow the hybrids with anti-AChE and anti-oxidant properties;19 benzothiazole (BTA) to promote the interaction with Aβ and anti-Aβ aggregation properties; on the other hand, an adequate linkage of TAC with BTA to warrant a dual binding mode within the active site of AChE and increase the enzymatic inhibition.14 Based on previous studies, an alkyl-amide linker could avoid linker strain and the length was selected based on docking simulation of the ligand into the enzyme, TcAChE. These studies also explained the interaction modes of the trihybrids with AChE. Preliminary docking results showed that three to five methylenes could provide the linker with adequate length to enable the dual mode interaction with the two molecular fragments (TAC, BTA) within the AChE gorge. A more detailed analysis of the simulation results shows that the trihybrids could simultaneously bind to both the catalytic anionic site (CAS) and the peripherical anionic site (PAS) (see representative results of the docking studies in Fig. 3).
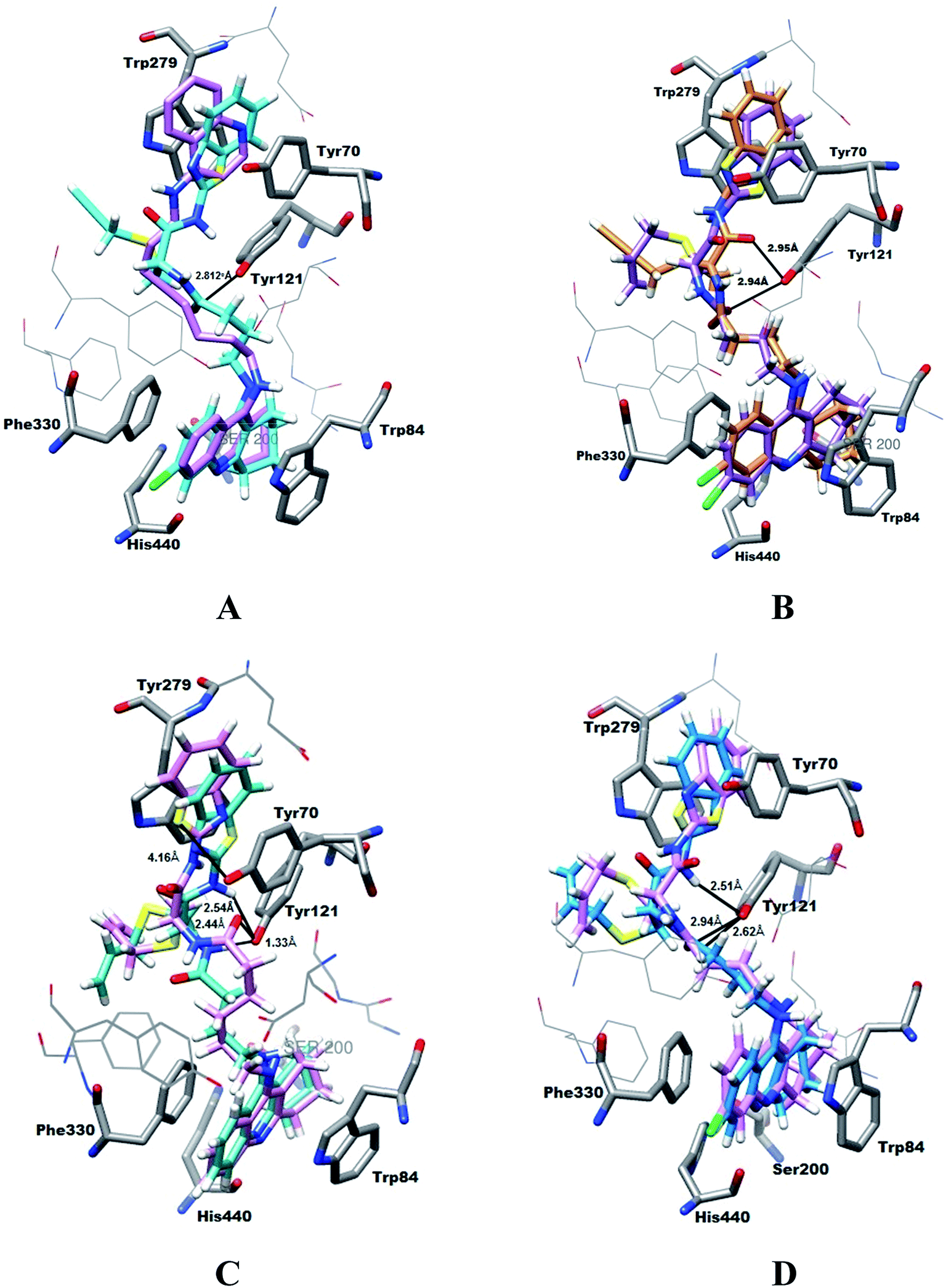 | ||
| Fig. 3 Docking results for the TAC–SAC–BTA and TAC–SPRC–BTA trihybrids with TcAChE: (A) superimposition of the original ligand (PDB entry 1ODC) (pink) and 10g (cyan); (B) superimposition of 10c (purple) and 10g (brown); (C) superimposition of 10b (pink) and 10a (cyan); (D) superimposition of 10c (pink) and 10a (blue). | ||
The tacrine appears inserted in the bottom of the enzyme gorge, adopting a parallel π–π stacking interaction with Trp84 and Phe330 of CAS, and blocking the accessibility of substract and water to the active site (formed by the “catalytic triad” Ser200, His440 and Glu327);23 the benzothiazole moiety occupies the PAS having a π–π interaction with Trp279 and Tyr70. The structure found for these hybrid–enzyme complex models is quite similar to the reported crystal structure of TcAChE complexed with a tacrine-quinoline inhibitor (N-4′-quinolyl-N′-9′′-(1′′,2′′,3′′,4′′-tetrahydroacridinyl)-1,8-diaminooctane; PDB entry 1ODC),24 as shown for 10g (Fig. 3A). These simulations also indicated the capacity of the linker amide group to establish H-bond interactions between its NH and the phenolic group of Tyr121. Interestingly, in opposition to that observed previously for the corresponding bihybrids (TAC–SAC and TAC–SPRC),19 all the trihybrids could accommodate the allyl and propargyl groups outwards the lipophylic part of PAS, which is left free for the stronger interaction with the BTA moiety. Furthermore, these allyl and propargyl groups do not seem to result in considerable differences on the ligand–enzyme interaction (see Fig. 3B for 10c and 10g). These simulations also illustrate that compounds with longer chains (n = 5), as 10b, have the BTA moiety slightly shifted outwards the PAS aromatic residues, which may result in somehow lower π–π interaction with this site than the shorter compounds (n = 3), as 10a (see Fig. 3C). Lastly, although 6-choro-tacrine is known for its higher activity, as compared with the corresponding tacrine derivatives, this was not clearly evidenced from the docking results, namely superimposition of 10c and 10a (see Fig. 3D). Notably, this chloro atom (green coloured) appears to be well accommodated in close distance of His440, which is thought to be the rationale for that enhancement on inhibitory activity, namely due to an H-bond interaction between its carbonyl oxygen and the tacrine pyridinium group.25
Overall, the modeling studies give some support to the expected ability of the trihybrids to interact with both the CAS and PAS of AChE, thus explaining some of the stronger calculated inhibitory activities, as compared with the corresponding bihybrids. However, since the binding modes found for these compounds were so similar to each other, their relative potency as inhibitors would be difficult to predict with confidence.
2.2. Chemistry
The designed tacrine–S-allylcysteine–benzothiazole (TAC–SAC–BTA) and tacrine–S-propargylcysteine–benzothiazole (TAC–SPRC–BTA) hybrids were synthesized according to a convergent synthetic approach shown in Scheme 1. The 9-chloro-1,2,3,4-tetrahydroacridine (3) was prepared from commercially available anthranilic acid as previously reported.26,27 Amino alkyl acids were attached to the 3a and 3b, using a catalytic amount of KI and phenol to obtain the intermediates 2-(1,2,3,4-tetrahydroacridine-9-ylamino) acids 4(a–d). The synthesis of the BTA–cysteine intermediates 9(a,b) involved four steps: firstly, L-cysteine (5) was S-alkylated with allyl bromide and propargyl bromide in the presence of NH4OH to yield the 3-allylsulfanyl-2-amino-propionic acid (6a) and 2-amino-3-prop-ynylsulfanyl-propionic acid (6b), respectively; their amino group was Boc-protected by reaction with di-tert-butyl dicarbonate (Boc) to yield compounds 7(a,b); these compounds were coupled with a BTA unit, through the condensation of the benzothiazo-2-amine (8) to their carboxylic group (T3P-activated), and subsequently the Boc group was released from the amine group with TFA to obtain the intermediates 9(a,b). Lastly the condensation of both intermediate compounds 4(a–d) and 9(a,b) in the presence of T3P and NMM afforded the ultimate novel hybrids 10(a–h). | ||
| Scheme 1 Reagents and conditions (i) POCl3, 180 °C, reflux, 3 h; (ii) 2 equiv. H2N(CH2)nCO2H, phenol, KI, reflux, 4 h; (iii) allyl bromide (iii-1) or propargyl bromide (iii-2), NH4OH (2 M), r.t., 3 h; (iv) di-tert-butyl dicarbonate and Na2CO3, THF, r.t., 5 h; (v) 1.5 equiv. T3P, 2.5 equiv. NMM, CH2Cl2, r.t., 3 h; (vi) TFA, CH2Cl2, r.t., 2 h; (vii) 1.5 equiv. T3P, 2.5 equiv. NMM, CH2Cl2, r.t., 3 h. | ||
2.3. Biological activity
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Compound code | R1 | R2 | n | AChE inhibition IC50a (μM) | MAO A inhibition IC50d (μM) | MAO B inhibition IC50d (μM) | Aβ42 self-aggr. inhib.b,c (%) | Aβ42 Cu-induc. aggr. inhib. (%) |
| a The values are mean of five independent experiments ±SD.b Inhibition of Aβ42 aggregation (in %) without and with copper (40 μM). The thioflavin-T fluorescence method was used, and the measurements were carried out in the presence of inhibitor (80 μM).c The values are the mean of two independent measurements in duplicate (SEM < 10%).d The values are the mean of two independent measurements each with 21 points (SEM < 15%). | ||||||||
| 10a | H |  |
3 | 0.37 ± 0.08 | 16.2 | 63 ± 8 | 57.8 | — |
| 10b | H |  |
5 | 0.37 ± 0.08 | 78.2 | — | ||
| 10c | Cl |  |
3 | 0.28 ± 0.07 | 10.8 | 55 | 31.6 | 56.6 |
| 10d | Cl | 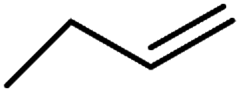 |
5 | 0.28 ± 0.07 | 14.0 | — | ||
| 10e | H |  |
3 | 0.35 ± 0.08 | 77.2 | — | ||
| 10f | H | 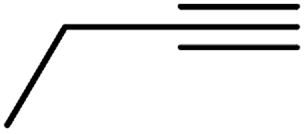 |
5 | 0.35 ± 0.08 | 56.3 | — | ||
| 10g | Cl | 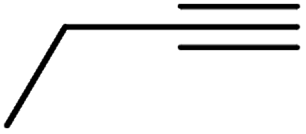 |
3 | 0.25 ± 0.06 | 27.7 | 85 | 24.1 | 37.6 |
| 10h | Cl | 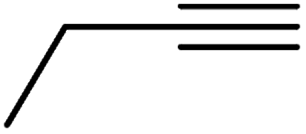 |
5 | 0.27 ± 0.06 | 77.1 | 95 | 19.3 | — |
| Tacrine | — | — | — | 0.46 ± 0.09 | 19.5 | — | ||
| Clorgyline | 0.42 × 10−3 | 10.6 | ||||||

Overall, comparison of the AChE inhibitory activity obtained for this series of trihybrids (TAC–SAC/SPRC–BTA) with that obtained previously for the corresponding bihybrid analogues (TAC–SAC/SPRC, see Table S1 in ESI†),19 showed that the inclusion of the third moiety (BTA) resulted in a considerable enhancement (up to one order of magnitude, with IC50 in sub-μM range), attributable to its stronger dual-binding mode with both the CAS and PAS of AChE, specifically due to the strong BTA-PAS π–π interaction.
The inhibition studies were carried out upon incubating Aβ42 in the presence and absence of the ligands. The results, summarized in Table 1, showed that all the compounds induced a decrease of the ThT fluorescence associated with the Aβ fibril binding, thus suggesting an inhibition of the Aβ42 self-aggregation process. In particular, all of them exhibited very good-to-moderate aggregation inhibitory capacity. Compounds 10b and 10e showed very good inhibitory capacity (78.2% and 77.2%, respectively), while other sets of compounds (10a, 10c, 10f) and (10d, 10g, 10h) exhibited, respectively, good and moderate activities. Comparison between the anti-Aβ aggregation capacities found for these trihybrids (TAC–SAC–BTA and TAC–SPRC–BTA) and the previously reported values for corresponding bihybrids (TAC–SAC and TAC–SPRC, see Table S1†)19 or tacrine itself, showed a general increase of up to 5-fold the percentage of inhibition for the same experimental conditions, which is clearly attributed to the further inclusion of the BTA moiety in these hybrids. Among the set of compounds studied herein, the chloro substitution resulted in the decrease of the interaction with Aβ, with apparent slightly stronger effect for compounds with longer chain size. In general, the allylic derivatives led to a somewhat better inhibition than the corresponding propargylic ones, which may be due to higher rigidity and increased lipophilicity (some water solubility limitations).
A survey on the effect of compounds on the inhibition of copper-induced Aβ aggregation was carried out for 10c and 10g. It was observed a higher inhibitory activity for the incubation of Aβ42 in the co-presence of copper (56.6, 37.6%) than that in its absence (31.6, 24.1%), thus suggesting that the ligands may modulate the metal-induced aggregation of Aβ42, similarly to previously reported for the analogues tacrine–SAC and TAC–SPRC.20 This can be rationalized by the metal-chelating properties of compounds which may compete with the amyloid peptide for the copper and decrease their aggregation ability, thus resulting in higher inhibition percentage. However, the enhancement of Aβ42 inhibitory activity due to the co-presence of copper observed for the trihybrids is lower than that previously observed for dihybrids (ca. 3.8 fold, see Table S1†),20 which may be attributed to the substitution of one N-amine by N-amide donor groups.
Without preincubation, the inhibition of MAO A was less, giving IC50 values as follows: 10a, 48.4 μM; 10c, 25.9 μM; 10g, 42.1 μM; 10h, 87.5 μM. Thus, preincubation decreased the IC50 only slightly (<3-fold) in contrast to the standard MAO A inhibitor, clorgyline, where the IC50 decreases 100-fold with 30 min preincubation.35 The very small difference in IC50 after 30 min preincubation was the same for both the allyl and propargyl compounds suggesting that the new propargyl derivatives do not inactivate MAO to any great extent. The high IC50 values obtained when inhibitor and substrate are added at the same time indicate low affinity binding, so it is likely that the propargyl group is not close enough to the N5 of the FAD in the active site of MAO to undergo enzyme-catalyzed activation.
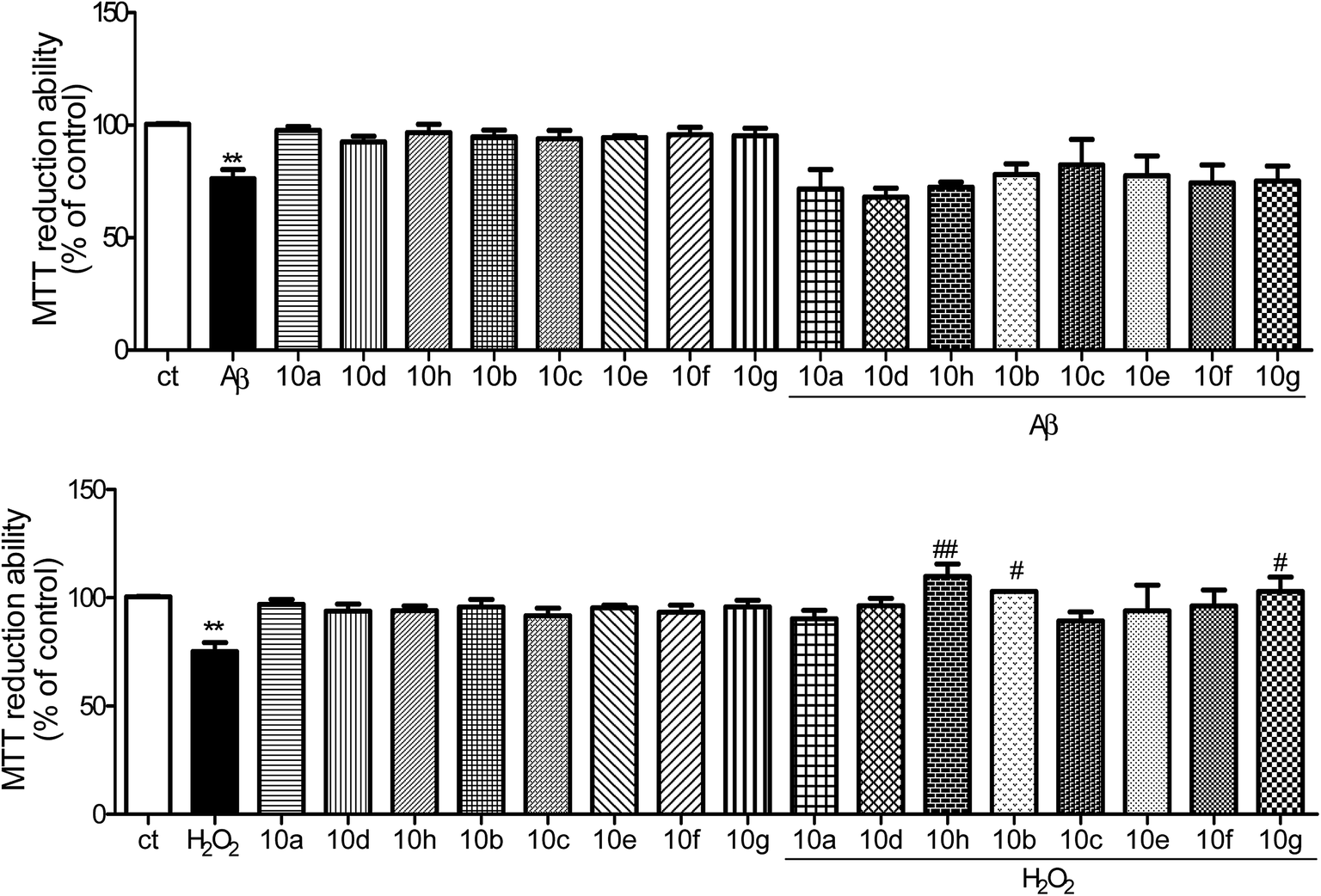 | ||
| Fig. 4 Effect of the described compounds on Aβ42 peptide or H2O2 toxicity in SH-SY5Y cells. SH-SY5Y cells were treated with Aβ42 (1 μM) or H2O2 (100 μM) for 24 h, in the absence or the presence of the compounds. Evaluation of cell viability was performed by using MTT reduction test. Results are expressed as the percentage of SH-SY5Y untreated cells, with the mean ± SEM derived from 3 different experiments. **p < 0.01, significantly different when compared with SH-SY5Y untreated cells; #p < 0.05; ##p < 0.01, significantly different when compared with H2O2 treated cells. | ||
Additionally, it is generally accepted that AD-related neurodegenerative processes involve oxidative stress that may be induced by H2O2. Indeed, H2O2 (100 μM, 24 h) induced a decrease in cell viability in SHSY-5Y cells (see Fig. 4B). Among the tacrine hybrids tested 10(b,g,h) were the most effective in preventing H2O2-mediated toxicity. These results are similar to those previously obtained with other tacrine hybrid compounds.19
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P), the ability to cross the BBB (logBB), the capacity to be absorbed through the intestinal tract to the blood (Caco-2 cell permeability), and the verification of Lipinski's rule of five, may give an idea of their drug-likeness for being orally active as anti-AD agents.
P), the ability to cross the BBB (logBB), the capacity to be absorbed through the intestinal tract to the blood (Caco-2 cell permeability), and the verification of Lipinski's rule of five, may give an idea of their drug-likeness for being orally active as anti-AD agents.All the compounds presented clogP values greater than five (Table S1†), thus with higher lipophilic character than recommended by Lipinski's rule.37 The compounds also have molecular weights higher than 500, which accounts for two violations of this rule. The high lipophilic character and the low BBB permeability (logBB) indicate that the compounds are not eligible as drug candidates for oral administration. However, they exhibited very good results in Caco-2 permeability, ranging from ca. 600 to 1400 nm s−1 (higher than 500 nm s−1 is considered good)36 with compounds 10e and 10g having lower cell permeabilities (according to the standard value). The other compounds presented greater values, indicating that the absorption through the intestinal tract to the blood is possible.
3. Conclusion
The conjugation of several functional moieties in one molecular entity has recently emerged as an important drug development strategy to combat the complex multifactorial Alzheimer's disease (AD). Following this strategy in this work, a series of eight novel trihybrids has been designed, prepared and bioassayed aimed at gathering information about potential anti-AD drug candidates. They incorporate three main moieties, tacrine (TAC), S-allyl- or S-propargyl-cysteine (SAC/SPRC) and benzothiazole (BTA), for inhibition/modulation of three main important AD targets: acetylcholinesterase inhibition, anti-oxidant activity and inhibition of amyloid β (Aβ) aggregation, with additional possible inhibition of the MAO enzyme. Analysis of the biological properties shown for these trihybrids was made in comparison with those recently reported for identical chemical library of bihybrids, lacking the BTA moiety. As hypothesized, the inclusion of the third moiety resulted in a considerable enhancement of the AChE inhibitory activity (up to one order of magnitude, with IC50 in sub-μM range, below that of the tacrine drug), attributed to its stronger dual-binding mode with both the CAS and PAS of AChE, specifically due to the strong BTA-PAS π–π interaction. Also, insertion of the BTA moiety on the trihybrids led to an enhancement of up to ca. 5-fold the percentage of self-Aβ aggregation inhibition, attributed to the well-known affinity of BTA derivatives for Aβ. Nevertheless, antioxidant activity of the trihybrids is lower than the bi-hybrids and similar to that of TAC. In addition, a preliminary study with some of these new trihybrids revealed only weak capacity for MAO inhibition, another known drug target for AD.Overall, a comparative analysis of some relevant anti-AD properties of the present trihybrids and those of the previous bihybrid analogues shows that conjugation of these three structural scaffolds in a molecular unit can provide a basis for the development of potential multifunctional drug candidates for Alzheimer's disease.
4. Experimental part
4.1. Chemistry
4.1.2.1. 4-(1,2,3,4-Tetrahydroacridin-9-ylamino)butanoic acid (4a). 9-Chloro-1,2,3,4-tetrahydroacridine (3a) and 4-aminobuytric acid afforded the pure title product as a light yellow colour solid; yield 62%; mp 157–160 °C; 1H NMR (MeOD) δ: 8.45 (d, J = 8.6 Hz, 1H, H-8), 7.85–7.73 (m, 2H, H-5 & H-6), 7.59–7.53 (m, 1H, H-7), 4.00 (t, J = 7.0 Hz, 2H, H-1′), 3.00 (brs, 2H, H-4), 2.69 (brs, 2H, H-1), 2.50 (t, J = 6.7 Hz, 2H, H-3′), 2.16–2.07 (m, 2H, H-2′), 1.96–1.93 (m, 4H, H-2 & H-3); MS-ESI (m/z): 285 (M + 1)+, 286 (M + 2)+.
4.1.2.2. 6-(1,2,3,4-Tetrahydroacridin-9-ylamino)hexanoic acid (4b). 9-Chloro-1,2,3,4-tetrahydroacridine (3a) and 6-aminohexanoic acid afforded the pure title product as a light yellow colour solid; yield 67%; mp 181–183 °C; 1H NMR (300 MHz, MeOD) δ 8.19 (d, J = 8.5 Hz, 1H, H-8), 8.04 (d, J = 8.4 Hz, 1H, H-5), 7.68–7.62 (m, 1H, H-6), 7.44–7.39 (m, 1H, H-7), 3.85 (t, J = 7.0 Hz, 2H, H-1′), 3.00 (brs, 2H, H-4), 2.59 (brs, 2H, H-1), 2.24 (t, J = 7.0 Hz, 2H, H-5′), 1.85–1.79 (m, 6H, H-2, H-3, & H-2′), 1.61–1.59 (m, 2H, H-4′), 1.42–1.39 (m, 2H, H-3′); MS-ESI (m/z): 313 ((M + 1)+)+, 314 (M + 2)+.
4.1.2.3. 4-(6-Chloro-1,2,3,4-tetrahydroacridin-9-ylamino)butanoic acid (4c). 6,9-Chloro-1,2,3,4-tetrahydroacridine (3b) and 4-aminobuytric acid afforded the pure title product as a light yellow colour solid; yield 59%; mp 230–232 °C; 1H NMR (MeOD) δ: 8.45 (d, J = 8.7 Hz, 1H, H-8), 7.76 (d, J = 1.9 Hz, 1H, H-5), 7.52 (dd, J = 7.2 &1.9 Hz, 1H, H-7), 4.00 (t, J = 6.9 Hz, 2H, H-1′), 3.00 (brs, 2H, H-4), 2.71 (brs, 2H, H-1), 2.50 (t, J = 8.1 Hz, 2H, H-3′), 2.20–2.13 (m, 2H, H-2′), 1.99–1.96 (m, 4H, H-2 & H-3); MS-ESI (m/z): 318 (M)+, 319 (M + 1)+.
4.1.2.4. 6-(6-Chloro-1,2,3,4-tetrahydroacridin-9-ylamino)hexanoic acid (4d). 6,9-Chloro-1,2,3,4-tetrahydroacridine (3b) and 6-aminohexanoic acid afforded the pure title product as a light yellow colour solid; yield 69%; mp 155–157 °C; 1H NMR (300 MHz, MeOD) δ 8.39 (d, J = 8.9 Hz, H-8), 7.82 (d, J = 2.1 Hz, 1H, H-5), 7.55 (dd, 1H, J = 7.0 & 2.1 Hz, H-7), 3.98 (t, J = 7.2 Hz, 2H, H-1′), 3.00 (br, 2H, H-4), 2.72 (br, 2H, H-3), 2.35 (t, J = 7.2 Hz, 2H, H-5′), 2.00–1.98 (m, 4H, H-2 & H-3); 1.94–1.86 (m, 2H, H-2′), 1.73–1.65 (m, 2H, H-4′), 1.55–1.49 (m, 2H, H-3′); MS-ESI (m/z): 346 (M+), 347 (M + 1)+.
4.1.3.1. (R)-3-Allylsulfanyl-2-amino-propionic acid (6a). L-Cysteine (5) and allyl bromide afforded the pure title product as a white powder; yield 75%; mp 220–221 °C; 1H NMR (300 MHz, D2O) δ: 5.96–5.79 (m, 1H, HC
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 5.29–5.19 (m, 2H, CH2CH), 3.94–3.82 (m, 1H, CHNH2), 3.21 (d, J = 7.2 Hz, 2H, CH2-allyl), 3.00–2.94 (m, 2H, CH2CHN); MS-ESI (m/z): 160 (M − 1)+, 162 (M + 1)+.
C), 5.29–5.19 (m, 2H, CH2CH), 3.94–3.82 (m, 1H, CHNH2), 3.21 (d, J = 7.2 Hz, 2H, CH2-allyl), 3.00–2.94 (m, 2H, CH2CHN); MS-ESI (m/z): 160 (M − 1)+, 162 (M + 1)+.
4.1.3.2. (R)-2-Amino-3-prop-ynylsulfanyl-propionic acid (6b). L-Cysteine (5) and propargyl bromide afforded the pure title product as a white shiny solids; yield 72%; mp 294–296 °C; 1H NMR (300 MHz, D2O) δ: 3.83–3.90 (m, 1H, CHNH2), 3.32–3.30 (m, 2H, CH2-propargyl), 3.24–3.01 (m, 2H, CH2CHN), 2.61 (t, J = 2.5 Hz, 1H, HCC); MS-ESI (m/z): 160 (M + 1)+.
4.1.4.1. (S)-3-(Allylthio)-2-(tert-butoxycarbonylamino)propanoic acid (7a). 3-Allylsulfanyl-2-amino-propionic acid (6a) was treated with Boc anhydride to afford the pure title product as semi solid; yield 90%; 1H NMR (300 MHz, DMSO-d6) δ: 7.1 (d, J = 8.3 Hz, 1H, HNCH), 5.79–5.69 (m, 1H, HC
C), 5.14–5.06 (m, 2H, CH2HC), 4.06–4.01 (m, 1H, CHNH), 3.15 (d, J = 7.1 Hz, 2H, CH2-allyl), 2.82–2.61 (m, 2H, CH2CHN), 1.38 (s. 9H, tBu![[H with combining low line]](https://www.rsc.org/images/entities/i_char_0048_0332.gif) ); MS-ESI (m/z): 261 (M+).
); MS-ESI (m/z): 261 (M+).
4.1.4.2. (S)-2-(tert-Butoxycarbonylamino)-3-(prop-2-ynylthio)propanoic acid (7b). 2-Amino-3-prop-ynylsulfanyl-propionic acid (6b) and Boc anhydride afforded the pure title product as semi solid; yield 83%; 1H NMR (300 MHz, DMSO) δ: 7.15 (d, J = 8.4 Hz, 1H, HNCH), 4.15–4.08 (1H, m, HCNH), 3.41–3.39 (m, 2H, CH2-propargyl), 3.08–3.81 (m, 2H, CH2CHN), 2.67 (t, J = 2.5 Hz, 1H, HCC), 1.41 (s. 9H, tBu
![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) ); MS-ESI (m/z): 259 (M+).
); MS-ESI (m/z): 259 (M+).
4.1.5.1. tert-Butyl-3-(allylthio)-1-(benzo[d]thiazol-2-ylamino)-1-oxopropan-2-yl-carbamate (8a). 2-Aminobenzothiazole (8) and N-Boc protected SAC (7a) afforded the pure title product as an colorless solid with 78% yield; mp 83–85 °C; 1H NMR (300 MHz, CDCl3) δ: 7.82–7.83 (m, 2H, H-4 & H-7), 7.43 (t, J = 8.0 Hz, 1H, H-5 or H-6), 7.31 (t, J = 8.0 Hz, 1H, H-6 or H-5), 5.73–5.65 (m, 1H, HC
C), 5.11–5.07 (m, 2H, H2CCH), 4.66–4.55 (m, 1H, HCNH), 3.08 (d, J = 7.1 Hz, 2H, CH2-allyl), 2.98–2.95 (m, 2H, CH2CHN), 1.46 (s. 9H, tBu); MS-ESI (m/z): 393 (M+).
4.1.5.2. tert-Butyl-1-(benzo[d]thiazol-2-ylamino)-1-oxo-3-(prop-2-ynylthio)propan-2-yl-carbamate (8b). 2-Aminobenzothiazole (8) and N-Boc protected SPRC (7b) afforded the pure title product as an colourless solid; yield 83%; mp 133–135 °C; 1H NMR (300 MHz, CDCl3) δ: 7.82–7.85 (m, 2H, H-4 & H-7), 7.45 (t, J = 8.0 Hz, 1H, H-5 or H-6), 7.31 (t, J = 8.0 Hz, 1H, H-6 or H-5), 4.82–4.80 (1H, m, HCNH), 3.26–3.22 (m, 4H, CH2-propargyl, & CH2CHN), 2.26 (s, 1H, HCC), 1.41 (s, 9H, tBu
); MS-ESI (m/z): 391 (M+).
4.1.6.1. 3-(Allylthio)-2-amino-N-(benzo[d]thiazol-2-yl)propanamide (9a). tert-Butyl-3-(allylthio)-1-(benzo[d]thiazol-2-ylamino)-1-oxopropan-2-yl-carbamate (8a) and TFA afforded the pure title product as a colourless solid; yield 86%; mp 105–107 °C; 1H NMR (CDCl3) δ: 7.78–7.82 (m, 2H, H-4 & H-7), 7.44 (t, J = 8.0 Hz, 1H, H-5 or H-6), 7.29 (t, J = 8.0 Hz, 1H, H-6 or H-5), 5.71–5.22 (m, 1H, HC
C), 5.13–5.20 (m, 2H, H2CCH), 3.79 (brs, 1H, HCNH), 3.08–3.15 (m, 3H, H2C-allyl & H2CCHN), 3.09–2.80 (m, 1H, H2CCHN); MS-ESI (m/z): 293 (M+).
4.1.6.2. 2-Amino-N-(benzo[d]thiazol-2-yl)-3-(prop-2-ynylthio)propanamide (9b). tert-Butyl-1-(benzo[d]thiazol-2-ylamino)-1-oxo-3-(prop-2-ynylthio)propan-2-yl-carbamate (8b) and TFA afforded the pure title product as a colourless solid; yield 89%; mp 112–114 °C; 1H NMR (DMSO) δ: 7.98 (d, J = 7.8 Hz, 1H, H-4) 7.75 (d, J = 7.8 Hz, 1H, H-7), 7.45 (t, J = 8.0 Hz, 1H, H-5 or H-6), 7.32 (t, 1H, J = 8.0 Hz, H-6 or H-5), 3.76 (t, J = 6.7 Hz, 1H, HCNH), 3.19 (s, J = 6.7 Hz, 2H, CH2-propargyl), 2.97–2.86 (m, 3H, –CH2CHN & HCC); MS-ESI (m/z): 291 (M+).
4.1.7.1. N-(3-(Allylthio)-1-(benzo[d]thiazol-2-ylamino)-1-oxopropan-2-yl)-4-(1,2,3,4-tetrahydroacridin-9-ylamino)butanamide (10a). 3-(Allylthio)-2-amino-N-(benzo[d]thiazol-2-yl)propanamide (9a) and 4-(1,2,3,4-tetrahydroacridin-9-ylamino)butanoic acid (4a) afforded the pure title product as a light yellow color solid; yield 65%; mp 95–102 °C; 1H NMR (400 MHz, MeOD-d4) δ: 8.33–8.34 (m, 1H, H-8), 7.75–7.78 (m, 2H, H-4′′′ & H-7′′′), 7.60–7.64 (m, 2H, H-5 & H-6), 7.47–7.52 (m, 1H, H-5′′′ or H-6′′′), 7.40–7.41 (m, 1H, H-6′′′ or H-5′′′), 7.29–7.30 (m, 1H, H-7), 5.70–5.80 (m, 1H, CH-allyl), 5.14 (d, 1H, J = 16.0 Hz, CH2-allyl), 5.08 (d, 1H, J = 8.0 Hz, CH2-allyl), 4.72 (t, J = 8.0 Hz, 1H, H-2′′), 3.88–3.90 (m, 2H, H-1′), 3.11–3.18 (m, 3H, –SCH2-allyl & H-3′′), 2.94–2.99 (m, 3H, H-4 & H-3′′), 2.74 (brs, 2H, H-1), 2.55 (t, J = 8.0 Hz, 2H, H-3′), 2.10–2.15 (m, 2H, H-2′), 1.85 (brs, 4H, H-2 & H-3); 13C NMR (100.5 MHz, DMSO-d6) δ: 172.8, 171.0, 158.1, 157.3, 151.6, 148.9, 145.9, 134.6, 131.9, 129.0, 126.6, 124.1, 123.9, 123.8, 122.2, 121.0, 120.0, 117.9, 115.7, 53.0, 47.9, 34.4, 34.2, 33.1, 32.8, 31.9, 31.7, 26.8, 25.3, 22.9, 22.5; MS-ESI (m/z): 558 (M − 1)+, 559 (M+), 560 (M + 1)+; analysis calc. for C30H33N5O2S2·0.52EtOH: C 62.83, H 6.06, N 12.01, S 11.00%; found: C 63.05, H 6.21, N 11.61%.
4.1.7.2. N-(3-(Allylthio)-1-(benzo[d]thiazol-2-ylamino)-1-oxopropan-2-yl)-6-(1,2,3,4-tetrahydroacridin-9-ylamino)hexanamide (10b). 3-(Allylthio)-2-amino-N-(benzo[d]thiazol-2-yl)propanamide (9a) and 6-(1,2,3,4-tetrahydroacridin-9-ylamino)hexanoic acid (4b) afforded the pure title product as a light yellow color solid; yield 71%; mp 96–100 °C; 1H NMR (400 MHz, MeOD-d4) δ: 8.13 (d, J = 8.0 Hz, 1H, H-8), 7.67–7.75 (m, 2H, H-4′′′ & H-7′′′), 7.59 (t, J = 8.0 Hz, 1H, H-5), 7.39 (t, J = 8.0 Hz, 1H, H-6), 7.36–7.37 (m, 2H, H-5′′′ & H-6′′′), 7.25 (t, J = 8.0 Hz, 1H, H-7), 5.50–5.78 (m, 1H, CH-allyl), 5.13 (d, 1H, J = 20.0 Hz,
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/i_char_e001.gif) CH2-allyl), 5.06 (d, 1H, J = 8.0 Hz, CH2-allyl), 4.75 (t, 1H, J = 8.0 Hz, H-2′′), 3.61 (t, 2H, J = 8.0 Hz, H-1′), 3.15 (d, 2H, J = 8.0 Hz, CH2-allyl), 2.93–2.96 (m, 3H, H-4 & H-3′′), 2.86–2.88 (m, 1H, H-3′′), 2.68 (brs, 2H, H-1), 2.30 (t, J = 8.0 Hz, 2H, H-5′), 1.87 (brs, 4H, H-2 & H-3), 1.66–1.73 (m, 4H, H-2′ & H-4′), 1.42–1.45 (m, 2H, H-3′); 13C NMR (100.5 MHz, MeOD-d4) δ: 174.7, 170.9, 159.1, 155.5, 153.1, 148.4, 144.1, 133.9, 131.9, 129.5, 125.8, 124.3, 123.7, 123.6, 123.4, 120.8, 120.2, 116.6, 53.1, 51.8, 35.0, 34.2, 31.6, 31.4, 30.3, 29.3, 25.9, 24.9, 24.3, 22.4, 21.7; MS-ESI (m/z): 588 (M+), 589 (M + 1)+, 590 (M + 2)+; analysis calc. for C32H37N5O2S2·0.20EtOH·0.5H2O: C 63.42, H 6.65, N 11.56%; found: C 63.01, H 6.62, N 11.37%.
CH2-allyl), 5.06 (d, 1H, J = 8.0 Hz, CH2-allyl), 4.75 (t, 1H, J = 8.0 Hz, H-2′′), 3.61 (t, 2H, J = 8.0 Hz, H-1′), 3.15 (d, 2H, J = 8.0 Hz, CH2-allyl), 2.93–2.96 (m, 3H, H-4 & H-3′′), 2.86–2.88 (m, 1H, H-3′′), 2.68 (brs, 2H, H-1), 2.30 (t, J = 8.0 Hz, 2H, H-5′), 1.87 (brs, 4H, H-2 & H-3), 1.66–1.73 (m, 4H, H-2′ & H-4′), 1.42–1.45 (m, 2H, H-3′); 13C NMR (100.5 MHz, MeOD-d4) δ: 174.7, 170.9, 159.1, 155.5, 153.1, 148.4, 144.1, 133.9, 131.9, 129.5, 125.8, 124.3, 123.7, 123.6, 123.4, 120.8, 120.2, 116.6, 53.1, 51.8, 35.0, 34.2, 31.6, 31.4, 30.3, 29.3, 25.9, 24.9, 24.3, 22.4, 21.7; MS-ESI (m/z): 588 (M+), 589 (M + 1)+, 590 (M + 2)+; analysis calc. for C32H37N5O2S2·0.20EtOH·0.5H2O: C 63.42, H 6.65, N 11.56%; found: C 63.01, H 6.62, N 11.37%.
4.1.7.3. N-(3-(Allylthio)-1-(benzo[d]thiazol-2-ylamino)-1-oxopropan-2-yl)-4-(6-chloro-1,2,3,4-tetrahydroacridin-9-ylamino)butanamide (10c). 3-(Allylthio)-2-amino-N-(benzo[d]thiazol-2-yl)propanamide (9a) and 4-(6-chloro-1,2,3,4-tetrahydroacridin-9-ylamino)butanoic acid (4c) afforded the pure title product as a light yellow color solid; yield 61%; mp 195–198 °C; 1H NMR (400 MHz, MeOD-d4) δ: 8.15 (d, J = 8.0 Hz, 1H, H-8), 7.80 (d, J = 8.0 Hz, 1H, H-5) 7.62–7.67 (m, 2H, H-4′′′ & H-7′′′), 7.40 (t, J = 8.0 Hz, 1H, H-7), 7.27–7.33 (m, 2H, H-5′′′ & H-6′′′), 5.70–5.80 (m, 1H, CH-allyl), 5.13 (d, 1H, J = 20.0 Hz, CH2-allyl), 5.07 (d, 1H, J = 8.0 Hz, CH2-allyl), 4.73 (t, J = 8.0 Hz, 1H, H-2′′), 3.68 (t, 2H, J = 8.0 Hz, H-1′), 3.14 (d, 2H, J = 8.0 Hz, –SCH2-allyl), 2.91–294 (m, 1H, H-3′′), 2.83–2.86 (m, 3H, H-4 & H-3′′), 2.67 (brs, 2H, H-1), 2.47 (t, J = 8.0 Hz, 2H, H-3′), 2.05–2.06 (m, 2H, H-2′), 1.84 (brs, 4H, H-2 & H-3); 13C NMR (100.5 MHz, DMSO-d6) δ: 172.8, 171.0, 158.6, 158.0, 151.6, 148.9, 147.6, 134.6, 133.5, 131.9, 126.7, 126.3, 124.2124.1, 122.2, 121.0, 117.9, 115.9, 53.0, 47.9, 34.2, 33.2, 32.8, 31.9, 26.8, 25.3, 22.8, 22.4; MS-ESI (m/z): 594 (M+), 595 (M + 1)+, 596 (M + 2)+; analysis calc. for C30H32N5O2S2Cl·0.70H2O: C 59.38, H 5.55, N 11.54%; found: C 59.07, H 5.55, N 11.36%.
4.1.7.4. N-(3-(Allylthio)-1-(benzo[d]thiazol-2-ylamino)-1-oxopropan-2-yl)-6-(6-chloro-1,2,3,4-tetrahydroacridin-9-ylamino)hexanamide (10d). 3-(Allylthio)-2-amino-N-(benzo[d]thiazol-2-yl)propanamide (9a) and 6-(6-chloro-1,2,3,4-tetrahydroacridin-9-ylamino)hexanoic acid (4d) afforded the pure title product as a light yellow color solid; yield 68%; mp 94–98 °C; 1H NMR (400 MHz, MeOD-d4) δ: 8.11 (d, J = 8.0 Hz, 1H, H-8), 7.76 (d, J = 8.0 Hz, 1H, H-4′′′) 7.69–7.71 (m, 2H, H-5 & H-7′′′), 7.40 (t, J = 8.0 Hz, 1H, H-5′′′ or H-6′′′), 7.32–7.35 (m, 1H, H-7), 7.27 (t, J = 8.0 Hz, 1H, H-6′′′ or H-5′′′), 5.72–5.79 (m, 1H, CH-allyl), 5.14 (d, 1H, J = 20.0 Hz,
CH2-allyl), 5.08 (d, 1H, J = 12.0 Hz, CH2-allyl), 4.75 (t, J = 8.0 Hz, 1H, H-2′′), 3.59 (t, 2H, J = 8.0 Hz, H-1′), 3.15 (d, 1H, J = 4.0 Hz, CH2-allyl), 2.92–2.98 (m, 3H, H-4 & H-3′′), 2.80–2.83 (m, 1H, H-3′′), 2.67 (brs, 2H, H-1), 2.30 (t, J = 8.0 Hz, 2H, H-3′), 1.88 (brs, 4H, H-2 & H-3), 1.66–1.73 (m, 4H, H-2′ & H-4′), 1.40–1.48 (m, 2H, H-3′); 13C NMR (100.5 MHz, DMSO-d6) δ: 172.9, 171.2, 159.7, 158.3, 150.9, 148.9, 148.0, 134.6, 132.9, 131.9, 127.1, 126.5, 125.9, 123.8, 121.0, 119.0, 117.9, 116.3, 53.0, 48.3, 35.3, 34.2, 33.9, 32.0, 30.7, 26.3, 25.4, 25.3, 23.0, 22.7; MS-ESI (m/z): 622 (M+), 623 (M + 1)+, 624 (M + 2)+; analysis calc. for C32H36N5O2S2Cl·0.79 EtOH: C 61.24, H 6.23, N 10.63%; found: C 61.08, H 6.38, N 10.30%.
4.1.7.5. N-(1-(Benzo[d]thiazol-2-ylamino)-1-oxo-3-(prop-2-ynylthio)propan-2-yl)-4-(1,2,3,4-tetrahydroacridin-9-ylamino)butanamide (10e). 2-Amino-N-(benzo[d]thiazol-2-yl)-3-(prop-2-ynylthio)propanamide (9b) and 4-(1,2,3,4-tetrahydroacridin-9-ylamino)butanoic acid (4a) afforded the pure title product as a light yellow color solid; yield 63%; mp 105–110 °C; 1H NMR (400 MHz, MeOD-d4) δ: 8.17 (d, J = 8.0 Hz, 1H, H-8), 7.78 (d, J = 8.0 Hz, H-4′′′) 7.64–7.70 (m, 2H, H-5 & H-7′′′), 7.56 (t, J = 8.0 Hz, 1H, H-6), 7.39 (t, 2H, J = 8.0 Hz, H-5′′′ & H-6′′′), 7.27 (t, J = 8.0 Hz, 1H, H-7), 4.82 (t, 1H, J = 8.0 Hz, H-2′′), 3.69 (t, 2H, J = 8.0 Hz, H-1′), 3.35 (s, 2H, –SCH2-propargyl), 3.21–3.26 (m, 1H, H-3′′), 3.01–3.06 (m, 1H, H-3′′), 2.90 (brs, 2H, H-4), 2.72 (brs, 2H, H-1), 2.60 (s, 1H, –CH-propargyl), 2.48 (t, J = 8.0 Hz, 2H, H-3′), 2.04–2.09 (m, 2H, H-3′), 1.86 (brs, 4H, H-2 & H-3); 13C NMR (100.5 MHz, DMSO-d6) δ: 172.8, 171.0, 158.2, 157.7, 151.1, 148.9, 146.6, 131.9, 128.6, 127.7, 126.6, 124.1, 123.8, 123.6, 122.19, 121.0, 120.3, 116.1, 80.6, 74.4, 52.8, 48.0, 33.4, 32.8, 26.9, 25.4, 23.1, 22.7, 19.1; MS-ESI (m/z): 556 (M − 1)+, 557 (M+), 558 (M + 1)+; analysis calc. for C30H31N5O2S2·0.23 EtOH: C 63.00, H 5.66, N 12.32, S 11.28%; found: C 64.02, H 5.79, N 12.18%.
4.1.7.6. N-(1-(benzo[d]thiazol-2-ylamino)-1-oxo-3-(prop-2-ynylthio)propan-2-yl)-6-(1,2,3,4-tetrahydroacridin-9-ylamino)hexanamide (10f). 2-Amino-N-(benzo[d]thiazol-2-yl)-3-(prop-2-ynylthio)propanamide (9b) and 6-(1,2,3,4-tetrahydroacridin-9-ylamino)hexanoic acid (4b) afforded the pure title product as a light yellow color solid; yield 75%; mp 96–100 °C; 1H NMR (400 MHz, MeOD-d4) δ: 8.30 (d, J = 8.0 Hz, 1H, H-8), 7.78 (m, 2H, H-4′′′ & H-7′′′), 7.50–7.54 (m, 1H, H-5), 7.39 (t, 2H, J = 8.0 Hz, H-6), 7.26 (t, J = 8.0 Hz, 1H, H-5′′′ or H-6′′′), 7.15 (t, J = 8.0 Hz, 1H, H-6′′′ or H-5′′′), 6.75–6.80 (m, 1H, H-7), 4.82 (t, 1H, J = 8.0 Hz, H-2′′), 3.86 (t, 2H, J = 8.0 Hz, H-1′), 3.37 (s, 2H, –SCH2-propargyl), 3.18–3.23 (m, 1H, H-3′′), 3.01–3.07 (m, 1H, H-3′′), 2.93 (brs, 2H, H-4), 2.63–2.64 (m, 3H, H-1 & –CH-propargyl), 2.35 (t, J = 8.0 Hz, 2H, H-5′), 1.91 (brs, 4H, H-2 & H-3), 1.79–187 (m, 2H, H-2′), 1.69–1.73 (m, 2H, H-4′), 1.48–1.53 (m, 2H, H-3′); 13C NMR (100.5 MHz, DMSO-d6) δ: 173.0, 171.1, 158.3, 158.0, 151.0, 148.9, 146.9, 131.9, 128.5, 128.3, 126.6, 124.0, 123.7, 123.6, 122.1, 121.0, 120.4, 116.0, 80.6, 74.3, 52.8, 48.3, 35.3, 33.7, 32.9, 30.8, 26.3, 25.4, 25.3, 23.1, 22.8, 19.1; MS-ESI (m/z): 584 (M − 1)+, 585 (M+), 586 (M + 1)+; analysis calc. for C30H31N5O2S2·0.51EtOH: C 64.06, H 6.13, N 11.49, S 10.52%; found: C 64.01, H 5.85, N 11.20%.
4.1.7.7. N-(1-(Benzo[d]thiazol-2-ylamino)-1-oxo-3-(prop-2-ynylthio)propan-2-yl)-4-(6-chloro-1,2,3,4-tetrahydroacridin-9-ylamino)butanamide (10g). 2-Amino-N-(benzo[d]thiazol-2-yl)-3-(prop-2-ynylthio)propanamide (9b) and 4-(6-chloro-1,2,3,4-tetrahydroacridin-9-ylamino)butanoic acid (4c) afforded the pure title product as a light yellow color solid; yield 63%; mp 119–122 °C; 1H NMR (400 MHz, MeOD-d4) δ: 8.19 (d, J = 8.0 Hz, 1H, H-8), 7.80 (d, J = 8.0 Hz, 1H, H-4′′′), 7.61–7.65 (m, 2H, H-5 & H-7′′′), 7.41 (t, J = 8.0 Hz, 1H, H-5′′′ or H-6′′′), 7.35 (d, 1H, J = 8.0 Hz, H-7), 7.30 (t, J = 8.0 Hz, 1H, H-6′′′ or H-5′′′), 4.80 (t, 1H, J = 8.0 Hz, H-2′′), 3.73 (t, 2H, J = 8.0 Hz, H-1′), 3.36 (s, 2H, –SCH2-propargyl), 3.18–3.23 (m, 1H, H-3′′), 3.03–3.07 (m, 1H, H-3′′), 2.84–2.86 (m, 2H, H-4), 2.67 (brs, 2H, H-1), 2.62 (s, 1H, –CH-propargyl), 2.49 (t, J = 8.0 Hz, 2H, H-3′), 2.07–2.10 (m, 2H, H-3′), 1.85 (brs, 4H, H-2 & H-3); 13C NMR (100.5 MHz, MeOD-d4) δ: 174.4, 170.3, 158.1, 155.7, 153.4, 148.3, 144.1, 135.7, 131.9, 126.0, 125.8, 124.3, 123.6, 122.3, 120.8, 120.3, 116.6, 114.2, 78.9, 71.6, 53.0, 49.3, 32.6, 32.1, 30.9, 29.2, 25.8, 24.1, 22.1, 18.5; MS-ESI (m/z): 592 (M+), 593 (M + 1)+, 594 (M + 2)+; analysis calc. for C30H30N5O2S2Cl·0.98H2O·0.9EtOH: C 58.64, H 5.78, N 10.75%; found: C 58.06, H 5.34, N 11.07%.
4.1.7.8. N-(1-(Benzo[d]thiazol-2-ylamino)-1-oxo-3-(prop-2-ynylthio)propan-2-yl)-6-(6-chloro-1,2,3,4-tetrahydroacridin-9-ylamino)hexanamide (10h). 2-Amino-N-(benzo[d]thiazol-2-yl)-3-(prop-2-ynylthio)propanamide (9b) and 6-(6-chloro-1,2,3,4-tetrahydroacridin-9-ylamino)hexanoic acid (4d) afforded the pure title product as a light yellow color solid; yield 70%; mp 98–106 °C; 1H NMR (400 MHz, MeOD-d4) δ: 8.10 (d, J = 8.0 Hz, 1H, H-8), 7.75 (d, J = 8.0 Hz, 1H, H-4′′′), 7.68–7.70 (m, 2H, H-5 & H-7′′′), 7.39 (t, J = 8.0 Hz, 1H, H-5′′′ or H-6′′′), 7.33 (d, 1H, J = 8.0 Hz, H-7), 7.27 (t, J = 8.0 Hz, 1H, H-6′′′ or H-5′′′), 4.83 (t, 1H, J = 8.0 Hz, H-2′′), 3.59 (t, 2H, J = 8.0 Hz, H-1′), 3.36 (s, 2H, –SCH2-propargyl), 3.18–3.23 (m, 1H, H-3′′), 3.03–3.06 (m, 1H, H-3′′), 2.92 (brs, 2H, H-4), 2.66 (brs, 2H, H-1), 2.62 (m, 1H, –CH-propargyl), 2.32 (t, J = 8.0 Hz, 2H, H-5′), 1.88 (brs, 4H, H-2 & H-3), 1.67–1.73 (m, 4H, H-2′ & H-4′), 1.42–1.48 (m, 2H, H-3′); 13C NMR (100.5 MHz, DMSO-d6) δ: 173.0, 170.9, 158.2, 158.0, 151.8, 148.8, 146.4, 133.7, 131.9, 126.6, 126.3, 125.5, 124.1, 122.1, 121.0, 118.2, 115.5, 80.6, 74.3, 52.8, 48.1, 35.2, 32.9, 30.6, 26.3, 25.3, 25.2, 22.8, 22.3, 19.1; MS-ESI (m/z): 620 (M+), 620 (M + 1)+, 621 (M + 2)+; analysis calc. for C32H34N5O2S2Cl·1.1EtOH: C 61.23, H 6.10, N 10.44%; found: C 61.57, H 6.15, N 9.87%.
4.2. Molecular modeling
To perform our docking studies the X-ray crystallographic structure of Torpedo californica-AChE (TcAChE) complexed with an inhibitor was taken from RCSB Protein Data Bank (PDB entry 1ODC), in order to be used as receptor. This structure was chosen because of the similarity between its original inhibitor (N-4′-quinolyl-N′-9′′-(1′′,2′′,3′′,4′′-tetrahydroacridinyl)-1,8-diaminooctane) and our ligands: all have a tacrine moiety and an aromatic group linked through an alkyl chain.39 The original structure was treated using Maestro v. 9.3 (ref. 40) by removing the original ligand, solvent, and cocrystallization molecules, and then adding the hydrogen atoms. The ligands were built using Maestro, and then, using Ghemical v. 2.0 (ref. 41) they were submitted to random conformational search (RCS) of 1000 cycles, and 2500 optimization steps using Tripos 5.2 force field.42 The minimized ligands were docked into the AChE structure with GOLD software v. 5.1,43 and the zone of interest was defined as the residues within 10 Å from the original position of the ligand in the crystal structure. The ‘allow early termination’ option was deactivated, and the remaining default parameters of Gold were used. The ligands were subjected to 100 genetic algorithm steps using ASP as fitness function.4.3. Prediction of pharmacokinetic properties
To analyze the potential of the new compounds as new anti-AD drugs, a brief prediction on pharmacokinetic proprieties was performed in silico. Parameters such as the lipo-hydrophilic character (clogP), blood–brain barrier partition coefficient (logBB), the ability to be absorbed through the intestinal tract (Caco-2 cell permeability) and CNS activity were calculated.
The ligands were built and minimized as previously mentioned for the docking studies. The structures were submitted to the calculation of these relevant pharmacokinetic proprieties and descriptors using QikProp v. 2.5.36 These predictions are for orally delivered drugs and assume nonactive transport.
4.4. Antioxidant activity
The antioxidant activity was evaluated by the DPPH method previously described.31 To a 2.5 mL solution of DPPH (0.002%) in methanol, four samples of each compound in solution were added in different volumes to obtain different concentrations in a 3.5 mL final volume. The samples were incubated for 30 min at room temperature.The absorbance was measured at 517 nm against the corresponding blank (methanol). The antioxidant activity was calculated by an eqn (1).44
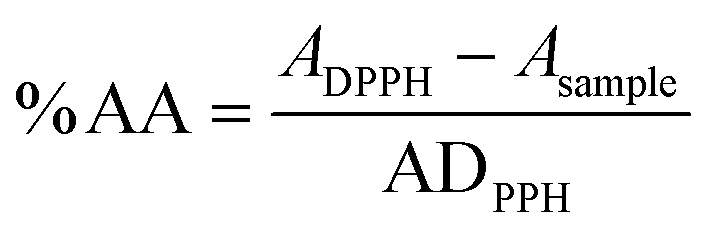 | (1) |
The tests were carried out in triplicate. The compound concentrations providing 50% of antioxidant activity (EC50) were obtained by plotting the antioxidant activity against the compound concentration.
4.5. Acetylcholinesterase activity
The enzymatic activity TcACHE was measured using an adaptation of the method previously described.15 The assay solution contained 374 μL of Hepes buffer (50 mM and pH8.0), a variable volume (10–50 μL) of the stock solution of each compound in methanol (1 mg mL−1), 25 μL of AChE stock solution, and the necessary amount of methanol to attain 0.925 mL of the sample mixture in a 1 mL cuvette. The samples were left to incubate for 15 min, and then, 75 μL of acetylthiocholine iodide (AChI) solution (16 mM) and 476 μL of DTNB (3 mM), were added. The reaction was monitored for 5 min at 405 nm. Assays were run with a blank containing all the components except AChE, which was replaced by Hepes buffer. The velocities of the reaction were calculated as well as the enzyme activity. A control reaction was carried out using the sample solvent (methanol) in the absence of any tested compound, and it was considered as 100% activity. The percentage inhibition of the enzyme activity due to the presence of increasing test compound concentration was calculated by the following eqn (2)| % I = 100 − (vi/vo × 100) | (2) |
4.6. Inhibition of self-mediated and Cu(II)-induced Aβ (1–42) aggregation
Amyloid-β peptide (1–42) (Aβ42) was purchased from Aldrich as a lyophilized powder and stored at −20 °C. The samples were treated with 1,1,1,3,3,3-hexafluoropropan-2-ol (HFIP) to avoid self-aggregation and reserved. HFIP pretreated Aβ42 samples were re-solubilized with a CH3CN/Na2CO3 (300 μM)/NaOH (250 μM) (48.3:48.3:4.3, v/v/v) solvent mixture in order to have a stable stock solution. This Aβ42 alkaline solution (500 μM) was diluted in phosphate buffer (0.215 M, pH 8.0) to obtain a 40 μM solution. Due to the hydrophobic nature of the compounds under study, they were first dissolved in methanol (1 mg mL−1) and then further diluted in phosphate buffer to a final concentration of 480 μM.
To study the Aβ42 aggregation inhibition, a reported method, based on the fluorescence emission of thioflavin T (ThT), was followed.16,28,29 It is based on the assumption that reduction of the ThT fluorescence, due to competition of the compounds for the peptide interaction, imply reduced Aβ aggregation. To study the effects of the compounds on copper-induced Aβ1–42 aggregation, solutions of Cu(II) (200 μM) in phosphate buffer (0.2 M, pH 8.0) were prepared from a CuCl2 stock solution (0.015 M). The assay consisted on the incubation of Aβ1–42 (40 μM) with or without Cu(II) (40 μM) in phosphate buffer with or without the ligands (80 μM). The incubation was performed at 37 °C, for 24 h, and for the control, a sample of the peptide was incubated under identical conditions but without the inhibitor. After incubation, the samples were added to a 96-well plate (BD Falcon) with 180 μL of 5 μM ThT in 50 mM glycine-NaOH (pH 8.5) buffer. Blank samples were prepared for each concentration in a similar way, devoid of peptide. After 5 min incubation with the dye, the ThT fluorescence was measured using a Spectramax Gemini EM (Molecular Devices) at the following wavelengths: 446 nm (excitation) and 485 nm (emission). The percent inhibition of aggregation due to the presence of the test compound was calculated by eqn (3), in which IFi and IF0 corresponded to the fluorescence intensities, in the presence and the absence of the test compound, respectively, minus the fluorescence intensities due to the respective blanks. The reported values were obtained as the mean ± SEM of duplicate of two different experiments.
| I% = 100 − (IFi/IF0 × 100) | (3) |
4.7. MAOs inhibition
MAO activity was measured in 96-well plates (triplicate wells) by coupling the production of hydrogen peroxide is via horseradish peroxidase to a dye producing the fluorescent resorufin.35,45 Membrane-bound human MAO A and MAO B, horseradish peroxidase, and Ampliflu Red were obtained from Sigma-Aldrich (UK). For reversible inhibition, MAO was added last to the assay mixture containing (at final concentrations) 20 μM Ampliflu Red, 1 U mL−1 horseradish peroxidase, 1 mM tyramine. To see whether there was irreversible inhibition; MAO and inhibitor were preincubated in 100 μL phosphate buffer pH 7.5 at 30 °C for 30 min before adding 100 μL containing the rest of the assay components to start the reaction. Solubility limited the maximum concentration of inhibitor to 100 μM. The data were analyzed in Graphpad PRISM 4 using the sigmoidal equation constrained to a bottom value of zero.4.8. Cells and treatments
Human neuroblastoma SH-SY5Y cell line was purchased from ATCC-CRL-2266. Briefly, the cells were grown in Dulbecco's modified Eagle's medium (DMEM) obtained from Gibco-Invitrogen (Life Technologies Ltd, UK) with 10% heat inactivated fetal calf serum, containing 50 U mL−1 penicillin, and 50 μg mL−1 streptomycin, under a humidified atmosphere of 95% air–5% CO2 at 37 °C. Cells were plated at 0.1 × 106 cells per mL for MTT cell proliferation assay. Medium was changed after 24 h and immediately before treatments.The tested compounds (10a, 10b, 10c, 10d, 10e, 10f, 10g, 10h) were dissolved in DMSO at a concentration of 25 mM and aliquots were stored at −20 °C. The compounds 10a, 10d and 10h were added to the medium at 2.5 μM final concentration; the compounds 10b, 10c, 10e, 10f, 10g were added to the medium at 1 μM final concentration; KC1 was added to the medium at 10 nM final concentration. The final concentration of DMSO in culture media did not exceed 0.05% (v/v) and no alterations on cells were observed. Cells were pre-incubated for 1 h with the compounds and then incubated with Aβ42 or H2O2 for another 24 h. Aβ42 was prepared as 276.9 μM stocks in sterile water and added to the medium to 1 μM final concentration. H2O2 was freshly prepared as 5 mM stocks in water and added to the medium to 100 μM final concentrations. Aβ42 was purchased from American Peptide (Sunnyvale, CA, USA) and H2O2 from Sigma Chemical Co (St. Louis, MO, USA). For all conditions tested, control experiments were performed in which the compounds tested, Aβ42 or H2O2 were not added.
4.9. MTT cell proliferation assay
Cell proliferation was determined by the colorimetric MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay.46 In viable cells, MTT is metabolized into a formazan that absorbs light at 570 nm. Following the cell treatment protocol the medium was aspirated and 0.5 mL MTT (0.5 mg mL−1) was added to each well. The plate was then incubated at 37 °C for 3 h. At the end of the incubation period the formazan precipitates were solubilized with 0.5 mL of acidic isopropanol (0.04 M HCl/isopropanol). The absorbance was measured at 570 nm. Cell reduction ability was expressed as a percentage of the untreated cells.All data result from the analysis of duplicates per experimental condition in at least three independent experiments and are expressed as the mean ± SEM. Statistical analyses were performed using GraphPad Prism 5 (GraphPad Software, San Diego, CA, USA). Statistical tests between multiple data sets and conditions were carried out using a one-way analysis of variance (ANOVA) with pairwise multiple comparison procedures using the post hoc Bonferroni's test to determine statistical significance, as appropriate. A p value < 0.05 was considered statistically significant.
Acknowledgements
The authors acknowledge the Portuguese Fundação para a Ciência e Tecnologia (FCT) for the project UID/QUI/00100/2013, postdoctoral fellowships (RSK and KC), and also the Portuguese NMR (IST-UL Center) and Mass Spectrometry Networks (Node IST-CTN) for providing access to their facilities. We also thank the doctoral fellowship from Erasmus Namaste program (AH) and P.-L. Joffrin for his contribution to the MAO bioassays at the University of St Andrews.References
- Alzheimer's Association, Alzheimer's Dement., 2014, 10, 47 Search PubMed.
- S. Oddo, A. Caccamo, J. D. Shepherd, M. P. Murphy, T. E. Golde, R. Kayed, R. Metherate, M. P. Mattson, Y. Akbari and F. M. LaFerla, Neuron, 2003, 39, 409 CrossRef CAS PubMed.
- S. Ayton, P. Lei and A. I. Bush, Free Radical Biol. Med., 2013, 62, 76 CrossRef CAS PubMed.
- J. L. Cummings, M. Travis and K. Zhong, Alzheimer's Res. Ther., 2014, 6, 37 CrossRef PubMed.
- A. Cavalli, M. L. Bolognesi, A. Minarini, M. Rosini, V. Tumiatti, M. Recanatini and C. Melchiorre, J. Med. Chem., 2008, 51, 347 CrossRef CAS PubMed.
- H. Zheng, T. Amit, O. Bar-Am, M. Fridkin, M. B. H. Youdim and S. A. Mandel, J. Alzheimer's Dis., 2012, 30, 1 CrossRef CAS PubMed.
- A. Mattevi, C. Binda, M. Li and F. Hubálek, Curr. Med. Chem., 2004, 11, 1983 CrossRef.
- M. B. Youdim and J. J. Buccafusco, Trends Pharmacol. Sci., 2005, 26, 27 CrossRef CAS PubMed.
- E. Nepovimova, E. Uliassi, J. Korabecny, L. E. Peña-Altamira, S. Samez, A. Pesaresi, G. E. Garcia, M. Bartolini, V. Andrisano, C. Bergamini, R. Fato, D. Lamba, M. Roberti, K. Kuca, B. Monti and M. L. Bolognesi, J. Med. Chem., 2014, 57, 8576 CrossRef CAS PubMed.
- R. Leon, A. G. Garcia and J. Marco-Contelles, Med. Res. Rev., 2013, 33, 139 CrossRef CAS PubMed.
- G. Small and R. Bullock, Alzheimer's Dement., 2011, 7, 177 CrossRef CAS PubMed.
- M. L. Crismon, Ann. Pharmacother., 1994, 28, 744 CAS.
- A. Romero, R. Cacabelos, M. J. Oset-Gasque, A. Samadi and J. Marco-Contelles, Bioorg. Med. Chem. Lett., 2013, 23, 1916 CrossRef CAS PubMed.
- R. S. Keri, C. Quintanova, S. M. Marques, A. R. Esteves, S. M. Cardoso and M. A. Santos, Bioorg. Med. Chem., 2013, 21, 4559 CrossRef CAS PubMed.
- A. Nunes, S. M. Marques, C. Quintanova, D. F. Silva, S. M. Cardoso, S. Chaves and M. A. Santos, Dalton Trans., 2013, 42, 6058 RSC.
- C. Quintanova, R. S. Keri, S. M. Marques, M. G-Fernandes, S. M. Cardoso, M. L. Serralheiro and M. A. Santos, Med. Chem. Commun., 2015, 6, 1969 RSC.
- A. Dairam, R. Fogel, S. Daya and J. L. Limson, J. Agric. Food Chem., 2008, 56, 3350 CrossRef CAS PubMed.
- J. M. Kim, H. J. Chang, W. K. Kim, N. Chang and H. S. Chun, J. Agric. Food Chem., 2006, 54, 6547 CrossRef CAS PubMed.
- R. S. Keri, C. Quintanova, S. Chaves, D. F. Silva, S. M. Cardoso and M. A. Santos, Chem. Biol. Drug Des., 2016, 87, 101 CAS.
- C. Quintanova, R. S. Keri, S. Chaves and M. A. Santos, J. Inorg. Biochem., 2015, 151, 58 CrossRef CAS PubMed.
- C. Lu, Q. Zhou, J. Yan, Z. Du, L. Huang and X. Li, Eur. J. Med. Chem., 2013, 62, 745 CrossRef CAS PubMed.
- S. Noel, S. Cadet, E. Gras and C. Hureau, Chem. Soc. Rev., 2013, 42, 7747 RSC.
- H. Dvir, I. Silman, M. Harel, T. L. Rosenberry and J. L. Sussman, Chem.–Biol. Interact., 2010, 187, 10 CrossRef CAS PubMed.
- PDB, entry 1ODC, http://www.rcsb.org/pdb/explore/explore.do?structureId=1ODC.
- E. H. Rydberg, B. Brumshtein, H. M. Greenblatt, D. M. Wong, D. Shaya, L. D. Williams, P. R. Carlier, Y. Pang, I. Silman and J. L. Sussman, J. Med. Chem., 2006, 49, 5491 CrossRef CAS PubMed.
- L. J. Sargent and L. Small, J. Org. Chem., 1946, 11, 359 CrossRef CAS PubMed.
- M. K. Hu and C. F. Lu, Tetrahedron Lett., 2000, 41, 1815 CrossRef CAS.
- M. Bartolini, C. Bertucci, M. L. Bolognesi, A. Cavalli, C. Melchiorre and V. Andrisano, ChemBioChem, 2007, 8, 2152 CrossRef CAS PubMed.
- X. Chao, X. He, Y. Yang, X. Zhou, M. Jin, S. Liu, Z. Cheng, P. Liu, Y. Wang, J. Yu, Y. Tan, Y. Huang, J. Qin and S. Rapposelli, Bioorg. Med. Chem. Lett., 2012, 22, 6498 CrossRef CAS PubMed.
- A. Hawe, M. Sutter and W. Jiskoot, Pharm. Res., 2007, 25, 1487 CrossRef PubMed.
- B. Tepe, D. Daferera, A. Sokmen, M. Sokmen and M. Polissiou, J. Agric. Food Chem., 2005, 90, 333 CAS.
- Q. H. Gong, Q. Wang, L. L. Pan, X. H. Liu, H. Xin and Y. Z. Zhu, Brain, Behav., Immun., 2011, 25, 110 CrossRef CAS PubMed.
- S. M. Denecke, Curr. Top. Cell. Regul., 2000, 36, 151 Search PubMed.
- M. J. Zhou, Z. J. Diwu, N. PanchukVoloshina and R. P. Haugland, Anal. Biochem., 1997, 253, 162 CrossRef CAS PubMed.
- G. Esteban, J. Allan, A. Samadi, A. Mattevi, M. Unzeta, J. Marco-Contelles, C. Binda and R. R. Ramsay, Biochim. Biophys. Acta, Proteins Proteomics, 2014, 1844, 1104 CrossRef CAS PubMed.
- QikProp, version 2.5, Schrödinger, LLC, New York, NY, 2005 Search PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3 CrossRef CAS.
- W. L. F. Armarego and D. D. Perrin, Purification of Laboratory Chemicals, Butterworth-Heinemann, Oxford, 4th edn, 1999 Search PubMed.
- E. H. Rydberg, B. Brumshtein, H. M. Greenblatt, D. M. Wong, D. Shaya, L. D. Williams, P. R. Carlier, Y. Pang, I. Silman and J. L. Sussman, J. Med. Chem., 2006, 49, 5491 CrossRef CAS PubMed.
- Maestro, version 9.3, Schrödinger Inc., Portland, OR, 2012 Search PubMed.
- T. Hassinen and M. J. Peräkylä, Comput. Chem., 2001, 22, 1229 CrossRef CAS.
- M. Clark, R. D. Cramer III and N. Van Opdenbosch, J. Comput. Chem., 2004, 10, 982 CrossRef.
- G. Jones, P. Willett, R. C. Glen, A. R. Leach and R. Taylor, J. Mol. Biol., 1997, 67, 727 CrossRef PubMed.
- J. Sebestík, S. M. Marques, P. L. Falé, S. Santos, D. M. Arduíno, S. M. Cardoso, C. R. Oliveira, M. L. Serralheiro and M. A. Santos, J. Enzyme Inhib. Med. Chem., 2011, 26, 485 CrossRef PubMed.
- R. R. Ramsay, A. Olivieri and A. Holt, J. Neural Transm., 2011, 118, 1003 CrossRef CAS PubMed.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra03455a |
| This journal is © The Royal Society of Chemistry 2016 |