
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotocatalytic hydroalkylation of 3-methyleneisoindolin-1-ones with unactivated alkyl iodides†
Abhishek
Kumar
,
Shrutheka
G. S.
and
Veera Reddy
Yatham
 *
*
School of Chemistry, Indian Institute of Science Education and Research, Thiruvananthapuram 695551, India. E-mail: reddy@iisertvm.ac.in
First published on 20th March 2025
Abstract
We report herein a simple method for hydroalkylation of 3-methyleneisoindolin-1-ones with unactivated iodoalkanes using visible light photocatalysis and a halogen atom transfer (XAT) process. This operationally simple method exhibits broad substrate scope and allows late-stage modifications of iodoalkanes derived from either active pharmaceutical ingredients or natural products, producing a range of structurally diverse and valuable corresponding hydroalkylation products in decent yields. The generation of alkyl radicals and carbanion intermediates was directly proven in the catalytic cycle through radical trapping/radical clock and isotope labeling studies, respectively.
Haloalkanes are ubiquitous, found in many synthetic intermediates and natural products and commonly employed as coupling partners in transition metal catalysis forging carbon–carbon and carbon–heteroatom bonds.1 Also, the utilization of haloalkanes as alkyl radical intermediates to achieve valuable and structurally diverse complex molecules is of great importance in synthetic organic chemistry.2 Based on the mechanism, alkyl radicals can be generated from haloalkanes by two major processes: (i) reduction through the single-electron transfer (SET) process; (ii) homolytic cleavage of a C–X bond through a halogen-atom transfer (XAT) process. Transition metal catalysis and photoredox catalysis are reliable systems for single electron reduction of haloalkanes to alkyl radicals.3 Also, significant efforts have been made towards the XAT-process of haloalkanes using tin, silicon, and trialkylborane-O2-based reagents.4 However, considering certain disadvantages associated with early methods recently, the Doyle and Leonori groups developed an efficient strategy for the XAT-process under visible light photocatalysis. Under mild reaction conditions, simple trialkyl amines are converted to nucleophilic α-aminoalkyl radicals, which can generate alkyl radicals through homolytic cleavage of alkyl C–X bonds.5 Furthermore, a few groups have also reported employing amine-boranes and boryl radicals as XAT-reagents that can generate alkyl radicals from haloalkanes.6
Isoindolinone is an important heterocyclic compound, found in many natural products and medicinally relevant molecules.7 Specifically, the 3-substituted-isoindolin-1-ones are some of the most privileged scaffolds, received well in the field of pharmaceutical and material chemistry.8 Due to their potential applications, various protocols for their synthesis have been reported using organometallic reagents, metal catalysis, or metal-free conditions.9 Notably, very recently, a few groups independently reported photoinduced reductive 1,5-HAT intramolecular cyclization of aryl halides, which leads to 3-substituted-isoindolin-1-one derivatives (Scheme 1a).10 All the reported methods use either metals, organometallic reagents, or pre-functionalized substrates. In the present work, we report a metal-free redox-neutral α-amino alkyl radical-mediated hydroalkylation of 3-methylene-isoindolin-1-ones (Scheme 1b).
 | ||
| Scheme 1 Known strategies for photoinduced reductive 1,5-HAT intramolecular cyclization of aryl halides (a); photocatalytic hydroalkylation of 3-methyleneisoindolin-1-one with unactivated alkyl iodides (b). | ||
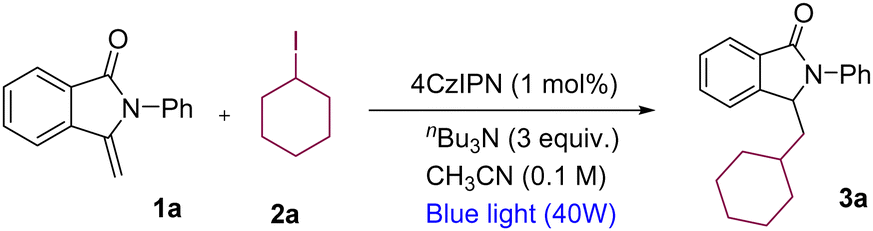
The initial investigation started with 3-methylene-2-phenylisoindolin-1-one 1a and iodocyclohexane 2a as an alkylating reagent in the presence of organic dye (4CzIPN) and nBu3N as an XAT-reagent in acetonitrile (as reaction medium) under 40 W blue LED irradiation at 45–50 °C. Gratifyingly, 3-(cyclohexylmethyl)-2-phenylisoindolin-1-one product 3a could be delivered in 72% yield using 0.1 mmol of 1a, 0.2 mmol of 2a, 3 equiv. of nBu3N and 1 mol% of 4CzIPN in acetonitrile (1 mL) (Table 1, entry 1). Several other commonly available organophotocatalysts and metal-based photocatalysts did not give better results than 4CzIPN (see ESI†). Moderate yields of the product were observed when employing other solvents (EtOAc, DMSO, DMF and 1,4-dioxane) as the reaction medium and amines (DIPEA and Et3N) as the XAT-reagent (Table 1, entries 2–7). Low yields of the products were observed upon lowering the amount of amine and iodocyclohexane (Table 1, entries 8 and 9). Employing amines that are not able to generate α-aminoalkyl radicals in our reaction conditions did not produce any product (Table 1, entry 10). Finally, controlled reactions were conducted in the absence of light, photocatalyst, and amine, which signified the importance of these components in our reaction conditions (Table 1, entries 11–13).
|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | 3a (%) |
| a NMR yields using 1,3,5-trimethoxy benzene as an internal standard. b Isolated yield. | ||
| 1 | None | 72 (66)b |
| 2 | Using EtOAc | 42 |
| 3 | Using DMSO | 51 |
| 4 | Using DMF | 51 |
| 5 | Using 1,4-dioxane | 56 |
| 6 | Using DIPEA | 31 |
| 7 | Using Et3N | 44 |
| 8 | Using nBu3N (2 equiv.) | 48 |
| 9 | Using 2a (1.5 equiv.) | 61 |
| 10 | Using DBU/HE/TMP/DABCO | 0 |
| 11 | No light | 0 |
| 12 | No amine | 0 |
| 13 | No 4CzIPN | 0 |

|
||
With the best conditions in hand, further studies focused on the scope of unactivated iodoalkanes with 3-methyleneisoindolin-1-one derivatives (Scheme 2). Various secondary six-membered ring unactivated iodoalkanes with and without heteroatoms reacted with 3-methylene-2-phenylisoindolin-1-one 1a to produce the corresponding hydroalkylation products (3a–3e) in moderate to good yields (45–66%). A variety of N-protected (N-Boc, N-Cbz, N-Ts, N-Bz) 4-iodopiperidines were smoothly converted to the corresponding hydroalkylated products (3f–3i) in good yields (53–83%). 5-Membered and 4-membered ring and bicyclic secondary iodoalkanes were well-tolerated under our conditions (3j–3m, 50–75%). Acyclic secondary iodoalkanes and tertiary iodoalkanes produced the corresponding hydroalkylation products (3n–3q, 57–64%) in good yields. A variety of simple primary alkyl iodides containing different functionalities (ether and silyl) worked well and afforded the corresponding hydroalkylation products (3r–3u) in good yields (54–68%). Next, testing different polyhaloalkanes such as CHCl3, CDCl3, and bromodifluoroacetate, bromodifluoroethanol and its derivative reacted smoothly with 1a and afforded the hydroalkylation products (3v–3z) in good yields (40–73%). To further showcase the potential applications of this methodology, we carried late-stage modification of a variety iodoalkanes derived from either active pharmaceutical ingredients or natural products. Indeed, a large variety of such molecules, such as ciprofibrate, gemfibrozil, naproxen and ibuprofen-derived iodoalkanes containing different functionalities, were converted to the corresponding alkyl radicals and participated in the hydro alkylation of 3-methyleneisoindolin-1-one (1a) in decent yields (3aa–3ad, 49–65%). To our surprise, employing isoxepac-derived alkyl iodide in our reaction conditions resulted in a low yield (30%) of the unexpected product 3ae.
 | ||
Scheme 2 Substrate scope of a variety of unactivated alkyliodides. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1 (0.2 mmol), 2 (0.4 mmol) and CH3CN (2 mL) at 45–50 °C, 10 h. bReaction was conducted using 1 mmol of 1a and 2 mmol of 2a for 48 h. cReaction time 15 h. dUsing the corresponding bromoalkane. eWith the corresponding iodoalkane. 1 (0.2 mmol), 2 (0.4 mmol) and CH3CN (2 mL) at 45–50 °C, 10 h. bReaction was conducted using 1 mmol of 1a and 2 mmol of 2a for 48 h. cReaction time 15 h. dUsing the corresponding bromoalkane. eWith the corresponding iodoalkane. | ||
Next, we tested the generality of the 3-methylene isoindolin-1-one derivatives (Scheme 3). Electron donating and electron-withdrawing N-aryl-substituted 3-methyleneisoindolin-1-ones containing different functionalities and sterically hindered N-aryl-substituted 3-methyleneisoindolin-1-ones reacted well with 4-iodotetrahydro-2H-pyran (2c), affording the hydroalkylation products (3af–3an) in moderate to good yields (51–86%). N-Alkyl, N-benzyl, and N-cycloalkyl-substituted 3-methyleneisoindolin-1-ones smoothly converted to the corresponding hydroalkylation products (3ao–3as) in good yields (45–75%). Finally, a heterocyclic amine (benzothiazol-2-amine), a medicinally relevant amine (leelamine) and 1-phenylethan-1-amine-derived 3-methyleneisoindolin-1-ones worked well in our reaction conditions and afforded the corresponding hydroalkylation products (3at–3av) in good yields.
 | ||
| Scheme 3 Substrate scope of 3-methyleneisoindolin-1-one derivatives. a1a (0.2 mmol), 2c (0.4 mmol), and CH3CN (2 mL) at 45–50 °C, 10 h. | ||
The efficiency of our hydroalkylation of 3-methyleneisoindolin-1-ones with a variety of alkyliodides prompted us to carry out further experiments to reveal the mechanism of the reaction (Scheme 4). First, we carried out UV-visible spectroscopy to check the feasibility of the formation of an electron–donor–acceptor (EDA) complex between 3-methylene-2-phenylisoindolin-1-one (1a) and iodocyclohexane (2a) and nBu3N (see ESI,† Fig. S7). The absorption spectrum of the individual and combined reactants does not support the formation of an EDA complex (see ESI,† Fig. S7). As anticipated, the “light–dark” experiment confirmed that our reaction required continuous light irradiation (see ESI†). Photoluminescence quenching studies indicated that nBu3N quenched the photoexcited state of 4CzIPN, but not the alkyl iodide and 3-methyleneisoindolin-1-one (see, ESI†). Next, a radical trapping and radical clock experiment was carried out (Scheme 4a and b). When the reaction was conducted in the presence of 3 equiv. of TEMPO, it did not produce any product (3a); instead, a radical trapping adduct (4) was identified by HRMS (Scheme 4a). Employing cyclopropyl methyl iodide as a source of alkyl radical resulted in a ring-opening product (3aw) under the standard reaction conditions (Scheme 4b). These two experiments indicate that the generation of alkyl radicals occurs in our reaction conditions.
 | ||
| Scheme 4 Preliminary mechanistic studies. (a) Radical scavenger; (b) radical clock experiment; (c) isotope labeling experiment using D2O. TEMPO = 2,2,6,6-tetramethylpiperidinyloxy. | ||
Based on our control experiments, radical trapping, radical clock experiments, and previous known literature precedents11,12 we proposed a tentative reaction mechanism for hydroalkylation of 3-methyleneisoindolin-1-ones with unactivated alkyl iodides, as shown in Scheme 5. Initially, the alkylamine transformed to a nucleophilic α-aminoalkyl radical (B) in the presence of the photoexcited photocatalyst (4CzIPN*) through stepwise single electron oxidation, followed by deprotonation. The subsequent halogen atom transfer process between radical intermediate B and the iodoalkane generates an alkyl radical (C). Next, trapping of the alkyl radical to 3-methyleneisoindolin-1-one produces another benzylic radical intermediate (D), which is further converted to a benzylic anion intermediate (E) via SET from the reduced photocatalyst13 to generate the ground state photocatalyst. Finally, protonation of the benzylic anion intermediate with H2O would deliver the product (3). Furthermore, the generation of the benzylic anion intermediate was indirectly confirmed by isotope-labeling studies (Scheme 4c). Specifically, product 5 (99%-D) was exclusively obtained upon the addition of D2O (5 equiv.) to the reaction medium (Scheme 4c).
 | ||
| Scheme 5 Mechanistic hypothesis. | ||
In conclusion, we have demonstrated a catalytic strategy for intermolecular hydroalkylation of 3-methyleneisoindolin-1-ones with inert iodoalkanes under visible light photocatalysis. A variety of primary, secondary, and tertiary iodoalkanes with diverse functional groups reacted well with electronically and sterically different 3-methyleneisoindolin-1-ones and afforded the corresponding products in good to moderate yields. Furthermore, this strategy can be scaled up and allows the late-stage modification of iodoalkanes derived from pharmaceutically relevant molecules. Preliminary mechanistic studies suggested that the reaction proceeds via the generation of radical intermediates and a benzylic anion overall in a redox-neutral manner.
Financial support by the Science and Engineering Research Board (SERB), Government of India (File Number: SRG/2021/000834) is greatly acknowledged. A. B. thanks UGC for the JRF fellowship.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) A. Yamamoto, Y. Nishimura and Y. Nishihara, Recent Advances in Cross-Coupling Reactions with Alkyl Halides, Applied Cross-Coupling Reactions, ed. Y. Nishihara, Springer, Berlin, Heidelberg, 2013, vol. 80, pp. 205–209 Search PubMed; (b) G. W. Gribble, Mar. Drugs, 2015, 13, 4044–4136 CrossRef CAS PubMed.
- (a) D. J. Weix, Acc. Chem. Res., 2015, 48, 1767–1775 CrossRef CAS PubMed; (b) M. R. Kwiatkowski and E. J. Alexanian, Acc. Chem. Res., 2019, 52, 1134–1144 CrossRef CAS PubMed; (c) K. P. S. Cheung, S. Sarkar and V. Gevorgyan, Chem. Rev., 2022, 122, 1543–1625 CrossRef; (d) A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B.-X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Chem. Rev., 2022, 122, 1485–1542 CrossRef CAS.
- (a) J. Zhou and G. C. Fu, J. Am. Chem. Soc., 2003, 125, 14726–14727 CrossRef CAS; (b) X. Wu, W. Hao, K.-Y. Ye, B. Jiang, G. Pombar, Z. Song and S. Lin, J. Am. Chem. Soc., 2018, 140, 14836–14843 CrossRef CAS PubMed; (c) S. Bera, R. Mao and X. Hu, Nat. Chem., 2021, 13, 270–277 CrossRef CAS PubMed; (d) A. Cai, W. Yan, C. Wang and W. Liu, Angew. Chem., Int. Ed., 2021, 60, 27070–27077 CrossRef CAS PubMed; (e) Y. Shen, J. Cornella, F. Juliá-Hernández and R. Martin, ACS Catal., 2017, 7, 409–412 Search PubMed; (f) H. Kim and C. Lee, Angew. Chem., Int. Ed., 2012, 51, 12303–12306 Search PubMed; (g) J. D. Nguyen, E. M. D’Amato, J. M. R. Narayanam and C. R. J. Stephenson, Nat. Chem., 2012, 4, 854–859 CrossRef CAS PubMed.
- (a) D. P. Curran and D. M. Rakiewicz, J. Am. Chem. Soc., 1985, 107, 1448–1449 CrossRef CAS; (b) W. P. Neumann, Synthesis, 1987, 665–683 Search PubMed; (c) K. Miura, Y. Ichinose, K. Nozaki, K. Fugami, K. Oshima and K. Utimoto, Bull. Chem. Soc. Jpn., 1989, 62, 143–147 CrossRef CAS; (d) D. P. Curran and A. I. Keller, J. Am. Chem. Soc., 2006, 128, 13706–13707 CrossRef CAS; (e) M. R. Medeiros, L. N. Schacherer, D. A. Spiegel and J. L. Wood, Org. Lett., 2007, 9, 4427–4429 Search PubMed; (f) J. J. Devery, J. D. Nguyen, C. Dai and C. R. J. Stephenson, ACS Catal., 2016, 6, 5962–5967 CAS.
- (a) R. K. Neff, Y. L. Su, S. Liu, M. Rosado, X. Zhang and M. P. Doyle, J. Am. Chem. Soc., 2019, 141, 16643–16650 Search PubMed; (b) Y. L. Su, L. Tram, D. Wherritt, H. Arman, W. P. Griffith and M. P. Doyle, ACS Catal., 2020, 10, 13682–13687 CAS; (c) T. Constantin, M. Zanini, A. Regni, N. S. Sheikh, F. Juliá and D. Leonori, Science, 2020, 367, 1021–1026 Search PubMed . For a recent comprehensive review, see: ; (d) F. Juliá, T. Constantin and D. Leonori, Chem. Rev., 2022, 122(2), 2292–2352 Search PubMed.
- (a) Z.-Q. Zhang, Y.-Q. Sang, C.-Q. Wang, P. Dai, X.-S. Xue, J. L. Piper, Z.-H. Peng, J.-A. Ma, F.-G. Zhang and J. Wu, J. Am. Chem. Soc., 2022, 144, 14288–14296 Search PubMed; (b) J. Koo, W. Kim, B. H. Jhun, S. Park, D. Song, Y. You and H. G. Lee, J. Am. Chem. Soc., 2024, 146, 22874–22880 CAS; (c) K. Li, X.-C. He, J. Gao, Y.-L. Liu, H.-B. Chen, H.-Y. Xiang, K. Chen and H. Yang, J. Org. Chem., 2024, 89, 12658–12667 Search PubMed.
- Selected reviews: (a) B. Sener, B. Goezler, R. D. Minard and M. Shamma, Phytochemistry, 1983, 22, 2073–2075 Search PubMed; (b) M. Efdi, S. Fujita, T. Inuzuka and M. Koketsu, Nat. Prod. Res., 2010, 24, 657–662 CAS; (c) G. Blaskó, D. J. Gula and M. Shamma, J. Nat. Prod., 1982, 45, 105–122 Search PubMed; (d) Y. C. Chia, F. R. Chang, C. M. Teng and Y. C. Wu, J. Nat. Prod., 2000, 63, 1160–1163 CAS; (e) K. Speck and T. Magauer, Beilstein J. Org. Chem., 2013, 9, 2048–2078 Search PubMed.
- (a) E. D. Clercq, J. Med. Chem., 1995, 38, 2491–2517 CrossRef; (b) I. Pendrak, S. Barney, R. Wittrock, D. M. Lambert and W. D. Kingsbury, J. Org. Chem., 1994, 59, 2623–2625 CrossRef CAS; (c) E. C. Taylor, P. Zhou, L. D. Jennings, Z. Mao, B. Hu and J.-G. Jun, Tetrahedron Lett., 1997, 38, 521–524 CrossRef CAS.
- (a) L. A. Paquette, R. D. Dura and I. Modolo, J. Org. Chem., 2009, 74, 1982–1987 CrossRef CAS PubMed; (b) J. B. Campbell, R. F. Dedinas and S. A. Trumbower-Walsh, J. Org. Chem., 1996, 61, 6205–6211 CrossRef CAS; (c) A. Couture, E. Deniau, D. Ionescu and P. Grandclaudon, Tetrahedron Lett., 1998, 39, 2319–2320 Search PubMed; (d) J. B. Campbell, R. F. Dedinas and S. Trumbower-Walsh, Synlett, 2010, 3008–3010 CrossRef CAS; (e) X. Gai, R. Grigg, T. Khamnaen, S. Rajviroongit, V. Sridharan, L. Zhang, S. Collard and A. Keep, Tetrahedron Lett., 2003, 44, 7441–7443 CAS; (f) R. Savela and C. Mendez-Galvez, Chem. – Eur. J., 2021, 27, 5344–5378 CAS; (g) S. Samanta, S. A. Ali, A. Bera, S. Giri and K. Samanta, New J. Chem., 2022, 46, 7780–7830 CAS; (h) R. Manoharan and M. Jeganmohan, Chem. Commun., 2015, 51, 2929–2932 CAS.
- (a) W.-X. Tang, K.-Q. Chen, D.-Q. Sun and X.-Y. Chen, Org. Biomol. Chem., 2023, 21, 715–718 RSC; (b) K.-Q. Chen, B.-B. Zhang, Z.-X. Wang and X.-Y. Chen, Org. Lett., 2022, 24, 4598–4602 CAS; (c) V. K. Simhadri, R. Sur and V. R. Yatham, J. Org. Chem., 2025, 90, 3557–3562 CAS.
- (a) G. S. Yedase, A. K. Jha and V. R. Yatham, J. Org. Chem., 2022, 87, 5442–5450 CrossRef CAS PubMed; (b) A. R. Tripathy, A. Kumar, R. Rahmathulla, A. K. Jha and V. R. Yatham, Org. Lett., 2022, 24, 5186–5191 CrossRef CAS; (c) A. R. Tripathy, A. Bisoyi, P. Arya, S. Venugopal and V. R. Yatham, ACS Org. Inorg. Au, 2024, 4, 229–234 CrossRef CAS . For mechanistic relevant photocatalystic hydroalkylation of diarylethylene derivatives and carbonylative hydroacylation of styrenes, see: ; (d) S. B. Cornelia, S. Michael and B.-S. Katharina, J. Org. Chem., 2022, 87, 11042–11047 CrossRef; (e) J. A. Forni, V. H. Gandhi and A. Polyzos, ACS Catal., 2022, 12, 10018–10027 CrossRef CAS.
- T.-Y. Shang, L.-H. Lu, Z. Cao, Y. Liu, W.-M. He and B. Yu, Chem. Commun., 2019, 55, 5408 Search PubMed.
- G.-Q. Wang, Y. Zhang, Y.-X. Zhou, D. Yang, P. Han, L.-H. Jing and K. Tang, J. Org. Chem., 2024, 89, 7899–7912 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and compound data. See DOI: https://doi.org/10.1039/d5cc00491h |
| This journal is © The Royal Society of Chemistry 2025 |