
Inducing local charge polarization by constructing isomeric covalent organic frameworks with different orientations of imine bonds for enhancing photocatalytic hydrogen evolution†
Huan
He
a,
Rongchen
Shen
a,
Peng
Zhang
 b,
Guijie
Liang
*c and
Xin
Li
*a
b,
Guijie
Liang
*c and
Xin
Li
*a
aInstitute of Biomass Engineering, Key Laboratory of Energy Plants Resource and Utilization, Ministry of Agriculture and Rural Affairs, South China Agricultural University, Guangzhou 510642, China. E-mail: Xinliscau@yahoo.com
bState Centre for International Cooperation on Designer Low-Carbon & Environmental Materials (CDLCEM), School of Materials Science and Engineering, Zhengzhou University, Zhengzhou, Henan 450001, P. R. China
cHubei Key Lab Low Dimens Optoelect Mat & Devices, Hubei University of Arts and Science, Xiangyang 441053, P. R. China. E-mail: guijie-liang@hotmail.com
First published on 21st November 2023
Abstract
Imine-linked covalent organic frameworks (COFs) have garnered significant attention in photocatalysis due to their ease of synthesis and excellent crystallinity. However, there is still limited research on the influence of imine bond orientation on the processes of exciton dissociation and charge carrier separation in COFs. This study presents the synthesis and characterization of two novel COFs with distinct imine bond orientations. Experimental and theoretical investigations reveal that though the structures of these COFs are similar, their performance in exciton dissociation and electron–hole pair separation varies significantly. The orientation of the imine bond plays a crucial role in inducing local charge polarization and delocalization, thus influencing the efficiency of exciton dissociation and charge separation. Notably, the photocatalytic hydrogen-evolution activity for the COFs with the N-atom orientation in imine bond towards the acceptor is three times higher than that for COFs with the N-atom orientation in imine bond towards the donor. This study provides a clear design strategy for imine-linked COF-based photocatalysts and advances the development of COFs in photocatalysis.
Introduction
Covalent organic frameworks (COFs), highly crystalline and porous organic frameworks composed of interconnected organic building blocks, have emerged as promising semiconductors for photocatalytic hydrogen evolution.1–7 The synthesis of COFs involves various organic reactions that offer precise control over their structure, porosity, and composition. This tunability optimizes their photocatalytic properties, leading to enhanced hydrogen evolution activity.8–12 However, despite their excellent performance as photocatalysts, COFs face challenges related to poor exciton dissociation and charge separation efficiency, which limit their further development. The high exciton binding energy can lead to exciton–exciton destruction and reduce the number of electron–hole pairs.13,14 To address these issues, the construction of donor–acceptor (D–A) type COFs has been explored to enhance unit polarity and promote conjugation, thereby facilitating exciton dissociation and the separation of electron–hole pairs.15–21 In the D–A system, excitons are typically generated at the donor moiety and transferred to the acceptor moiety at the D–A interface, where they dissociate into electron–hole pairs.22–24 The chemical bond linkers represent the D–A interface of COFs to connect the donor and acceptor, where the dissociation of the excitons occurs. Therefore, the chemical bond linker is crucial in determining exciton dissociation and charge carrier separation in COFs for photocatalytic applications.In this regard, functional and structural diversity is primarily introduced into COFs through various linkers. Recent years have seen several published studies focusing on the chemical bond connections in COFs.25–27 Investigating performance changes induced by different connection modes contributes to a better understanding of the reaction mechanisms involved in COF photocatalytic processes. For instance, Han et al. found that imine- and vinylene-linked COFs show different charge-transfer pathways, affecting exciton relaxation.28 Lu et al. found that the presence of linked amine groups in COFs, compared to imine groups, results in superior activation of reaction substrates, facilitation of bond cleavage, and decreased activation energy for the reaction.29 Jiang et al. found that enaminone-linked COFs display improved exciton dissociation and charge separation capabilities compared to imine-linked and quinoline-linked COFs.30 Yu and his co-workers found that cyano-substituted alkene-linkages will provide both strong electron acceptors and efficient π-conjugated transport.31 These findings demonstrate the crucial role of chemical bond linkers in the practical applications of COFs. Imine-linked COFs possess the advantages of ease of synthesis and excellent crystallinity over COFs connected by other chemical bonds in the field of photocatalysis. Imine-bond orientation in COFs may result in the isomerism of covalent organic frameworks. However, it is rare to find reports of the structure–activity relationship between imine-bond orientation and photocatalytic performance in practical application. Recently, Thomas et al. discovered that imine-bonded D–A COFs with different orientations exhibit significant differences in their photophysical properties, resulting in notable differences in their photocatalytic performance.32 Li et al. found that COFs with imine-bond orientation displayed similar structures but significantly different photoelectric properties.33 These findings underscore the importance of imine-bond orientation in tailoring the photocatalytic performance of imine-linked COFs. However, there is currently a lack of reports investigating the impact of imine bond orientations on exciton dissociation and charge carrier separation in COFs, which are crucial challenges in COF photocatalysis.
In this study, we report the synthesis of two novel COFs (denoted as PyAl-TpbAm-COF and PyAm-TpbAl-COF) connected by imine bonds formed through the Schiff-base polymerization reactions. The pyrene molecule is electron enriched, and serves as a donor building block. In contrast, benzene serves as the acceptor building block. The photocatalytic hydrogen evolution performance of the PyAl-TpbAm-COF, is three times higher than that of PyAm-TpbAl-COF. Theoretical and experimental results demonstrated that imine-bond orientation may induce local charge polarization and delocalization, thereby affecting exciton dissociation and charge separation. This represents the crucial preliminary step of activating COFs as photocatalysts for photocatalytic hydrogen evolution. Moreover, besides providing detailed insights into the structure–activity relationship, this work also offers valuable insights for optimizing the performance of COFs.
Results and discussion
PyAl-TpbAm-COF and PyAm-TpbAl-COF were constructed by Schiff base reaction. We used 4,4′,4′′,4′′′-(pyrene-1,3,6,8-tetrayl)tetrabenzaldehyde(PyAl) and 4′,5′-bis(4-aminophenyl)-[1,1′:2′,1′′-terphenyl]-4,4′′-diamine, (TpbAm) as the monomers for PyAl-TpbAm-COF. We also selected 4,4′,4′′,4′′′-(pyrene-1,3,6,8-tetrayl)tetraaniline(PyAm) and 1,2,4,5-tetrakis-(4-formylphenyl)benzene(TpbAl) to produce PyAm-TpbAl-COF (Fig. 1A). For these COFs, the only difference was the directionality of the imine bond. The PXRD patterns of PyAl-TpbAm-COF and PyAm-TpbAl-COF exhibit prominent diffraction peaks. Experimental diffraction patterns (black), refined (red) and residual (gray), and patterns simulated from the structural model (blue) for PyAl-TpbAm-COF and PyAm-TpbAl-COF are shown. For these COFs, the Pawley-refined PXRD patterns (Material Studio 2020) agree with the experimental results well. Rwp and Rp of PyAl-TpbAm-COF are 6.73 and 4.98%, respectively, and Pawley-refined cell parameters are a = 18.2834 Å, b = 21.3144 Å, c = 5.9711 Å, α = β = γ = 90°. The Pawley-refined cell parameters for PyAm-TpbAl-COF are almost identical because the only difference is the directionality of the imine bond. Rwp and Rp of PyAm-TpbAl-COF are 8.87 and 7.55%, respectively, and its Pawley-refined cell parameters are consistent with those of PyAl-TpbAm-COF a = 18.2834 Å, b = 21.3144 Å, c = 5.9711 Å, α = β = γ = 90° (Fig. 1B, C and Table S1†). Both COFs have equally high crystallinity, but PyAl-TpbAm-COF has a minor deviation from the theoretical value and a higher agreement. Then, the chemical structures of PyAl-TpbAm-COF and PyAm-TpbAl-COF were confirmed by FTIR spectroscopy. Compared to monomers, the peaks of –NH2 and –CHO at about 3300 and 1750 cm−1 decreased obviously. In contrast, a new characteristic signal belonging to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N at 1650 cm−1 was observed (Fig. 1F and G).34 In the C 1s XPS spectrum for PyAl-TpbAm-COF and PyAm-TpbAl-COF, the 284.8, 286.2 and 289.5 eV peaks correspond to the CC, CN and C–O, respectively. The peaks at about 398.4, 398.8 and 401.4 eV in the N 1s spectrum for PyAl-TpbAm-COF and PyAm-TpbAl-COF are attributed to the CN, C–N and N–H (Fig. S1 and S2†). Nitrogen sorption measurements were used to assess the porosity of these COFs at 77.3 K. The Brunauer–Emmett–Teller (BET) surface areas of PyAl-TpbAm-COF and PyAm-TpbAl-COF were found to be 1237 and 1281 m2 g−1, respectively (Fig. 1D and E). This isothermal desorption curve is a type IV isotherm, indicating a mesoporous material feature.
N at 1650 cm−1 was observed (Fig. 1F and G).34 In the C 1s XPS spectrum for PyAl-TpbAm-COF and PyAm-TpbAl-COF, the 284.8, 286.2 and 289.5 eV peaks correspond to the CC, CN and C–O, respectively. The peaks at about 398.4, 398.8 and 401.4 eV in the N 1s spectrum for PyAl-TpbAm-COF and PyAm-TpbAl-COF are attributed to the CN, C–N and N–H (Fig. S1 and S2†). Nitrogen sorption measurements were used to assess the porosity of these COFs at 77.3 K. The Brunauer–Emmett–Teller (BET) surface areas of PyAl-TpbAm-COF and PyAm-TpbAl-COF were found to be 1237 and 1281 m2 g−1, respectively (Fig. 1D and E). This isothermal desorption curve is a type IV isotherm, indicating a mesoporous material feature.
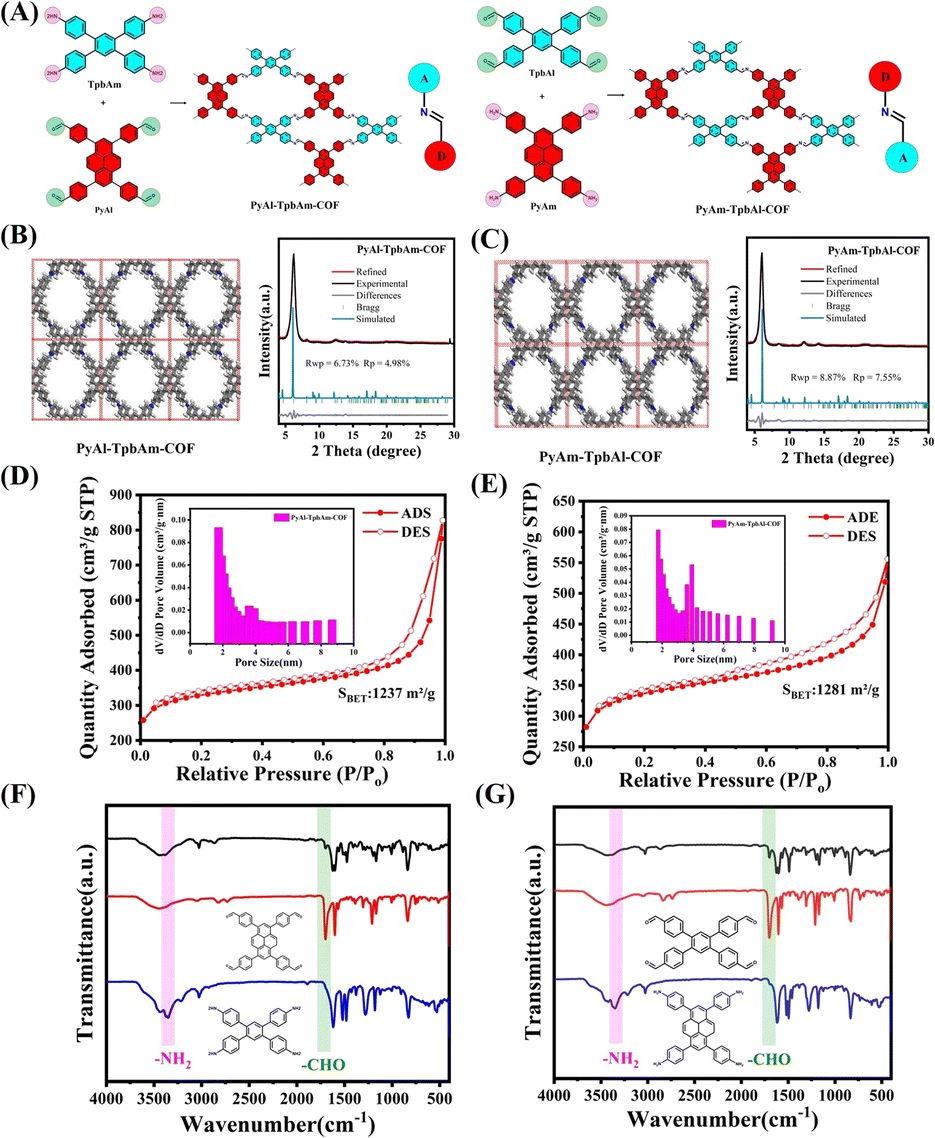 | ||
| Fig. 1 (A) Synthesis route of the PyAl-TpbAm-COF and PyAm-TpbAl-COF; crystal structures and experimental, Pawley-refined and simulated powder X-ray diffraction patterns of (B) PyAl-TpbAm-COF and (C) PyAm-TpbAl-COF; N2 adsorption and desorption isotherms of (D) PyAl-TpbAm-COF and (E) PyAm-TpbAl-COF; FTIR spectra of (F) PyAl-TpbAm-COF and (G) PyAm-TpbAl-COF. | ||
The properties of mesopores are also reflected in the pore size distribution of COFs. SEM and TEM were used to measure the morphology of PyAl-TpbAm-COF and PyAm-TpbAl-COF. Fig. 2A, D and S3† display TEM images of both PyAl-TpbAm-COF and PyAm-TpbAl-COF, showing porous nanosheet morphology. Meanwhile, clear lattice stripes could be observed in the TEM and SAED images, which suggests that both PyAl-TpbAm-COF and PyAm-TpbAl-COF are highly crystalline (Fig. 2A, B, D and E). The observed channel width of approximately 1.4 nm is consistent with the size of the pore size distribution (Fig. S13†). Through the Fourier transform, the distorted hexagonal channel pores of COFs could be found (Fig. 2C and F).
 | ||
| Fig. 2 (A) TEM images, (B) SAED and (C) FFT-image of PyAl-TpbAm-COF. (D) TEM images, (E) SAED and (F) FFT-image of PyAm-TpbAl-COF. | ||
Ultraviolet-visible (UV-vis) absorption spectroscopy can be used to analyze the photophysical properties of PyAl-TpbAm-COF and PyAm-TpbAl-COF. PyAl-TpbAm-COF and PyAm-TpbAl-COF show similar light absorption up to 550 nm (Fig. 3A). Therefore, the bandgaps of PyAl-TpbAm-COF and PyAm-TpbAl-COF were about 2.29 and 2.31 eV (Fig. S4†), respectively. Mott–Schottky plots were used to characterize the CB levels of PyAl-TpbAm-COF and PyAm-TpbAl-COF (Fig. S5 and S6†). The flat band levels of PyAl-TpbAm-COF and PyAm-TpbAl-COF were −0.67 and −0.65 eV, respectively. Therefore, the CB levels of PyAl-TpbAm-COF and PyAm-TpbAl-COF were −0.47 and −0.45 eV (vs. NHE), respectively. Combined with the bandgaps of the as-prepared COFs, the VB levels of PyAl-TpbAm-COF and PyAm-TpbAl-COF were 1.84 and 1.84 eV, respectively (Fig. 3B). This result suggests that both PyAl-TpbAm-COF and PyAm-TpbAl-COF meet the conditions to transfer photoexcited electrons to H+ for hydrogen evolution thermodynamically.35 Meanwhile, both PyAl-TpbAm-COF and PyAm-TpbAl-COF displayed a positive slope in the Mott–Schottky plots, indicating an n-type semiconductor characteristic.
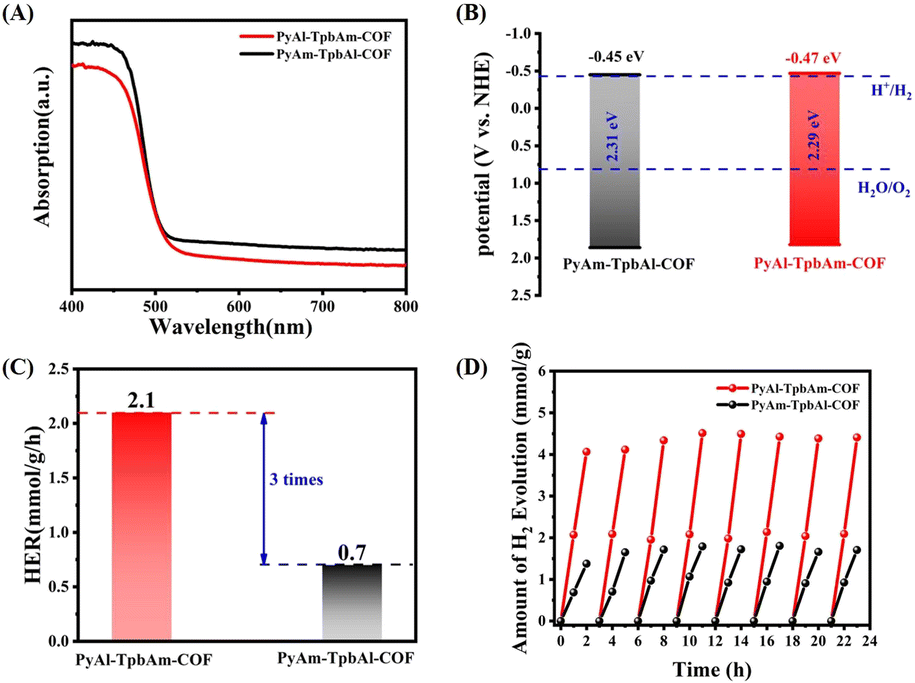 | ||
| Fig. 3 (A) UV-vis spectrum and (B) band levels of PyAl-TpbAm-COF and PyAm-TpbAl-COF; (C) average hydrogen evolution rate and (D) cyclic hydrogen evolution of PyAl-TpbAm-COF and PyAm-TpbAl-COF. | ||
The photocatalytic hydrogen evolution performances of PyAl-TpbAm-COF and PyAm-TpbAl-COF were measured under visible light irradiation with 3% wt Pt as a cocatalyst and ascorbic acid as a sacrificial agent (Fig. 3C). Both PyAl-TpbAm-COF and PyAm-TpbAl-COF show linear increases during the hydrogen evolution process. The average hydrogen evolution rate of PyAl-TpbAm-COF was 2.1 mmol g−1 h−1, which was much higher than that of the PyAm-TpbAl-COF (0.7 mmol g−1 h−1). This result indicates that the orientation of imine bonds plays a key role in improving the photocatalytic hydrogen evolution. Three groups of different orientation imine bond COF activity comparison were provided to avoid the experimental specificity (Fig. S10†). Both PyAl-TpbAm-COF and PyAm-TpbAl-COF show almost no photocatalytic performance without Pt serving as a cocatalyst, which indicates that both PyAl-TpbAm-COF and PyAm-TpbAl-COF lack active sites. Besides efficient hydrogen evolution performance, excellent stability is also observed for two photocatalysts in actual use. Clearly, both PyAl-TpbAm-COF and PyAm-TpbAl-COF exhibit excellent stability during the 8-cycle hydrogen reaction (Fig. 3D).
In order to understand the role of the orientation of imine bonds, we analyze the exciton and charge-carrier properties of PyAl-TpbAm-COF and PyAm-TpbAl-COF. We measured temperature-dependent PL measurements to detect the exciton binding energies of prepared COFs. It could be found that PyAl-TpbAm-COF with the N-atom orientation of imine bonds towards the acceptor shows lower exciton binding energies. The exciton binding energies of PyAm-TpbAl-COF were 73.8 meV, while the exciton binding energies of PyAl-TpbAm-COF decreased to 57.0 meV. Accordingly, switching the N-atom orientation of imine bonds could significantly accelerate the exciton dissociation (Fig. 4A–D).36 We analyze the exciton dissociation processes by steady-state delayed phosphorescence. The intensity of steady-state delayed phosphorescence could give the amount of radiative decay of triplet excitons. As shown in Fig. 4E, the intensity for PyAl-TpbAm-COF is remarkably lower than that of PyAm-TpbAl-COF, which indicates the significantly reduced amount of triplet excitons. The delayed phosphorescence lifetimes of PyAl-TpbAm-COF and PyAm-TpbAl-COF are 33.00 and 29.92 ns, respectively (Fig. 4F). The remarkably reduced delayed phosphorescence lifetime suggests the significantly improved triple exciton dissociation.13,37 These results indicate that the excitons in PyAl-TpbAm-COF are more prone to dissociation than those in PyAm-TpbAl-COF. The increased concentration of free charge carriers in PyAl-TpbAm-COF further contributes to its high photocatalytic performance. We performed fs-TA spectroscopy to detect the charge-transfer mechanism. Both PyAl-TpbAm-COF and PyAm-TpbAl-COF displayed a negative peak at 480–600 nm and a positive peak at 600–750 nm, which are attributed to the ground-state bleach (GSB) and excited-state absorption (ESA), respectively (Fig. S14†). Herein, we utilize a pump–probe configuration with a 400 nm pump and a white-light probe (450–750 nm) to track the photogenerated electron kinetics. When ethylene glycol is used as a dispersant, the height of the positive absorption band of COFs increases when N atom is oriented towards the electron acceptor at around 700 nm, which corresponds to the ultrafast separation of carriers and the rapid capture of electrons. To understand the delayed dynamics of photogenerated charge carriers for PyAl-TpbAm-COF and PyAm-TpbAl-COF, we used bi-exponential fitting to analyze the decay trace. The τ1 and τ2 of PyAl-TpbAm-COF were 11.80 and 578.91 ps, respectively, corresponding to the trapped electrons of surface states and electron trapping. The τ1 and τ2 of PyAm-TpbAl-COF were 13.19 and 583.93 ps, respectively. Therefore, when N atom is oriented towards A, the COF is more beneficial to the separation and transport of electrons and holes. This result can be inferred from the chemical structure of the COFs. In the D–A system, excitons are generally generated on the D moiety and transferred to the A moiety. At the D–A interface, they dissociate into electron–hole pairs. The D–A interface of COFs is represented by the imine bond, where the excitons dissociate. When the nitrogen atom of the imine bond is oriented towards D compared to being oriented towards A, the distance between it and D in the coplanar plane is shorter. This non-coplanar alignment hinders exciton dissociation (Fig. S15†).38
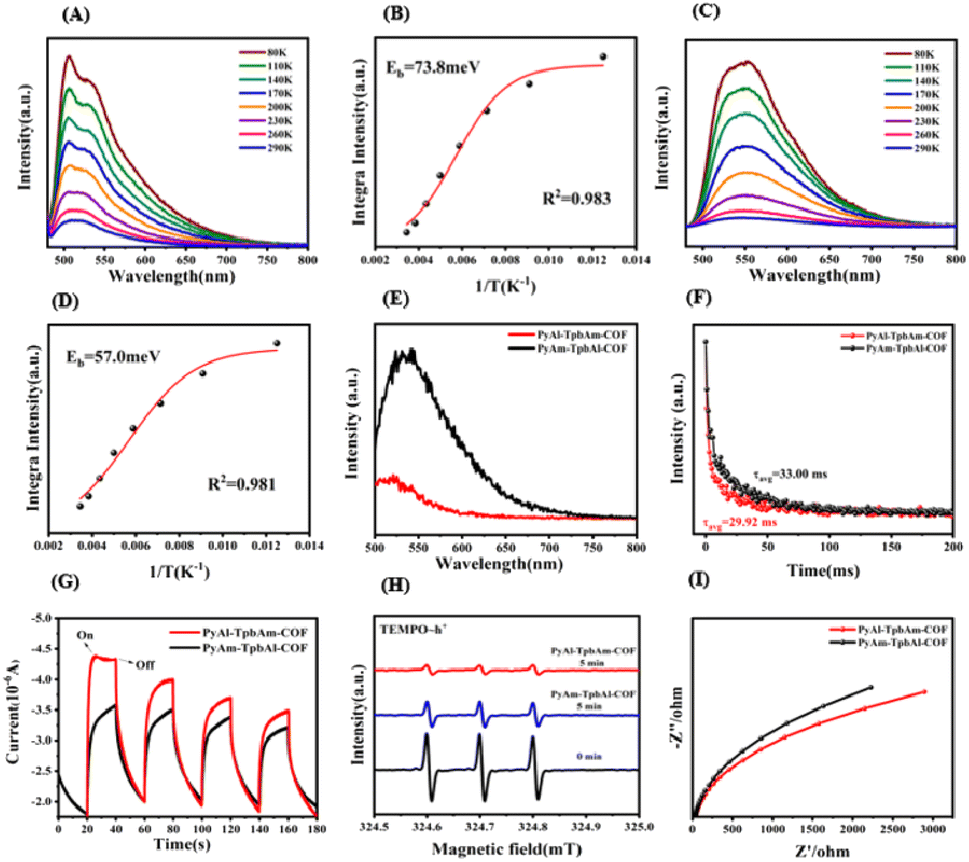 | ||
| Fig. 4 (A) Temperature-dependent PL spectra with excitation wavelength at 360 nm and (B) exciton binding energies of PyAm-TpbAl-COF; (C) temperature-dependent PL spectra with excitation wavelength at 360 nm and (D) exciton binding energies of PyAl-TpbAm-COF; (E) low-temperature steady-state phosphorescence recorded at a delayed time of 10 μs and (F) time-resolved phosphorescence kinetics for PyAl-TpbAm-COF and PyAm-TpbAl-COF. (G) Photocurrent, (H) EIS spectra and (I) EPR spectra TEMPO˙ h+ for PyAl-TpbAm-COF and PyAm-TpbAl-COF. | ||
We further analyze the origins of the difference in the charge-carrier separation and migration properties of PyAl-TpbAm-COF and PyAm-TpbAl-COF by photocurrent and electrochemical impedance (EIS).39,40 It could be found that PyAl-TpbAm-COF shows higher photocurrent intensity than PyAm-TpbAl-COF, which suggests that more photogenerated charge carriers are captured on the surface of PyAl-TpbAm-COF (Fig. 4G).41,42 The same results can be observed in the EPR spectra. The intensity variation of TEMPO, resulting from its reaction with photogenerated holes to form TEMPO−˙ h+ adducts, can serve as an indicator of changes in the content of photogenerated holes. The observation of a strong TEMPO signal under dark conditions indicates a high concentration of TEMPO. Under visible light irradiation, both PyAl-TpbAm-COF and PyAm-TpbAl-COF exhibit a characteristic decrease in the intensity signal of TEMPO. This can be attributed to the consumption of TEMPO by photogenerated holes.43 After 5 minutes of light irradiation, it is evident that the TEMPO signal intensity of PyAl-TpbAm-COF is significantly lower than that of PyAm-TpbAl-COF (Fig. 4H and S7†). The EPR results indicate a higher abundance of photogenerated holes captured by TEMPO on the surface of PyAl-TpbAm-COF. In the EIS spectrum (Fig. 4I), the lowest electrochemical impedance and the smallest charge transfer resistance were realized by PyAl-TpbAm-COF, further suggesting that the N-atom orientation of imine bonds towards the acceptor boosts the electron–hole separation rate. The separation efficiency of photogenerated electron–hole pairs was detected by the PL test. The results demonstrate that PyAl-TpbAm-COF exhibits a lower recombination rate of electron–hole pairs, thus effectively separating photogenerated electron–hole pairs (Fig. S12†).44
Density functional theory (DFT) was utilized to observe and compare the local polarization and carrier separation behavior of the as-synthesized COFs.45–48 As schematically presented in Fig. 5A and B, PyAl-TpbAm-COF shows a dipole moment of 2.48 D, which is larger than that of PyAm-TpbAl-COF (2.39 D for the PyAm-TpbAl-COF), indicating that the N-atom orientation of the imine bond changes the polarity of the system and promotes the charge redistribution of the molecular skeleton.49,50 The frontier molecular orbitals (HOMO–LUMO) and energy levels of the smallest repeating units of PyAl-TpbAm-COF and PyAm-TpbAl-COF are calculated by a well-defined DFT approach in order to understand the photocatalytic mechanism. The band gaps of the two COFs are calculated to be 3.23 eV and 3.25 eV, respectively (Fig. S8†). The HOMO and LUMO energy level diagram for the monomer unit is shown in Fig. S11.† The calculated integrals of Sr (Sr index) for PyAl-TpbAm-COF and PyAm-TpbAl-COF were 0.64997 and 0.67391, respectively (Fig. 5C and D). A lower Sr index indicates more efficient charge carrier separation. Moreover, the electron delocalization index (EDI) results are as follows: PyAl-TpbAm-COF (5.34) > PyAm-TpbAl-COF (4.74), which is consistent with the Sr index results.51 The calculated UV-vis spectra in Fig. S11† are similar to the experimental results. The filter being used in this study is an ultraviolet filter, which means that only the visible portion of the spectrum is considered. For PyAl-TpbAm-COF, the maximum absorption peak in visible light comes from the transition of S0 → S1. The excited state S1 could be described as a linear addition of main configurations HOMO → LUMO and HOMO → LUMO+1. For PyAm-TpbAl-COF, the maximum absorption peak comes from the transition of S0 → S1. The excited state S1 is mainly contributed by main configurations HOMO → LUMO (Fig. 5E and F). It is possible to increase the excitation pathways in visible light and the excitation frequency by controlling the different N-atom orientations of the imine bonds, beneficial for exciton dissociation.
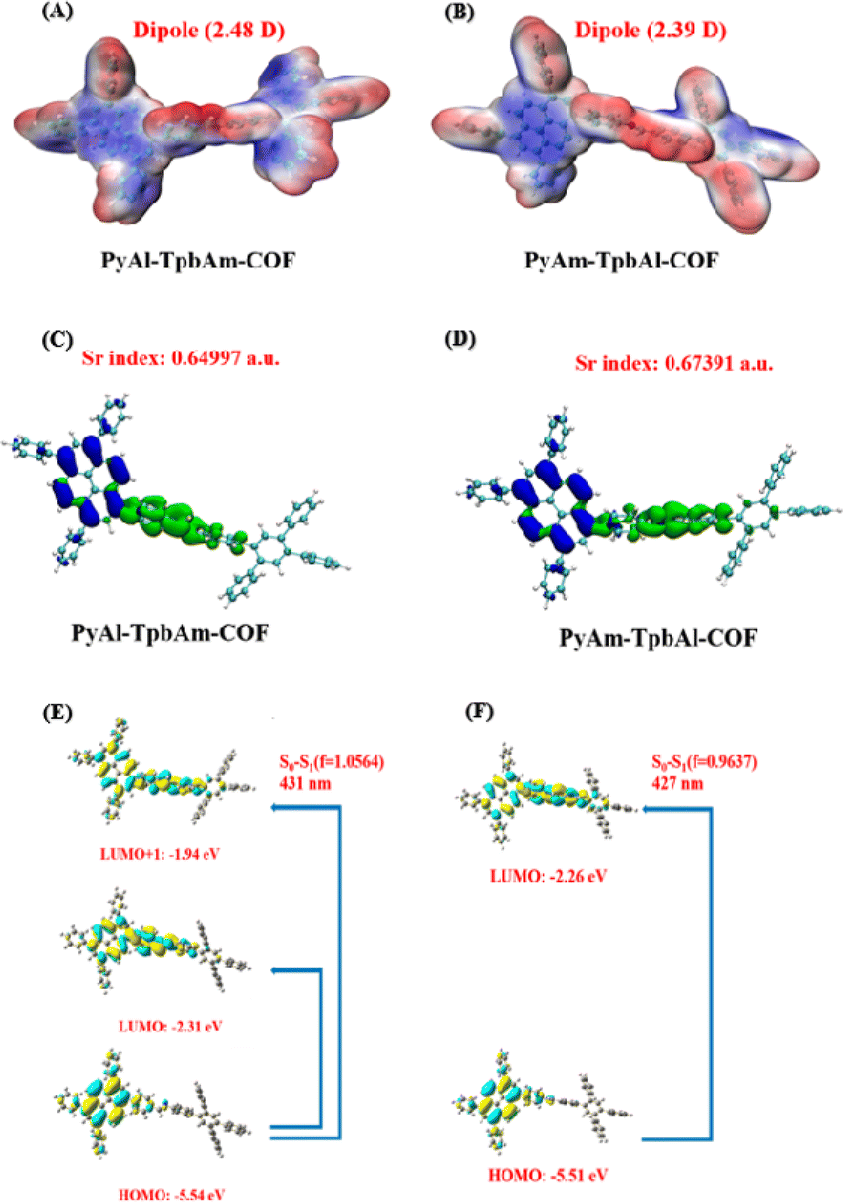 | ||
| Fig. 5 (A) PyAl-TpbAm-COF and (B) PyAm-TpbAl-COF in the ground state, blue and red represent electron accumulation and depletion, respectively; the excited state electronic structures of COF photocatalysts highlighting electron–hole distribution in (C) PyAl-TpbAm-COF and (D) PyAm-TpbAl-COF; the TD-DFT calculated electronic transition of (E) PyAl-TpbAm-COF and (F) PyAm-TpbAl-COF, respectively. | ||
Conclusions
In summary, we have successfully synthesized two novel imine-linked COFs: PyAl-TpbAm-COF and PyAm-TpbAl-COF. These COFs exhibit isomeric structures with different N-atom orientations of their imine bonds. Despite having similar compositions and pore sizes, they demonstrate distinct exciton dissociation and charge carrier separation properties. The N-atom orientation of the imine bond significantly influences local charge polarization and delocalization, thereby impacting the efficiency of exciton dissociation and charge separation processes. This research has the potential to be applied to other two-dimensional COFs for fine-tuning exciton dissociation and charge carrier separation, serving as a guide for the design of COFs with superior performance.Author contributions
Huan He, Rongchen Shen and Xin Li designed the systems, synthesized the photocatalysts, performed the experimental measurement, analyzed the data, and wrote the manuscript. Guijie Liang carried out transient absorption spectroscopy experiments and temperature-dependent PL spectra. Peng Zhang analyzed experimental data.Conflicts of interest
There are no conflicts to declare.Acknowledgements
X. Li thanks the National Natural Science Foundation of China (21975084 and 51672089) and Natural Science Foundation of Guangdong Province (2021A1515010075) for their support.References
- Z. Liang, R. Shen, Y. H. Ng, Y. Fu, T. Ma, P. Zhang, Y. Li and X. Li, Chem Catal., 2022, 2, 2157–2228 CrossRef CAS.
- R. Shen, C. Qin, L. Hao, X. Li, P. Zhang and X. Li, Adv. Mater., 2023, 35, 2305397 CrossRef CAS.
- R. Shen, G. Liang, L. Hao, P. Zhang and X. Li, Adv. Mater., 2023, 35, 2303649 CrossRef CAS.
- L. Sun, L. Li, J. Yang, J. Fan and Q. Xu, Chin. J. Catal., 2022, 43, 350–358 CrossRef CAS.
- G.-B. Wang, S. Li, C.-X. Yan, F.-C. Zhu, Q.-Q. Lin, K.-H. Xie, Y. Geng and Y.-B. Dong, J. Mater. Chem. A, 2020, 8, 6957–6983 RSC.
- L.-L. Li, S. Liu, Q. Zhang, N.-T. Hu, L.-M. Wei, Z. Yang and H. Wei, Acta Phys.-Chim. Sin., 2017, 33, 1960–1977 CAS.
- Z. Zhou, Y. Xiao, J. Tian, N. Nan, R. Song and J. Li, J. Mater. Chem. A, 2023, 11, 3245–3261 RSC.
- S.-Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- Y. Chen, D. Yang, B. Shi, W. Dai, H. Ren, K. An, Z. Zhou, Z. Zhao, W. Wang and Z. Jiang, J. Mater. Chem. A, 2020, 8, 7724–7732 RSC.
- Q. Niu, L. Mi, W. Chen, Q. Li, S. Zhong, Y. Yu and L. Li, Chin. J. Catal., 2023, 50, 45–82 CrossRef CAS.
- S. Li, X. Chen and Y. Yuan, Acta Phys.-Chim. Sin., 2023, 39, 2303032 Search PubMed.
- Y. Zang, R. Wang, P.-P. Shao, X. Feng, S. Wang, S.-Q. Zang and T. C. W. Mak, J. Mater. Chem. A, 2020, 8, 25094–25100 RSC.
- Y. Qian, D. Li, Y. Han and H.-L. Jiang, J. Am. Chem. Soc., 2020, 142, 20763–20771 CrossRef CAS PubMed.
- Y. Cao, I. D. Parker, G. Yu, C. Zhang and A. J. Heeger, Nature, 1999, 397, 414–417 CrossRef CAS PubMed.
- Z. Xie, X. Yang, P. Zhang, X. Ke, X. Yuan, L. Zhai, W. Wang, N. Qin, C.-X. Cui, L. Qu and X. Chen, Chin. J. Catal., 2023, 47, 171–180 CrossRef CAS.
- J. Xu, C. Yang, S. Bi, W. Wang, Y. He, D. Wu, Q. Liang, X. Wang and F. Zhang, Angew. Chem., Int. Ed., 2020, 59, 23845–23853 CrossRef CAS PubMed.
- L. Guo, Y. Niu, S. Razzaque, B. Tan and S. Jin, ACS Catal., 2019, 9, 9438–9445 CrossRef CAS.
- C. Li, J. Liu, H. Li, K. Wu, J. Wang and Q. Yang, Nat. Commun., 2022, 13, 2357 CrossRef CAS PubMed.
- X. Wang, L. Chen, S. Y. Chong, M. A. Little, Y. Wu, W.-H. Zhu, R. Clowes, Y. Yan, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, Nat. Chem., 2018, 10, 1180–1189 CrossRef CAS.
- Z.-A. Lan, G. Zhang, X. Chen, Y. Zhang, K. A. I. Zhang and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 10236–10240 CrossRef CAS.
- R. Shen, X. Li, C. Qin, P. Zhang and X. Li, Adv. Energy Mater., 2023, 13, 2203695 CrossRef CAS.
- Y. Dong, H. Cha, H. L. Bristow, J. Lee, A. Kumar, P. S. Tuladhar, I. McCulloch, A. A. Bakulin and J. R. Durrant, J. Am. Chem. Soc., 2021, 143, 7599–7603 CrossRef CAS.
- Y. Qian, Y. Han, X. Zhang, G. Yang, G. Zhang and H.-L. Jiang, Nat. Commun., 2023, 14, 3083 CrossRef CAS.
- L. Hao, R. Shen, S. Chen, W. Bi, L. Wang, G. Liang, P. Zhang and X. Li, J. Mater. Chem. A, 2022, 10, 24064–24072 RSC.
- H. Wu, X. He, X. Du, D. Wang, W. Li, H. Chen, W. Fang and L. Zhao, Small, 2023, 2304367 CrossRef CAS PubMed.
- L. Hao, K. Huang, N. Wang, R. Shen, S. Chen, W. Bi, N. Li, P. Zhang, Y. Li and X. Li, Dalton Trans., 2022, 51, 14952–14959 RSC.
- M. Zhang, X. Wu, Y. Xie, X. Hao, Q. Wang, Y. Zhao, J. Wu and X. Pan, Mater. Chem. Front., 2023, 7, 5399–5405 RSC.
- Y. Wang, Y.-Z. Cheng, K.-M. Wu, D.-H. Yang, X.-F. Liu, X. Ding and B.-H. Han, Angew. Chem., Int. Ed., 2023, e202310794 CAS.
- S. Huang, B. Zhang, D. Wu, Y. Xu, H. Hu, F. Duan, H. Zhu, M. Du and S. Lu, Appl. Catal., B, 2024, 340, 123216 CrossRef CAS.
- X. Guan, Y. Qian, X. Zhang and H.-L. Jiang, Angew. Chem., Int. Ed., 2023, 62, e202306135 CrossRef PubMed.
- M. Yang, C. Mo, L. Fang, J. Li, Z. Yuan, Z. Chen, Q. Jiang, X. Chen and D. Yu, Adv. Funct. Mater., 2020, 30, 2000516 CrossRef CAS.
- J. Yang, S. Ghosh, J. Roeser, A. Acharjya, C. Penschke, Y. Tsutsui, J. Rabeah, T. Wang, S. Y. Djoko Tameu, M.-Y. Ye, J. Grüneberg, S. Li, C. Li, R. Schomäcker, R. Van De Krol, S. Seki, P. Saalfrank and A. Thomas, Nat. Commun., 2022, 13, 6317 CrossRef CAS.
- W. Dong, Z. Qin, K. Wang, Y. Xiao, X. Liu, S. Ren and L. Li, Angew. Chem., Int. Ed., 2023, 62, e202216073 CrossRef CAS PubMed.
- Y.-H. Yao, J. Li, H. Zhang, H.-L. Tang, L. Fang, G.-D. Niu, X.-J. Sun and F.-M. Zhang, J. Mater. Chem. A, 2020, 8, 8949–8956 RSC.
- J. Zou, G. Liao, J. Jiang, Z. Xiong, S. Bai, H. Wang, P. Wu, P. Zhang and X. Li, Chin. J. Struct. Chem., 2022, 41, 2201025–2201033 CAS.
- H. Wang, X. Sun, D. Li, X. Zhang, S. Chen, W. Shao, Y. Tian and Y. Xie, J. Am. Chem. Soc., 2017, 139, 2468–2473 CrossRef CAS.
- R. Shen, N. Li, C. Qin, X. Li, P. Zhang, X. Li and J. Tang, Adv. Funct. Mater., 2023, 33, 2301463 CrossRef CAS.
- Z.-A. Lan, M. Wu, Z. Fang, X. Chi, X. Chen, Y. Zhang and X. Wang, Angew. Chem., Int. Ed., 2021, 60, 16355–16359 CrossRef CAS.
- S. Cheng, Q. Xiong, C. Zhao and X. Yang, Chin. J. Struct. Chem., 2022, 41, 2208058–2208064 CAS.
- Y. Yang, J. Wu, B. Cheng, L. Zhang, A. A. Al-Ghamdi, S. Wageh and Y. Li, Chin. J. Struct. Chem., 2022, 41, 2206006–2206014 CAS.
- R. Gao, J. Bai, R. Shen, L. Hao, C. Huang, L. Wang, G. Liang, P. Zhang and X. Li, J. Mater. Sci. Technol., 2023, 137, 223–231 CrossRef CAS.
- Y. Hu, X. Li, W. Wang, F. Deng, L. Han, X. Gao, Z. Feng, Z. Chen, J. Huang, F. Zeng and F. Dong, Chin. J. Struct. Chem., 2022, 41, 2206069–2206078 CAS.
- Z.-A. Lan, G. Zhang, X. Chen, Y. Zhang, K. A. I. Zhang and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 10236–10240 CrossRef CAS.
- S. Tao, S. Wan, Q. Huang, C. Li, J. Yu and S. Cao, Chin. J. Struct. Chem., 2022, 41, 2206048–2206054 CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- Z. Liu, T. Lu and Q. Chen, Carbon, 2020, 165, 461–467 CrossRef CAS.
- J. Zhang and T. Lu, Phys. Chem. Chem. Phys., 2021, 23, 20323–20328 RSC.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- X. Yan, B. Wang, J. Ren, X. Long and D. Yang, Angew. Chem., Int. Ed., 2022, 61, e202209583 CrossRef CAS.
- H. Ben, G. Yan, H. Liu, C. Ling, Y. Fan and X. Zhang, Adv. Funct. Mater., 2022, 32, 2104519 CrossRef CAS.
- Z. Chen, J. Wang, M. Hao, Y. Xie, X. Liu, H. Yang, G. I. N. Waterhouse, X. Wang and S. Ma, Nat. Commun., 2023, 14, 1106 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta05173k |
| This journal is © The Royal Society of Chemistry 2024 |