
Microwave-assisted synthesis of ZIF-9@xGO composites as cooperative electrocatalysts for electro-oxidation of benzyl alcohols coupled with H2 production†
Sayantan
Chongdar‡
 a,
Anirban
Ghosh‡
a,
Rajaram
Bal
bc and
Asim
Bhaumik
*a
a,
Anirban
Ghosh‡
a,
Rajaram
Bal
bc and
Asim
Bhaumik
*a
aSchool of Materials Sciences, Indian Association for the Cultivation of Science, 2A & 2B Raja S. C. Mullick Road, Jadavpur, Kolkata 700 032, India. E-mail: msab@iacs.res.in
bLight Stock Processing Division, CSIR-Indian Institute of Petroleum, Dehradun 248005, India
cAcademy of Scientific and Innovative Research, Ghaziabad, Uttar Pradesh 201002, India
First published on 20th November 2023
Abstract
Selective electrochemical oxidation of alcohols to their corresponding aldehydes or acids utilizing water as the oxygen source is not only considered a green and sustainable approach to synthesize value added chemicals but also economically demanding as the overall process is coupled with green H2 production. However, restricted production due to low current density and the use of expensive noble-metal-based electrocatalysts has limited its industrial viability. On the other hand microwave-assisted synthesis enables a rapid, economical and effective way to fabricate various advanced porous materials. In this regard, herein we report microwave assisted rapid synthesis of a metal–organic framework materials ZIF-9 nanocrystals and ZIF-9@xGO composites by varying the wt% of graphene oxide (GO). Remarkably, upon GO loading the current density increases significantly from 75 mA cm−2 to 204 mA cm−2 at 1.6 V vs. RHE in an alkaline solution with 0.1 M benzyl alcohol, thereby imparting direct impact towards benzoic acid yield (∼84%) and faradaic efficiency (∼88%) of the process. Moreover, the optimised electrocatalyst (i.e. ZIF-9@10GO) exhibits a H2 evolution rate of 273 mmol g−1 h−1 in the presence of benzyl alcohol. Our results indicate that in situ generated Co(OH)2/CoOOH hybrid species over the ZIF-9 surface plays an active role in this electrocatalysis. Additionally, this electrocatalytic system shows good functional group tolerance, exhibiting wide scope for different substrates with high faradaic efficiency.
Introduction
The sluggish four electron transfer kinetics of anodic oxygen evolution reaction (OER) demands a high cell potential, making it quiet challenging for electrolyzing water on a larger scale.1 On the other hand electrochemical oxidation of organics with cell potential lower than that of the OER in a hybrid hydrolysis system is an alternative strategy, for developing an anodic reaction that can effectively lower the energy consumption towards water-splitting. Moreover, utilizing water as the oxygen source for oxidizing organics is a greener sustainable approach towards producing various value-added chemicals.2 However, the reported current densities for such anodic reactions have been lower than 200 mA cm−2, which hinders the overall efficiency and profitability in large-scale production.3 The traditional approach for alcohol oxidation requires high energy and pressure, often with the usage of oxidizing agents like peroxides,4 which is not economical and involves a complex work-up process. Hence, electrochemical alcohol oxidation serves as an eco-friendly and convenient approach under mild reaction conditions, to produce the corresponding aldehyde and acid, which are considered as value added chemicals, being widely utilized in pharmaceutical and chemical industries. Apart from the valuable organics, these electrochemical reactions are paired with H2 evolution, which is a promising alternative fuel that can abate the excessive use of fossil fuels, addressing the environmental problems with promotion of the carbon-neutral cycle.In comparison to other materials, metal organic frameworks (MOFs) and their composites have received exceptional attention as electrocatalysts and energy storage materials due to their large active surface area, tunable porosity and synergistic assembly of suitable metals and ligands.5 Zeolitic imidazolate frameworks (ZIFs) are a particular category of MOFs, which are topologically isostructural with zeolites, having a periodic M–N–C bond,6 that have served as effective electrocatalysts over a wide domain of electrochemical reactions.6 Among various ZIFs, Co based ones are extensively investigated as electrocatalysts owing to their redox chemistry (i.e. Co2+/Co3+).7 However, electrocatalyst ZIF-9 possessing a sodalite topology (SOD) with a benzimidazolate (Bim) ligand, has been sparsely investigated. Several downsides concerning electrochemical water splitting for ZIF-9, resulted due to its large over potential, sluggish kinetics and several other factors.8 As documented in previous literature, with a suitable combination of conductive carbon based material with these ZIFs, the aforesaid obstacles can be overcome.9 Graphene oxide (GO) is explored as an excellent carbon based conductive material, owing to its high effective surface area, facile charge transfer and its convenient synthesis.10 Thus, wrapping GO with various MOF materials can drastically change the electronic properties of the obtained composite material. GO contains several hydroxy and epoxy functionalities throughout the surface, which can transfer electron density from the oxygen atoms to the empty orbitals of unsaturated metal sites of MOFs.11 Thus, grafting GO over ZIFs can increase their ionic conductivity and thereby boost their electrocatalytic activity.
To reduce prolonged reaction time and produce uniform dimensionality of the particles, the microwave assisted synthetic strategy has taken over the traditional solvothermal technique, which is also beneficial in terms of the industrial outlook.12 Over the past few years, several attempts have been made to synthesize ZIF based materials via a microwave assisted strategy.13 Herein, we have shown that through microwave assisted synthesis, we can obtain phase pure ZIF-9 MOF within an hour of reaction time, on the same scale as that in the solvothermal method. Therefore, under the same period of time as in the case of the solvothermal method, one can put multiple batches in a microwave to scale up the synthesis. To the best of our knowledge, ZIF-9 MOF has not been reported using a microwave assisted synthetic route to date. This microwave assisted synthetic approach has also been executed to fabricate ZIF-9 based composite (ZIF-9@xGO) materials, for electrocatalytic oxidation of benzylic alcohols selectively into their corresponding acids with high faradaic efficiency and high yield H2 generation at the cathode (Fig. 1).
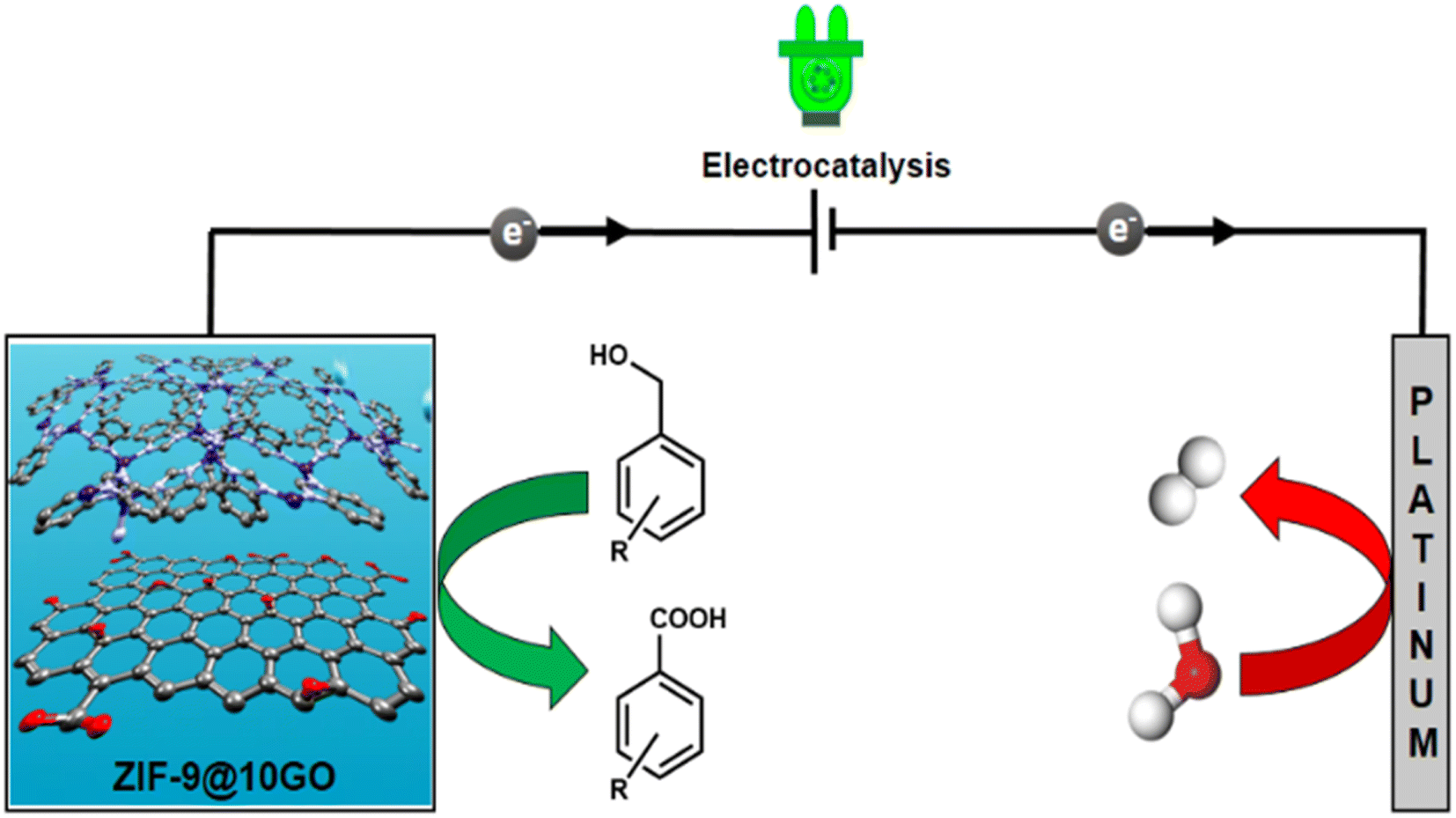 | ||
| Fig. 1 Schematic illustration of benzyl alcohol electro-oxidation coupled with H2 production over the ZIF-9@10GO catalyst. | ||
Results and discussion
Single crystals of ZIF-9 MOF with a chemical formula of (C3.82H2.73Co0.27N1.09), have been crystallized in a trigonal system with an R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) space group (Table S1†). As seen from Scheme 1, the asymmetric unit composed of a Co(II) ion was coordinated tetrahedrally with four nitrogen atoms of the Bim ligand. Next, through the microwave-assisted synthetic protocol we synthesized ZIF-9 nanoparticles in the bulk phase in one hour's time. The X-ray diffractograms of both the single crystal and the crystalline ZIF-9 powder, synthesized via the microwave assisted route, perfectly matched with the simulated XRD pattern of ZIF-9 (Fig. 2a), thereby confirming its bulk phase purity. Henceforth, the microwave assisted ZIF-9 was used for other characterization studies and further application. The synthesized GO exhibited characteristic PXRD peaks (Fig. 2a),14 and was later grafted onto ZIF-9 under microwave conditions, with varying weight percentages forming ZIF-9@xGO (where, x = 2.5, 5 and 10). It should be noted that even after the introduction of GO, the ZIF-9 phases were retained as evidenced from the PXRD pattern (Fig. 2b). The Raman spectra of GO showed characteristic D and G bands at 1353 cm−1 and 1597 cm−1 respectively.15 However, the pristine ZIF-9 MOF exhibited characteristic bands at 1586, 1365, 1347, 1273 and 1017.5 cm−1. It should be noted that due to the overlap of ZIF-9's 1586 cm−1 signal with the G band of GO, the G band appeared broader in the case of ZIF-9@xGO (Fig. 2c).15
space group (Table S1†). As seen from Scheme 1, the asymmetric unit composed of a Co(II) ion was coordinated tetrahedrally with four nitrogen atoms of the Bim ligand. Next, through the microwave-assisted synthetic protocol we synthesized ZIF-9 nanoparticles in the bulk phase in one hour's time. The X-ray diffractograms of both the single crystal and the crystalline ZIF-9 powder, synthesized via the microwave assisted route, perfectly matched with the simulated XRD pattern of ZIF-9 (Fig. 2a), thereby confirming its bulk phase purity. Henceforth, the microwave assisted ZIF-9 was used for other characterization studies and further application. The synthesized GO exhibited characteristic PXRD peaks (Fig. 2a),14 and was later grafted onto ZIF-9 under microwave conditions, with varying weight percentages forming ZIF-9@xGO (where, x = 2.5, 5 and 10). It should be noted that even after the introduction of GO, the ZIF-9 phases were retained as evidenced from the PXRD pattern (Fig. 2b). The Raman spectra of GO showed characteristic D and G bands at 1353 cm−1 and 1597 cm−1 respectively.15 However, the pristine ZIF-9 MOF exhibited characteristic bands at 1586, 1365, 1347, 1273 and 1017.5 cm−1. It should be noted that due to the overlap of ZIF-9's 1586 cm−1 signal with the G band of GO, the G band appeared broader in the case of ZIF-9@xGO (Fig. 2c).15
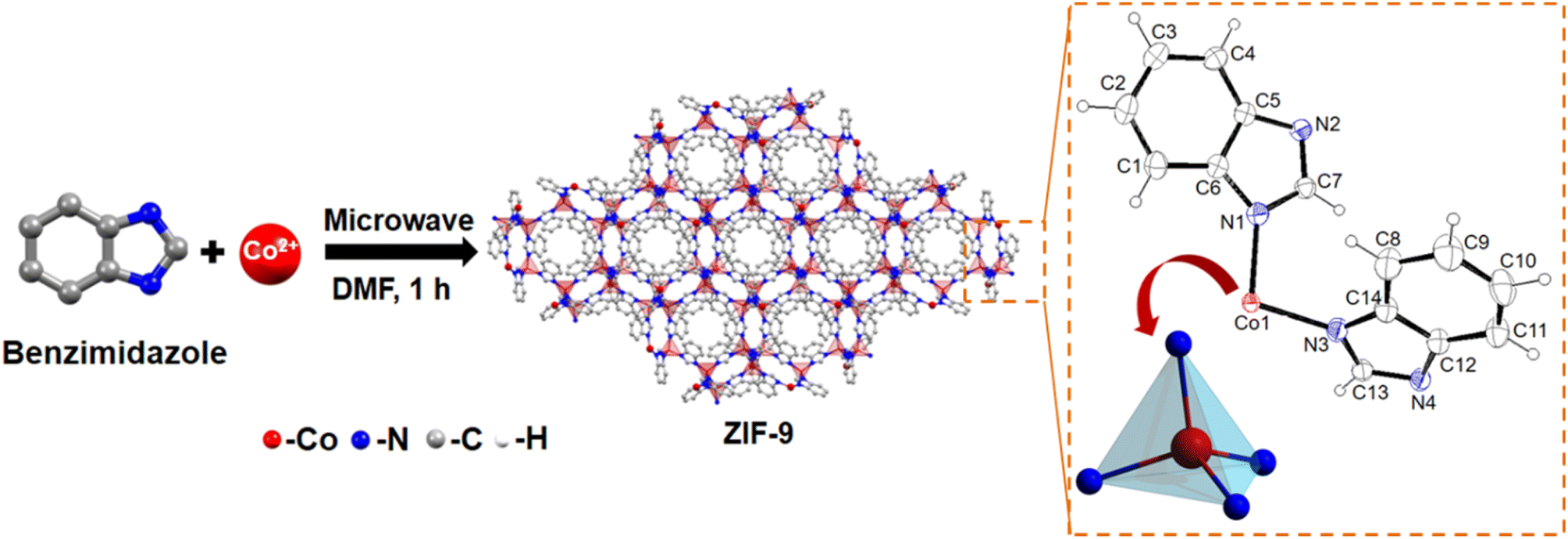 | ||
| Scheme 1 Schematic representation of the microwave assisted synthesis of ZIF-9. | ||
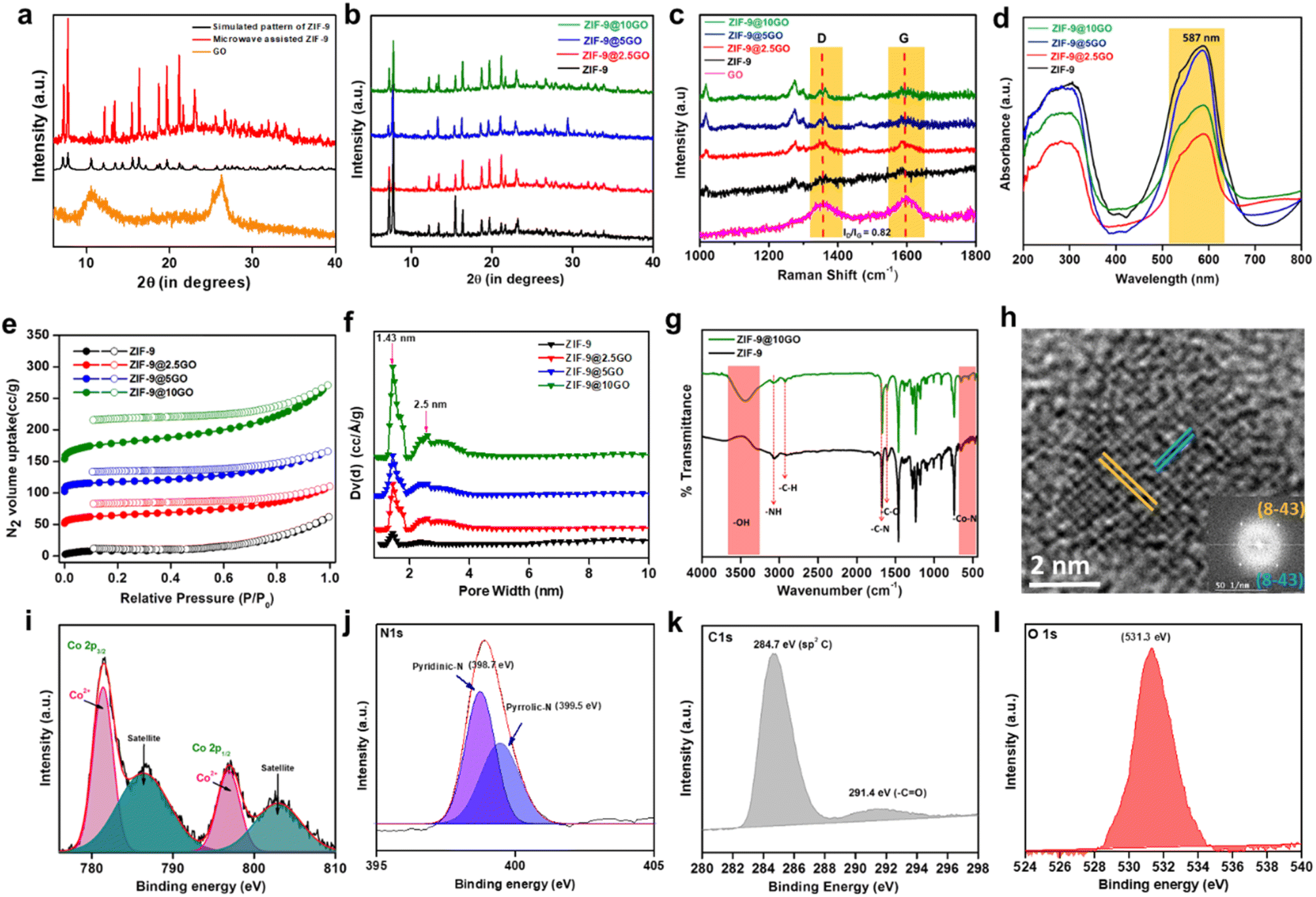 | ||
| Fig. 2 (a) PXRD pattern of ZIF-9 simulated from a single crystal (black), ZIF-9 synthesized using microwave (red), and graphene oxide (orange). (b) PXRD pattern of ZIF-9 (black), ZIF-9@2.5GO (red), ZIF-9@5GO (blue), and ZIF-9@10GO (green). (c) Raman spectra of graphene oxide (pink), ZIF-9 (black), ZIF-9@2.5GO (red), ZIF-9@5GO (blue), and ZIF-9@10GO (green). (d) UV-vis spectra of ZIF-9 (black), ZIF-9@2.5GO (red), ZIF-9@5GO (blue), and ZIF-9@10GO (green). (e) N2 sorption isotherms of ZIF-9 (black), ZIF-9@2.5GO (red), ZIF-9@5GO (blue), and ZIF-9@10GO (green), where filled circles represent adsorption, whereas open circles depict desorption. Y-axis values for isotherms of ZIF-9@2.5GO were enhanced by 50 units, those of ZIF-9@5GO were enhanced by 100 units whereas those of ZIF-9@10GO were enhanced by 150 units for clarity. (f) Pore size distribution curve of ZIF-9 (black), ZIF-9@2.5GO (red), ZIF-9@5GO (blue), and ZIF-9@10GO (green). (g) FTIR spectra of ZIF-9 (black) and ZIF-9@10GO (green). (h) HR-TEM image of ZIF-9 showing lattice fringes of the (8–4 3) lattice planes, (inset: the corresponding FFT pattern of ZIF-9). Narrow-range XPS spectrum of (i) Co 2p, (j) N 1s, (k) C 1s and (l) O 1s. | ||
Solid state UV-vis diffuse reflectance spectroscopy (DRS) analysis showed an absorption peak of ZIF-9 at 587 nm due to the d–d transition of tetrahedral Co(II) centers, which got retained in all possible combinations (Fig. 2d). In order to investigate the thermal stability of pristine ZIF-9 and ZIF-9@10GO, we performed thermogravimetric analysis (TGA) in air. As seen from Fig. S1,† a loss of about 15% at around 350 °C could be attributed to the removal of uncoordinated solvent molecules (DMF). Beyond 350 °C a sharp fall in weight% was seen due to the collapse of the ZIF-9 framework. However, in contrast to this, ZIF-9@10GO showed an early fall in weight% (∼20% at around 300 °C) due to the presence of carbonaceous material (i.e., GO) on the ZIF-9 framework (Fig. S2†). ZIF-9 possesses a low BET surface area because of the large benzimidazole moieties, which block the intrinsic pores, making it inaccessible to the N2 molecules.16 ZIF-9 exhibited a type-I BET isotherm at low P/P0 with a specific surface area of 37 m2 g−1 (Fig. 2e). However, upon GO modification the surface area of the composites increased considerably due to the inherent surface area of the carbonaceous material (i.e., GO). As anticipated, ZIF-9@2.5GO, ZIF-9@5GO and ZIF-9@10GO showed BET surface areas of 54, 64 and 108 m2 g−1, respectively (Fig. 2e). Now since catalysis is controlled by surface phenomena, it is expected that ZIF-9@10GO would show superior catalytic performance compared to its congeners. Pore size distribution (PSD) was calculated by the NLDFT method (Fig. 2f), where the pore size remains almost unchanged (1.43 nm) even after GO modification, suggesting no pore blocking. However, it is worth noting that upon GO addition, a bimodal PSD appeared, which accounted for the GO (Fig. 2f). FTIR spectra of both ZIF-9 and ZIF-9@10GO showed identical footprints as seen in Fig. 2g. A sharp peak at 1607 cm−1 was attributed to –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching mode, while all the sharp bands between 400 and 750 cm−1 were due to the Co–N bond stretching frequency.17 Additionally, the FTIR spectrum of ZIF-9@10GO showed enhancement of the –C–H stretching impression at 2850 and 2930 cm−1, which was due to the addition of GO. The high-resolution transmission electron microscopy (HR-TEM) image of the ZIF-9 nanocrystal (Fig. 2h) exhibited lattice fringes with a d-spacing of 0.25 nm corresponding to the (8 −4 3) lattice plane. The X-ray photoelectron spectroscopy (XPS) analysis confirmed that the Co centres in ZIF-9@10GO were present in the +2 oxidation state (Fig. 2i), with two corresponding peaks at 781.4 eV and 796.9 eV for 2p3/2 and 2p1/2 respectively.18 A peak at 398.9 eV was observed in N 1s spectra of the benzimidazolate moiety (Fig. 2j). The XPS spectra of the C atom showed a distinct peak at 284.7 eV corresponding to the sp2 hybridized C (1s) arising from the benzimidazolate moiety, while a peak at 291.4 eV was due to –CO, originating from GO (Fig. 2k). Again a significant impression of O 1s spectra was observed at 531.3 eV due to GO addition (Fig. 2l).
N stretching mode, while all the sharp bands between 400 and 750 cm−1 were due to the Co–N bond stretching frequency.17 Additionally, the FTIR spectrum of ZIF-9@10GO showed enhancement of the –C–H stretching impression at 2850 and 2930 cm−1, which was due to the addition of GO. The high-resolution transmission electron microscopy (HR-TEM) image of the ZIF-9 nanocrystal (Fig. 2h) exhibited lattice fringes with a d-spacing of 0.25 nm corresponding to the (8 −4 3) lattice plane. The X-ray photoelectron spectroscopy (XPS) analysis confirmed that the Co centres in ZIF-9@10GO were present in the +2 oxidation state (Fig. 2i), with two corresponding peaks at 781.4 eV and 796.9 eV for 2p3/2 and 2p1/2 respectively.18 A peak at 398.9 eV was observed in N 1s spectra of the benzimidazolate moiety (Fig. 2j). The XPS spectra of the C atom showed a distinct peak at 284.7 eV corresponding to the sp2 hybridized C (1s) arising from the benzimidazolate moiety, while a peak at 291.4 eV was due to –CO, originating from GO (Fig. 2k). Again a significant impression of O 1s spectra was observed at 531.3 eV due to GO addition (Fig. 2l).
High-resolution transmission electron microscopy (HRTEM) images (Fig. 3a and b) of the ZIF-9@10GO electrocatalyst revealed spherical particles with sizes ranging between (80 and 110) nm. The ZIF-9 particle morphology was similar to the one previously reported in the literature.19 Moreover, the selected area electron diffraction (SAED) pattern shown in Fig. 3c, gave further insight into the crystalline nature of ZIF-9 nanocrystals. The HRTEM images of ZIF-9, ZIF-9@2.5GO, and ZIF-9@5GO are shown in Fig. S3(a)–(c).† The field-emission scanning electron microscopy (FESEM) images (Fig. 3d–f) of ZIF-9@10GO also exhibited spherical morphology, corroborating the HRTEM images. Interestingly, (20–30) nm thick GO sheets were visible along with the nanospheres, suggesting uniform distribution of GO in the electrocatalyst. Fig. S3(d)–(f),† show FESEM images of ZIF-9, ZIF-9@2.5GO, and ZIF-9@5GO.
 | ||
| Fig. 3 (a) HRTEM images of the ZIF-9@10GO electrocatalyst. The marked area in (a) is zoomed in as (b). (c) SAED pattern of the ZIF-9@10GO electrocatalyst. (d)–(f) FESEM images of the ZIF-9@10GO electrocatalyst. The marked area in (d) is zoomed in as (e) and marked area in (e) is zoomed in as (f). | ||
Over the years, electrochemical oxidation over transition metal-oxyhydroxides (MOOHs), as a propitious anodic material has been well-documented.20 The “volcano plot” in an alkaline medium, depicts cobalt based oxides as an efficient electrocatalyst for water oxidation.21 In this sense, we synthesized cobalt-based zeolitic imidazolate frameworks (ZIF-9), which have been reported to generate cobalt oxyhydroxides (CoOOH) at a specific potential in an alkaline medium.8 In a three-electrode electrochemical cell, using a glassy carbon electrode (GCE) as the conducting substrate, we first investigated the redox nature of ZIF-9 in nitrogen-saturated 1 M KOH electrolyte, by cyclic voltammetry (CV), at a scan rate of 10 mV s−1 (Fig. 4a, inset). As illustrated in the anodic direction of Fig. 4a, ZIF-9 exhibited the first oxidation peak at ∼1.2 V vs. RHE, corresponding to the Co(II) to Co(III) oxidation in CoOOH, while the second peak for Co(III) to Co(IV) oxidation was at ∼1.45 V vs. RHE, consistent with previous report.22 The very idea of putting together graphene oxide (GO) into this ZIF-9, emerged considering GO’s capability to couple electroactive species onto its surface and favouring the electron transport at electrodes.23 Similar to the pristine ZIF-9 material, the CV of this composite material (ZIF-9@10GO) was looked into in nitrogen-saturated 1 M KOH electrolyte, at a scan rate of 10 mV s−1 (Fig. 4a). As shown in Fig. 4a, an enhancement in the anodic and cathodic current density was observed for the composite material (red curve) in comparison to that of the pristine material (black curve), with similar redox peaks.
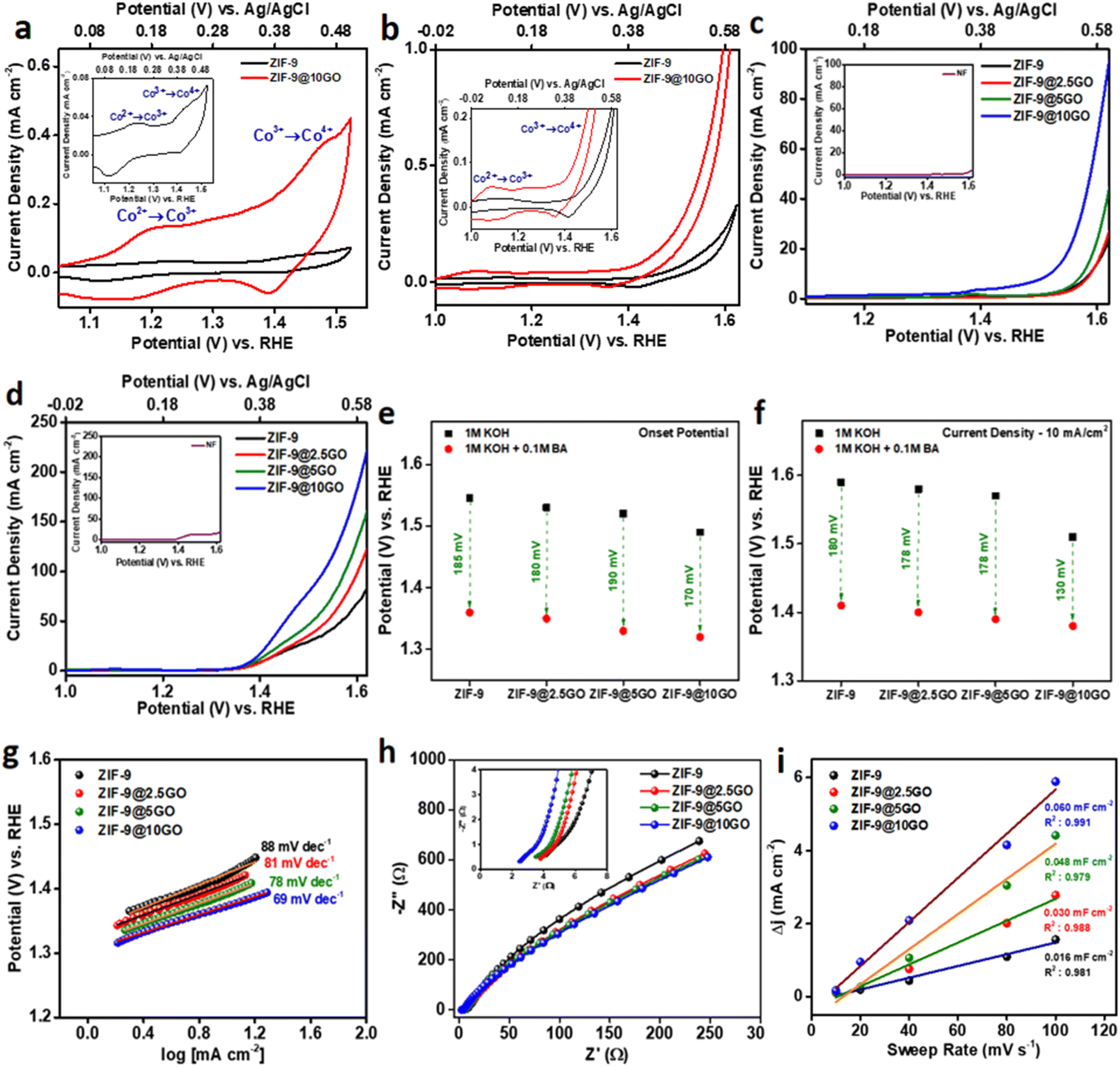 | ||
| Fig. 4 (a) CV curves of ZIF-9 and ZIF-9@10GO electrocatalysts over a glassy carbon electrode in 1 M KOH (inset: CV curve of ZIF-9). (b) CV curves of ZIF-9 and ZIF-9@10GO electrocatalysts over a glassy carbon electrode in 1 M KOH with 0.1 M BA (inset: CV curves of ZIF-9 and ZIF-9@10GO). (c) LSV curves of the electrocatalysts over NF in 1 M KOH (inset: LSV curve of bare NF). (d) LSV curves of the electrocatalysts over NF in 1 M KOH with 0.1 M BA (inset: LSV curve of bare NF). (e) Onset potential drop for the electrocatalysts after addition of 0.1 M BA. (f) Overpotential drop at a current density of 10 mA cm−2 for the electrocatalysts after addition of 0.1 M BA. (g) Tafel plots of the electrocatalysts in 1 M KOH with 0.1 M BA. (h) EIS Nyquist plots in 1 M KOH with 0.1 M BA (inset: magnified EIS Nyquist plots at higher frequencies). (i) Cdl plots for the electrocatalysts in 1 M KOH with 0.1 M BA. | ||
It is well documented that GO due to its specific 2D structure and presence of various oxygenated functional moieties, facilitates the electron/charge transfer between the electroactive species and electrode surface.10 Thus in order to shed light on the effect of GO on ZIF-9, the weight percentage of GO loading was varied. All the materials (ZIF-9, ZIF-9@2.5GO, ZIF-9@5GO and ZIF-9@10GO) were deposited in Ni foam (NF) to attain higher current density. Fig. 4c shows the linear sweep voltammetry (LSV) curves of the aforementioned materials in nitrogen-saturated 1 M KOH solution, at a scan rate of 10 mV s−1. As expected, the current density increased, as the loading percentage of GO increased from 2.5 to 10 wt%, along with the enhancement in oxidation peaks. With the same loaded sample, 10 wt% loaded GO, at 1.6 V vs. RHE, showed four times more current density than pristine ZIF-9, suggesting the effective role of GO in reducing the activation energy for the OER. However, on further increasing the GO wt% to 15, the observed current density decreased (Fig. S4a†). This might be because a higher percentage of GO loading, in turn reduced the percentage of ZIF-9, which happens to be the active site in this electrocatalysis. Moreover, a high percentage of GO loading might also result in stacking of GO and affect the distribution of active sites in the catalyst.24 In a control experiment we loaded only GO over NF, which resulted in very low current density in the LSV (Fig. S4c and d†), suggesting Co centres to be the active species in this electrocatalysis.
Previous reports have explained that oxidation of Co2+ ion generates electrophilic OH*, which shows catalytic activity towards alcohol oxidation.25 The CV curves with the addition of 0.1 M BA in 1 M KOH (on GCE), at a scan rate of 10 mV s−1 are shown in Fig. 4b. Compared to blank voltammetry in KOH, an enhancement in the current density was achieved for both the catalysts, with addition of BA at a higher potential (>1.45 V vs. RHE), indicating BA oxidation (Fig. S5†).26 Herein, Co3+ ions acted as a chemical oxidant for BA oxidation, and get reduced to Co2+ ions, as indicated by the reappearance of an oxidation peak for Co(II)/Co(III) after the addition of BA. In the presence of BA, it is worth noting that the oxidation peak for Co(II)/Co(III) was smaller compared to the one obtained in only KOH. This might be due to the fact that only a fraction of Co3+ ions obtained in the course of reversible electrochemical Co(II)/Co(III) oxidation can catalyse chemical oxidation of BA.27
As obtained in a 1 M KOH medium, the LSV curves of ZIF-9, ZIF-9@2.5GO, ZIF-9@5GO and ZIF-9@10GO, over NF exhibited similar trends in the presence of 0.1 M BA, with ZIF-9@10GO showing the highest current density (Fig. 4d). Note that there was no noticeable difference upon reducing the GO wt% to 1, while a 15 wt% GO loaded catalyst exhibited a similar trend as observed in 1 M KOH (Fig. S4b†). With the addition of BA, the in situ formed OH* on CoOOH oxidises the organic molecule, resulting in a significant increase in current density. Interestingly, with the addition of 0.1 M BA the overpotential at a current density of 10 mA cm−2, significantly got reduced compared to that of the OER (Fig. 4f). This finding was consistent for all the catalysts, indicating the acceleration of the anodic reaction through BA oxidation. As shown in Fig. 4e, the onset potential for all electrocatalysts was remarkably low in the case of a hybrid water electrolysis system as compared to a traditional alkaline water electrolysis system, insinuating superior activity towards BA oxidation.
By fitting polarization data to the Tafel equation η = a + b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log |J| (where η is the overpotential, b is the Tafel slope, and J is the current density), the Tafel slope for the electrocatalysts was derived. The reaction kinetics for electrocatalysis were studied by using the Tafel polarization curve (E vs. log |J|), shown in Fig. 4g. In the hybrid water electrolysis system, the electrocatalyst with the highest GO loading (i.e., ZIF-9@10GO) showed the smallest Tafel slope, suggesting faster electrocatalytic kinetics and higher electrocatalytic activity towards BA oxidation with an increase in GO wt%. To understand in depth the effect of GO on electronic properties and charge-transfer resistance upon addition into the pristine ZIF-9 material, we performed electrochemical impedance spectroscopy (EIS) in the hybrid aqueous system. An equivalent circuit was obtained based on the semicircle of EIS, which consisted of three components: the equivalent series resistance (Rs), the charge-transfer resistance (Rct) and constant phase element (CPE). The Rs was composed of the intrinsic resistance of electrolyte and electrocatalysts, which was found to be the least for the electrocatalyst with the highest loading of GO (Fig. 4h, inset), implying escalation of conductivity with addition of GO (Table S2†). The kinetic resistance of charge transfer, on the interface of the electrode and electrolyte can be acquired by using Rct. As shown in Fig. 4h a smaller semicircle at a lower frequency for ZIF-9@10GO suggested lower charge transfer resistance compared to that of other electrocatalysts. The minimum value of Rct for ZIF-9@10GO showed its maximum efficiency towards BA oxidation among the other congeners.
log |J| (where η is the overpotential, b is the Tafel slope, and J is the current density), the Tafel slope for the electrocatalysts was derived. The reaction kinetics for electrocatalysis were studied by using the Tafel polarization curve (E vs. log |J|), shown in Fig. 4g. In the hybrid water electrolysis system, the electrocatalyst with the highest GO loading (i.e., ZIF-9@10GO) showed the smallest Tafel slope, suggesting faster electrocatalytic kinetics and higher electrocatalytic activity towards BA oxidation with an increase in GO wt%. To understand in depth the effect of GO on electronic properties and charge-transfer resistance upon addition into the pristine ZIF-9 material, we performed electrochemical impedance spectroscopy (EIS) in the hybrid aqueous system. An equivalent circuit was obtained based on the semicircle of EIS, which consisted of three components: the equivalent series resistance (Rs), the charge-transfer resistance (Rct) and constant phase element (CPE). The Rs was composed of the intrinsic resistance of electrolyte and electrocatalysts, which was found to be the least for the electrocatalyst with the highest loading of GO (Fig. 4h, inset), implying escalation of conductivity with addition of GO (Table S2†). The kinetic resistance of charge transfer, on the interface of the electrode and electrolyte can be acquired by using Rct. As shown in Fig. 4h a smaller semicircle at a lower frequency for ZIF-9@10GO suggested lower charge transfer resistance compared to that of other electrocatalysts. The minimum value of Rct for ZIF-9@10GO showed its maximum efficiency towards BA oxidation among the other congeners.
Next the surface coverage (Γ) of each of these electrocatalysts was determined in the hybrid water electrolysis system by plotting the anodic and cathodic peak current density vs. the scan rate separately (Fig. S6†). A linear proportionality between the peak current density and scan rate was observed for all the materials. The average of slopes obtained from anodic and cathodic curves were used to determine the surface coverage, following the equation:
| Ip = n2F2AΓν/4RT |
The performance of an electrocatalyst is mainly governed by the available electrochemically active surface area (ECSA), which can be estimated from the electrochemical double-layer capacitance (Cdl). As shown in Fig. S7,† by performing CV at different scan rates in the non-faradaic region, we obtain the Cdl values for all the electrocatalysts (Fig. 4i). It was observed that ZIF-9@10GO exhibited the highest Cdl value, which was almost four times higher than that of pristine ZIF-9, manifesting more exposed surface area, thus accounting for the maximum current density.28 In order to evaluate the stability of the electrocatalysts, we performed chronoamperometry at a constant voltage of 1.45 V vs. RHE for 20000 s in 1 M KOH with 0.1 M BA (Fig. S8†). All the catalysts (ZIF-9, ZIF-9@2.5GO, ZIF-9@5GO and ZIF-9@10GO) exhibited a steady chronoamperometric curve, thus validating their amenability towards electro-oxidation of BA. Moreover, with each addition of 0.1 M BA over the ZIF-9@10GO/NF catalyst there was a sharp increase in the current density in the chronoamperometric curve, at a constant potential of 1.45 V vs. RHE, thereby elucidating its activity towards BA oxidation (Fig. S9†).
Once we optimized our electrocatalyst (i.e., ZIF-9@10GO), we compared its catalytic activity towards 0.1 M BA oxidation at different constant potentials (i.e., 1.45 V, 1.50 V and 1.55 V vs. RHE) in 1 M KOH. As seen in Fig. 5a, at 1.45 V the conversion was only ca. 21% after 12 h of reaction, with ∼78% selectivity and ∼90% faradaic efficiency (FE) for benzoic acid. This high FE% suggested that most of the charge passed during the electro-oxidation was consumed for the production of benzoic acid, indicating much less OER at the anode. A further increase in the potential to 1.50 V, increased the amount of charge passing through the system, which uplifted the conversion to ∼89% after 12 h, with ∼95% selectivity and ∼88% FE for the acid product (Fig. 5b). Interestingly at 1.55 V, the selectivity for the acid product increased to ca. 99%, suggesting favoured 4e− oxidation at higher potential. However, a drop in FE% at 1.55 V for the acid product indicated the occurrence of the OER at more positive potential (Fig. 5c). Note that even at 1.55 V, the conversion of BA was not inhibited considerably by the OER. Considering the above results, we chose 1.50 V vs. RHE as our optimized potential for the alcohol oxidation reaction.
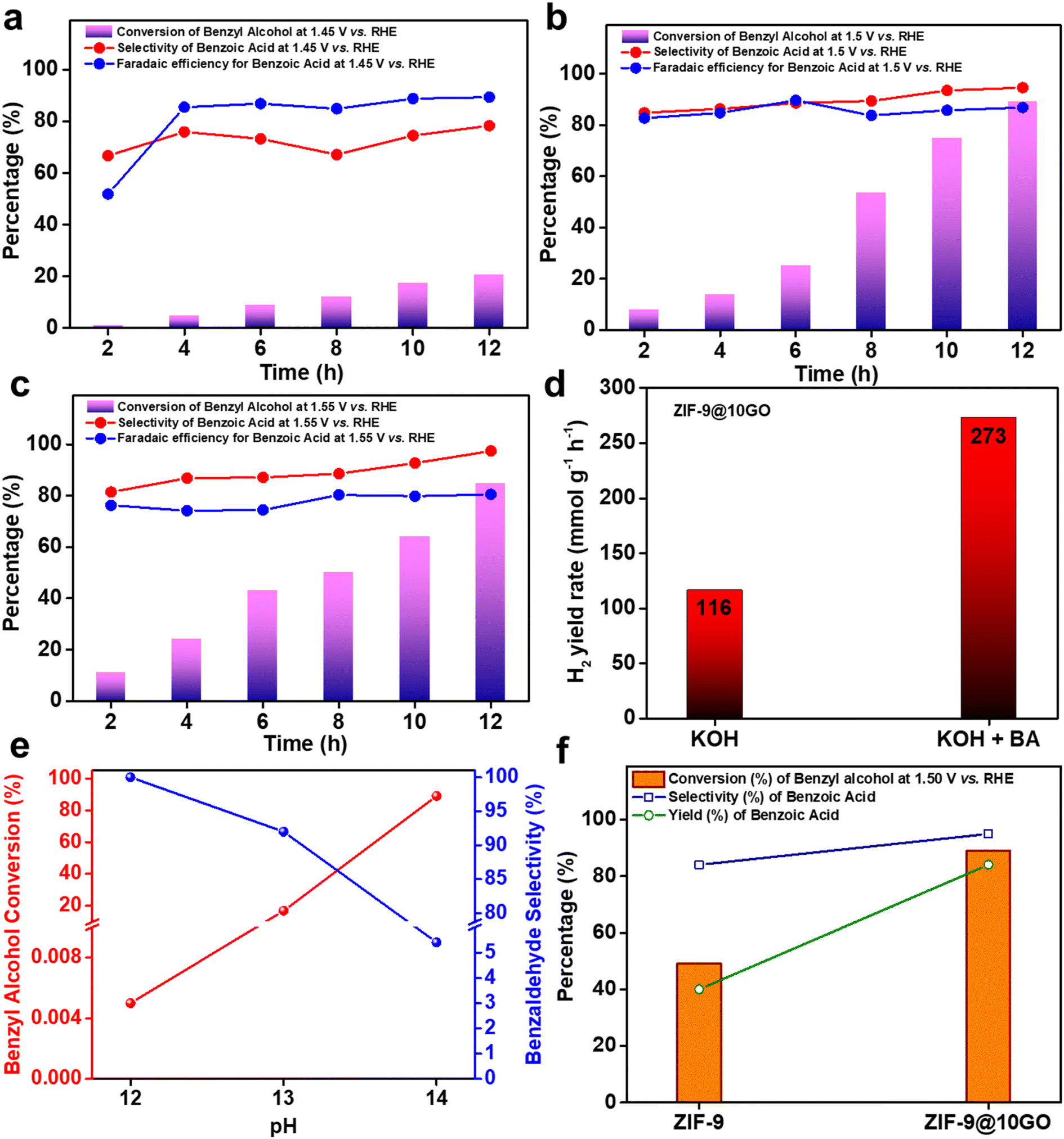 | ||
| Fig. 5 Time-dependent conversion of BA, selectivity of the formation of benzoic acid and faradaic efficiency of benzoic acid at constant applied potentials (a) 1.45 V vs. RHE. (b) 1.50 V vs. RHE. (c) 1.55 V vs. RHE. (d) Space-time yield of H2 in the presence and absence of benzyl alcohol. (e) Effect of pH on benzyl alcohol conversion and benzaldehyde selectivity. (f) Comparison of the electrocatalytic performance between pristine ZIF-9 and ZIF-9@10GO. | ||
We then investigated the influence of catalyst mass loading on BA oxidation at 1.50 V vs. RHE. As shown in Fig. S10,† the conversion of BA increased initially as the loading was increased from 2 mg cm−2 to 4 mg cm−2. This elevation in conversion might be due to the increase in the number of electrochemically accessible active catalytic sites upon higher loading. However, a further increment in the loading to 6 mg cm−2, decreased the BA conversion without affecting the FE% of the acid product. This observation could be rationalized considering the exchange current density (j0), which is used to measure the electrode's readiness to proceed with an electrochemical reaction. The higher the value of j0, the more active the electrode surface is towards a particular chemical reaction. By considering the standard redox potential, E0 for benzoic acid to be −0.23 V vs. Ag/AgCl (i.e., 0.793 V vs. RHE, at 25 °C and pH = 14),29 and by extrapolating the Tafel line, we obtain the j0 for the 4 mg cm−2 and 6 mg cm−2 loaded electrodes (Fig. S11†). The exchange current density for 4 mg cm−2 and 6 mg cm−2 loaded electrodes was 8.869 × 10−7 A cm−2 and 6.012 × 10−7 A cm−2 respectively. Henceforth, the 4 mg cm−2 loaded catalyst exhibited the best catalytic activity towards BA oxidation. Apart from anodic BA oxidation, at the counter Pt cathode, liberation of H2 gas took place. The rate of yield of H2 reaches around 273 mmol g−1 h−1 in the presence of BA (Fig. 5d), which was notably higher compared to that in only KOH solution (116 mmol g−1 h−1). It is pertinent to mention that this H2 evolution rate is much higher than that of related hybrid electrocatalytic systems: MoSx-graphene-Ni foam (13.47 mmol g−1 h−1), Cr–C hybrid (38.5 mmol g−1 h−1) or 12%Ni@COF-TPP-CB[6]-TiO2 (18.7 mmol g−1 h−1).30–32 To assess whether pH plays a role in the electrocatalytic activity of the catalyst, we carried out a series of experiments altering the pH of the electrolyte. A significant drop in the BA conversion was observed as we lowered the pH of the electrolyte, with almost insignificant conversion at pH = 12 (Fig. 5e). Interestingly, lowering the pH increased the selectivity of benzaldehyde (Fig. 5e), thus corroborating its sensitivity towards the electrolyte's pH.
The optimized catalytic system demonstrated a wide substrate scope for various substituted benzyl alcohols (Table 1). To begin with, various para-substituted BA (1b–1f) were examined possessing either an electron-donating or electron-withdrawing functionality. The electro-oxidation process proceeded smoothly with the electron rich alcohols (1b–1d), transforming them into their corresponding acids in good yields and high selectivity (88–99%). The only exception was 4-methoxybenzyl alcohol (1e), which showed a less satisfactory result (yield ∼33%) due to the stabilization of carbon-centred benzylic radical, which in turn favoured undesired reaction pathways.33 Furthermore, the oxidation of electron-withdrawing p-nitro BA (1f) showed an inferior result with ∼13% yield, which was consistent with previous findings.34 The meta-halogenated BAs (1g, 1h) were converted to their corresponding acids with high yield (81–91%) and selectivity (98%). Then we extended our investigation over an oxygen containing furan ring (1i) to afford 2-furoic acid with 90% yield and high selectivity. Nonetheless, for the allylic system we chose cinnamyl alcohol (1j) which got poorly oxidised to its corresponding acid.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Product | Conversion (%) | Selectivityb (%) | Yieldc (%) | FEd (%) |
| a Reaction conditions: during the reaction the room temperature of the laboratory was 30 °C, cell volume – 40 mL, and 0.1 M substrate was used. b Selectivity of the acid product, while the remaining (%) corresponds to aldehyde. c Yield calculated through NMR, taking mesitylene as the internal standard. d FE % for the acid product. | |||||
| 1a |
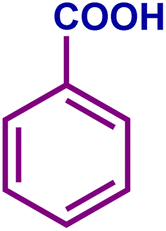
|
89 | 95 | 84 | 88 |
| 1b |
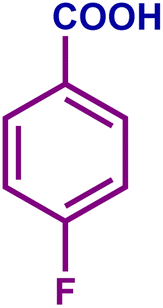
|
70 | 99 | 70 | 71 |
| 1c |
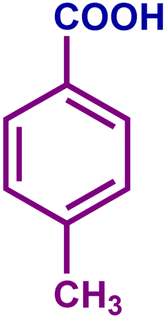
|
73 | 88 | 64 | 79 |
| 1d |
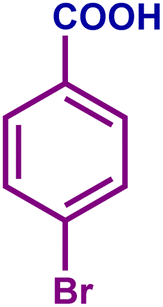
|
96 | 99 | 96 | 81 |
| 1e |
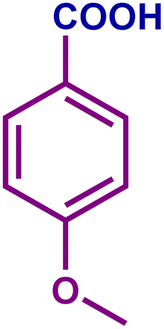
|
61 | 54 | 33 | 54 |
| 1f |
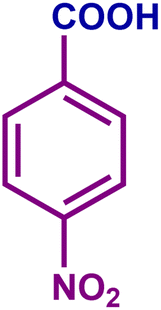
|
45 | 29 | 13 | 25 |
| 1g |
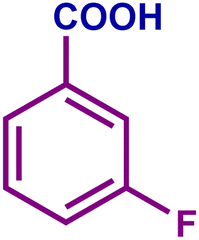
|
92 | 98 | 91 | 74 |
| 1h |
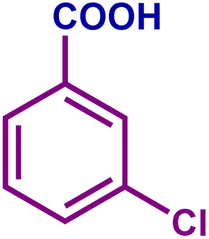
|
82 | 98 | 81 | 76 |
| 1i |
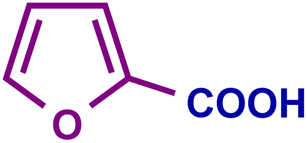
|
95 | 95 | 90 | 84 |
| 1j |
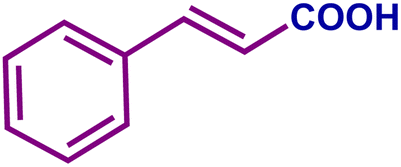
|
22 | 62 | 14 | 81 |
Based on the previous reported mechanisms of electrochemical oxidation,35,36 and our experimental studies, we hypothesized a reaction scheme (Fig. 6) in order to understand the plausible mechanistic pathway, the role of the electrocatalyst and electrolyte's pH and finally the fate of the electrocatalyst after electrochemical experiments. As soon as we expose our catalyst (ZIF-9@10GO) to the KOH medium, the benzimidazole linkers from the surface get replaced by the hydroxide ions (Fig. 6A–C) and are eventually transformed into Co(OH)42− or α-Co(OH)2 (D). Lee et al. previously explained that during an anodic sweep the OH− ions migrate into the pores of the ZIF structure and in time, replace the organic linkers to form Co(OH)3− ions.35 During the cathodic sweep these Co(OH)3− ions get removed from the framework there by destroying the pristine ZIF structure. Another anodic sweep would deposit these Co(OH)3− ions onto the anode, and eventually get transformed into Co(OH)2. A phase transfer from less stable α-Co(OH)2 to more stable octahedral β-Co(OH)2 (E) occurs.34 In the alkaline medium, during the anodic sweep the Co(OH)2 undergoes proton-coupled electron transfer (PCET) (in two steps), to get converted to higher valent Co3+ (2) and Co4+ (3) species, which were considered to be the active species during the electro-oxidation reactions. This explanation was confirmed from the two oxidation peaks obtained in the CV curve, corresponding to Co(II)/Co(III) and Co(III)/Co(IV) oxidation. Now in order to validate the PCET process involved in the oxidation steps, we studied the potential-pH plot (Fig. S12a†), also known as the Pourbaix diagram for the corresponding redox-active species. At room temperature, the pH-dependent Pourbaix expression can be expressed as:
| E1/2 = E°′ − 0.059 × (m/n) × pH |
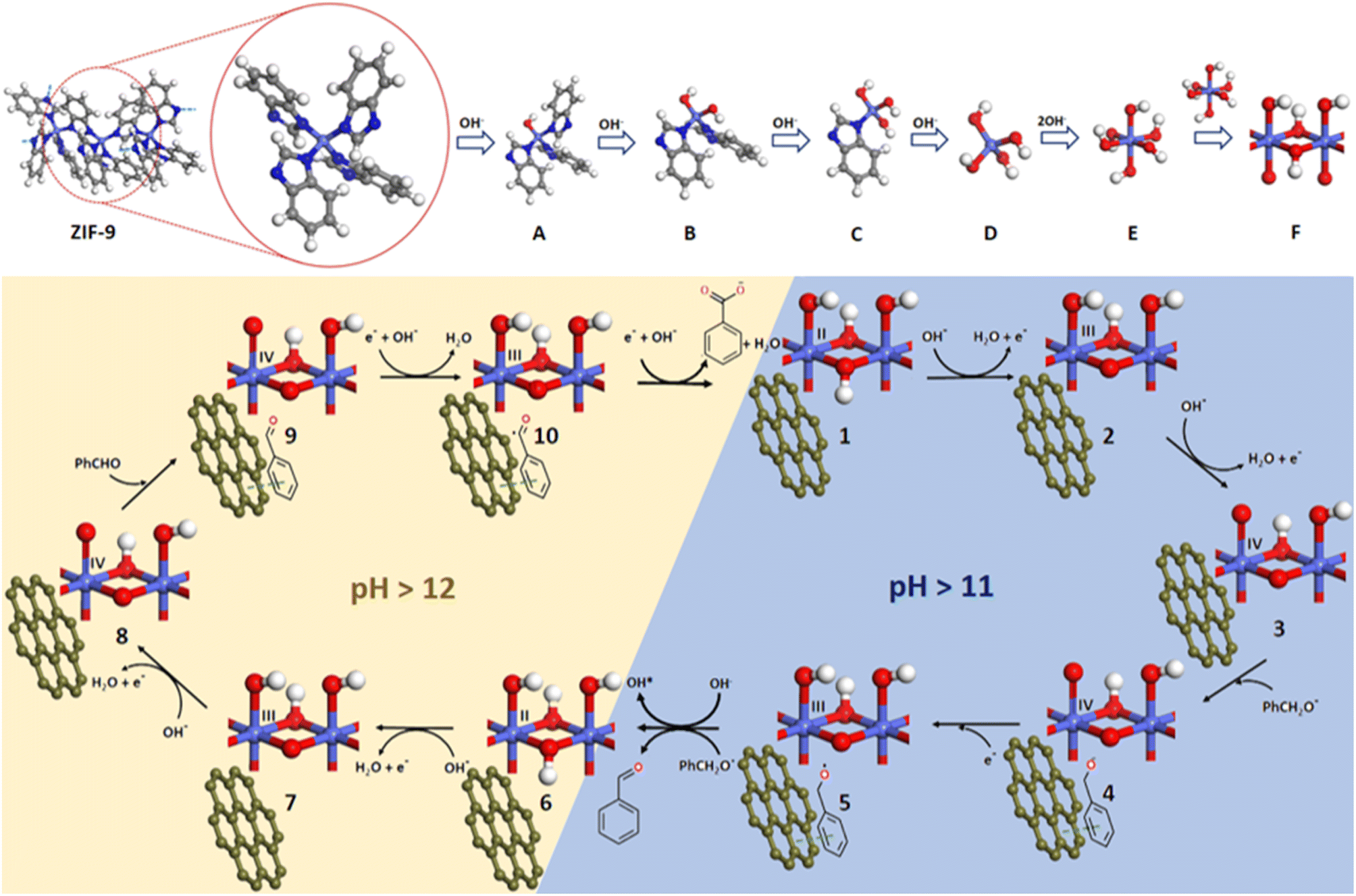 | ||
| Fig. 6 Schematic representation of the proposed reaction mechanism for benzyl alcohol electro-oxidation. (A: Co(PhIm)3OH; B: Co(PhIm)2(OH)2; C: Co(PhIm) (OH)3; D: α-Co(OH)2; E: β-Co(OH)2; F: CoOOH, where PhIm : benzimidazolate). | ||
The obtained slope from Fig. S12b,† suggested the stoichiometry of protons and electrons involved in the redox process, which was found to be close to 59 mV per pH (51 mV per pH) suggesting a 1H+/1e− process as shown in the aforementioned Fig. 6. Note that such a study for Co(III)/Co(IV) redox couple was not possible, as the Co(III)/Co(IV) oxidation signal overlapped with the OER catalytic current. Apart from facilitating the electron/charge transfer between the electroactive species and electrode surface, GO plays a crucial role in binding substrates, thus aiding its catalytic conversion. This claim was further verified considering the catalytic conversion of BA over pristine ZIF-9 and the ZIF-9@10GO electrocatalyst (Fig. 5f). Under similar catalytic conditions, ZIF-9 bereft of GO converted only 49% of BA, with an acid yield of only 41%, compared to 84% over ZIF-9@10GO. It must be noted that NF loaded with only GO showed much less BA conversion (Fig. S13†). The initial deprotonation of alcohol to its corresponding alkoxide occurs in an alkaline medium (ca. pH > 11).38 Henceforth, benzyl alkoxide was considered as the reactant molecule in this mechanism. Once benzyl alkoxide gets adsorbed over the catalyst (4), it gets oxidised by donating an electron to the Co(IV) center (4). Now, the in situ formed Co(III) species (5) further gets reduced to Co(II) species (6) at ∼1.1 V vs. RHE, producing electrophilic OH* from the aqueous alkaline medium, which in turn oxidises PhCH2O* to benzaldehyde. This scenario of BA oxidation is similar to the one suggested by Tao et al.39 where they probed the electrophilic OH* with alcohol, transforming it into the corresponding aldehyde/acid.
Previous reports have claimed that electrochemical alcohol oxidation proceeds through a cascade catalysis mechanism, where alcohol gets converted to aldehyde via 2e− oxidation, followed by another 2e− oxidation to acid.40 The benzoic acid selectivity vs. time plot shown in Fig. S14† was consistent with the abovementioned claim, as at constant potential of 1.50 V vs. RHE, oxidation of BA to benzaldehyde was observed prior to its conversion to benzoic acid. Koper et al.41 suggested that the aldehydes are prone towards oxidation in the presence of electron acceptors, in an alkaline medium. While Duan et al.42 demonstrated the role of pH (i.e., concentration of OH− ions) in benzoate selectivity, which matched with those of our pH dependent study (Fig. 5e). On the basis of these results, we argued that in the presence of enough concentration of OH− ions (i.e., pH > 12), the Co(II) species (6) further gets oxidized to Co(IV) species (8). Now, as the benzaldehyde gets adsorbed onto the catalytic system, it gets oxidised to PhCHO* by donating an electron to the Co(IV) species to form Co(III) species (10). This Co(III) species further gets reduced to Co(II) (1), producing electrophilic OH*, which then reacts with PhCHO* to give benzoic acid.
In order to understand the fate of the electrocatalyst, we performed FTIR, PXRD and XPS analysis of the post-electrocatalytic sample. As seen from Fig. 7a, the characteristic stretching frequency pattern of ZIF-9@10GO remains unchanged after electrochemical oxidation in both 1 M KOH and in 1 M KOH + 0.1 M BA, demonstrating its superior stability. Interestingly, only in the presence of a KOH medium, a new band at 582 cm−1 appeared, which could be assigned to the Co–O vibrational mode of Co3+ species,43 which supports our abovementioned claim in Fig. 6. Similarly, the appearance of a band at 703 cm−1 could be ascribed to δO–H vibration, which was absent in ZIF-9@10GO before electrocatalysis.44 Moreover, a wide band at around 1400 cm−1 corresponded to –OH deformation originating from the adsorbed water molecules over the catalytic surface of ZIF-9@10GO.45 It is to be noted that the pristine (dry) catalyst did not show this band. Moreover, in order to validate our claim, we performed the FT-IR analysis of the used catalyst (i.e., ZIF-9@10GO) after treating it at 100 °C (Fig. S15†). It was observed that the broad peak at around 1400 cm−1 decreased significantly, thereby justifying our claim. A new band at 831 cm−1 could be assigned to the CoIVO vibrational mode of species 3 (Fig. 6), similar to the one obtained by Frei et al. where they claimed that during the water oxidation reaction the formation of CoIVO species takes place via oxidation of surface CoIII–OH species.46 However, in the presence of 0.1 M BA, two new bands at 2715 and 2607 cm−1 appear which can be characterised as the C–H stretching vibration of CH2(νCH2) of physisorbed benzyl alcohol.47 Note that due to the presence of the benzimidazole ligand in ZIF-9, GO and hydroxyl groups, it is difficult to identify the other stretching bands of physisorbed BA. The chronoamperometric analysis of ZIF-9@10GO at a constant voltage of 1.50 V vs. RHE for 12 h in 1 M KOH with 0.1 M BA (Fig. S16†), demonstrated almost 82% retention of the current density. This drop in the current density is due to the formation of non-porous Co(OH)2/CoOOH on the surface of the catalyst. The catalytic recyclability of the ZIF-9@10GO electrocatalyst towards BA oxidation is shown in Fig. S17.† Moreover, the PXRD pattern of the used electrocatalyst after 12 h of chronoamperometry showed new peaks at 20.24°, 38.89°, 50.58° and 65.34° of 2θ, corresponding to the (003), (012), (015) and (110) planes of the β-CoOOH phase (JCPDS no. 73-1213). It is worth noting that few ZIF-9 peaks were retained (Fig. 7b, marked by an asterisk) with loss of crystallinity, implying that the formation of CoOOH species taking place mainly over the surface of ZIF-9. Moreover, the deconvoluted XPS spectra (Fig. 7c) of the used electrocatalyst exhibited two new peaks at 795.75 and 780.6 eV which could be assigned to the Co3+ valence state of CoOOH,48,49 formed during the electro-oxidation process, supporting the abovementioned XRD results. The FESEM images of the ZIF-9@10GO electrocatalyst after 12 h of electrocatalysis are shown in Fig. 8a–c. It is to be noted that after electrocatalysis formation of CoOOH nanosheets over the spherical ZIF-9 particles was clearly visible from the micrographs. These morphological features of CoOOH were similar to those previously reported in the literature.48 It is worth mentioning that these nanosheets were much thicker (100–150 nm) compared to the GO sheets observed in Fig. 3f. Moreover, as seen from the energy-dispersive X-ray (EDX) spectra (Fig. S19 and S20†), the atomic % of oxygen increased from 7.34% to 8.82% for the ZIF-9@10GO electrocatalyst after 12 h of electrocatalysis, thereby supporting our claim of partial replacement of benzimidazole linkers to form CoOOH species. Note that the atomic % of Co almost remains unchanged before and after electrocatalysis, propounding that there was no leaching of Co from the catalyst during electrocatalysis.
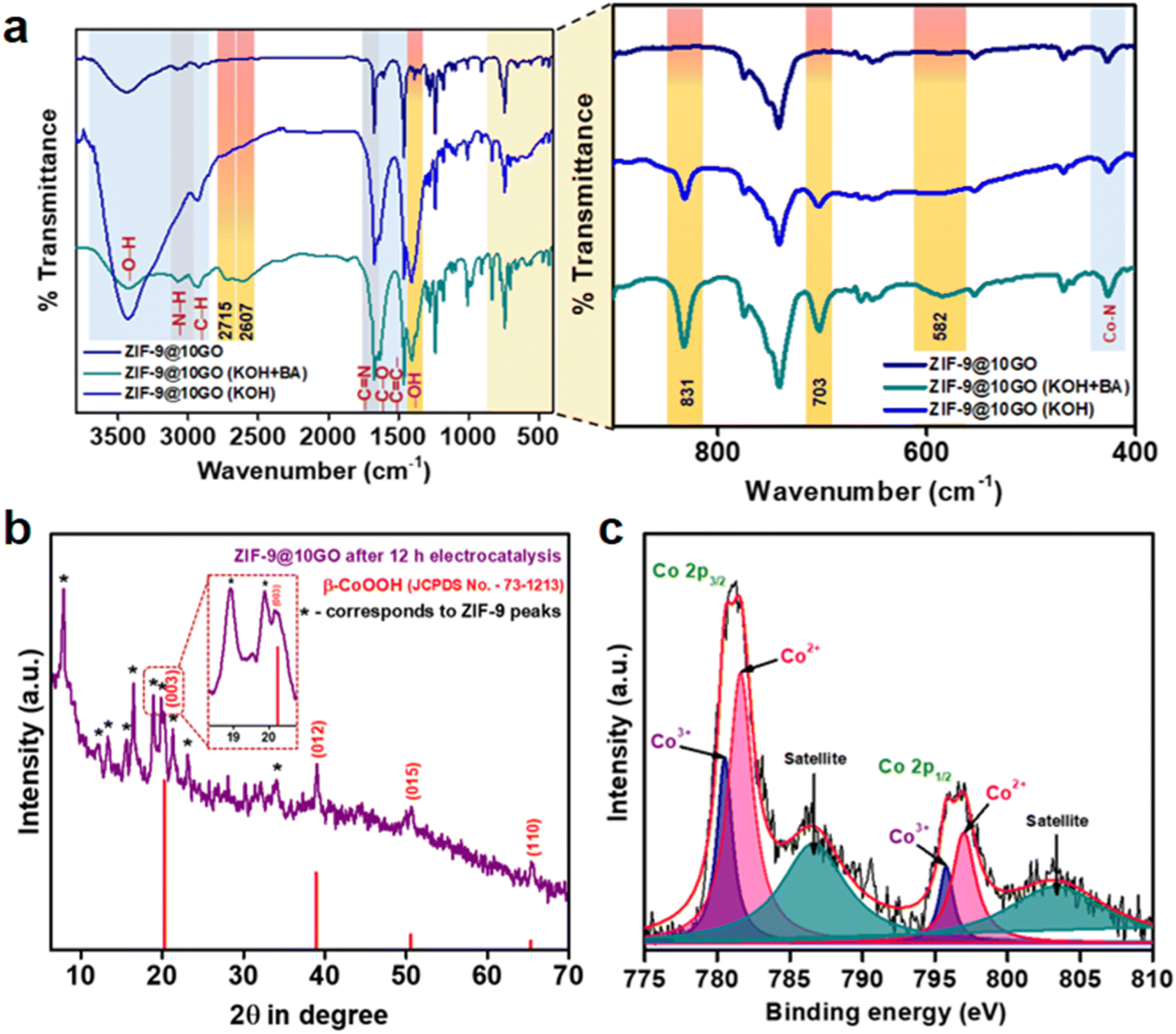 | ||
| Fig. 7 (a) FTIR spectra of the ZIF-9@10GO, ZIF-9@10GO (after electrochemical experiments in 1 M KOH) and ZIF-9@10GO (after electrochemical experiments in 1 M KOH with 0.1 M benzyl alcohol). (b) PXRD pattern of ZIF-9@10GO after 12 h of electrocatalysis. (c) Narrow-range XPS spectrum of Co 2p of the post-electrocatalytic ZIF-9@10GO. | ||
 | ||
| Fig. 8 (a–c) FESEM images of ZIF-9@10GO after 12 h of electrocatalysis. | ||
Now in order to substantiate the role of our catalyst ZIF-9@10GO towards the oxidation of BA, we performed a controlled experiment with β-Co(OH)2@10GO/NF (PXRD pattern of β-Co(OH)2 is shown in Fig. S14a,† which matched with the JCPDS card number 74-1057). After 12 h of electrocatalysis in 1 M KOH with 0.1 M BA, β-Co(OH)2@10GO/NF showed a BA conversion of only 6.6%, with 66.7% selectivity towards benzoic acid (Fig. S21†). This reduction in the catalytic activity could be ascribed to the reduced porosity in β-Co(OH)2 (Fig. S18b†). Henceforth, it is quite clear that higher porosity in the electrode enables greater diffusion and interaction of the electrolyte with the active sites of the catalyst, resulting in higher catalytic performance. Fig. S22† shows a comparison of current densities obtained by other reported non-noble based electrocatalysts in recent years, where notably ZIF-9@10GO showed an excellent performance. Moreover, during BA electro-oxidation, the coupled H2 production rate at the cathode was on par with that of the best catalysts reported in the literature (Table S4†).50–55
Conclusions
In summary, a microwave assisted synthetic protocol was utilized to synthesize phase pure ZIF-9 and ZIF-9@xGO composites, which were later employed for HER coupled electro-oxidation of benzyl alcohol to benzoic acid. At the catalytic surface, replacement of benzimidazole linkers by hydroxide ions took place forming α/β-Co(OH)2, which eventually got transformed into CoOOH during electro-oxidation. The in situ produced Co(OH)2/CoOOH hybrid species over the ZIF-9 surface served as the actual catalytic sites, and thereby ZIF-9 acted as the pre-catalyst. It is worth mentioning that normal Co(OH)2 hardly showed any electrocatalytic activity towards benzyl alcohol conversion to benzoic acid. Therefore, the in situ produced Co(OH)2/CoOOH species over ZIF-9 might introduce disorder through structural rearrangement, which consequently improved the electrocatalytic activity of the material. Additionally, GO played a crucial role in facilitating the transfer of electron/charge, thereby exhibiting direct impact on the obtained current density. Moreover, the catalyst exhibited high catalytic performance towards a wide range of substrates, including electron donating, withdrawing and allylic systems. All the experimental data were consistent with the explained model. FT-IR, PXRD and XPS analysis of the post-electrocatalytic catalyst depicted the formation of a Co(OH)2/CoOOH hybrid, which facilitated the electro-oxidation of benzyl alcohols. The enhanced electrocatalytic performance of the ZIF-9@10GO catalyst was attributed to the high specific surface area and exposed catalytic sites, which assisted greater ion diffusion and interactions at the electrode–electrolyte interface. This work not only showcased an example of selective oxidation of benzyl alcohol over a MOF/GO composite electrocatalyst with high FE%, but also offered ample opportunity to rationally design more effective electrocatalysts that could selectively oxidize organics along with coupled green H2 production.Data availability
All experimental procedures, instrumentation, crystal structure and refinement data, TGA analysis plots, additional CV data, surface coverage plots, ECSA, chronoamperometric data, and catalytic cycles are available in the ESI.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. C. would like to thank IACS for a Senior Research Fellowship. A. G. wants to thank CSIR for his Senior Research Fellowship. A. B. would like to thank DST-SERB for a core research grant (Project ID: CRG/2022/002812).Notes and references
- J. Shan, C. Ye, S. Chen, T. Sun, Y. Jiao, L. Liu, C. Zhu, L. Song, Y. Han, M. Jaroniec, Y. Zhu, Y. Zheng and S. Z. Qiao, J. Am. Chem. Soc., 2021, 143, 5201–5211 CrossRef CAS PubMed.
- Y. Song, Z. Li, K. Fan, Z. Ren, W. Xie, Y. Yang, M. Shao and M. Wei, Appl. Catal., B, 2021, 299, 120669 CrossRef CAS.
- Y. Zhang, F. Gao, C. Wang, Y. Shiraishi and Y. Du, ACS Appl. Mater. Interfaces, 2019, 11, 30880–30886 CrossRef CAS PubMed.
- A. Bhaumik and R. Kumar, J. Chem. Soc. Chem. Commun., 1995, 349–350 RSC.
- B. Wang, A. P. Cote, H. Furukawa, H. M. O'Keeffe and O. M. Yaghi, Nature, 2008, 453, 207–211 CrossRef CAS PubMed.
- C. Eßbach, I. Senkovska, T. Unmüssig, A. Fischer and S. Kaskel, ACS Appl. Mater. Interfaces, 2019, 11, 20915–20922 CrossRef.
- X. Chen, D. Liu, G. Cao, Y. Tang and C. Wu, ACS Appl. Mater. Interfaces, 2019, 11, 9374–9384 CrossRef CAS.
- S. Wang, Y. Hou, S. Lin and X. Wang, Nanoscale, 2014, 6, 9930–9934 RSC.
- S. S. Sankar, K. Karthick, K. Sangeetha, A. Karmakar, R. Madhu and S. Kundu, J. Mater. Chem. A, 2021, 9, 11961–12002 RSC.
- B. Seger and P. V. Kamat, J. Phys. Chem. C, 2009, 113, 7990–7995 CrossRef CAS.
- W. Wei, Z. Liu, R. Wei, G. C. Han and C. Liang, RSC Adv., 2020, 10, 29923–29934 RSC.
- F. Hillman, J. M. Zimmerman, S. M. Paek, M. R. Hamid, W. T. Lim and H. K. Jeong, J. Mater. Chem. A, 2017, 5, 6090–6099 RSC.
- H. T. Kwon and H. K. Jeong, Chem. Commun., 2013, 49, 3854–3856 RSC.
- K. W. Putz, O. C. Compton, C. Segar, Z. An, S. T. Nguyen and L. C. Brinson, ACS Nano, 2011, 5, 6601–6609 CrossRef CAS.
- D. López-Díaz, M. L. Holgado, J. L. García-Fierro and M. M. Velázquez, J. Phys. Chem. C, 2017, 121, 20489–20497 CrossRef.
- A. Noguera-Díaz, J. Villarroel-Rocha, V. P. Ting, N. Bimbo, K. Sapag and T. J. Mays, J. Chem. Technol. Biotechnol., 2019, 94, 3787–3792 CrossRef.
- P. Y. Zhou, L. M. Wang, J. J. Lv, R. S. Li, F. Y. Gao, X. B. Huang, Y. F. Lu and G. Wang, J. Mater. Chem. A, 2022, 10, 4981–4991 RSC.
- S. K. Das, M. K. Bhunia, M. M. Seikh, S. Dutta and A. Bhaumik, Dalton Trans., 2011, 40, 2932–2939 RSC.
- J. Dai, S. Xiao, J. Liu, J. He, J. Lei and L. Wang, RSC Adv., 2017, 7, 6288–6296 RSC.
- L. Trotochaud, J. K. Ranney, K. N. Williams and S. W. Boettcher, J. Am. Chem. Soc., 2012, 134, 17253–17261 CrossRef CAS.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS.
- A. Moysiadou, S. Lee, C. S. Hsu, H. M. Chen and X. Hu, J. Am. Chem. Soc., 2020, 142, 11901–11914 CrossRef CAS.
- X. Zuo, S. He, D. Li, C. Peng, Q. Huang, S. Song and C. Fan, Langmuir, 2010, 26, 1936–1939 CrossRef CAS PubMed.
- F. C. Shen, Y. Wang, Y. J. Tang, S. L. Li, Y. R. Wang, L. Z. Dong, Y.-F. Li, Y. Xu and Y. Q. Lan, ACS Energy Lett., 2017, 2, 1327–1333 CrossRef CAS.
- F. Dionigi, Z. Zeng, I. Sinev, T. Merzdorf, S. Deshpande, M. B. Lopez, S. Kunze, I. Zegkinoglou, H. Sarodnik, D. Fan, A. Bergmann, J. Drnec, J. Ferreira de Araujo, M. Gliech, D. Teschner, J. Zhu, W. X. Li, J. Greeley, B. R. Cuenya and P. Strasser, Nat. Commun., 2020, 11, 2522 CrossRef CAS.
- W. Zhang, S. Yin, X. Li, G. Xu and T. Xie, Electrochem. Commun., 2016, 63, 1–4 CrossRef CAS.
- B. J. Taitt, D. H. Nam and K. S. Choi, ACS Catal., 2019, 9, 660–670 CrossRef CAS.
- J. Wang, F. Xu, H. Jin, Y. Chen and Y. Wang, Adv. Mater., 2017, 29, 1605838 CrossRef PubMed.
- S. Hosseini, J. N. Janusz, M. Tanwar, A. D. Pendergast, M. Neurock and H. S. White, J. Am. Chem. Soc., 2022, 144, 21103–21115 CrossRef CAS PubMed.
- Y. H. Chang, C. T. Lin, T. Y. Chen, C. L. Hsu, Y. H. Lee, W. J. Zhang, K. H. Wei and L. J. Li, Adv. Mater., 2013, 25, 756–760 CrossRef CAS.
- Y. C. Zhou, W. J. Zhou, D. M. Hou, G. Q. Li, J. Q. Wan, C. H. Feng, Z. G. Tang and S. W. Chen, Small, 2016, 12, 2768–2774 CrossRef CAS.
- A. Khaligh, Y. Sheidaei and D. Tuncel, ACS Appl. Energy Mater., 2021, 4, 3535–3543 CrossRef CAS.
- M. R. Barone and A. M. Jones, Org. Biomol. Chem., 2017, 15, 10010–10015 RSC.
- C. Zhou, Z. Guo, Y. Dai, X. Jia, H. Yu and Y. Yang, Appl. Catal., B, 2016, 181, 118–126 CrossRef CAS.
- W. Zheng, M. Liu and L. Y. S. Lee, ACS Catal., 2019, 10, 81–92 CrossRef.
- H. Tian, Y. J. Zhang, Z. Y. Zhang, T. T. Lv, L. Bian, H. Wang, J. X. Li, Y. Yamauchi and Z. L. Wang, ChemCatChem, 2023, 15, e202201220 CrossRef CAS.
- M. Pourbaix, Atlas of Electrochemical Equilibria in Aqueous Solutions, Pergamon Press Ltd, New York, 1st edn, 1966 Search PubMed.
- B. N. Zope, D. D. Hibbitts, M. Neurock and R. J. Davis, Science, 2010, 330, 74–78 CrossRef CAS PubMed.
- H. B. Tao, Y. Xu, X. Huang, J. Chen, L. Pei, J. Zhang, J. G. Chen and B. Liu, Joule, 2019, 3, 1498–1509 CrossRef CAS.
- S. E. Michaud, M. M. Barber, K. E. Rivera Cruz and C. C. McCrory, ACS Catal., 2022, 13, 515–529 CrossRef.
- Y. Kwon, S. C. Lai, P. Rodriguez and M. T. Koper, J. Am. Chem. Soc., 2011, 133, 6914–6917 CrossRef CAS.
- H. Zhou, Z. Li, S. M. Xu, L. Lu, M. Xu, K. Ji, R. Ge, Y. Yan, L. Ma, X. Kong, L. Zheng and H. Duan, Angew. Chem., 2021, 133, 9058–9064 CrossRef.
- J. Gonzalez-Prior, J. I. Gutierrez-Ortiz, R. Lopez-Fonseca, E. Finocchio and B. de Rivas, Catal. Sci. Technol., 2016, 6, 5618–5630 RSC.
- P. Jeevanandam, Y. Koltypin, A. Gedanken and Y. Mastai, J. Mater. Chem., 2000, 10, 511–514 RSC.
- M. Jahan, Q. Bao, J. X. Yang and K. P. Loh, J. Am. Chem. Soc., 2010, 132, 14487–14495 CrossRef CAS.
- M. Zhang, M. De Respinis and H. Frei, Nat. Chem., 2014, 6, 362–367 CrossRef CAS PubMed.
- P. Zhu, X. Sun, Y. Wang, J. Zhang, X. Gu and Z. Zheng, J. Catal., 2021, 402, 208–217 CrossRef CAS.
- Y. Fu, L. Li, S. Ye, P. Yang, P. Liao, X. Ren, C. He, Q. Zhang and J. Liu, J. Mater. Chem. A, 2021, 9, 453–462 RSC.
- Z. Y. Wang, L. Wang, S. Liu, G. R. Li and X. P. Gao, Adv. Funct. Mater., 2019, 29, 1901051 CrossRef.
- G. Liu, Y. Chen, Y. Chen, Y. Shi, M. Zhang, G. Shen, P. Qi, J. Li, D. Ma, F. Yu and X. Huang, Adv. Mater., 2023, 2304716 CrossRef CAS.
- G. Li, G. Han, L. Wang, X. Cui, N. K. Moehring, P. R. Kidambi, D. Jiang and Y. Sun, Nat. Commun., 2023, 14, 525 CrossRef CAS PubMed.
- J. Wang, X. Guan, H. Li, S. Zeng, R. Li, Q. Yao, H. Chen, Y. Zheng and K. Qu, Nano Energy, 2022, 100, 107467 CrossRef CAS.
- Z. Chai, T. T. Zeng, Q. Li, L. Q. Lu, W. J. Xiao and D. Xu, J. Am. Chem. Soc., 2016, 138, 10128–10131 CrossRef CAS PubMed.
- Y. Song, W. Xie, Y. Song, H. Li, S. Li, S. Jiang, J. Y. Lee and M. Shao, Appl. Catal., B, 2022, 312, 121400 CrossRef CAS.
- F. Sun, J. Qin, Z. Wang, M. Yu, X. Wu, X. Sun and J. Qiu, Nat. Commun., 2021, 12, 4182 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2264516. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ta04894b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |