
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent progress and prospects in the organocatalytic Morita–Baylis–Hillman reaction
Mohammed Maneesha
a,
S. H. Haritha
b,
Thaipparambil Aneeja
a and
Gopinathan Anilkumar
 *ac
*ac
aSchool of Chemical Sciences, Mahatma Gandhi University, Priyadarsini Hills P. O., Kottayam, Kerala 686560, India. E-mail: anilgi1@yahoo.com; anil@mgu.ac.in
bPostgraduate Department of Chemistry, Sree Narayana College, Sivagiri, Sreenivasapuram P. O., Varkala, Thiruvananthapuram, Kerala 695145, India
cInstitute for Integrated Programs and Research in Basic Sciences (IIRBS), Mahatma Gandhi University, Priyadarsini Hills P. O., Kottayam, Kerala 686560, India
First published on 8th May 2024
Abstract
The organocatalytic asymmetric Morita–Baylis–Hillman (MBH) reaction produces a significant carbon–carbon bond formation that involves the addition of α,β-unsaturated carbonyl compounds to activated alkenes to give α-methylene-β-hydroxycarbonyl compounds. Commercially available starting materials, excellent atom economy, mild reaction conditions, and flexible products are the major highlights of this method. In this review, we discuss the recent developments in the organocatalytic MBH reaction, covering literature from 2018 to 2023.
 Mohammed Maneesha | Mohammed Maneesha was born in Vadanappally, Thrissur, Kerala, India. She obtained her BSc degree from Calicut University (Sree Narayana College, Nattika) in 2020 and her MSc degree from the School of Chemical Sciences, Mahatma Gandhi University, Kottayam in 2022. She passed the GATE (Gratuate Aptitude Test in Engineering) 2024. Her research interests are in the areas of transition metal catalysis and porous organic molecules. |
 S. H. Haritha | S. H. Haritha was born in Thiruvananthapuram, Kerala, India. She obtained her BSc degree in 2021 and MSc degree in 2023 from Kerala University (Sree Narayana College, Varkala). Her research interests revolve around transition metal catalysis and organic synthesis. |
 Thaipparambil Aneeja | T. Aneeja was born in Chittariparamba, Kannur, Kerala, India. She obtained her BSc degree from Kannur University (Nirmalagiri College, Kuthuparamba) in 2016 and her MSc degree from the Department of Applied Chemistry, CUSAT in 2018. She passed the CSIR-UGC National Eligibility Test 2019 for a research fellowship. Currently, she is pursuing her doctoral research under the guidance of Dr G. Anilkumar in the School of Chemical Sciences, Mahatma Gandhi University, Kottayam. |
 Gopinathan Anilkumar | Gopinathan Anilkumar was born in Kerala, India and received his PhD in 1996 from Regional Research Laboratory (renamed as the National Institute for Interdisciplinary Science and Technology NIIST-CSIR), Trivandrum with Dr Vijay Nair. He performed postdoctoral studies at the University of Nijmegen, The Netherlands (with Professor Binne Zwanenburg), Osaka University, Japan (with Professor Yasuyuki Kita), Temple University, USA (with Professor Franklin A. Davis), Leibniz-Institüt für Organische Katalyse (IfOK), Rostock, Germany (with Professor Matthias Beller), and Leibniz-Institüt für Katalyse (LIKAT), Rostock, Germany (with Professor Matthias Beller). He was a senior scientist at AstraZeneca (India). Currently, he is a Professor of Organic Chemistry at the School of Chemical Sciences, Mahatma Gandhi University in Kerala, India. His research interests are in the areas of organic synthesis, medicinal chemistry, heterocycles, and catalysis. He has published more than 190 papers in peer-reviewed journals, 7 patents, three book chapters, and edited two books entitled “Copper Catalysis in Organic Synthesis” (Wiley-VCH, 2020) and “Green Organic Reactions” (Springer, in press). He has received the Dr S. Vasudev Award from the Govt. of Kerala, India for best research (2016) and the Evonik research proposal competition award (second prize in 2016). His h-index is 32. |
1 Introduction
Carbon–carbon bond-forming reactions have emerged as a promising tool in synthetic organic chemistry.1,2 The Morita–Baylis–Hillman (MBH) reaction is one of the most widely used and useful carbon–carbon bond-forming reactions, and it involves the formation of α-methylene-β-hydroxycarbonyl compounds via the addition of α,β-unsaturated carbonyl compounds to activated alkenes.3,4 When imines are utilized in this reaction instead of aldehydes, it is known as the aza-Morita–Baylis–Hillman (aza-MBH) reaction.5,6In 1968, Morita was the first to perform the phosphine-catalyzed MBH reaction,7,8 and after that, an amine-catalyzed strategy was introduced by Baylis and Hillman in 1972 (Scheme 1).9 Unfortunately, this reaction was ignored by the scientific community for a decade and did not receive any attention until 1980.
 | ||
| Scheme 1 General scheme for Morita–Baylis–Hillman reaction. | ||
Later, organic chemists including Drewes,10 Perlmutter,11 Hoffmann,12 and Basavaiah13 recognized the importance of this reaction and began to explore its potential and scope. Thus, from the mid-1990s onwards, there has been tremendous progress in exploring the MBH reaction. The notable advantages of the MBH reaction include commercially available starting materials, flexible product with multifunctional groups, mild reaction conditions, and excellent atom economy. Moreover, this reaction usually employs a nucleophilic organocatalytic system, which does not generate heavy metal pollution.
Despite these advancements, several challenges remain in this field. For example, the discovery of a catalyst that is applicable to a broad range of substrates remains necessary. Even though some methodologies for the enantioselective MBH reaction have been established, the general conditions for performing the asymmetric MBH reaction have not yet been discovered.14
In recent years, several reviews on advancements in the MBH reaction have been published. In 2017, Pellissier published a review focusing on the progress in the organocatalytic MBH reaction.15 Several reviews subsequently appeared that focused on the biocatalytic asymmetric MBH reaction,16 application of DABCO as a catalyst for the MBH reaction,17 and an organocatalytic MBH-type domino reaction.18 Because of the wide importance and applicability of this reaction, we have summarized the recent advancements in the organocatalytic MBH reaction, from 2018 to 2023. For additional clarity, this review has been organized based on the organocatalyst utilized.
2 The Morita–Baylis–Hillman reaction using amines and amine derivatives as catalysts
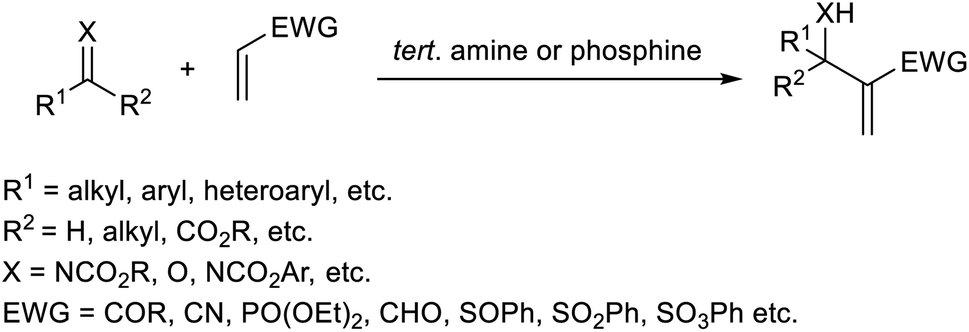
Trabocchi et al. provided an account of the organocatalytic MBH reaction employing dual iminium together with a Lewis base as the catalyst.19 They performed the reaction between cyclopent-2-enone and a broad range of aromatic as well as aliphatic aldehydes, and achieved the desired allylic alcohols in high yields. It was found that secondary amines function as co-catalysts in this reaction. There was a decreased yield of the MBH reaction when the amine was least basic. They observed poor enantiomeric excess when chiral amines were used in this reaction.The optimized conditions of this method were identified as 0.2 equiv. of pyrrolidine as an iminium ion generator in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 THF-1 M NaHCO3 at 25 °C for approximately 40 h (Scheme 2). Aromatic aldehydes with electron-withdrawing groups performed well in this reaction compared to those with electron-donating groups. This method was found feasible for heterocyclic aldehydes, and the desired products were obtained in excellent yields.
:4 THF-1 M NaHCO3 at 25 °C for approximately 40 h (Scheme 2). Aromatic aldehydes with electron-withdrawing groups performed well in this reaction compared to those with electron-donating groups. This method was found feasible for heterocyclic aldehydes, and the desired products were obtained in excellent yields.
 | ||
| Scheme 2 MBH reaction employing dual iminium together with a Lewis base catalyst. | ||
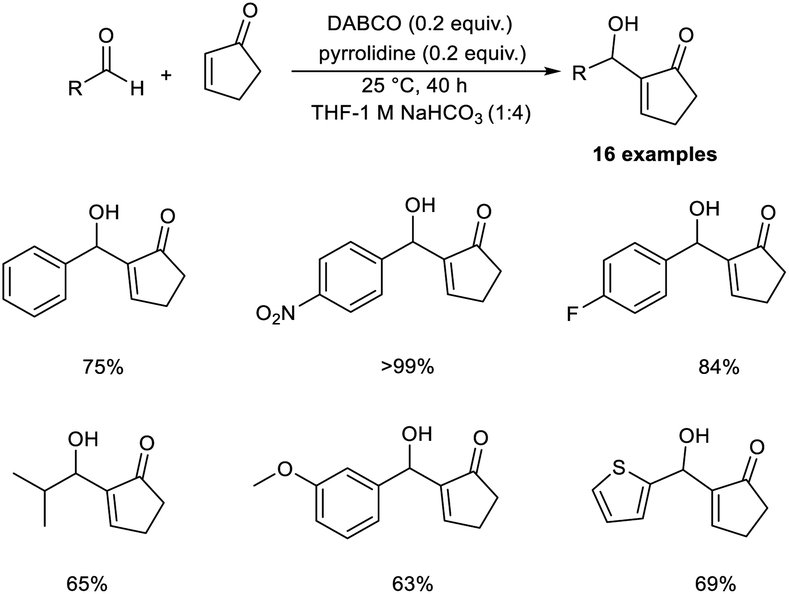
Bombonato et al. demonstrated that amino acid ionic liquid (AAIL) was capable of catalysing the MBH reaction.20 They prepared AAIL from ammonium salts, derivatives of glycerol, and natural amino acids. This method involves the reaction between methyl vinyl ketone and aromatic aldehydes in the presence of ionic liquids obtained from L-proline and L-histidine without the addition of additives or organic solvents. The structure of the amino acid ionic liquid was confirmed by 1H, 13C NMR, infrared spectroscopy, and elemental analysis.
In this method, p-nitrobenzaldehyde and methyl vinyl ketone were selected as model substrates, and the optimized conditions include the use of AAIL (20 mol%) at room temperature (Scheme 3). The synthesized MBH adducts were found to be optically inactive, even though AAIL contains chiral active sites. Aldehydes substituted with electron-withdrawing groups afforded MBH adducts in satisfactory yields in a short time compared to those with electron-donating groups.
 | ||
| Scheme 3 Amino acid ionic liquid-catalyzed MBH reactions. | ||
Coelho and co-workers established the MBH reaction employing vinyl-1,2,4-oxadiazoles as efficient nucleophilic partners.21 In this method, they studied the reaction of 5-vinyl-3-aryl-1,2,4-oxadiazoles with aromatic as well as aliphatic aldehydes (Scheme 4). This reaction proceeded well in the presence of 1.05 equiv. of DABCO. From the studies, it was found that a broad range of aldehydes, including heteroaromatic aldehydes, was well tolerated in this reaction. The rate of the MBH reaction can be altered by increasing the concentration of oxadiazole. Vinyl-1,2,4-oxadiazoles possessing an electron-donating group on the aromatic ring offer higher yields than those with electron-withdrawing groups. In this method, they utilized acetic acid, a weak acid, as an additive and did not use any additional solvent.
 | ||
| Scheme 4 MBH reaction employing vinyl-1,2,4-oxadiazoles as nucleophilic partners. | ||
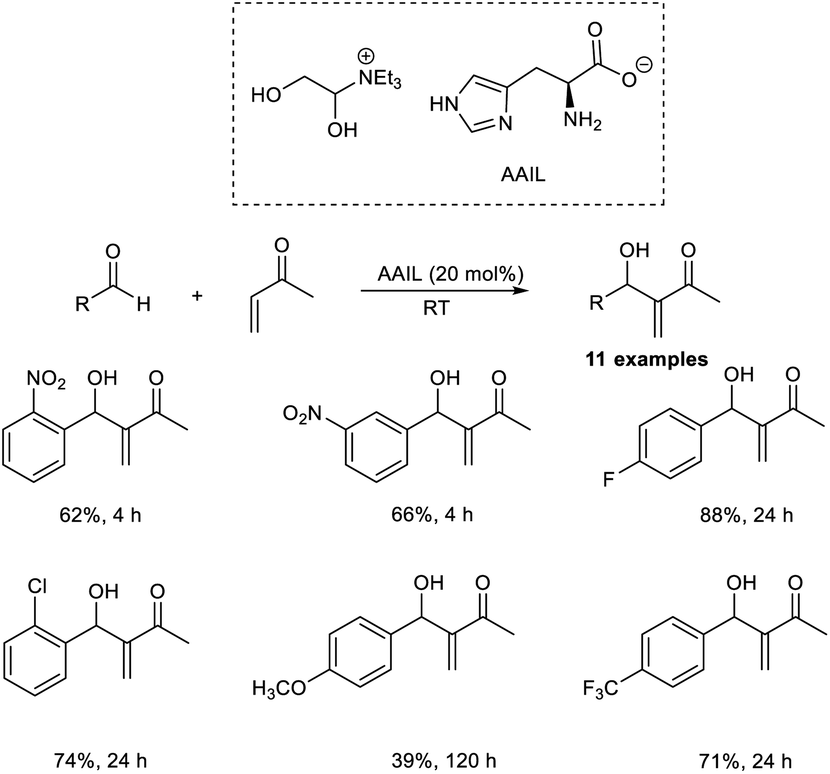
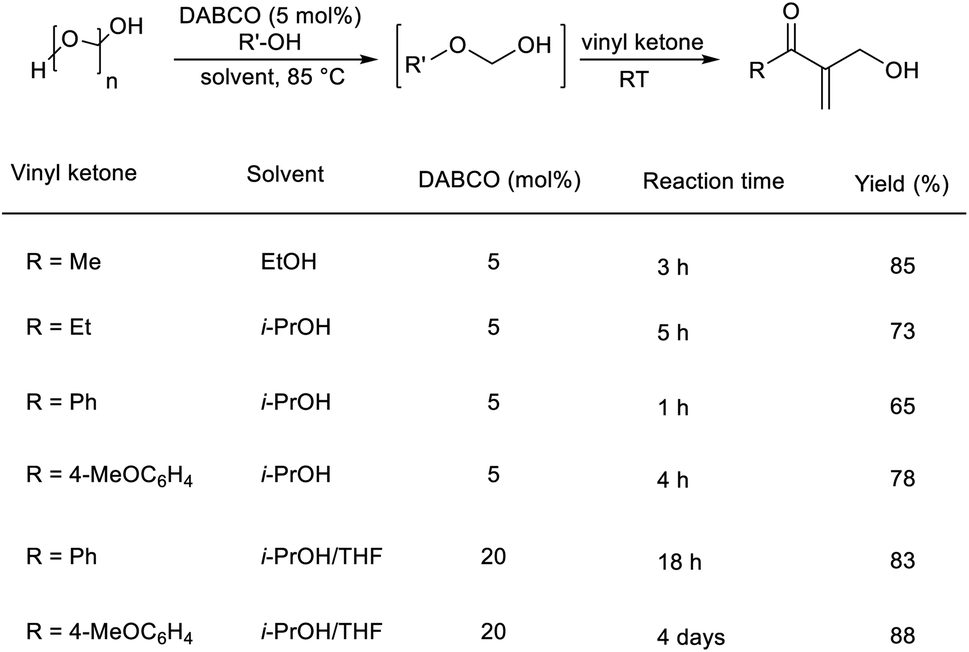
Pietruszka and co-workers disclosed the synthesis of α-hydroxymethylated vinyl ketones via an organocatalytic MBH reaction followed by one-pot etherification.22 In this reaction, aliphatic as well as aromatic vinyl ketones reacted well with formaldehyde in the presence of DABCO as a catalyst in alcoholic solution (Scheme 5). The major highlights of this reaction include excellent atom economy, shorter reaction time, inexpensive catalyst, and low catalyst loading.
 | ||
| Scheme 5 One-pot synthesis of α-hydroxymethylated vinyl ketones via organocatalytic MBH reaction followed by one-pot etherification. | ||
Portony et al. demonstrated the organocatalytic MBH reaction by utilizing Lewis acids to improve the chemoselectivity.23 The MBH reaction between methyl vinyl ketone and p-nitrobenzaldehyde leads to several byproducts due to the extreme reactivity of some reactants and products. It was found that the imidazole-based organocatalyst containing Lewis acids coordinated on diaminopyridine pockets enhanced the chemoselectivity of the MBH reaction to achieve the desired product in increased yields. Under optimized conditions (5 mol% of imidazole-based organocatalyst (C1 and C2) and 2 mol% silver acetate as a Lewis acid additive for 17 h), the required product was obtained with 87% conversion and 90% chemoselectivity (Scheme 6). This methodology was found to be suitable for electron-deficient aromatic aldehydes.
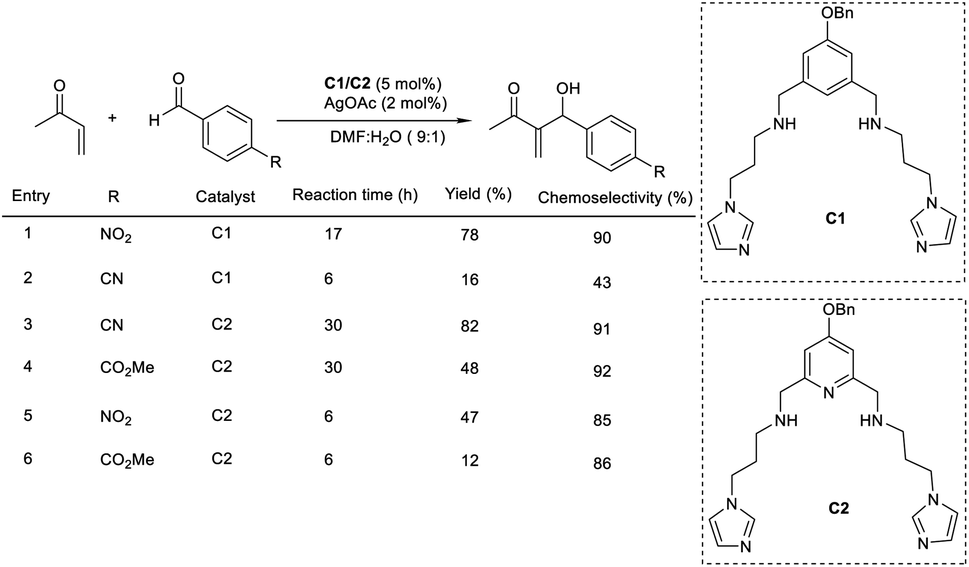 | ||
| Scheme 6 Organocatalytic Morita–Baylis–Hillman reaction using Lewis acids. | ||
Dechoux and co-workers illustrated the organocatalytic MBH reaction involving non-hazardous, renewable, and bio-based methyl coumalate.24 The reaction was found feasible with several imines and aldehydes, which favor the formation of C–C bonds with methyl coumalate. They carried out the reaction using p-nitrobenzaldehyde and methyl coumalate as a model substrate, and the optimized conditions were identified as 20 mol% triethylamine as the catalyst, and K2CO3 as an additive in ethanol at room temperature for 1.5 hours to obtain the desired product in 70% yield in one step without generating any undesirable outcomes (Scheme 7).
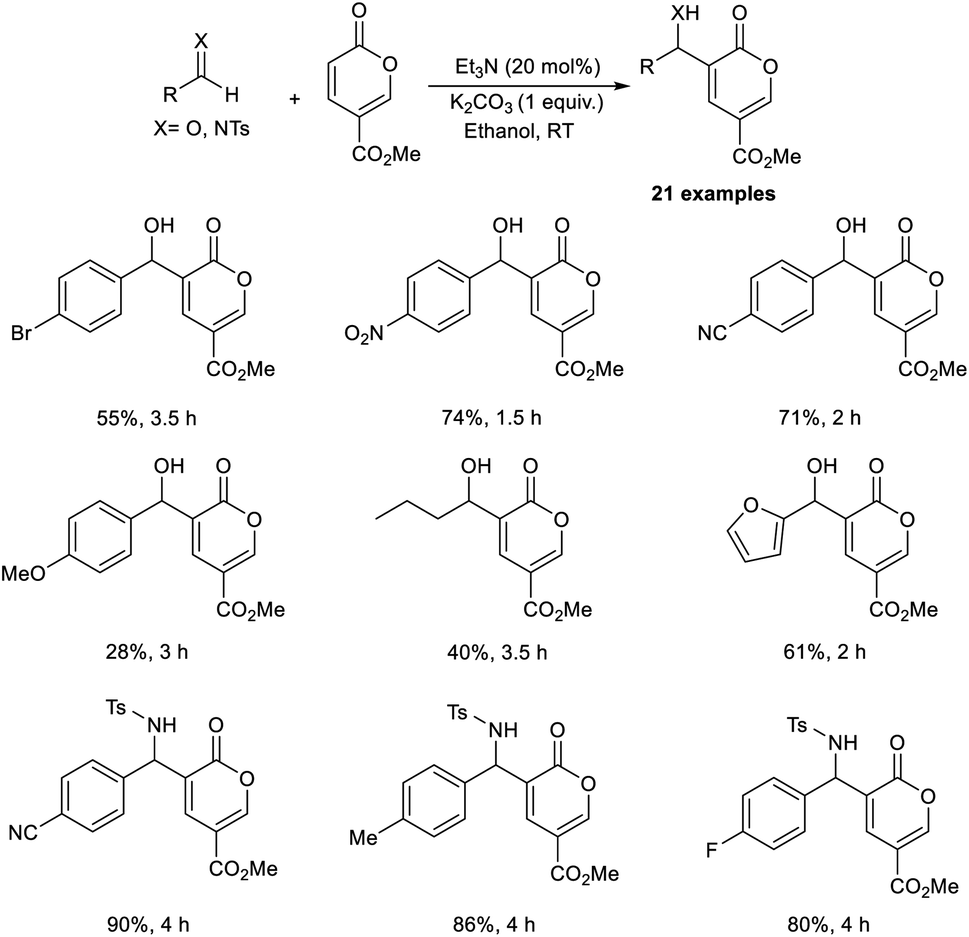 | ||
| Scheme 7 Morita–Baylis–Hillman reaction of methyl coumalate using triethylamine as the catalyst. | ||
This method was found applicable to a wide range of aliphatic as well as aromatic aldehydes. It was realized that aromatic aldehydes with electron-withdrawing groups were well tolerated in this reaction compared to those with electron-donating groups. Moreover, electron-rich imines performed well compared to electron-deficient imines. This method offers excellent atom economy and is solvent-free. The MBH adducts attained by this method were found to be applicable towards the synthesis of antihypertensive agents and diphenylmethanol derivatives.
Browne et al. established an organocatalytic aza-MBH reaction conducted by ball-milling that utilized tertiary amine as the catalyst.25 Their initial reactions commenced with tosyl-protected imine and ethyl acrylate as model substrates, and the optimized conditions for this reaction were found to be 20 mol% 3-hydroxyquinuclidine (3-HQD) as the tertiary amine catalyst, 3 mass equiv. NaCl as the grinding auxiliary, and toluene as a liquid additive under ball-milling for 3 h (Scheme 8). Aromatic aldimines substituted with halogens were well tolerated in this reaction. Sterically hindered aromatic aldimines also reacted via this method, but the required product was produced in low yield. Notable characteristics of this reaction include excellent atom economy, solvent-free condition, shorter reaction time, excellent enantioselectivity, and the reaction can be scaled up to 12-fold.
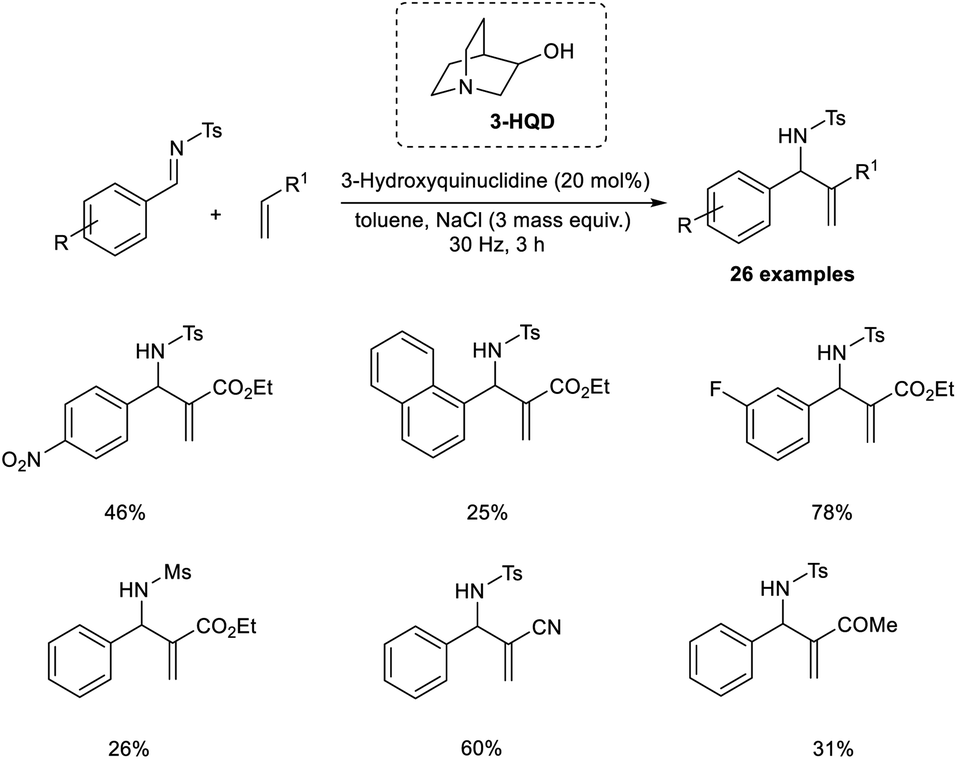 | ||
| Scheme 8 Ball-milling enabled this aza-MBH reaction utilizing a tertiary amine catalyst. | ||
The efficiency of a chiral pyrrolidine-based organocatalyst in the asymmetric MBH reaction through hydrogen bonding interaction was proposed by Bhusare and co-workers.26 This methodology involves the condensation of a broad variety of aromatic aldehydes with acrylate to access β-hydroxy acrylate as the product (Scheme 9). Initially, they carried out the reaction using p-nitrobenzaldehyde and methyl acrylate as model substrates. (S)-N-(2,4-Dinitrophenyl)pyrrolidine-2-carboximide C3 was found to be the most efficient catalyst for this reaction. The most optimal result was obtained in the presence of 10 mol% catalyst (C3), 10 mol% DMAP as the base in ethanol, and the desired product was obtained in excellent yield with high enantioselectivity. Using this method, aromatic aldehydes with electron-withdrawing groups gave the desired product within 14–17 h.
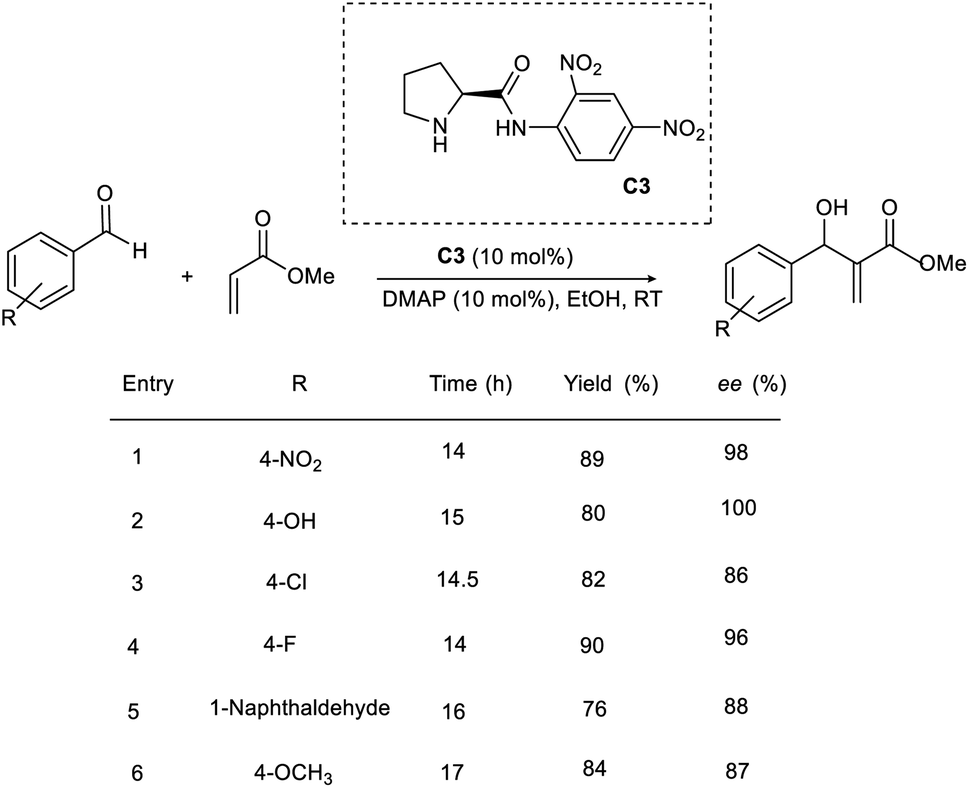 | ||
| Scheme 9 Organocatalytic synthesis of β-hydroxy acrylate via the Baylis–Hillman reaction. | ||
Sharifi and co-workers demonstrated a methodology for the synthesis of substituted allyl dithiocarbamates via a one-pot multicomponent reaction.27 This methodology initiates with the MBH reaction of activated alkenes and aromatic aldehydes employing DABCO as the catalyst, followed by acetylation using acetic anhydride in the presence of DMAP, and finally, nucleophilic substitution with dithiocarbamates (Scheme 10). This methodology exhibits high regioselectivity, i.e., when aldehydes were reacted with acrylonitrile, the E-stereoisomer was produced, while the reaction with methyl acrylate afforded the corresponding Z-isomer.
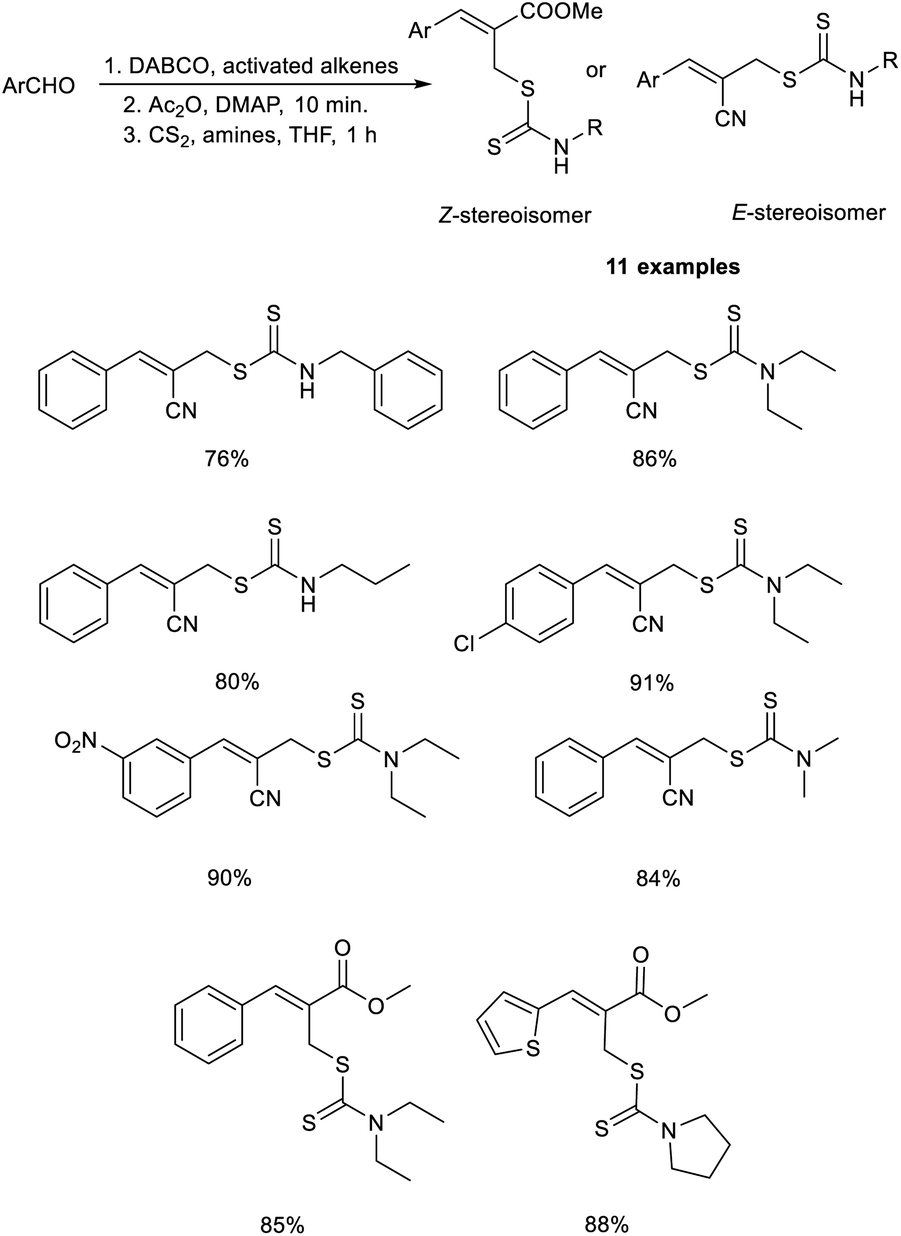 | ||
| Scheme 10 Synthesis of substituted allyl dithiocarbamates via a one-pot multicomponent reaction. | ||
The organocatalytic MBH reaction co-catalyzed by Lewis base and a Brønsted acid was put forward by Mancheno and co-workers.28 They designed the reaction between substituted 6-membered sulfamidate imines with a broad range of acrylate-type Michael acceptors to obtain α,β-unsaturated derivatives of cyclic sulfamidates as the desired outcome. The optimized conditions for this methodology were identified as 15 mol% DABCO as the Lewis base catalyst, and 15 mol% acetic acid as the Brønsted acid additive in 1,4-dioxane at room temperature.
Acrylate-containing electron-donating groups were well tolerated in this reaction (Scheme 11). Moreover, alkyl- or aryl-substituted sulfamidate imines smoothly underwent this reaction to afford the desired product in excellent yield. Sulfamidate imines with electron-rich as well as electron-withdrawing groups performed well in this reaction. However, bromo-substituted substrates were found to be less efficient using this method.
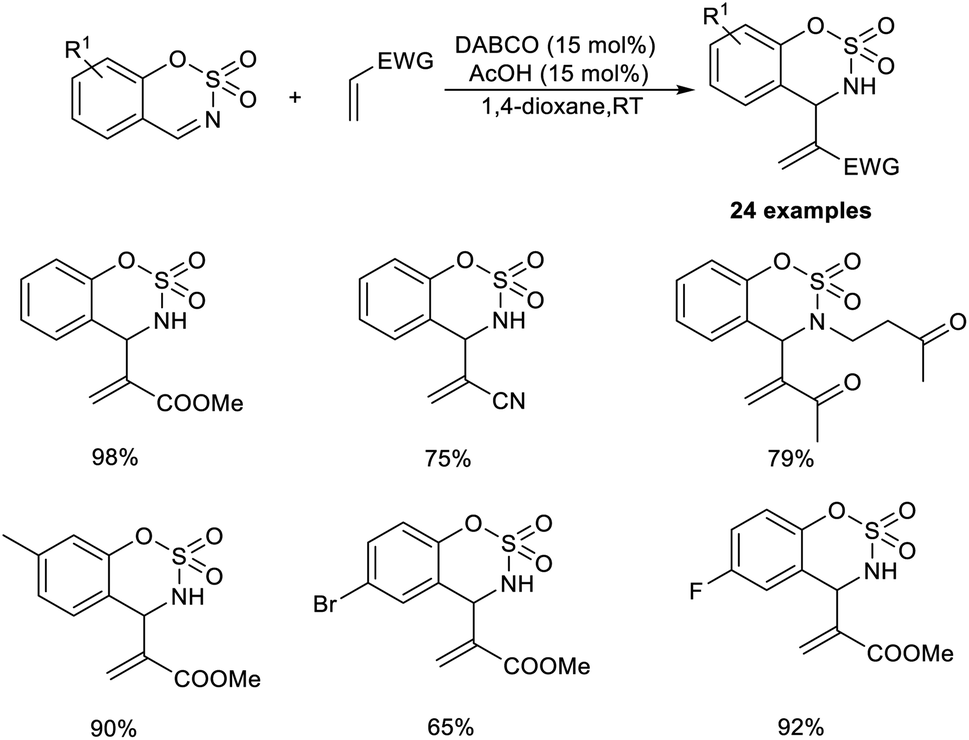 | ||
| Scheme 11 Synthesis of α,β-unsaturated derivatives of cyclic sulfamidates via the MBH reaction. | ||
Computational analysis by Yadav indicated that there was an increased rate for the organocatalytic MBH reaction catalyzed by N,N-dimethylhydroxylamine.29 It was identified that the increased nucleophilicity of nitrogen consequent to the α-effect of oxygen in Me2NOH was responsible for the lowering of activation energy in this reaction. The reaction was found to be feasible with an activated alkene such as 2-cyclopentanone and electrophiles such as CH3CHO.
Srivastava reported an efficient MBH reaction using active pyridine-based ionic liquid-linked quinuclidine as a catalyst.30 He synthesised three types of ionic crystals that possessed 3-quinuclidinone support. The reaction was performed with various aldehydes and acrylates, and a higher yield was obtained as compared with the usual procedure at room temperature. Among three catalysts, Py-catalyst-3 proved to be more efficient for this reaction (Scheme 12). From these studies, it is understood that the solvent has no significant effect on the yield of the reaction. Compounds such as p-methoxy benzaldehyde, cyclic enones, and Garner aldehyde (Scheme 13), which are usually inert to the MBH reaction, participated well in this method. The major advantage of this reaction is that the products can be isolated without any chromatographic techniques, and there is higher recyclability of the catalyst.
 | ||
| Scheme 12 MBH reaction using active pyridine-based ionic liquid linked quinuclidine catalyst. | ||
 | ||
| Scheme 13 MBH reaction of Garner aldehyde. | ||
Tai and co-workers developed an efficient MBH reaction using brucine diol (BD) as the catalyst.31 When 20 mol% BD was utilized as the catalyst, they obtained the desired MBH adduct in moderate enantiomeric excess (ee) value and yield. They were able to achieve higher yields using a co-catalytic BD/brucine N-oxide (BNO) system (Scheme 14).
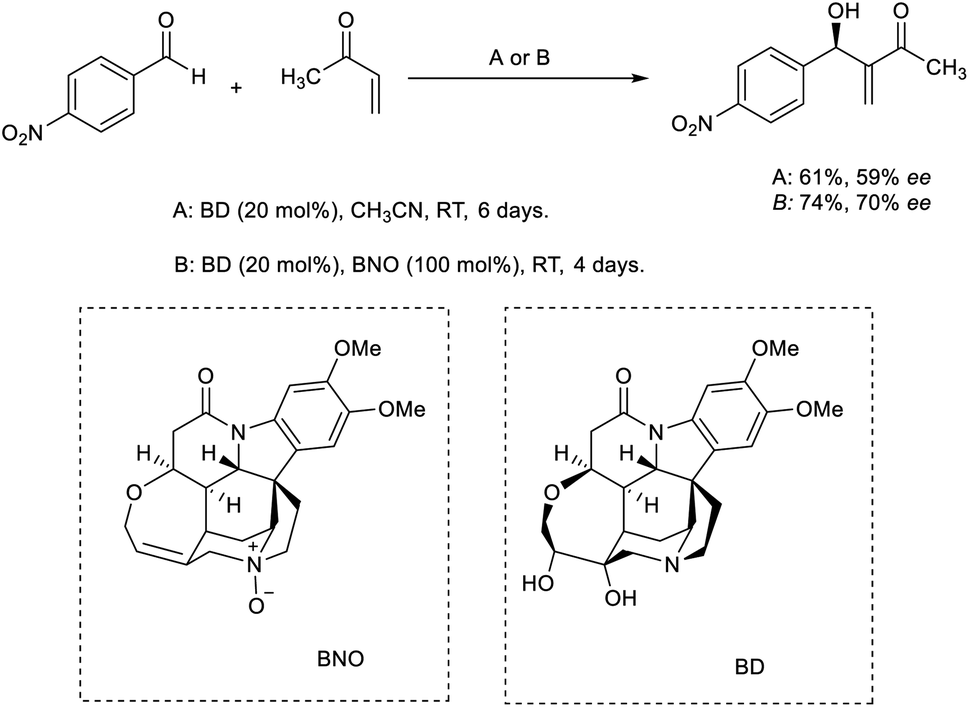 | ||
| Scheme 14 MBH reaction using BD or the co-catalytic BD/BNO system. | ||
Coelho et al. deduced an easy, inexpensive, and improved protocol for the MBH reaction with unusual substrate scope.32 This method was found to operate through an acid-catalyzed mechanism to increase the reaction rate and yield. In this reaction, they utilized piperonal and methyl acrylate as the model substrates, and the optimized conditions were identified as 0.1 mole equivalent of DABCO as the catalyst and 0.1 mole equivalent of acetic acid as the Bronsted-acid co-catalyst under solvent-free conditions at room temperature (Scheme 15).
 | ||
| Scheme 15 DABCO-catalyzed MBH reaction without the use of solvent. | ||
From the previous reports, it is clear that the MBH reaction of electron-rich aldehydes was difficult and always occurred at a slow reaction rate. This protocol was found to be successful in overcoming this limitation, as electron-rich aldehydes smoothly underwent the MBH reaction via this method. The synthesis of the natural product (±)-sitophilure from MBH adducts prepared by this method further enhances the significance of this method. The major highlights of this reaction include excellent atom economy without the use of solvent.
The MBH reaction of N-alkylquinolinium salts was described by Kalek and Pareek.33 In this method, the authors utilized DBU, which serves as a base as well as a catalyst, to activate electron-poor olefin. A wide range of α-(1,2-dihydroquinolin-2-yl)vinyl esters, sulfones, and ketones were obtained via regioselective dearomatization at the C-2 position. The reaction was performed at a lower temperature (0 °C) in DCM (Scheme 16). In the case of N-alkylpyridinium with an activating ester group, regioselective functionalisation at the C-4 position was observed.
 | ||
| Scheme 16 MBH reaction of N-alkyl quinolidium salt and N-alkylpyridinium with activating ester groups. | ||
In 2022, Chen et al. described a new methodology for the synthesis of C7-carbasugars via intramolecular MBH reaction.34 In this strategy, substituted dual precursor obtained from D-mannose underwent cyclization to give the required carbasugars in excellent yield with high stereoselectivity. DMAP was identified as the catalyst of choice for this protocol (Scheme 17). This method was successful for the preparation of different carbasugars such as epicorepoxydon A, epoxydines B and C, (−)-MK7607, (−)-streptol, and (−)-gabosine E.
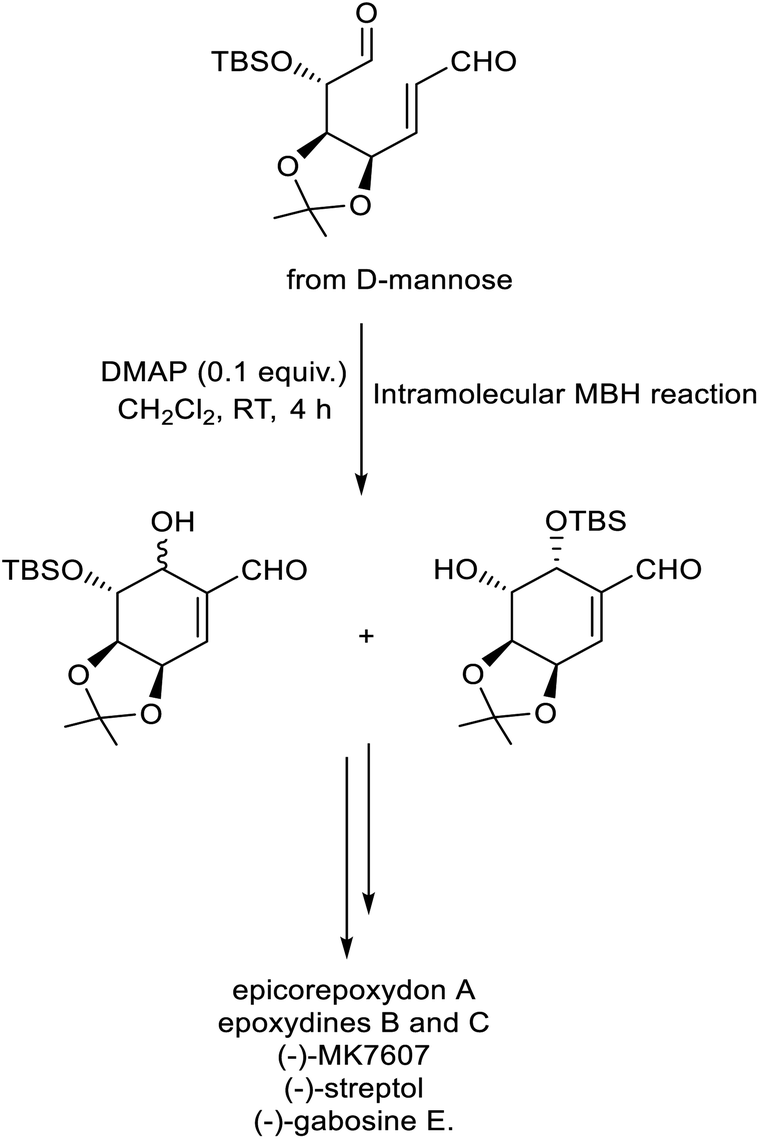 | ||
| Scheme 17 Synthesis of C7-carbasugars via the MBH reaction. | ||
Chen and co-workers reported the synthesis of chiral sulfones containing 3-hydroxyloxindoles via an organocatalytic asymmetric MBH reaction.35 β-Isocupreidine was chosen as an efficient catalyst for this reaction. The initial reaction was carried out using isatin and vinyl methyl sulfone as model substrates, and the optimized conditions included β-isocupreidine (10 mol%) as the catalyst in 1,4-dioxane at 40 °C under an Ar atmosphere for two weeks (Scheme 18). It was observed that N-substituted isatin with electron-donating groups performed well in this reaction compared to those with electron-withdrawing groups. The reaction was found to be feasible with N-allyl-substituted isatins, and the desired product was obtained in satisfactory yield with high enantioselectivity.
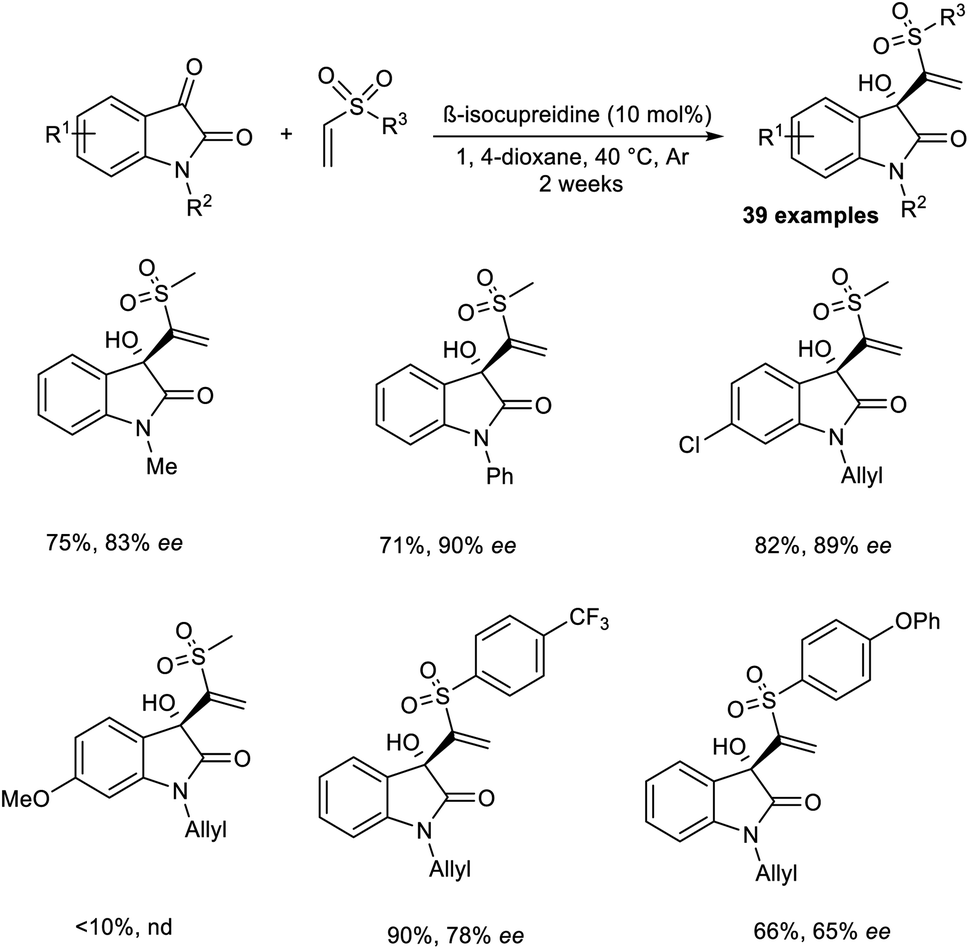 | ||
| Scheme 18 Synthesis of chiral sulfones containing 3-hydroxyloxindoles using an organocatalyst. | ||
Aryl vinyl sulfones with electron-withdrawing groups were well tolerated in this reaction. However, this reaction was found to be incompatible for substrates with electron-donating groups. This reaction was also attempted using other electrophiles such as acetaldehyde and 3-methyl butanal, but the desired product was obtained in trace amounts. Notable characteristics of this reaction include wide substrate scope, excellent yield, and high enantioselectivity.
Coelho and co-workers introduced a new approach for the preparation of tricyclic indolizines from cyclic enones and 2-pyridinecarbaldehydes through an organocatalytic MBH reaction followed by intramolecular cyclization and dehydration.36 Bicyclic imidazolyl alcohol (BIA) was found to be an effective catalyst for this aqueous MBH reaction. Higher yields of the required product were obtained in the case of cycloheptenone as compared to cyclohexanone. Steric factors play a significant role in this reaction, as 5,5-dimethyl-2-cyclohexenone failed to give the expected product. The substrate scope studies indicated that the feasibility of the cyclization step was greatly influenced by the ring size of cycloalkenone. Computational studies were also performed to justify the reactivity trends (Scheme 19).
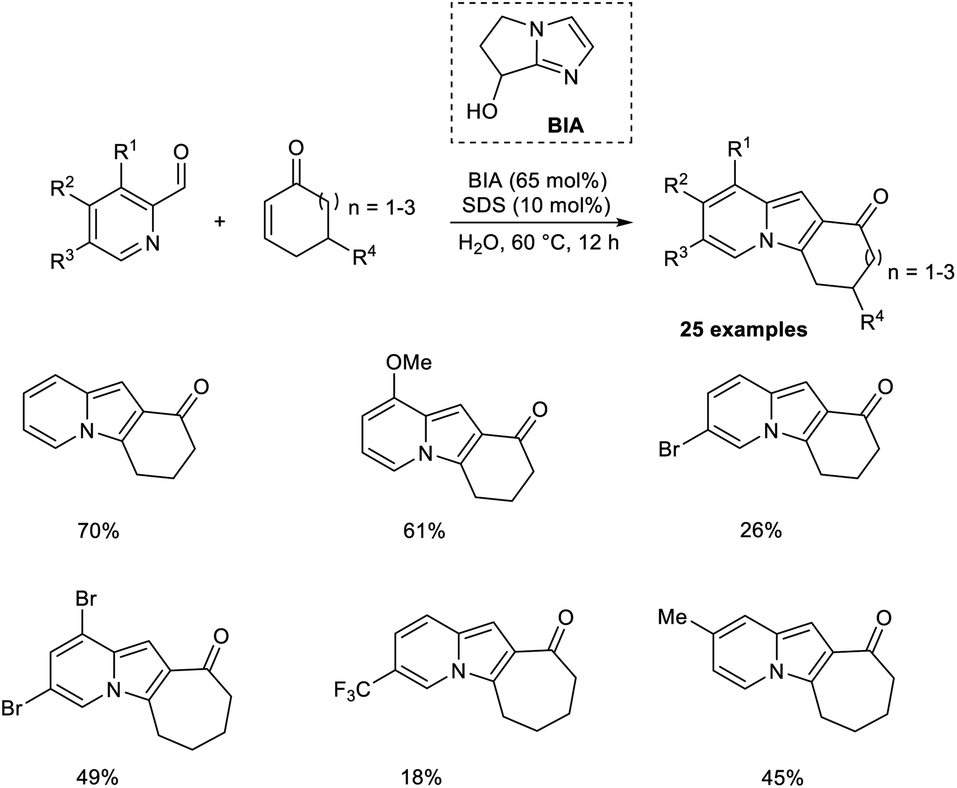 | ||
| Scheme 19 Synthesis of tricyclic indolizines via organocatalytic MBH reaction. | ||
The first MBH reaction between acrylates and 5-substituted-2-furaldehydes was reported by Dutta and co-workers.37 They used 1,4-diazabicyclo[2.2.2]octane (DABCO) as the organocatalyst (Scheme 20). The reaction was carried out in a solvent-free environment at room temperature, and satisfactory to exceptional yields of the required products were obtained. Different 5-substituted-2-furaldehydes were found suitable for this method.
 | ||
| Scheme 20 MBH reaction between acrylates and 5-substituted-2-furaldehydes. | ||
Zhao and co-workers introduced a new methodology for the synthesis of enantiopure 2-alkenyl-2-phenyl-1,2-dihydro-3H-indol-3-ones via enantioselective organocatalytic aza-MBH reaction between cyclic ketimines and α,β-unsaturated γ-butyrolactams.38 This method was found to be successful for the aza-MBH reaction of cyclic ketimines, which is less explored and challenging. They selected 2-Ph-3H-indol-3-one and α,β-unsaturated γ-butyrolactam as the model substrates, and the optimized conditions included 20 mol% of organocatalyst C4 in DCE at 30 °C under a nitrogen atmosphere (Scheme 21).
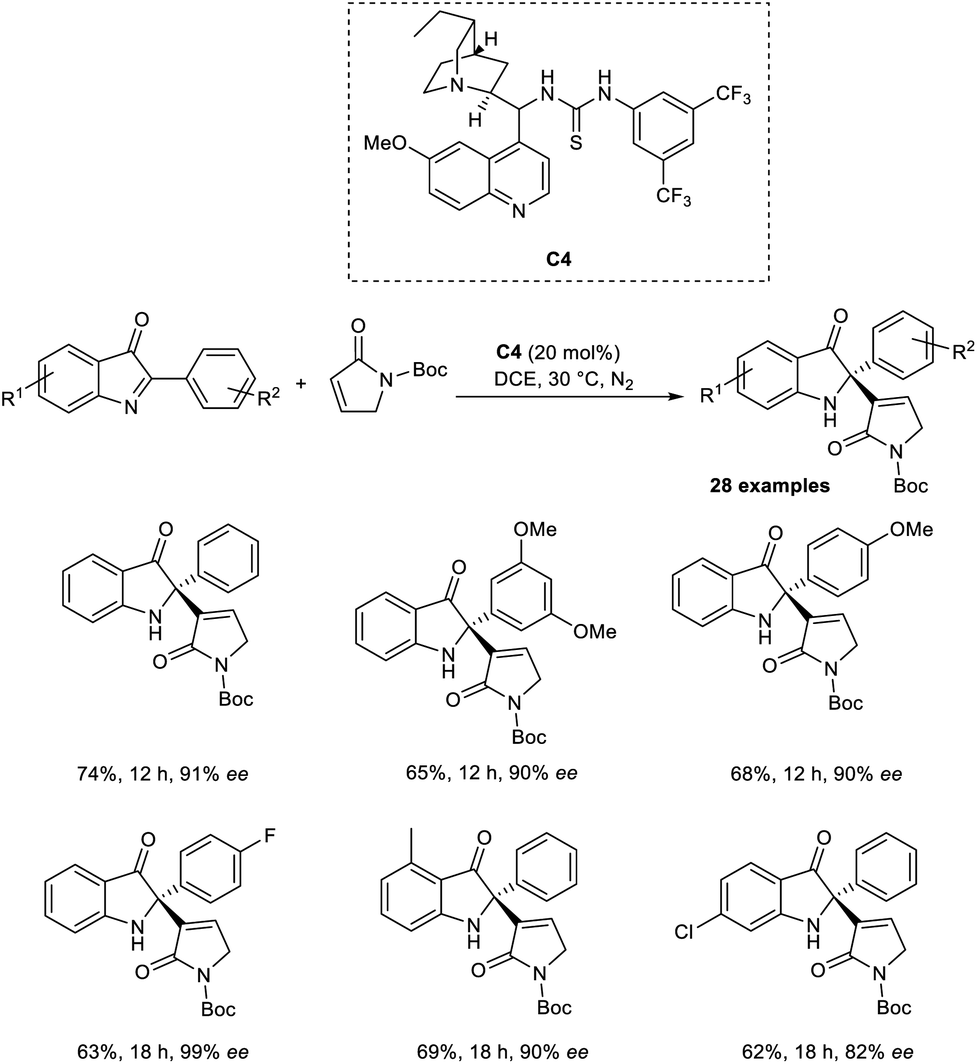 | ||
| Scheme 21 Organocatalytic aza-MBH reaction of cyclic ketimines with α,β-unsaturated γ-butyrolactam. | ||
From the substrate scope studies, it is understood that this method was found suitable for 2-aryl-3H-indol-3-ones substituted with electron-rich and electron-deficient groups at the para position of the phenyl ring. Under optimized conditions, cyclic α,β-unsaturated ketones such as cyclohexanone and cyclopentenone failed to achieve the desired product. Notable characteristics of this methodology include satisfactory yields, high enantioselectivity, and excellent selectivity.
3 The Morita–Baylis–Hillman reaction using phosphines as catalysts
In 2018, Du and Chen introduced the MBH reaction for α-substituted activated olefins, such as 3-olefinic oxindoles using S-phosphines as catalysts.39 In this method, 3-olefinic oxindoles with β-ester groups were selected as the model substrate and reacted with various electrophiles. Phosphine with a bifunctional thiourea group C5 or Bu3P was used as the catalyst with K2CO3 additive in toluene at room temperature for 48 h (Scheme 22). No Wittig reaction was observed in the presence of P(Bu)3, but dephosphoration was observed when HCHO was used as the electrophile. Likewise, no additive was required when trifluroacetaldehye was used as the electrophile, because the CF3 group it contains assists in producing diastereoselective products. | ||
| Scheme 22 MBH reaction for α-substituted activated olefins such as 3-olefinic oxindoles using S-phosphines as catalysts. | ||
Synthesis of heteroarenes, cyclohepta-, and cyclopenta-fused arenes via an MBH-type reaction of α,β-ynones was developed.40 In this strategy, 10 mol% was readily available, and cost-effective PPh3 was utilized as the catalyst (Scheme 23). Wide varieties of 2-arylidine-1,3-indanediones and 2-alkylidines with electron-donating and electron-withdrawing substituents were achieved in moderate to satisfactory yields via this method. They also extended this reaction to biaryl-ynone and observed a higher yield of the desired product when PCy3 was utilized as the catalyst (Scheme 24).
 | ||
| Scheme 23 Phosphine-catalysed MBH-type reaction of α,β-ynones. | ||
 | ||
| Scheme 24 MBH reaction of biaryl-ynones using PCy3 as the catalyst. | ||
Vicario and co-workers developed the MBH reaction by utilizing enantioselective transannular catalysts.41 This technique employs the enantiopure chiral bifunctional phosphine as a Lewis base catalyst to acquire bicyclic carbo-and heterocyclic systems with different ring sizes. This reaction was found to be highly efficient for the synthesis of adducts with bicyclo[5.4.0]undecane and bicyclo[5.3.0]decane systems. Initially, they performed this reaction using ketoenone as a model substrate, and the optimized conditions were identified as 10 mol% bifunctional phosphine C6 as the catalyst, and phenol as the additive in CHCl3 at 25 °C.
Under the optimized conditions, substrates that gave products with a bicyclo[5.3.0]decane core reacted well to achieve the desired product with high enantioselectivity and in excellent yields. Substrates with differently substituted fused aromatic systems performed well in this reaction. This method was found suitable for substrates with different heteroatoms. The reaction was found to be less efficient when α,β-unsaturated lactones were used as substrates (Scheme 25). The synthesis of the natural product, (−)-γ-gurjunene, via this method enhanced the significance of this reaction.
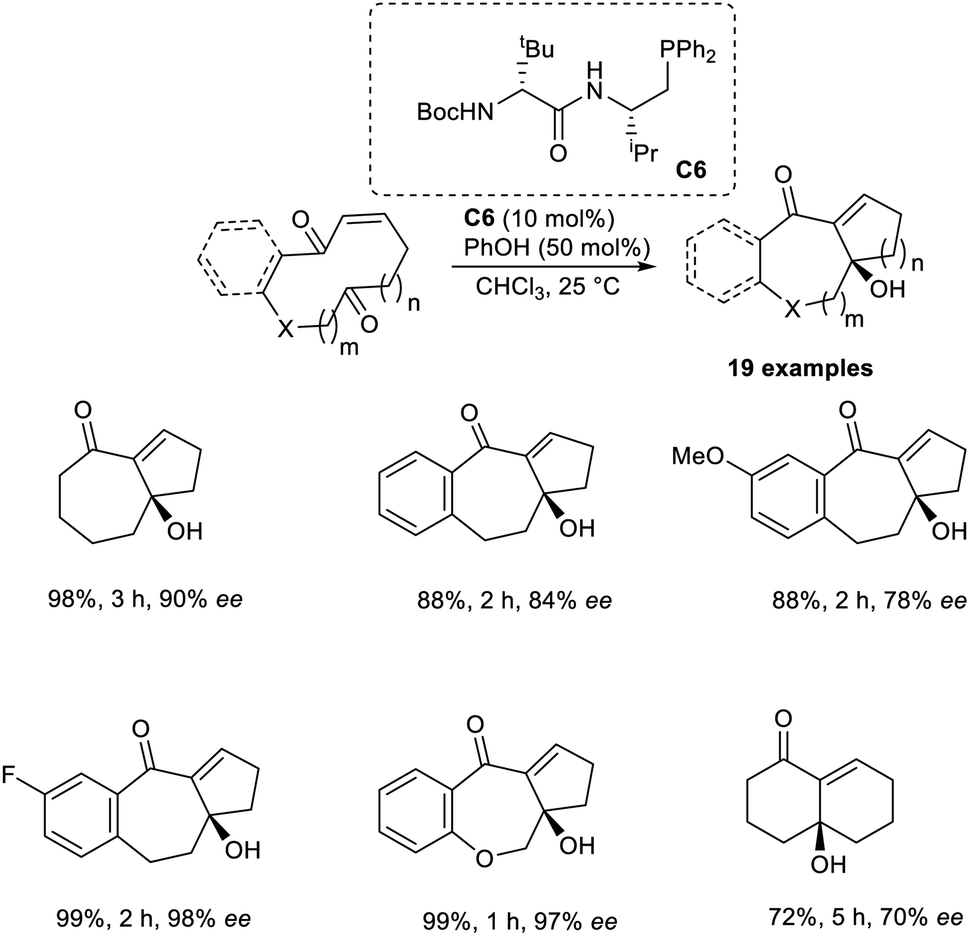 | ||
| Scheme 25 Enantiopure chiral bifunctional phosphine-catalyzed MBH reaction. | ||
Extremely functionalized chiral bibenzocycloheptanes were synthesized through an annulative intramolecular organocatalytic MBH reaction.42 In this work, the authors selected enone-aldehydes as the model substrate, and the most optimal results were obtained when 20 mol% of PBu3 was employed as a Lewis-base catalyst in DMF at room temperature (Scheme 26). It was found that the reaction preferred a metal-free condition and required phosphines as the catalyst. Seven-membered carbocycles were synthesized by this method. Enone core-accommodating electron-withdrawing and electron-donating groups were well tolerated in this reaction. The realization of various bioactive molecules with a dibenzocycloheptene skeleton offers proof that this a fascinating methodology.
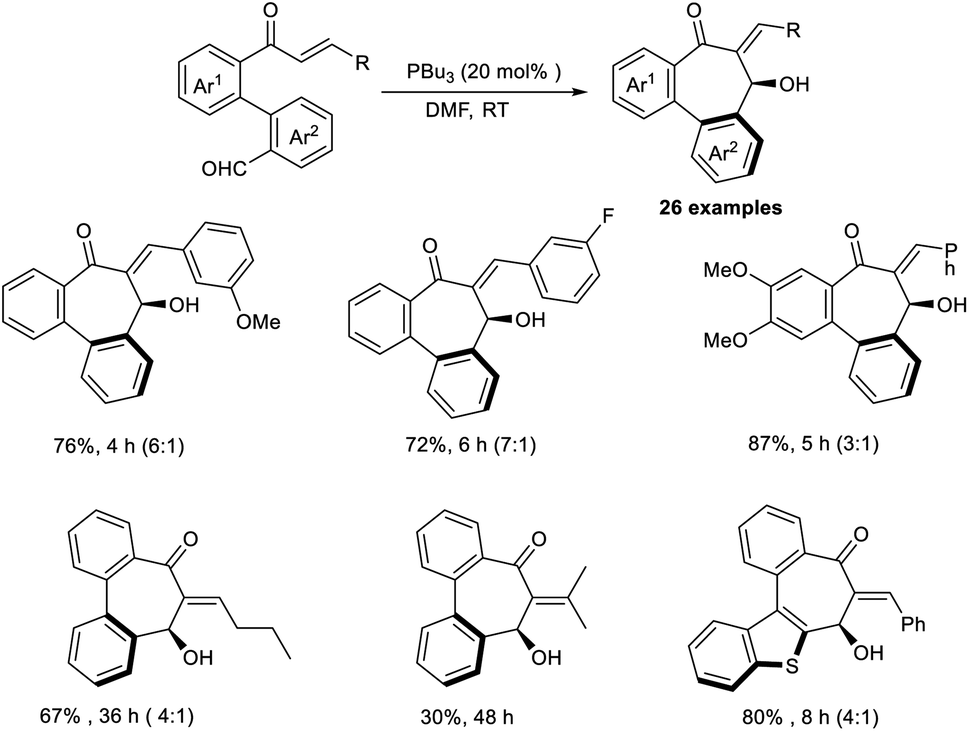 | ||
| Scheme 26 Synthesis of chiral dibenzocycloheptanes through the MBH reaction. | ||
Rachwalski and co-workers gave an account of a highly effective asymmetric MBH reaction catalyzed by chiral aziridine phosphines.43 They used this method to study the reaction of a wide range of aromatic aldehydes with methyl vinyl ketones or methyl acrylate to obtain allylic alcohols as the desired product with satisfactory yields and excellent enantioselectivity. Aziridine phosphine with an S-configuration (C7) provided the desired allylic alcohols with the same configuration (Scheme 27). Aromatic aldehydes with substituents such as CH3 and Br were well tolerated in this reaction. However, there was no advantage to using 4-methoxybenzaldehyde with this method in an attempt to obtain a better outcome.
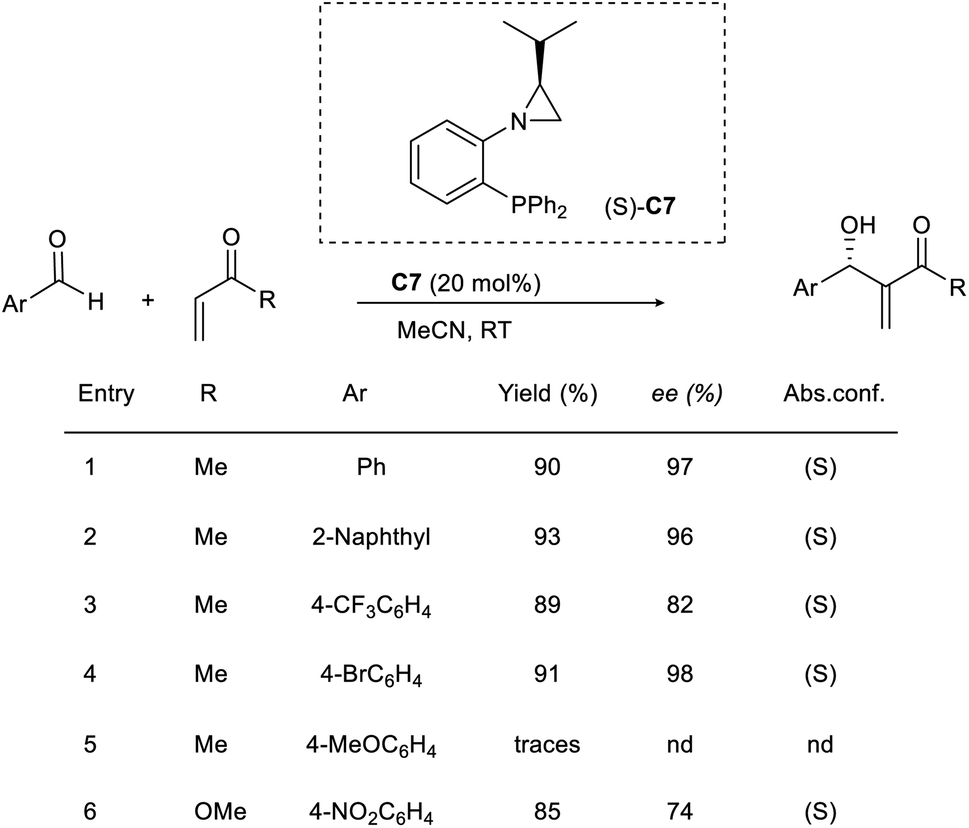 | ||
| Scheme 27 MBH reaction catalyzed by chiral aziridine phosphines. | ||
An efficient strategy for the synthesis of 3-aminomethylated maleimides through the MBH reaction has been established.44 The MBH reaction between maleimides and 1,3,5-triazinanes was achieved under optimized conditions (20 mol% PPh3 as the catalyst, 20 mol% PhCO2H as the promoter in DCE at 70 °C for 15 min) (Scheme 28). In this method, N-phenylmaleimide derivatives with electron-donating and electron-withdrawing substituents participated to create the desired product in satisfactory yields. Under the optimized conditions, incomplete conversion was observed in the case of 1,3,5-triazinanes with electron-withdrawing substituents. However, satisfactory yields of the desired product were achieved in the case of 1,3,5-triazinane-bearing groups such as Br, CN, OCF3, and CO2Et under modified conditions (1 equiv. of PhCO2H).
 | ||
| Scheme 28 Synthesis of 3-aminomethylated maleimides via the MBH reaction. | ||
4 Vinylogous Morita–Baylis–Hillman reaction or the Rauhut–Currier reaction
An organocatalytic enantioselective cross-vinylogous MBH-type reaction of methyl coumalate with α,β-unsaturated aldehydes in the presence of chiral amine organocatalyst was reported by Zu and co-workers.45 The methyl coumalate was used as an activated diene to generate enolate, and enals were activated by an imminium catalyst to form a Michael acceptor. The optimised conditions include the use of 10 mol% diphenylprolinol TBS ether organocatalyst C8, 10 mol% 2CF3-PhCOOH as an additive in EtOH solvent at room temperature for 24 h (Scheme 29). The steric and electronic properties of the alcoholic solvent play a role in determining the yield of the products. This reaction was found feasible for nearly all enal substrates except pyrone groups with an activating group at C-6 or a blocking group at C-5. The reaction can be proceeded by three alternate pathways: the usual addition elimination, Friedel–Crafts alkylation, or the open-close model reaction.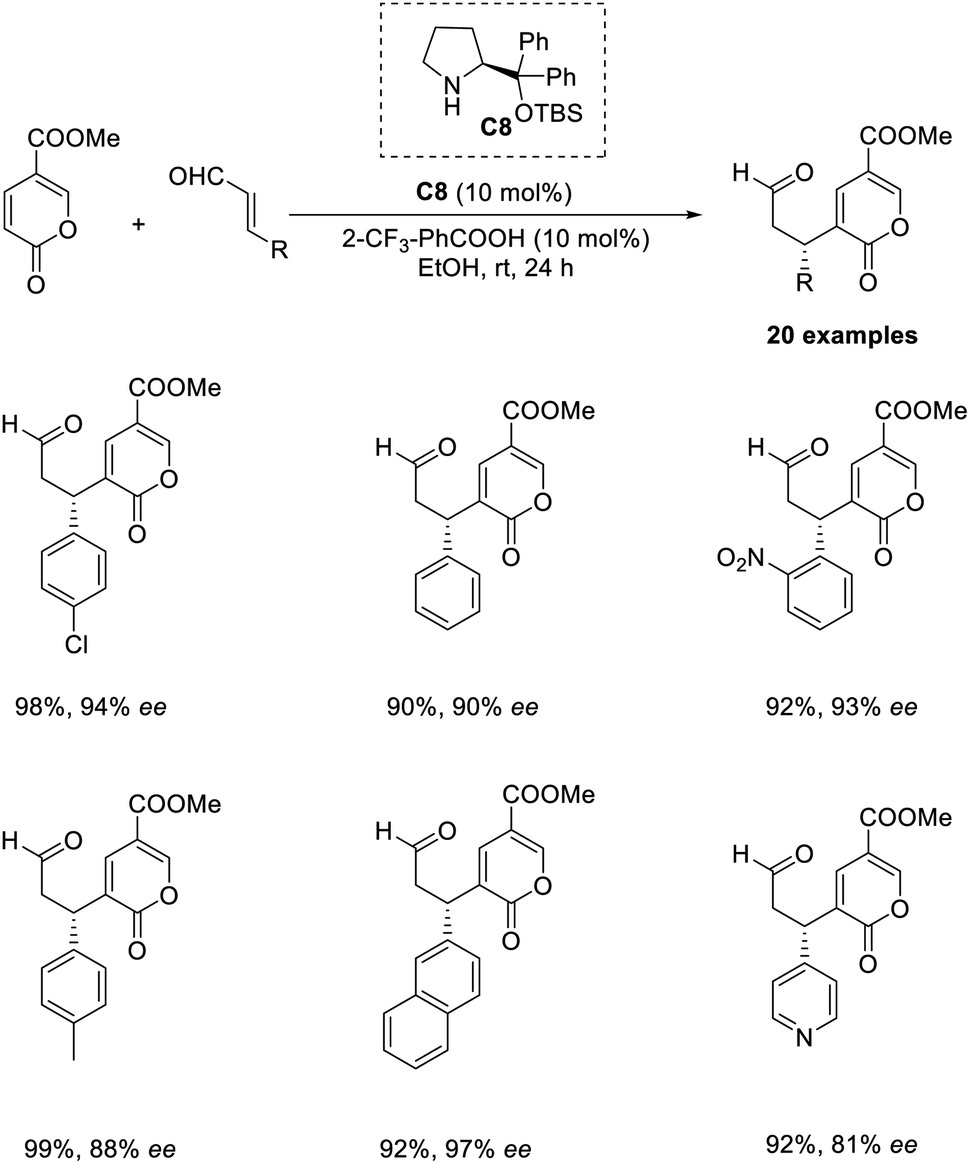 | ||
| Scheme 29 Cross vinylogous MBH-type reaction of methyl coumalate with α,β-unsaturated aldehydes. | ||
Wang and co-workers proposed an efficient methodology for the α-functionalization of 2-vinylpyridines via cross-Rauhut–Currier reaction between 2-vinylpyridines with 2-ene-1,4-diones and 3-aroyl acrylates. The optimized conditions were 20 mol% chiral phosphine catalysts C9 in DCM at −40 °C for 36 h (Scheme 30).46 Vinylpyridine-containing electron-withdrawing groups promoted the reaction and gave the desired products with high selectivity. The reaction was found suitable for 3-aroyl acrylates with electron-donating and electron-withdrawing substituents. There was higher enantioselectivity for sterically demanding substrates bearing ortho-substituted aromatic groups.
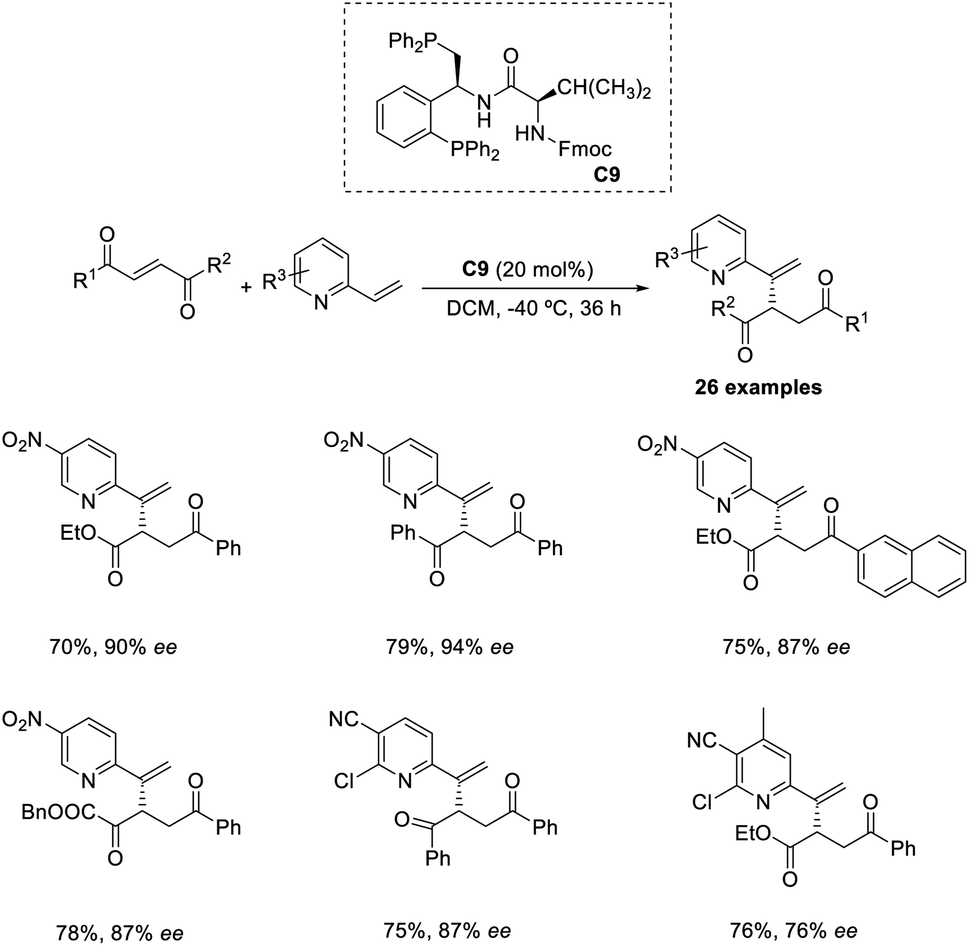 | ||
| Scheme 30 Synthesis of functional pyridines via cross-Rauhut–Currier reaction. | ||
Synthesis of differently substituted vinyl diarylmethane derivatives from α,β-unsaturated carbonyl compounds and p-quinone methides (p-QM) via the intermolecular Rauhut–Currier reaction has been previously reported.47 In this method, bis(amino)-cyclopropenylidene C10 was identified as an efficient catalyst (Scheme 31). The electronic properties of the substituents on the aromatic ring of p-QM did not greatly influence the yield of the reaction. Sterically hindered p-QMs were also well tolerated in this method. A major limitation of this method is that this protocol was applicable only to cyclic enones. Under optimized conditions, acyclic enones such as acrylates, vinylketones, or acrylamides failed to afford the required product.
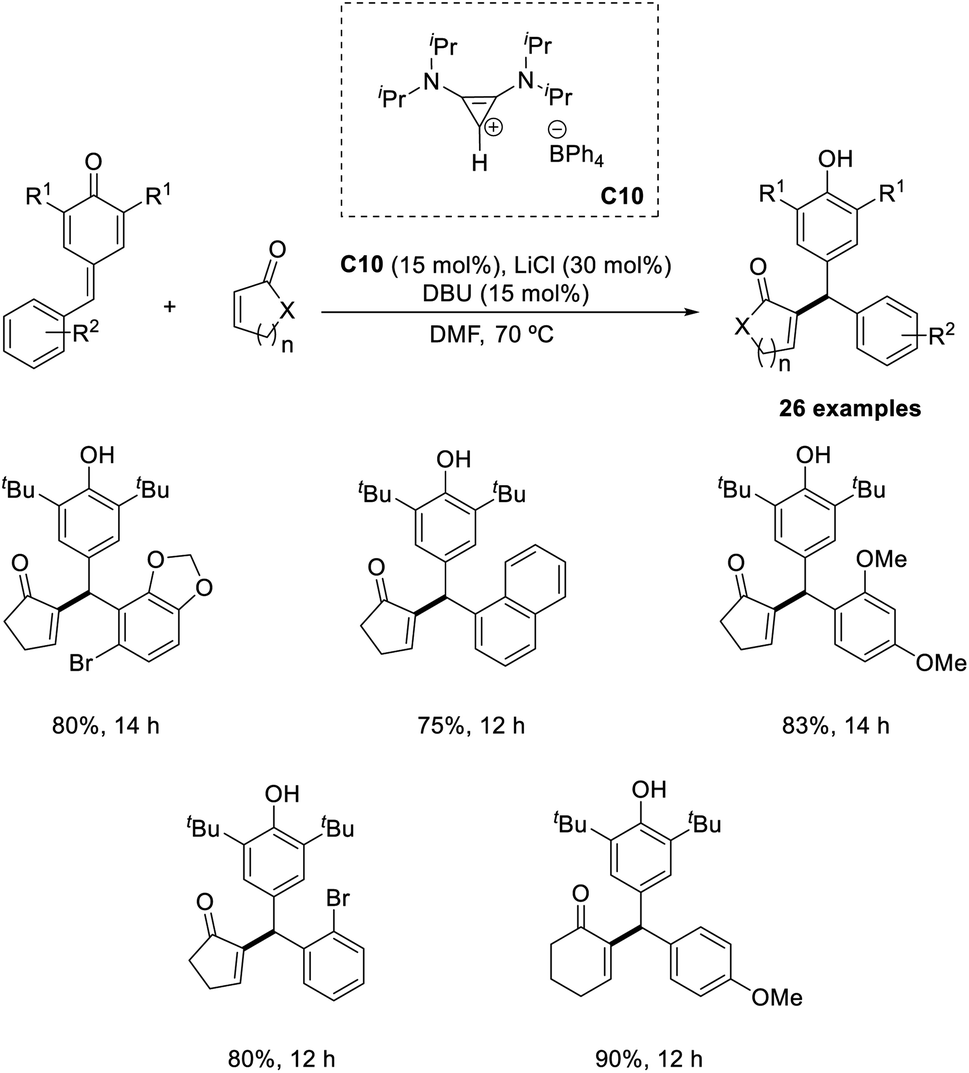 | ||
| Scheme 31 Reaction of α,β-unsaturated carbonyl compounds with p-quinone methides. | ||
Chandra and co-workers described a vinylogous Rauhut–Currier reaction between zwitterionic allenoates and p-quinine methides.48 In this strategy, they synthesised diarylmethane-substituted allenoate using DMAP as the catalyst in DCM at 25 °C (Scheme 32). The proposed mechanism includes three major steps: (i) zwitterionic formation of allenoate A, (ii) the addition of an ion to p-QM in a 1,6 pattern, followed by (iii) the transfer of protons and the removal of the catalyst (Scheme 33).
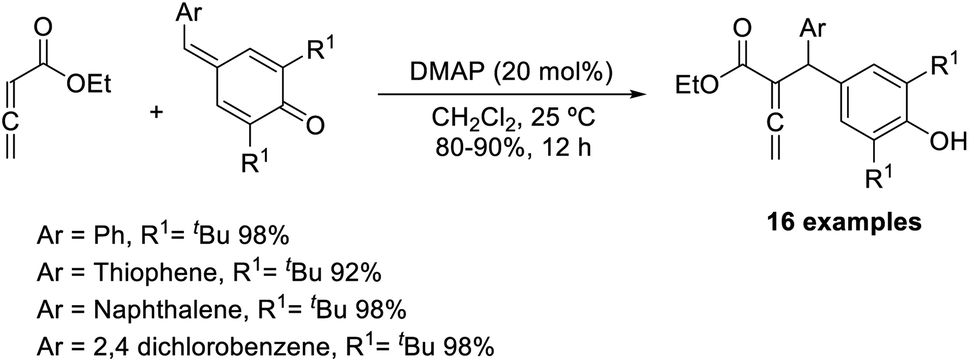 | ||
| Scheme 32 Vinylogous Rauhut–Currier reaction between p-quinone methides with allenoates. | ||
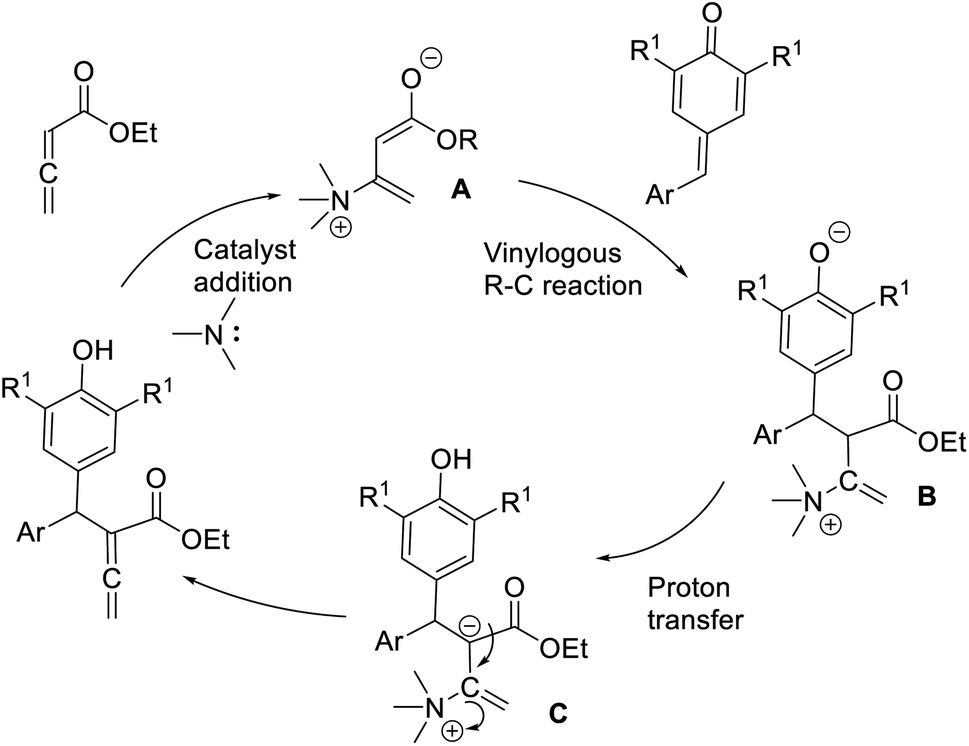 | ||
| Scheme 33 Proposed mechanism of the reaction of p-quinone methides with allenoates. | ||
Kawabata and co-workers disclosed the selective synthesis of γ-adducts from vinyl cyclopentenone and N-Boc imines through a vinylogous aza-MBH reaction.49 Their studies revealed the fact that the nature of the protecting group of aldimine nitrogen strongly influences the regiochemistry. While the reaction of vinyl cyclopentenone with N-Ts imines affords γ-adducts/α-adducts depending on the nature of the catalyst, the reaction with N-Boc protection selectively furnishes a γ-adduct, irrespective of the catalyst. This reaction was found suitable for ortho-, meta-, and para-substituted N-Boc aromatic aldimines, and the required products were obtained in moderate to satisfactory yield with high regioselectivity. The reaction was found equally compatible for N-Boc aldimines with electron-donating and electron-withdrawing substituents (Scheme 34).
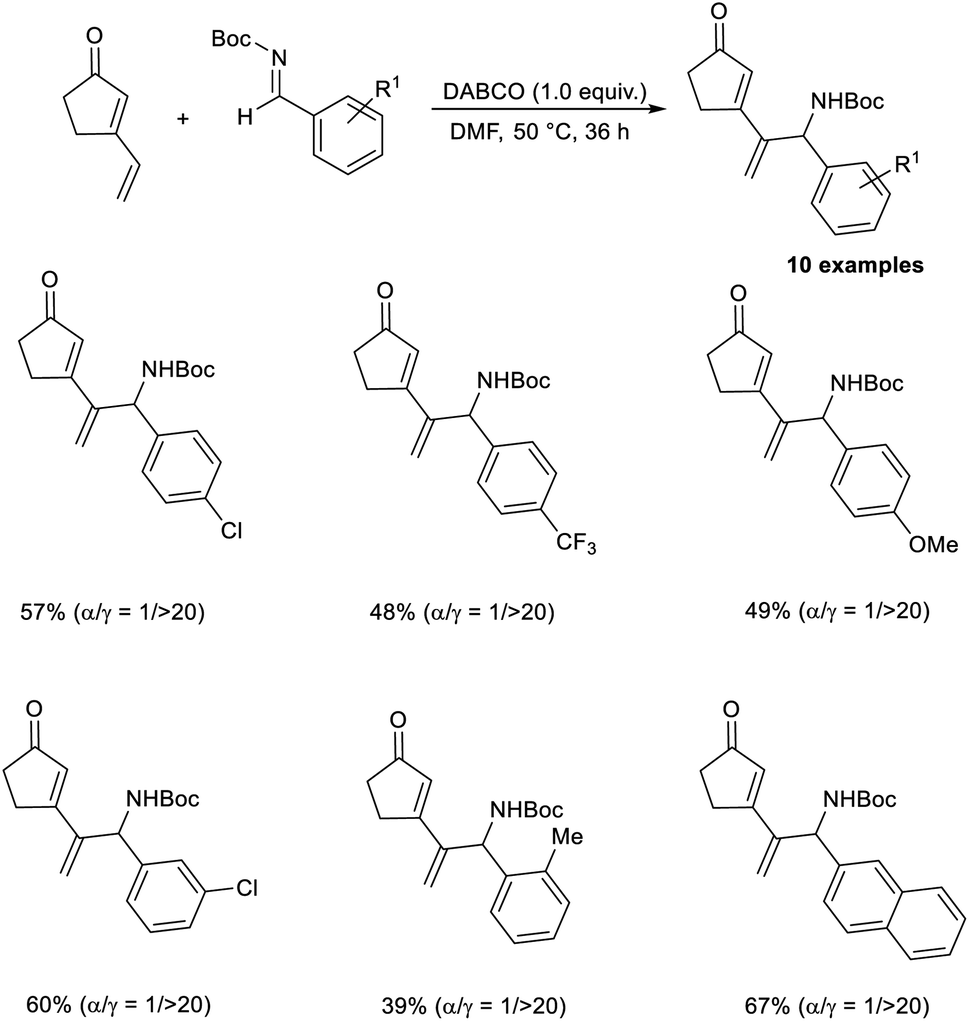 | ||
| Scheme 34 Synthesis of γ-adducts from vinyl cyclopentenone and N-Boc imines through a vinylogous aza-Morita–Baylis–Hillman reaction. | ||
Kawabata established the asymmetric region as well as the enantioselective vinylogous aza-MBH reaction by utilizing an organocatalyst.50 This methodology involves the formation of γ-adducts by the reaction between vinylcyclopentenone and several aldimines (Scheme 35). Chiral nucleophilic 4-pyrrolidino pyridine was employed as the catalyst in this reaction.
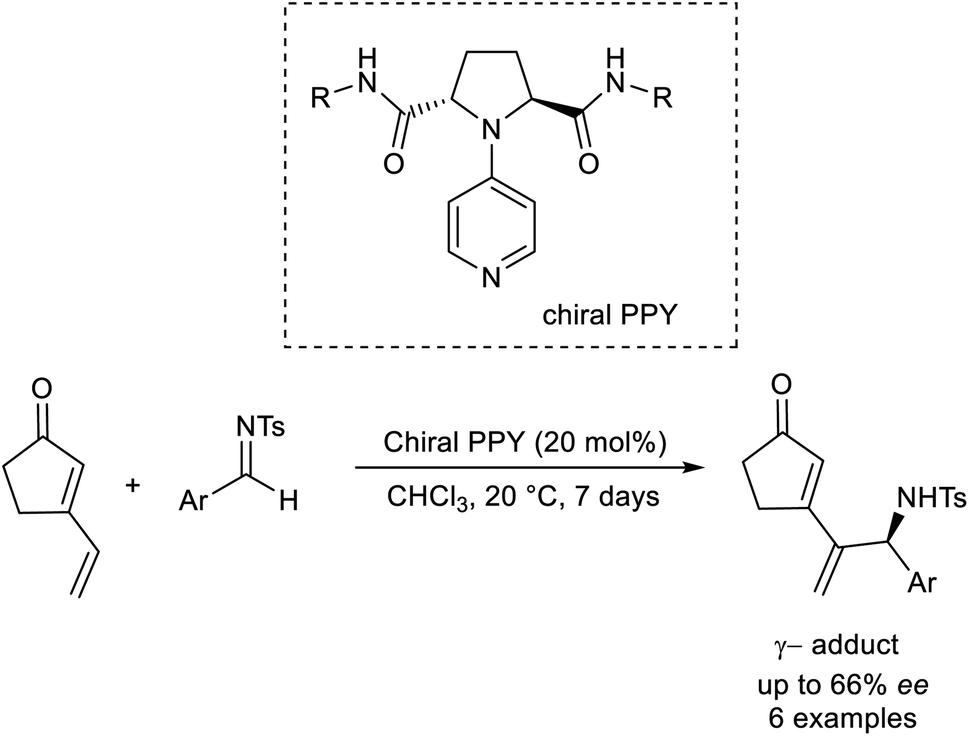 | ||
| Scheme 35 Organocatalytic synthesis of γ-adducts by reaction between vinylcyclopentenone and several aldimines. | ||
Huang et al. demonstrated a new methodology for the synthesis of trisubstituted cyclopentenes through the Rauhut–Currier/Wittig domino reaction.51 The optimized conditions consisted of P(i-Bu)3 (100 mol%) acting as the catalyst in the presence of 2 equiv. of AcOH in dioxane at 100 °C (Scheme 36). Aryl vinyl ketones with electron-donating and electron-withdrawing substituents performed well in this reaction. A wide range of functional groups such as bromo, chloro, fluoro, methyl, and trifluoromethyl on the phenyl ring in chalcones was well tolerated in this strategy. Notable features of this method include high efficiency, eco-friendliness, and mild reaction conditions.
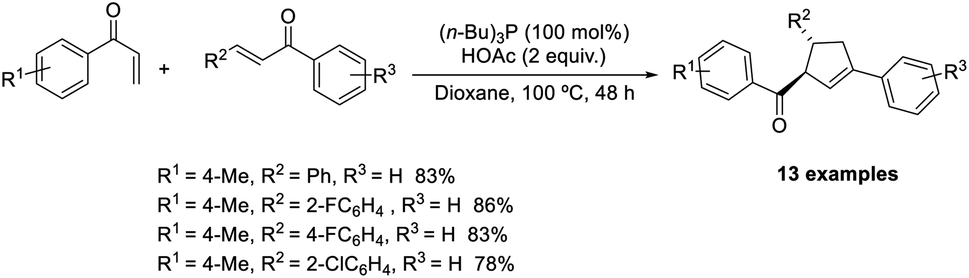 | ||
| Scheme 36 Cross–Rauhut–Currier reaction for the synthesis of trisubstituted cyclopentenes. | ||
Similarly, unsaturated pyrazolones can be produced via the Rauhut–Currier reaction, using pyrazolone as the starting material.52 Tetrahydropyranopyrazoles, a crucial compound in drug development, are formed when pyrazolone reacts with nitrostyrene. It was found that DMAP (20 mol%) was an effective catalyst for this reaction in toluene at room temperature for five days (Scheme 37). A higher yield of the required product was obtained in the case of pyrazolones with para-substituted aryl rings. This is the first report that describes the application of nitrostyrenes in the Rauhut–Currier reaction.
 | ||
| Scheme 37 Synthesis of tetrahydropyranopyrazoles using the Rauhut–Currier reaction. | ||
5 Miscellaneous
Bharadwaj suggested a faster and more effective intramolecular MBH reaction using Na2S as the catalyst.53 In this methodology, the reaction time can be decreased up to fifteen minutes. A dual electrophilic site template was synthesized, and under ideal conditions, an aza-IMBH adduct was produced. Conventional catalysts such as TPP, DABCO, and DBU do not produce products at a faster rate of reaction (Scheme 38).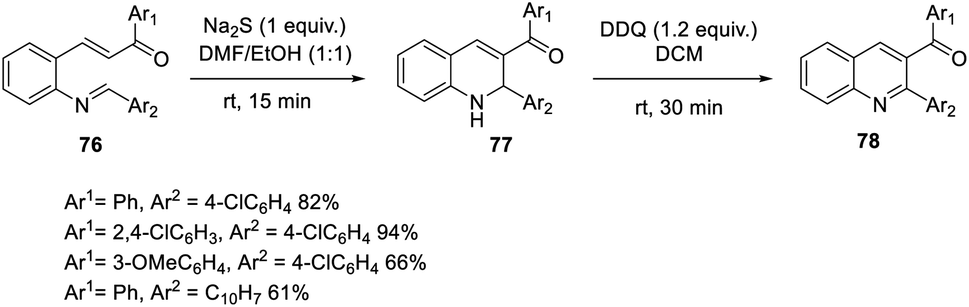 | ||
| Scheme 38 Intramolecular MBH-type reaction using Na2S as the catalyst. | ||
Hocker and co-workers developed an efficient artificial cofactor embedded in a fabricated protein host, streptavidin, which was used as the catalyst in the Baylis–Hillman reaction.54 In this method, the authors selected streptavidin as the protein host due to its facile crystallization procedure.
The synthesis of highly functionalized allylic alcohols via an organocatalytic asymmetric MBH reaction employing sodium phosphate buffer as the reaction medium was proposed by Zheng et al.55 In this study, they utilized recombinant Candida antarctica lipase B (rCalBs) as the catalyst, but the obtained products were not enantio-enriched. They also performed this reaction using enoate reductase (ER) to obtain asymmetric products with 36.9% conversion and 6.8% enantiomeric excess. Then, they investigated the efficiency of using a single amino acid as a catalyst in the MBH reaction. They found that in the presence of single L-proline and D-proline as catalysts, the desired (R) and (S) MBH products were obtained with 63.9% and 62.1% enantiomeric excess, respectively.
The MBH reaction between various aldehydes and methyl acrylate or acrylonitrile has been previously described.56 In this methodology, efficient and environmentally benign ethylcarbodiimide (EDC) hydrochloride salt was employed as a catalyst at room temperature (Scheme 39). Benzaldehydes with electron-donating substituents were well tolerated in this reaction. A lower yield of the desired product was obtained in the case of aliphatic aldehyde such as acetaldehyde. Mild reaction conditions, no byproduct formation, short reaction time, and excellent product yields are the major highlights of this method.
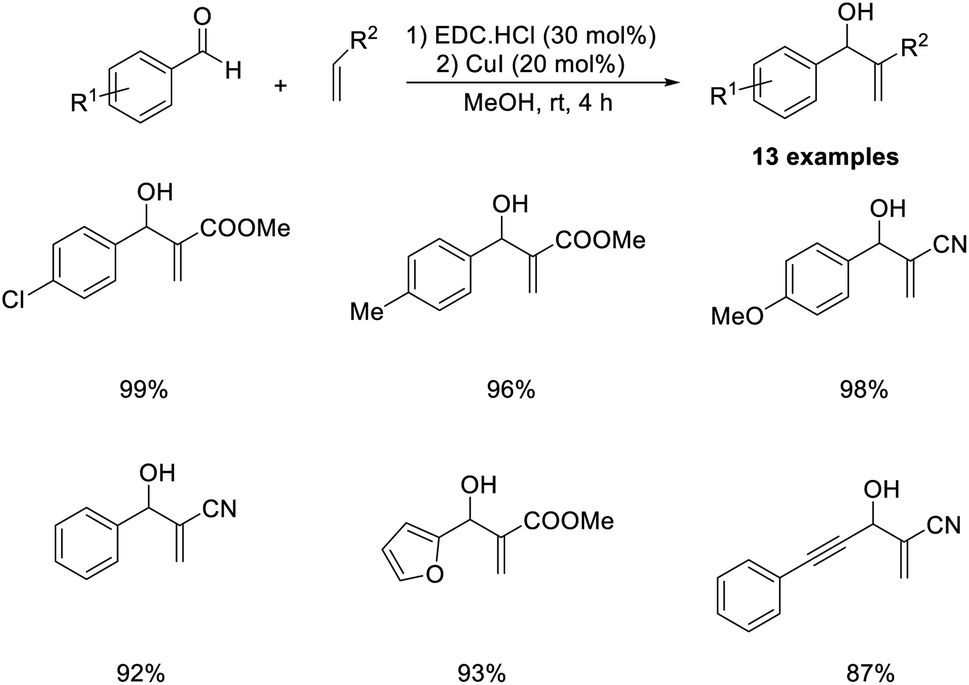 | ||
| Scheme 39 MBH reaction using ethylcarbodiimide (EDC) hydrochloride salt as a catalyst. | ||
Nitroalkenes were identified as efficient substrates in the MBH reaction.57
It is possible to perform the MBH reaction on β-aminonitroalkenes without the need of a base or catalyst.58 β-Aminonitroalkene was treated with various electrophiles, including ninhydrin, vinyl sulphone, trifluropyrate, ethyl glyoxylate, and N-tosylazadienes in acetonitrile at room temperature (Scheme 40). Solvent plays a sole role in this reaction. Aminonitrostyrenes with different electron-donating substituents such as Me and OMe gave the expected products in high yields. However, yields were reduced when electron-withdrawing groups were present. The nitronate moiety is intrinsically nucleophilic due to the push–pull characteristic of nitroenamine. When vinyl sulfone and azadiene were involved, it was spontaneously added to the electrophile in a Michael fashion. However, when glyoxylate and ninhydrin were involved, it did not do so in an aldol fashion. The nitroenamine moiety was restored in the intermediates through intramolecular proton transfer. The MBH adduct then underwent spontaneous intramolecular cyclization to produce hemiaminal.
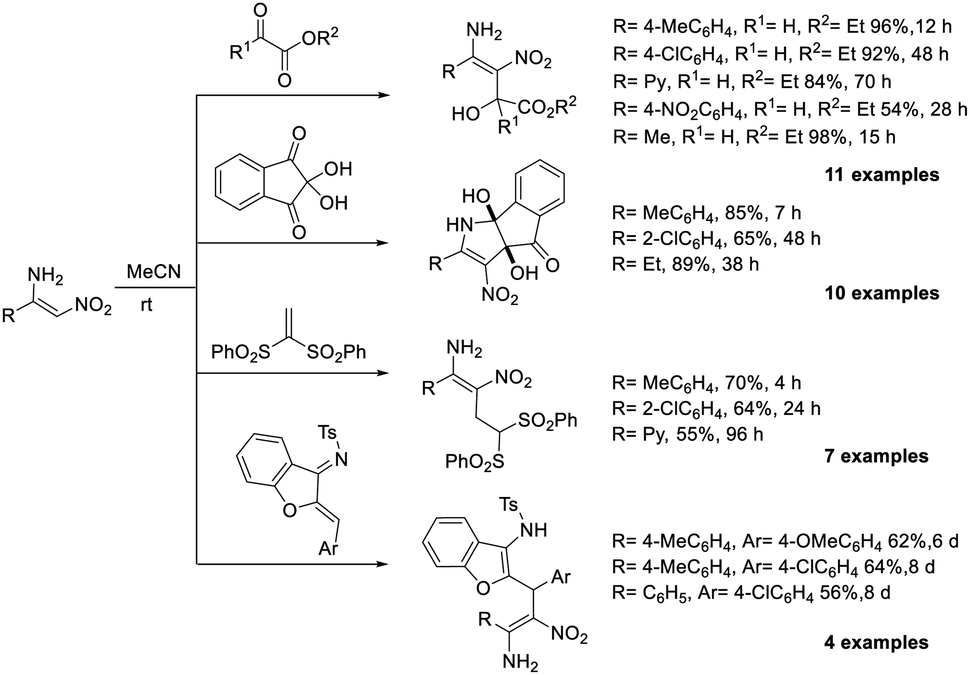 | ||
| Scheme 40 MBH reaction of β-aminonitroalkenes. | ||
6 Conclusion
Morita–Baylis–Hillman reactions are notable C–C coupling reactions due to their excellent atom economy and green perspective. In the past decades, different organocatalysts such as chiral phosphines and amines were widely exploited as catalysts in the MBH reaction.Most of the reported methods utilized aldehydes and imines as electrophiles. Recently, other electrophiles such as isatins, sulfonated imines, and indole-derived imines were identified as suitable partners in the MBH reaction. Comparing the catalytic activity of tertiary amines and phosphines, a lower reaction time was observed in the case of tertiary amines.
Recently, activated alkenes, such as acrylamides and vinyl sulfones/sulfoxides, have been used as one of the substrates. This review also discusses some reports involving activated electrophiles such as alkyl halides and ketones. Novel catalytic systems such as chiral bicyclic entities, synthetic protein hosts, and ionic liquids have been exploited for use in the MBH reaction.
Even though there has been tremendous progress with the MBH reaction, a catalyst that is suitable for all substrates has yet to be discovered. Despite these significant developments, the scope of the MBH reaction has not been fully explored, and much focus will be needed in the future to establish additional environmentally benign protocols with highly efficient catalytic systems.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
TA thanks the Council of Scientific and Industrial Research (CSIR, New Delhi) for a senior research fellowship.References
- N. G. Schmidt, E. Eger and W. Kroutil, ACS Catal., 2016, 6, 4286–4311 CrossRef CAS PubMed
.
- D. Ravelli, S. Protti and M. Fagnoni, Chem. Rev., 2016, 116, 9850–9913 CrossRef CAS PubMed
- Y. Wei and M. Shi, Chem. Rev., 2013, 113, 6659–6690 CrossRef CAS PubMed
- V. C. Sanchez, M. J. Simirgiotis and L. S. Santos, Molecules, 2009, 14, 3989–4021 CrossRef PubMed
- V. Declerck, J. Martinez and F. Lamaty, Chem. Rev., 2009, 109, 1–48 CrossRef CAS PubMed
- F. Le Hu and M. Shi, Org. Chem. Front., 2014, 1, 587–595 RSC
- K. Morita, Z. Suzuki and H. Hirose, Bull. Chem. Soc. Jpn., 1968, 41, 2815 CrossRef CAS
-
(a) K. Morita, Japan Patent, 6803364, 1968, Chem. Abstr., 1968, 69, 58828s Search PubMed
- A. Baylis and B. Hillman, German Patent, 2155113, 1972, Chem. Abstr., 1972, 77, 34174 Search PubMed
- S. E. Drewes and G. H. P. Roos, Tetrahedron, 1988, 44, 4653–4670 CrossRef CAS
- P. Perlmutter and C. C. Teo, Tetrahedron Lett., 1984, 25, 5951–5952 CrossRef CAS
- H. M. R. Hoffmann and J. Rabe, J. Org. Chem., 1985, 50, 3849–3859 CrossRef CAS
- D. Basavaiah, T. Satyanarayana and A. J. Rao, Chem. Rev., 2003, 103, 811–892 CrossRef CAS PubMed
- H. Santos, L. A. Zeoly, M. T. Rodrigues Jr, F. S. Fernandes, R. C. Gomes, W. P. Almeida and F. Coelho, ACS Catal., 2023, 13, 3864–3895 CrossRef CAS
- H. Pellissier, Tetrahedron, 2017, 23, 2831–2861 CrossRef
- W. P. Juma, D. Nyoni, D. Brady and M. L. Bode, ChemBioChem, 2022, 23, e202100527 CrossRef CAS PubMed
- D. I. Bugaenko, A. V. Karchava and M. A. Yurovskaya, Chem. Heterocycl. Compd., 2020, 56, 128–144 CrossRef CAS
- S. Takizawa, Chem. Pharm. Bull., 2020, 68, 299–315 CrossRef PubMed
- R. Innocenti, G. Menchi and A. Trabocchi, Synlett, 2018, 29, 820–824 CrossRef CAS
- M. P. Pereira, R. S. Martins, M. A. L. Oliveira and F. I. Bombonato, RSC Adv., 2018, 8, 23903–23914 RSC
- F. S. Fernandes, M. T. Rodrigues Jr, L. A. Zeoly, C. Conti, C. F. F. Angolini, M. N. Eberlin and F. Coelho, J. Org. Chem., 2018, 83, 15118–15127 CrossRef CAS PubMed
- M. Mantel, M. Guder and J. Pietruszka, Tetrahedron, 2018, 74, 5442–5450 CrossRef CAS
- A. Fallek and M. Portnoy, ChemistrySelect, 2019, 4, 3175–3179 CrossRef CAS
- L. Chang, S. Thorimbert and L. Dechoux, Org. Biomol. Chem., 2019, 17, 2784–2791 RSC
- M. T. J. Williams, L. C. Morrill and D. L. Browne, ACS Sustain. Chem. Eng., 2020, 8, 17876–17881 CrossRef CAS PubMed
- M. S. Menkudle, S. S. Pendalwar, S. V. Goswami, W. N. Jadhav and S. R. Bhusar, SN Appl. Sci., 2020, 2, 672–677 CrossRef CAS
- A. Z. Halimehjani, Y.
L. Nosood and M. Sharifi, Synth. Commun., 2020, 50, 966–972 CrossRef
- G. Khassenova and O. G. Mancheno, Eur. J. Org Chem., 2021, 2021, 2752–2755 CrossRef CAS
- V. K. Yadav, ChemRxiv, 2021, preprint, pp. 1–10, DOI:10.26434/chemrxiv.14672691.v1.
- V. Srivastava, Lett. Org. Chem., 2021, 18, 668–676 CrossRef
- V. Angamuthu, C.-H. Lee and D.-F. Tai, Catalysts, 2021, 11, 237 CrossRef CAS
- N. S. Camilo, H. Santos, L. A. Zeoly, F. S. Fernandes, M. T. Rodrigues Jr, T. S. Silva, S. R. Lima, J. C. Serafim, A. S. B. de Oliveira, A. G. Carpanez, G. W. Amarante and F. Coelho, Eur. J. Org Chem., 2022, 2022, e202101448 CrossRef CAS
- A. Pareek and M. Kalek, Adv. Synth. Catal., 2022, 364, 2846–2851 CrossRef CAS
- Y. Jia, M. Wang, F. Wu, Y. Wang and X. Chen, J. Org. Chem., 2022, 87, 14636–14645 CrossRef CAS PubMed
- H.-J. He, R.-Q. Wang, L.-X. Wan, L.-Y. Zhou, H.-Y. Li, G.-B. Li, Y.-C. Xiao and F.-E. Chen, J. Org. Chem., 2023, 88, 3802–3807 CrossRef CAS PubMed
- L. A. Zeoly, L. A. Acconcia, M. T. Rodrigues, H. Santos, R. A. Cormanich, J. C. Paniagua, A. Moyano and F. Coelho, Org. Biomol. Chem., 2023, 21, 3567–3581 RSC
- H. N. Anchan and S. Dutta, ChemistrySelect, 2023, 8, e202300264 CrossRef CAS
- X.-X. Wu, Y. He, X.-X. Qiao, T. Ma, C.-P. Zou, G. Li and X.-J. Zhao, J. Org. Chem., 2023, 88, 6599–6610 CrossRef CAS PubMed
- J. Gu, B.-X. Xiao, Y.-R. Chen, Q.-Z. Li, Q. Ouyang, W. Du and Y.-C. Chen, Org. Lett., 2018, 20, 2088–2091 CrossRef CAS PubMed
- A. Mondal, R. Hazra, J. Grover, M. Raghu and S. S. V. Ramasastry, ACS Catal., 2018, 8, 2748–2753 CrossRef CAS
- R. Mato, R. Manzano, E. Reyes, L. Carrillo, U. Uria and J. L. Vicario, J. Am. Chem. Soc., 2019, 141, 9495–9499 CrossRef CAS PubMed
- A. Mondal, Shivangi, P. Tung, S. V. Wagulde and S. S. V. Ramasastry, Chem. Commun., 2021, 57, 9260–9263 RSC
- A. B. Szychowska, A. Zawisza, S. Lesniak and M. Rachwalski, Catalysts, 2022, 12, 394–402 CrossRef
- W.-K. Wang, F.-Y. Bao, S.-T. Wang and S.-Y. Zhao, J. Org. Chem., 2023, 88, 7489–7497 CrossRef CAS PubMed
- Q. Liu and L. Zu, Angew. Chem., Int. Ed., 2018, 57, 9505–9509 CrossRef CAS PubMed
- C. Qin, Y. Liu, Y. Yu, Y. Fu, H. Li and W. Wang, Org. Lett., 2018, 20, 1304–1307 CrossRef CAS PubMed
- P. Goswami, S. Sharma, G. Singh and R. Vijaya Anand, J. Org. Chem., 2018, 83, 4213–4220 CrossRef CAS PubMed
- V. R. Reddy, N. Maripally, R. Mutyala, J. B. Nanubolu and R. Chandra, Tetrahedron Lett., 2018, 59, 2631–2635 CrossRef CAS
- N. Gondo, Y. Tanigaki, Y. Ueda, U. Yoshihiro and T. Kawabata, Synlett, 2020, 31, 398–402 CrossRef CAS
- N. Gondo, K. Fujimura, R. Hyakutake, Y. Ueda and T. Kawabata, Tetrahedron Lett., 2023, 115, 154306–154311 CrossRef CAS
- Y. Li, G. Xu and Z. Huang, Org. Biomol. Chem., 2021, 19, 2487–2491 RSC
- N. Bania, B. Mondal, S. Ghosh and C. P. Subhas, J. Org. Chem., 2021, 86, 4304–4312 CrossRef CAS PubMed
- K. C. Bharadwaj, J. Org. Chem., 2018, 83, 14498–14506 CrossRef CAS PubMed
- H. Lechner, V. R. Emann, M. Breuning and B. Hocker, ChemBioChem, 2021, 22, 1573–1577 CrossRef CAS PubMed
- X. Du, X. Cui, X. Gaob and L. Zheng, J. Chem. Technol. Biotechnol., 2022, 97, 2912–2920 CrossRef CAS
- P. Shukla, A. Asati, D. Patel, M. Singh, V. K. Rai and A. Rai, ChemistrySelect, 2023, 8, e202202747 CrossRef CAS
- S. T. Sivanandan, D. K. Nair and I. N. N. Namboothiri, Org. Biomol. Chem., 2023, 21, 6243–6262 RSC
- S. Pednekar, S. T. Sivanandan, D. Kumar, R. B. Krishna and I. N. N. Namboothiri, J. Org. Chem., 2023, 88, 4799–4808 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2024 |