
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA nonsolvolytic fluorine/LiNO3-containing electrolyte for stabilizing dynamic interfaces in Li||LiMn2O4 batteries†
Tian Tangab,
Nyalaliska W. Utomo b,
J. X. Kent Zhengc and
Lynden A. Archer*b
b,
J. X. Kent Zhengc and
Lynden A. Archer*b
aDepartment of Materials Science and Engineering, Cornell University, Ithaca, NY 14853, USA
bRobert Frederick Smith School of Chemical and Biomolecular Engineering, Cornell University, Ithaca, NY 14853, USA. E-mail: laa25@cornell.edu
cMcKetta Department of Chemical Engineering, The University of Texas at Austin, Austin, Texas 78712, USA
First published on 10th May 2024
Abstract
Mn-based high voltage cathodes, e.g., spinel LiMn2O4, are considered among the most promising materials for cost-effective, next generation energy storage. When paired with a Li metal anode, secondary batteries based on Li||LiMn2O4 in principle offer a straightforward, scalable approach for achieving cost-effective and high energy density storage demanded in applications. In practice, however, such batteries fail to live up to their promise. Rapid capacity fading caused by irreversible Mn dissolution at the cathode coupled with mossy/dendritic Li deposition at the anode limit their useful life. In this study, we report on the design of electrolytes based on a binary blend of two widely available salts, LiNO3 and LiTFSI, in ethylene carbonate (EC), which simultaneously overcome failure modes at both the cathode and anode of Li||LiMn2O4 batteries. The electrolyte design is motivated by a recent finding that compared with their linear counterparts (e.g., dimethyl carbonate), cyclic carbonates like EC dissolve considerably larger amount of LiNO3, which markedly improves anode reversibility. On the other hand, it is known that nonsolvolytic fluorine-containing Li salts like LiTFSI, lowers the electrolyte's susceptibility to solvolysis, which generates HF species responsible for Mn leaching at the cathode. In particular, we report instead that fluorine groups in the TFSI salt, promote formation of a favorable, fluorine-rich interphase on the Li metal anode. Electrochemical measurements show that the electrolytes enable remarkably improved charge–discharge cycling stability (>1000 charge–discharge cycles) of Li||LiMn2O4 batteries. In-depth atomic-resolution electron microscopy and X-ray/synchrotron diffraction experiments reveal the fundamental source of the improvements. The measurements show that crystallographic degradation of Mn-based cathodes (e.g., surface Mn leaching and bulk defect generation) upon cycling in conventional electrolytes is dramatically lowered in the LiNO3 + LiTFSI/EC electrolyte system. It is shown further that the reduction of Mn dissolution not only improves the cathode stability but improves the reversibility of the Li metal anode via a unique re-deposition mechanism in which Li and Mn co-deposit on the anode. Taken together, our findings show that the LiNO3 + LiTFSI/EC electrolyte system holds promise for accelerating progress towards practical Li||LiMn2O4 batteries because it stabilizes the dynamic interfaces required for long-term stability at both the Li anode and the LiMn2O4 cathode.
Developing energy storage technologies using low-cost, earth abundant materials are key to successful energy transition in the transportation and electric power generation sectors in the 21st century. Electrochemical energy storage in rechargeable batteries is regarded as one route towards this goal owing to the flexibility, modulability and relatively high aggregate energy density storage possible over the lifetime of rechargeable batteries,2 in comparison with single-use alternatives. Among the contemporary battery chemistries being considered, Li-ion technology has achieved market dominance.3 One aspect that limits the sustainability of these batteries originates from usage of materials to which access is constrained, either by natural or geopolitical factors; both can be illustrated by the material's market price evolution over time.4,5 For example, Co, an essential material used in many state-of-the-art Li-ion battery cathodes, has increased in price from $20k per metric ton in 2014 to $80k per ton in 2022, according to data from the London Metal Exchange. These price increases are expected to become worse as electrification penetrates more fully in the transportation sector and as increased use of storage to manage intermittent supplies of renewable power on the grid begin to tax the finite world-wide Co supply.
Cobalt-free Li-ion battery cathodes have emerged recently as a priority technology, particularly when Co is replaced by in-expensive earth abundant elements like Mn, which is currently priced at $2k–5k per metric ton (in the form of pure metal). The LiMn2O4 cathode is therefore of much lower cost than other commercial cathodes such as LiCoO2, see Fig. 1.6,7 In particular, when a high-Mn content cathode is paired with a Li metal anode, the Li||LiMn2O4 battery could offer a higher voltage and a greater energy density in comparison with traditional graphite||LiMn2O4. Unfortunately, a battery of this kind would suffer from well-known interfacial instability at both electrodes, namely, dendritic Li deposition at the anode8,9 and transition metal dissolution at the cathode.3,10–17 Task-specific chemical design of electrolyte systems that meet the oftentimes different needs at the two electrodes of distinct natures is needed to enable electrochemical stability over extended cycling.
 | ||
| Fig. 1 Key properties (cost,1 voltage, and energy density) of representative cathode materials for lithium batteries. The energy density is calculated based on the anode-free condition ideally, excluding the weight of other cell parts. LiMn2O4 stands out for its remarkably lower cost and high voltage, in comparison with battery cathode materials of contemporary interest. | ||
A large volume of work has been done to understand and improve the reversibility of Li metal plating/stripping in various electrolyte media. LiNO3 salt additives in ether-based electrolytes have for instance been shown to markedly improve galvanostatic cycling of Li–S batteries both by promoting formation of a stable solid-electrolyte interphase on the Li metal anode17,18 and by lowering the thermodynamic activity of the ether solvent, enhancing its electrochemical stability.18 Carbonate-based electrolytes feature wider electrochemical stability windows than ethers and are known to be a requirement for long term stability of rechargeable batteries based cathodes that operate at nominal voltages above approximately 3 V. Unfortunately, the solubility of LiNO3 is much lower in carbonate electrolytes typically used in lithium batteries, in comparison to ethers. Recent studies report that the solubility of LiNO3 in carbonates is elevated markedly for cyclic molecules (e.g., ethylene carbonate (EC)) versus linear molecules (e.g., dimethyl carbonate (DMC)) commonly used in lithium batteries because of their lower melting point temperatures and generally superior ion transport properties at room temperature.19 This finding opens up multiple avenues for designing an electrolyte for Li||LiMn2O4 batteries that takes advantage of beneficial effects of LiNO3 in stabilizing the interphase on Li metal (e.g., formation of Li3N and LiNxOy), and also suggests that traditionally overlooked, high-boiling point carbonate solvents like EC, could see new uses as stand-alone electrolyte solvents for enhancing battery safety.
Salt blends composed of two or more molecular species are now commonplace in battery electrolyte design because each component may provide task-specific advantages for enhancing anode or cathode reversibility. The simultaneous use of EC as a stand-alone electrolyte solvent and focus on batteries based on high-voltage LiMn2O4 cathodes make the choice of the second salt (LiNO3 is here designated as the first salt because of its known benefits for improving stability of the Li anode) non-trivial. These choices nonetheless also constrain the range of anion chemistries that one might select. In particular, the need for the second salt to dissolve well in the EC/LiNO3 electrolyte and at the same time undergo electroreduction at the Li anode to generate a self-limited, fluorine rich SEI,20 further constrains the anion choices. Additionally, we discuss later the deleterious effects of HF on the stability of Mn-based cathodes. Fluorine-containing salts that facilitate formation of a stable F-containing interphase on Li metal anode, but which are immune to solvolysis to generate HF are therefore of greatest interest. These considerations, lead to the basic electrolyte design concept proposed and evaluated in the study. Briefly, a well-designed electrolyte for the Li||LiMn2O4 cell should have three principal attributes: (i) it must be able to dissolve large amounts of LiNO3; (ii) it should remain oxidatively stable at the high voltages at the cathode and should undergo only localized electroreduction at the Li anode to form a self-limiting fluorine-rich SEI on the anode; (iii) none of the components should be susceptible to solvolytic degradation to produce HF in the battery cell. These considerations are all met by the choice of lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) as a second salt to pair with LiNO3, in an EC-based electrolyte. We note further that while lithium bis(fluorosulfonyl)imide (LiFSI) meets some of these requirements, it has been reported to be less oxidatively stable at high voltages.21
In-depth electrochemical and structural analysis of Li||LiMn2O4 batteries adopting the proposed electrolyte design provide insights into their advantages and disadvantages relative to conventional electrolytes. These studies largely confirm the effectiveness of the task-specific electrolyte design concept proposed in the previous section. A key additional finding is that electrolyte design plays a dominant role in the failure of Mn cathode materials. Specifically, lattice defects that are pronounced in some electrolytes can be essentially eliminated in others, as determined by atomic-resolution characterization of the cycled cathode structure. In addition to the (de)/stabilization of the surface facets, it is found that the bulk defect content of LiMn2O4 materials demonstrates a strong dependence on electrolyte chemistry. Fast electrochemical and structural degradation is observed on LiMn2O4 cycled in electrolytes with F-containing salts able to readily generate HF by solvolysis (including hydrolysis),19,20 whereas both the surface facet and the bulk crystal structures of LiMn2O4 are preserved when cycled in electrolytes that are not prone to HF-generating solvolysis. The results underscore the role played by the dynamic chemical environments developed inside the battery electrodes and provide insights into electrolyte design strategies for Mn-based cathodes and other transition metal oxide electrode materials that suffer from similar dissolution issues.22
Fig. 2 reports the cycling stability of Li||LiMn2O4 batteries measured in carbonate-based electrolytes prepared with LiNO3 and a series of different Li-ion salts, namely, LiTFSI, LiClO4, LiBF4 and LiPF6; see Materials and methods for details. The results in Fig. 2A reveal an obvious declining trend of capacity retention with salt chemistry in the order LiTFSI > LiClO4> LiBF4 ≈ LiPF6. Motivated by these observations, we single out the LiNO3-containing electrolytes reinforced with LiTFSI and LiPF6 for in-depth investigation. We note that electrolytes containing these salts yield batteries with the highest and lowest specific capacity retention (also reflected in the voltage profiles in Fig. 2B). Additionally, these electrolytes are being actively studied in contemporary Li battery research.21–24
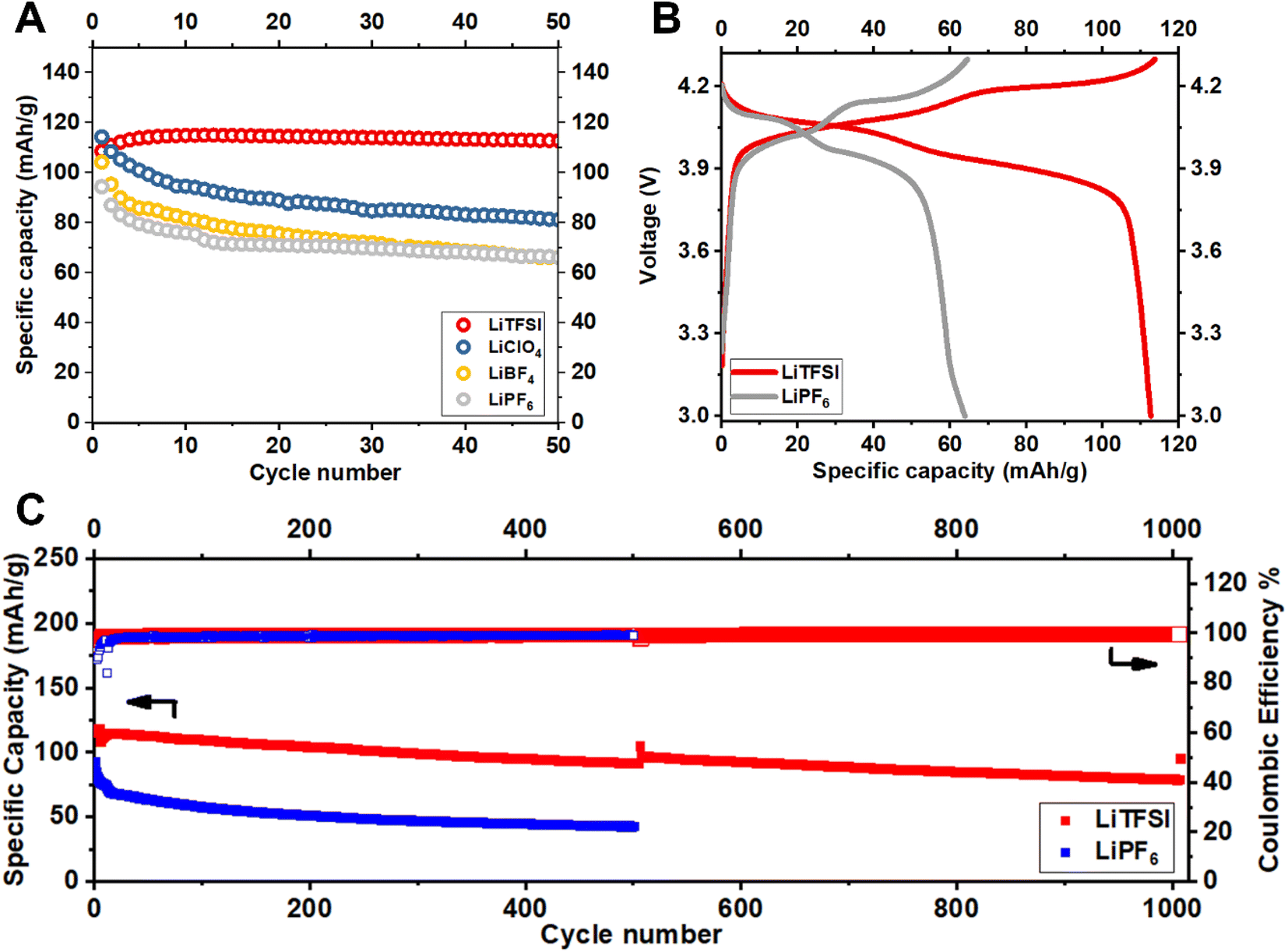 | ||
| Fig. 2 Galvanostatic charge–discharge cycling of Li||LiMn2O4 electrochemical cells in representative electrolytes. (A) Effect of Li salt on cycling stability and capacity retention of Li||LiMn2O4 battery cells. (B) Comparison of the charge–discharge voltage profiles over 50 cycles for Li||LiMn2O4 batteries using electrolytes containing LiTFSI and LiPF6 electrolyte, respectively. (C) Long-term cycling stability of the batteries. | ||
Fig. 2C reports the long-term cycling performance of Li||LiMn2O4 batteries in these two electrolytes. The results show that battery cells containing LiPF6-based electrolyte hardly maintains a 50 mA h g−1 specific capacity over 250 cycles, whereas those using LiTFSI-based electrolyte claims a specific capacity of 118 mA h g−1 and a retention of 81% over 1000 cycles. The cell is intentionally set to charge–discharge at a slow rate of 0.1C for 1 cycle after each 500 cycles at a normal rate of 1C in order to evaluate whether the capacity fading is caused by irreversible materials loss or resistance buildup (i.e., the gradually increasing iR overpotential, which reduces the effective range of cycling)—capacity loss via irreversible material degradation cannot be achieved at reduced cycling rates, but capacity loss due to the latter should be strongly dependent on the applied current i. Noteworthy is that, the majority of the capacity fading observed for LiMn2O4 cycled in the LiTFSI electrolyte can be attributed to resistance buildup, as opposed to irreversible materials loss, since the capacity difference between 0.1C and 1C becomes increasingly pronounced from the 1st, the 500th, to the 1000th cycle. In contrast, the cell using the LiPF6 electrolyte exhibits a so-called endless charging behavior at 0.1C (Fig. S1†) even in the first cycle, which is indicative of electrolyte decomposition.25 The measurements in Fig. 2 raise the question that why and how electrolyte chemistry imposes such significant influences on the cycling stability of LiMn2O4 particularly.
We performed aberration-corrected scanning transmission electron microscopy (STEM) to interrogate the atomic-scale structural evolution of LiMn2O4 taking place during cycling in these two representative electrolytes (Fig. 3; see also Fig. S2† for STEM/EDS analysis). The LiMn2O4 cycled in the two electrolytes, respectively, show distinct crystallographic features both at the particle level and at the atomic level. The LiMn2O4 cycled in the LiPF6 electrolyte show poorly-defined surface termination (Fig. 3A and B), in stark contrast to the well-defined facets observed on LiMn2O4 particles cycled in the LiTFSI electrolyte (Fig. 3C and D), where surface terminations along low-index crystallographic facets are observed, e.g., (110) and (311) families; see Fig. 4A–C for the crystal structures of LiMn2O4. This comparison at the particle level means that the LiMn2O4 underwent significant Mn dissolution through the surface, creating the observed meandering morphology in the LiPF6 electrolyte, whereas the surface of LiMn2O4 particles cycled in LiTFSI electrolyte is intact, suggesting negligible Mn dissolution, which is consistent with the specific capacity retention measured in Fig. 2. We would further note that, the Mn dissolution and surface roughening process is in theory self-reinforcing, as it creates fresh, high-index crystal facets that are more susceptible to unfavorable reactions, including but not limited to Mn dissolution.22,26
 | ||
| Fig. 3 Atomic resolution scanning transmission electron microscopy characterization of the crystallographic stability of the LiMn2O4 electrodes after 100 charge–discharge cycles. (A) and (B) High angle annular dark field (HAADF) image and bright field (BF) image of LiMn2O4 cycled in LiPF6 electrolyte, respectively. (C) and (D) High angle annular dark field (HAADF) image and bright field (BF) image of LiMn2O4 cycled in LiTFSI electrolyte, respectively. | ||
Additionally, we find that the LiMn2O4 cycled in the LiPF6 electrolyte contains a significantly higher level of lattice defects, such as stacking faults and the associated dislocations, in the inner part of the particle (Fig. 3A and B; see also Fig. S3† for enlarged images). These should be compared to the lattice of LiMn2O4 cycled in the LiTFSI electrolyte, where no such defects are observable within a reasonably large range of interest (i.e., tens of nm; see also Fig. S4†). These bulk lattice defects are known to be another source of cathode materials degradation via—for example—physical cracks, etc.27,28 This STEM observation on the local structures is consistent with X-ray diffraction (XRD) patterns as a statistical measurement of the samples from a global point of view (Fig. 4D and E). Despite the similarity of the XRD patterns in Fig. 4D, the close-up plots of the three representative samples (i.e., pristine before cycling, cycled in LiPF6, and cycled in LiTFSI, respectively) reflect the crystallographic evolution occurring in LiMn2O4's bulk lattice over cycling (Fig. 4E).
 | ||
| Fig. 4 Crystal structure of LiMn2O4 and X-ray diffraction characterization of the materials. (A) 3D crystal model of LiMn2O4. Projected views of the crystal from (B) 〈112〉 direction, and (C) 〈110〉 direction, respectively. (D) X-ray diffraction patterns of LiMn2O4; black: pristine, blue: cycled in LiPF6 electrolyte, red: cycled in LiTFSI electrolyte, respectively. (E) Close-up plot of the LiMn2O4 (400) peak under these conditions. | ||
The peaks from LiMn2O4 particles cycled in the LiPF6 electrolyte exhibit pronounced shifting, broadening, and possibly splitting. Such features indicate irreversible structural transition from the original cubic spinel LiMn2O4 lattice before cycling. These transitions are significantly suppressed in LiMn2O4 particles cycled in the LiTFSI electrolyte as a comparison. The crystallographic stability of LiMn2O4 in the LiTFSI electrolyte is further revealed by synchrotron powder diffraction (Fig. S5†); no discernible peak shift is observed. The peak shift found in LiMn2O4 particles cycled in LiPF6 towards higher value suggests a smaller interplanar distance according to Bragg's law (2d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sinθ = nλ). Since all the electrodes are in a discharged state, that lattice shrinkage suggests that the electrode can only be lithiated/discharged to a limited extent. The STEM—as a local characterization—and the XRD—as a global measurement—consistently demonstrate that the crystallographic stability of LiMn2O4 has a strong dependence on the chemical environments, even with the inherent property of the lattice with Jahn–Teller (J–T) distortion. Based on the XRD results, we also conclude that the cause for different performance is not different quality of the pristine electrode materials.
sinθ = nλ). Since all the electrodes are in a discharged state, that lattice shrinkage suggests that the electrode can only be lithiated/discharged to a limited extent. The STEM—as a local characterization—and the XRD—as a global measurement—consistently demonstrate that the crystallographic stability of LiMn2O4 has a strong dependence on the chemical environments, even with the inherent property of the lattice with Jahn–Teller (J–T) distortion. Based on the XRD results, we also conclude that the cause for different performance is not different quality of the pristine electrode materials.
The critical role played by electrolyte salts on the cycling stability of LiMn2O4 could be understood by scrutinizing the chemical properties of the salts. It has been shown in multiple prior reports that the complex anions formed by the Lewis acid–base reaction between F− anion and a strong Lewis acid, e.g., PF5, AsF5, BF3, are highly susceptible to solvolytic reactions with electrolyte components such as the organic solvent or the trace amount of water in storage or under dynamic electrochemical conditions—for example—high voltage.20,29–31 Such reactions generate hydrofluoric acid molecules (HF) as a product. In a separate context of ionic liquid manufacturing, this phenomenon has been regarded as a serious issue for this group of solvolytic anions, whereas ClO4− and TFSI− exhibit excellent chemical stability against solvolytic reactions.32 HF is highly corrosive, and is capable of destabilizing the crystallographic facets of LiMn2O4—for example—by transition metal leaching. The chemical corrosivity of the solvolysis-generated HF leads to the collapse of the well-defined crystallographic facets and to the generation of bulk lattice defects. The slightly better capacity retention in LiBF4 electrolyte may be ascribed to the higher H2O tolerance than LiPF6 as acknowledged in prior studies.33 These observations and analyses together reveal that the solvolytic HF is the major source of capacity fading observed on LiMn2O4 (see the right panel of Fig. 5), and that such a trend could be readily suppressed by using a non-solvolytic salt, such as LiTFSI.
 | ||
| Fig. 5 Schematic diagram illustrating the solvolytic degradation of transition metal oxide cathodes. The right panel: when the Li-ion salt in the electrolyte is formed by the Lewis acid–base reaction between F− and a strong Lewis acid, such as PF5, BF3, etc., the resultant F-coordinated anion exhibit a high susceptibility to solvolytic decomposition either with the organic solvent or the trace amount of water. Such solvolytic reactions produce free hydrofluoric acid that strongly etches the surface of the transition metal (TM) oxide cathode material, causing irreversible dissolution of TM into electrolyte in the form of cations. The dissolved TM cations are transferred to the anode side driven by concentration gradient field and the electric field alternating during charge–discharge cycles and are deposited onto the surface of the anode. This parasitic TM deposition reaction introduces significant heterogeneity into the solid-electrolyte interphase (SEI) and triggers outward, nonuniform Li metal plating morphology. As such, the solvolytic instability of the electrolyte poses critical challenges to both the cathodes and the anodes, whereas these detrimental processes are completely absent if this type of chemical instability is eliminated by rational design of the electrolyte chemistry (the left panel). | ||
Comparing the performance of LiClO4 and LiTFSI, the inferior performance of LiClO4 is attributable to the inability of forming a good F-containing solid-electrolyte interphase (SEI) on the Li anode, causing Li anode's rapid deterioration.34–37 The Li plating/stripping coulombic efficiency measurements in Fig. S6–S8† evidently demonstrate the LiTFSI-based electrolyte's capability of stabilizing the interfaces formed on the Li metal surface. The LiTFSI-based electrolyte exhibits stable plating/stripping behaviors even after 500 cycles, whereas the LiClO4-based electrolyte only allows ∼30 Li plating/stripping cycles. The stabilization effect of LiTFSI is attributed to the formation of a favored, LiF-rich SEI via the decomposition of LiTFSI.38,39 The LiF-rich SEI formed in LiTFSI-containing electrolytes has been widely reported and studied in the existing literature by a variety of characterization tools, e.g., X-ray photoelectron spectroscopy.40
As a further verification of the HF-induced Mn dissolution mechanism and to study the dynamic interface at the Li metal anode, we performed microscopy and spectroscopy characterization of the Li metal anodes. As is obvious from comparing Fig. 6A and B, the Li has a remarkably more uniform morphology when cycled in the LiTFSI electrolyte than the LiPF6 electrolyte. In addition, pronounced Mn accumulation has been detected on the surface of Li metal anode after cycling in the LiPF6 electrolyte, but not on Li anode cycled in the LiTFSI electrolyte (Fig. 6C and D). A well-defined Mn Kα peak is seen on Li metal cycled in the LiPF6 electrolyte, whereas this peak is completely absent on Li metal cycled in the LiTFSI electrolyte (Fig. 6D). It has been reported that the dissolved TM cations, e.g., Mn2+/3+, could be electro-/chemically reduced and thereby deposited onto the anode in the form of elemental metal particles. This heterogeneous TM deposition is notorious for producing outward, aggressive growth of the Li metal.41 See also optical photos of the cycled Li metal anodes in the insets of Fig. 6A and B. Based on the evidence, we conclude that the solvolysis-induced TM dissolution from the cathode imposes critical negative effects on the operation of both the cathode itself, and also the anode—for example—Li metal in this case, or even more traditional graphite electrodes.42,43 To further investigate the physical properties of the electrolyte, we also performed Differential Scanning Calorimetry (DSC); see Fig. S9† for the measurement.
 | ||
| Fig. 6 Scanning electron microscopy and energy dispersive spectroscopy characterization of Li metal anodes after cycling. SEM images of Li metal anodes cycled in (A) LiPF6 electrolyte and (B) LiTFSI electrolyte, respectively. Insets are optical photos of cycled Li metal anodes. The blackening of the Li surface observed in LiPF6 suggests serious degradation. (C) Mn content detected by EDS on Li metal after 500 charge–discharge cycles in these two electrolytes. (D) Characteristic EDS spectra of Mn measured on these two electrodes. | ||
In summary, aided by electrochemical, structural, and chemical analyses, we find that electrolytes composed of LiTFSI + LiNO3 in an ethylene carbonate solvent, are remarkably effective in enabling long-duration cycling of Li||LiMn2O4 batteries. The electrolytes appear to achieve this feat by stabilizing the dynamic interfaces formed at both electrodes and by limiting HF generation and associated Mn dissolution from the cathode. Our results also show that the instability of LiMn2O4 cathodes is primarily a result of the chemical environment surrounding the LiMn2O4 particles. The TM dissolution induced by solvolytic HF not only directly introduces irreversible capacity loss in the cathode, but also indirectly promotes heterogeneity on the anode through unexpected Mn co-deposition. These effects eventually result in capacity fading or even more fatal failures such as internal shorts because of outwards growth of the metal. The importance of a fluorine-containing salt is highlighted by the stark contrast between the performance of the LiTFSI, and the LiClO4 cells, respectively; the non-solvolytic fluorine in TFSI is shown to play a critical role in ensuring stable Li plating/stripping hypothetically via the formation of a fluorine-enriched interphase. We conclude that the combination of LiTFSI, LiNO3, and EC represents a balanced solution that meets the different needs as required by the two distinct electrodes in Li||LiMn2O4. The findings evidently provide key insights for stabilizing next-generation Li-based batteries using sustainable materials via task-specific, electrolyte design.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported as part of the Center for Mesoscale Transport Properties, an Energy Frontier Research Center supported by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, under award #DE-SC0012673. The work made use of the Cornell Center for Materials Research Shared Facilities which are supported through the NSF MRSEC program (DMR-1719875).References
- TrendForce, Li-Ion Battery Industry Chain Price, 2024 Search PubMed.
- Z. Yang, et al., Electrochemical energy storage for green grid, Chem. Rev., 2011, 111, 3577–3613 CrossRef CAS PubMed.
- H. Yaghoobnejad Asl and A. Manthiram, Toward sustainable batteries, Nat Sustainability, 2021, 4, 379–380 CrossRef.
- N. R. Rachidi, G. T. Nwaila, S. E. Zhang, J. E. Bourdeau and Y. Ghorbani, Assessing cobalt supply sustainability through production forecasting and implications for green energy policies, Resour. Policy, 2021, 74, 102423 CrossRef.
- G. S. Seck, E. Hache and C. Barnet, Potential bottleneck in the energy transition: the case of cobalt in an accelerating electro-mobility world, Resour. Policy, 2022, 75, 102516 CrossRef.
- C. Bauer, et al., Charging sustainable batteries, Nat. Sustain., 2022, 1–3, 176–178 CrossRef.
- M. M. Thackeray and K. Amine, LiMn2O4 spinel and substituted cathodes, Nat. Energy, 2021, 6, 566 CrossRef CAS.
- J. X. K. Zheng, J. Yin, T. Tang and L. A. Archer, Moss-like Growth of Metal Electrodes: On the Role of Competing Faradaic Reactions and Fast Charging, ACS Energy Lett., 2023, 8, 2113–2121 CrossRef CAS.
- J. X. K. Zheng, Perspective—Reversibility of Electro-Plating/Stripping Reactions: Metal Anodes for Rechargeable Batteries, J. Electrochem. Soc., 2022, 169, 100532 CrossRef CAS.
- F. K. Shokoohi, J. M. Tarascon and B. J. Wilkens, Fabrication of thin-film LiMn2O4 cathodes for rechargeable microbatteries, Appl. Phys. Lett., 1991, 59, 1260–1262 CrossRef CAS.
- Z. Liu, A. Yu and J. Y. Lee, Cycle life improvement of LiMn2O4 cathode in rechargeable lithium batteries, J. Power Sources, 1998, 74, 228–233 CrossRef CAS.
- X. Zhu, et al., LiMnO2 cathode stabilized by interfacial orbital ordering for sustainable lithium-ion batteries, Nat Sustainability, 2021, 4, 392–401 CrossRef.
- A. Yamada, M. Tanaka, K. Tanaka and K. Sekai, Jahn–Teller instability in spinel Li–Mn–O, J. Power Sources, 1999, 81, 73–78 CrossRef.
- K. Chudzik, et al., Improving the performance of sulphur doped LiMn2O4 by carbon coating, J. Power Sources, 2019, 434, 226725 CrossRef CAS.
- M. Michalska, et al., Improved electrochemical performance of LiMn2O4 cathode material by Ce doping, Electrochim. Acta, 2018, 276, 37–46 CrossRef CAS.
- J. Zheng, R. Garcia-Mendez and L. A. Archer, Engineering Multiscale Coupled Electron/Ion Transport in Battery Electrodes, ACS Nano, 2021, 15, 19014–19025 CrossRef CAS PubMed.
- O. Yamada, M. Ishikawa and M. Morita, Charge/discharge cycling and impedance response of LiMn2O4 electrode in organic electrolyte solutions with different compositions, Electrochim. Acta, 2000, 45, 2197–2201 CrossRef CAS.
- Q. Zhao, J. Zheng and L. Archer, Interphases in lithium–sulfur batteries: toward deployable devices with competitive energy density and stability, ACS Energy Lett., 2018, 3, 2104–2113 CrossRef CAS.
- M. Stich, M. Göttlinger, M. Kurniawan, U. Schmidt and A. Bund, Hydrolysis of LiPF6 in carbonate-based electrolytes for lithium-ion batteries and in aqueous media, J. Phys. Chem. C, 2018, 122, 8836–8842 CrossRef CAS.
- M. Liu, et al., Hydrolysis of LiPF6-containing electrolyte at high voltage, ACS Energy Lett., 2021, 6, 2096–2102 CrossRef CAS.
- Q. Zhao, et al., Upgrading Carbonate Electrolytes for Ultra-Stable Practical Lithium Metal Batteries, Angew. Chem., 2022, 134, e202116214 CrossRef.
- S. J. Hong, et al., Efficient scalable hydrothermal synthesis of MnO2 with controlled polymorphs and morphologies for enhanced battery cathodes, ACS Energy Lett., 2023, 8, 1744–1751 CrossRef CAS.
- J. Zheng, et al., Physical Orphaning versus Chemical Instability: Is Dendritic Electrodeposition of Li Fatal?, ACS Energy Lett., 2019, 4, 1349–1355 CrossRef CAS.
- J. Zheng, et al., Nonplanar electrode architectures for ultrahigh areal capacity batteries, ACS Energy Lett., 2018, 4, 271–275 CrossRef.
- E. G. Leggesse, K.-H. Tsau, Y.-T. Liu, S. Nachimuthu and J.-C. Jiang, Adsorption and decomposition of ethylene carbonate on LiMn2O4 cathode surface, Electrochim. Acta, 2016, 210, 61–70 CrossRef CAS.
- R. E. Warburton, H. Iddir, L. A. Curtiss and J. Greeley, Thermodynamic stability of low-and high-index spinel LiMn2O4 surface terminations, ACS Appl. Mater. Interfaces, 2016, 8, 11108–11121 CrossRef CAS PubMed.
- A. Singer, et al., Nucleation of dislocations and their dynamics in layered oxide cathode materials during battery charging, Nat. Energy, 2018, 3, 641–647 CrossRef CAS.
- A. Ulvestad, et al., Topological defect dynamics in operando battery nanoparticles, Science, 2015, 348, 1344–1347 CrossRef CAS PubMed.
- A. Stoppa, J. Hunger and R. Buchner, Conductivities of binary mixtures of ionic liquids with polar solvents, J. Chem. Eng. Data, 2009, 54, 472–479 CrossRef CAS.
- A. E. Gebala and M. M. Jones, The acid catalyzed hydrolysis of hexafluorophosphate, J. Inorg. Nucl. Chem., 1969, 31, 771–776 CrossRef CAS.
- S. J. Kuhn, Nitronium salts: I. New methods for the preparation of NO2+ BF4−, NO2+ PF6−, NO2+ AsF6−1, Can. J. Chem., 1962, 40, 1660–1663 CrossRef CAS.
- R. P. Swatloski, J. D. Holbrey and R. D. Rogers, Ionic liquids are not always green: hydrolysis of 1-butyl-3-methylimidazolium hexafluorophosphate, Green Chem., 2003, 5, 361–363 RSC.
- S. S. Zhang, K. Xu and T. R. Jow, Study of LiBF4 as an electrolyte salt for a Li-ion battery, J. Electrochem. Soc., 2002, 149, A586 CrossRef CAS.
- R. D. Rauh, T. F. Reise and S. B. Brummer, Efficiencies of cycling lithium on a lithium substrate in propylene carbonate, J. Electrochem. Soc., 1978, 125, 186 CrossRef CAS.
- K.-H. Chen, et al., Dead lithium: mass transport effects on voltage, capacity, and failure of lithium metal anodes, J. Mater. Chem. A, 2017, 5, 11671–11681 RSC.
- L. Suo, et al., Fluorine-donating electrolytes enable highly reversible 5-V-class Li metal batteries, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 1156–1161 CrossRef CAS PubMed.
- J. Zheng, et al., Regulating electrodeposition morphology of lithium: towards commercially relevant secondary Li metal batteries, Chem. Soc. Rev., 2020, 49, 2701–2750 RSC.
- H. Zhang, C. Shen, Y. Huang and Z. Liu, Spontaneously formation of SEI layers on lithium metal from LiFSI/DME and LiTFSI/DME electrolytes, Appl. Surf. Sci., 2021, 537, 147983 CrossRef CAS.
- Z. Shadike, et al., Identification of LiH and nanocrystalline LiF in the solid–electrolyte interphase of lithium metal anodes, Nat. Nanotechnol., 2021, 16, 549–554 CrossRef CAS PubMed.
- M. Nie and B. L. Lucht, Role of lithium salt on solid electrolyte interface (SEI) formation and structure in lithium ion batteries, J. Electrochem. Soc., 2014, 161, A1001 CrossRef CAS.
- W. Li, U.-H. Kim, A. Dolocan, Y.-K. Sun and A. Manthiram, Formation and inhibition of metallic lithium microstructures in lithium batteries driven by chemical crossover, ACS Nano, 2017, 11, 5853–5863 CrossRef CAS PubMed.
- J. Wandt, et al., Transition metal dissolution and deposition in Li-ion batteries investigated by operando X-ray absorption spectroscopy, J. Mater. Chem. A, 2016, 4, 18300–18305 RSC.
- C. Delacourt, et al., Effect of manganese contamination on the solid-electrolyte-interphase properties in Li-ion batteries, J. Electrochem. Soc., 2013, 160, A1099–A1107 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra08016a |
| This journal is © The Royal Society of Chemistry 2024 |