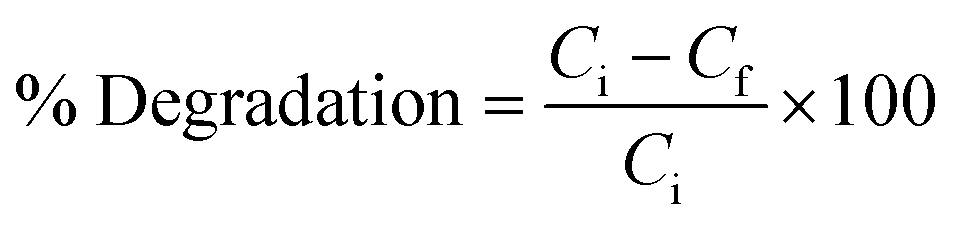Highly efficient sunlight-driven photo-adsorptive degradation of organic pollutants by green synthesized Z-scheme heterojunction CeO2@ZnO nanocomposite†
Received
21st August 2023
, Accepted 13th November 2023
First published on 13th November 2023
Abstract
Creating a high-efficiency heterojunction with enhanced photocatalytic properties is considered a promising approach to wastewater decontamination. Herein, Sapindus mukorossi seed extract was used to act as capping and reducing agent due to the presence of saponins and polyphenols during the synthesis of ZnO and CeO2 nanoparticles. Sharp PXRD peaks confirmed that the spherical nanocomposite had great crystallinity and purity. The CeO2@ZnO nanocomposite efficiently removes eriochrome black T (EBT) dye (98%) and endosulfan (ES) pesticide (96%). In addition to improved redox capacity, the heterojunction system exhibits quick transfer, long lifetime of photoinduced charge carriers, high-efficiency separation, and long-lived charge carriers. The band gap of ZnO observed was 3.1 eV and that of CeO2 was 2.8 eV which decreased after doping to 2.6 eV which showed the Z-scheme of CeO2@ZnO nanoparticles. The flow of electrons and holes followed the unique Z-scheme heterojunction mechanism between hierarchical ZnO and CeO2 which produced active radical species. First-order kinetics followed by initial Langmuir adsorption constituted the degradation process. From experiments using different radical quenchers (t-BuOH, p-BZQ, Na2EDTA), it was concluded that peroxide radical plays a significant role in the degradation of toxic EBT and ES. The green-fabricated nanocomposite also showed excellent efficiency in the degradation of ES and EBT pollutants in actual wastewater samples. LC-MS analysis confirmed the formation of safer metabolites after the degradation of both pollutants. This study offers a fresh and green methodology for building Z-scheme heterojunctions of modified ZnO in photocatalysis application.
Environmental significance
The present research article deals with synthesizing Z-scheme CeO2@ZnO nanocomposite semiconductors via an environmentally friendly green approach utilizing Sapindus mukorossi seed extract as a natural surfactant. This work utilized the synthesized nanocomposite, and its constituent nanoparticles, to remediate toxic organic pollutants from real wastewater samples under direct sunlight. The synthesized nanocomposite has excellent surface activity compared with the native nanoparticles. It is found to be reusable for up to six consecutive cycles without significant loss in activity, ensuring its sustainability for large-scale industrial applications, which is highly encouraging. Since this work is related to the generation of nanomaterials via a green approach, the work complements the green chemistry principles. Further, the synthesized nanomaterials of this work, again utilized for environment remediation under natural sunlight, show great significance for the environment.
|
1. Introduction
Significant issues are brought on by freshwater scarcity, environmental changes, and rapid industrialization changes, especially in rising nations.1 Due to the presence of hazardous colorants, substituted phenols, pesticides, toxic heavy metals and recalcitrant effluents generated by numerous businesses and human activities, almost one-third of surface drinking water is contaminated.2,3
A widely used organochlorine insecticide for managing pests in agriculture is endosulfan (ES), which has a broad spectrum of activity.4 Due to their potent endocrine disruptive effects, which cause human carcinogenicity, organ dysfunction, reproductive harm, and mutation, ES isomers (α- and β-, 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratio) have been categorized as persistent micro-pollutants.5 ES half-life in water varies depending on the pH, with values ranging from 5 weeks at pH 7.0 to 5 months at pH 5.5.6 It has been acknowledged that ES long-term persistence and bioaccumulation in the food chain are key contributors to its acute toxicity in both aquatic and terrestrial life forms.7 ES has proved to be a potent endocrine disruptor due to high solubility in lipids which causes reproductive damage, mutation, carcinogenicity, and organ dysfunction in living beings.8 As a result, substantial research has been focused on the speciation and mineralization of ES from aqueous sources. In various countries, the use of ES is restricted or banned, and the Stockholm Convention added to the list of persistent organic pollutants to eliminate the use of ES.7 Hence, the permissible limit of ES in drinking water has been fixed at 20 g L−1 (WHO) and 0.4 μg L−1 (Bureau of Indian Standards).9
:3 ratio) have been categorized as persistent micro-pollutants.5 ES half-life in water varies depending on the pH, with values ranging from 5 weeks at pH 7.0 to 5 months at pH 5.5.6 It has been acknowledged that ES long-term persistence and bioaccumulation in the food chain are key contributors to its acute toxicity in both aquatic and terrestrial life forms.7 ES has proved to be a potent endocrine disruptor due to high solubility in lipids which causes reproductive damage, mutation, carcinogenicity, and organ dysfunction in living beings.8 As a result, substantial research has been focused on the speciation and mineralization of ES from aqueous sources. In various countries, the use of ES is restricted or banned, and the Stockholm Convention added to the list of persistent organic pollutants to eliminate the use of ES.7 Hence, the permissible limit of ES in drinking water has been fixed at 20 g L−1 (WHO) and 0.4 μg L−1 (Bureau of Indian Standards).9
Another major class of pollutants is dyes used in manufacturing clothes, papers, carpets, leather goods, and other materials to give them vibrant colors. Around the world, the dye industry produces 800000 tonnes of synthetic dyes annually.10 Many businesses favor azo dyes above all other synthetic dyes because they offer a variety of colors, superior color fastness, and a lower price. Nearly 70% of the 10000 dyes utilized across diverse industries are azo dyes.11 Eriochrome black T (EBT) is a commonly used dye in the carpet and textile manufacturing industries that has a mono azo group (N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), giving wool or yarn for weaving a dark blue/black color.12 EBT is extensively used for silk textiles and in various institutional laboratories to identify the hardness of water (0.05–0.08 g as an indicator).13
N), giving wool or yarn for weaving a dark blue/black color.12 EBT is extensively used for silk textiles and in various institutional laboratories to identify the hardness of water (0.05–0.08 g as an indicator).13
The physicochemical properties of ES and EBT are mentioned in Table 1. In view of the toxicity of ES and EBT, it is necessary to remove both pollutants from environmental matrices by using cost-effective and environmentally friendly approaches. Table 2 lists various materials utilized for the removal of ES and EBT dye via various methodologies. An interesting area of study with prospective applications in the generation of renewable energy and environmental cleanup is the rational design of semiconductor heterostructure photocatalysts with broad spectrum response in the visible region.14 Zinc oxide (ZnO) has been intensively researched for photodegradation because of its noteworthy properties including low cost, nontoxicity, high photosensitivity, and redox potential. With a wide bandgap (Eg = 3.37 eV), the photocatalytic activity of ZnO is usually limited by inefficient exploitation of solar energy.15 Besides, the fast recombination of photoinduced electron–hole pairs is another issue to hinder the improvement of its photocatalytic activity.16 In comparison to a pure semiconductor, heterostructure materials exhibit enhanced optical absorption, simple charge transfer characteristics, and resistance to charge recombination.17 Many innovative heterostructure photocatalysts have been developed recently that exhibit outstanding activity towards liquid- and vapor-phase photodegradation of micropollutants using an appropriate preparative technique.18 Numerous binary heterostructure materials have been reported with various electron migration schemes, such as type-I, type-II, S-scheme, and Z-scheme heterostructure, to create innovative photocatalysts with improved optoelectronic properties.19 Particularly, it has been thought that creating a straight Z-scheme heterojunction is more efficient than creating a normal type-II heterojunction.20 The Z-scheme photocatalytic heterojunction achieves a high redox potential and efficient spatial separation of photoinduced charge carriers.21 ZnO stands out as a prominent metal oxide photocatalyst, alongside other diverse options such as SiO2, SnO2, TiO2 and Fe2O3. ZnO boasts unique physicochemical characteristics, including a substantial surface area, sensitivity, and stability.22–26 Because of the aforementioned benefits, ZnO-based Z-scheme heterojunctions have drawn more interest in the process of eliminating organic dyes from wastewater under light irradiation.
Table 1 Physicochemical property of ES and EBT
| Pollutant |
Formula |
M. wt. (g mol−1) |
CAS number |
M. pt (°C) |
Solubility in water (g L−1) |
Half life (days) |
| ES |
C9H6Cl6O4S |
422.92 |
1031-07-8 |
178–184 |
0.15 |
35–150 |
| EBT |
C20H12N3NaO7S |
461.381 |
1787-61-7 |
— |
0.5 |
— |
Table 2 Comparison data of ES and EBT degraded reported by other studies
| S. No. |
Name |
Material used |
Characterization |
Result |
Reference |
| 1 |
CdS/Bi20TiO32/Bi4Ti3O12 |
Endosulfan |
Photocatalytic degradation in visible light |
92.9% in 90 min |
69
|
| 2 |
Wood charcoal |
Endosulfan |
Adsorption |
Catalytic dose: 0.53 mg g−1; pollutant conc: 2 mg L−1; 93% in 24 h |
70
|
| 3 |
TiO2 |
Endosulfan |
Photocatalytic degradation |
pH: 7; pollutant conc: 7.5 ppm; 95% |
71
|
| 4 |
Carbon slurry |
Endosulfan |
Adsorption |
pH 6.5; catalytic dose 0.02 g L−1, 34.11 mg g−1 in 90 min |
72
|
| 5 |
Faujasite zeolites and protonic beta type zeolites |
Endosulfan |
Adsorption |
793.65 mg g−1 |
73
|
| 6 |
g-C3N4/Cu/TiO2 |
Endosulfan |
Photocatalytic degradation |
pH: 6.8; pollutant conc: 15 ppm; 60% in 80 min |
74
|
| 7 |
EBT (25 mg L−1) |
ZnO–CuHCF (15 mg) |
PXRD, FT-IR, FESEM-EDS, TEM, BET |
Degradation (97–99%) in sunlight, follow first order kinetics |
75
|
| 8 |
EBT |
β-Cyclodextrin coated Fe3O4 (10 ± 2 nm), (60 mg) |
PXRD, FTIR, XPS, HR-TEM, SAED, VSM |
Degradation (90%) with hydrogen peroxide |
76
|
| 9 |
EBT (20 mg L−1) |
Tridoped TiO2/CNT (0.1 g) |
FE-SEM, EDS, FT-IR, Raman, XRD, UV-visible |
66% removal observed by TOC due to degradation and followed first order kinetics |
77
|
| 10 |
EBT (0.08 mM) |
ZnO (100 mg) |
XRD, HR-TEM, SAED, STEM, EDS, UV–vis, BET |
Degradation (92%) in UV light |
78
|
| 11 |
EBT (20 mg L−1) |
NiO–ZnO on nanozeolite X (1 g L−1) |
XRD, UV/vis-DRS, FT-IR, TEM and BET |
Degradation (85%) in UV sunlight |
79
|
| 12 |
EBT (100 mg L−1) |
Eu-doped TiO2 (3 g L−1) |
XRD, SEM, FT-IR, UV-vis |
Degradation (98%) in sunlight |
80
|
| 13 |
Present study |
CeO2@ZnO (35 mg L−1) |
XRD, SEM, EDS, XPS, FTIR and UV/vis |
ES: 96% in 360 min |
— |
| EBT: 98% in 300 min |
Aside from the benefits of high stability, abundance, nontoxicity, and low cost, cerium oxide (CeO2), an active rare earth oxide, has been widely recognized as a good electron acceptor and a high oxygen storage carrier.27 Materials made of CeO2 are frequently used in oxygen sensors, solid oxide fuel cells, automobile three-way catalysts, and water gas shift processes.35,36 CeO2 also exhibits outstanding photocatalytic activities in water splitting for hydrogen evolution and in wastewater treatment.28,29 More significantly, prior reports indicate that the CeO2 energy level structure and the ZnO energy level structure fit rather well.30 Because of their compatible band structures, it is therefore reasonable to assume that an interfacial Z-scheme heterojunction between CeO2 and ZnO will be achieved.29 This is expected to result in a highly effective photocatalyst with a high redox capacity and efficient separation of photoinduced charge carriers. Despite the fact that certain CeO2@ZnO composites have been created for photocatalytic applications, it is still very difficult to fully understand and persuasively establish their photocatalytic mechanism in the removal of pollutant molecules. The synthesis of ZnO and CeO2 nanoparticles employs several methods and encounters challenges like rapid recombination of electron–hole pairs and reliance on UV exposure due to their wide bandgap which makes ZnO less favored for applications under sunlight.31–33 To overcome such problems, in this work, a facile, cost-effective and eco-friendly methodology is adapted to synthesize CeO2@ZnO nanocomposite by using seed extract of Sapindus mukorossi (as a natural material). The juice from the plant serves as reducing and capping agents for the synthesis of various types of nanoparticles.34,35 Because of their compatible band structures, it is therefore reasonable to expect that an interfacial Z-scheme heterojunction between CeO2 and ZnO would be achieved.36–38 This heterojunction is anticipated to be a highly efficient photocatalyst with effective separation of photoinduced charge carriers and high redox capacity.39 As per the literature, this is the first study of the green synthesis of CeO2@ZnO employed for the removal of ES pesticide and EBT dye. Literature revealed that visible light active CeO2@ZnO has been mainly synthesized by using the hydrothermal method and hybrid electrochemical–thermal technique. This is the first report related to the green synthesis methodology of CeO2@ZnO nanocomposite that is efficiently more reusable for the removal of organic pollutants as compared to chemically synthesized CeO2@ZnO. The photocatalytic effectiveness is increased due to the creation of a heterojunction between CeO2 and ZnO nanoparticles. Adopting a green synthesis methodology makes the synthesis more biocompatible, facile and cost-effective. For the maximum removal efficiency, various optimal parameters including amount of targeted pollutant, catalytic dose, source of irradiation, time, and pH were investigated. Green-synthesized CeO2@ZnO nanocomposite show enhancement in the active sites, improving the charge transport properties, along with charge separation efficiency when employed for the photocatalytic degradation of ES and EBT. Reusability and scavenger analysis were looked at in order to establish the durability of the synthesized nanocomposite, the significant role of free radicals during the degradation and real-life application.
2. Experimental methodology
2.1 Materials
Zinc nitrate, cerium nitrate, hydrochloric acid, and sodium hydroxide were purchased from Loba Chemie, India. ES and EBT were purchased from Merck. The whole experiment was conducted using deionized water as the solvent. The Sapindus mukorossi was purchased from a local market of Jalandhar, Punjab. The solar intensity and temperature were measured throughout the experiment using pyranometers and thermometers, respectively.
2.2 Green synthesis of ZnO, CeO2 and CeO2@ZnO nanocomposite
ZnO.
Zinc nitrate solution (0.1 M,100 mL) was prepared in distilled water, and 2 mL Sapindus mukorossi extract was added with regular stirring. To obtain pH 9, a solution of NaOH (0.1 M, 100 mL) was then added and stirred for half an hour at room temperature. After that, the solution was left alone until a larger yield of sediments had been formed. Filtered white precipitates were dried for five hours at 70 °C before being muffled at 500 °C.
CeO2.
An aqueous solution of cerium nitrate (0.1 M, 100 mL) was prepared, and 2 mL Sapindus mukorossi extract was added with continuous addition of sodium hydroxide (0.1 M, 100 mL) to reach the pH 9. The system was stirred for one hour continuously to get a homogeneous mixture and kept untouched for over 12 hours. The obtained precipitates were filtered, washed, dried, and muffled at 500 °C.
CeO2@ZnO.
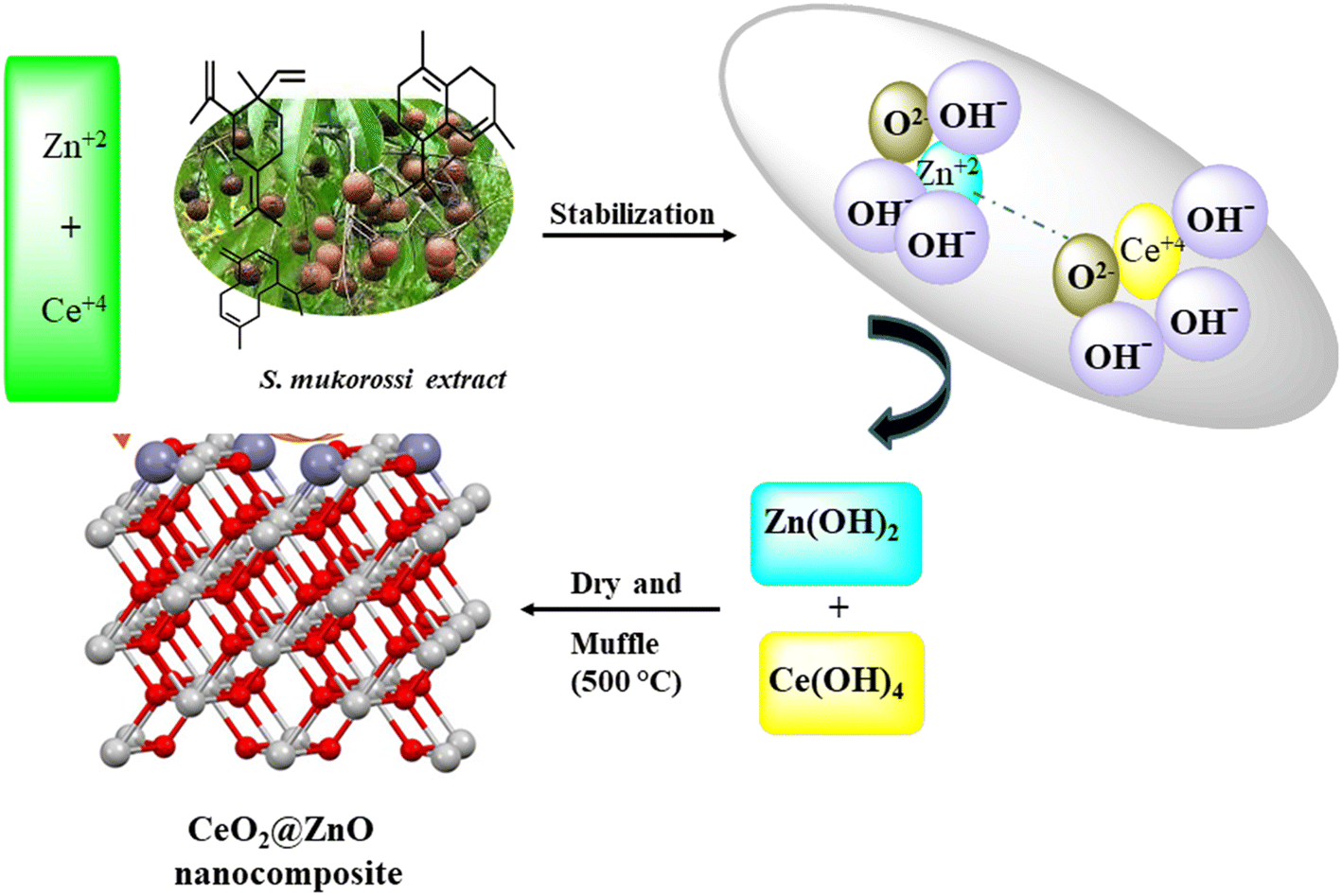
Nanocomposites of CeO2@ZnO were prepared by mixing equal molar concentrations of CeO2 and ZnO in a porcelain dish. The mixture was maintained at 500 °C for five hours in a muffle furnace. After cooling the mixture, the CeO2@ZnO nanocomposite crystals took on their yellowish color. Fig. 1 displays a schematic diagram of the nanocomposite synthesis.
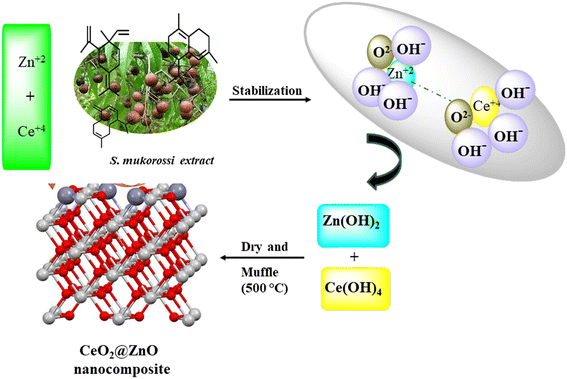 |
| | Fig. 1 Pictorial representation for the green synthesis of CeO2@ZnO nanocomposite by using S. mukorossi leaf extract. | |
2.3 Characterization techniques
Various instrumentation techniques were used for the characterization of the synthesized nanoparticles and degradation studies: powder X-ray diffraction (Panalytical X Pert Pro), Fourier transform infrared spectrophotometry (Perkin Elmer), field emission scanning electron microscopy (Nova Nano FE-SEM 450 (FEI)) of gold-coated samples with energy dispersive X-ray spectroscopy (EDS), X-ray photoelectron spectroscopy (ESCA+ Omicron Nanotechnology), UV spectrophotometry (Agilent Carry 100), zeta potential measurements (Nano ZSP (ZEN 5600)) and transmission electron microscopy (Tecnai G2 20 S-TWIN [FEI]). In addition, a CPA-225D Sartorius analytical balance was used for weighing purpose and a LABWAN-PH-61WW instrument was used for the preparation of pH solutions.
2.4 Setup for the photocatalytic degradation of ES and EBT from spiked water by using a nanocomposite of CeO2@ZnO
To remove toxic ES and EBT dye from spiked water under sunlight irradiation, the photoactivity capabilities of the CeO2@ZnO heterojunction and parent nanoparticles (ZnO and CeO2) were investigated. The amount of toxic pollutants ES (50–90 mg L−1) and EBT (2–10 mg L−1), photocatalytic dosage (25–45 mg), pH (3 to 11), and duration were optimized to achieve maximal breakdown. Throughout the experiment of the present research work, borax (0.1 M NaOH + 0.025 M borax) and citrate buffer (0.1 M trisodium citrate + 0.1 M citric acid) were used to maintain the pH of the solutions. Simulated samples were exposed to sunlight under optimal conditions for six hours (11 am to 5 pm). The solar intensity and temperature were measured throughout the experiment using pyranometers and thermometers, respectively. The average ambient conditions that were observed were 420 ± 176 lux and 25 ± 1.2, respectively. The reaction mixtures were also examined in complete darkness to assess the impact of irradiation sources. Blank samples were also examined for in-depth study of the CeO2@ZnO removal efficiency. Eqn (1), where Ci is the pollutant beginning concentration and Cf the final concentration, was used to compute the synthetic photocatalysts' percent degradation efficiency:
3. Characterization of photocatalyst
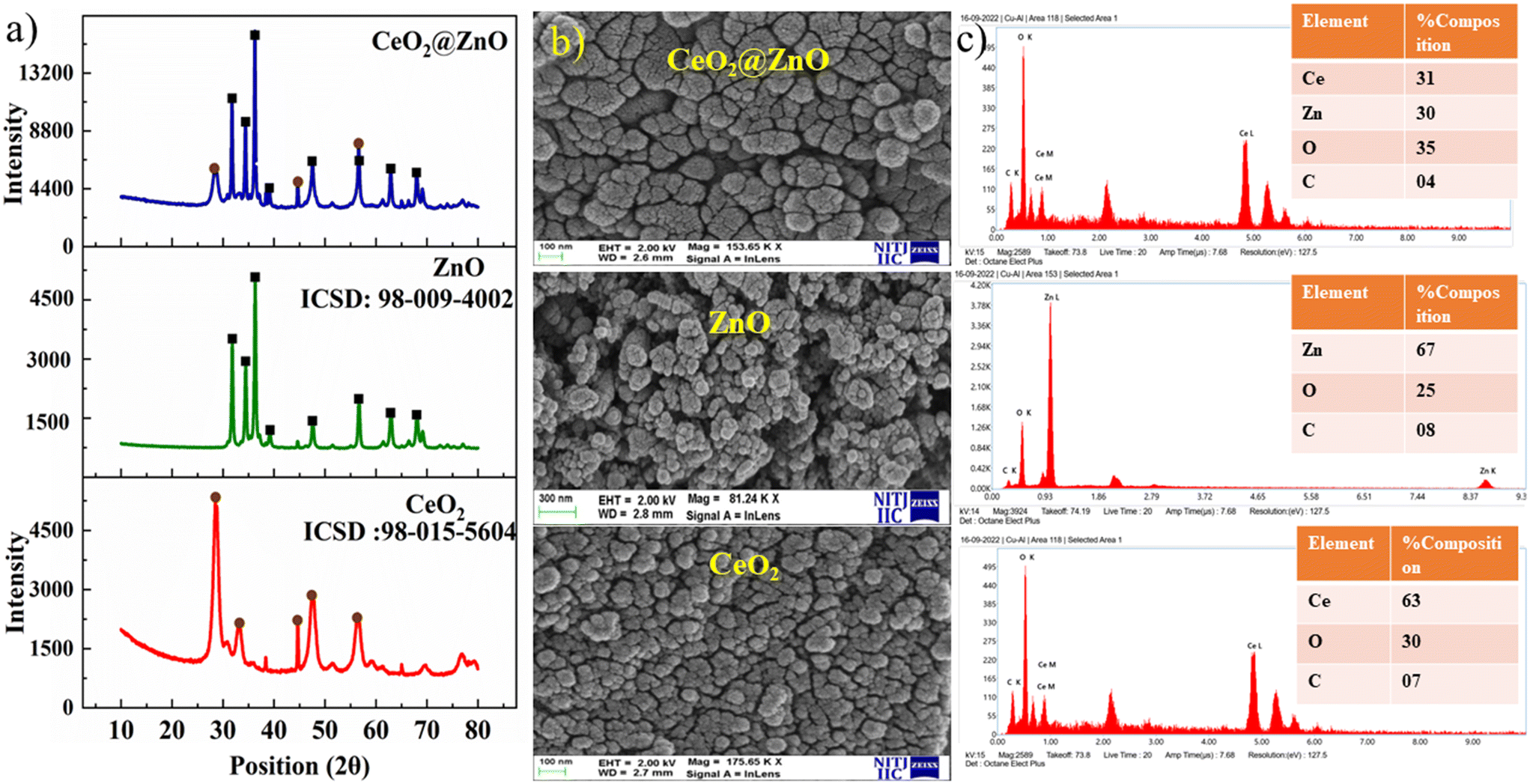
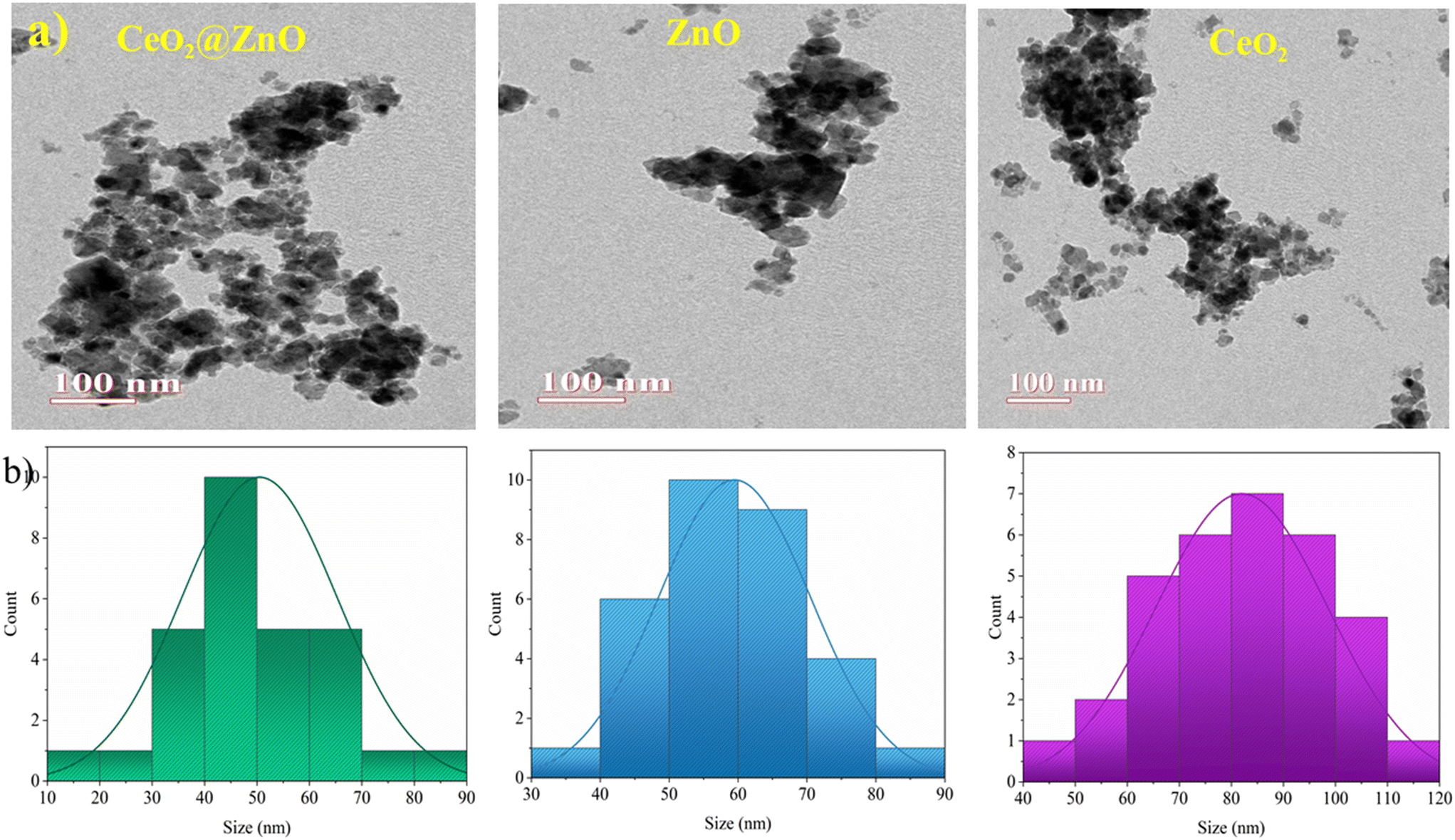
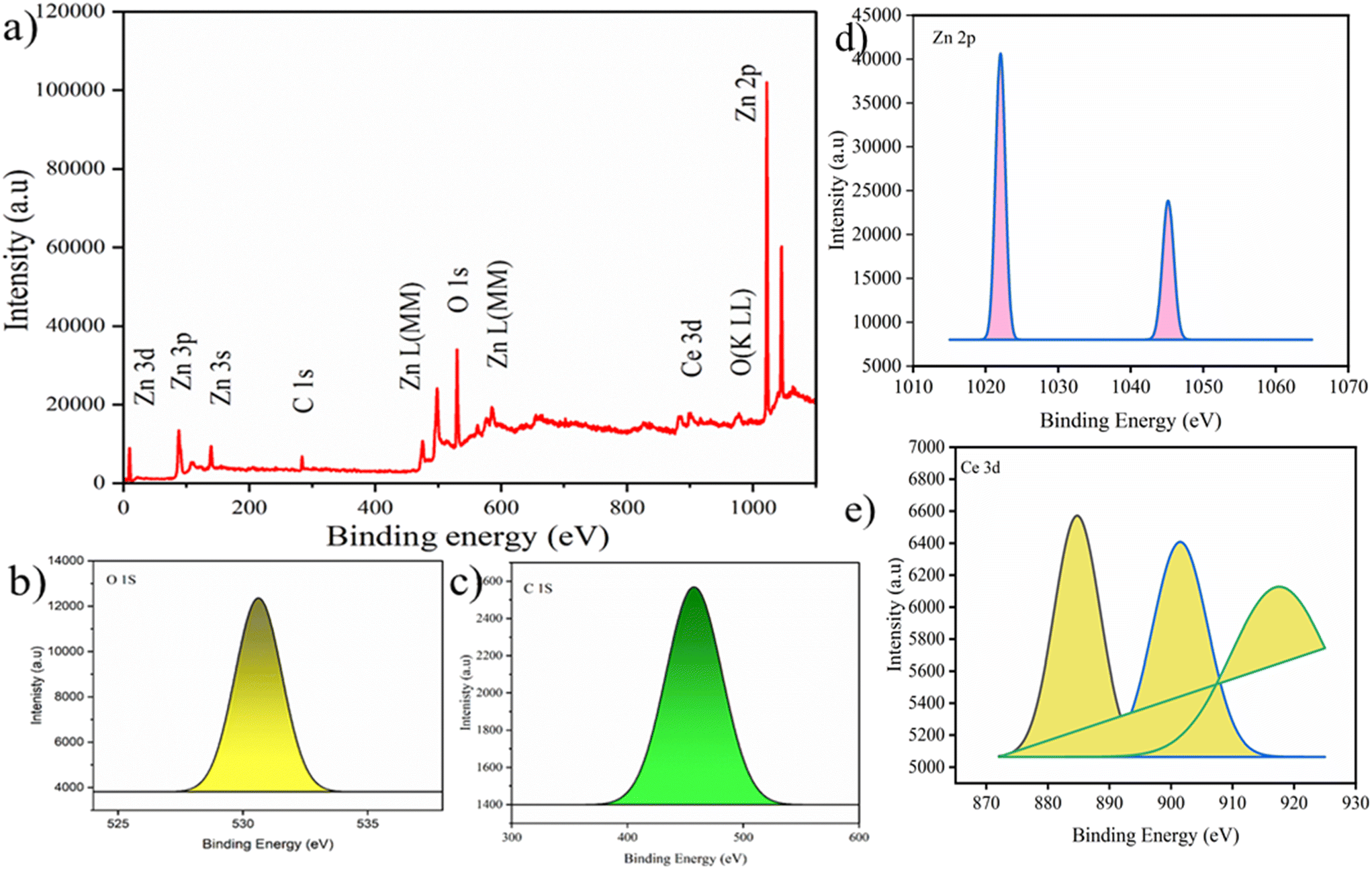
The structural characteristics and phase composition of the nanocatalysts were examined using powder X-ray diffraction (PXRD), field emission scanning electron microscopy (FE-SEM), and X-ray photoelectron spectroscopy (XPS) analyses. Hexagonal ZnO showed P63mc space group with lattice parameters a = b = 0.32 nm; c = 0.52 nm and α = β = 90°; γ = 120° with ICSD card number 98-09-4002. The density and volume of ZnO cells were calculated at 5.6 g cm−3 and 47 × 106 pm3. ZnO showed a maximum intensity peak at an angle 36.29° (d spacing: 0.24 nm; Miller indices: (011)) followed by 31.83° (d spacing: 0.28 nm; Miller indices: (010)) and 34.47° (d spacing: 0.26; Miller indices: (002)).40,41 No additional peaks confirmed the purity, highly crystalline nature, and secondary phase.42 Meanwhile, CeO2 showed cubic phase with ICSD card number 98-015-5604 having face group Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m with lattice parameters a = b = c = 0.54 nm and α = β = γ = 90°. The density and volume of the CeO2 cell were calculated at 7.2 g cm−3 and 159 × 106 pm3. The maximum intensity peaks were observed at angles 28. 59°, 47.71°, and 56.53° with Miller indices (111), (022), and (113), respectively (Fig. 2a). In the XRD pattern of CeO2@ZnO nanocomposite, a weak peak at 28.5° that corresponds to the Miller index (111) of the cubic geometry CeO2 component can also be seen. The diffraction peak's gradually rising intensity indicates that the composite CeO2 content has increased. The XRD patterns show that ZnO and CeO2 were successfully incorporated.22 The common peaks of both parent materials (ZnO and CeO2) in the CeO2@ZnO pattern confirmed the effective doping. The ratio of CeO2 and ZnO in CeO2@ZnO was 3:1, determined by using high-score software. In CeO2@ZnO nanocomposite, greater molecular mass nanoparticles typically cover most of the area with determined symmetry. In the FT-IR spectrum of CeO2@ZnO nanocomposite, the band attributed to the stretching vibration of –OH due to absorption of water molecules was observed at 3419 cm−1. The strong absorption stretching vibration peak at 435 cm−1 corresponds to Sn–O–Zn. The shifting of peaks observed in the CeO2@ZnO nanocomposite spectrum as compared to spectra of parent species (ZnO and CeO2) confirmed the effective doping of both parent nanomaterials (Fig. S1†). Spherical particles of ZnO and CeO2 were observed in FE-SEM images while the CeO2@ZnO nanocomposite showed distorted morphology (Fig. 2b). The EDS spectrum of CeO2@ZnO nanocomposite (Ce: 31%, Zn: 30%, O: 35%, C: 4%) confirmed the maximum percentage of CeO2 as compared to ZnO. Data for the parent nanoparticles ZnO (Zn: 67%, O: 25%, C: 8%) and CeO2 (Ce: 63%, O: 30%, C: 7%) were also obtained (Fig. 2c). The presence of phytochemicals in the plant extract used during the synthesis was found on the surface of nanomaterials, which gives a significant contribution for the reduction phenomenon and provided more stability to the CeO2@ZnO nanocomposite. The presence of carbon and a higher percentage of oxygen in the nanomaterial spectra may be due to presence of phytochemicals found in plant extracts. The nanoparticle EDS data exhibited good agreement with the data of PXRD in that the CeO2 contained in the CeO2@ZnO nanocomposite was present at a greater level than ZnO. TEM analysis was used for in-depth study related to morphology and internal structure of the nanocatalysts. Spherical geometry was observed for CeO2, ZnO, and CeO2@ZnO nanocomposite in TEM images. The average particle size of CeO2 (60–100 nm), ZnO (50–70 nm), and CeO2@ZnO (40–50 nm) was calculated by using ImageJ software (Fig. 3). In the XPS survey spectrum of the CeO2@ZnO nanocomposite, all the constituent elements were confirmed by the characteristic peaks of each element (Fig. 4). The XPS spectrum of the CeO2@ZnO nanocomposite revealed the presence of the scoring levels C (1s), Zn (2p), O (1s) and Ce (3d). The two peaks, which have binding energies of 1021.2 and 1044.3 eV, respectively, show the interaction at the interface between CeO2 and ZnO.22 The peaks due to Ce4+ are confirmed by the XPS spectrum of Ce (3d). The XPS finding showed good agreement with PXRD as well as EDS results in that CeO2 was present in superior amount as compared to ZnO in CeO2@ZnO nanomaterials. It has been documented in the literature that adding CeO2 nanoparticles to hierarchical ZnO alters the chemical state of surface Ce ions. According to those findings, ZnO and CeO2 formed a heterojunction as a result of electrons moving from ZnO to CeO2. The band gap of the produced CeO2@ZnO nanomaterials was estimated using UV-visible reflectance spectra. The assumption behind the Tauc equation is that energy is dependent on the absorption coefficient (α), described in the ESI.†42 The results demonstrated that the CeO2@ZnO nanocomposite showed a band gap of 2.6 eV, while that of parent nanoparticles ZnO was 3.1 eV and CeO2 was 2.8 eV (Fig. S2†). Because of the synergistic effects and variable changes in adsorption caused by doping, the band gap is shifted in the visible region. The larger band gap of ZnO restricted its environmental application in the visible region.43 The energy difference between valence band (VB) and conduction band (CB) of ZnO nanoparticles may lead to absorption of more visible light and exhibiting enhanced photocatalytic activity as compared to ZnO doped with CeO2. Literature revealed the electrical structures of ZnO and CeO2; density functional theory (DFT) computations were performed that showed, particularly, the O 2p and Zn 3d orbitals dominate the VB of ZnO while the Zn 3s and 3p orbitals dominate the CB. The O 2p orbital dominates the VB of CeO2, while the Ce 4f orbital dominates the CB. More significantly, it is clear that ZnO has a band gap of 3.08 eV and is a direct-gap semiconductor; CeO2 is an indirect-gap semiconductor with a 2.77 eV estimated band gap. Because of the limitations of DFT calculations, the calculated band gap is slightly less than the measured data.22 The wavelength increases when the band gap narrows due to strain.43 A similar change was also observed in N-Bi2O3@SnO2, ZnS@UF (1.8 eV), ZnO@CdS (1.67 eV), and ZnO@UF (1.3 eV) after doping.42–45 The band gap shrinks due to strain creation during doping, increasing catalyst activity.46 Gas isotherms of BET were used to evaluate the surface area and properties of the CeO2@ZnO nanohybrid. The nitrogen adsorption–desorption isotherm revealed that the CeO2@ZnO nanohybrid (80 m2 g−1) had a higher surface area than ZnO (20 m2 g−1) and CeO2 (40 m2 g−1) (Fig. S3†). Due to doping between ZnO and CeO2 nanoparticles, surface area, dissolving rate, and porosity increased in the CeO2@ZnO nanohybrid suggesting that the photocatalytic activity increases in proportion to the specific surface area of semiconductor photocatalysts. The hysteresis loop corresponded to the type IV category, indicating that the synthesised material had a mesoporous structure having pore size for CeO2@ZnO nanocomposite of 1.3 nm, CeO2 of 1.2 nm and ZnO of 0.98 nm. Furthermore, more surface area means greater number of active sites for adsorption of pollutants on the surface of the catalyst resulting in a greater percentage removal of pollutants.47 It indicates that the electron–hole recombination rate is much higher than the surface reaction rate and the recombination rate is presumed to be almost the same for each photocatalyst. Hence, the larger surface area of the CeO2@ZnO nanohybrid leads to strong photoactivity. To know the stability and charge of the material, zeta potential analysis was employed.48 The zeta potential values for CeO2@ZnO (−18.0 mV), ZnO (−10.0 mV), and CeO2 (−12.0 mV) were determined (Fig. S4†). The zeta potential was measured at neutral pH ∼ 7, and the result becomes more negative when CeO2 is doped with ZnO. The neutral nanoparticles' charged surface and the nanocomposite's greater negative zeta potential values revealed a phenolic group from plant juice that is anionically capped.49 The more negative value of the particle size analysis results in the probability of lesser accumulation and stabilization of nanoparticles.46–48 Similar results were also reported in the literature for N-Bi2O3@SnO2, ZnS@UF, ZnO@CdS and ZnO@UF after doping.42–45
m with lattice parameters a = b = c = 0.54 nm and α = β = γ = 90°. The density and volume of the CeO2 cell were calculated at 7.2 g cm−3 and 159 × 106 pm3. The maximum intensity peaks were observed at angles 28. 59°, 47.71°, and 56.53° with Miller indices (111), (022), and (113), respectively (Fig. 2a). In the XRD pattern of CeO2@ZnO nanocomposite, a weak peak at 28.5° that corresponds to the Miller index (111) of the cubic geometry CeO2 component can also be seen. The diffraction peak's gradually rising intensity indicates that the composite CeO2 content has increased. The XRD patterns show that ZnO and CeO2 were successfully incorporated.22 The common peaks of both parent materials (ZnO and CeO2) in the CeO2@ZnO pattern confirmed the effective doping. The ratio of CeO2 and ZnO in CeO2@ZnO was 3:1, determined by using high-score software. In CeO2@ZnO nanocomposite, greater molecular mass nanoparticles typically cover most of the area with determined symmetry. In the FT-IR spectrum of CeO2@ZnO nanocomposite, the band attributed to the stretching vibration of –OH due to absorption of water molecules was observed at 3419 cm−1. The strong absorption stretching vibration peak at 435 cm−1 corresponds to Sn–O–Zn. The shifting of peaks observed in the CeO2@ZnO nanocomposite spectrum as compared to spectra of parent species (ZnO and CeO2) confirmed the effective doping of both parent nanomaterials (Fig. S1†). Spherical particles of ZnO and CeO2 were observed in FE-SEM images while the CeO2@ZnO nanocomposite showed distorted morphology (Fig. 2b). The EDS spectrum of CeO2@ZnO nanocomposite (Ce: 31%, Zn: 30%, O: 35%, C: 4%) confirmed the maximum percentage of CeO2 as compared to ZnO. Data for the parent nanoparticles ZnO (Zn: 67%, O: 25%, C: 8%) and CeO2 (Ce: 63%, O: 30%, C: 7%) were also obtained (Fig. 2c). The presence of phytochemicals in the plant extract used during the synthesis was found on the surface of nanomaterials, which gives a significant contribution for the reduction phenomenon and provided more stability to the CeO2@ZnO nanocomposite. The presence of carbon and a higher percentage of oxygen in the nanomaterial spectra may be due to presence of phytochemicals found in plant extracts. The nanoparticle EDS data exhibited good agreement with the data of PXRD in that the CeO2 contained in the CeO2@ZnO nanocomposite was present at a greater level than ZnO. TEM analysis was used for in-depth study related to morphology and internal structure of the nanocatalysts. Spherical geometry was observed for CeO2, ZnO, and CeO2@ZnO nanocomposite in TEM images. The average particle size of CeO2 (60–100 nm), ZnO (50–70 nm), and CeO2@ZnO (40–50 nm) was calculated by using ImageJ software (Fig. 3). In the XPS survey spectrum of the CeO2@ZnO nanocomposite, all the constituent elements were confirmed by the characteristic peaks of each element (Fig. 4). The XPS spectrum of the CeO2@ZnO nanocomposite revealed the presence of the scoring levels C (1s), Zn (2p), O (1s) and Ce (3d). The two peaks, which have binding energies of 1021.2 and 1044.3 eV, respectively, show the interaction at the interface between CeO2 and ZnO.22 The peaks due to Ce4+ are confirmed by the XPS spectrum of Ce (3d). The XPS finding showed good agreement with PXRD as well as EDS results in that CeO2 was present in superior amount as compared to ZnO in CeO2@ZnO nanomaterials. It has been documented in the literature that adding CeO2 nanoparticles to hierarchical ZnO alters the chemical state of surface Ce ions. According to those findings, ZnO and CeO2 formed a heterojunction as a result of electrons moving from ZnO to CeO2. The band gap of the produced CeO2@ZnO nanomaterials was estimated using UV-visible reflectance spectra. The assumption behind the Tauc equation is that energy is dependent on the absorption coefficient (α), described in the ESI.†42 The results demonstrated that the CeO2@ZnO nanocomposite showed a band gap of 2.6 eV, while that of parent nanoparticles ZnO was 3.1 eV and CeO2 was 2.8 eV (Fig. S2†). Because of the synergistic effects and variable changes in adsorption caused by doping, the band gap is shifted in the visible region. The larger band gap of ZnO restricted its environmental application in the visible region.43 The energy difference between valence band (VB) and conduction band (CB) of ZnO nanoparticles may lead to absorption of more visible light and exhibiting enhanced photocatalytic activity as compared to ZnO doped with CeO2. Literature revealed the electrical structures of ZnO and CeO2; density functional theory (DFT) computations were performed that showed, particularly, the O 2p and Zn 3d orbitals dominate the VB of ZnO while the Zn 3s and 3p orbitals dominate the CB. The O 2p orbital dominates the VB of CeO2, while the Ce 4f orbital dominates the CB. More significantly, it is clear that ZnO has a band gap of 3.08 eV and is a direct-gap semiconductor; CeO2 is an indirect-gap semiconductor with a 2.77 eV estimated band gap. Because of the limitations of DFT calculations, the calculated band gap is slightly less than the measured data.22 The wavelength increases when the band gap narrows due to strain.43 A similar change was also observed in N-Bi2O3@SnO2, ZnS@UF (1.8 eV), ZnO@CdS (1.67 eV), and ZnO@UF (1.3 eV) after doping.42–45 The band gap shrinks due to strain creation during doping, increasing catalyst activity.46 Gas isotherms of BET were used to evaluate the surface area and properties of the CeO2@ZnO nanohybrid. The nitrogen adsorption–desorption isotherm revealed that the CeO2@ZnO nanohybrid (80 m2 g−1) had a higher surface area than ZnO (20 m2 g−1) and CeO2 (40 m2 g−1) (Fig. S3†). Due to doping between ZnO and CeO2 nanoparticles, surface area, dissolving rate, and porosity increased in the CeO2@ZnO nanohybrid suggesting that the photocatalytic activity increases in proportion to the specific surface area of semiconductor photocatalysts. The hysteresis loop corresponded to the type IV category, indicating that the synthesised material had a mesoporous structure having pore size for CeO2@ZnO nanocomposite of 1.3 nm, CeO2 of 1.2 nm and ZnO of 0.98 nm. Furthermore, more surface area means greater number of active sites for adsorption of pollutants on the surface of the catalyst resulting in a greater percentage removal of pollutants.47 It indicates that the electron–hole recombination rate is much higher than the surface reaction rate and the recombination rate is presumed to be almost the same for each photocatalyst. Hence, the larger surface area of the CeO2@ZnO nanohybrid leads to strong photoactivity. To know the stability and charge of the material, zeta potential analysis was employed.48 The zeta potential values for CeO2@ZnO (−18.0 mV), ZnO (−10.0 mV), and CeO2 (−12.0 mV) were determined (Fig. S4†). The zeta potential was measured at neutral pH ∼ 7, and the result becomes more negative when CeO2 is doped with ZnO. The neutral nanoparticles' charged surface and the nanocomposite's greater negative zeta potential values revealed a phenolic group from plant juice that is anionically capped.49 The more negative value of the particle size analysis results in the probability of lesser accumulation and stabilization of nanoparticles.46–48 Similar results were also reported in the literature for N-Bi2O3@SnO2, ZnS@UF, ZnO@CdS and ZnO@UF after doping.42–45
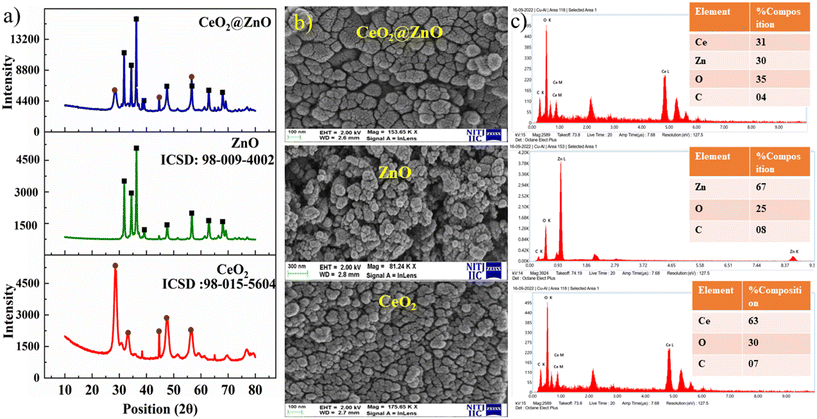 |
| | Fig. 2 (a) Powder X-ray diffraction (PXRD) patterns, (b) field emission scanning electron microscopy (FE-SEM) images and (c) elemental detection spectroscopy (EDS) of CeO2@ZnO nanocomposite, ZnO and CeO2. | |
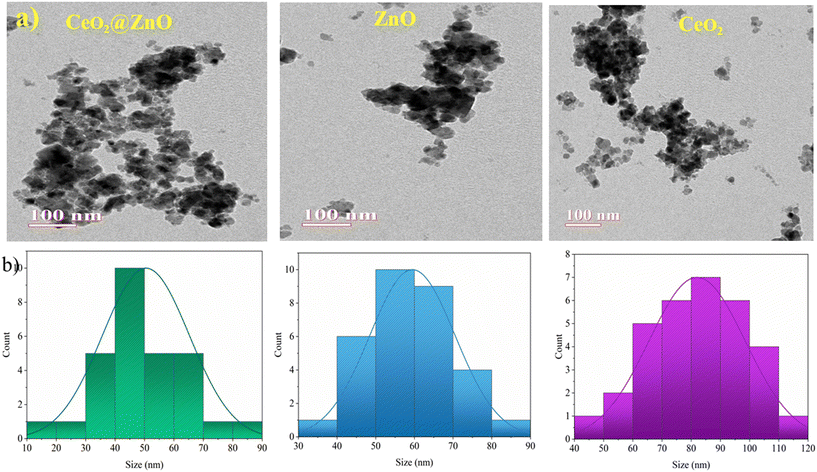 |
| | Fig. 3 (a) Transmission electron microscopy images and (b) size histograms of CeO2@ZnO nanocomposite and native CeO2 nanoparticles as well as ZnO. | |
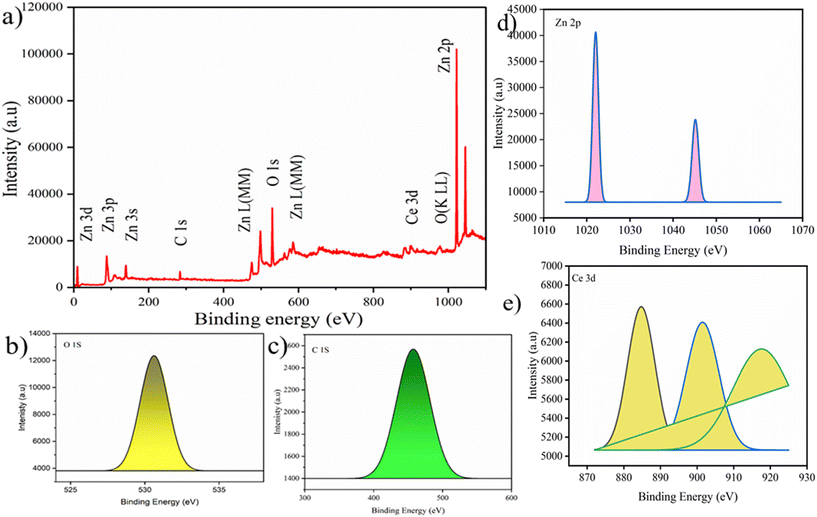 |
| | Fig. 4 (a) Survey surface scan and (b–e) elemental scan of XPS of CeO2@ZnO nanocomposite. | |
4. Procedure for the photocatalytic degradation of ES and EBT
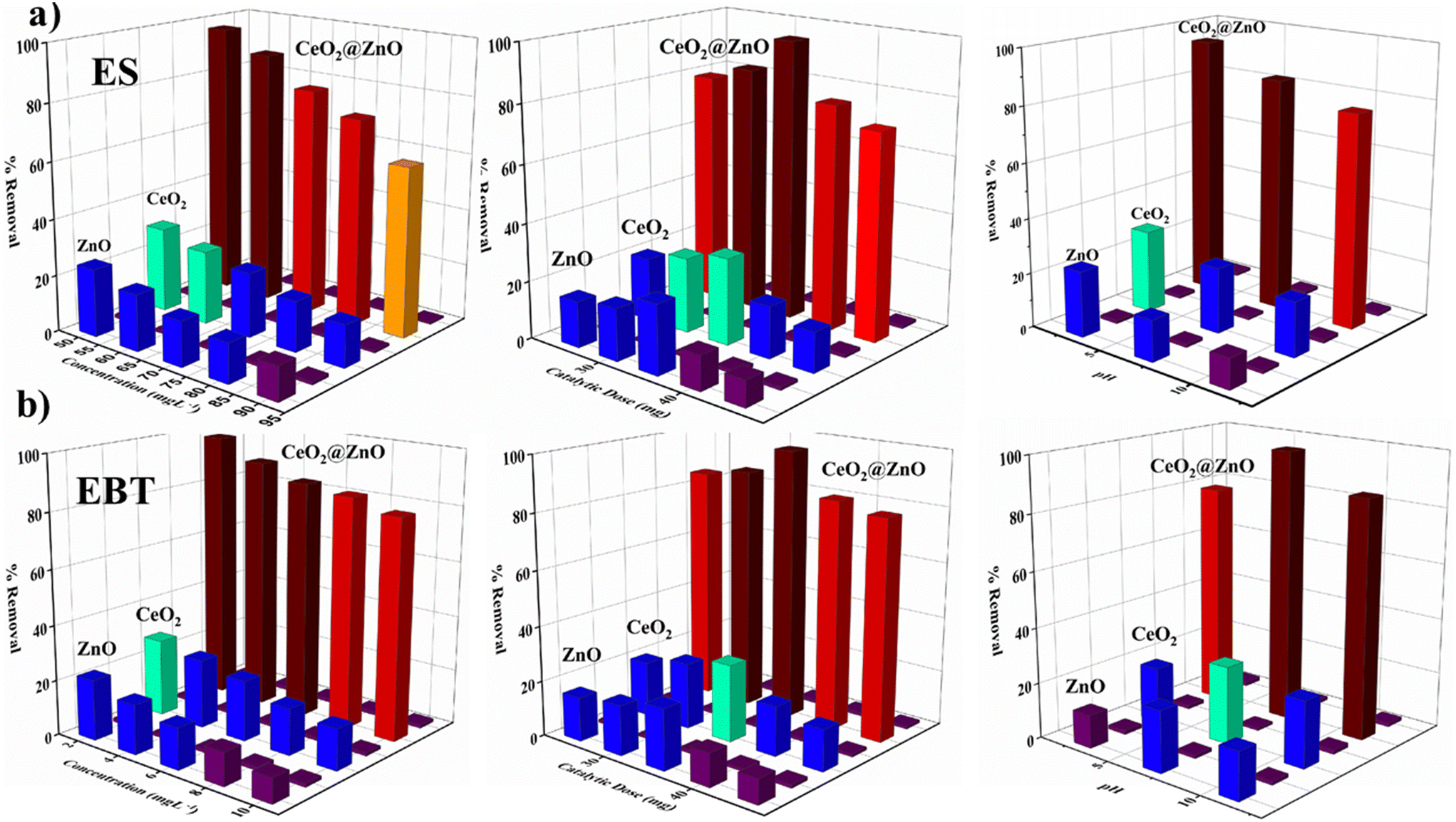
In the presence of natural sunlight, the photocatalytic efficiency of CeO2@ZnO and parent nanoparticles (CeO2 and ZnO) was investigated against harmful ES and EBT dye in spiked water. The elimination of ES and EBT was studied by varying the concentration of ES (50–90 mg L−1) and EBT (2–10 mg L−1), nanocatalyst amount (25–45 mg), and pH (3–11) (Fig. 5). It is significant to note that all variables were held constant except for one. Variable concentration for ES and EBT was selected because a spectrophotometer was used to evaluate their linear absorbance and occurrence in environment matrices. The maximum degradation of both pesticide ES (96%) and dye EBT (98%) by employing photocatalyst (35 mg) was achieved at a starting concentration of toxic pesticide of 50 mg L−1 and 2 mg L−1 at neutral pH for EBT and acidic pH (3) for ES under sunlight irradiation. As the concentration of ES and EBT increased, the degradation percentage declined with all nanocatalysts. For the removal of more pollutant molecules of ES and EBT, more active sites of the photocatalyst are required. But the limited/fixed amount of catalysts cannot remove so many pollutant molecules. Another primary reason for the decline in degradation is due to the overload of contaminated molecules of ES and EBT in a small volume of the glass vials that reduced the path length of photons entering the solution, thus degradation further decreasing with an increase in ES and EBT. Additionally, more active species were required for the extra pollutant molecules, but it was anticipated that the amount of free radicals produced would remain constant for a given catalytic dose, sunlight intensity, and time duration of sunlight irradiation.50 Additionally, a portion of the active radicals produced during the breakdown of the ES and EBT metabolites may be consumed. As a result, there were not enough active radicals in the specific reaction mixture to effectively destroy a lot of contaminants. The nanocomposite's considerable number of surface active sites justified a faster rate of pollutant degradation than increasing catalyst concentrations up to the optimum scale. Later, a progressive decline in the curve with an increase in the amount of catalysts was thought to be caused by the molecules colliding with the nano photocatalyst and becoming inactive in a small vessel.51 Additionally, the buildup of nanoparticles and the screening effect prevented light rays from entering the contaminated fluid. Due to the change in surface characteristics of the nanocomposite at variable pH, it is essential to study the pH effect. Maximum degradation was achieved at neutral pH for EBT and acidic pH (3) for ES. Pesticide sorption and dechlorination took place at the original pH of the solution. While basic pH causes the nanocatalyst to passivate, acidic pH accelerates oxidation.52 The production of secondary reductants on the catalyst's surface, which prevents pesticide interaction with its active sites, caused the removal capacity of the nanocomposite to drop at pH levels over 4. The nanocatalyst passivating coating is removed by a weak acidic environment, which activates the nanocatalyst for the dichlorination of ES. Additionally, at pH 4 or extremely low pH, a higher concentration of H+ ions harms carbon, and metal oxidation slows the breakdown of pesticides. While in the case of the azo anionic EBT dye, active species were present in equal amounts and canceled one another out in neutral conditions. A large concentration of active species adsorb on the photocatalyst surface and occupy its active sites in alkaline conditions. The irradiation source (sunlight) proved to be a significant parameter in photocatalytic degradation, due to which optimized parameters were also studied in sunlight for 6 hours. The parameters relating to sunlight have already been mentioned in section 3. However, a glass vial with optimized pollutant and catalytic dosage parameters was covered with black tape and placed in a black sealed chamber box for 5.5 hours. The removal of both contaminants (ES and EBT) by photocatalyst was found to be higher in natural sunlight (96–98%) followed by bulb (20–28%) and the dark (10–19%) as shown Fig. S5.† The dark blue gets decolorized within 300 minutes, showing effective degradation of EBT dye. The results showed that the nanocomposite had significant photoactivity due to its reduced bandgap, allowing easy electron excitation over the absorption phenomenon of a suitable quantity of energy from sunlight. Furthermore, the lower optical power pushed the spectra to the visible region, resulting in higher pollutant breakdown under sunlight.
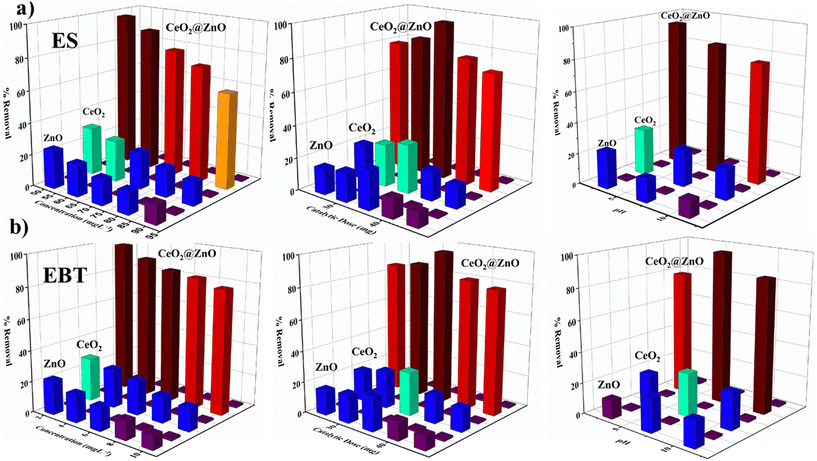 |
| | Fig. 5 Concentration-, catalytic dose- and pH-dependent removal of (a) endosulfan (ES) and (b) eriochrome black T (EBT) dye by using synthesized CeO2@ZnO nanocomposite, CeO2 and ZnO. | |
5. Investigation of the mechanism of photocatalytic degradation of ES and EBT
The removal process typically entails two steps: initial pollutant adsorption on nanocatalyst surfaces, followed by photocatalytic destruction by oxidative species created by the semiconducting properties of the nanocatalyst.53 Adsorption typically goes through four stages: medium resistance, particle diffusion, mass transfer, and adherence to the photocatalyst surface.54 The removal process consisted of mass transfer, which was limited by diffusion resistance, and adsorption on the active sites of the nanocatalyst. Adsorption, light absorption, and charge transfer are the processes in photocatalytic degradation that result in dynamic species like hydroxyl radicals.55 The adsorption capacity of nanoparticles was enhanced due to the high surface area, which also made it possible to transfer ES and EBT molecules to the active sites quickly.
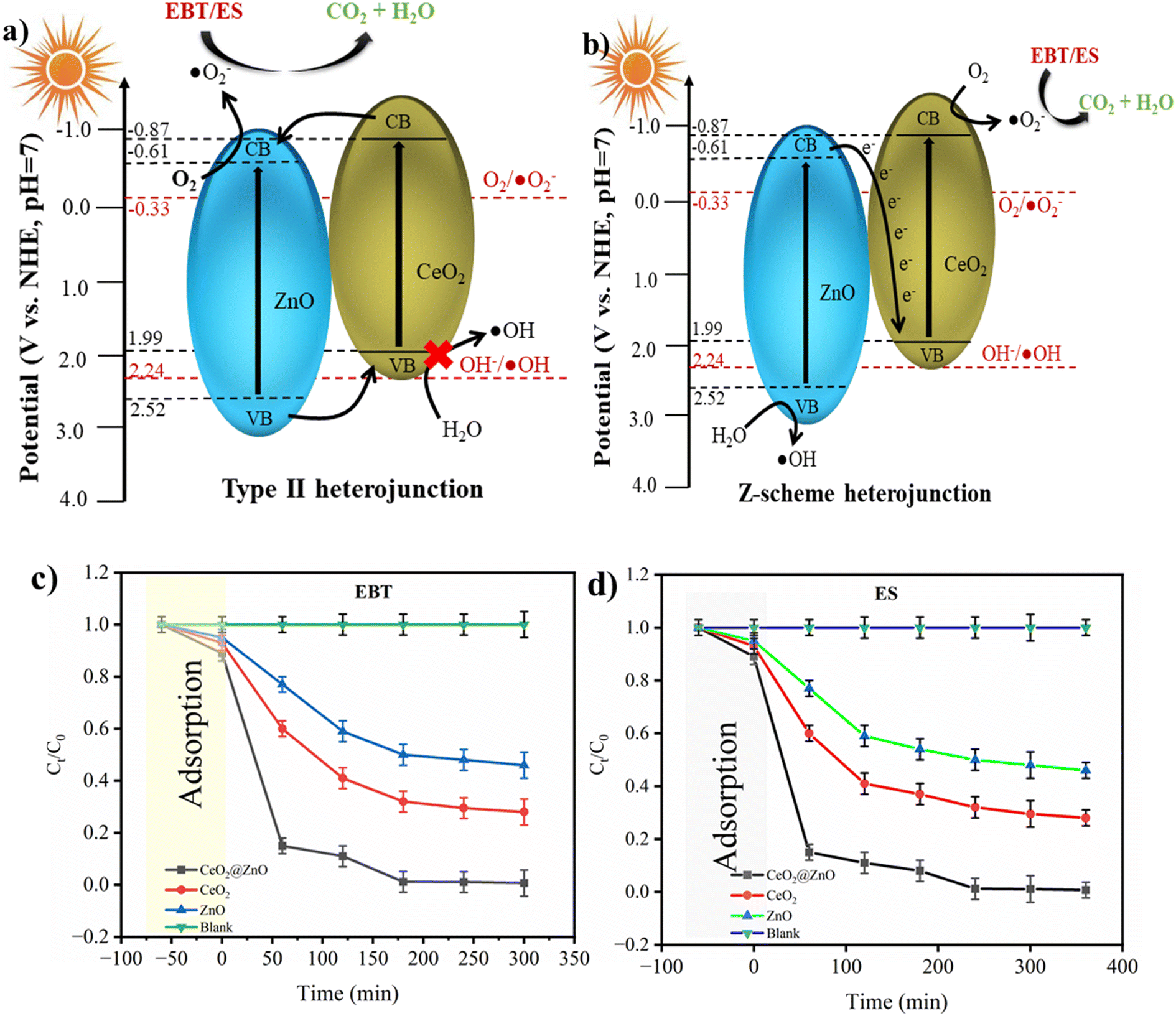
The band gap energies of the photocatalyst's VB and CB are equal to or higher than that of the photons that are absorbed by the photocatalytic materials.56 Photon absorption results in the separation of charge by causing the excitation of electrons from the VB to the CB and creating positive h+ in the VB.56 A sizable amount of photons was produced when sunlight was present, and the hybrid connection would determine the electron and hole life spans or charge separation in the CB and VB, respectively. The literature revealed that the flat-band potential (Vfb) of CeO2 and ZnO is −0.87 and −0.61 V, respectively.59 It is known that the CB bottoms of n-type semiconductor Vfb positions are roughly equivalent. Therefore, it is estimated that the CB bottoms of CeO2 and ZnO are −0.87 and −0.61 eV, respectively. The thermodynamic restrictions state that CeO2 can only form ˙O2− radicals, while ZnO can produce hydroxyl and ˙O2− radicals given its acceptable band edges.57
Basically, two types of photoinduced charge carrier migrations are produced in the presence of sunlight on the CeO2@ZnO heterojunction based on band gap studies (Fig. 6a and b). Suppose the migratory behaviors of photogenerated charge carriers in the CeO2@ZnO composites are compatible with the mechanism of the type II heterojunction. In that case, the photoinduced electrons will migrate from the CB of CeO2 to the CB of ZnO. The significant Coulomb repulsion that could result from the photoelectrons in their CBs makes this transfer of photoelectrons challenging to accomplish.58 The observed band gap of ZnO was 3.1 eV (VB: 2.52; CB: −0.61 eV; Eg = 2.52 − (−0.61) = 3.1 eV) and CeO2 showed a band gap of 2.8 eV (VB: 1.99 eV; CB: −0.87 eV; Eg = 1.99 − (−0.87) = 2.86 eV) which show the same relation with band gap energy. However, after incorporation of CeO2 into ZnO, a decrease in band gap is observed, i.e. 2.6 eV (VB: 1.99 eV; CB: −0.61 eV; Eg = 1.99 − (−0.61) = 2.6 eV), indicative of the Z-scheme nature of CeO2@ZnO nanoparticles. According to the type II mechanism, the photoinduced holes in the VB of ZnO would migrate to the VB of CeO2. The VB edge potential of CeO2 is more negative than the potential of OH/˙OH (2.24 V), as shown in Fig. 6; therefore, the holes in the VB of CeO2 are unable to oxidize OH to form hydroxyl radicals.
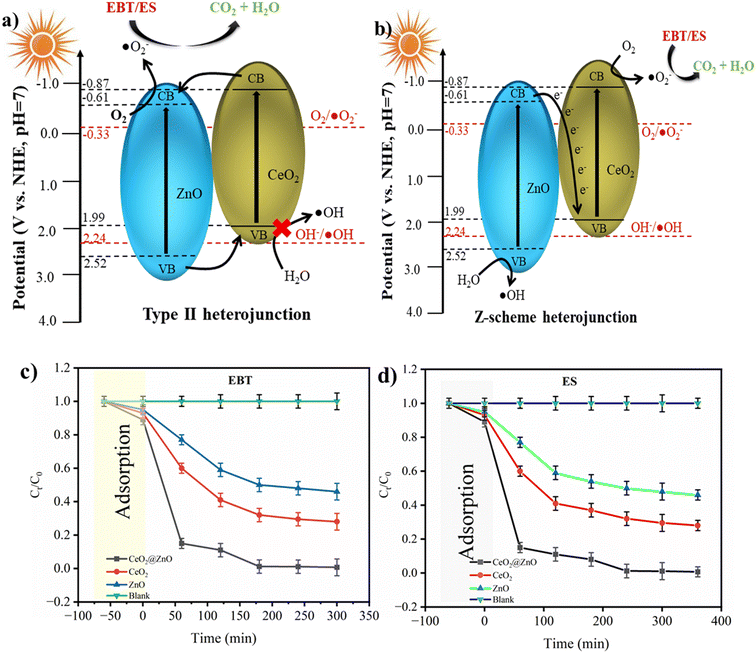 |
| | Fig. 6 Schematic diagram of the photogenerated electron–hole separation process for CeO2@ZnO composite: (a) type II heterojunction and (b) Z-scheme heterojunction. Chemical kinetics of (c) EBT dye and (d) ES pesticide. | |
Given that the oxidation reactions take place on CeO2, which has a lower oxidation potential, and the reduction reactions take place on ZnO, which has a lower reduction potential, it should be noted that this type II process might simultaneously reduce photoelectrons' ability to be reduced and photoinduced holes' ability to be oxidized. Hence, the capacity to produce active species can be compromised in this type II photocatalytic system. The literature revealed that CeO2@ZnO nanocomposites make Z-type heterojunctions.59 In this study, the lifespan of active species carrying charge controlled the photocatalytic breakdown of ES and EBT. Charge recombination capability is prevented by the reduced photoluminescence (PL) intensity of the nanocomposite. Better charge carrier separation and faster transit were shown with lower PL emission intensity. PL is employed for the determination of proficiency of trapping, recombination probability of charge carriers and transmission in a semiconductor. The charge separation and possible mechanism of photodegradation might be understood in terms of the PL spectra. A lower intensity in PL analysis indicated a decrease in the electron–hole recombination which in turn increased the separation efficiency of generated charge carriers. The emission spectra https://www.sciencedirect.com/topics/earth-and-planetary-sciences/emission-spectra of nanocatalysts were obtained at 350 nm excitation wavelength keeping in view that the wavelength should be greater than the band gap energy. The CeO2@ZnO nanocomposite displayed a lower PL intensity in comparison to the native nanoparticles of CeO2 and ZnO (Fig. S6†). All the nanocatalysts exhibited PL signal in the range from 200 nm to 400 nm. Higher PL intensity of native materials initiated the higher recombination between photoinduced charge carriers (e− in the CB and h+ in the VB).60 Due to insufficient intensity observation, the CeO2@ZnO nanocomposite's capacity to recombine electron and hole pairs was reduced. As a result of their interaction, resistivity went up, the amount of n-type donor carriers decreased, and charge recombination was suppressed. An interfacial electric field was generated at the point where CeO2 and ZnO came together. Doping ZnO lengthens the charged separation span to prevent e−/h+ recombination. When highly reactive holes and water interact, the result is a hydroxyl species radical, whereas electrons create peroxide radicals. In their interactions with dissolved oxygen, ˙O2− is produced.61 After that, oxygen molecules and holes can interact to form hydroperoxyl radica ls. By the superoxide anion, OH radicals can also be produced in addition to HOO˙ radicals.62 The general mechanism of active radical formation and degradation can be described by the equations below:
| h+ + ES/EBT → DegradedProducts |
| OH˙ + ES/EBT → DegradedProducts |
| ˙O2− + ES/EBT → DegradedProducts |
| ˙O2− + H2O → OH˙ + OH− + 1/2O2 |
6. Proposed chemical kinetics of the removal of ES and EBT
At optimized conditions (pollutant concentration: 50 mg L−1 for ES, 2 mg L−1 for EBT; nanocatalyst: 35 mg; pH ∼ 3 for ES, pH ∼ 7 for EBT; and sunlight), the CeO2@ZnO nanocatalyst showed rapid degradation of ES (96%) and EBT (98%) following first-order kinetics (faster reduction at initial stage observed with slower reduction over time) (Fig. 6c and d). With the lower band gap of CeO2@ZnO nanocatalyst, when exposed to sunlight, electron excitation begins. The synergism phenomenon altered the lifetime of electrons. As a result of the oxidation of ES and EBT, enormous amounts of hydroxyl were produced.63 Throughout the entire process, a large number of electrons were produced, which ultimately caused the breakdown of hazardous and carcinogenic pollutants into oxidant intermediate products and safer metabolites. To explain the adsorption phenomenon of ES and EBT, discrete adsorption models were also studied, those of Langmuir, Dubinin–Radushkevich (DRK), Sips, Temkin, and Freundlich (Fig. S7 and S8†). For the DRK, Slips and Freundlich models, statistical validity was not demonstrated. Negative lnCe values, on the other hand, rendered the Temkin model inappropriate. More details on adsorption model curves are available in the ESI.† The statistical significance of p-value and regression (R2) were computed for each adsorption model, and it was found that the Langmuir isotherm was followed during the adsorption process for all adsorption models (Table S1†). The half-life (t1/2) and rate constant of the parent nanomaterials and linked nanoparticles are also shown in Table 3. The activation energy has been found by using the Arrhenius equation: k = Ae−Ea/RT, where k is the rate constant, T is temperature (298–300 K), A is the pre-exponential factor, Ea is the activation energy for the reaction, and R is the universal gas constant. The Ea for ES is found to be 45–47 kJ mol−1 and for EBT is 48–50 kJ mol−1. Because the rest of the adsorption models have unsatisfactory statistical parameters and the negative slope value made them undesirable, CeO2@ZnO, with the lowest t1/2 and highest k-value, followed by CeO2 and ZnO nanoparticles, exhibited quicker removal of selected pollutants. The maximum amounts of ES and EBT were adsorbed by CeO2@ZnO, followed by CeO2 and ZnO.
Table 3 Rate constant (h−1) and half-life of ES and EBT at optimized conditions
| Catalyst |
CeO2@ZnO |
CeO2 |
ZnO |
Blanka |
|
k (h−1) |
t
1/2 (h) |
R
2
|
k (h−1) |
t
1/2 (h) |
R
2
|
k (h−1) |
t
1/2 (h) |
R
2
|
k (h−1) |
t
1/2 (h) |
R
2
|
| Note: triplicate experiments (n = 3) were evaluated for estimation of error bar. Blank (15 mL of each pollutant with definite concentration without catalyst). |
| ES |
0.50 |
1.3 |
0.99 |
0.27 |
2.5 |
0.99 |
0.15 |
4.6 |
0.98 |
0.01 |
69.3 |
0.99 |
| EBT |
0.80 |
0.8 |
0.99 |
0.48 |
1.4 |
0.98 |
0.21 |
3.3 |
0.99 |
0.08 |
68.3 |
0.98 |
7. Scavenger analysis
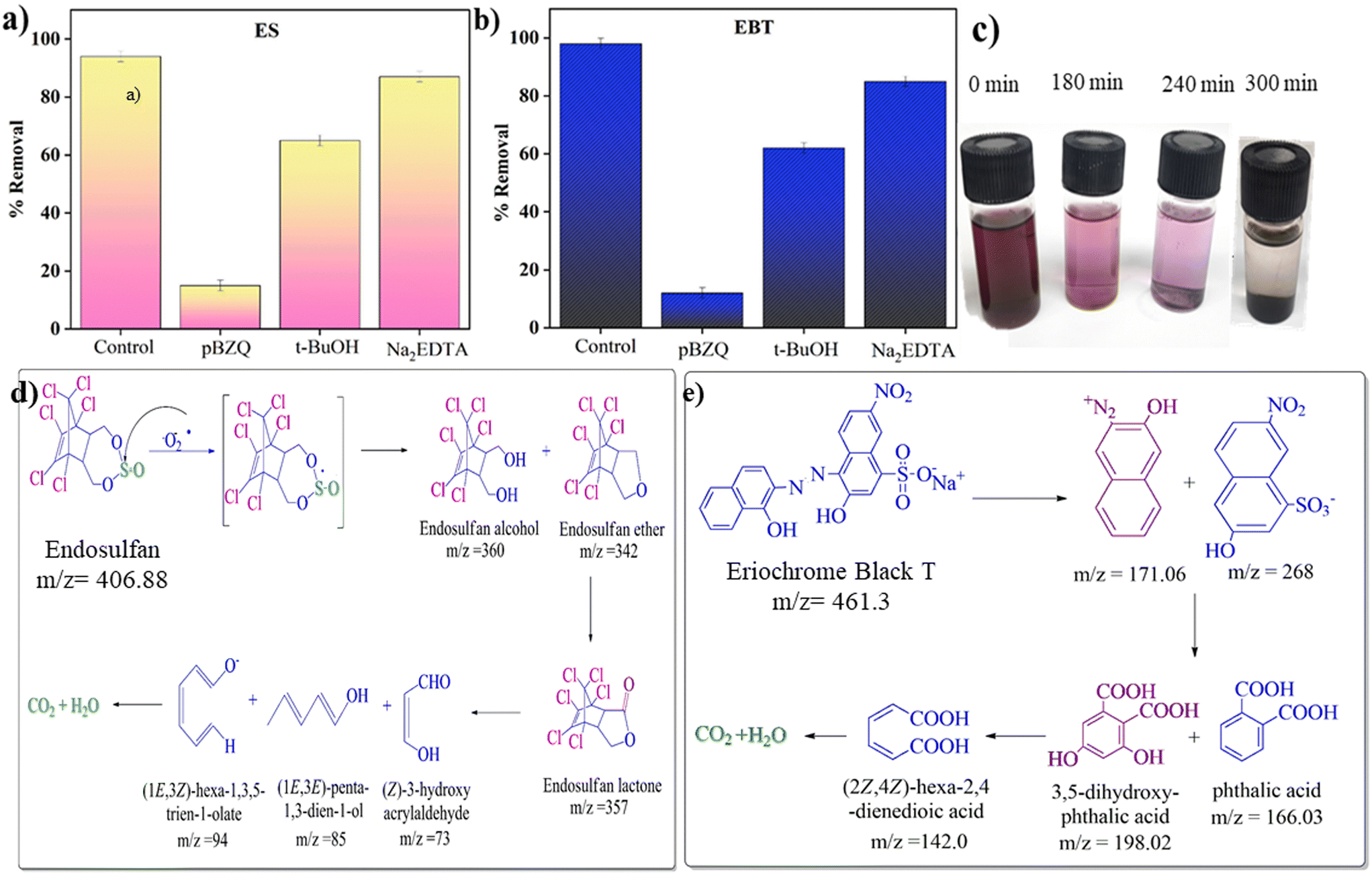
This work aimed to identify the reactive species that cause the degradation of ES and EBT. Charge separation was observed due to the excitation of e− from a lower energy level to a higher one following the generation of holes in lower energy levels in the presence of sunlight. Hydroxyl radicals are produced when extremely active h+ reacts with H2O, and ˙O2− is produced when e− interact with dissolved O2 molecules in water. Afterward, the hydroperoxyl radical (HO2˙) is formed when ˙O2− reacts with H+ and superoxide anion formation due to interaction with ˙OH radicals.54–56 For the quenching of holes, ˙OH radical and ˙O2−, different quenchers were used, namely ethylenediaminetetraacetic acid disodium salt (Na2EDTA), tert-butyl alcohol (t-BuOH) and para-benzoquinone (pBZQ).63 Na2EDTA or any hole scavenger can interact with a photocatalyst and ensure reduction of a target pollutant either by direct injection of electrons or by indirect injection of electrons. In the direct injection process, the electrons from Na2EDTA are directly filled in the holes in the VB, thereby reducing the electron–hole recombination rate. However, the organic species (t-BuOH) accepts holes from the VB either directly or indirectly, and subsequently is oxidized, suppressing the electron–hole recombination as well as increasing the reduction efficiency. The interaction involves a redox reaction where pBZQ undergoes reduction by the active species, thereby neutralizing them and preventing further degradation of pollutants.64 In a 10 mL vial containing 2 mg L−1 of EBT, 50 mg L−1 of ES, photocatalytic dose (35 mg) at neutral pH (7) for EBT and acidic pH (3) for ES, 2 mL of quenchers was added and placed in sunlight for 6 hours. The same sample was prepared for comparison without adding quencher as a control sample. Accordingly, hydroxyl (˙OH) and holes (h+) play complementary roles, while superoxide ions (˙O2−) play a dominant role as the active species in the photocatalytic degradation.54–56 A significantly lower degradation rate was observed in the case of pBZQ. Next, the percentage degradation of both pollutants is shown as a bar diagram in Fig. 7a and b. The control sample showed maximum removal of ES and EBT. However, samples having Na2EDTA and t-BuOH also showed effective removal of the targeted pollutants. Scavenger trapping studies concluded that superoxide ions (˙O2−) are responsible for removing ES and EBT. The literature revealed that electron paramagnetic resonance (EPR) spectra were measured to analyze those active species generated in the photodegradation systems of ZnO, CeO2, and ZnO@CeO2. The weak EPR signal attributed to DMPO–˙O2− can be observed for the pure ZnO photocatalytic system, suggesting that ZnO is unfavorable for producing the active species of ˙O2− radicals. Meanwhile, CeO2 shows a relatively stronger EPR signal revealing that CeO2 could produce more active species of ˙O2− radicals than ZnO. The EPR results suggest that the pure CeO2 component cannot produce ˙OH radicals and the ZnO component can produce a small amount of ˙OH radicals, most probably due to different oxidation abilities of the photogenerated holes in their VBs. On the basis of the above results, constructing a heterojunction between ZnO and CeO2 can obviously promote the production of ˙O2− and ˙OH radicals, which are considered as the main reactive species to degrade the pollutant molecules during the photodegradation process.22 Similar results show that active radical species play a significant role in the degradation of organic pollutants also found for various other nanocomposites such as TiO2-based, TiO2–ZnO–Fe3O4, Ag@AgCl/ZnAl-LDH and BC-CdS.65–67
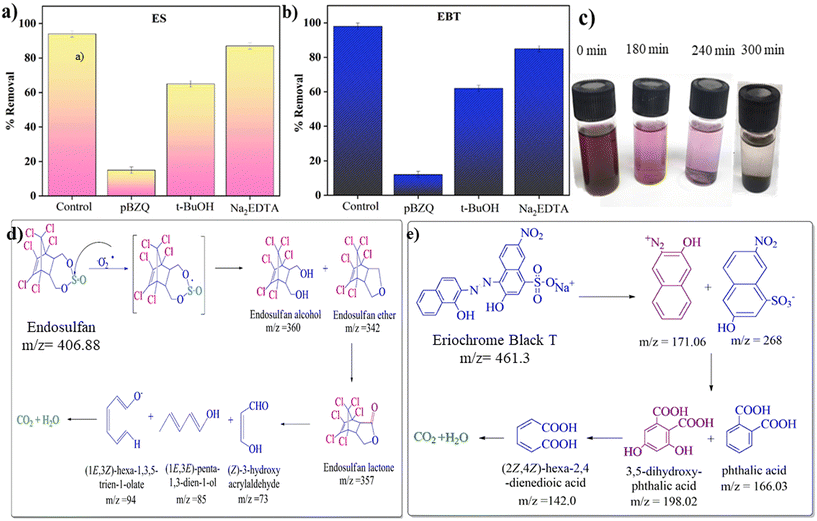 |
| | Fig. 7 Bar diagrams showing the percentage of (a) ES and (b) EBT dye degradation under sunlight in the presence of radical scavengers. (c) Decolorization of EBT dye with time. Proposed degradation pathways for the degradation of (d) ES and (e) EBT dye over CeO2@ZnO nanocomposite. | |
8. Photodegradation pathways of ES and EBT by using CeO2@ZnO nanocomposite
Total ion chromatogram (TIC) and mass spectrum (MS) data from gas chromatography-mass spectroscopy (GC-MS) were used to identify the proposed degradation routes for the specific pesticide and dye (Fig. S9 and S10†). The decolorization of EBT dye with time is shown in Fig. 7c. The attack of peroxide active radical on SO of ES generated the radical cation that rapidly hydrolyzed via oxygen to produce an oxygen-centered radical intermediate. The beta-elimination results in the formation of endosulfan ether (m/z = 406) via an oxygen-centered radical intermediary. The oxidative conversion of endosulfan ether to endosulfan alcohol (m/z = 360) and lactone (m/z = 357) occurs. In the hydrogen abstraction procedure, peroxide radicals attack a lactone byproduct to produce a carbon-centered radical.68 TIC showed that very short-lived species, such as endosulfan sulfate, ether, alcohol, and lactone, formed during degradation. Additionally, ring opening and oxidation transformed 2,3,4,5-tetrachlorophenylmethanol (m/z = 243) into smaller organic molecules. Numerous minor metabolites discovered along the proposed paths were produced by reductive degradation and, after that, were mineralized (Fig. 7d). According to the degradation route of EBT dye, 2-nitronaphthalene (m/z = 73) and naphthalene-1-ol (m/z = 141) are produced when peroxide radicals attack the azo group present in the EBT dye. A byproduct of advanced oxidation was benzene-1,2,3-triol (m/z = 128). However, the TIC showed the short-lived species benzene-1,2,3-triol (m/z = 128). The product (1E,3Z)-hexa-1,3,5-trien-1-ol (m/z = 98) was produced as a result of the ring-opening and oxidation process, and it later broke down into (E)-prop-1-en-ol (m/z = 59) (Fig. 7e). In addition, too many peroxide radicals cause the end products to mineralize from hazardous compounds.68
9. Recyclability performance of the photocatalyst
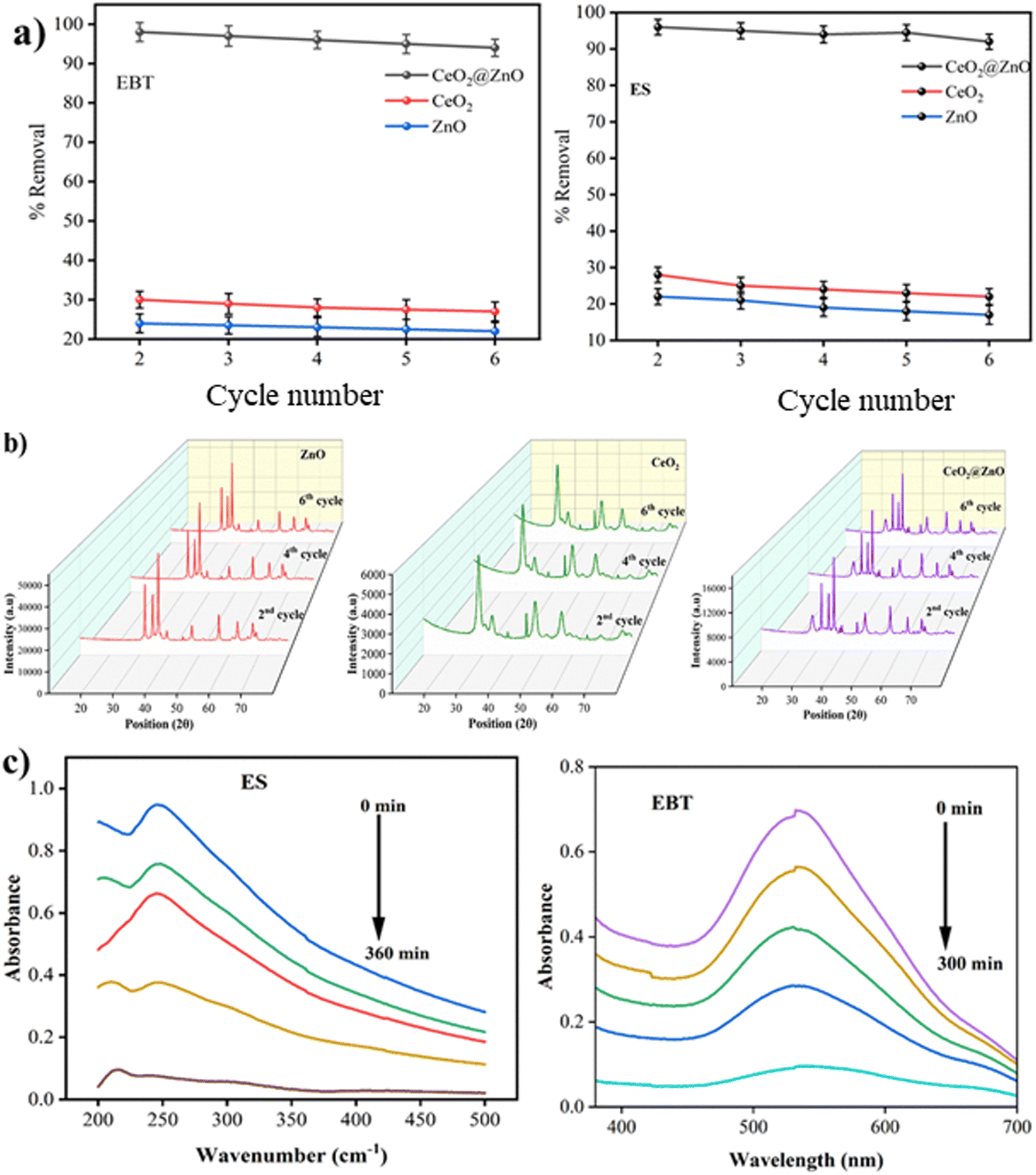
The doped CeO2@ZnO nanocomposite efficiently removes ES and EBT in natural sunlight. To assess the catalyst's reusability (35 mg), fresh samples were centrifuged, cleaned with distilled water and solvent (ethanol), and dried in a hot-air oven for 10 hours. The efficiency of the CeO2@ZnO nanocomposite declined from 96% to 86% for ES; 98% to 90% for EBT, showing practical reusability (Fig. 8a). The hybrid nanocatalyst was found to be recyclable for the same reaction scenario at least six times while retaining photocatalytic activity and formation of active species. The slight drop in the percentage removal of ES and EBT in every consecutive run may be attributed to the recovery methodology of the nanocomposite caused by the resistance between nanoparticles. A conglomerate nanoparticle's surface may also be coated with ES and EBT, preventing the major active positions for the absorption of photons from functioning. Until the sixth run, the CeO2@ZnO nanocomposite was employed efficiently, supported by the PXRD results (Fig. 8b).
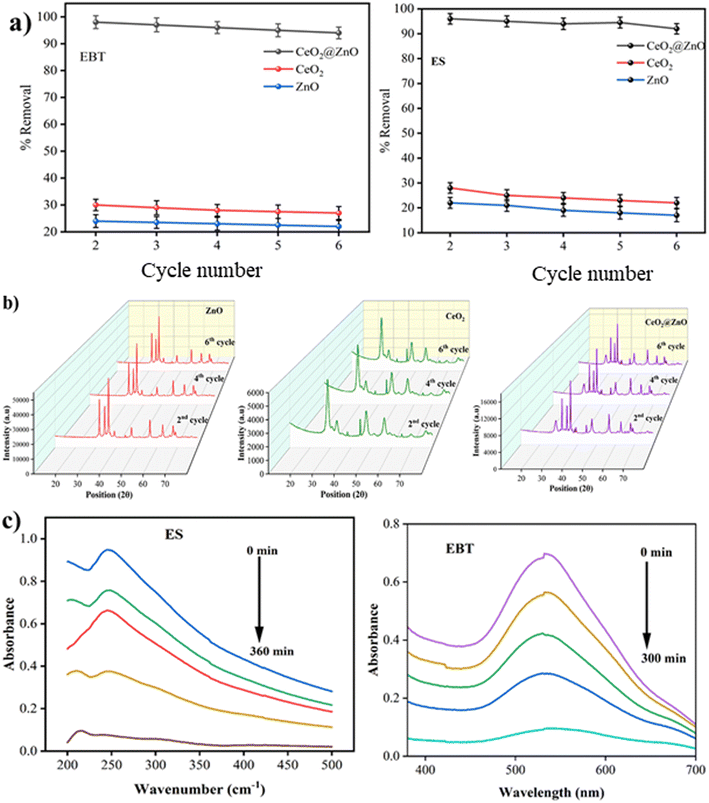 |
| | Fig. 8 (a) Reusability analysis of nanocomposite for the removal of EBT dye and ES up to 6 cycles. (b) PXRD data for reusability of ZnO, CeO2 and CeO2@ZnO nanocomposite up to 6 cycles. (c) UV-visible spectra of degradation of ES pesticide and EBT dye from waste water. | |
10. Degradation of pollutants (ES and EBT) from real field sample analysis
To investigate the practical potential of the CeO2@ZnO nanocomposite for photocatalytic removal of ES pesticide and EBT dye in different wastewater samples, 50 mL sample was collected from an agricultural site in Jalandhar, Punjab (WWS-1), and centrifuged to remove solid impurities. For comparison, a standard solution of ES (2 mL; 50 mg L−1) spiked in 8 mL (WWS-2) was used (Fig. 8c). The optimized CeO2@ZnO nanocomposite (35 mg) was added to WWS-1 and placed under sunlight for 6 hours. The absorbance of ES suppressed at 246 nm confirming 86% removal (calculated by using eqn (1)) was observed in the real field sample analysis. The EBT dye real field sample was collected from the experimental lab of NIT, Jalandhar. The EBT dye is used as an indicator in the experimental lab to determine the hardness of water samples. The effluent was collected after the completion of the experiment and treated with the CeO2@ZnO nanocomposite under sunlight irradiation for 5 hours. The absorbance of EBT suppressed at 533 nm confirming 90% removal (calculated using eqn (1)) was observed (Fig. 8c).
11. Conclusion
An extract of Sapindus mukorossi was employed to synthesize photoactive catalyst CeO2@ZnO nanocomposite using facile and eco-friendly methodology. PXRD and various microscopic techniques confirm the effective doping of both parent nanoparticles (CeO2 and ZnO). The higher negative zeta potential value (−18 eV), higher surface area (80 m2 g−1) and lower band gap (2.6 eV) of the CeO2@ZnO nanocomposite were favorable characteristics for efficient removal of ES and EBT under sunlight irradiation. The maximum removal was seen in sunlight rather than an artificial light source and in the dark when using optimum parameters. First-order kinetic models describe how quickly both targeted pollutants degrade by photocatalysis. Additionally, in the presence of sunshine, significant quantities of peroxide radicals are formed in water due to e−–h+ pairs, which target harmful pollutants (ES and EBT) at their least-hindered sites and transform them into safer metabolites, as shown by GC-MS investigations. Furthermore, the CeO2@ZnO nanocomposite could be recycled up to six times, emphasizing its sustainability. Overall, the CeO2@ZnO nanocomposite is a potential alternative material that might be used in practical applications and is anticipated to lessen environmental stress due to its good stability and reusability.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Dr Manviri Rani thanks DST-SERB, New Delhi (sanction order no. SRG/2019/000114) and TEQIP-III, MNIT Jaipur, India for financial assistance. Dr Uma Shanker thanks TEQIP-III, NIT Jalandhar, for financial support. Ms Keshu thanks the Ministry of Education (MoE), New Delhi, for the research fellowship.
References
- B. Tian, T. Tong, F. Chen and J. Zhang, Wuli Huaxue Xuebao, 2007, 23(7), 978–982, DOI:10.1016/S1872-1508(07)60052-7.
- A. Choudhry, A. Sharma, S. I. Siddiqui, I. Ahamad, M. Sajid, T. A. Khan and S. A. Chaudhry, Environ. Res., 2023, 220, 115193, DOI:10.1016/j.envres.2022.115193.
- T. Manasfi, K. Lebaron, M. Verlande, J. Dron, C. Demelas, L. Vassalo and J. Boudenne, Int. J. Hyg. Environ. Health, 2019, 222(1), 1–8, DOI:10.1016/j.ijheh.2018.06.008.
- P. Sathishkumar, K. Mohan, A. R. Ganesan, M. Govarthanan, A. R. M. Yusoff and F. L. Gu, J. Hazard. Mater., 2021, 416, 125779, DOI:10.1016/j.jhazmat.2021.125779.
- A. Mudhoo, A. Bhatnagar, M. Rantalankila, V. Srivastava and M. Sillanpää, Chem. Eng. J., 2019, 360, 912–928, DOI:10.1016/j.cej.2018.12.055.
- P. A. Greve, Meded. Fac. Landbouwwet., Univ. Gent, 1971, 36, 439–447 CAS.
- S. Kumar, S. K. Sharma, R. D. Kaushik and L. P. Purohit, Mater. Today Chem., 2021, 20, 100464, DOI:10.1016/j.mtchem.2021.100464.
- X. Y. Yu and X. W. Lou, Appl. Catal., B, 2018, 8(3), 1701592, DOI:10.1002/aenm.201701592.
- V. K. Gupta and I. Ali, Environ. Sci. Technol., 2008, 42(3), 766–770, DOI:10.1021/es7025032.
- R. Jamee and R. Siddique, Eur. J. Microbiol. Immunol., 2019, 9, 114–118, DOI:10.1556/1886.2019.00018.
- I. De, M. Pahuja, H. M. ud din Wani, A. Dey, T. Dube, R. Ghosh and M. Singh, Ecotoxicol. Environ. Saf., 2022, 243, 113985, DOI:10.1016/j.ecoenv.2022.113985.
- M. T. Yagub, T. K. Sen, S. Afroze and H. M. Ang, Adv. Colloid Interface Sci., 2014, 209, 172–184, DOI:10.1016/j.cis.2014.04.002.
- N. Barka and M. M. E. L. Abdennouri, J. Taiwan Inst. Chem. Eng., 2011, 42, 320–326, DOI:10.1016/j.jtice.2010.07.004.
- H. Li, B. Sun, T. Gao, H. Li, Y. Ren and G. Zhou, Chin. J. Catal., 2022, 43(2), 461–471, DOI:10.1016/S1872-2067(21)63915-3.
- C. Tian, Q. Zhang, A. Wu, M. Jiang, Z. Liang, B. Jiang and H. Fu, Chem. Commun., 2012, 48, 2858–2860, 10.1039/C2CC16434E.
- D. Chen, Z. Wang, T. Ren, H. Ding, W. Yao, R. Zong and Y. Zhu, J. Phys. Chem. C, 2014, 118, 15300–15307, DOI:10.1016/j.hazadv.2022.100081.
- L. Liu, T. Hu, K. Dai, J. Zhang and C. Liang, Chin. J. Catal., 2022, 43(1), 46–55, DOI:10.1016/S1872-2067(20)63560-4.
- G. Liu, L. Han, J. Wang, Y. Yang, Z. Chen, B. Liu and X. An, J. Mater. Sci. Technol., 2024, 174, 188–194, DOI:10.1016/j.jmst.2023.08.012.
- Z. Gao, J. Wang, Y. Muhammad, Y. Zhang, S. J. Shah, Y. Hu, Z. Chu and Z. Zhao, J. Chem. Eng., 2020, 388, 124389, DOI:10.1016/j.cej.2020.124389.
- C. Liu, J. Xu, J. Niu, M. Chen and Y. Zhou, Sep. Purif. Technol., 2020, 241, 116622, DOI:10.1016/j.seppur.2020.116622.
- M. Ebrahimi, M. Samadi, S. Yousefzadeh, M. Soltani, A. Rahimi and A. Z. Moshfegh, ACS Sustainable Chem. Eng., 2017, 5(1), 367–375, DOI:10.1021/acssuschemeng.6b01738.
- R. Ullah and J. Dutta, J. Hazard. Mater., 2008, 156(1–3), 194–200, DOI:10.1016/j.jhazmat.2007.12.033.
- W. Yu, J. Zhang and T. Peng, Appl. Catal., B, 2016, 181, 220–227, DOI:10.1016/j.apcatb.2015.07.031.
- Y. Wang, H. B. Fang, Y. Z. Zheng, R. Ye, X. Tao and J. F. Chen, Nanoscale, 2015, 7(45), 19118–19128 RSC.
- R. Zha, R. Nadimicherla and X. Guo, J. Mater. Chem., 2015, 3(12), 6565–6574, 10.1039/C5TA00764J.
- M. Alavi and M. Moradi, Inorg. Chem. Commun., 2022, 142, 109590, DOI:10.1016/j.inoche.2022.109590.
- G. Zhang, J. Ao, Y. Guo, Z. Zhang, M. Shao, L. Wang, L. Zhou and J. Shao, RSC Adv., 2014, 4, 20131–20135, 10.1039/C4RA01004C.
- M. Liu, S. Wang, T. Chen, C. Yuan, Y. Zhou, S. Wang and J. Huang, J. Power Sources, 2015, 274, 730–735, DOI:10.1016/j.jpowsour.2014.10.122.
- P. Zhou, J. Yu and M. Jaroniec, J. Adv. Mater., 2014, 26(29), 4920–4935, DOI:10.1002/adma.201400288.
- M. Zubair, A. Razzaq, C. A. Grimes and S.-I. In, Cu2ZnSnS4(CZTS)-ZnO: A Noble Metal-Free Hybrid Z-Scheme Photocatalyst for Enhanced Solar-Spectrum Photocatalytic Conversion of CO2 to CH4, J. CO2 Util., 2017, 20, 301–311, 10.1039/C6RA02763F.
- N. Tian, H. Huang, C. Liu, F. Dong, T. Zhang, X. Du, S. Yu and Y. Zhang, J. Mater. Chem. A, 2015, 3, 17120–17129, 10.1039/C5TA03669K.
- L. Zhu, H. Li, Z. Liu, P. Xia, Y. Xie and D. Xiong, J. Phys. Chem. C, 2018, 122(17), 9531–9539, DOI:10.1021/acs.jpcc.8b01933.
- C. Tian, Q. Zhang, A. Wu, M. Jiang, Z. Liang, B. Jiang and H. Fu, Chem. Commun., 2012, 48(23), 2858–2860, 10.1039/C2CC16434E.
- R. Ullah and J. Dutta, J. Hazard. Mater., 2008, 156(1–3), 194–200, DOI:10.1016/j.jhazmat.2007.12.033.
- W. Yu, J. Zhang and T. Peng, Appl. Catal., B, 2016, 181, 220–227, DOI:10.1016/j.apcatb.2015.07.031.
- J. F. Muñoz-Chilito and J. E. Rodríguez-Páez, J. Environ. Chem. Eng., 2021, 9(1), 104890, DOI:10.1016/j.jece.2020.104890.
- M. S. Pujar and S. M. Hunagund,
et al.
, J. Sol-Gel Sci. Technol., 2022, 101, 356–369, DOI:10.1007/s10971-021-05715-7.
- L. Ma, X. Ai, W. Jiang, P. Liu, Y. Chen and K. Lu,
et al.
, Colloid Interface Sci. Commun., 2022, 49, 100636, DOI:10.1016/j.colcom.2022.100636.
- H. Pan, F. Wang, J. Huang and N. Chen, Wuli Huaxue Xuebao, 2008, 24(6), 992–996, DOI:10.1016/S1872-1508(08)60045-5.
- A. K. Zak, W. A. Majid, M. E. Abrishami and R. Yousefi, Solid State Sci., 2011, 13, 251–256, DOI:10.1016/j.solidstatesciences.2010.11.024.
- S. Jaiswal and P. Mishra, Med. Microbiol. Immunol., 2018, 207, 39–53, DOI:10.1007/s00430-017-0525-y.
- M. Rani, Keshu and U. Shanker, Ceram. Int., 2023, 49(23), 38183–38195, DOI:10.1016/j.ceramint.2023.09.149.
- M. Rani, J. Yadav, Keshu and U. Shanker, J. Colloid Interface Sci., 2021, 60, 689–703, DOI:10.1016/j.jcis.2021.05.152.
- S. Choudhary, M. Rani, Keshu and U. Shanker, Environ. Nanotechnol., Monit. Manage., 2022, 18, 100746, DOI:10.1016/j.enmm.2022.100746.
- J. Yadav, M. Rani and U. Shanker, J. Environ. Chem. Eng., 2022, 10(3), 107452, DOI:10.1016/j.jece.2022.107452.
- Keshu, M. Rani and U. Shanker, J. Mol. Liq., 2023, 387, 122611, DOI:10.1016/j.molliq.2023.122611.
- M. Rani, Keshu, S. Pandey, Rishabh, S. Sharma and U. Shanker, J. Photochem. Photobiol., A, 2023, 115160, DOI:10.1016/j.jphotochem.2023.115160.
- A. Kumar, P. Choudhary, A. Kumar, P. H. Camargo and V. Krishnan, Small, 2022, 18(1), 2101638, DOI:10.1002/smll.202101638.
- B. Manikandan, K. R. Murali and R. John, Iran. J. Catal., 2021, 11(1), 1–11 CAS.
- M. Grote, J. L. Boudenne, J. P. Croué, B. I. Escher, U. von Gunten, J. Hahn, T. Höfer, H. Jenner, J. Jiang, T. Karanfil and M. Khalanski, Water Res., 2022, 217, 118383, DOI:10.1016/j.watres.2022.118383.
- S. Jaiswal and P. Mishra, Med. Microbiol. Immunol., 2018, 207, 39–53, DOI:10.1007/s00430-017-0525-y.
- P. Sarrami, S. Karbasi, Z. Farahbakhsh, A. Bigham and M. Rafienia, Int. J. Biol. Macromol., 2022, 220, 1368–1389, DOI:10.1016/j.ijbiomac.2022.09.117.
- K. Xu, J. Shen, S. Zhang, D. Xu and X. Chen, J. Mater. Sci. Technol., 2022, 121, 236–244, DOI:10.1016/j.jmst.2021.12.070.
- M. Rani, Keshu, Ankit and U. Shanker, ChemistrySelect, 2023, 8(20), e202300270, DOI:10.1002/slct.202300270.
- M. Rani, Ankit, Keshu and U. Shanker, Biomass Convers. Biorefin., 2023, 2023, 1–16, DOI:10.1007/s13399-023-04374-8.
- M. Rani, Keshu and U. Shanker, J. Environ. Sci. Technol., 2023, 20, 5491–5508, DOI:10.1007/s13762-022-04255-z.
- J. Zhang, Y. Ma, Y. Du, H. Jiang, D. Zhou and S. Dong, Appl. Catal., B, 2017, 209, 253–264, DOI:10.1016/j.apcatb.2017.03.017.
- Y. Hu, X. Li, W. Wang, F. Deng and L. Han,
et al.
, Chin. J. Chem., 2022, 41(6), 2206069–2206078, DOI:10.14102/j.cnki.0254-5861.2022-0103.
- C. He, S. Wu, N. Zhao, C. Shi, E. Liu and J. Li, ACS Nano, 2013, 7(5), 4459–4469, DOI:10.1021/nn401059h.
- A. Rezaei, M. R. Rezaei and M. H. Sayadi, J. Taiwan Inst. Chem. Eng., 2021, 121, 154–167, DOI:10.1016/j.jtice.2021.03.048.
- Y. Yao, Y. Zhou, L. Liu, Y. Xu, Q. Chen, Y. Wang, S. Wu, Y. Deng, J. Zhang and A. Shao, Front. Mol. Biosci., 2020, 7, 193, DOI:10.3389/fmolb.2020.00193.
- M. H. Sayadi, M. D. Pavlaki, R. Martins, B. Mansouri, C. R. Tyler, J. Kharkan and H. Shekari, Chemosphere, 2021, 269, 128689, DOI:10.1016/j.chemosphere.2020.128689.
- M. Rani, M. Keshu and U. Shanker, J. Environ. Manage., 2021, 300, 113777, DOI:10.1016/j.jenvman.2021.113777.
- O. Fonagy, E. Szabo-Bardos and O. Horvath, J. Photochem. Photobiol., A, 2021, 407, 113057, DOI:10.1016/j.jphotochem.2020.113057.
- S. Kanan, M. A. Moyet, R. B. Arthur and H. H. Patterson, Catal. Rev., 2020, 62(1), 1–65, DOI:10.1080/01614940.2019.1613323.
- A. Saljooqi, T. Shamspur and A. Mostafavi, Environ. Sci. Pollut. Res., 2021, 28, 9146–9156, DOI:10.1007/s11356-020-11122-2.
- P. Sarrami, S. Karbasi, Z. Farahbakhsh, A. Bigham and M. Rafienia, Int. J. Biol. Macromol., 2022, 220, 1368–1389, DOI:10.1016/j.ijbiomac.2022.09.117.
- Keshu, M. Rani, J. Yadav, S. Chaudhary and U. Shanker, J. Environ. Chem. Eng., 2021, 9(6), 106763, DOI:10.1016/j.jece.2021.106763.
- K. Das, R. Bariki, D. Majhi, A. Mishra, K. K. Das, R. Dhiman and B. G. Mishra, Appl. Catal., B, 2022, 303, 120902, DOI:10.1016/j.apcatb.2021.120902.
- S. Yedla and A. K. Dikshit, Appl. Catal., B, 2008, 134(2), 102–109, DOI:10.1016/j.apcatb.2021.120902.
- A. Begum and S. K. Gautam, Indian J. Environ. Prot., 2011, 31, 658–665, DOI:10.1016/j.cej.2018.12.055.
- V. K. Gupta, R. Jain, A. Mittal, M. Mathur and S. J. Sikarwar, J. Colloid Interface Sci., 2007, 309(2), 464–469, DOI:10.1016/j.jcis.2006.12.010.
- A. H. Yonli, I. Batonneau-Gener and J. Koulidiati, J. Hazard. Mater., 2012, 203, 357–362, DOI:10.1016/j.jhazmat.2011.12.042.
- K. Wang, Z. Xing, D. Meng, S. Zhang, Z. Li, K. Pan and W. Zhou, Appl. Catal., B, 2021, 281, 119482, DOI:10.1016/j.apcatb.2020.119482.
- Rachna, M. Rani and U. Shanker, J. Environ. Chem. Eng., 2020, 8(4), 104040, DOI:10.1016/j.jece.2020.104040.
- A. Gogoi, M. Navgire, K. C. Sarma and P. Gogoi, Mater. Chem. Phys., 2019, 231, 233–243, DOI:10.1016/j.matchemphys.2019.04.013.
- G. Mamba, X. Y. Mbianda and A. K. Mishra, Mater. Chem. Phys., 2015, 149, 734–742, DOI:10.1016/j.matchemphys.2014.11.035.
- J. Kaur and S. Singhal, Superlattices Microstruct., 2015, 83, 9–21, DOI:10.1016/j.spmi.2015.03.022.
- M. Karimi-Shamsabadi, M. Behpour, A. K. Babaheidari and Z. Saberi, J. Photochem. Photobiol., A, 2017, 346, 133–143, DOI:10.1016/j.jphotochem.2017.05.038.
- P. Franco, O. Sacco, I. De Marco, D. Sannino and V. Vaiano, Top. Catal., 2020, 63, 1193–1205 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *b
*b