
The retinal chromophore environment in an inward light-driven proton pump studied by solid-state NMR and hydrogen-bond network analysis†
Marie
Pinto
 a,
Maryam
Saliminasab
a,
Andrew
Harris
a,
Michalis
Lazaratos
b,
Ana-Nicoleta
Bondar
cd,
Vladimir
Ladizhansky
*a and
Leonid S.
Brown
*a
a,
Maryam
Saliminasab
a,
Andrew
Harris
a,
Michalis
Lazaratos
b,
Ana-Nicoleta
Bondar
cd,
Vladimir
Ladizhansky
*a and
Leonid S.
Brown
*a
aDepartment of Physics and Biophysics Interdepartmental Group, University of Guelph, Guelph, Ontario N1G 2W1, Canada. E-mail: vladizha@uoguelph.ca; lebrown@uoguelph.ca
bFreie Universität Berlin, Physics Department, Theoretical Molecular Biophysics Group, D-14195 Berlin, Germany
cUniversity of Bucharest, Faculty of Physics, Măgurele 077125, Romania
dForschungszentrum Jülich, Institute for Computational Biomedicine (IAS-5/INM-9), 52428 Jülich, Germany
First published on 23rd August 2024
Abstract
Inward proton pumping is a relatively new function for microbial rhodopsins, retinal-binding light-driven membrane proteins. So far, it has been demonstrated for two unrelated subgroups of microbial rhodopsins, xenorhodopsins and schizorhodopsins. A number of recent studies suggest unique retinal–protein interactions as being responsible for the reversed direction of proton transport in the latter group. Here, we use solid-state NMR to analyze the retinal chromophore environment and configuration in an inward proton-pumping Antarctic schizorhodopsin. Using fully 13C-labeled retinal, we have assigned chemical shifts for every carbon atom and, assisted by structure modelling and molecular dynamics simulations, made a comparison with well-studied outward proton pumps, identifying locations of the unique protein–chromophore interactions for this functional subclass of microbial rhodopsins. Both the NMR results and molecular dynamics simulations point to the distinctive polar environment in the proximal part of the retinal, which may result in a hydration pattern dramatically different from that of the outward proton pumps, causing the reversed proton transport.
Introduction
Microbial rhodopsins, light-activated retinal-binding helical membrane proteins, display a large variety of functions. Among these proteins, outward light-driven proton pumping is most likely the best-studied, mainly due to many years of research done on the prototypical archaeal (bacteriorhodopsin, BR) and eubacterial (green proteorhodopsin, GPR) representatives of this group along with their close homologs.1–12 The wealth of structural, kinetic, and spectroscopic information obtained for these proteins allowed reconstructing almost a complete mechanism of outward proton transport, identifying its main players and the key steps. The interplay of retinal isomerization, proton affinity changes of donors and acceptors, protein conformational changes, and water dynamics is now known in detail.The situation is different for recently discovered inward proton pumps, such as xenorhodopsins (XeRs) and schizorhodopsins (SzRs),13,14 where mechanisms responsible for the reversal of the proton transport direction are still being investigated and debated. One common structural feature of these two divergent groups of inward proton pumps is an incomplete (compared to the classical outward proton pumps) retinal Schiff base counterion. While in BR and its homologs a positively charged protonated Schiff base interacts with two negatively charged aspartates (on helix C and helix G) and a strongly bound water molecule, both XeRs and SzRs possess only one aspartate in this region, on helix C or on helix G, respectively. It was suggested that this modified counterion allows for the Schiff base reorientation towards the cytoplasm, to which the proton is released.5,15–17 To reorient the Schiff base to the extracellular side for reprotonation, an additional retinal isomerization was invoked, either anti–syn isomerization of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond or cis–trans isomerization of the C13C14 bond.15,17–19 In contrast to the outward proton pumps, where retinal reisomerization occurs after the Schiff base reprotonation, it was suggested that the C13C14 isomerization occurs in the M state with a deprotonated Schiff base, at least in the case of SzRs. Several recent studies suggested that the main factor responsible for the inward proton transport in SzRs is their unique retinal–protein interactions, originating from close contacts with the sole carboxylic counterion on helix G and the neighbouring aromatic amino acid sidechains.16–22 Such altered retinal–protein interactions can affect the retinal conformation (such as planarity) and π-electron distribution in the dark and/or photointermediate states, and influence the orientation of the Schiff base along with the relative rates of retinal reisomerization and reprotonation.17
N bond or cis–trans isomerization of the C13C14 bond.15,17–19 In contrast to the outward proton pumps, where retinal reisomerization occurs after the Schiff base reprotonation, it was suggested that the C13C14 isomerization occurs in the M state with a deprotonated Schiff base, at least in the case of SzRs. Several recent studies suggested that the main factor responsible for the inward proton transport in SzRs is their unique retinal–protein interactions, originating from close contacts with the sole carboxylic counterion on helix G and the neighbouring aromatic amino acid sidechains.16–22 Such altered retinal–protein interactions can affect the retinal conformation (such as planarity) and π-electron distribution in the dark and/or photointermediate states, and influence the orientation of the Schiff base along with the relative rates of retinal reisomerization and reprotonation.17
Solid-state NMR (ssNMR) is a powerful tool to investigate retinal–protein interactions and the exquisite details of retinal conformation in both animal and microbial rhodopsins as well as their mutants and photointermediates.23–33 Assisted by the uniform or specific 13C, 15N, or 2H labelling of retinal, ssNMR can report on the chemical environment of its specific atoms, its isomeric state, polyene chain twist, β-ionone ring conformation, and retinal–protein contacts, including the Schiff base interactions.34–40 Complete 13C retinal chemical shift assignments are available for both isomeric forms of BR and bovine visual rhodopsin,24,39 with fairly extensive assignments obtained for GPR, its blue-light absorbing mutant L105Q, and light-driven sodium pump KR2,28,33,38 with only a few carbon atom assignments available for a number of other microbial rhodopsins.29,40–42 Comparison of retinal chemical shifts between different proteins, their mutants and photointermediates, as well as various model retinoid compounds may yield interesting insights into the details of the chromophore structure and interactions in different functional classes of rhodopsins.
Here, we employ ssNMR to study the retinal chromophore environment in a previously uncharacterized inward proton-pumping Antarctic SzR, with an unusual helix C motif. While most of the studied SzRs have an F/Y–S–E/D/H helix C motif (as opposed to the classic D–T–D triad of BR, corresponding to Asp85, Thr89, and Asp96),13,17,18 Antarctic SzRs (AntRs) showed even more variability, displaying an F/L/M–S–E/Q motif.22 More recently, we found that some Antarctic SzRs show even less conservation and possess a G–S–S/T/A/Y motif (Fig. S1, ESI†). The lack of conservation of the cytoplasmic glutamate is consistent with our earlier mutagenesis data on the AntR with an FSE motif, in which the E81Q mutant was fully functional.22
In this work, we reconstituted one of such unusual Antarctic SzRs (which we call GSS AntR) with a biosynthetically 13C-labeled retinal38 and studied its chromophore structure and environment by analysing the complete carbon chemical shifts of the retinal. Comparison of retinal chemical shifts obtained for GSS AntR with those known for other rhodopsins was assisted by structure modelling and MD simulations and revealed significant differences in the chromophore environment which may contribute to the unique function of this protein.
Experimental
Protein expression and purification, and retinal and lipid reconstitution
Most of the procedures in this and the next section follow our protocols established for AntR and Proteo-SRs,22,43 with minor modifications as follows. The gene encoding GSS AntR (Department of Energy, Joint Genome Institute, Integrated Microbial Genomes & Microbiomes,44 Gene ID Ga0307935_10093322) was cloned into the pET21a(+) vector (EMD Millipore, Billerica, MA) by GenScript (Piscataway, NJ) using NdeI–XhoI restriction sites, which encodes the C-terminal 6× His-tag after the LE insert. This vector was transformed into E. coli C41 (DE3) OverExpress Chemically Competent Cells (Lucigen, Middleton, WI) for expression using Lucigen's heat-shock transformation protocol. To prepare the competent cells, 5 mL of sterile 2xYT medium (1% yeast extract, 1.6% tryptone, 1% NaCl) was inoculated with 20 μL of frozen competent cell stock and grown overnight at 37 °C in an orbital shaker at 240 rpm. The following day, 30 mL of sterile 2xYT medium was combined with 0.3 mL of the overnight cell culture in a 250 mL baffle flask. The flask was placed in an orbital shaker under the same conditions until the desired growth of OD600 = 0.4 was achieved. The culture was centrifuged at 4680 × g, 4 °C for 5 min. The supernatant was removed, and the cell pellet was gently resuspended in 9 mL of frozen storage buffer (FSB) (10 mM KCl, 50 mM CaCl2·2H2O, 10 mM potassium acetate, and 10% (w/v) glycerol). The resuspended cells were incubated on ice for 10 minutes before being centrifuged again at 4680 × g, 4 °C for 5 min. The supernatant was removed, and the process of adding FSB, incubating on ice, and centrifuging was repeated two more times. After the final round of centrifugation, the supernatant was discarded, cell pellet was resuspended in 2.4 mL of FSB, and 100 μL aliquots were placed in sterile microfuge tubes for storage at −80 °C. A 100 μL aliquot of competent cells and the GSS AntR plasmid were thawed on wet ice and gently homogenized. 5 μL of the plasmid stock was diluted in 995 μL of Milli-Q water and the absorbance at 260 nm was measured. This value was used to calculate the volume of DNA solution required to collect 50 ng of DNA which was added to the competent cells, stirred gently, then incubated on wet ice for 30 minutes. Cells were heat-shocked by placing in a water bath heated to 42 °C for 45 seconds. The sample was returned to the wet ice for 2 minutes before being collected in a microfuge tube with 2xYT medium. The tube was placed in an orbital shaker for 1 hour at 37 °C and 250 rpm. 100 μL of the transformed cell culture was spread onto 2xYT medium agar plates (2xYT medium with 1.5% agar and 0.1 mg mL−1 ampicillin at pH 7.0) and incubated overnight at 37 °C. For small-scale colony screening, eight isolated bacterial colonies were selected from plates to inoculate 2 mL of 2xYT medium with 0.1 mg mL−1 ampicillin for incubation at 37 °C and 240 rpm overnight and transferred to 25 mL 2xYT medium. When the 25 mL culture reached an OD600 of 0.4, it was induced by 1 mM isopropyl-β-D-thiogalactoside (IPTG) and 7.5 μM all-trans-retinal. The induced cell culture was incubated for 4 h at 37 °C and 275 rpm. The cells were collected through low-speed centrifugation at 4680 × g and 4 °C. The colony with the most intense pink colour and largest pellet was selected as the optimal colony for large-scale protein expression.For a large-scale expression of GSS AntR, 2 mL of 2xYT medium with 0.1 mg mL−1 ampicillin was inoculated with 20 μL of concentrated cell stock and placed in an orbital shaker overnight (16–24 hours) at 240 rpm and 30 °C. The overnight cell culture was transferred to 25 mL of 2xYT and returned to the shaker again for overnight growth. The culture was diluted to an OD600 of 0.1 in 1 L of 2xYT and allowed to grow in the shaker at 275 rpm until the target OD600 of 0.35–0.4 was reached. The cell cultures for retinal-free GSS AntR production were induced with 1 mM IPTG only but supplemented with 7.5 μM all-trans-retinal in the case of the natural abundance (NA) retinal-bound sample and returned to the shaker to grow for 4 hours. Once completed, the cell culture was centrifuged at 4680 × g for 15 min and the supernatant was discarded. The cells were washed with 150 mM NaCl and stored at −20 °C before protein purification.
To purify retinal-bound or retinal-free GSS AntR, the cell pellet was thawed and resuspended in 30 mL of lysis buffer (0.15 M NaCl, 0.05 M Tris, 1 mM MgCl2·7H2O) with 15 mg lysozyme and 6 μL of DNAse and mixed (IKA VIBRAX) in the dark at 5000 rpm for 4 hours. The cells were broken into fragments using an ultrasonic probe sonicator (Fisher Model 500 Sonic Dismembrator, 1 min 45 s at 100% amplitude, followed by five rounds of 2 min and 30 s, with 30 s on and 30 s off, at 45% amplitude) and then centrifuged at 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g for 50 min to separate the membrane fragments from the supernatant. The supernatant was discarded, and the membrane pellets were resuspended in solubilization buffer (5 mM Tris, 1% n-dodecyl-β-D-maltopyranoside (DDM), pH 7.5) and allowed to mix overnight at 4 °C. The solubilized sample was, again, centrifuged at 150000 × g for 50 min. The supernatant was retained and combined with Ni2+-NTA-agarose resin and 10× binding buffer (3 M NaCl, 0.5 M Tris, 1% DDM, 10 mM imidazole, pH 8) and gently rocked at 4 °C overnight.
000 × g for 50 min to separate the membrane fragments from the supernatant. The supernatant was discarded, and the membrane pellets were resuspended in solubilization buffer (5 mM Tris, 1% n-dodecyl-β-D-maltopyranoside (DDM), pH 7.5) and allowed to mix overnight at 4 °C. The solubilized sample was, again, centrifuged at 150000 × g for 50 min. The supernatant was retained and combined with Ni2+-NTA-agarose resin and 10× binding buffer (3 M NaCl, 0.5 M Tris, 1% DDM, 10 mM imidazole, pH 8) and gently rocked at 4 °C overnight.
The resin was added to a column filter and the contents that did not bind to the resin flowed through to be discarded. The resin was washed several times with washing buffer (0.3 M NaCl, 0.05 M Tris, 35 mM imidazole, 0.5% DDM, pH 8) to remove any residual contaminants from the column. With each addition of the washing buffer, the contents that flowed through the column were assayed using a UV-Vis spectrophotometer (Cary 50) and the absorption peak at 280 nm was monitored. When the peak amplitude fell below an OD of 0.05, the resin was considered sufficiently clean. At this point, the bottom of the column was capped and 30 mL of elution buffer (0.3 M NaCl, 0.05 M Tris, 0.5 M imidazole, 0.5% DDM, pH 8) was added to the resin and allowed to sit for 10 min. Next, the cap was removed and the contents that flowed through the filter column were collected. The sample was added to a 10000 MW Amicon concentrator centrifuge tube and centrifuged at 3500 × g and 4 °C to concentrate the sample and remove the elution buffer. The sample was flushed with reconstitution buffer (5 mM NaCl, 10 mM Tris, 0.05% DDM, pH 8) to ensure all imidazole was removed. In the case of retinal-bound GSS AntR, the protein yield was ∼6.2 mg of purified protein per 1 L culture. In the case of retinal-free GSS AntR, the yield had to be verified by titrating a small aliquot of the purified protein with a 0.6 mM isopropanol solution of all-trans-retinal and the 382 nm peak of free retinal and the 550 nm peak of GSS AntR had to be monitored to ensure the saturation. The yield of retinal-free GSS AntR was found to be ∼2.5 mg of purified protein per 1 L culture, and it was reconstituted with an equimolar amount of biosynthetically produced 13C-labeled all-trans-retinal, produced as described below. To improve the optical properties of the sample and to remove insoluble impurities, the 13C-retinal-reconstituted GSS AntR was filtered using a 0.22 μm syringe filter.
To prepare lipids for proteoliposome reconstitution, 10 mg of 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) and 1.1 mg of 1, 2-dimyristoyl-sn-glycero-3-phosphate (DMPA) with chloroform were added to a round bottom flask and mixed with a stir bar for 2 hours. The chloroform was evaporated using a gentle stream of air and the lipids were dried under a vacuum overnight. Once dried, 1 mL of reconstitution buffer without DDM was added to the lipid film and resuspended using a stir bar and a water sonication bath. The protein samples were reconstituted into liposomes comprising DMPC/DMPA lipids (Avanti) (9:1) (w/w) at a protein-to-lipid ratio of 2:1 (w/w). The rhodopsin sample was added to the lipid mixture with additional Triton X-100. The target protein concentration was 3.74 A550/mL, the target lipid concentration was 1.16 mg mL−1, and the target Triton X-100 concentration was 0.8 mg mL−1. The detergent was removed by incubating the sample with 0.8 mg of Biobeads (SM2, Bio-Rad) per 1 mL of sample for 24 hours. The proteoliposomes were removed from the Biobeads using a syringe and centrifuged at 150000 × g for 30 min at 4 °C.
Samples and experimental set-ups for functional and initial spectroscopic characterization of GSS AntR
Ion transport assays were performed on whole E. coli cells from 1 L culture collected at 4680 × g and 4 °C for 10 min. The cells were washed three times with unbuffered solution (10 mM KCl, 10 mM MgSO4, and 100 μM CaCl2). One third of the cell pellet was resuspended in 25 mL of the unbuffered solution. Gently stirred cell suspensions were illuminated with yellow light (Cole Parmer 9741-50 illuminator) using a glass filter (>460 nm), and the pH changes were monitored using a glass electrode (Accumet Microprobe extra long calomel combo electrode) and recorded using a digital oscilloscope (Agilent Technologies DSO 1052B Digital Storage Oscilloscope). 10 μM of the proton uncoupler CCCP (from ethanol stock) was added in control experiments.For spectroscopy in the visible range, the cells were sonicated (Fisher Sonic Dismembrator Model 500) to produce fragments of membranes small enough to minimize light scattering, as described previously.45 The sonicated sample was spun-down at 4680 × g and 4 °C for 10 min and the supernatant was collected. The membrane fragments were then sedimented using ultracentrifugation at 150000 × g and 4 °C for 50 min, and a soft upper part of the pellet containing smaller membrane fragments was collected and centrifuged at 30000 × g for 10 min to remove larger membrane fragments. Flash photolysis experiments were run on these smaller membrane fragments encased in polyacrylamide gels. The gels were prepared using 700 μL of the membrane suspension, 300 μL of the 33% acrylamide and 1% bisacrylamide solution mixture, 2.4 μL of 10% ammonium persulfate, and 3 μL of N,N,N′,N′-tetramethylethylenediamine. After solidification, the gels were washed with 1 L of distilled water for at least 4 h at room temperature and stored at 4 °C in distilled water. If different pH and salt conditions were required, the storage buffer was replaced by soaking the gel in an appropriate buffer (100 mL or more) for at least 90 minutes prior to the measurement.
Static spectroscopy was performed on a Cary 50 spectrophotometer (Varian). Flash-photolysis spectroscopy was performed using a custom-built single-wavelength spectrometer described elsewhere.46 In short, the photocycle was initiated with 7 ns pulses of the second harmonic of an Nd-YAG laser at 532 nm (Continuum Minilite II). Absorption changes of the monochromatic light (provided by an Oriel QTH source and two monochromators) were observed using an Oriel photomultiplier, an amplifier with a 350 MHz bandwidth, and a Gage AD converter (CompuScope 12100-64M). Kinetic traces were averaged (normally 200–1000 traces) and converted into a quasi-logarithmic time scale using in-house software.
For Raman spectroscopy, the proteoliposome pellet of GSS AntR with natural abundance retinal was hydrated with a buffer of desired pH and 5 μL of wet paste was added to the well on the slide. For the pH 9 measurements, the sample was hydrated with NMR buffer (10 mM NaCl, 25 mM CHES, pH 9) and spun-down to remove excess buffer to form the wet paste. For the pH 4 measurements, the sample was hydrated with a buffer comprising 5 mM NaCl and 10 mM potassium acetate. The Raman spectra were obtained using the FRA106/s accessory to a Bruker IFS66vs spectrometer, with Nd-YAG laser excitation at 1064 nm, at a 4 cm−1 resolution with OPUS software. For each experiment, a minimum of 1000 scans were averaged.
13C-all-trans-retinal biosynthesis
The biosynthetic production of 13C-all-trans-retinal in E. coli followed the previously established protocol38 with minor improvements and modifications. Briefly, we used BL21 Gold E. coli cells (Agilent Technologies) transformed with two plasmids: chloramphenicol-selectable pAC-Beta-ipi (Addgene) and ampicillin-selectable pKA103 (custom-made in K.-H. Jung's lab, Sogang University, Seoul, South Korea). The plasmid pAC-Beta-ipi encodes the CrtE, B, I, Y, and isopentenyl diphosphate isomerase (ipi) genes which synthesize β-carotene from FPP precursor under a constitutively active promoter.47,48 During growth, the accumulation of β-carotene gives the cells an orange colour. The plasmid pKA103 is derived from plasmid pKJ900 and contains genes for C-terminally 6xHis-tagged GPR and 15′-15′-β-carotene dioxygenase, the enzyme responsible for cleaving β-carotene into two retinal molecules.49 Expression of the β-carotene cleavage enzyme is regulated by L-arabinose. The addition of 0.2% (w/v) L-arabinose causes the orange colour to transition to white as the β-carotene is consumed and converted to retinal. The production of GPR is regulated by a lac operon promoter inducible by IPTG. With the addition of IPTG to the cells, the bacteria begin producing GPR, which will combine with the endogenously produced retinal giving an observable pink colour. The intensity of the colour can be used for the small-scale colony re-screening, which is required for optimal production of retinal from the old frozen cell stocks.10 μL of the thawed cell stock from storage at −80 °C was added to 1 mL of 2xYT medium to produce three dilutions of 1:100, 1:10000, and 1:1000000. 20 μL of each dilution was added to cell culture plates containing 2xYT with 1.5% (w/v) agar, 50 μg mL−1 chloramphenicol, and 100 μg mL−1 ampicillin. Plates were placed upside-down in an incubator at 37 °C overnight. Eight of the best colonies were selected based on size and grown independently in 2 mL of 2xYT medium overnight in an orbital shaker at 37 °C. A 20 μL aliquot of the overnight cell culture was added to 2 mL of a minimal medium, M9 (0.1% (w/v) NH4Cl, 0.6% (w/v) Na2HPO4, 0.3% (w/v) KH2PO4, 0.05% (w/v) NaCl, 1 mM MgSO4, 0.1 mM CaCl2, 0.5 mM trace metals, 0.4% glucose, and 100× BME vitamins) with antibiotics (50 μg mL−1 chloramphenicol and 100 μg mL−1 ampicillin) overnight at 30 °C. The culture was diluted to an OD600 of 0.1 and returned to the orbital shaker to grow at 275 rpm until the OD600 between 0.35 and 0.4 is reached. At this point, cells were induced with 0.2% (w/v) L-arabinose and 1 mM IPTG and continued to grow in the orbital shaker for 21 hours. The cell cultures were centrifuged at 5000 × g for 10 min at 4 °C. The resulting pellets were compared by size and the intensity of pink color, and the optimal cell colony was selected.
For the large-scale 13C-all-trans-retinal production, the selected colony was grown in M9 minimal medium with 13C-glucose and ammonium chloride as the sole carbon and nitrogen sources, respectively. Ten 2 mL aliquots of M9 were inoculated with 20 μL of the cell stock and placed in an orbital shaker at 30 °C and 240 rpm for 16 hours. Each 2 mL culture was added to its own 100 mL of M9 and grown to an OD600 of 0.35. Each flask was induced with 0.2% (w/v) L-arabinose, and 25 mL of n-octane was layered on top for retinal extraction and placed back in the orbital shaker for 48 hours. The cells were combined into FEP bottles and centrifuged at 4680 × g for 10 min at 4 °C. Centrifugation results in phase separation of the cell culture based on density and hydrophobicity.50 The upper layer contains the less dense octane, the lipophilic retinal, and other hydrophobic solutes. The lower layer contains the aqueous medium and cells. The top layer was carefully extracted and filtered, first through a medium porosity sintered glass funnel under vacuum and then through a 0.22 μm PVDF syringe filter fitted to a glass syringe. The filtered medium was added to a round-bottom flask and secured under a fume hood where the octane was evaporated using a gentle stream of air. Next, 5 mL of 2-propanol was added and the retinal was resuspended using a gentle swishing motion. A 5 μL aliquot of the sample was diluted in 2-propanol and the absorbance of the sample at 382 nm was measured. After correcting the spectrum for scattering, the yield of the 13C-retinal was estimated as ∼0.25 mg per liter of culture.
Solid-state NMR
To prepare the samples for ssNMR, the proteoliposomes of GSS AntR with biosynthetic 13C-all-trans-retinal were centrifuged at 900000 × g for 6 hours to remove excess water. The pellet was packed into a 3.2 mm thin-walled ssNMR rotor (Bruker) for analysis. All NMR experiments were carried out on a Bruker Avance III 800 MHz spectrometer using a Bruker EFREE 3.2 mm MAS triple resonance 1H/13C/15N probe. One-dimensional 13C spectra were recorded at MAS rates of 14.3 and 13.3 kHz and at a sample temperature of ∼5 °C using cross-polarization (CP) for excitation with a 50 kHz radiofrequency field applied on the carbon channel, and with the proton radiofrequency field ramped linearly around the n = 1 Hartman–Hahn condition.51 Protons were decoupled during 13C acquisition using 83 kHz SPINAL64 decoupling.52 Two two-dimensional (2D) 13C–13C correlation spectra were recorded at 14.3 kHz with 10 ms and 30 ms dipolar-assisted rotational resonance (DARR) mixing.53 A third spectrum was collected at 14.3 kHz using radiofrequency driven recoupling (RFDR) with a mixing time of 16 rotor cycles.54 2D spectra were collected with spectral widths of 296 and 300 ppm in the direct (t2) and indirect (t1) dimensions, respectively, with acquisition lengths of 20 ms in t2 and 12.72 ms in t1. 40 scans per increment were collected with a recycle delay of 1.7 s. The data were processed using Bruker BioSpin TopSpin (version 4.0.9) and analyzed using CARA software.55 Carbon chemical shifts were indirectly referenced to DSS through the 13C adamantane downfield peak resonating at 40.48 ppm.56
Molecular dynamics simulations
To dock the retinal molecule to the protein, we used Modeller 10.458,59 to prepare homology models of retinal-bound GSS AntR based on (i) the structure of M. repens proton-pumping mutant (6wp8), which is among the structural templates used to generate GSS_1 and GSS_2 (Table S1, ESI†); (ii) the structure of SzR4, which was used as a template for GSS_3. In each of these two sets of homology modeling computations, we generated ten models and selected the models with the best DOPE score, Model_i and Model_ii. We then used VMD to overlap Model_i onto the ColabFold structural models GSS_1 and GSS_2, and Model_ii onto GSS_3.
The resulting retinal-bound structural models were then subjected to coordinate driving computations to set retinal in an all-trans geometry, followed by geometry minimizations using CHARMM. During these CHARMM computations, we allowed changes of the Cartesian coordinates of the retinal molecule, the covalently bound Lys, and 25 amino acid residues from the retinal binding pocket (498 atoms in total); all other protein coordinates were kept fixed.
610 water molecules, with chloride ions added for charge neutrality. We used the CHARMM36 force-field parameters for the protein, lipid molecules, and ions,64,65 the retinal force-field parameters,66,67 and the TIP3P water model.68 We treated short-range non-bonded interactions with a switch function between 10 and 12 Å, and Coulomb interactions, with the smooth particle mesh Ewald summation.69,70
For all MD simulations we used NAMD.71–73 During geometry optimization of the simulation system, heating to 300 K, and the first 1 ns of equilibration, we placed soft harmonic restraints of 4 kcal mol−1 Å−2 on the backbone hetero-atoms, 2 kcal mol−1 Å−2 on sidechains, and 1 kcal mol−1 Å−2 on water oxygen atoms and lipid headgroups. Four additional equilibration steps, 1 ns each, were performed with progressively weaker harmonic restraints on hetero-atoms: restraints were first halved; in the next step, we used harmonic restraints of 1 kcal mol−1 Å−2 on the hetero-atoms of the protein backbone, retinal, water, and ions; all these restraints were then reduced to 0.5 kcal mol−1 Å−2; one last 1 ns equilibration step was performed with all harmonic restraints switched off. Simulations were continued with production runs (free of any harmonic restraint), and we consider as the origin of time (t = 0) the start of the production runs.
We used the NVT ensemble (constant number of atoms N, constant volume V, and constant temperature T) for heating the simulation systems, and the NPT ensemble (constant N, constant P, and constant T) for all subsequent equilibration and production runs. For the NPT simulations, we used a Langevin dynamics74 scheme and a Nosé–Hoover piston. We used an integration step of 1 fs during heating, equilibration, and the first 1 ns of the production runs; for the remainder of the production runs, we used a multiple time integration scheme75 with 1 fs for the bonded forces, 2 fs for short-range non-bonded, and 4 fs for long-range non-bonded forces. We kept fixed the lengths of all covalent bonds to H atoms.76
The production runs were extended to 399 ns for the reference simulation of GSS AntR with default ColabFold selection of multiple templates, and to 311 ns each for the GSS AntR modeled using manual selection of multiple templates (labeled as repeat #1) and using the structure of SzR4 as a template (labeled as repeat #2).
Earlier computations on BR indicated that the retinal C15 atom could H-bond to a cytoplasmic water molecule79 and, indeed, upon visual inspection of the simulations we report here, we identified transient water H-bonding with the C15–H15 group. Consequently, both the Schiff base nitrogen and C15 retinal atoms were included in the H-bond graph computations.
The graph of hydrophobic contacts between protein sidechains was computed with the Bridge/Bridge2 interface with a distance criterion of 4.5 Å between carbon atoms of the hydrophobic sidechains.
All graph computations were performed using ∼20000 equally spaced coordinate snapshots from the last ∼200 ns of each simulation. Upon close inspection of the H-bond graphs, we chose to show in the H-bond graphs the H-bonds that have a minimum occupancy of 10%, i.e., which are present in at least ∼2000 coordinate sets. This H-bond occupancy threshold allows us to illustrate the transient water bridging of the retinal Schiff base C15–H15 group and Ser69 via a cytoplasmic water molecule.
Time series for selected interatomic distances were monitored from the entire length of the production runs.
Results and discussion
Initial characterization of GSS AntR
The previously uncharacterized Antarctic SzR (GSS AntR, IMG Gene ID: Ga0307935_10093322) has an unusual helix C motif, in which the leading Phe (homolog of BR's D85) is replaced by Gly and the terminal Glu (homolog of BR's D96) is replaced by Ser, converting the typical FSE motif of SzRs into GSS (Fig. S1, ESI†). Despite having a unique helix C motif and a number of other differences in the primary structure with other SzRs/AntRs, GSS AntR conserves the residues deemed to be central for inward proton transport in AntR with the FSE motif (which we will call FSE AntR to avoid any confusion), such as Arg67, Ser74, Cys75, Asp185, and Tyr193 (FSE AntR numbering).22 Accordingly, illumination of E. coli cells expressing GSS AntR showed robust alkalinization of the unbuffered external medium, which is removed by the protonophore CCCP, indicating active inward proton transport (Fig. S2A, ESI†). This result is consistent with both the lack of strict conservation of the helix C motif (other than the central Ser) in SzRs and with the functional phenotype of the E81Q mutant of FSE AntR.13,18,22 The robust inward proton pumping shown by GSS AntR is consistent with its rather fast photocycle (Fig. S2C, ESI†), which displays about 3× faster deprotonation and reprotonation of the Schiff base (M-like intermediate observed at 400 nm) than FSE AntR.22 It also shows a significantly faster photocycle turnover, as judged from the last sub-second/second photocycle phase, than that observed for FSE AntR, fully relaxing by about 400 ms.Similar to FSE AntR and some other SzRs,22,80 GSS AntR shows acid-induced blue-shift of the absorption spectrum maximum (Fig. S2B, ESI†), presumably due to protonation of the helix G counterion. While FSE AntR showed a shift from 558 to 538 nm between pH 9 and 4,22 GSS AntR shows a smaller shift from 544 to 534 nm. We previously showed that the spectral blue-shift of FSE AntR at low pH is associated not only with the counterion protonation but also with retinal isomerization from all-trans to 13-cis-15-syn.22 Interestingly, such retinal isomerization at low pH is not observed in GSS AntR, as obvious from nearly identical Raman spectra at pH 9 and 4, showing predominantly all-trans-retinal at both pH values (Fig. S2D, ESI†). It is thus possible that the smaller acid-induced spectral shift is associated with the lack of trans–cis isomerization, which is normally accompanied by a blue-shift of the visible spectrum. Besides the characteristic fingerprint C–C stretching bands of all-trans-retinal (at 1200, 1211, and 1163 cm−1),81 we note the CN stretching vibration of the Schiff base at 1640 cm−1 (almost identical to that in FSE AntR)22 and CC ethylenic stretch at 1528 cm−1. The latter shifts to 1530 cm−1 at pH 4 in accordance with the blue-shift in the visible absorption.82 Finally, one needs to note two prominent hydrogen out-of-plane (HOOP) vibrations, at 990 cm−1 (observed as a shoulder of the methyl rocking peak at 1005 cm−1) and 961 cm−1. Both HOOP bands are typical for SzRs and have been assigned to the C15H and C11HC12H wagging modes of the retinal (Fig. S3, ESI†), respectively, indicating a significant twist of the polyene chain in these areas.16,20
Structural characterization of GSS AntR based on atomic-level MD simulations and graph analyses of H-bond and hydrophobic network interactions
During all three independent simulations we performed, bulk water molecules enter the inter-helical region of the protein, where they participate in a dynamic protein–water H-bond network (Fig. 1, 2 and Fig. S4–S6, ESI†). On average, the H-bond network of GSS AntR has about 45 H-bond connections between the protein sidechains (these can be direct H-bonds between the sidechains, or water bridges with up to three water molecules between the sidechains; Fig. 2C); when water bridging is excluded from the H-bond computations, GSS AntR has only about 15 H-bonds (Fig. 2D). The finding that more than half of the protein's H-bond network is mediated by water is compatible with recent computations based on a high-resolution structure of bovine rhodopsin.83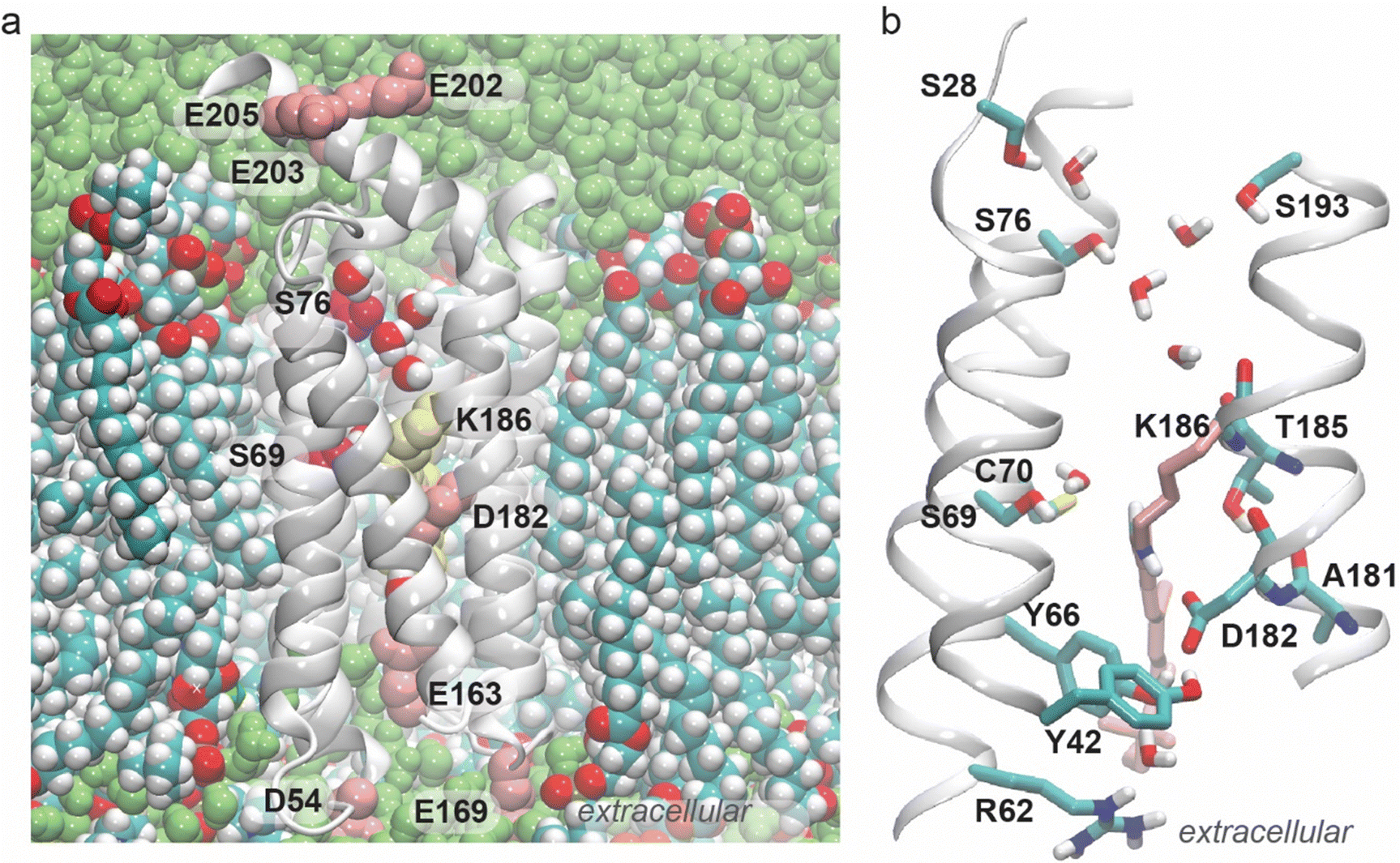 | ||
| Fig. 1 Water-mediated H-bonding of GSS AntR. During the simulations, water molecules enter the inter-helical region of the protein, where they mediate H-bond networks in both halves of the protein. The C15H retinal bond forms a water bridge to Ser69, whereas the Schiff base connects directly to Asp182. (a) Cut-away view of GSS AntR in a hydrated lipid membrane. The retinal molecule and selected protein sidechains are shown as van der Waals spheres. Note the water chain between Ser76 and the vicinity of the Lys186 backbone. (b) Close view of the retinal and of the internal water-mediated H-bond network; the retinal and Lys186 are shown as pink bonds, with the exception of the Schiff base NH and C15H bonds, which are shown in atom colors. | ||
 | ||
| Fig. 2 H-bond network computed from the main simulation of GSS AntR. The minimum H-bond occupancy used is 10%. Nodes that are filled with light green are residues potentially important for function. H-bond network (a) and time series of the number of H-bonds in the graph (c) obtained for direct H-bonds between sidechains, and water bridges with up to three water molecules. The corresponding H-bond graphs computed only for the water bridges, without direct H-bonds between the sidechains, are presented in Fig. S7 and S8 (ESI†). H-bond network (b) and time series of the number of H-bonds (d) obtained when only direct H-bonds between the sidechains are included in computations. (e) Time series of the H-bond contact between the retinal and Ser69 in the H-bond graph from panel (a), extracted with a Connected Component Analysis in Bridge2. (f) Time series of the number of water molecules within 3.5 Å distance from the retinal C15 atom along the entire simulation. Note that the H-bond graph computation from panels (a–e) identify H-bonds based on both distance and angle criteria, and thus are more restrictive. | ||
In all three simulations performed, we found that several water molecules visit the cytoplasmic half of the protein, and overall the H-bond networks computed from the three simulations are very similar. For clarity, we discuss the H-bond network of the reference simulation, which is that of GSS AntR modelled using the standard ColabFold selection of the multiple templates. We then compare the H-bond networks computed from the reference simulation with the two repeats (Fig. S4–S6, ESI†).
At the cytoplasmic side of the retinal, one water molecule comes to within the H-bond distance (3.5 Å) of the retinal C15 atom in ∼100 ns of the simulation (Fig. 2F); one water molecule, very rarely two, is close to retinal C15 during the trajectory segment from ∼200 ns to ∼300 ns, but none for the next ∼50 ns, and then a water molecule, and, very infrequently, two, is close to the retinal C15 atom during the last ∼30 ns of the simulation (Fig. 1B and 2F).
The stricter H-bond criteria used for the H-bond graph computations we present include both the distance and the H-bond angle. According to these, H-bond contacts between the retinal and Ser69 are sampled at the same time intervals as those in which a water molecule is found within the H-bond distance from retinal C15 (Fig. 2E and F), and the average length of the water bridge between the retinal and Ser69 is one H-bonding water molecule. The H-bond connection between the retinal and Ser69 disappears when the H-bond graph is computed only for direct H-bonds between sidechains (Fig. 2B) – in that graph, the retinal Schiff base NH group forms a direct H-bond to Asp182 (Fig. 1B, 2B and Fig. S4B, ESI†). Likewise, the water-mediated bridge with Ser69 is the only connection found for the retinal when the H-bond network is computed only for water-mediated bridges, without contributions from direct H-bonds (Fig. S7 and S8, ESI†).
The water-mediated H-bonding between the retinal C15H group and the hydroxyl group of Ser69 is particularly important because Ser69 is the first Ser of the GSS motif (Fig. 1 and Fig. S1, ESI†). It corresponds to BR's Thr89 (Fig. S1, ESI†), which plays a key role as an intermediate carrier for the transfer of protons from the retinal Schiff base to Asp85, according to quantum mechanical molecular mechanical computations (QM/MM) and experiments.84–86
The one-water-mediated bridge between the retinal C15H group and Ser69 is somewhat infrequent, with an average H-bond occupancy of ∼14% (Fig. S4, ESI†). The mostly direct inter-helical H-bond between Ser69 and Thr38 is sampled frequently, ∼89% of the time (Fig. S4A (ESI†), average water bridge length = 0.1), whereas C70 (Fig. 2B) makes only extremely infrequent (<1% of the time) H-bond contacts such that it is absent from the H-bond graphs shown in Fig. 2. One helical turn from Thr38 towards the cytoplasmic side, Ser35 forms direct, frequent inter-helical H-bonds with Tyr190 and Ser15 (H-bond occupancies >50%; Fig. 2B and Fig. S4, ESI†). Infrequently, Tyr190 can connect to Ser76 (the second serine of the GSS motif), mostly via one water molecule (Fig. S4, ESI†). A chain of 2–3 water molecules can transiently bridge the hydroxyl group of Ser76 to the backbone carbonyl group of Thr185 (Fig. 1B).
In addition to Tyr190, Ser76 forms water bridges to Ser28, Ser193, and Asn200. Asn200 is close in the amino acid sequence to three carboxylic groups – Glu202, Glu203, and Glu205 – and it forms water bridges to Glu203. Each of the three carboxylic sidechains forms water bridges to each other and to other nearby polar and positively charged groups, while Glu203 connects to His29 via water and Arg204 (Fig. 2A, Fig. S4 and S5, ESI†). The overall organization of the H-bond cluster of Ser76, Glu202, Glu203, and Glu205 remains largely the same when the H-bond graph is computed only for water-mediated bridges between sidechains (compare Fig. 2A with Fig. S7 and S8, ESI†), but it is largely reduced to a handful of single H-bonds and small localized clusters when the graph is computed without water-mediated bridges, and only for direct H-bonds between sidechains (Fig. 2B). Moreover, when H-bonded water molecules are not part of the H-bond graph computation, Ser76 is absent from the H-bond network (Fig. 2B), which further indicates that water-mediated H-bond bridges between protein sidechains are largely responsible for the extended H-bond network at the cytoplasmic side of GSS AntR.
The local cluster of Glu sidechains with a nearby His is reminiscent of the clusters of carboxylic (and, sometimes, histidine) sidechains that have been hypothesized to function as a proton antenna cluster in BR,87–90 cytochrome c oxidase,91,92 and the soluble PsbO subunit of photosystem II;93,94 in such a cluster, interactions of the Glu sidechains with Arg/Lys, and dynamic water-mediated bridges between protein sidechains, help to control the relative orientation of the carboxylic sidechains.95 Based on these similarities, we suggest that the three carboxylic Glu sidechains collectively help collect the proton released from the retinal Schiff base.
The all-trans-retinal Schiff base (formed by Lys186) remains connected to the extracellular H-bond network via Asp182, a residue that also has direct H-bonds to Tyr42 and Tyr66, and a water-mediated bridge to Arg62 (Fig. 2 and Fig. S4, ESI†). Through the direct H-bond between Arg62 and Glu163, it further connects to the cluster of direct H-bonds mediated by Glu163 (corresponding to BR's Glu194, Fig. S1, ESI†); when only water-mediated bridges between sidechains are included in the H-bond graph computation, the Asp182 H-bond cluster is disconnected from both the retinal Schiff base and the extracellular side (Fig. S7 and S8, ESI†).
The backbone carbonyl of Asp182 samples infrequent H-bonding with the Thr185 hydroxyl group, which prefers to H-bond to the backbone carbonyl group of Ala181 (Fig. 1B and Fig. S9, ESI†). Intra-helical H-bonds between a Thr hydroxyl group and the backbone carbonyl of the i-4 residue, as found here for Thr185 and Ala181 in all three simulations performed (Fig. S9, ESI†), are compatible with the high propensity of Ser and Thr hydroxyl groups for intrahelical H-bonds.96,97 Such H-bonds compete with amide groups for H-bonding to carbonyl groups. The competition between the hydroxyl and amide groups for backbone carbonyl H-bonding can associate with a locally more dynamic helical segment.98 The persistent H-bond distance between the Thr185 sidechain and the Ala181 backbone carbonyl group explains why Thr185 is absent from the H-bond graphs presented in Fig. 2 and Fig. S4–S6 (ESI†): Thr185 prefers to H-bond to the backbone, whereas with the H-bond graphs we focus on the H-bonds between sidechains and water-mediated bridges between sidechains.
In addition to their highly stable H-bonding to Asp182, Tyr42 and Tyr66 form water bridges to each other and to Arg62 (Fig. S4 and S5, ESI†); as it reaches across the inter-helical space to H-bond to Asp182, the Tyr66 sidechain effectively occupies the cavity near the Schiff base. Within about one helical turn of both Tyr42 and Tyr66, there are residues that contribute more persistent H-bonds: close to Tyr42, Thr38 H-bonds to Ser69 and, close to Tyr66, Arg62 forms a direct, persistent H-bond with Glu163. The latter residue also has persistent H-bonds to Trp63, Ser161, and Tyr178 (Fig. 2 and Fig. S4, ESI†).
As noted above, we obtained similar H-bond networks from the independent simulations of GSS AntR started from the other two structural models we derived (Fig. S5 and S6, ESI†). The water-mediated H-bond between the retinal C15H bond and Ser69, which is sampled infrequently in the reference simulation, is sampled, also infrequently, in these two additional simulations (average occupancies of 10% and 25%; Fig. S4A and S5A, ESI†). This suggests that such water-mediated bridging between the retinal and Ser69 could indeed be a signature of GSS AntR, rather than an artefact of the structural model. Energetically favourable interactions between the C15H bond of the all-trans-retinal and a cytoplasmic water molecule have been predicted by early computations on BR.79 Moreover, Ser69 corresponds to BR's Thr89, and the water molecule that bridges the C15H retinal bond in GSS AntR and Ser69 (Fig. 1B) would correspond to the cytoplasmic water molecule that bridges the Schiff base NH bond to Thr89 in the BR's L intermediate, for which experiments and computations found a key role as an intermediate proton carrier.84,85 The fact that the crystal structure of SzR4 lacks the cytoplasmic water molecule, yet such water molecule transiently visits GSS AntR in all three independent simulations we performed, including that based solely on the SzR4 structure as a template, is compatible with the finding that the water molecule comes to within H-bond distance from the retinal only infrequently (Fig. 2F), and the water bridge has low occupancy in all simulations (Fig. 2 and Fig. S4–S6, ESI†). Mobile water molecules that are not tightly bound to the protein can be invisible to structural biology;99 moreover, the structure of SzR4 was solved at a cryogenic temperature in a crystal environment, whereas our simulations are performed at room temperature in a fluid lipid membrane environment. In BR's ground state with the all-trans-retinal, mutation of Asp85 to Asn suffices for water molecules to rapidly enter the cytoplasmic half of the protein during atomic-level simulations, such that an H-bonded water chain can connect Asp96 to the Schiff base NH bond.100 In Gloeobacter rhodopsin (GR), a sextuple mutant with Thr125 mutated to Asp and Glu132 to Gln (BR T89 and D96, respectively) water bridges the Schiff base and Ser77 at the cytoplasmic side.101
We suggest that, within the limitations of our structural models and MD simulations, the remarkably stable H-bond network at the extracellular side of the Schiff base, particularly of the Tyr42–Tyr66–Asp182 cluster, explains why the Schiff base lacks H-bonding with water. Asp182, with its three direct sidechain H-bonds present at all times during the simulations, appears to be a poor attractant for water molecules to visit the Schiff base region, and the Tyr66–Asp182 H-bond functions as a gate that hinders water access (Fig. 1).
Taken together, the ensemble of the H-bond graph analyses we present here (Fig. 2 and Fig. S4–S6, ESI†) indicates that the Schiff base region is rather polar, and that its H-bond connections are qualitatively distinct in the two halves of the pump. The retinal connects to the extracellular side via a direct H-bond between the Schiff base and Asp182, where Asp182 has highly stable (occupancies ∼89–100%) direct H-bonds with Tyr42 and Tyr66 (Fig. 1B, 2A, B and Fig. S4, ESI†). In contrast, H-bonding between the retinal C15H and Ser69, and, more generally, H-bonding at the cytoplasmic side, is mediated by water bridges (Fig. 1B, 2A and Fig. S4–S8, ESI†).
The retinal polyene chain is largely surrounded by hydrophobic residues that connect non-polar residues to the retinal and residues of the H-bond network. The bulky sidechain of Trp63, which is part of the highly stable H-bonds of the extracellular Glu163 cluster (Fig. 2A, C and Fig. S4, ESI†), connects to a network of persistent hydrophobic contacts that essentially spans the entire protein (Fig. S10, ESI†). Ser161 of the same H-bond cluster (see the H-bond networks in Fig. 2A and B) is within 1.5 helical turns from Phe158 – which is within hydrophobic contact from the retinal, and part of a cluster of hydrophobic sidechains in contact with each other and with the retinal (see Trp63, Ile98, Met99, Phe118, Met122, Phe125, Phe154, Pro155, and Phe158 in Fig. S10, ESI†). Val34 and Leu73, which are close to Ser35 and Ser69, respectively, are also part of the hydrophobic contact network near the retinal (Fig. S10, ESI†). Adjacent in the sequence to Thr185, whose sidechain is near the Schiff base (Fig. 2A), Phe184 is part of a hydrophobic cluster with Val188, Val183 and Ile187 that reaches to Phe12 and, from here, to Val39 (Fig. S10C, ESI†), that is, to a sidechain adjacent in the sequence to Thr38 – which is part of the water-mediated H-bond cluster of the retinal C15H bond and Ser69 (Fig. 2A and Fig. S4, ESI†).
We suggest that the peculiar H-bond network we find from three independent simulations of GSS AntR has a key role in function. At the extracellular side of the Schiff base, Asp182 and Glu163 are central to local H-bond clusters that contain mainly direct H-bonds: the network that extends from the Schiff base to Glu163 (Fig. 1A) has 9 sidechains and just about 4–5 water molecules in the Arg62–Asp182 H-bond cluster (Fig. S4B, ESI†). The GSS AntR counterion Asp182 thus appears to be a poor water attractant, such that there is no water in direct contact with the Schiff base NH bond. Instead, a water molecule can transiently bridge the retinal C15H bond to Ser69. Nearby, at the cytoplasmic side of the retinal Schiff base, a chain of H-bonded water molecules can transiently connect the helix G backbone to Ser76, which is part of an extended H-bond cluster with a stretch of three carboxylic sidechains (see Glu202, Glu203, and Glu205 in Fig. 1B). Upon photoisomerization, a cytoplasmically oriented Schiff base NH bond could help transform the cytoplasmic water cluster into a proton-conducting wire, ultimately transferring the Schiff base proton to the cytoplasmic bulk. The Thr185 hydroxyl sidechain which, when retinal is all-trans, H-bonds to the backbone of Ala181 and Asp182, could become an H-bond partner to the carboxyl group of Asp182, thus compensating for the broken H-bond between Asp182 and the NH bond of the isomerized retinal.
Assignment of the carbon resonances of retinal in GSS AntR
Reconstitution of NA GSS AntR with biosynthetically produced 13C-all-trans-retinal allows for a complete carbon resonance assignment using 2D MAS carbon spectroscopy. This is in contrast to our previous experiments with GPR where both the protein and the retinal were 13C-labelled, resulting in a significant overlap of some ring and methyl resonances of the retinal with protein signals and preventing their assignments.38 The 1D CP-MAS ssNMR spectrum of retinal in GSS AntR (Fig. 3) shows many well-resolved carbon peaks, but there are also many peaks that result from either the natural abundance signals, e.g., the most obvious carbonyl resonances in the 170–180 ppm region, and possibly from 13C-labelled impurities co-extracted by octane. | ||
| Fig. 3 1D 13C CP-MAS ssNMR spectrum of NA GSS AntR with 13C-all-trans-retinal. The experiment was performed at a spinning rate of 14.3 kHz at 5 °C, and 4096 scans were collected. Peak assignments for specific carbon atoms, assigned using the 2D spectra shown below, included in red. Refer to Fig. S3 (ESI†) for the retinal structure and carbon atom numbering. Spinning side bands identified from a comparison with the spectrum collected at a spinning rate of 13.3 kHz are marked with asterisks. | ||
To minimize contributions from the NA signals and obtain retinal-specific spectral correlations for spectroscopic assignments, we collected a set of three through-space 2D ssNMR 13C–13C correlation spectra. The first two were obtained with 10 ms and 30 ms DARR mixing, where the shorter mixing time allows for correlation between atoms within 1–2 bond lengths, while the increased 30 ms mixing time additionally facilitates more distant correlations, within, roughly, 3–4 bonds. Chemical shifts for each carbon atom were assigned by identifying cross-peaks between two interacting nuclei and using the transfer of magnetization through space to “walk” from one atom to another through connectivities in the 2D spectra, assisted by previously known typical values of retinal 13C chemical shifts in BR and GPR.24,38,102 DARR enables long-range polarization transfers using through-space interactions between carbon spins. As this technique is combined with the CP excitation of carbons from neighboring protons, the signal may be attenuated for fully deprotonated carbon atoms within the retinal molecule (Fig. S3, ESI†). This also explains the asymmetrical intensity of some peaks generated using DARR (Fig. 4).
 | ||
| Fig. 4 2D DARR MAS ssNMR spectra of NA GSS AntR with 13C all-trans-retinal. The red spectrum was collected with DARR mixing of 10 ms, and the blue spectrum was collected with DARR mixing of 30 ms. The cross-peaks seen only at the longer mixing time are annotated in red. | ||
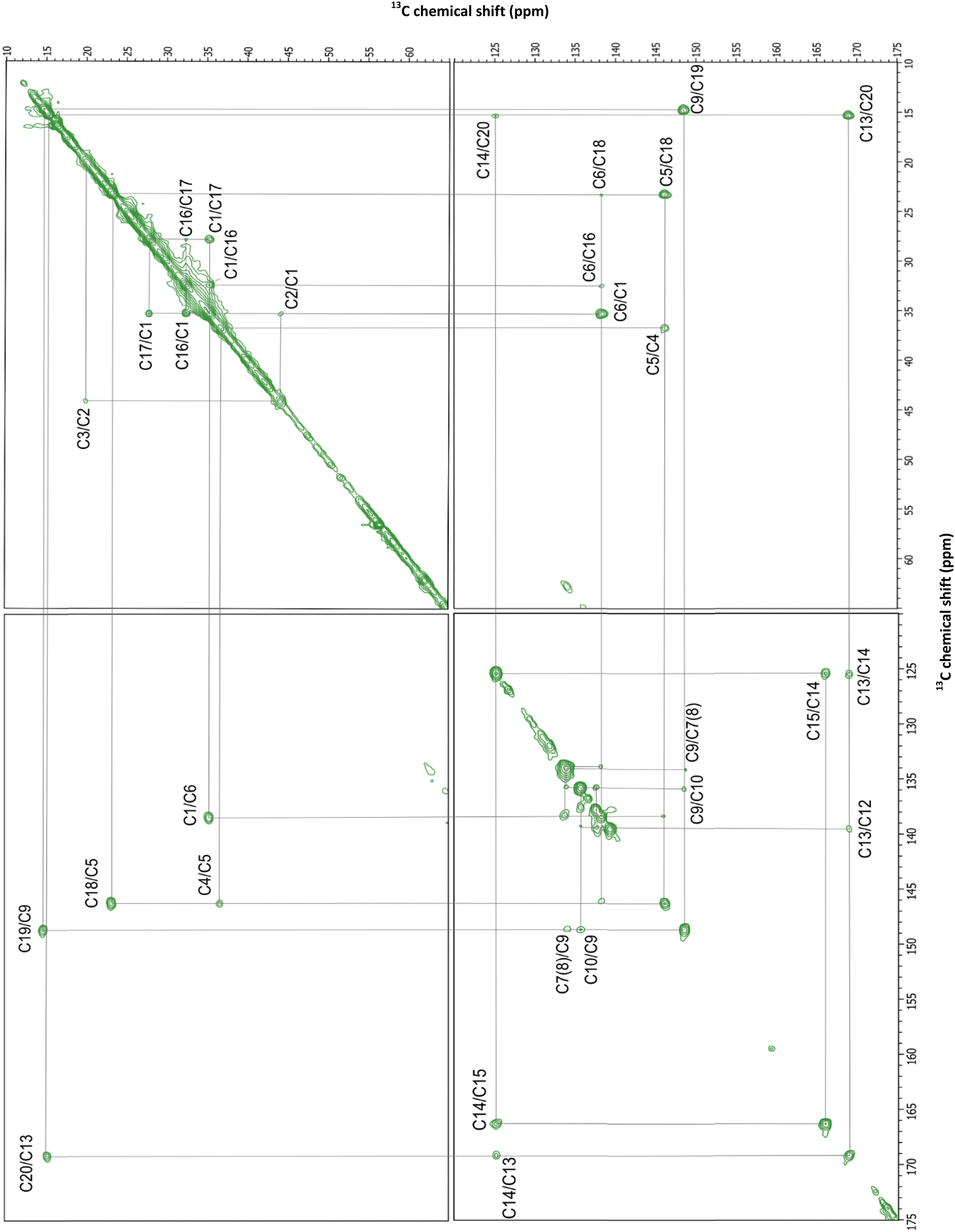
A strong cross-peak at 166.1/125.2 ppm corresponding to the correlation of C15 and C14 was used as the starting point on the DARR spectrum walk (Fig. 4). These values were close to the 163.7 and 121.8 ppm chemical shifts of C15 and C14 previously assigned in GPR.38 Both C14 and C15 demonstrate a two- and three-bond correlation to the C20 methyl carbon at 125.2/15.3 and 166.1/15.3 ppm, respectively. The observed chemical shift of 15.3 ppm for C20 closely aligns with the 15.7 and 15.1 ppm chemical shifts of C20 in GPR and BR (Table S2, ESI†).24,38 The methyl C20 is directly bonded to C13 and shows a strong correlation cross-peak at 15.3/169.1 ppm along with a reverse correlation cross-peak of similar intensity at 169.1/15.3 ppm. Additional cross-peaks at 15.3/139.3 and 139.3/15.3 ppm connect C20 to the next closest carbon atom through space, C12. Both C12 and C14 in turn show C13/C12 and C13/C14 correlations at 169.1/139.3 and 169.1/125.2 ppm, respectively, as well as the correlation between themselves at 139.3/125.2 ppm (Fig. 4 and 5).
 | ||
| Fig. 5 Selected region of the 2D DARR MAS ssNMR spectra of NA GSS AntR with 13C all-trans-retinal spanning 150–125 ppm, focusing on the cross-peaks of C6 to C11 atoms. The red and blue spectra correspond to mixing times of 10 ms and 30 ms, respectively. The cross-peaks seen only at a longer mixing time are annotated in red. | ||
C12 (139.3 ppm) is connected by a double bond to C11, which results in two strong and symmetric cross-peaks at 139.3/137.6 and 137.6/139.3 ppm (Fig. 4 and 5). In turn, C12 is separated by two bonds from C10, producing a cross-peak at 139.3/135.7 ppm. C10 demonstrated the strongest correlation with C11, generating similar intensity cross-peaks at 135.7/137.6 and 137.6/135.7 ppm. C10 also produces a cross-peak with C9 (at 135.7/148.6 ppm). C9 is directly bonded to the C19 methyl group which produces strong cross-peaks at 148.6/14.8 ppm and 14.8/148.6 ppm. Likewise, a cross-peak between directly bonded C9 and C8 (at 148.6/134.0 ppm) is clearly visible. To solidify these assignments, correlations of the methyl C19 with C8 and C10 can be observed (Fig. 4), at 14.8/134.0 and 14.8/135.7 ppm. A weak correlation of C10 and C8 can be also found at a longer mixing time (Fig. 5).
Due to the potential degeneracy of C8/C7/C6 resonances, walking further from C8 was ambiguous, so a second starting point at the C5 resonance was used, based on its strong correlation to two distinct atoms, β-ionone ring C4 and methyl C18, forming cross-peaks at 146.2/36.8 ppm and 146.2/23.3 ppm, respectively (Fig. 4). The chemical shift of the C18 methyl carbon and the shifts for C4 and C5 align well with values previously established in BR24 and thus were assigned with confidence. C4 and C18 also produce an isolated and intense correlation at 36.8/23.3 ppm. Finding the chemical shift of C4 provided a foundation for assigning values to other carbon atoms within the β-ionone ring. C4 is separated from C3 and C2 by one- and two-bonds, respectively, forming a resolved and intense peak with C3 at 36.8/20.0 ppm and 20.0/36.8 ppm, and an isolated but less intense peak with C2 at 36.8/44.1 and 44.1/36.8 ppm. Accordingly, C2 and C3 display a strong correlation at 44.1/20.0 ppm, and a weak correlation of C3 with C5 can be observed at a longer mixing time. In turn, C2 interacts strongly with C1 (35.3 ppm) and to a lower degree with C1's attached methyl groups, C16 and C17. These methyl groups form a distinct correlation cross-peak with other carbon atoms but the specific assignment of “C16” and “C17” is arbitrary. Both C16 (32.4 ppm) and C17 (27.8 ppm) interact with their attachment point C1 with equally strong intensity. In further support of these assignments, there are symmetrical cross-peaks at 27.8/32.4 and 32.4/27.8 ppm between the C16 and C17 methyl carbons, as well as weaker cross-peaks for C1/C3, C3/C17, and C3/C18 carbon pairs (Fig. 4).
While the chemical shift values were confidently assigned to seventeen of the twenty carbon atoms of retinal using the short and long mixing time DARR ssNMR spectra (Fig. 4 and 5), the chemical shifts of C6, C7, and C8 remained ambiguous due to the potential overlap and weaker signals from fully deprotonated C6. To obtain reliable assignments for these carbon atoms, an additional 2D carbon–carbon correlation experiment (Fig. 6 and 7) was conducted with RFDR mixing.54,103 In contrast to a second-order DARR recoupling which is mediated by protons, RFDR is the first-order recoupling method which relies on the direct carbon–carbon dipolar interaction and does not require the presence of protons. As such, it is more suitable for establishing correlations within low proton density moieties, such as the vicinity of deprotonated C6. In addition to the one-bond correlations obtained in the 2D DARR spectra, the 2D RFDR experiment permitted assignment of the previously ambiguous chemical shift of C6 (138.3 ppm) by observing the strong C6/C1 cross-peak at 138.3/35.3 ppm and by the presence of correlation cross-peaks with C16 and C18 at 138.8/32.4 and 138.8/23.3 ppm, respectively (Fig. 6). One can also note a weaker C5/C6 correlation (Fig. 7) at 146.2/138.3 ppm. C7 and C8 appear to have overlapping resonances that cannot be easily distinguished, but we tentatively assign C7 to 133.8 ppm and C8 to 134.0 ppm based on several of the observed correlations. First, the C7/C6 correlation at 133.8/138.3 ppm is seen in both DARR and RFDR spectra, clearly shifted from the previously mentioned C8/C9 and C8/C10 correlations at 134/148.6 and 134/135.7 ppm (Fig. 5 and 7). Likewise, one can see weak but distinct correlations of C7 with the methyl carbons of C16 and C17 in the longer mixing time DARR spectra, at 133.8/32.4 and 133.8/27.8 ppm (Fig. 4). Table S2 (ESI†) summarizes the chemical shift values obtained for all-trans-retinal within GSS AntR compared to the chemical shifts known for other microbial rhodopsins. Overall, the chemical shifts are consistent with the all-trans-configuration of retinal suggested by the Raman data (Fig. S2D, ESI†),24,81 and the 6-s-trans-configuration of the β-ionone ring (as judged from the values of C5 and C8 resonances).25
 | ||
| Fig. 6 2D RFDR carbon–carbon correlation spectrum of NA GSS AntR with 13C all-trans-retinal. A short RFDR mixing time of ∼1.119 ms (16 rotor cycles) was used to ensure that only short range, one- and sometimes two-bond correlations are observed. | ||
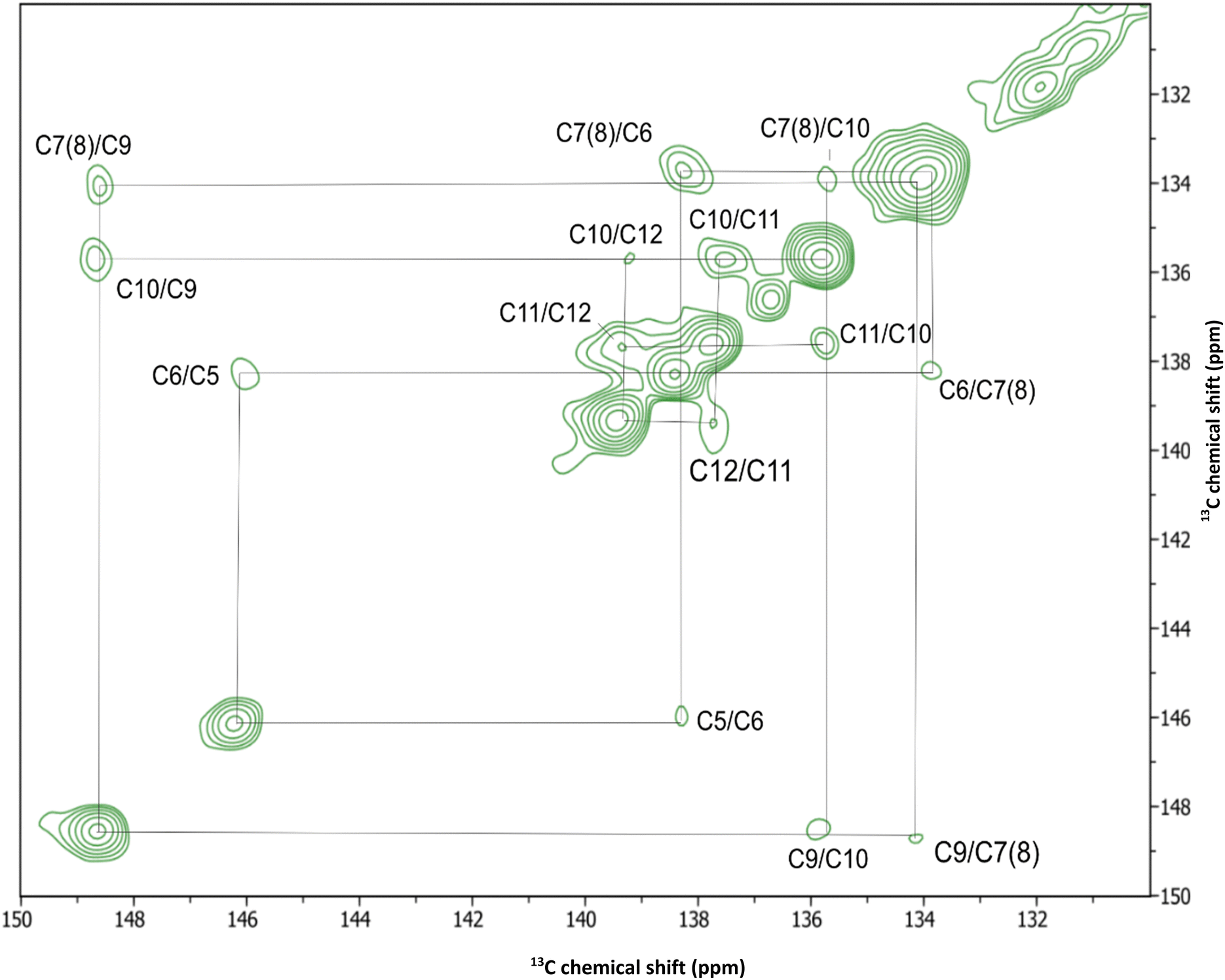 | ||
| Fig. 7 Selected region of the 2D RFDR carbon–carbon correlation spectrum of NA GSS AntR with 13C all-trans-retinal spanning 150–125 ppm, focusing on the cross-peaks of C6 to C11 atoms. | ||
Comparison of the retinal carbon resonances of GSS AntR and outward proton-pumping microbial rhodopsins
Fig. 8 highlights the differences between the chemical shifts of retinal in inward proton-pumping GSS AntR and outward proton-pumping microbial rhodopsins BR24 and GPR33,38 (based on the absolute values listed in Table S2, ESI†). While the retinal carbon chemical shifts among microbial rhodopsins have been fully assigned only for BR, it is instructive to take the incomplete dataset for GPR into consideration, in order to verify if the observed chemical shift differences may be related to the difference in function and not to the more subtle structure variations in the retinal binding pocket. Using this criterion, significant chemical shift differences consistently observed between GSS AntR and both BR and GPR are mainly localized in the proximal part of the polyene chain (C11–C15) and C7. Below, we discuss the possible causes of these differences using structure modelling and MD simulations along with the documented retinal chemical shift alterations known for other rhodopsins.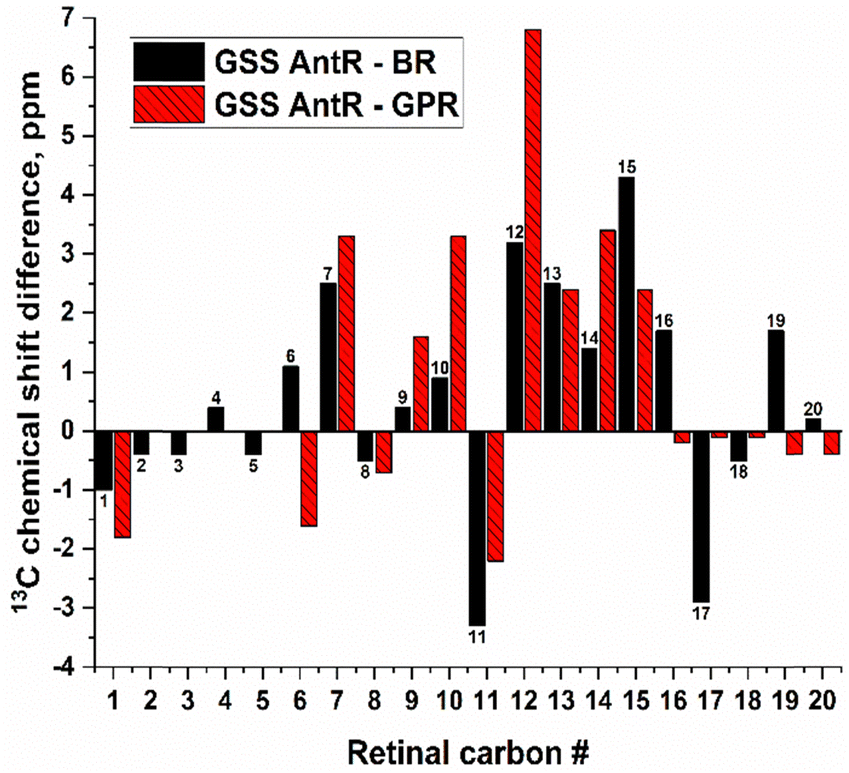 | ||
| Fig. 8 Differences of the retinal carbon chemical shifts in GSS AntR and BR (black), and GSS AntR and GPR (red). See the absolute values and sources in Table S2 (ESI†), and note that the GPR dataset is incomplete. | ||
Overall, the higher chemical shifts for the C12–C15 fragment in GSS AntR are consistent with the more polar environment in this region.25 This could be potentially contributed by the much higher proximity of the helix G aspartic counterion (Asp182, homolog of BR's Asp212) to the Schiff base and C15 (Fig. S10 and S11, ESI†) than in BR and GPR, the geometry well documented for other SzRs.16,18,20,22 Additionally, the proximity of the polar Thr185 sidechain (homolog of Ala215 of BR) to C15 (Fig. S11, ESI†) as well as the water molecule bridging its proton to Ser69 (Fig. 1 and 2) could contribute to its chemical shift increase as well. Proximity of negatively charged groups is well known to increase specific carbon chemical shifts of retinal, as was demonstrated for the C12 and C14 nuclei of the D85N mutant of BR and the C12 atom of the E181Q mutant of bovine rhodopsin.25,31 Consistent with that tendency, the C15 and C13 chemical shifts of KR2 (C15 close to Asp116) and 13-cis-15-syn-BR (C15 close to Asp212) are elevated (Table S2, ESI†). It should be noted that twisting of the polyene chain caused by a strong Schiff base–Asp182 interaction and suggested by the Raman data (Fig. S2D, ESI†) could contribute as well.28,104–106 Elevated C15 resonance in channelrhodopsin-2 may follow the same trend,42 as its helix G counterion distance is much shorter than in BR.107 Interestingly, the presence of polar non-charged residues can also significantly affect the carbon shifts, as was convincingly demonstrated for the L105Q mutant of GPR, in which C15 resonance was strongly upshifted, while C11 resonance was moderately downshifted,33 in parallel with our results (Fig. 8 and Table S2, ESI†). One should also note tight hydrophobic interactions suggested from the MD simulations, which may contribute to the elevated chemical shift of C7 (with the sidechain of Ile98) and C12 (with the sidechain of Phe154) (Fig. S10 and S11, ESI†).
An additional interesting feature of GSS AntR worthy of structural analysis is the asymmetry of C16/C17 ring methyl groups suggested by their vastly different carbon resonances (32.4 and 27.8 ppm). This is in stark contrast to BR (Fig. 8 and Table S2, ESI†) where these methyl carbons have identical chemical shifts.24 Until recently, such methyl asymmetry had been only observed for bovine visual rhodopsin (which has 6-s-cis-conformation of the β-ionone ring) and was thought to originate from a steric conflict with neighbouring sidechains, forcing one of the methyls into a proximity with C3 proton (so-called gamma effect).25,39,108 More recently, similar asymmetry was observed for two microbial rhodopsins, GPR and KR2, which possess 6-s-trans-conformation of the β-ionone ring (Fig. 8 and Table S2, ESI†).28,33 Even though the C16/C17 ring methyl asymmetry observed for GSS AntR cannot be considered a distinctive feature of inward proton pumps in view of a similar pattern found for GPR, it may give valuable information on its retinal binding pocket and β-ionone ring's fine geometry.
The difference in chemical shifts of C16 and C17 methyl carbons in GSS AntR is mirrored by the uneven pattern of their cross-peaks. One of the methyl groups, assigned C17, produces a cross-peak with C3, while the other methyl does not (Fig. 4). This is consistent with the interatomic C3–C16 and C3–C17 distances measured from the MD simulations (Fig. S11, ESI†). In addition, the other methyl group, C16, produces a more intense cross-peak with C7 than C17. The intensities of the cross-peaks relevant to C16 and C17 methyls as determined by their signal-to-noise ratio are compared in Table S3 (ESI†). In the RFDR spectrum (Fig. 6), C6 produces cross-peaks with C16 and C18, but not C17, despite all having the same two-bond separation from C6. These NMR data suggest that the C16 and C17 methyl groups may not sit perfectly symmetrical in relation to the polyene chain and the β-ionone ring plane, in addition to having different interactions with neighboring sidechains. For example, MD simulations suggest that C16 has a fairly tight hydrophobic contact with Phe158, which is not observed for C17 (Fig. S10 and S11, ESI†).
Conclusions
The reversal of proton transport direction in some microbial rhodopsins is an intriguing phenomenon yet to be fully understood. Here, we studied the retinal chromophore environment in GSS AntR, a previously uncharacterized SzR. Our ssNMR data and MD simulations suggest a number of unique retinal–protein and retinal–water interactions, which may be responsible for the changed vectoriality of proton transport. While the proximal part of retinal seems to reside in a much more polar milieu than that in the outward proton pumps, there are also tight hydrophobic interactions in the distal part. Strikingly, these unique polar interactions may result in the reversed hydration pattern, where the extracellular side of the Schiff base lacks water, but the cytoplasmic side of the retinal is more hydrated than in BR and GPR, and the C15 atom H-bonds to the water molecule. It is tempting to suggest that the altered polarity and hydration of the proximal part of the retinal are the culprit of proton transport inversion.Author contributions
Conceptualization: L. S. B., V. L., and A.-N. B.; investigation: M. P., M. S., A. H., M. L., V. L., and A.-N. B.; formal analysis: M. P., L. S. B., V. L., and A.-N. B.; supervision: L. S. B., V. L., and A.-N. B.; writing – original draft: M. P., M. S., L. S. B., V. L., and A.-N. B.; writing – review & editing: all authors; funding acquisition: L. S. B., V. L., and A.-N. B.Data availability
The data supporting this article have been included in the main text and as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC) grants to V. L. (RGPIN-2020-04489) and L. S. B. (RGPIN-2018-04397), as well as Project C4 within the Collaborative Research Center SFB 198 (Protonation Dynamics in Protein Function), funded by the German Research Foundation (DFG), to A.-N. B. Computations were performed using computing resources at the RWTH-Aachen University and at the JURECA-DC supercomputing center in the Forschungszentrum Jülich. The authors gratefully acknowledge computing time on the supercomputer JURECA109 at the Forschungszentrum Jülich under grant no. DYNAMICNETWORKS. Preliminary test simulations were performed using computers of the Physics Department at the Freie Universität Berlin, and a computing time allocation from the HLRN, the North-German Supercomputing Alliance. We thank Dr Elena Govorunova and Dr John Spudich (University of Texas Health Science Center at Houston, TX, USA) for the attempt to better characterize the transport properties of AntRs in HEK cells. We thank Raoul Vaz, Sajjad Salimi Nanehkarani, Ciaran Henry, and Dr Sameer Al-Abdul-Wahid (University of Guelph) for helping optimize retinal isolation and collect and interpret the NMR spectra.Notes and references
- A. Rozenberg, K. Inoue, H. Kandori and O. Beja, Annu. Rev. Microbiol., 2021, 75, 427–447 CrossRef CAS PubMed.
- O. P. Ernst, D. T. Lodowski, M. Elstner, P. Hegemann, L. S. Brown and H. Kandori, Chem. Rev., 2014, 114, 126–163 CrossRef CAS PubMed.
- E. G. Govorunova, O. A. Sineshchekov, H. Li and J. L. Spudich, Annu. Rev. Biochem., 2017, 86, 845–872 CrossRef CAS PubMed.
- V. Gordeliy, K. Kovalev, E. Bamberg, F. Rodriguez-Valera, E. Zinovev, D. Zabelskii, A. Alekseev, R. Rosselli, I. Gushchin and I. Okhrimenko, Methods Mol. Biol., 2022, 2501, 1–52 CrossRef CAS PubMed.
- L. S. Brown, Biochim. Biophys. Acta, Biomembr., 2022, 1864, 183867 CrossRef CAS PubMed.
- J. K. Lanyi, Annu. Rev. Physiol., 2004, 66, 665–688 CrossRef CAS PubMed.
- J. Herzfeld and J. C. Lansing, Annu. Rev. Biophys. Biomol. Struct., 2002, 31, 73–95 CrossRef CAS PubMed.
- J. P. Klare, I. Chizhov and M. Engelhard, Results Probl. Cell Differ., 2008, 45, 73–122 CAS.
- U. Haupts, J. Tittor and D. Oesterhelt, Annu. Rev. Biophys. Biomol. Struct., 1999, 28, 367–399 CrossRef CAS PubMed.
- C. Bamann, E. Bamberg, J. Wachtveitl and C. Glaubitz, Biochim Biophys Acta, 2014, 1837, 614–625 CrossRef CAS PubMed.
- J. K. Lanyi and S. P. Balashov, in Halophiles and Hypersaline Environments, ed. A. Ventosa, Springer-Verlag, 2011, ch. 17, pp. 319–340 Search PubMed.
- C. Wickstrand, P. Nogly, E. Nango, S. Iwata, J. Standfuss and R. Neutze, Annu. Rev. Biochem., 2019, 88, 59–83 CrossRef CAS PubMed.
- K. Inoue, S. P. Tsunoda, M. Singh, S. Tomida, S. Hososhima, M. Konno, R. Nakamura, H. Watanabe, P. A. Bulzu, H. L. Banciu, A. S. Andrei, T. Uchihashi, R. Ghai, O. Beja and H. Kandori, Sci. Adv., 2020, 6, eaaz2441 CrossRef CAS PubMed.
- K. Inoue, S. Ito, Y. Kato, Y. Nomura, M. Shibata, T. Uchihashi, S. P. Tsunoda and H. Kandori, Nat. Commun., 2016, 7, 13415 CrossRef PubMed.
- K. Inoue, S. Tahara, Y. Kato, S. Takeuchi, T. Tahara and H. Kandori, J. Phys. Chem. B, 2018, 122, 6453–6461 CrossRef CAS PubMed.
- T. Shionoya, M. Singh, M. Mizuno, H. Kandori and Y. Mizutani, Biochemistry, 2021, 60, 3050–3057 CrossRef CAS PubMed.
- T. Urui, K. Hayashi, M. Mizuno, K. Inoue, H. Kandori and Y. Mizutani, J. Phys. Chem. B, 2024, 128, 744–754 CrossRef CAS PubMed.
- A. Higuchi, W. Shihoya, M. Konno, T. Ikuta, H. Kandori, K. Inoue and O. Nureki, Proc. Natl. Acad. Sci. U. S. A., 2021, 118 Search PubMed.
- K. Hayashi, M. Mizuno, H. Kandori and Y. Mizutani, Angew. Chem., Int. Ed., 2022, 61, e202203149 CrossRef CAS PubMed.
- T. Urui, T. Shionoya, M. Mizuno, K. Inoue, H. Kandori and Y. Mizutani, J. Phys. Chem. B, 2024, 128, 2389–2397 CrossRef CAS PubMed.
- C. F. Chang, M. Konno, K. Inoue and T. Tahara, J. Phys. Chem. Lett., 2023, 14, 7083–7091 CrossRef CAS PubMed.
- A. Harris, M. Lazaratos, M. Siemers, E. Watt, A. Hoang, S. Tomida, L. Schubert, M. Saita, J. Heberle, Y. Furutani, H. Kandori, A. N. Bondar and L. S. Brown, J. Phys. Chem. B, 2020, 124, 4851–4872 CrossRef CAS PubMed.
- G. S. Harbison, S. O. Smith, J. A. Pardoen, J. M. Courtin, J. Lugtenburg, J. Herzfeld, R. A. Mathies and R. G. Griffin, Biochemistry, 1985, 24, 6955–6962 CrossRef CAS PubMed.
- S. O. Smith, H. J. de Groot, R. Gebhard, J. M. Courtin, J. Lugtenburg, J. Herzfeld and R. G. Griffin, Biochemistry, 1989, 28, 8897–8904 CrossRef CAS PubMed.
- S. Ahuja, M. Eilers, A. Hirshfeld, E. C. Yan, M. Ziliox, T. P. Sakmar, M. Sheves and S. O. Smith, J. Am. Chem. Soc., 2009, 131, 15160–15169 CrossRef CAS PubMed.
- P. J. R. Spooner, J. M. Sharples, S. C. Goodall, P. H. M. Bovee-Geurts, M. A. Verhoeven, J. Lugtenburg, A. M. A. Pistorius, W. J. DeGrip and A. Watts, J. Mol. Biol., 2004, 343, 719–730 CrossRef CAS PubMed.
- P. J. E. Verdegem, P. H. M. Bovee-Geurts, W. J. de Grip, J. Lugtenburg and H. J. M. de Groot, Biochemistry, 1999, 38, 11316–11324 CrossRef CAS PubMed.
- J. Kaur, C. N. Kriebel, P. Eberhardt, O. Jakdetchai, A. J. Leeder, I. Weber, L. J. Brown, R. C. D. Brown, J. Becker-Baldus, C. Bamann, J. Wachtveitl and C. Glaubitz, J. Struct. Biol., 2019, 206, 55–65 CrossRef CAS PubMed.
- J. Becker-Baldus, C. Bamann, K. Saxena, H. Gustmann, L. J. Brown, R. C. Brown, C. Reiter, E. Bamberg, J. Wachtveitl, H. Schwalbe and C. Glaubitz, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 9896–9901 CrossRef CAS PubMed.
- V. S. Bajaj, M. L. Mak-Jurkauskas, M. Belenky, J. Herzfeld and R. G. Griffin, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 9244–9249 CrossRef CAS PubMed.
- M. E. Hatcher, J. G. G. Hu, M. Belenky, P. Verdegem, J. Lugtenburg, R. G. Griffin and J. Herzfeld, Biophys. J., 2002, 82, 1017–1029 CrossRef CAS PubMed.
- A. Naito, Y. Makino, A. Shigeta and I. Kawamura, Biophys. Rev., 2019, 11, 167–181 CrossRef CAS PubMed.
- J. Mao, X. Jin, M. Shi, D. Heidenreich, L. J. Brown, R. C. D. Brown, M. Lelli, X. He and C. Glaubitz, Sci. Adv., 2024, 10, eadj0384 CrossRef CAS PubMed.
- A. V. Struts, U. Chawla, S. M. Perera and M. F. Brown, Methods Mol. Biol., 2015, 1271, 133–158 CrossRef CAS PubMed.
- A. F. L. Creemers and J. Lugtenburg, J. Am. Chem. Soc., 2002, 124, 6324–6334 CrossRef CAS PubMed.
- J. Lugtenburg, A. F. L. Creemers, M. A. Verhoeven, A. A. C. van Wijk, P. J. E. Verdegem, M. C. F. Monnee and F. J. H. M. Jansen, Pure Appl. Chem., 1999, 71, 2245–2251 CrossRef CAS.
- A. J. Leeder, L. J. Brown, J. Becker-Baldus, M. Mehler, C. Glaubitz and R. C. D. Brown, J Labelled Comp. Radiopharm., 2018, 61, 922–933 CrossRef CAS PubMed.
- R. A. Munro, J. de Vlugt, M. E. Ward, S. Y. Kim, K. A. Lee, K. H. Jung, V. Ladizhansky and L. S. Brown, J. Biomol. NMR, 2019, 73, 49–58 CrossRef CAS PubMed.
- A. F. L. Creemers, S. Kiihne, P. H. M. Bovee-Geurts, W. J. DeGrip, J. Lugtenburg and H. J. M. de Groot, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 9101–9106 CrossRef CAS PubMed.
- S. Suzuki, S. Kumagai, T. Nagashima, T. Yamazaki, T. Okitsu, A. Wada, A. Naito, K. Katayama, K. Inoue, H. Kandori and I. Kawamura, Biophys. Chem., 2023, 296, 106991 CrossRef CAS PubMed.
- S. Arikawa, T. Sugimoto, T. Okitsu, A. Wada, K. Katayama, H. Kandori and I. Kawamura, Biophys Physicobiol, 2023, 20, e201017 CAS.
- J. Becker-Baldus, A. Leeder, L. J. Brown, R. C. D. Brown, C. Bamann and C. Glaubitz, Angew. Chem., Int. Ed., 2021, 60, 16442–16447 CrossRef CAS PubMed.
- M. Saliminasab, Y. Yamazaki, A. Palmateer, A. Harris, L. Schubert, P. Langner, J. Heberle, A. N. Bondar and L. S. Brown, J. Phys. Chem. B, 2023, 127, 7872–7886 CrossRef CAS PubMed.
- I. A. Chen, K. Chu, K. Palaniappan, M. Pillay, A. Ratner, J. Huang, M. Huntemann, N. Varghese, J. R. White, R. Seshadri, T. Smirnova, E. Kirton, S. P. Jungbluth, T. Woyke, E. A. Eloe-Fadrosh, N. N. Ivanova and N. C. Kyrpides, Nucleic Acids Res., 2019, 47, D666–D677 CrossRef CAS PubMed.
- A. Harris, M. Saita, T. Resler, A. Hughes-Visentin, R. Maia, F. Pranga-Sellnau, A. N. Bondar, J. Heberle and L. S. Brown, Phys. Chem. Chem. Phys., 2018, 20, 3184–3199 RSC.
- S. A. Waschuk, A. G. Bezerra, L. Shi and L. S. Brown, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 6879–6883 CrossRef CAS PubMed.
- G. Sabehi, A. Loy, K. H. Jung, R. Partha, J. L. Spudich, T. Isaacson, J. Hirschberg, M. Wagner and O. Beja, PLoS Biol., 2005, 3, 1409–1417 CrossRef CAS PubMed.
- F. X. Cunningham, Jr. and E. Gantt, Plant J., 2005, 41, 478–492 CrossRef PubMed.
- S. Y. Kim, S. A. Waschuk, L. S. Brown and K. H. Jung, Biochim Biophys Acta, 2008, 1777, 504–513 CrossRef CAS PubMed.
- H. J. Jang, B. K. Ha, J. W. Kim, K. H. Jung, J. Ahn, S. H. Yoon and S. W. Kim, Biotechnol. Lett., 2014, 36, 497–505 CrossRef CAS PubMed.
- S. R. Hartmann and E. L. Hahn, Phys. Rev., 1962, 128, 2042–2053 CrossRef CAS.
- B. M. Fung, A. K. Khitrin and K. Ermolaev, J. Magn. Reson., 2000, 142, 97–101 CrossRef CAS PubMed.
- K. Takegoshi, S. Nakamura and T. Terao, Chem. Phys. Lett., 2001, 344, 631–637 CrossRef CAS.
- G. J. Boender, S. Vega and H. J. M. de Groot, J. Chem. Phys., 2000, 112, 1096–1106 CrossRef CAS.
- R. Keller, The Computer Aided Resonance Assignment Tutorial, CANTINA Verlag Goldau, 1st edn, 2004 Search PubMed.
- C. R. Morcombe and K. W. Zilm, J. Magn. Reson., 2003, 162, 479–486 CrossRef CAS PubMed.
- M. Mirdita, K. Schütze, Y. Moriwaki, L. Heo, S. Ovchinnikov and M. Steinegger, Nat. Methods, 2022, 19, 679–682 CrossRef CAS PubMed.
- A. Sali and T. L. Blundell, J. Mol. Biol., 1993, 234, 779–815 CrossRef CAS PubMed.
- M. A. Marti-Renom, A. Stuart, A. Fiser, R. Sanchez, F. Melo and A. Sali, Annu. Rev. Biomol. Struct., 2000, 29, 291–325 CrossRef CAS PubMed.
- W. Humphrey, W. Dalke and K. Schulten, J. Mol. Graph., 1996, 14, 33–38 CrossRef CAS PubMed.
- B. R. Brooks, R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan and M. Karplus, J. Comput. Chem., 1983, 4, 187–217 CrossRef CAS.
- B. R. Brooks, C. L. I. Brooks, A. D. MacKerell Jr., L. Nilsson, R. J. Petrella, B. Roux, Y. Won, G. Archontis, C. Bartels, S. Boresch, A. Caflisch, L. Caves, Q. Cui, A. D. Dinner, M. Feig, S. Fischer, J. Gao, M. Hodoscek, W. Im, K. Kuczera, T. Lazaridis, J. Ma, V. Ovchinnikov, E. Paci, R. W. Pastor, C. B. Post, J. Z. Pu, M. Schaefer, B. Tidor, R. M. Venable, H. L. Woodcock, X. Wu, W. Yang, D. M. York and M. Karplus, J. Comput. Chem., 2009, 30, 1545–1614 CrossRef CAS PubMed.
- M. Lomize, I. D. Pogozheva, H. Joo, H. I. Mosberg and A. L. Lomize, Nucleic Acid Res., 2011, 40, D370–D376 CrossRef PubMed.
- A. D. MacKerell Jr., D. Bashford, M. Bellot, R. L. Dunbrack, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. I. Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, D. Yin and M. Karplus, J. Phys. Chem. B, 1998, 102, 3586–3616 CrossRef PubMed.
- A. D. MacKerell Jr., M. Feig and C. L. I. Brooks, J. Comput. Chem., 2004, 25, 1400–1415 CrossRef PubMed.
- A. D. Gruia, A.-N. Bondar, J. C. Smith and S. Fischer, Structure, 2005, 13, 617–627 CrossRef CAS PubMed.
- E. Tajkhorshid, J. Baudry, K. Schulten and S. Suhai, Biophys. J., 2000, 78, 683–693 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- L. Kalé, R. Skeel, M. Bhandarkar, R. Brunner, A. Gursoy, N. Krawetz, J. Phillips, A. Shinozaki, K. Varadarajan and K. Schulten, J. Comput. Phys., 1999, 151, 283–312 CrossRef.
- J. C. Phillips, B. Braun, W. Wang, J. Gumbart, E. Takjkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kale and K. Schulten, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS PubMed.
- J. C. Phillips, D. J. Hardy, J. D. C. Maia, J. E. Stone, J. V. Ribeiro, R. C. Bernardi, R. Buch, G. Fiorin, J. Henin, W. Jiang, R. McGreevy, M. C. R. Melo, B. K. Radak, R. D. Skeel, A. Singahoroy, Y. Wang, B. Roux, A. Aksimentiev, Z. Luthey-Schulten, L. V. Kale, K. Schulten, C. Chipot and E. Tajkhorshid, J. Chem. Phys., 2020, 153, 044130 CrossRef CAS PubMed.
- G. J. Martyna, D. J. Tobias and M. L. Klein, J. Chem. Phys., 1994, 101, 4177–4189 CrossRef CAS.
- M. Tuckermann, B. J. Berne and G. J. Martyna, J. Chem. Phys., 1992, 97, 1990–2001 CrossRef.
- J.-P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- M. Siemers, M. Lazaratos, K. Karathanou, F. Guerra, L. S. Brown and A.-N. Bondar, J. Chem. Theory Comput., 2019, 15, 6781–6798 CrossRef CAS PubMed.
- M. Siemers and A.-N. Bondar, J. Chem. Inf. Model., 2021, 61, 2998–3014 CrossRef CAS PubMed.
- B. Roux, M. Nina, R. Pomes and J. C. Smith, Biophys. J., 1996, 71, 670–681 CrossRef CAS PubMed.
- K. Kojima, S. Yoshizawa, M. Hasegawa, M. Nakama, M. Kurihara, T. Kikukawa and Y. Sudo, Sci. Rep., 2020, 10, 20857 CrossRef CAS PubMed.
- S. O. Smith, M. S. Braiman, A. B. Myers, J. A. Pardoen, J. M. L. Courtin, C. Winkel, J. Lugtenburg and R. A. Mathies, J. Am. Chem. Soc., 1987, 109, 3108–3125 CrossRef CAS.
- H. Kakitani, T. Kakitani, H. Rodman, B. Honig and R. Callender, J Phys Chem-Us, 1983, 87, 3620–3628 CrossRef CAS.
- E. Bertalan, M. J. Rodrigues, G. F. X. Schertler and A. N. Bondar, Br. J. Pharmacol., 2024 DOI:10.1111/bph.16387.
- A. N. Bondar, J. Baudry, S. Suhai, S. Fischer and J. C. Smith, J. Phys. Chem. B, 2008, 112, 14729–14741 CrossRef CAS PubMed.
- E. Nango, A. Royant, M. Kubo, T. Nakane, C. Wickstrand, T. Kimura, T. Tanaka, K. Tono, C. Song, R. Tanaka, T. Arima, A. Yamashita, J. Kobayashi, T. Hosaka, E. Mizohata, P. Nogly, M. Sugahara, D. Nam, T. Nomura, T. Shimamura, D. Im, T. Fujiwara, Y. Yamanaka, B. Jeon, T. Nishizawa, K. Oda, M. Fukuda, R. Andersson, P. Bath, R. Dods, J. Davidsson, S. Matsuoka, S. Kawatake, M. Murata, O. Nureki, S. Owada, T. Kameshima, T. Hatsui, Y. Joti, G. Schertler, M. Yabashi, A. N. Bondar, J. Standfuss, R. Neutze and S. Iwata, Science, 2016, 354, 1552–1557 CrossRef CAS PubMed.
- S. Hayashi, E. Tajkhorshid and K. Schulten, Biophys. J., 2002, 83, 1281–1297 CrossRef CAS PubMed.
- S. Checover, Y. Marantz, E. Nachliel, M. Gutman, M. Pfeiffer, J. Tittor, D. Oesterhelt and N. A. Dencher, Biochemistry, 2001, 40, 4281–4292 CrossRef CAS PubMed.
- E. Nachliel, M. Gutman, S. Kiryati and N. A. Dencher, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 10747–10752 CrossRef CAS PubMed.
- M. Gutman and E. Nachliel, Annu. Rev. Phys. Chem., 1997, 48, 329–356 CrossRef CAS PubMed.
- M. Gutman, E. Nachliel and R. Friedman, Biochim. Biophys. Acta, 2006, 1757, 931–941 CrossRef CAS PubMed.
- Y. Marantz, E. Nachliel, A. Aagaard, P. Brzezinski and M. Gutman, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 8590–8595 CrossRef CAS PubMed.
- P. Adelroth and P. Brzezinski, Biochim. Biophys. Acta, 2004, 1655, 102–115 CrossRef CAS PubMed.
- A. N. Bondar and H. Dau, Biochim. Biophys. Acta, 2012, 1817, 1177–1190 CrossRef CAS PubMed.
- T. Shutova, V. V. Klimov, B. Andersson and G. Samuelsson, Biochim. Biophys. Acta, 2007, 1767, 434–440 CrossRef CAS PubMed.
- A. N. Bondar, Biochim. Biophys. Acta, Biomembr., 2022, 1864, 184052 CrossRef CAS PubMed.
- T. M. Gray and B. W. Matthews, J. Mol. Biol., 1984, 175, 75–81 CrossRef CAS PubMed.
- L. G. Presta and G. D. Rose, Science, 1988, 240, 1632–1641 CrossRef CAS PubMed.
- C. del Val, S. H. White and A.-N. Bondar, J. Membr. Biol., 2012, 245, 717–730 CrossRef PubMed.
- M. Levitt and B. H. Park, Structure, 1993, 1, 223–226 CrossRef CAS PubMed.
- C. del Val, L. Bondar and A.-N. Bondar, J. Struct. Biol., 2014, 186, 95–111 CrossRef CAS PubMed.
- E. Bertalan, M. Konno, M. Del Carmen Marin, R. Bagherzadeh, T. Nagata, L. Brown, K. Inoue and A. N. Bondar, J. Phys. Chem. B, 2024, 128, 7407–7426 CrossRef CAS PubMed.
- G. S. Harbison, P. P. J. Mulder, H. Pardoen, J. Lugtenburg, J. Herzfeld and R. G. Griffin, J. Am. Chem. Soc., 1985, 107, 4810–4816 Search PubMed.
- A. E. Bennett, J. H. Ok, R. G. Griffin and S. Vega, J. Chem. Phys., 1992, 96, 8624–8627 CrossRef CAS.
- S. O. Smith, J. Courtin, E. van den Berg, C. Winkel, J. Lugtenburg, J. Herzfeld and R. G. Griffin, Biochemistry, 1989, 28, 237–243 CrossRef CAS PubMed.
- I. Gushchin, V. Shevchenko, V. Polovinkin, K. Kovalev, A. Alekseev, E. Round, V. Borshchevskiy, T. Balandin, A. Popov, T. Gensch, C. Fahlke, C. Bamann, D. Willbold, G. Buldt, E. Bamberg and V. Gordeliy, Nat. Struct. Mol. Biol., 2015, 22, 390–395 CrossRef CAS PubMed.
- T. Nishikawa, M. Murakami and T. Kouyama, J. Mol. Biol., 2005, 352, 319–328 CrossRef CAS PubMed.
- H. E. Kato, F. Zhang, O. Yizhar, C. Ramakrishnan, T. Nishizawa, K. Hirata, J. Ito, Y. Aita, T. Tsukazaki, S. Hayashi, P. Hegemann, A. D. Maturana, R. Ishitani, K. Deisseroth and O. Nureki, Nature, 2012, 482, 369–374 CrossRef CAS PubMed.
- P. J. R. Spooner, J. M. Sharples, S. C. Goodall, H. Seedorf, M. A. Verhoeven, J. Lugtenburg, P. H. M. Bovee-Geurts, W. J. DeGrip and A. Watts, Biochemistry-Us, 2003, 42, 13371–13378 CrossRef CAS PubMed.
- Jülich_Supercomputing_Centre, J. Large-Scale Res. Facil., 2021, 7, A182 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp02611j |
| This journal is © the Owner Societies 2024 |