
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalyzing sustainability: phytic acid as a green precursor for metal-free carbon electrocatalysts in ORR†
Sergio
García-Dalí
 *ab,
Javier
Quílez-Bermejo
a,
Jimena
Castro-Gutiérrez
a,
María T.
Izquierdo
c,
Alain
Celzard
ad and
Vanessa
Fierro
*a
*ab,
Javier
Quílez-Bermejo
a,
Jimena
Castro-Gutiérrez
a,
María T.
Izquierdo
c,
Alain
Celzard
ad and
Vanessa
Fierro
*a
aUniversité de Lorraine, Centre National de la Recherche Scientifique (CNRS), Institut Jean Lamour (IJL), F-88000, Épinal, France. E-mail: Vanessa.Fierro@univ-lorraine.fr
bDepartamento de Ciencia de los Materiales e Ingeniería Metalúrgica, Universidad de Oviedo, 33004, Oviedo, Spain
cInstituto de Carboquímica (ICB-CSIQ), Miguel Luesma Castán 4, E-50018, Zaragoza, Spain
dInstitut Universitaire de France (IUF), Épinal, France
First published on 9th June 2023
Abstract
The sluggish kinetics and high overpotential of the oxygen reduction reaction (ORR) in fuel cells and metal–air batteries are currently resolved through the use of expensive and scarce platinum-based electrocatalysts. Here, a green and straightforward synthesis method for preparing metal-free phosphorus-doped carbon (PDC) materials via carbonization of phytic acid at temperatures ranging from 700 to 1000 °C is reported. This study provides valuable insights into the effect of carbonization temperature on the surface chemistry and catalytic performance of PDCs. The PDC electrocatalysts exhibit ORR electrocatalytic activity equivalent to that of the state-of-the-art P-doped carbons and show a correlation between the content of C–P species and the electrocatalytic activity for the ORR. The study suggests that C–P-type species are responsible for the improved ORR. These findings not only demonstrate a practical and low-cost approach for preparing highly efficient metal-free electrocatalysts for the ORR, but also highlight their potential for the development of sustainable energy technologies.
Sustainability spotlightThis study reports a sustainable and significant breakthrough in the development of metal-free electrocatalysts for the oxygen reduction reaction (ORR) in fuel cells and metal-air batteries. A green and simple synthesis method was used to prepare phosphorus-doped carbon (PDC) materials by carbonizing phytic acid (PA). These PDC materials exhibited highly efficient ORR electrocatalytic activity, equivalent to that of expensive platinum-based electrocatalysts. The study also provided valuable insights into the surface chemistry and catalytic performance of PDCs, revealing the potential of PA-derived carbon materials in energy conversion. Overall, this low-cost and practical approach for producing metal-free electrocatalysts demonstrates their potential for sustainable energy technology development. |
Introduction
Due to the increasing global energy demand, significant efforts have been made to mitigate the environmental impact of our dependence on fossil fuels in the economic system.1 Green technologies such as fuel cells and metal-air batteries have shown great potential as sustainable and environmentally friendly alternatives to traditional fossil fuel-based devices.2,3 The oxygen reduction reaction (ORR) is a fundamental electrochemical process that plays a crucial role in the energy conversion mechanism of these devices.4 However, the sluggish kinetics and high overpotential of the ORR are major challenges that need to be addressed to improve the efficiency of energy conversion systems.5In addition, the cost of ORR electrodes is a major concern6,7 as platinum (Pt) and other noble metals deposited onto carbon supports are the state-of-the-art electrocatalysts currently used in commercial ORR devices.8,9 Despite its effectiveness, the use of Pt-based electrocatalysts is not a sustainable solution on a global scale due to the high cost of Pt, its scarcity and low stability under working conditions.10,11 Therefore, there is a need to find alternative electrocatalysts that are both cost-effective and environmentally friendly for widespread implementation of these electrochemical technologies. For instance, iron (Fe), nickel (Ni), and cobalt (Co) have shown promise as alternative electrocatalysts for ORR due to their high electrocatalytic performance.12–16 However, these metals pose their own set of issues. Under reaction conditions, the leaching of metal nanoparticles can occur, which can reduce the durability of the electrocatalyst and raise environmental concerns.17,18 In response to the limitations of metal-containing electrocatalysts, a new alternative based on metal-free carbon electrocatalysts is emerging as an efficient solution for ORR, particularly due to the low cost associated with their preparation.19–23 However, one of the main challenges associated with the use of carbon materials as electrocatalysts is their low electrocatalytic activity due to the homogeneous distribution of electron density along the carbon layers. To overcome this limitation, carbon materials are often doped with heteroatoms to tailor their properties and increase their electrocatalytic performance. Of all the heteroatoms commonly used for carbon doping, such as nitrogen (N), oxygen (O), sulfur (S), boron (B) and halogens, nitrogen (N) has been the most studied due to its well-known effect on electrocatalytic activity.21–35 In comparison, phosphorus (P) has received relatively less attention as a doping atom for ORR electrocatalysts. However, in recent years, there has been increasing interest in using P as a heteroatom to produce high-performance carbons.36 As a result, P has been considered the “next heteroatom to be explored” by the scientific community.37
The main methods used for the production of P-doped carbons (PDCs),38 such as carbonization of the precursor mixture containing carbon and P,39 post-functionalization with a P compound of a prefabricated carbon,40 or co-deposition of carbon and P from a gas phase,41 rely on the use of environmentally unfriendly precursors, phosphoric acid being the main example. There is therefore a need for an alternative method to synthesize highly active PDCs using biosourced precursors. To this end, phytic acid (PA), a biosourced compound commonly found in nature that contains both carbon and phosphorus, has shown promise as a precursor for the production of carbonaceous materials with high P content without requiring additional reagents.42 Nevertheless, questions remain unanswered regarding the impact of heat treatment during the carbonization of PA and its effects on ORR performance.
In order to optimize the properties of the PDCs to be used as electrocatalysts and to better understand the effect of P species on ORR, PA was carbonized at different temperatures to change the chemical state of P in the carbon material. The PA-derived PDCs were thoroughly characterized, concluding that the oxidation state of P heteroatoms is a critical factor in the ORR catalytic activity. Additionally, this study presents a simple and eco-friendly method for the production of PDCs using a biosourced precursor. The resulting PDCs exhibited excellent electrocatalytic performance for ORR.
Experimental details
Chemical and reagents
Phytic acid (PA) solution (50 wt% in H2O), Pluronic® F-127, and Nafion® (5 wt%) were supplied by Sigma Aldrich. Potassium hydroxide (KOH, 85%) and isopropanol (99.9%) were purchased from VWR. All reactants were used as received.Synthesis of PA-derived PDCs
To prepare an easy-to-handle paste, 4 g of PA solution was combined with 2 g of Pluronic® F127 and mixed in an agate bowl (50 mL) with 10 agate balls (1 cm diameter) using a planetary ball mill (PM100, Retsch) at a speed of 500 rpm for 1 hour. The resulting paste was then subjected to heat treatment in a high-temperature tubular furnace at temperatures ranging from 700 to 1000 °C for 1 hour. The furnace was purged with and N2 flow of 150 mL min−1 at room temperature for 1 hour prior to heat treatment. The heating rate was set at 5 °C min−1. The obtained carbon samples were labeled PDC-T, where T represents the heat treatment temperature.Instrumentation
PDC-T materials were characterized by elemental analysis (EA), N2 adsorption–desorption isotherms, X-ray diffraction (XRD), Raman spectroscopy, transmission electron microscopy (TEM) and X-ray photoelectron spectroscopy (XPS).The carbon, hydrogen, nitrogen, sulfur, and oxygen content of each sample was directly measured using a Vario EL cube analyzer (Elementar). About 2 mg of the sample was subjected to heat treatment at 1700 °C in an oxygen-containing helium atmosphere. The analyzer used a thermal conductivity detector to analyze the gases generated during sample combustion, which were separated through a chromatographic column. The P content (wt%) was determined considering P as the only element apart from C, H, O, N and S, and calculating the difference to 100.
N2 adsorption–desorption isotherms were acquired using a fully automated ASAP2020 manometric adsorption unit (Micromeritics) at −196 °C. Prior to the adsorption experiments, the samples were outgassed at 90 °C for at least 24 hours under high-vacuum conditions (2–4 × 10−4 Pa) in the degassing port. Further degassing was performed in the analysis port for at least 6 hours before the first dosing of N2, which started with the samples under high vacuum. The warm and cold volumes were measured after N2 adsorption–desorption to prevent helium entrapment in the narrowest pores. The BET area (ABET) of the materials was determined in accordance with the guidelines recommended by IUPAC43 using MicroActive® (Micromeritics) software. The pore size distributions (PSDs) were obtained using SAIEUS® (Micromeritics) software to apply the 2D non-local density functional theory for heterogeneous surfaces (2D-NLDFT-HS) to the N2 adsorption isotherm of the PDC-700 material. For the materials synthesized at higher temperatures (800–1000 °C), i.e., those with more developed microporosity, H2 adsorption–desorption isotherms at −196 °C were also obtained following the procedure described above for N2 adsorption. Then, the PSDs of these PDCs treated at high temperature were obtained by simultaneously applying the 2D-NLDFT-HS to the N2 and H2 adsorption isotherms.
The structural order of all carbon samples was determined using the Bruker D8 Advance A25 polycrystalline powder X-ray diffractometer. This device is equipped with a Cu anode X-ray source operating at 40 kV and 40 mA, a scintillation detector, and a graphite monochromator. Silver behenate was used as a reference for height correction. Furthermore, Raman spectra were obtained using a Horiba Scientific XploRa Raman spectrometer, which used a 50× objective and a green laser light (535 nm wavelength, circularly polarized, and filtered to 10% of its nominal power) dispersed using a holographic grating (1200 lines mm−1). Each spectrum was obtained by accumulating two spectra measured from 800 to 2200 cm−1 over 180 s.
TEM images were captured using a JEM - ARM 200 F Cold FEG TEM/STEM equipped with probe and image spherical aberration correctors. To prepare the samples for imaging, the powdered material was dispersed in ethanol using low-power sonication. A drop of the resulting suspension was deposited onto a carbon-coated copper TEM grid (200 mesh) and allowed to air-dry.
An ESCAPlus OMICROM spectrometer was used to obtain XPS spectra, equipped with a non-monochromatized MgKα X-ray source. Shirley-type background subtraction, peak fitting, and quantification were conducted using CASA software.
Electrochemical measurements
PDC-T materials were electrochemically characterized in a three-electrode cell with a rotating ring-disk electrode (RRDE) connected to a PGSTAT302N bi-potentiostat (Metrohm). The working electrode comprised a glassy carbon disk with an area of 0.196 cm2 and a platinum ring as a second working electrode. The reference electrode was a reversible hydrogen electrode (RHE) and the counter electrode was a glassy carbon rod (8 mm diameter). Electrochemical measurements were carried out at room temperature in aqueous alkaline medium (0.1 M KOH). To prepare the working electrodes, PDC-T materials were dispersed (4 mg mL−1) in an isopropanol/water solution (20/80, v/v) containing 0.2 wt% of Nafion®. 4 aliquots of 8.42 μL of the resulting ink were drop-casted onto the glassy carbon disk to give a carbon load of 0.68 mg cm−2. The prepared electrodes were immersed in the working solution under vacuum for a few minutes to achieve maximum wettability.44 Then, the electrode was introduced into the working solution under continuous nitrogen bubbling. Prior to the electrocatalytic study, to stabilize the electrode, cyclic voltammetry (CV) scans were recorded from 1.00 to 0.00 V vs. RHE at a scan rate of 50 mV s−1 (20 cycles). Next, linear sweep voltammetry (LSV) measurements were carried out under O2 bubbling at 1600 rpm with a scan rate of 5 mV s−1 from 1.00 V to 0.00 V vs. RHE. The geometric area of the disk (0.196 cm−2) was used to normalize the applied currents. The platinum ring potential was set at 1.5 V vs. RHE to calculate the yield of hydrogen peroxide (H2O2) during the ORR measurements. Tafel slopes were calculated from LSV measurements of each sample.Results and discussion
Physicochemical properties of PDC-T materials
Fig. 1 illustrates the green synthesis process used to produce the PDC-T series. The corresponding carbonization yields are listed in Table 1 and range from 33% at 700 °C to 7% at 1000 °C. Such a decrease could be related to the removal of phosphorus and oxygen groups from PA. To shed light on that, elemental analysis results for carbon, oxygen, phosphorus, and hydrogen contents are shown in Table 1. These data reveal a significant reduction in phosphorus and oxygen content, indicating that the removal of phosphate groups is likely responsible for the lower carbonization yield. The presence of six units of phosphate-type moieties per PA molecule can account for the diminished carbonization yield associated with this reagent. Moreover, the data indicate that the heat treatment temperature has a significant impact on the heteroatom content of the PDC materials, as evidenced by the decrease in the proportions of oxygen, phosphorus, and hydrogen with increasing temperature. This is accompanied by a corresponding increase in carbon content as other heteroatoms are removed during the heat treatment.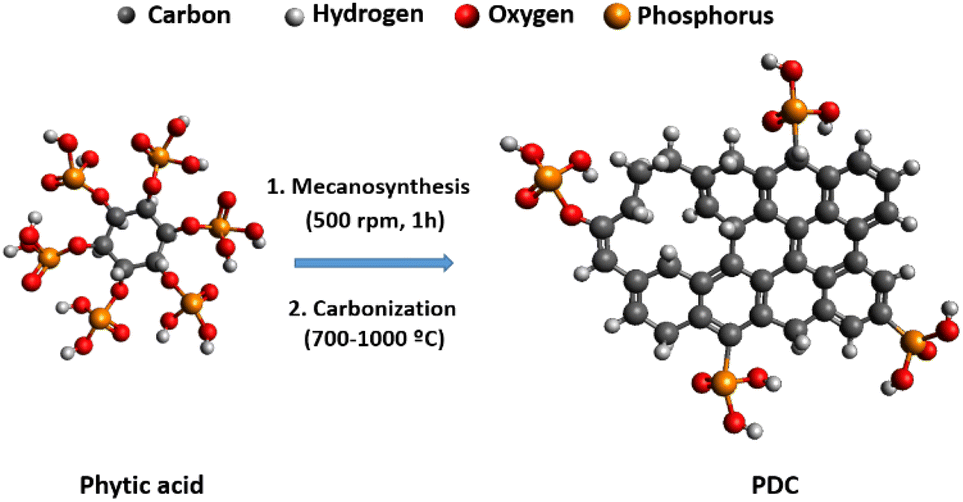 | ||
| Fig. 1 Schematic representation of the synthesis process of the PDC-T series materials. | ||
| Sample | Yield (%) | C (wt%) | O (wt%) | H (wt%) | P (wt%) |
|---|---|---|---|---|---|

|
33 | 35.7 | 34.6 | 2.0 | 27.7 |

|
13 | 51.8 | 30.5 | 1.7 | 16.0 |

|
10 | 64.3 | 23.3 | 1.2 | 11.2 |

|
7 | 77.0 | 15.1 | 0.6 | 7.3 |
The ABET values obtained from the N2 adsorption isotherms (Fig. 2a) exhibit a notable variance depending on the carbonization temperature (Fig. 2b). At 700 °C, the material shows an N2 isotherm typical of low-surface area or non-porous materials, as supported by the values shown in Fig. 2b. Nonetheless, there is a considerable and gradual rise in ABET with increasing temperature. The values are 211 m2 g−1 for PDC-800, 723 m2 g−1 for PDC-900, and 960 m2 g−1 for PDC-1000.
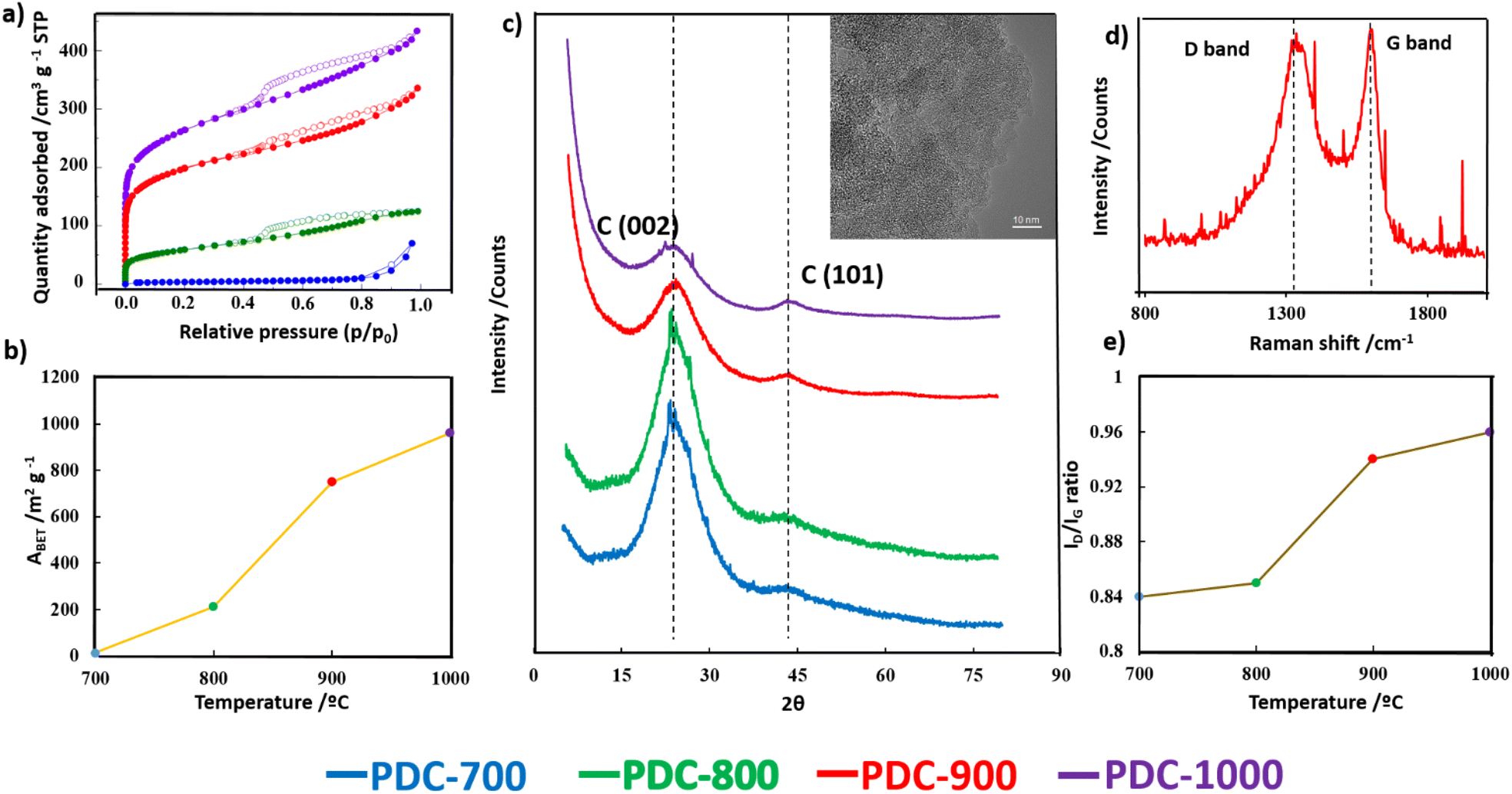 | ||
| Fig. 2 (a) N2 adsorption (full symbols) – desorption (empty symbols) isotherms of the PDC-T series; (b) plot of ABET obtained from N2 adsorption isotherms; (c) XRD patterns of the PDC-T series and TEM image of PDC-900 (inset); (d) Raman spectra of PDC-900; and (e) plot of ID/IG ratio obtained from Raman spectra. | ||
The gas release during heat treatment leads to the development of a microporous structure in the PDC materials (see the pore size distributions, PSDs, in Fig. S1†).
This, in turn, explains the observed increase in ABET. The widening of the knee in the N2 isotherms at p/p0 > 0.05 indicates the presence of supermicropores, which range from 0.7 to 2 nm in width, with increasing carbonization temperature. This is also reflected in the widening of the PSDs, which should facilitate the diffusion of ions and gases to and from the surface.
Transmission Electron Microscopy (TEM) is a powerful imaging technique used to examine the structure and morphology of materials at the nanoscale. In the case of the PDC-T material, TEM images (see the inset in Fig. 2c for PDC-900) reveal the characteristic morphology of a highly disordered pore network. These pores are assumed to form during the carbonization process and have sizes in the range of a few nanometers, consistent with the results of the N2 adsorption characterization. It has been shown that such a type of complex and interconnected network with a wide range of pore sizes could facilitate the diffusion of gases or ions within the material.45
The PDC-T materials were also analyzed by XRD and the results are presented in Fig. 2c. A broad (002) diffraction band centered at 2θ = 23°, indicative of amorphous carbons in which the parallel aromatic carbon sheets are randomly twisted with respect to each other, was observed in all samples. This is consistent with the high degree of disorder observed in the TEM image (inset of Fig. 2c). However, the higher the heat treatment temperature, the higher the intensity of the diffraction band centered at 2θ = 43°, referred to as the (101) carbon plane. The increase in the intensity of this second band suggests that high-temperature heat treatment promotes the formation of larger graphitic domains in the carbon structure, leading to an increase in the order along the planar direction.46
The Raman spectrum of PDC-900 in Fig. 2d displays the two characteristic bands commonly found in carbon materials. The D band, which appears around 1350 cm−1, is related to defects in the carbon layers and to heteroatoms, while the G band, which appears at 1590 cm−1, corresponds to the elongation mode of C–C in the aromatic rings and to sp2 carbon vibrations. The (ID/IG) ratio is calculated based on the intensities of the D and G bands. ID/IG is a widely recognized as a quantitative measure of the degree of graphitization in carbon-based materials. In amorphous carbons, the development of the D band indicates ordering, exactly opposite from the case of graphite.47 A higher the ID/IG ratio signifies a decreased presence of defects, disorder, or amorphous carbon regions within the material, indicating a more organized carbon structure.
Conversely, lower values of the ID/IG ratio suggest a higher level of disordered carbon structure. The increasing ID/IG ratio with heat treatment temperature, shown in Fig. 2e for all PDC-T materials, indicates an increase in the degree of order at the nanoscale, consistent with the second stage of the ordering trajectory described by Ferrari and Robertson,47,48 which is also in line with the XRD results.
To investigate the surface chemical changes occurring during heat treatment, X-ray photoelectron spectroscopy (XPS) was performed on all PDC-T materials. Table S1† summarizes the carbon, oxygen, and phosphorus contents on the surface of these materials. Fig. 3a displays the P 2p XPS spectra. As expected, the P content on the carbon surface decreases with the temperature of the heat treatment (Fig. 3b). All carbon materials exhibit a peak at 134.1 eV, which is attributed to C–O–P species, namely C–O–PO3-type groups, coming from the initial PA structure.49 Another peak appears at 133.1 eV, which is attributed to C–P species bonded with one or two carbon atoms, C2-PO2 or C-PO3. Notably, Fig. 3c shows that the higher the temperature of the heat treatment, the higher the contribution of the C–P peak, reaching a maximum at 900 °C. These results suggest a conversion of the C–O–P groups into C–P species at high temperatures. However, for PDC-1000, the C–P-type species do not seem to be stable, as there is a significant decrease in the contribution of the C–P peak compared to that of PDC-900. In Fig. S2a,† the intense peak observed at 284.7 eV in the C 1s spectra is attributed to the presence of graphitic carbon with sp2 hybridization,50 which supports the formation of graphitic domains also evidenced by the Raman and XRD experiments. Additionally, the C 1s spectra exhibit peaks corresponding to different functional groups, such as alcohol or C–O–P groups at 286.3 eV, carbonyl groups at 287.4 eV, and carboxylic or ester groups at 289.1 eV.51 The contribution of C–O-type species (blue line) decreases above 800 °C, whereas the contribution of C–P species increases at this temperature, which is consistent with the P 2p spectra.52 The O 1s XPS spectra (Fig. S2b†) show characteristic peaks of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O or PO bonds at 531.1 eV, as well as C–O–H or C–O–P groups at 533.0 eV and aromatic CO or P–O–P groups at 534.6 eV.53
O or PO bonds at 531.1 eV, as well as C–O–H or C–O–P groups at 533.0 eV and aromatic CO or P–O–P groups at 534.6 eV.53
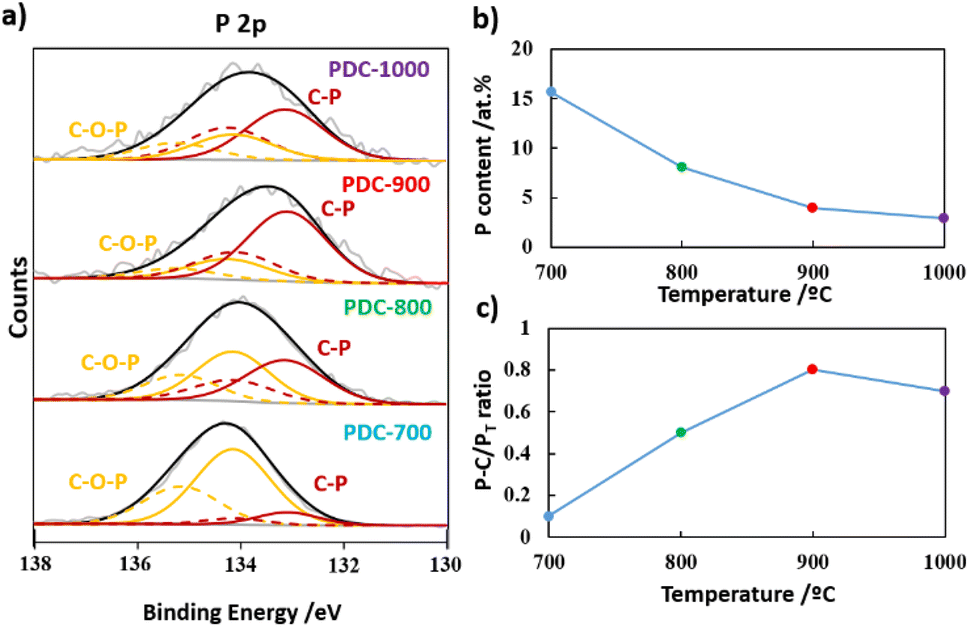 | ||
| Fig. 3 (a) P 2p XPS spectra, (b) P content, and (c) P–C/PT ratio of the PDC-T series. | ||
Electrochemical properties of PDC-T materials
The electrocatalytic performance of the as-prepared PDC-T materials towards the ORR was evaluated in a 0.1 M KOH solution. Fig. 4a shows the CV profiles recorded in an aqueous, N2-saturated, 0.1 M KOH solution at 50 mV s−1, prior to electrocatalytic testing. A nearly rectangular CV profile, which is a typical feature of carbon materials with excellent electrical conductivity and well-developed textures, was observed in all materials. The improved electrochemical double layer capacities found in PA-derived carbons obtained at high temperatures can be attributed to enhanced electrical conductivity resulting from condensation reactions occurring at elevated temperatures. These reactions frequently lead to a higher degree of graphitization in carbon materials and increased electron mobility.54 | ||
| Fig. 4 (a) Cyclic voltammetry in 0.1 M KOH, N2-saturated solution at 50 mV s−1; (b) linear sweep voltammetry in 0.1 M KOH, O2-saturated solution at 1600 rpm; (c) H2O2 formation; (d) Tafel slopes of all PDC-T materials; (e) stability of PDC-900 under continuous LSV cycling; (f) literature comparison of the ORR performance of other P-doped materials (unfilled circles) to PCD-900 (red solid circle). | ||
However, the notable variations in electrochemical double layer behavior are primarily attributed to the surface area development of carbon materials through heat treatment. Indeed, pyrolysis at higher temperatures promotes the development of micropores, which increases the surface area and, consequently, enhances the formation of the double-layer. In order to evaluate the electrocatalytic activity of the PDC-T materials for the ORR, linear sweep voltammetry (LSV) tests were performed under O2-saturated, alkaline conditions (0.1 M KOH). The results are presented in Fig. 4b, from which the potential required to reach −0.1 mA cm−2 (EONSET) was determined and used as a descriptor to evaluate the electrocatalytic activity in the ORR. The electrocatalytic behavior of the materials improved with increasing the heat treatment temperature. The EONSET value of PDC-700 was found to be 0.77 V vs. RHE, while PDC-800 exhibited a similar EONSET value of 0.78 V vs. RHE. In contrast, the EONSET values for PDC-900 and PDC-1000 were 0.83 V and 0.82 V vs. RHE, respectively. This suggests that temperatures above 900 °C are required to form the most active sites for ORR in P-doped carbon materials.
The active sites appear to be attributed to C–P-type groups produced from C–O–P groups. Although this transformation slightly begins at 700 °C, it occurs mainly during high-temperature treatments (800–1000 °C), as observed by XPS.
The ORR performance of the electrode materials is not only related to the electrocatalytic activity but also to the number of transferred electrons, which is an indication of the selectivity of the reaction. The closer the number of electrons transferred during the ORR is to four, the higher the energy produced in fuel cells or metal-air batteries. In addition, a 2-electron pathway indicates the production of hydrogen peroxide, which is corrosive and can easily damage most components of these electrochemical devices. Therefore, to assess the mechanisms involved in the ORR for each material, it is also necessary to quantify the production of H2O2 during the reaction. Fig. 4c shows the H2O2 formation for all materials, which mainly exhibit moderate H2O2 production, around 50%. Nevertheless, the samples obtained at the highest temperatures show the lowest H2O2 production, especially PDC-900, which also suggests the presence of different active sites than those observed in the materials treated at different temperatures. Additionally, the materials obtained at temperatures above 900 °C exhibit a larger ABET that promotes the retention of the H2O2 in the micropores. A higher retention time increases the possibility of reaching new active sites, where H2O2 can be reduced further to water molecules through a 2 + 2 electron mechanism:55
| O2 + 2H2O +2e− → H2O2 + 2OH− |
| H2O2 + 2e− → 2OH− |
Tafel slopes were obtained to investigate further the kinetics and mechanisms of electron transfer at the active sites of these PDCs.
As shown in Fig. 4d, the Tafel slopes were very similar, from 110 to 80 mV dec−1, suggesting that the electron transferred to the OH on the surface to produce OH− in the last step of the process is the rate-limiting stage for the ORR in all materials.56
The operational stability of the sample with the most positive EONSET, PDC-900, was tested by comparing the LSV curve before and after 1000 cycles. The data presented in Fig. 4e revealed that PDC-900 exhibits high stability, with a decay of only 0.02 V after 1000 cycles. Therefore, PDC-900 was found to be a promising electrocatalyst for the ORR due to its high electrocatalytic activity, with low H2O2 production and great stability under the working conditions. This behavior of PDC-900 could be attributed to the presence of C–P bonds. Theoretical studies have demonstrated that phosphorus atoms present in C–P-type models exhibit a significant positive charge density.57 This positive charge density serves as active sites capable of chemisorbing the dioxygen molecule, thereby initiating subsequent ORR reactions.58 Furthermore, the introduction of phosphorus heteroatoms can induce the creation of metallic states within the carbon material,59 thereby enhancing the electrical conductivity of the carbon materials.
This, combined with the large ABET, promotes greater accessibility to active sites to reduce H2O2 molecules to water via a 2 + 2 electron pathway. Furthermore, this material, which exhibits competitive electrocatalytic performance compared to the most recent PDCs found in the literature (Fig. 4f and Table S2†), is obtained by a simple and ecological method.
It is indeed worth mentioning that the PDC materials in the literature are prepared by more complex, costly or polluting syntheses, which highlights the relevance of the method proposed of this study. Specifically, the material with the highest performance (yellow circle in Fig. 4f) was synthesized using a high-cost template,60 while the other PDCs in the literature were prepared by multi-step synthesis methods (green circle),61 using expensive P-containing precursors (violet circle),62 organic solvents (brown circle)63 or the synthesis of carbon nanotubes as the carbon source (blue circle).64
Conclusions
A highly efficient P-doped carbon (PDC) for the ORR has been successfully synthesized through a green and simple method involving the carbonization of phytic acid (PA) at 900 °C. The resulting sample exhibits an EONSET of 0.83 V vs. RHE, which is comparable or better than state-of-the-art P-doped carbon-based electrocatalysts. Extensive characterization of the PDCs prepared by heat treatment at temperatures ranging from 700 to 1000 °C provides insights into the evolution of the surface chemistry of the materials. In particular, this study shows that the C–P content reached its maximum value at 900 °C, in agreement with the highest electrocatalytic activity, suggesting that C–P-species are responsible for the improved ORR. These interesting results open up possibilities for developing new ORR electrodes based on phosphorus as a doping agent in carbon materials. Overall, this study demonstrates the potential of PA-derived carbon materials in energy conversion, with a specific emphasis on their usefulness in ORR electrocatalysis.Author contributions
SGD & JQB: conceptualization, investigation, methodology, writing – original draft; JCG & MTI: investigation, writing – original draft; AC: validation, writing – review & editing, resources, supervision; VF: conceptualization, validation, writing – review & editing, resources, supervision, project administration, funding acquisition.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This study was partially supported by the French PIA project “Lorraine Université d’Excellence”, reference ANR-15-IDEX-04-LUE, and the TALiSMAN and TALiSMAN2 projects funded by ERDF. SGD thanks the Ministerio de Universidades, the European Union, and the University of Oviedo for their financial support (MU-21-UP2021-030 3026715).Notes and references
- J. Quílez-Bermejo, S. Pérez-Rodríguez, A. Celzard and V. Fierro, Front. Mater., 2022, 9, 1–18 CrossRef.
- M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245–4269 CrossRef CAS PubMed.
- X. Li, L. Zhao, J. Yu, X. Liu, X. Zhang, H. Liu and W. Zhou, Nano-Micro Lett., 2020, 12, 1–29 CrossRef CAS PubMed.
- A. M. Gómez-Marín and J. M. Feliu, Encycl. Interfacial Chem., 2018, 820–830 Search PubMed.
- L. Li, P. Wang, Q. Shao and X. Huang, Adv. Mater., 2021, 33, 1–24 Search PubMed.
- F. Cheng and J. Chen, Chem. Soc. Rev., 2012, 2172–2192 RSC.
- Z.-L. Wang, D. Xu, J.-J. Xu and X.-B. Zhang, Chem. Soc. Rev., 2013, 43, 7746–7786 RSC.
- J. Stacy, Y. N. Regmi, B. Leonard and M. Fan, Renewable Sustainable Energy Rev., 2017, 69, 401–414 CrossRef CAS.
- A. Morozan, B. Jousselme and S. Palacin, Energy Environ. Sci., 2011, 4, 1238–1254 RSC.
- E. J. Popczun, C. G. Read, C. W. Roske, N. S. Lewis and R. E. Schaak, Angew. Chem., 2014, 5531–5534 CrossRef.
- B. Hinnemann, P. G. Moses, J. Bonde, K. P. Jørgensen, J. H. Nielsen, S. Horch, I. Chorkendorff and J. K. Nørskov, J. Am. Chem. Soc., 2005, 5308–5309 CrossRef CAS PubMed.
- K. Zhang, Y. Zhang, Q. Zhang, Z. Liang, L. Gu, W. Guo, B. Zhu, S. Guo and R. Zou, Carbon Energy, 2020, 2, 283–293 CrossRef CAS.
- H. Li, Q. Li, P. Wen, T. B. Williams, S. Adhikari, C. Dun, C. Lu, D. Itanze, L. Jiang, D. L. Carroll, G. L. Donati, P. M. Lundin, Y. Qiu and S. M. Geyer, Adv. Mater., 2018, 30, 1705796 CrossRef PubMed.
- S. Li, C. Cheng, X. Zhao, J. Schmidt and A. Thomas, Angew. Chem., Int. Ed., 2018, 57, 1856–1862 CrossRef CAS PubMed.
- J. Quílez-Bermejo, S. García-Dalí, A. Daouli, A. Zitolo, R. L. S. Canevesi, M. Emo, M. T. Izquierdo, M. Badawi, A. Celzard and V. Fierro, Adv. Funct. Mater., 2023, 2300405 CrossRef.
- C. X. Zhao, J. N. Liu, J. Wang, D. Ren, B. Q. Li and Q. Zhang, Chem. Soc. Rev., 2021, 50, 7745–7778 RSC.
- J. Masa, W. Xia, M. Muhler and W. Schuhmann, Angew. Chem., Int. Ed., 2015, 54, 10102–10120 CrossRef CAS PubMed.
- H. T. Chung, D. A. Cullen, D. Higgins, B. T. Sneed, E. F. Holby, K. L. More and P. Zelenay, Science, 2017, 357, 479–484 CrossRef CAS PubMed.
- Z. Zhao, Z. Yuan, Z. Fang, J. Jian, J. Li, M. Yang, C. Mo, Y. Zhang, X. Hu, P. Li, S. Wang, W. Hong, Z. Zheng, G. Ouyang, X. Chen and D. Yu, Adv. Sci., 2018, 5, 1800760 CrossRef PubMed.
- R. Paul, Q. Dai, C. Hu and L. Dai, Carbon Energy, 2019, 1, 19–31 CrossRef.
- A. Dessalle, J. Quílez-Bermejo, V. Fierro, F. Xu and A. Celzard, Carbon, 2023, 203, 237–260 CrossRef CAS.
- Y. Zhu and B. Zhang, J. Energy Chem., 2021, 58, 610–628 CrossRef CAS.
- A. Shah, H. Singh, P. Prajongtat, M. C. Joshi, S. Hannongbua, N. Chattham, Y. K. Kim, S. Kumar and D. P. Singh, New J. Chem., 2022, 47, 1360–1370 RSC.
- J. Quílez-Bermejo, M. Melle-Franco, E. San-Fabián, E. Morallón and D. Cazorla-Amorós, J. Mater. Chem. A, 2019, 7, 24239–24250 RSC.
- Z. Duan and G. Henkelman, J. Phys. Chem. C, 2020, 124, 12016–12023 CrossRef CAS.
- J. Quílez-Bermejo, K. Strutyński, M. Melle-Franco, E. Morallón and D. Cazorla-Amorós, ACS Appl. Mater. Interfaces, 2020, 12, 54815–54823 CrossRef PubMed.
- J. Quílez-Bermejo, E. Morallón and D. Cazorla-Amorós, Carbon, 2022, 189, 548–560 CrossRef.
- A. R. Ferens, R. D. Weinstein, R. Giuliano and J. A. Hull, Carbon, 2011, 50, 192–200 CrossRef.
- F. Rodriguez-Reinoso, M. Molina-Sabio and M. A. Munecas, J. Phys. Chem., 1992, 2707–2713 CrossRef CAS.
- W. Kiciński, M. Szala and M. Bystrzejewski, Carbon, 2014, 68, 1–32 CrossRef.
- T. J. Bandosz and T. Z. Ren, Carbon, 2017, 118, 561–577 CrossRef CAS.
- T. Nakajima, Surface Modification of Carbon Anodes for Lithium Ion Batteries by Fluorine and Chlorine, Elsevier, 2015 Search PubMed.
- P. Barpanda, G. Fanchini and G. G. Amatucci, Carbon, 2011, 49, 2538–2548 CrossRef CAS.
- J. Quílez-Bermejo, E. Morallón and D. Cazorla-Amorós, Carbon, 2020, 165, 434–454 CrossRef.
- J. Quílez-Bermejo, S. Pérez-Rodríguez, R. Canevesi, D. Torres, E. Morallón, D. Cazorla-Amorós, A. Celzard and V. Fierro, Carbon, 2022, 196, 708–717 CrossRef.
- E. H. Ramírez-Soria, S. García-Dalí, J. M. Munuera, D. F. Carrasco, S. Villar-rodil, J. M. D. Tascón, J. I. Paredes and J. Bonilla-cruz, ACS Appl. Mater. Interfaces, 2021, 13, 54860–54873 CrossRef PubMed.
- M. Enterría and J. L. Figueiredo, Carbon, 2016, 108, 79–102 CrossRef.
- A. M. Puziy, O. I. Poddubnaya, B. Gawdzik and J. M. D. Tascón, Carbon, 2020, 157, 796–846 CrossRef CAS.
- M. S. Solum, R. J. Pugmire, M. Jagtoyen and F. Derbyshire, Carbon, 1995, 33, 1247–1254 CrossRef CAS.
- R. T. Mayes, P. F. Fulvio, Z. Ma and S. Dai, Phys. Chem. Chem. Phys., 2011, 13, 2492–2494 RSC.
- Z.-W. Liu, F. Peng, H.-J. Wang, H. Yu, W.-X. Zheng and J. Yang, Angew. Chem., Int. Ed., 2011, 50, 3091–3324 CrossRef.
- M. A. Patel, F. Luo, M. R. Khoshi, E. Rabie, Q. Zhang, C. R. Flach, R. Mendelsohn, E. Garfunkel, M. Szostak and H. He, ACS Nano, 2016, 10, 2305–2315 CrossRef CAS PubMed.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- L. Bouleau, S. Pérez-Rodríguez, J. Quílez-Bermejo, M. T. Izquierdo, F. Xu, V. Fierro and A. Celzard, Carbon, 2022, 189, 349–361 CrossRef CAS.
- J. Castro-Gutiérrez, N. Díez, M. Sevilla, M. T. Izquierdo, A. Celzard and V. Fierro, Renewable Sustainable Energy Rev., 2021, 151 Search PubMed.
- M. Okamura, A. Takagaki, M. Toda, J. N. Kondo, K. Domen, T. Tatsumi, M. Hara and S. Hayashi, Chem. Mater., 2006, 18, 3039–3045 CrossRef CAS.
- A. C. Ferrari and J. Robertson, Phys. Rev. B, 1999, 61, 14095–14107 CrossRef.
- S. Bernard, O. Beyssac, K. Benzerara, N. Findling, G. Tzvetkov and G. E. Brown, Carbon, 2010, 48, 2506–2516 CrossRef CAS.
- X. Wu and L. R. Radovic, Carbon, 2006, 44, 141–151 CrossRef CAS.
- S. K. Jerng, D. S. Yu, J. H. Lee, C. Kim, S. Yoon and S. H. Chun, Nanoscale Res. Lett., 2011, 6, 1–6 CrossRef PubMed.
- J. Xu, L. Chen, H. Qu, Y. Jiao and J. Xie, Appl. Surf. Sci., 2014, 320, 674–680 CrossRef CAS.
- J. Wu, X. Zheng, C. Jin, J. Tian and R. Yang, Carbon, 2015, 92, 327–338 CrossRef CAS.
- V. Sydorchuk, O. I. Poddubnaya, M. M. Tsyba, O. Zakutevskyy, O. Khyzhun, S. V Khalameida and A. M. Puziy, Appl. Surf. Sci., 2021, 535, 147667 CrossRef CAS.
- Y. R. Rhim, D. Zhang, D. H. Fairbrother, K. A. Wepasnick, K. J. Livi, R. J. Bodnar and D. C. Nagle, Carbon, 2010, 48, 1012–1024 CrossRef CAS.
- A. Gabe, R. Ruiz-Rosas, C. González-Gaitán, E. Morallón and D. Cazorla-Amorós, J. Power Sources, 2019, 412, 451–464 CrossRef CAS.
- D. S. Gnanamuthu and J. V. Petrocelli, J. Electrochem. Soc., 1967, 114, 1036 CrossRef CAS.
- P. Le Floch, Coord. Chem. Rev., 2006, 250, 627–681 CrossRef CAS.
- X. Bai, E. Zhao, K. Li, Y. Wang, M. Jiao, F. He, X. Sun, H. Sun and Z. Wu, Carbon, 2016, 105, 214–223 CrossRef CAS.
- N. Yang, X. Zheng, L. Li, J. Li and Z. Wei, J. Phys. Chem. C, 2017, 121, 19321–19328 CrossRef CAS.
- D. Yang, D. Bhattacharjya, S. Inamdar, J. Park and J. Yu, J. Am. Chem. Soc., 2012, 134, 16127–16130 CrossRef CAS PubMed.
- J. Wu, Z. Yang, X. Li, Q. Sun, C. Jin, P. Strasser and R. Yang, J. Mater. Chem. A, 2013, 1, 9889–9896 RSC.
- V. C. Hoang, V. G. Gomes and K. N. Dinh, Electrochim. Acta, 2019, 314, 49–60 CrossRef CAS.
- Y. Zhang, N. Huang, S. Qiao, J. Zhang, Z. Gao, Z. Qiu, S. Zhou and X. Jiang, Mod. Phys. Lett. B, 2019, 33, 1–8 CAS.
- M. Q. Guo, J. Q. Huang, X. Y. Kong, H. J. Peng, H. Shui, F. Y. Qian, L. Zhu, W. C. Zhu and Q. Zhang, Xinxing Tan Cailiao., 2016, 31, 352–362 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3su00119a |
| This journal is © The Royal Society of Chemistry 2023 |