
Aldehyde end-capped CO2-based polycarbonates: a green synthetic platform for site-specific functionalization†
Molin
Wang
ab,
Shunjie
Liu
 *ab,
Xuesi
Chen
ab,
Xianhong
Wang
*ab and
Fosong
Wang
ab
*ab,
Xuesi
Chen
ab,
Xianhong
Wang
*ab and
Fosong
Wang
ab
aKey Laboratory of Polymer Ecomaterials, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, 130022, China. E-mail: sjliu@ciac.ac.cn; xhwang@ciac.ac.cn
bUniversity of Science and Technology of China, Hefei, 230026, China
First published on 7th March 2022
Abstract
Aldehyde end-capped polymers are unique for their quantitative functionalization degree, and are useful in drug delivery, diagnosis and surface modification. However, relevant research studies have mainly focused on polyolefins as main materials, whose non-biodegradable characters may impede their further applications. To address this issue, we developed a strategy to synthesize aldehyde end-capped CO2-based polycarbonates, namely the copolymerization of propylene epoxide and CO2 in the presence of 4-formylbenzoic acid as the chain transfer agent. Well-defined polycarbonates were obtained with controllable Mn in the range of 3.7–19.0 kg mol−1 and Đ of ∼1.1. The high reactivity aldehyde end groups of polymers permit further post-polymerization. As a proof-of-concept, several typical post-polymerization reactions were performed to diversify functionalities, including regulating hydrophilicity, changing thermal properties, and causing aggregation-induced emission and amino acid conjugation, which suggested the potential of aldehyde end-capped polycarbonates as a green platform to prepare functional materials.
Introduction
Building polymers bearing highly reactive groups is considered as an effective approach to prepare diverse functional materials due to their desirable potential in further post-polymerization.1 Through the strategy of functional group transformations, a broad range of functional sections are allowed to be introduced into polymer scaffolds, especially those groups incompatible with the polymerization process.2,3 Commonly, modifiable polymer scaffolds can be classified into two types according to the position of reactive groups (pendant and chain-end).4 Thereinto, terminally functionalized polymers attract increasing interest on account of their distinctive characteristics. First of all, site-specific functional groups realize the quantitative end-functionalization,3,5 which is helpful in comprehending the structure–property relationships of polymers at the microscopic level.6–8 Secondly, the intrinsic nature of these polymers will be retained while obtaining new properties.9 Finally, the precise location of specific groups is beneficial for studying chain dynamics, conformational transitions, and intra- and inter-molecular associations.4,10The aldehyde group is fascinating for its active reactivity with amines, hydrazines and alcohols even under mild conditions, which enables it to conjugate with disparate functional entities, especially bioactive substances.11–16 Based on these advantages, aldehyde end-capped polymers are promising for the applications of drug delivery, diagnosis, surface modification and construction of supramolecular architectures.17–19 For instance, Haddleton et al. demonstrated the synthesis of aldehyde end-capped methacrylic polymers by living radical polymerization for protein conjugation.13 Brookhart et al. developed a strategy to prepare aldehyde end-functionalized polyolefins using an unsaturated alcohol as the chain-transfer agent (CTA).20 These contributions suggested the great potential of aldehyde end-capped polymers as functional materials. However, most studies mainly concentrated on polyolefins as main materials, whose non-biodegradable characters may impede their further applications.
The ring-opening copolymerization (ROCOP) of CO2 and epoxides is an environmentally benign approach to prepare biodegradable polycarbonates, which has received extensive attention due to the expectation of substitution of petroleum-based polymers. Since the pioneering work of Inoue et al. in 1969,21,22 this reaction has achieved enormous development thanks to the breakthrough in catalytic systems,23–31 which has successfully led to a leap from laboratory preparation to industrialization, especially for poly(propylene carbonates) (PPCs).32 However, their inert nature and lack of functionalities limited their further applications in high value-added materials fields. Therefore, great efforts have been made to synthesize modifiable CO2-based polycarbonates.1,33 Nevertheless, most of the reported modifiable polycarbonate scaffolds are pendant type and the reactive groups are limited to C–C unsaturated bonds, including alkenyl,34–37 alkynyl,38 furfuryl,39,40etc. (Fig. 1a). Hence, it is appealing to develop site-specific functional CO2-based polycarbonates.41
 | ||
| Fig. 1 CO2-Based polycarbonates for post-functionalization. (a) Polycarbonates with reactive side-chains. (b) Aldehyde end-capped polycarbonates. | ||
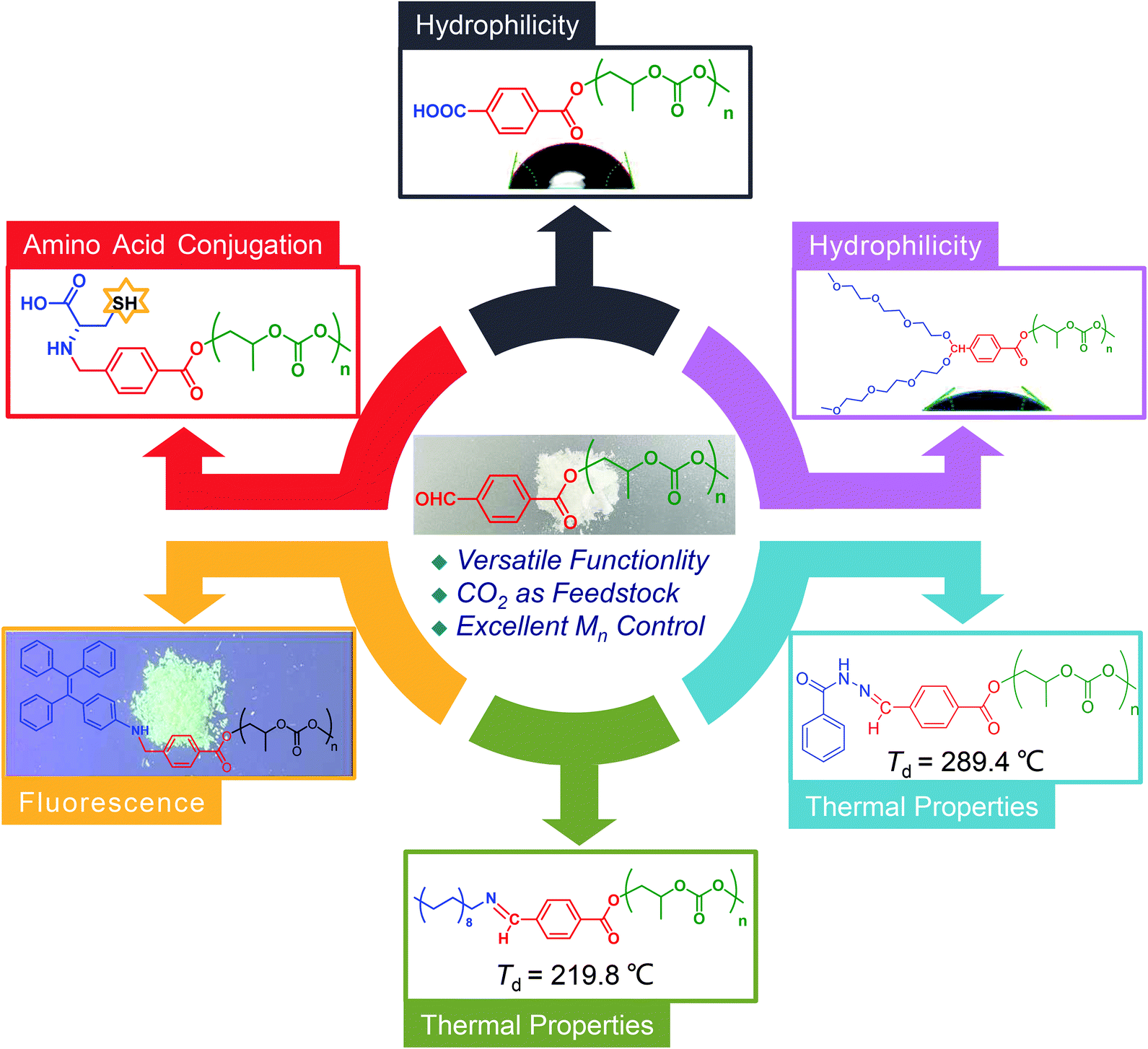
For the above-mentioned reasons, we developed a strategy to synthesize aldehyde end-capped CO2-based polycarbonates through the immortal copolymerization of propylene epoxide (PO) and CO2 in the presence of an aldehyde-containing CTA (Fig. 1b). Notably, the “immortal” nature of the ROCOP of CO2/PO shows high tolerance to the proton-containing CTA, which assures the achievement of polymers in a controlled way.42–44 As a proof-of-concept, we performed the copolymerization of CO2/PO catalyzed by the binary (salen)Co(III)Cl/PPNCl complex using 4-formylbenzoic acid (4-FBA) as the CTA. The role of 4-FBA is of great importance. 4-FBA could initiate the polymerization reaction by the carboxylic group but simultaneously keep the aldehyde group intact, which avoids the tedious process of protection and deprotection. Alternatively, the quantitative feeding of the CTA leads to well-controlled number-average molecular weight (Mn) and narrow molar mass dispersity (Đ) of the resulting polymers. As a result, aldehyde end-capped CO2-based PPCs were obtained with a controllable Mn of 3.7–19.0 kg mol−1 and a Đ of ∼1.1. Furthermore, several examples of the post-polymerization functionalization of the as-prepared polycarbonates were conducted based on the coupling of aldehydes with amines, hydrazines and alcohols (Scheme 1). The modification of end groups leads to versatility including regulating hydrophilicity, changing thermal properties, aggregation-induced emission (AIE) and amino acid conjugation. We expect that the aldehyde end-capped CO2-based polycarbonate could serve as a green platform for the construction of diverse functional polymers.
 | ||
| Scheme 1 Various post-polymerization functionalization routes for aldehyde end-capped CO2-based polycarbonates. | ||
Results and discussion
Design of the polymerization system
The immortal polymerization of epoxides and CO2 is an effective pathway to prepare aliphatic polycarbonates with well-defined chain end functionalities in the presence of CTA.45 For the purpose of producing aldehyde end-capped polycarbonates, the copolymerization of PO/CO2 catalyzed by salenCo(III)Cl/PPNCl was conducted in the presence of 4-FBA. SalenCo(III)Cl is easy to synthesize (Fig. S1–S3†) and has exhibited acceptable activity and selectivity in immortal polymerization. At the same time, the resulting polymers possess a completely alternating structure that ensures biodegradability.46,47 Based on the reaction mechanism of the immortal polymerization, the residue of the initiator is usually retained at the end of the polymer chain. During the process of polymerization, rapid and reversible chain transfer reactions occur between the active anion of the growing chain and abundant protic compound (4-FBA), leading to the aldehyde end-capped PPC with narrow molecular weight distribution.43Synthesis and characterization of aldehyde end-capped PPC
The copolymerization results are shown in Table 1. Through adjusting the monomer-to-initiator (4-FBA + salenCo(III)Cl + PPNCl) molar ratios (M/I), we obtained a series of aldehyde end-capped PPCs with various Mn. With the increasing 4-FBA loading from 30 to 100 equiv. of catalyst, the Mn of PPC can be accurately regulated from 19.0 to 3.7 kg mol−1 while maintaining a narrow Đ ∼ 1.1, implying a controllable chain transfer behavior. The polymerization time was enhanced from 48 h to 72 h with the increase of CTA loading to ensure high monomer conversion, as the competitive incorporation of carboxyl and PO to the metal center led to the deceleration of polymerization.48,49 For entries 1–3, the conversion remained above 90% when the CTA loading was lower than 40 equiv. of the catalyst. The residual PO could be attributed to the difficulty in stirring caused by the increased viscosity in the late stage of bulk polymerization. Further improving the CTA loading to more than 50 equiv. of catalyst resulted in a decrease in conversion (∼80%) even though the reaction time was prolonged to 72 h (entries 4–6), which confirmed the restriction of the CTA to the activity of the catalyst. In all cases, a high carbonate unit content (CU, 99%) and polymer selectivity over cyclic carbonate (98%) were obtained, manifesting the high efficiency of salenCo(III)Cl/PPNCl. On the other hand, the good linear relationship shown in Fig. 2a between the Mn of PPC and M/I indicated the precise molecular-weight controllability of 4-FBA. The GPC traces of the resulting polymers are also displayed in Fig. 2b. It is worth noting that the GPC curves presented a bimodal distribution when the Mn was greater than 10.6 kg mol−1, which could be attributed to the chain transfer caused by the diols formed from contaminant water and PO during immortal polymerization.42,50 The increasing additional CTA feed gradually covered the chain transfer effect of water, ultimately resulting in a singlet distribution in GPC curves. | ||
| Fig. 2 (a) The relationship of the Mn of aldehyde-terminated polycarbonates with M/I. I = salenCo(III)Cl + PPNCl + CTA. (b) The GPC traces of aldehyde-terminated polycarbonates with different molecular weights. | ||
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entrya | Feedb | t (h) | Conv.c (%) | Polymerd (%) | CUe (%) | M n (kg mol−1)f | Đ |
a The polymerization reactions were carried out in 4.0 mL epoxides in 10 mL autoclaves, at 30 °C and 3.0 MPa CO2.
b The molar ratio of PO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :salenCo(III)Cl:PPNCl:CTA.
c Monomer conversion (conv.) is determined by 1H NMR spectroscopy.
d Selectivity of polycarbonate over cyclic carbonate, determined by 1H NMR spectroscopy.
e Carbonate unit content, determined by 1H NMR spectroscopy.
f Determined by gel-permeation chromatography (GPC) in CH2Cl2 at 35 °C calibrated with polystyrene standards. :salenCo(III)Cl:PPNCl:CTA.
c Monomer conversion (conv.) is determined by 1H NMR spectroscopy.
d Selectivity of polycarbonate over cyclic carbonate, determined by 1H NMR spectroscopy.
e Carbonate unit content, determined by 1H NMR spectroscopy.
f Determined by gel-permeation chromatography (GPC) in CH2Cl2 at 35 °C calibrated with polystyrene standards.
|
|||||||
| 1 | 5000/1/1/30 | 48 | 92 | 98 | 99 | 19.0 | 1.17 |
| 2 | 5000/1/1/35 | 48 | 92 | 98 | 99 | 15.7 | 1.14 |
| 3 | 5000/1/1/40 | 48 | 96 | 98 | 99 | 14.7 | 1.15 |
| 4 | 5000/1/1/50 | 72 | 89 | 98 | 99 | 10.6 | 1.13 |
| 5 | 5000/1/1/75 | 72 | 82 | 98 | 99 | 6.0 | 1.14 |
| 6 | 5000/1/1/100 | 72 | 79 | 98 | 99 | 3.7 | 1.12 |

To verify the introduction of the terminal aldehyde group, the chemical structures of the obtained polymers were investigated using 1H NMR spectra (Fig. 3a). The obvious characteristic peaks at 5.01, 4.21 and 1.35 ppm belonged to CH, CH2 and CH3, respectively, in carbonate units. The tiny but distinct signal at 10.12 ppm was ascribed to the aldehyde group derived from 4-FBA. The peaks of aromatic protons were also observed at 8.20 and 7.99 ppm with a consistent integral area, which suggested the stability of the aldehyde group during the immortal polymerization process. Furthermore, the alternating structure of the resulting polycarbonates was proved by the absence of signals of the ether linkage (homopolymer of epoxides, at 3.30–3.60 ppm). Matrix-assisted laser desorption/ionization time-of-flight mass spectroscopy (MALDI-TOF) was performed to further analyze the composition of the polymer end groups. Fig. 3b only reveals one species corresponding to the aldehyde end-capped PPC with another hydroxyl terminal group. In fact, the nature of the immortal polymerization revealed that the initiating groups were derived from not only the CTA, but also the catalyst and cocatalyst.48,51 Hence, the Cl-terminated PPC was unavoidable but could be ignored under high CTA loading. To sum up, thanks to the good compatibility of 4-FBA for immortal polymerization, aldehyde end-capped CO2-based polycarbonates with controllable molecular weight were synthesized through an efficient and straightforward pathway, in favor of further research on post-polymerization.
 | ||
| Fig. 3 (a) 1H NMR spectrum of aldehyde-end-capped PPC (Mn = 10.6 kg mol−1) (300 MHz, CDCl3, 25 °C). Inset: Enlarged proton peaks of end groups. (b) MALDI-TOF-MS spectrum of the same sample (Mn = 10.6 kg mol−1). | ||
Abundant post-polymerization functionalization candidates
Based on the successful preparation of well-defined aldehyde end-capped PPCs, we explored diverse routes of post-polymerization guided by the strategy of functional group transformation. The high reactivity of the aldehyde group allows it to participate in various chemical reactions, including oxidation, condensation with amines or hydrazines, acetalation with alcohols, etc.11,20Fig. 4a displays an overview of the scheme for post-polymerization. For simplicity, aldehyde end-capped PPC is abbreviated as P-CHO. P-CHO with an Mn of 10.6 kg mol−1 was taken as the substrate for further modification due to the moderate content of aldehyde end groups. The molecular weights of P-CHO and its derivatives were determined by GPC and are summarized in Fig. S4 and Table S1.† | ||
| Fig. 4 (a) The synthetic methods of end-functionalized polymers from aldehyde end-capped polycarbonates. (b) The contact angle test of P–CHO, P–COOH and P–OEG. (c) The glass transition temperature (Tg) and thermal decomposition temperature (Td) of P–CHO and its post-modified polymers. | ||
PPC has potential applications as biodegradable and biocompatible materials, but its high hydrophobic nature impairs biodegradability and cell adhesion.34,52,53 Hence, we first attempted to improve the hydrophilicity by the introduction of hydrophilic groups through two approaches. One of them is the oxidation of –CHO to –COOH using m-chloroperbenzoic acid (m-CPBA) to obtain carboxyl end-capped polycarbonate P–COOH (Fig. S5†). Excessive m-CPBA ensured the complete transformation of aldehyde groups, which was confirmed by the absence of signals at 10.12 ppm related to –CHO in the 1H NMR spectrum (Fig. S6†). Meanwhile, MALDI-TOF spectroscopy only displayed one species ascribed to PPC with a carboxyl end group (Fig. S7†). All these data suggested the formation of P–COOH. The other approach is an acetalation between 1 equiv. –CHO and 2 equiv. triethylene glycol monomethyl ether for the purpose of linking two oligoethylene glycol (OEG) chains (Fig. S8†). Different from the mild temperature of the above one, this reaction needs 100 °C to promote the process by azeotropic distillation of water using methylbenzene as the carrier solvent. Fortunately, the structure of the resulting polymer P–OEG was verified by 1H NMR spectroscopy (Fig. S9†) and MALDI-TOF spectroscopy (Fig. S10†), manifesting the complete conversion of P–CHO. Fig. 4b shows that the contact angles to water of P–CHO (76°), P–COOH (61°) and P–OEG (38°) decreased gradually, indicating the improved hydrophicility.53,54
On the other hand, to improve the thermal properties, P–CHO was post-functionalized with aromatic or aliphatic groups. As shown in Fig. 4a and Fig. S11,† phenyl terminated P–CHO (P–BEN) was synthesized by the condensation of –CHO and hydrazine. This reaction proceeded efficiently and completely at room temperature without any catalyst, which exhibited the characteristic of “click” reaction.55,56 Diphenyl terminated P–CHO (P–BPH) was prepared through a similar pathway but required p-toluenesulfonic acid (TsOH) as the catalyst (Fig. S12†). Finally, P–OA, the PPC bearing an aliphatic chain end group of 18 carbons, was synthesized via the condensation of –CHO and –NH2 (Fig. S13†). This reaction was carried out at room temperature with CH3COOH as the catalyst. Likewise, the results of 1H NMR spectroscopy and MALDI-TOF spectroscopy were in line with the expected structures of the obtained polymers (Fig. S14–S19†). Fig. 4c shows the glass-transition temperature (Tg) and thermal decomposition temperature (Td) of the modified PPC. Compared with P–CHO (Tg = 35.0 °C), P–BEN and P–BPH only showed a slightly improved Tg (∼36 °C). However, P–OA exhibited a relatively obvious decline in Tg (30 °C), owing to the introduction of flexible alkyl chains. On the other hand, the Td of P–BEN (289 °C) and P–BPH (266 °C) increased significantly compared to that of P–CHO (242 °C), while the Td of P–OA decreased to 214 °C, which indicated the great influence of the robust end group on the thermal stability of polymers.57
Finally, we prepared two kinds of unique functional polymers derived from P–CHO to expand its application field. Among them, tetraphenylethylene (TPE) terminated P–CHO (P–TPE) was generated from the reaction of P–CHO and TPE–NH2 (Fig. S20†). In contrast to the above-mentioned reaction, sodium triacetoxyborohydride (STAB) was employed to reduce the imine and provide stable amino bonds, while promoting the complete conversion of –CHO.13 The conjunction of TPE with P–CHO was confirmed by 1H NMR spectroscopy and MALDI-TOF spectroscopy (Fig. S21 and S22†). P–TPE has typical AIE attributes as shown in Fig. 5.58–60 Under the excitation of 321 nm, P–TPE showed a fluorescence emission peak at ∼414 nm in THF/water mixtures. Significantly, the fluorescence intensity gradually increased with the increase of the water fraction, and showed a similar linear relationship in Fig. 5b. According to the previous reports, with the increasing water fraction, hydrophobic P–TPE tended to aggregate and triggered the restriction of the intramolecular motion mechanism of TPE, leading to a stronger fluorescence emission.58,61 Another strategy is associating P–CHO with amino acid through reductive amination reaction (Fig. S23†). Under mild conditions, P–CHO selectively reacted with the carboxyl group of L-cysteine and generated amino acid end-capped P–Cy with authenticated structures (Fig. S24 and S25†). It was worth noting that P–Cy inherited sulfhydryl and carboxyl groups from L-cysteine, which was of significance for further modification through the click reaction and deprotonation reaction.37 Finally, yet importantly, this result suggested the possibility of P–CHO connecting with biologically relevant substances, including peptides, proteins and drugs.
 | ||
| Fig. 5 (a) Fluorescence spectra of P–TPE in THF/water mixtures with different water fractions. (b) Trends of maximum FL intensity (wavelength = 414 nm) with different water fractions in THF/water mixtures. Concentration : 2.5 mg mL−1. λex = 321 nm. | ||
Conclusion
In summary, aldehyde end-capped CO2-based PPCs were prepared through an easily accessible immortal polymerization with high efficiency. The Mn of the resulting polymers could be precisely controlled in the range of 3.7–19.0 kg mol−1 with a Đ of ∼1.1 depending on the monomer-to-initiator molar ratios. 4-FBA acted as a CTA to initiate the polymerization while introducing the aldehyde terminal group simultaneously. In development, aldehyde end-capped PPC presented extensive modifiability for deriving end-functionalized polymers with various properties, including hydrophilicity regulation, thermal-property adjustment, AIE and amino acid conjugation. This study offered a flexible strategy to prepare site-specific functional polymers.Experimental
All the experimental details are provided in the ESI.†Author contributions
Molin Wang – Conceptualization, data curation, formal analysis, investigation, methodology, writing – original draft and writing – review & editing. Shunjie Liu – Conceptualization, supervision and writing – original draft and writing – review & editing. Xuesi Chen – Supervision. Xianhong Wang – Resources, supervision and writing – review & editing. Fosong Wang – Supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Fundamental Science Center projector in the National Natural Science Foundation of China (Grant No. 51988102).Notes and references
- Y. Y. Wang and D. J. Darensbourg, Coord. Chem. Rev., 2018, 372, 85–100 CrossRef CAS
.
- G. Sun, C. Cheng and K. L. Wooley, Macromolecules, 2007, 40, 793–795 CrossRef CAS PubMed
- A. Hirao and M. Hayashi, Macromolecules, 1999, 32, 6450–6460 CrossRef CAS
- M. Beija, M. T. Charreyre and J. M. G. Martinho, Prog. Polym. Sci., 2011, 36, 568–602 CrossRef CAS
- S. Liu, X. Zhao, H. Guo, Y. Qin, X. Wang and F. Wang, Macromol. Rapid Commun., 2017, 38, 1600754 CrossRef PubMed
- M. Chen, K. P. Ghiggino, S. H. Thang and G. J. Wilson, Angew. Chem., Int. Ed., 2005, 44, 4368–4372 CrossRef CAS PubMed
- F. Ercole, T. P. Davis and R. A. Evans, Macromolecules, 2009, 42, 1500–1511 CrossRef CAS
- F. Ercole, N. Malic, S. Harrisson, T. P. Davis and R. A. Evans, Macromolecules, 2010, 43, 249–261 CrossRef CAS
- E. Wang, S. Liu, H. Cao, C. Zhuo, X. Wang and F. Wang, Sci. China: Chem., 2021, 65, 162–169 CrossRef
- J. Duhamel, Acc. Chem. Res., 2006, 39, 953–960 CrossRef CAS PubMed
- G. S. Heo, S. Cho and K. L. Wooley, Polym. Chem., 2014, 5, 3555–3558 RSC
- X. Feng, D. Taton, E. L. Chaikof and Y. Gnanou, Biomacromolecules, 2007, 8, 2374–2378 CrossRef CAS PubMed
- L. Tao, G. Mantovani, F. Lecolley and D. M. Haddleton, J. Am. Chem. Soc., 2004, 126, 13220–13221 CrossRef CAS PubMed
- M. Shi, A. L. Li, H. Liang and J. Lu, Macromolecules, 2007, 40, 1891–1896 CrossRef CAS
- J. Hwang, R. C. Li and H. D. Maynard, J. Controlled Release, 2007, 122, 279–286 CrossRef CAS PubMed
- Y. Song, D. Li, J. He, M. Zhang and P. Ni, Chin. Chem. Lett., 2019, 30, 2027–2031 CrossRef CAS
- Y. Nagasaki, T. Okada, C. Scholz, M. Iijima, M. Kato and K. Kataoka, Macromolecules, 1998, 31, 1473–1479 CrossRef CAS
- J. Li, S. Yang, L. Wang, X. Wang and L. Liu, Macromolecules, 2013, 46, 6832–6842 CrossRef CAS
- X. W. Han, O. Daugulis and M. Brookhart, Organometallics, 2021, 40, 2709–2715 CrossRef CAS
- X. W. Han, O. Daugulis and M. Brookhart, J. Am. Chem. Soc., 2020, 142, 15431–15437 CrossRef CAS PubMed
- S. Inoue, H. Koinuma and T. Tsuruta, Makromol. Chem., 1969, 130, 210–220 CrossRef CAS
- S. Inoue, H. Koinuma and T. Tsuruta, J. Polym. Sci., Part B: Polym. Lett., 1969, 7, 287–292 CrossRef CAS
- M. Cheng, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 1998, 120, 11018–11019 CrossRef CAS
- Z. Qin, C. M. Thomas, S. Lee and G. W. Coates, Angew. Chem., Int. Ed., 2003, 42, 5484–5487 CrossRef CAS PubMed
- X. H. Zhang, S. Chen, X. M. Wu, X. K. Sun, F. Liu and G. R. Qi, Chin. Chem. Lett., 2007, 18, 887–890 CrossRef CAS
- E. K. Noh, S. J. Na, S. Sujith, S. W. Kim and B. Y. Lee, J. Am. Chem. Soc., 2007, 129, 8082–8083 CrossRef CAS PubMed
- Y. Qin, X. Wang, X. Zhao and F. Wang, Chin. J. Polym. Sci., 2008, 26, 241–247 CrossRef CAS
- D. Zhang, S. K. Boopathi, N. Hadjichristidis, Y. Gnanou and X. Feng, J. Am. Chem. Soc., 2016, 138, 11117–11120 CrossRef CAS PubMed
- B. Grignard, S. Gennen, C. Jerome, A. W. Kleij and C. Detrembleur, Chem. Soc. Rev., 2019, 48, 4466–4514 RSC
- G. W. Yang, Y. Y. Zhang, R. Xie and G. P. Wu, J. Am. Chem. Soc., 2020, 142, 12245–12255 CrossRef CAS PubMed
- X. B. Lu, Acta Polym. Sin., 2016, 1166–1178 CAS
- H. Cao, Y. Qin, C. Zhuo, X. Wang and F. Wang, ACS Catal., 2019, 9, 8669–8676 CrossRef CAS
- M. Scharfenberg, J. Hilf and H. Frey, Adv. Funct. Mater., 2018, 28, 1704302 CrossRef
- O. Hauenstein, S. Agarwal and A. Greiner, Nat. Commun., 2016, 7, 11862 CrossRef CAS PubMed
- J. F. Zhang, W. M. Ren, X. K. Sun, Y. Meng, B. Y. Du and X. H. Zhang, Macromolecules, 2011, 44, 9882–9886 CrossRef CAS
- D. J. Darensbourg and F. T. Tsai, Macromolecules, 2014, 47, 3806–3813 CrossRef CAS
- Y. Wang, J. Fan and D. J. Darensbourg, Angew. Chem., Int. Ed., 2015, 54, 10206–10210 CrossRef CAS PubMed
- J. Hilf and H. Frey, Macromol. Rapid Commun., 2013, 34, 1395–1400 CrossRef CAS PubMed
- Y. Hu, L. Qiao, Y. Qin, X. Zhao, X. Chen, X. Wang and F. Wang, Macromolecules, 2009, 42, 9251–9254 CrossRef CAS
- Y. Hu, L. Qiao, Y. Qin, X. Wang, X. Zhao and F. Wang, Acta Polym. Sin., 2011, 11, 1336–1340 CrossRef
- Y. Y. Zhang, G. W. Yang and G. P. Wu, Macromolecules, 2018, 51, 3640–3646 CrossRef CAS
- A. Cyriac, S. H. Lee, J. K. Varghese, E. S. Park, J. H. Park and B. Y. Lee, Macromolecules, 2010, 43, 7398–7401 CrossRef CAS
- A. Cyriac, S. H. Lee, J. K. Varghese, J. H. Park, J. Y. Jeon, S. J. Kim and B. Y. Lee, Green Chem., 2011, 13, 3469–3475 RSC
- S. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2861–2871 CrossRef CAS
- S. Asano, T. Aida and S. Inoue, J. Chem. Soc., Chem. Commun., 1985, 1148–1149 RSC
-
H. Cao and X. Wang, in Synthetic Polymer Chemistry: Innovations and Outlook, ed. Z. Zhao, R. Hu, A. Qin and B. Z. Tang, Carbon Dioxide Copolymer From Delicate Metal Catalyst: New Structure Leading to Practical Performance, Royal Society of Chemistry, Cambridge, 2020, pp. 197–242 Search PubMed
- C. Zhuo, Y. Qin, X. Wang and F. Wang, Chin. J. Polym. Sci., 2017, 36, 252–260 CrossRef
- C. A. L. Lidston, B. A. Abel and G. W. Coates, J. Am. Chem. Soc., 2020, 142, 20161–20169 CrossRef CAS PubMed
- H. Cao, R. Zhang, Z. Zhou, S. Liu, Y. Tao, F. Wang and X. Wang, ACS Catal., 2022, 12, 481–490 CrossRef CAS
- G. P. Wu and D. J. Darensbourg, Macromolecules, 2016, 49, 807–814 CrossRef CAS
- C. T. Cohen, T. Chu and G. W. Coates, J. Am. Chem. Soc., 2005, 127, 10869–10878 CrossRef CAS PubMed
- Y. Qin, L. Gu and X. Wang, Acta Polym. Sin., 2013, 13, 600–608 CrossRef
- L. Gu, Y. Qin, Y. Gao, X. Wang and F. Wang, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2834–2840 CrossRef CAS
- M. Wang, E. Wang, H. Cao, S. Liu, X. Wang and F. Wang, Chin. J. Chem., 2021, 39, 3037–3043 CrossRef CAS
- J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249–1262 RSC
- R. K. Iha, K. L. Wooley, A. M. Nystrom, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620–5686 CrossRef CAS PubMed
- J. Tang and E. Y. X. Chen, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 2271–2279 CrossRef CAS
- S. Liu, Y. Cheng, H. Zhang, Z. Qiu, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Angew. Chem., Int. Ed., 2018, 57, 6274–6278 CrossRef CAS PubMed
- J. Luo, Z. Xie, J. W. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC
- E. Wang, S. Liu, J. W. Y. Lam, B. Z. Tang, X. Wang and F. Wang, Macromolecules, 2020, 53, 5839–5846 CrossRef CAS
- H. Q. Peng, B. Liu, P. Wei, P. Zhang, H. Zhang, J. Zhang, K. Li, Y. Li, Y. Cheng, J. W. Y. Lam, W. Zhang, C. S. Lee and B. Z. Tang, ACS Nano, 2019, 13, 839–846 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2py00129b |
| This journal is © The Royal Society of Chemistry 2022 |