
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A monomeric (trimethylsilyl)methyl lithium complex: synthesis, structure, decomposition and preliminary reactivity studies†
Nathan
Davison
,
Paul G.
Waddell
,
Casey
Dixon
,
Corinne
Wills
,
Thomas J.
Penfold
* and
Erli
Lu
 *
*
Chemistry-School of Natural and Environmental Sciences, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK. E-mail: Tom.Penfold@newcastle.ac.uk; erli.lu@newcastle.ac.uk
First published on 26th November 2021
Abstract
Monomeric organolithium (LiR) complexes could provide enhanced Li–C bond reactivity and suggest mechanisms for a plethora of LiR-mediated reactions. They are highly sought-after but remain a synthetic challenge for organometallic chemists. In this work, we report the synthesis and characterisation of a monomeric (trimethylsilyl)methyl lithium complex, namely [Li(CH2SiMe3)(κ3-N,N′,N′′-Me6Tren)] (1), where Me6Tren is a tetradentate neutral amine ligand. The structure of 1 was comprehensively examined by single-crystal X-ray diffraction, variable temperature NMR spectroscopy and electron absorption spectroscopy. Complex 1 decomposes via ligand C–H and C–N activations to produce a Li amide complex 2. Preliminary reactivity studies of 1 reveal C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O insertion and C–H activation reaction patterns.
O insertion and C–H activation reaction patterns.
Introduction
Organolithium complexes (LiR; where R is alkyl or aryl) have been an indispensable tool for synthetic chemists for over a century.1–3 In solutions, LiR complexes form aggregates via bridging Li–C bonds:4 although this bridging behaviour stabilises the highly polar Li–C bond, it also diminishes the Li–C bond reactivity. Consequently, from the perspective of reactivity, a monomeric LiR complex with a terminal unsupported Li–C bond ought to be the most reactive version of an organolithium reagent, although exceptions have been reported.5 From the structure–reactivity relationship point of view, the LiR monomer is hypothesised as a key intermediate in the mechanisms of LiR-mediated reactions.4 Isolating the LiR monomers and studying their structure and reactivity would shed light on the mechanisms of the ubiquitous LiR-mediated reactions. For these reasons, synthetic organometallic chemists have devoted decades of effort to synthesising the LiR monomers and studying their structures and reactivity.5–18The most successful strategy to break the LiR aggregates and isolate LiR monomers is to employ neutral multidentate amines,4 which coordinate to the Li and kinetically stabilise the LiR monomers. Since the 1980s, this multidentate amine strategy has been successfully implemented by several groups to isolate a series of [Li(R)(L)] monomers (L = multi-amine ligands; R = CH2SiMe3,5,6 CH(SiMe3)2,7,8tBu,9,10sBu,8iPr,11 benzyl12–17). Recently, we reported the first methyllithium monomer [Li(CH3)(DETAN)], enabled by a bespoke hexa-dentate ligand [{Et2NCH2CH2N(CH2CH2)}3], namely N,N′,N′′-tris-(2-N-diethylaminoEthyl)-1,4,7-triAza-cycloNonane (DETAN).18
Despite the successes, the LiR monomer is still a challenging synthetic target: most LiR monomers are highly reactive and decompose at room temperature. This is reflected by their scarcity. For instance, (trimethylsilyl)methyl lithium (LiCH2SiMe3) is a widely used and commercially available reagent. Still, there are only two reports of its monomers5,6 (Scheme 1), with a tridentate and a bidentate amine ligand, respectively. Developing more LiR monomers is desirable to establish a comprehensive structure–reactivity relationship.
 | ||
| Scheme 1 The two reported LiCH2SiMe3 monomers5,6 and the new LiCH2SiMe3 monomer in this work. | ||
Another knowledge gap in this field is the decomposition pathways of the LiR monomers. In most cases, the monomeric Li–C bond is reactive and could decompose at or above room temperature. However, their decomposition pathways and mechanisms are poorly understood. Our recent MeLi monomer work postulated two decomposition pathways, involving N–C/C–H cleavages and de-coordination, respectively.18 However, due to the low solubility/stability of the decomposition products, these hypothesised decomposition pathways were lacking in direct evidence; in particular, no Li-containing decomposition products were isolated and characterised. An in-depth understanding of the LiR monomer decomposition mechanisms would help synthetic chemists to design their reactions by employing appropriate ligands/conditions to suppress undesired side reactions and tune the selectivity of products.
In this work, we report the synthesis and characterisation of a new LiCH2SiMe3 monomer, [Li(CH2SiMe3)(Me6Tren)] (1), including SCXRD and comprehensive NMR studies. Complex 1's decomposition and preliminary reactivity studies are also reported herein.
Results and discussion
Synthesis and characterisation of [Li(CH2SiMe3)(Me6Tren)] (1)
Complex 1 was synthesised via a reaction between the hexamer [LiCH2SiMe3]6 and Me6Tren in hexanes at −35 °C (Scheme 2). 1 is readily soluble in hexanes, and was isolated as a white crystalline solid in 81% yield by crystallisation from concentrated hexanes solution at −35 °C. 1 is stable in hexanes and benzene/d6-benzene at room temperature for several hours, with observable but slow decomposition (vide infra). However, dissolving in toluene at room temperature leads to an immediate colour change from colourless to red, indicating chemical transformation (vide infra).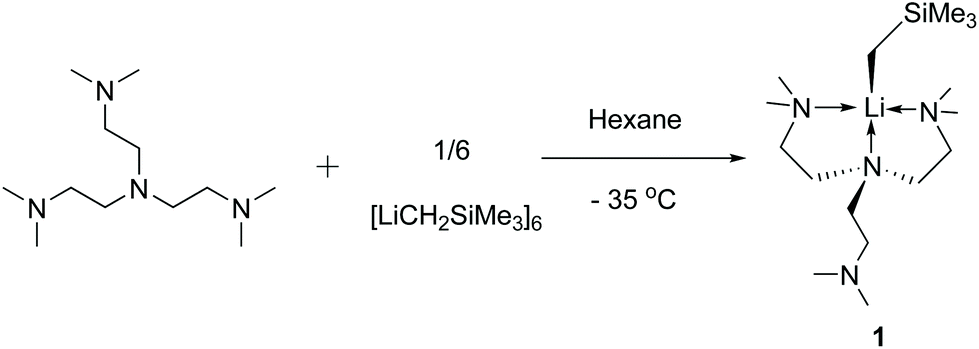 | ||
| Scheme 2 Synthesis of the LiCH2SiMe3 monomer [Li(CH2SiMe3)(κ3-N,N′,N′′-Me6Tren)] (1). | ||
The 1H NMR spectrum in C6D6 of 1 at 25 °C exhibits two singlets at −1.61 ppm and 0.51 ppm, with an integration of 2 and 9 protons, respectively. The singlet at −1.61 ppm (2Hs) is assigned to be the two protons on the lithiated methyl LiCH2SiMe3, while the 0.51 ppm singlet (9Hs) is the protons on the trimethylsilyl LiCH2SiMe3. The NMe2 and NCH2CH2 protons exhibit as a set of broad peaks in the range of 2.3 to 1.9 ppm, with a distinguishable shape and intense signal for the NMe2 functional groups, and broader signals for the NCH2CH2 groups. These 1H signals at room temperature indicate a C3v symmetric coordination mode of the Me6Tren ligand, corroborated by a single 13C environment of –NMe2 groups in the 13C{1H} NMR spectrum. The symmetric NMR signals intuitively suggest that all the three sidearms of the Me6Tren ligand coordinate to the Li, forming a C3v structure. This is contrary to 1's solid-state structure from the single-crystal X-ray diffraction (SCXRD) study (vide infra). The 7Li NMR spectrum of 1 features a singlet at 2.11 ppm.
Single crystals of 1 suitable for SCXRD study were grown from concentrated hexanes solutions at −35 °C. The single-crystal diffraction data was collected at −123 °C. The solid-state molecular structure of 1 is displayed in Fig. 1 with key bond lengths and angles listed in the caption. The Li–C bond length in 1 (2.122(5) Å) is close to the [Li(CH2SiMe3)(PMDTA)] (2.113(2) Å6) but significantly longer than the [Li(CH2SiMe3)(TMCDA)] (2.069(3) Å5). In the solid-state structure, 1 features a partially coordinated Me6Tren ligand in a κ3-N,N′,N′′ coordination mode: two of the three sidearms coordinate to Li, while the other is coordination free. The coordination geometry around Li1 is best described as a distorted triangular pyramid: Li1, N2, N3 and C13 are nearly co-planar (∡C13–Li1–N2 + ∡C13–Li1–N3 + ∡N3–Li1–N2 = 354.6°). It is intriguing that the –CH2SiMe3 alkyl group locates in a cis-position to the coordination-free sidearm with a ∡C13–Li1–N1 angle of 121.3(2)°. Similar cis-geometry was observed in the [Li(CH2SiMe3)(PMDTA)] complex reported by Stalke and co-workers, explained by weak intermolecular interactions (intermolecular Li⋯CH2SiMe3 distance: 4.6 Å).6 However, we could not find such weak intermolecular interactions in 1's lattice.
 | ||
| Fig. 1 (a) X-ray crystal structure of [Li(CH2SiMe3)(κ3-N,N′,N′′-Me6Tren)] (1) at −123 °C with 50% probability ellipsoids. Hydrogen atoms are omitted and only the disordered component with the highest occupancy is shown for clarity. (b) Space filling presentation of 1 (grey: carbon; green: lithium; blue: nitrogen; coral: silicon). The selected bond distances (Å) and angles (°) are: Li1–C13 2.122(5); Li1–N1 2.177(4); Li1—N2 2.167(4); Li1–N3 2.189(4); Si1–C13–Li1 129.77(17); C13–Li1–N1 121.3(2); C13–Li1–N2 127.4(2); C13–Li1–N3 112.91(19); N2–Li1–N3 114.29(19). | ||
To study if the κ3-N,N′,N′′ coordination mode of 1 persists in solution, where such weak intermolecular interactions in crystal lattice would be cancelled, we conducted a variable-temperature (VT) 1H NMR study of 1 in d8-THF. The 1H NMR spectrum of 1 at room temperature, apparently, suggests that all the three sidearms are chemically equivalent (Fig. 2 25 °C), indicated by only one set of the NMe2 (Fig. 2 25 °C, b) and the NCH2CH2 (Fig. 2 25 °C, a) signals. However, when cooled to −103 °C, the signals split into two sets with a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 integral ratio (Fig. 2 –103 °C), which matches the solid-state structure in the κ3-N,N′,N′′ coordination mode. Therefore, we conclude that the κ3-N,N′,N′′ coordination mode persists in solution. The room temperature 1H NMR spectrum probably results from a fast fluxional coordination and de-coordination process at the NMR time scale. The decoordination of one sidearm in the Me6Tren ligand has been observed with bulky Li-substituents, such as [2-tBu-C5H5N]−1 (ref. 19) or [N(SiMe3)2]−1.20 The –CH2SiMe3 alkyl group in 1 could rotate around the Li–C bond and features a big dynamic radius (Fig. 1(b) space-filling model), it could therefore push one of the three sidearms away and forms the observed structure of 1.
:1 integral ratio (Fig. 2 –103 °C), which matches the solid-state structure in the κ3-N,N′,N′′ coordination mode. Therefore, we conclude that the κ3-N,N′,N′′ coordination mode persists in solution. The room temperature 1H NMR spectrum probably results from a fast fluxional coordination and de-coordination process at the NMR time scale. The decoordination of one sidearm in the Me6Tren ligand has been observed with bulky Li-substituents, such as [2-tBu-C5H5N]−1 (ref. 19) or [N(SiMe3)2]−1.20 The –CH2SiMe3 alkyl group in 1 could rotate around the Li–C bond and features a big dynamic radius (Fig. 1(b) space-filling model), it could therefore push one of the three sidearms away and forms the observed structure of 1.
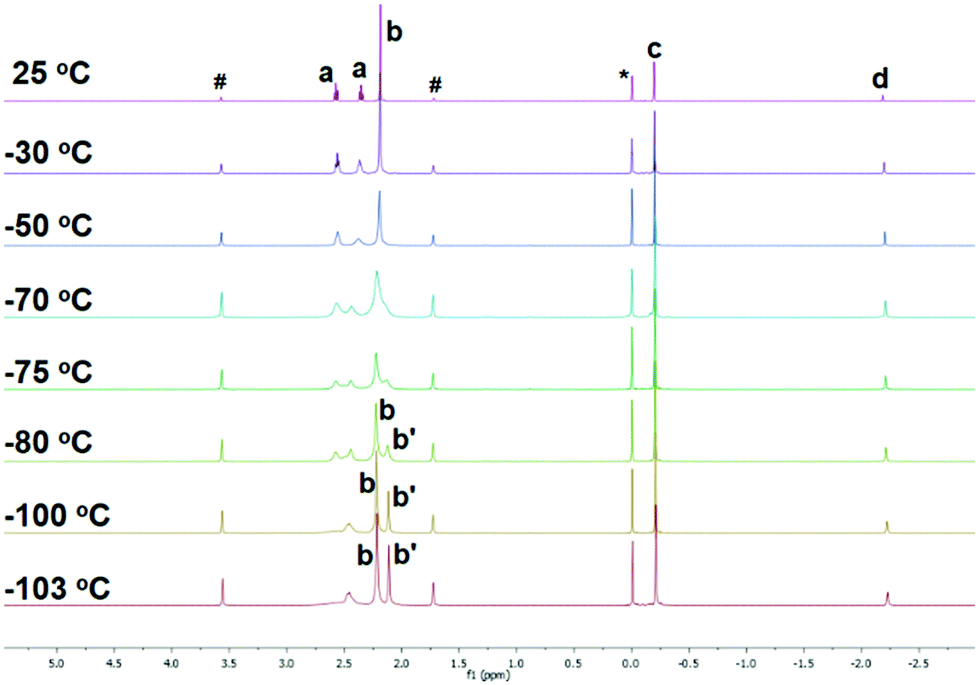 | ||
| Fig. 2 Variable temperature (VT) 1H NMR of complex 1 in d8-THF. (a: NCH2CH2; b and b′: NMe2; c: CH2SiMe3; d: CH2SiMe3; *: Me4Si; #: d8-THF). | ||
The UV/Vis spectra of organolithium complexes has been studied in the 1960s–1980s,21–29 in the context of photoexcited states of organic anions.30 However, in more recent works, the UV/Vis spectra are missing for the isolated monomeric lithium alkyl complexes.4–11 We examined the UV/Vis spectrum of 1 in hexanes, and made a comparison with the parent [LiCH2SiMe3]6 hexamer and the Me6Tren ligand (Fig. S11†). Both complex 1 and the Me6Tren ligand feature a strong (>6000 L mol−1 cm−1) absorption in the deep UV region (∼220 nm). These absorptions are probably due to electron density transitions based on the saturated amine (Me6Tren),31 but a possible Li–C → N–CH3/Si–CH3 charge transfer was also identified by time-dependent DFT (TD-DFT) calculations (Fig. S12†). The strong UV absorption of the Me6Tren ligand highlights that, precaution must be taken when interpreting the UV spectrum of complexes bearing such saturated amine ligands, where the ligand-based absorption could interfere and overlap with other charge transfers.
Decomposition of [Li(CH2SiMe3)(Me6Tren)] (1)
Complex 1 decomposes in C6D6 or hexanes solutions over two days. In situ1H/7Li NMR studies reveal the production of vinyl dimethyl amine (H2CCH–NMe2), Me4Si and a Li-containing product [Li(μ-N-κ2-N,N′-N(CH2CH2NMe2)2]2 (2) (Scheme 3). Similar decomposition was also observed for a reaction between tBuLi and Me6Tren (Scheme 3) (see ESI†), which is faster (completed in 15 minutes at room temperature), in line with the stronger Brønsted basicity of tBuLi compared to LiCH2SiMe3. The solid-state structure of 2 was elucidated by SCXRD (Fig. 3), and is the same as reported by Klumpp and co-workers via a different synthetic route.32
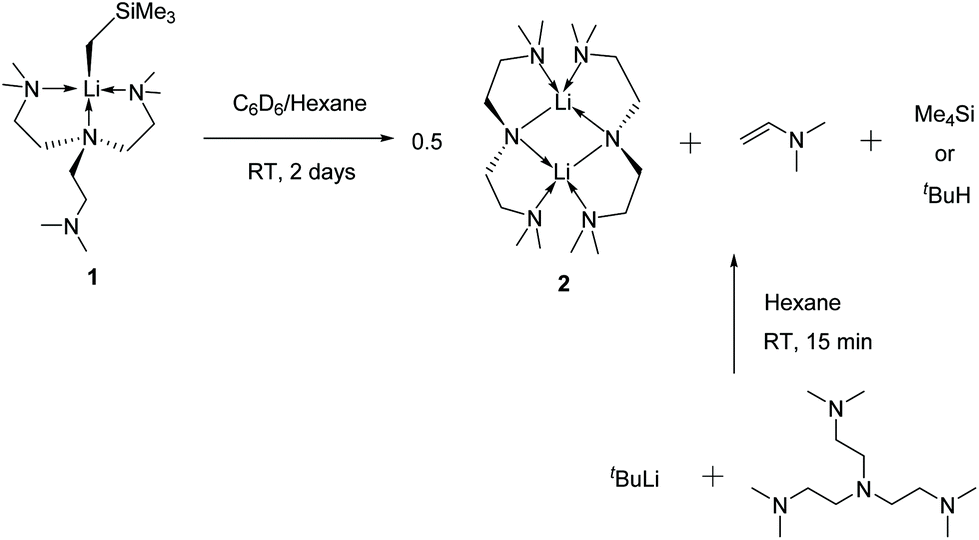 | ||
| Scheme 3 Decomposition of 1 to form 2, vinyl dimethyl amine and tetramethylsilane. | ||
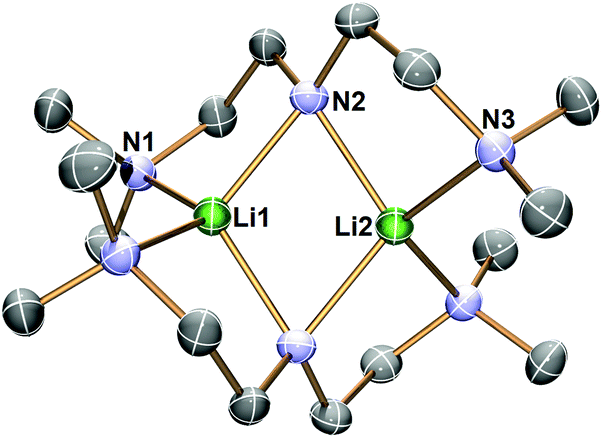 | ||
| Fig. 3 X-ray crystal structure of [Li(μ-N-κ2-N,N′-N(CH2CH2NMe2)2]2 (2) at −123 °C with 50% probability ellipsoids. Hydrogen atoms are omitted for clarity. The selected bond distances (Å) and angles (°) are: Li1–N1 2.0903(18); Li1–N2 2.0286(19); Li2–N2 2.0192(18); Li2–N3 2.0864(17); L1⋯Li2 2.376(4); Li1–N2–Li2 71.89(9). | ||
The production of 2 occurs via C–H and N–C bond cleavages of one sidearm of the Me6Tren ligand. Similar decomposition pathways were postulated by us for a MeLi monomer,18 and by Chen and co-workers for a scandium imido complex.33 By 1H NMR monitoring of 1's decomposition for 2 days, it is obvious that 1 is involved in the rate-determining step of the decomposition, as suggested by a non-linear concentration–time decay ([1]–t) curve (Fig. 4 orange). More information is revealed by examining the products concentration increase curves (Fig. 4 blue, green and lilac). It is evident that Me4Si's concentration increases more rapidly at the initial stage of the reaction, with a steeper [c]–t curve from 0 to circa 2000 minutes. The vinyl dimethyl amine and complex 2 follow a similar increasing pattern, considering that 2 is a dimer therefore, its concentration is halved. From the data, we postulate that the decomposition of 1 follows a stepwise mechanism (Fig. 4). The step-1 is the deprotonation of the coordination-free sidearm, producing Me4Si and an intermediate, which was not isolated. The postulated intermediate was not observed in the 1H NMR spectra, possibly because its signals overlap with the Me6Tren ligand signals of 1. However, the 1H NMR signals of the postulated intermediate do not interfere with the signals used for acquiring integrals and concentrations, namely −1.66 ppm (s, 2H, Li–CH2 for 1); 0.00 ppm (s, 12H for SiMe4); 3.81 (d, 1H, for Me2N-CHCH2); 3.44 (m, br, 8H, NCH2CH2N for 2). Therefore, the accuracies of the integrals and concentrations are not compromised. Step-2 was a combination of rearrangement, cleavage of an N–C bond, formation of CC and Li–N bonds, and dimerisation into complex 2.
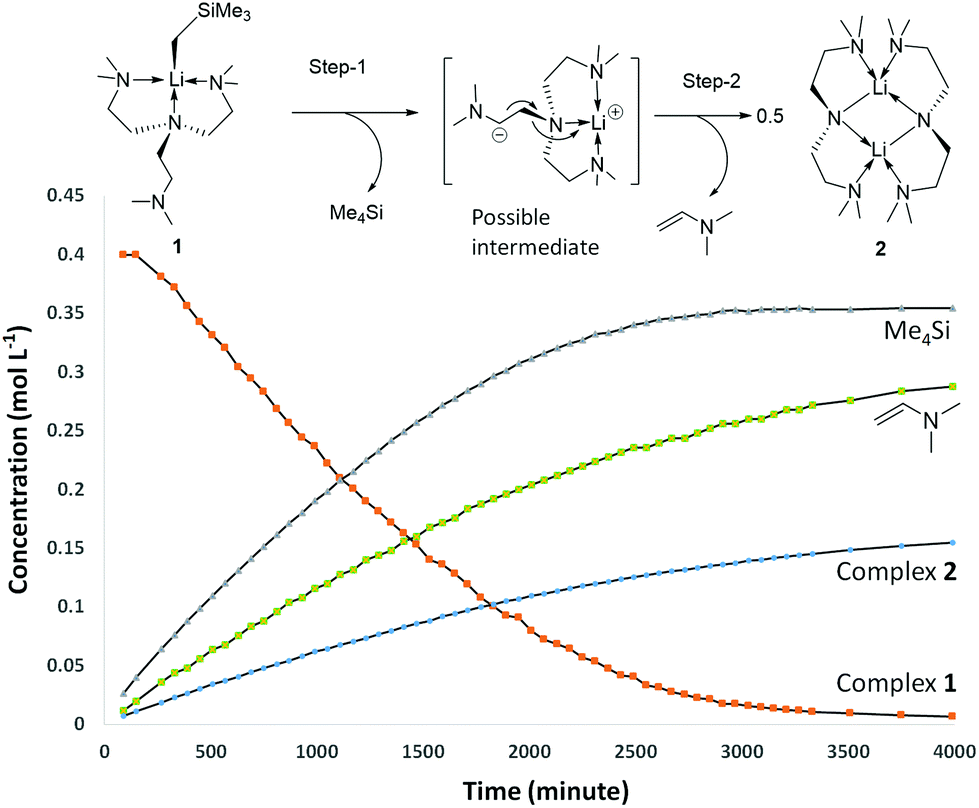 | ||
| Fig. 4 Substrate concentration decay curve and products concentration increase curves for 1's decomposition at 25 °C (initial concentration [1]0 = 0.4 mol L−1). The solid lines are for visual guidance only. The Me4Si and vinyl dimethyl amine are volatiles, therefore their concentrations did not arrive the theoretic value of 0.4 mol L−1 upon 1's full decomposition. The postulated stepwise decomposition mechanism is presented at the top. | ||
The Li–Namide bonds in complex 2 are reactive. 2 reacts with silicone grease to produce [Li{κ3-O,N,N′-OSiMe2N(CH2CH2NMe2)2}]2 (3) (Scheme 4) (see ESI† for details). 3 was characterised by SCXRD (Fig. 5). Silicone grease acts as a serendipitous reactant in organometallic chemistry, which could be activated by highly reactive species.34–36 A noticeable structural feature of 3 is the planar tertiary amine fragment (N1) (∑∡ = 360°). A planar amine is hypothesised to be the transition state of amine pyramidal inversion,37 which plays an essential role in N-nucleophilic reaction, N-substitution and N2-to-NH3 conversion (Haber–Bosch process). Therefore, a planar amine is sought-after by organic chemists as a model compound to understand the reaction mechanisms.38 The formation of a planar amine in 3 is probably due to hyperconjugation between the N lone pair and the silicon atom.39 In spite of the serendipitous formation of 3, deliberate reactions between 2 and an excess amount of grease did not increase the yield of 3, which is always a minor by-product. We conducted the stoichiometric reaction between 2 and one equivalent of hexamethyldisiloxane (Me3Si–O–SiMe3, HMDSO), which is a small molecular analogue of silicone grease, in C6D6 in a grease-free environment (Teflon J. Young tap NMR tube). The reaction was monitored by 1H and 7Li NMR spectra: we observed no reaction at room temperature (48 hours) or 60 °C (48 hours).
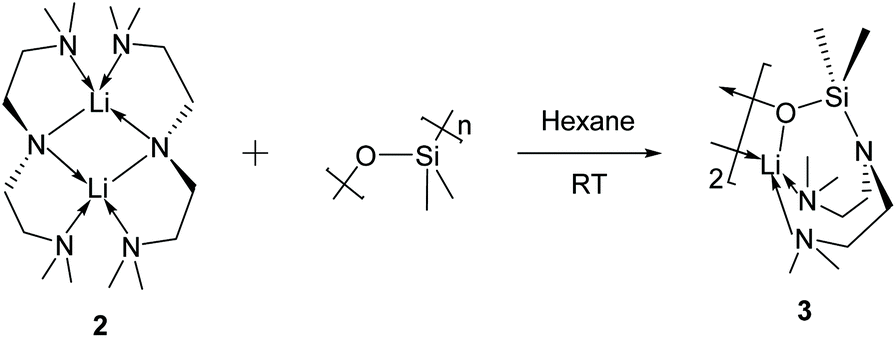 | ||
| Scheme 4 Reaction between silicone grease and complex 2. | ||
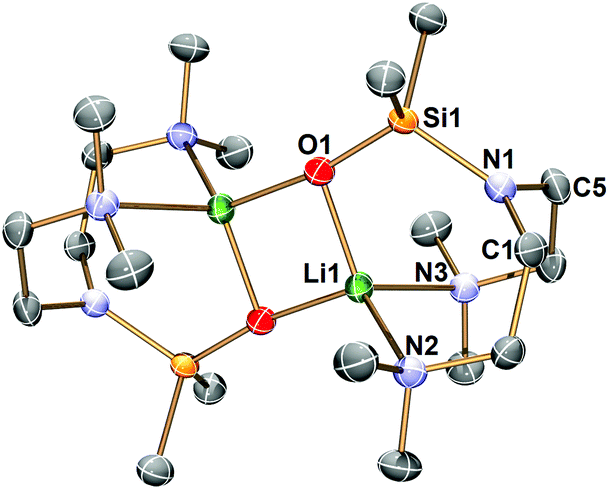 | ||
| Fig. 5 X-ray crystal structure of [Li{κ3-O,N,N′-OSiMe2N(CH2CH2NMe2)2}]2 (3) at −123 °C with 50% probability ellipsoids. Hydrogen atoms are omitted for clarity. The selected bond distances (Å) and angles (°) are: Li1–O1 1.851(3); Li1–N2 2.213; Li1–N3 2.232; Si1–O1 1.5794(13); Si1–N1 1.7439(16); N1–C1 1.454(2); N1–C5 1.454(2); Si1–N1–C5 121.65(12); Si1–N1–C1 123.14(12); C1–N1–C5 115.21(15). | ||
Preliminary reactivity studies of [Li(CH2SiMe3)(Me6Tren)] (1)
Complex 1 reacts with benzophenone to produce an alkyloxo lithium complex [Li(OC(CH2SiMe3)Ph2)]4 (4) as a tetramer (Scheme 5). The SCXRD structure of 4 is exhibited in Fig. 6. 4 is the result of CO bond insertion into the Li–C bond and Me6Tren ligand de-coordination. The free Me6Tren ligand was observed in the 1H NMR spectrum of the crude reaction products (see ESI†). 4 features a cubic Li4O4 core structure.
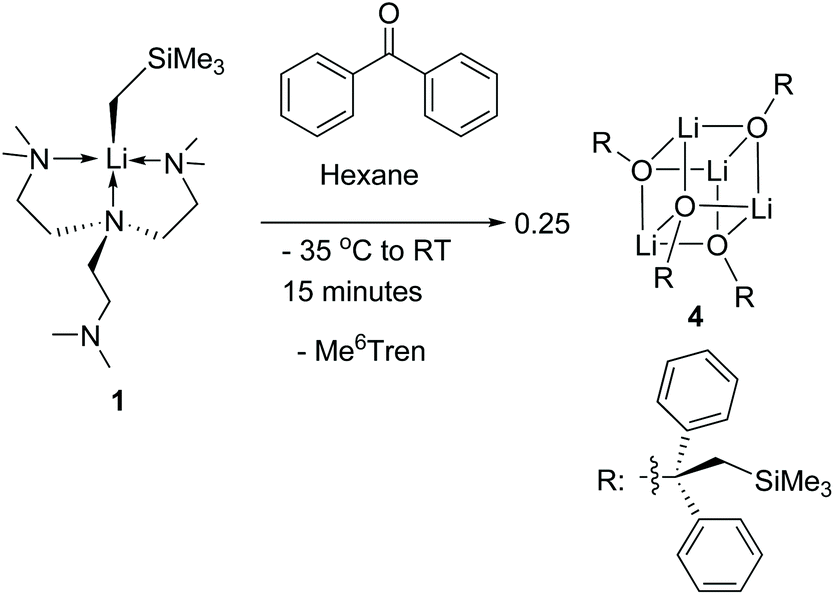 | ||
| Scheme 5 The reaction between 1 and benzophenone to produce 4. | ||
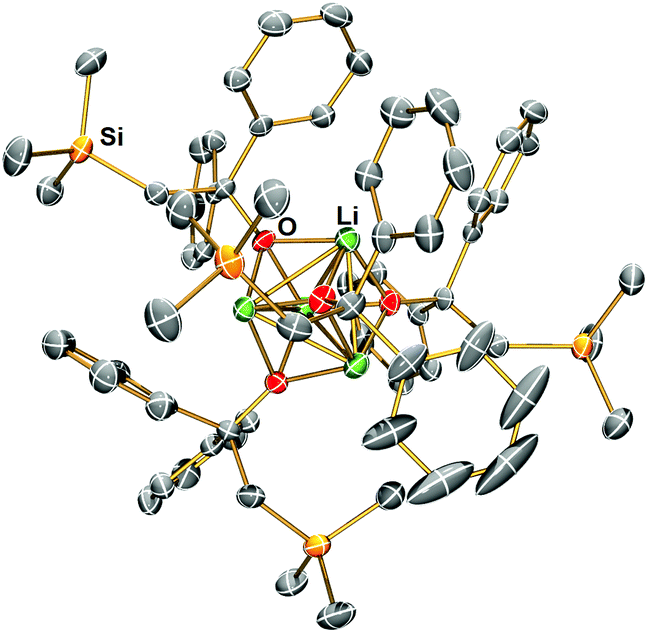 | ||
| Fig. 6 X-ray crystal structure of [Li(OC(CH2SiMe3)Ph2)]4 (4) at −123 °C with 50% probability ellipsoids. Hydrogen atoms are omitted for clarity. | ||
Complex 1 readily reacts with toluene to produce [Li(CH2Ph)(Me6Tren)] (5) (Scheme 6). The reaction was characteristic of its colour changing: upon adding toluene, the solution turned from colourless (1) to red within 15 minutes at −78 °C. Complex 5 was identified by comparing its 1H and 13C{1H} NMR spectra with the authenticated spectra reported by Mulvey and co-workers in 2011.12 In contrast with the facile C–H activation mediated by the monomer 1, the parent [LiCH2SiMe3]6 hexamer only reacts sluggishly with toluene at room temperature. We attribute the enhanced C–H activation reactivity of 1 to its monomeric structure. Similar facile toluene C–H activation, induced by a LiCH2SiMe3 dimer [{Me2N(CH2)2OMe}·(LiCH2SiMe3)]2, was reported by Stalke and co-workers.14
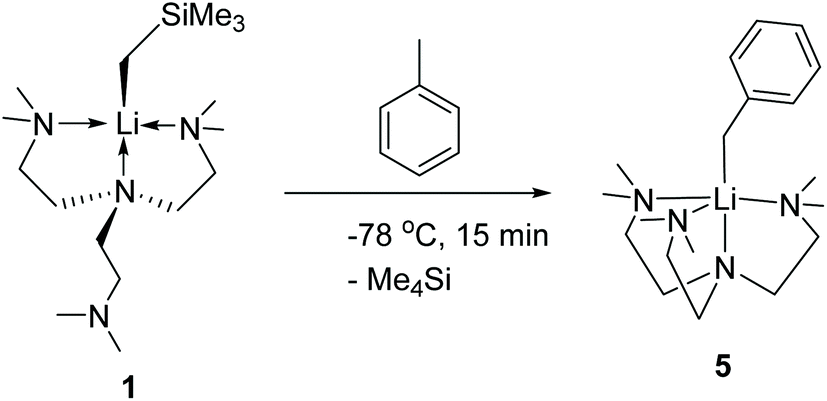 | ||
| Scheme 6 C sp3–H activation of toluene by the LiCH2SiMe3 monomer 1 to produce 5. | ||
Conclusions
This work reports the synthesis and characterisation of a monomeric (trimethylsilyl)methyl lithium complex 1, which is only the third member of its class. 1's coordination mode was elucidated by SCXRD and variable-temperature NMR spectroscopy. 1 exhibits moderate thermal stability. Its decomposition occurs via stepwise C–H and N–C bond cleavage, leading to a lithium amide complex 2. The resultant lithium amide complex 2 features a highly reactive Li–Namide bond, which undergoes an insertion reaction with silicone grease to produce a silyl-oxo complex 3 with a planar tertiary amine centre. Preliminary reactivity studies reveals enhanced C–H activation reactivity towards the C–H bond in toluene compared to its parent [LiR]6 hexamer, as well as benzophenone's CO insertion into the Li–C bond.
This work adds a new member to the small class of structurally characterised monomeric organolithium complexes. The decomposition and reactivity studies could provide insights for organic/organometallic synthetic chemists to assist their design of reaction conditions, to achieve a fine balance between enhanced reactivity and stability.
Experimental
General procedures
All manipulations were carried out using Schlenk techniques, or a Vigor glovebox equipped with a −35 °C freezer and a cold well, under an atmosphere of dry argon. Solvents were dried by sodium metal and sodium/potassium alloy, then distilled under vacuum. All solvents were stored in the glovebox over potassium mirrors except for ethers, which were stored over activated 4 Å molecular sieves. The deuterated solvents were distilled from sodium/potassium alloy, degassed by three freeze–pump–thaw cycles and stored under argon. (Trimethylsilyl)methyl lithium (LiCH2SiMe3) was prepared from refluxing chloromethyl trimethyl silane (ClCH2SiMe3) and excess lithium metal in hexanes.40 Tris[2-(dimethylamino)ethyl]amine (Me6Tren) was purchased from Merck and dried over 4 Å molecular sieves prior to use.1H, 13C{1H} and 7Li NMR spectra were recorded on a Bruker 300 Avance III spectrometer operating at 300.13, 75.48 and 116.64 MHz respectively. Variable temperature and kinetic 1H NMR experiments were carried out on a Bruker 500 Avance III HD spectrometer operating at 500.15 MHz. Chemical shifts are quoted in ppm and are relative to SiMe4 (1H and 13C) or external 0.1 M LiCl in D2O (7Li). The UV/Vis absorption spectrum of 1 was recorded on a Shimadzu UV-1800 spectrometer in the wavelength range of 190 to 1100 nm using a 10 mm path-length J. Young tap quartz cuvette.
Synthesis of [Li(CH2SiMe3)(κ3-N,N′,N′′-Me6Tren)] (1)
LiCH2SiMe3 (0.0942 g, 1 mmol) was dissolved in hexanes (1 ml). Me6Tren (0.230 g, 1 mmol) was dissolved in hexanes (1 ml) and cooled to −35 °C. The solution of LiCH2SiMe3 was added to the solution of Me6Tren in one portion. The resulting pale yellow solution was left at room temperature for 5 minutes, before being placed in a −35 °C freezer. After 3 days, colourless crystals suitable for single crystal X-ray diffraction resulted. The mother liquor was removed and the solid was dried in vacuo (81% yield). 1H NMR (300 MHz, d6-benzene, 25 °C): δ (ppm) 2.20–1.90 (br, 12H, NCH2CH2N), 2.05 (s, 18H, N(CH3)2), 0.51 (s, 9H, Si(CH3)3), −1.61 (s, 2H, LiCH2Si). 13C{1H} NMR (75 MHz, d6-benzene, 25 °C): δ (ppm) 57.7 (NCH2CH2N), 52.1 (NCH2CH2N), 45.9 (N(CH3)2), 7.1 (Si(CH3)3), −4.8 (LiCH2Si). 7Li NMR (117 MHz, d6-benzene, 25 °C): δ (ppm) 2.10.Synthesis of [Li(μ-N-κ2-N,N′-N(CH2CH2NMe2)2]2 (2) and [Li{κ3-O,N,N′-OSiMe2N(CH2CH2NMe2)2}]2 (3)
LiCH2SiMe3 (0.188 g, 2 mmol) was partially dissolved in hexanes (1 ml). Me6Tren (0.461 g, 2 mmol) was dissolved in hexanes (1 ml). The solution of Me6Tren was added in one-portion to the LiCH2SiMe3 solution. The resulting pale yellow solution was left at room temperature for 3 days. The volatiles were removed in vacuo. A pale yellow solid resulted. The solid was dissolved in hexanes (2 ml) and the solution was filtered then concentrated to 0.5 ml. The solution was placed in a −35 °C freezer. After 3 days, an off-white crystalline solid resulted. The mother liquor was removed and the solid was dried in vacuo (60% yield).Alternative synthesis of 2
t BuLi (0.128 g, 2 mmol) was partially dissolved in hexanes (1 ml). Me6Tren (0.461 g, 2 mmol) was dissolved in hexanes (1 ml). The solution of Me6Tren was added in one-portion to the tBuLi solution at room temperature. Immediately, a bright orange solution resulted that turned very pale within 1 minute. The solution was left at room temperature for 1 hour. The volatiles were removed in vacuo. A pale yellow solid resulted. The solid was dissolved in hexanes (2 ml) and the solution was filtered then concentrated to 0.5 ml. The solution was placed in a −35 °C freezer. After 3 days, an off-white crystalline solid resulted. The mother liquor was removed and the solid was dried in vacuo (57% yield). Single crystals of 2 suitable for single crystals X-ray diffraction were grown from dissolving the crude solid in hexanes (2 ml). The solution was filtered and placed in a −35 °C freezer. After 3 days, large colourless blocks resulted. 1H NMR (300 MHz, d6-benzene, 25 °C): δ (ppm) 3.44 (m, br, 8H, NCH2CH2N), 3.09 (m, br, 4H, NCH2CH2N), 2.25 (m, br, 4H, NCH2CH2N), 2.04 (s, br, 24H, NCH3)2. 13C{1H} NMR (75 MHz, d6-benzene, 25 °C): δ (ppm) 63.8 (NCH2CH2N), 58.3 (NCH2CH2N), 47.4 (br, N(CH3)2), 41.9 (br, N(CH3)2). 7Li NMR (117 MHz, d6-benzene, 25 °C): δ (ppm) 1.51.Synthesis of [Li{κ3-O,N,N′-OSiMe2N(CH2CH2NMe2)2}]2 (3)
3 was produced as a minor side product from the reaction producing 2 when silicon grease was applied. 3 co-crystallises with 2 with similar crystal habits: both are pale yellow prismatic crystals. Due to their similar solubility in common inorganic solvents and crystallisation behaviours, it is impossible to isolate 3 from 2 by fractional crystallisation. The NMR spectra (1H, 7Li and 13C) of the mixture suggest that 2 is the dominant ingredient with no observable 3. Therefore, we postulate that 3 is a minor serendipitous product from 2 reacting with silicon grease. Deliberate treatment of 2 with excess amount of silicon grease did not result in the increased yield of 3. In a grease-free environment (J. Young tap NMR tube), complex 2 does not react with one equivalent of hexamethyldisiloxane (HMDSO) in C6D6 solution at room temperature or 60 °C within 48 hours.Synthesis of [Li(OC(CH2SiMe3)Ph2)]4 (4)
At −35 °C, a solution of 1 (0.649 g, 2 mmol) in 5 mL of hexanes was added to a solution of benzophenone (0.364 g, 2 mmol) in 5 mL of hexanes. The solution was allowed to stay at room temperature for 15 minutes, then all volatiles were removed under vacuum, to afford a white solid, which is a mixture of 4 and Me6Tren (see the 1H NMR spectrum, Fig. S7 in ESI†). Attempts were made to remove the Me6Tren by washing the mixture with hexanes. However, the solubilities of 4 and Me6Tren in hexanes are similar, even at −35 °C. Due to the persisting presence of Me6Tren, a meaningful yield of 4 is not available. However, the 1H and 7Li NMR spectra of the mixture suggest a clean conversion. 1H NMR (300 MHz, d6-benzene, 25 °C): δ (ppm) 7.38–7.27 (m, 2H, ArH), 7.22–7.15 (m, 3H, ArH), 1.49 (s, 2H, OC(Ph2)(CH2SiMe3)), −0.19 (s, 9H, OC(Ph2)(CH2SiMe3)). 7Li NMR (117 MHz, d6-benzene, 25 °C): δ (ppm) 0.43.Synthesis of [Li(CH2Ph)(Me6Tren)] (5)
Toluene (30 ml) was added to 1 (3.8904 g, 12 mmol) at room temperature. Immediately, a bright yellow solution resulted, which was stirred for 1 hour. The resulting dark orange solution was placed in a −20 °C freezer. After three days, yellow needle-shaped crystals resulted. The mothor liquor was removed and the crystals dried in vacuo to afford 5 (77% yield). 1H and 13C{1H} NMR spectra matched the spectra reported by Mulvey and co-workers in 2011.12Author contributions
N. D. and E. L. designed and conducted the experiments, synthesised and characterised the complexes. P. G. W collected, solved, refined and analysed the crystallographic data. C. D. and C. W. designed the VT NMR and kinetic NMR experiments, collected and analysed the data. T. J. P. conducted the calculations. E. L. conceptualised the central idea, supervised the work, analysed the data and wrote the manuscript with contributions from all the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Chemistry Technical Support Team (Dr Laura McCorkindale, Dr Amy Roberts, Ms Alexandra Rotariu) at Newcastle University for supporting our research. N. D. and E. L. thank Professor Andrew C. Benniston and Dr Keith Izod (both are from Newcastle University) for their support in laboratory infrastructure. E. L. thanks Emeritus Professor William Clegg (Newcastle University) for the insightful discussions. E. L. thanks the Newcastle University Academic Track (NUAcT) Fellowship and The Royal Society of Chemistry Research Enablement Grants (E20-5153) for financial support. N. D. thanks Newcastle University for a NUAcT PhD studentship. T. J. P. thanks the EPSRC for funding (EP/R021503/1).Notes and references
- W. Schlenk and J. Holtz, Ber. Dtsch. Chem. Ges., 1917, 50, 262 CrossRef CAS.
- U. Wietelmann and J. Klett, Z. Anorg. Allg. Chem., 2018, 644, 194 CrossRef CAS PubMed.
- Z. Rappoport and I. Mare, The chemistry of organolithium compounds, John Wiley & Sons, Ltd, Chichester, West Sussex, England, 2004 Search PubMed.
- H. J. Reich, Chem. Rev., 2013, 113, 7130 CrossRef CAS PubMed.
- L. Knauer, J. Wattenberg, U. Kroesen and C. Strohmann, Dalton Trans., 2019, 48, 11285 RSC.
- T. Tatic, H. Ott and D. Stalke, Eur. J. Inorg. Chem., 2008, 3765 CrossRef CAS.
- M. F. Lappert, L. M. Engelhardt, C. L. Raston and A. H. White, J. Chem. Soc., Chem. Commun., 1982, 1323 RSC.
- C. Strohmann and V. H. Gessner, J. Am. Chem. Soc., 2007, 129, 8952 CrossRef CAS PubMed.
- C. Strohmann, T. Seibel and K. Strohfeldt, Angew. Chem., Int. Ed., 2003, 42, 4531 CrossRef CAS PubMed.
- C. Strohmann and V. H. Gessner, Angew. Chem., Int. Ed., 2007, 46, 8281 CrossRef CAS PubMed.
- C. Strohmann, V. H. Gessner and A. Damme, Chem. Commun., 2008, 3381 RSC.
- M. G. Davidson, D. Garcia-Vivo, A. R. Kennedy, R. E. Mulvey and S. D. Robertson, Chem. – Eur. J., 2011, 17, 3364 CrossRef CAS.
- A. Lennartson, J. Sundberg, T. Wiklund, G. Hilmersson and M. Hakansson, Eur. J. Inorg. Chem., 2010, 3029 CrossRef CAS.
- T. Tatic, S. Hermann, M. John, A. Loquet, A. Lange and D. Stalke, Angew. Chem., Int. Ed., 2011, 50, 6666 CrossRef CAS.
- L. M. Engelhardt, W.-P. Leung, C. L. Raston, G. Salem, P. Twiss and A. H. White, J. Chem. Soc., Dalton Trans., 1988, 2403 RSC.
- J. Arnold, V. Knapp, J. A. R. Schmidt and A. Shafir, J. Chem. Soc., Dalton Trans., 2002, 3273 RSC.
- D. R. Armstrong, M. G. Davidson, D. Garcia-Vivo, A. R. Kennedy, R. E. Mulvey and S. D. Robertson, Inorg. Chem., 2013, 52, 12023 CrossRef CAS.
- N. Davison, E. Falbo, P. G. Waddell, T. J. Penfold and E. Lu, Chem. Commun., 2021, 57, 6205 RSC.
- S. D. Robertson, A. R. Kennedy, J. J. Liggat and R. E. Mulvey, Chem. Commun., 2015, 51, 5452 RSC.
- D. M. Cousin, M. G. Davidson, C. J. Frankis, D. García-Vivó and M. F. Mahon, Dalton Trans., 2010, 39, 8278 RSC.
- R. Waack and M. Doran, J. Am. Chem. Soc., 1963, 85, 1651 CrossRef CAS.
- E. E. van Tamelen, J. I. Brauman and L. E. Ellis, J. Am. Chem. Soc., 1965, 87, 4964 CrossRef CAS.
- H. J. S. Winkler, R. Bollinger and H. Winkle, J. Org. Chem., 1967, 32, 1700 CrossRef CAS.
- W. H. Glaze and T. L. Brewer, J. Am. Chem. Soc., 1969, 91, 4490 CrossRef CAS.
- E. E. van Tamelen, J. I. Brauman and L. E. Ellis, J. Am. Chem. Soc., 1971, 93, 6141 CrossRef CAS.
- E. E. van Tamelen, J. I. Brauman and L. E. Ellis, J. Am. Chem. Soc., 1971, 93, 6145 CrossRef CAS.
- W. A. Nugent, F. Bertini and J. K. Kochi, J. Am. Chem. Soc., 1974, 96, 4945 CrossRef CAS.
- L. M. Tolbert, J. Am. Chem. Soc., 1980, 102, 6808 CrossRef CAS.
- M. A. Fox, C.-C. Chen and K. A. Campbell, J. Org. Chem., 1983, 48, 321 CrossRef CAS.
- M. A. Fox, Chem. Rev., 1979, 79, 253 CrossRef CAS.
- E. Tanenbaum, E. M. Coffin and A. J. Harrison, J. Chem. Phys., 1953, 21, 311 CrossRef.
- G. L. J. van Vliet, F. J. J. de Kanter, M. Schakel, G. W. Klumpp, A. L. Spek and M. Lutz, Chem. – Eur. J., 1999, 5, 1091 CrossRef CAS.
- J.-X. Chu, Q.-H. Zhou, Y.-X. Li, X.-B. Leng and Y.-F. Chen, Sci. China: Chem., 2014, 57, 1098 CrossRef CAS.
- I. Haiduc, Organometallics, 2004, 23, 3 CrossRef CAS.
- L.-C. Pop and M. Saito, Coord. Chem. Rev., 2016, 314, 64 CrossRef CAS.
- V. Nahrstedt, A. Raauf, C. Hegemann, V. Brune, J. Schläfer and S. Mathur, Z. Anorg. Allg. Chem., 2021, 1102 CrossRef CAS.
- V. Wolfart, R. Gleiter, C. Krieger and H. Pritzkow, Tetrahedron Lett., 1998, 39, 513 CrossRef CAS.
- N. Mandal, A. K. Pal, P. Gain, A. Zohaib and A. Datta, J. Am. Chem. Soc., 2020, 142, 5331 CrossRef CAS PubMed.
- S. G. Wierschke, J. Chandrasekhar and W. L. Jorgensen, J. Am. Chem. Soc., 1985, 107, 1496 CrossRef CAS.
- P. J. Davidson, D. H. Harris and M. F. Lappert, J. Chem. Soc., Dalton Trans., 1976, 2268 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2114458–2114460 (1–3) and 2120419 (4). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt03532k |
| This journal is © The Royal Society of Chemistry 2022 |