
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSite-specific role of bifunctional graphitic carbon nitride catalyst for the sustainable synthesis of 3,3-spirocyclic oxindoles in aqueous media†
Anshu Dandia*a,
Dinesh Kumar Mahawara,
Pratibha Sainia,
Surendra Sainia,
Shyam L. Guptaab,
Kuldeep S. Rathorec and
Vijay Parewa *a
*a
aCentre of Advanced Studies, Department of Chemistry, University of Rajasthan, Jaipur, India. E-mail: dranshudandia@yahoo.co.in; parewavijay@uniraj.ac.in; parewavijay.parewa@gmail.com
bGovernment Polytechnic College, Near Itarana Fly Over, Kalimori, Alwar, Rajasthan 301001, India
cDepartment of Physics, Arya College of Engineering and IT, Jaipur, India. E-mail: kuldeep_ssr@yahoo.com
First published on 23rd August 2021
Abstract
Functionalized graphitic carbon nitride (Sg-C3N4) has been manufactured and used as a reusable catalyst for the one-pot production of various spiro-pyrano chromenes and spiro indole-3,1′-naphthalene tetracyclic systems in aqueous media. An ultrasound-assisted method has been used for the functionalization of g-C3N4. The catalytic functionalities and the structural integrity of the catalyst were characterized via different analytical tools. The catalytic site-specific role of Sg-C3N4 was confirmed via various control experiments in one-pot reaction sequences. We recognized that Sg-C3N4 acts as a bifunctional acid–base catalyst for the first reaction sequence whereas it is an acidic catalyst for the second reaction sequence during the one-pot production of various spiro-pyrano chromenes. In addition, the bifunctional acid–base catalytic role of Sg-C3N4 has been confirmed for the first reaction sequence whereas it has a basic catalytic role for the second reaction sequence during the one-pot production of spiro indole-3,1′-naphthalene tetracyclic systems. Diverse C–C, C–O, and C–N bonds, six-membered cycles, stereogenic centers, and spiro frameworks were formed in a single reaction, enhancing the biocidal profile and possibly resulting in the discovery of new medicinal properties. The mild reaction environment, simple workup, easy separation, low cost, heterogeneity, and recyclability of Sg-C3N4 are some rewards of this approach.
1 Introduction
Carbon is one of the most multifaceted elements in the periodic table. Carbon-based substances have achieved insightful consequences in many areas of science and technology because of their noteworthy properties.1 In previous decades, many scientists worked on developing inventive carbon-based compounds for wide-ranging applications.2 Carbon materials have a splendid ability to provide at the nano-scale because they have fabulous thermal and electrical conductivity as well as lightness and elevated mechanical strength that traditional bulk substances cannot possess.3 With the multiplicity of their nanostructures, these distinctive values can be attained over an enormously broad range of environments. Therefore, they are exhaustively investigated for innumerable applications in optoelectronics and photonics, nanomedicine and biotechnology, advanced electrodes and polymer composites.4 It is the chemical genius of carbon that it can create different nanostructures with entirely different properties.5 Amongst carbon-based substances, graphitic carbon nitride (g-C3N4), has elicited a huge focus in organic transformation because of its robust nature. g-C3N4 as an abundant and nontoxic substance can be effortlessly synthesized from carbon constituents, by the one-step polymerization of N-rich chemical compounds.6 Due to their distinctive properties they are potential candidates for a variety of applications in numerous fields.7 The significance of g-C3N4 as a catalyst or catalytic support has also been extensively recognized for numerous chemical reactions.8 However, the preparation of bifunctional g-C3N4 with acid and base sites is still a difficult task for the chemist. Enormous efforts9 have been made to introduce acidic groups into basic g-C3N4. But in these methods the inherent reasonable basic groups of g-C3N4 have been destroyed and the resulting material has very low or even no basicity. Therefore, there is a still a need for a mild and efficient protocol for the concurrent introduction of acid and base sites in g-C3N4 to make it a bifunctional catalyst.Organic compounds with a spiro heterocyclic moiety are receiving wide scientific interest because of their distinctive conformational and chemical characteristics as well as the biological properties that are often coupled with the asymmetric spiro carbon atom.10 More specifically, the spiro-oxindole nucleus is found in a variety of biologically active heterocyclic compounds and natural products and can be of interest in the development of innovative clinically significant heterocyclic motifs.11 For example, a novel class of marine toxins isolated from dinoflagellates and shellfish, like pteriatoxin and pinnatoxins,12 shows that a spiro aza system is responsible for the biological activity. Synthetic derivatives of spiro-oxindole ring systems, for example spirotryprostatin B, elacomine, alstonisine and horsfiline, have become imperative synthetic targets as these structural frameworks form the central units of numerous naturally occurring compounds with important biological activities.13
Several spiro-oxindole derivatives show interesting biological activities (Fig. 1): for example, as progesterone receptor modulators,14 potent nonpeptide inhibitors of the p53–MDM2 interaction, and antitubercular,15 anticancer,16 anti-HIV,17 and antimalarial18 agents. Notably, the spiro-oxindole subunit is also found in spirotryprostatin A and B, which have been recognized as innovative inhibitors of microtubule assembly.19 In addition such spiro-oxindole heterocycles have been reported to be potent therapeutic agents against malaria. Isopteropodine and pteropodine have been found to modulate the utility of muscarinic serotonin receptors.20
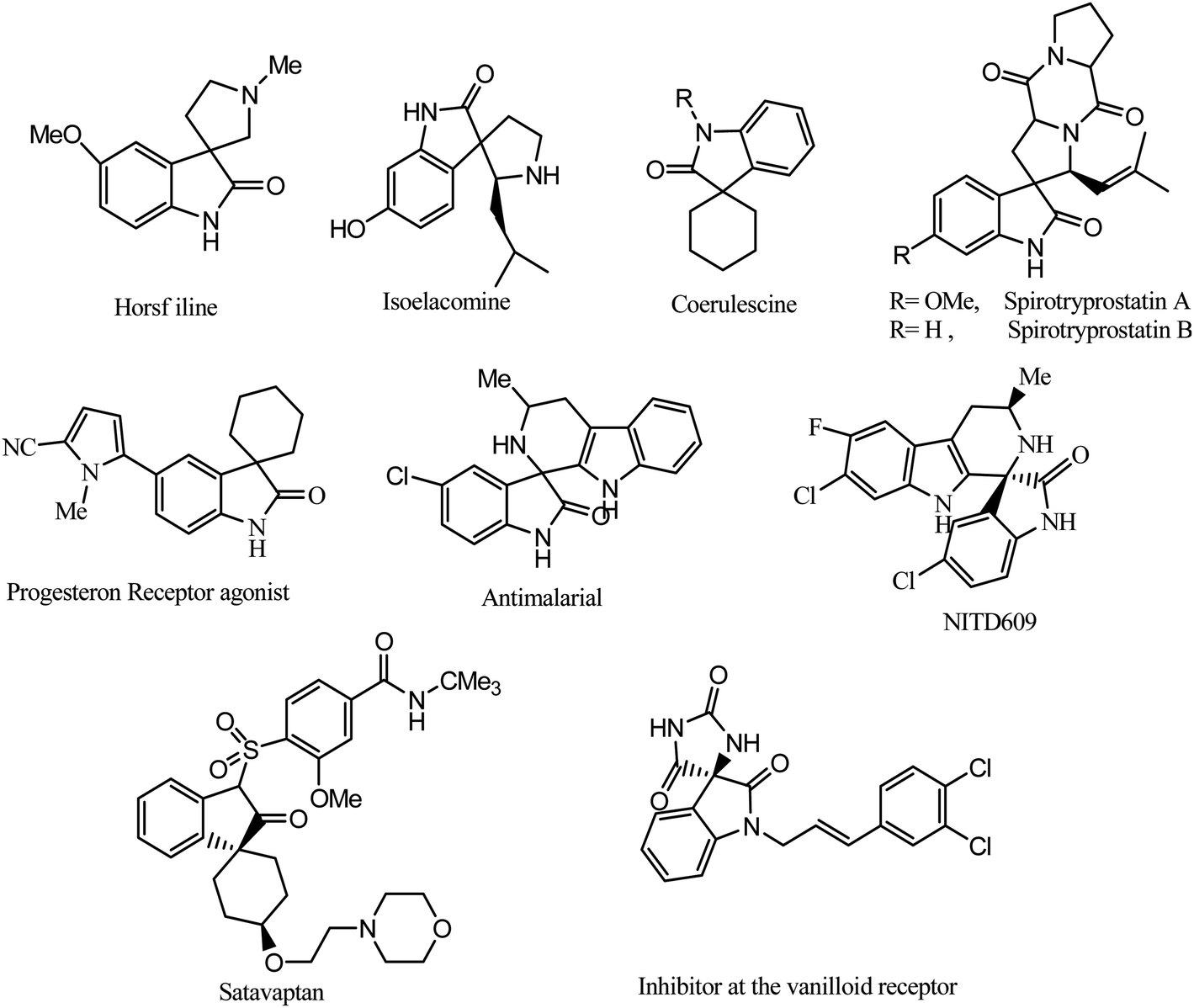 | ||
| Fig. 1 Examples of spirocyclic oxindoles in natural products and pharmaceutical applications. | ||
As a result of the extensive variety of the biological relevance of a 3,3-spirocyclic oxindole ring system with a quaternary center, enormous endeavors have been made to construct these bioactive spiro-oxindole derivatives by synthetic chemists. Numerous solvents and catalysts have been used to achieve this transformation.21 Recently, Shirini et al. described the synthesis of spiro-oxindoles using an Fe3O4/g-C3N4 nanocomposite.21k Albeit with a subtle boost in reaction conditions, somewhere down the line they still fall short of the required flexibility. During our investigations on the synthesis of spiro derivatives and a detailed literature survey, it was observed that traditional methods suffer from many disadvantages: a multi-step process with a tedious workup procedure, prolonged refluxing in elevated harsh conditions using volatile or corrosive solvents and reactants with strong acidic/basic catalysts which may lead to the formation of the target product in lower yields. Furthermore, indigenous overheating can result in substrate, product or reagent decomposition, with a decrease in the yield of the required product, the formation of a mixture of products and a complicated isolation process with further purification using chromatographic techniques. Furthermore, the major shortcoming of nearly all current methods is that the catalysts are damaged in the workup process and cannot be recovered or recycled, reducing the turn over number (TON) or turn over frequency (TOF). In these environmentally conscious days, for reasons of economy and pollution, environ-economic green chemical procedures have to be developed to eliminate pollution problems, and replacing or reducing corrosive acids or volatile organic solvents in the reaction medium are among the most promising ways to reach these goals. Therefore, in an extension of our attempts towards the enlargement of novel synthetic methodologies for heterocyclic frameworks and nanoparticle synthesis,22,23 we have developed a bifunctional g-C3N4 (by the simultaneous incorporation of –SO3H and –NH2 groups) by the reaction of melamine-derived g-C3N4 and aqueous H2SO4 under ultrasound irradiation. The bifunctional g-C3N4 (Sg-C3N4) has been characterized by diverse analytical techniques and used as a metal-free catalyst for the synthesis of various spiro-oxindole derivatives. Due to the admirable assistance between acid and base sites, the catalyst demonstrates a very high reactivity and selectivity for the synthesis of spiro-oxindole derivatives. By comparable experiments, we have identified the responsible catalytic functionality in this Sg-C3N4 for the construction of various spiro-oxindole derivatives (Scheme 1).
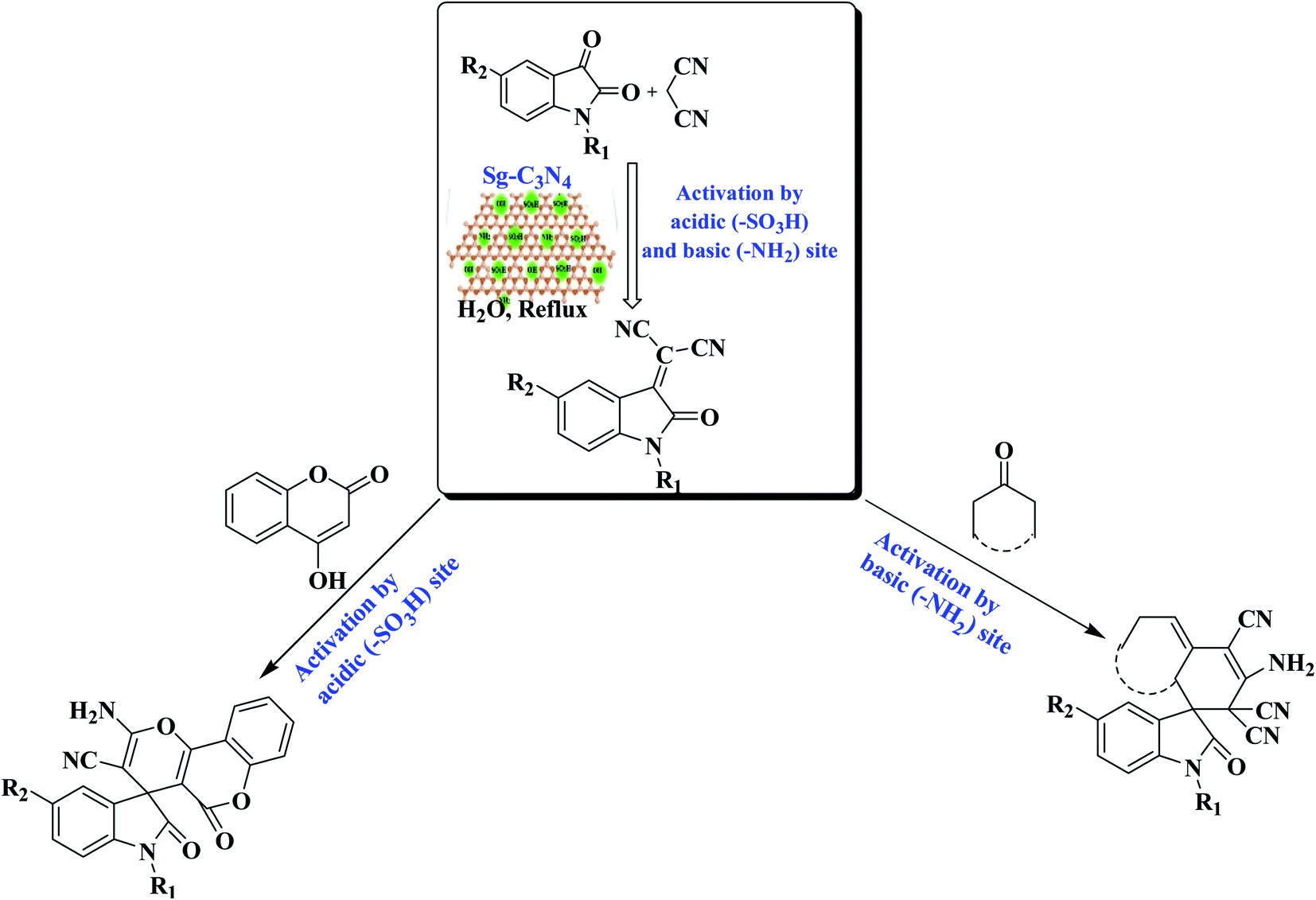 | ||
| Scheme 1 The synthesis of various spiro-oxindole derivatives. | ||
2 Results and discussion
To achieve the goal of the convenient synthesis of bifunctional g-C3N4, we have firstly prepared pristine g-C3N4 from the easily available starting material melamine. Bifunctionalization of g-C3N4 has been accomplished by a simple ultrasound-assisted method through the reaction of pristine g-C3N4 and aqueous H2SO4 (30%). The as-prepared nanomaterials were characterized by XRD, SEM, EDAX, XPS and FT-IR analyses.The crystal or amorphous structures and phase purity levels of the prepared g-C3N4 and Sg-C3N4 catalysts were investigated with X-ray diffraction (XRD) patterns (Fig. 2). There are two sharp diffraction peaks at 12.4° and 27.4°, corresponding to the (100) and (002) reflection of graphitic carbon of synthesized g-C3N4. The diffraction peaks of the XRD pattern of the synthesized Sg-C3N4 catalyst (H2SO4-treated sample) are similar to those of the prepared catalyst g-C3N4. Despite the intensity of the (002) peak of Sg-C3N4 being higher with a minor change in its 2θ value compared to g-C3N4, this result indicates that the graphitic-like structures of the prepared catalyst still remained after the acid treatment. The extra exposed graphitic-like structural units of Sg-C3N4 are responsible for the increase in the intensity.
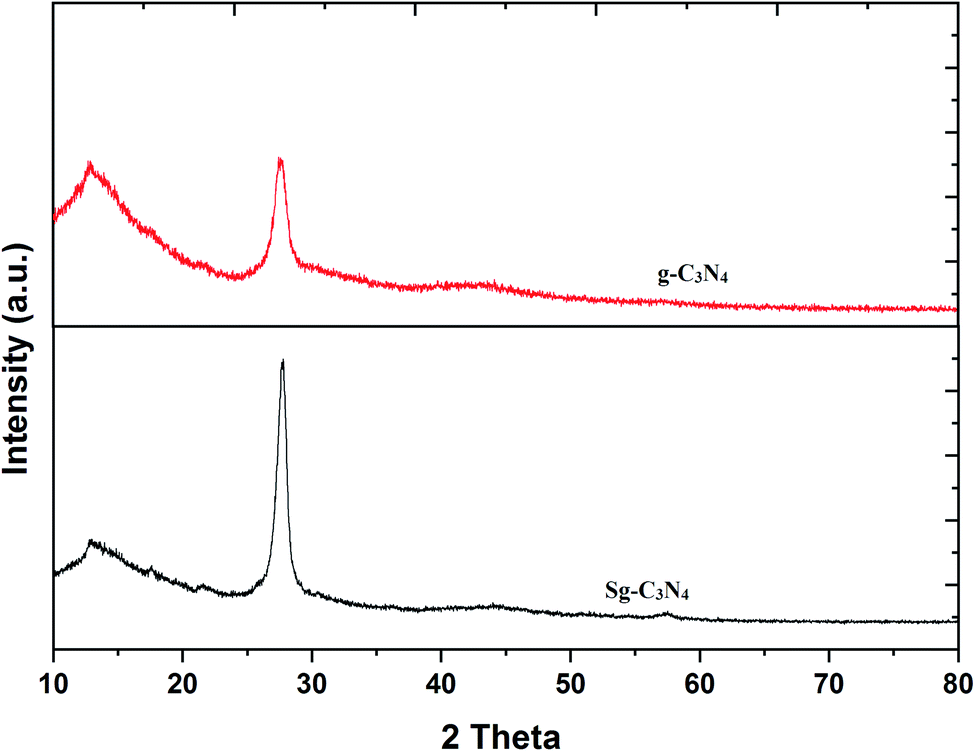 | ||
| Fig. 2 XRD patterns of g-C3N4 and Sg-C3N4. | ||
The micro-structures of g-C3N4, as revealed in the SEM images (Fig. 3) indicate the large aggregation of g-C3N4 units. The SEM image of the acid-treated sample (Sg-C3N4) reveals that there are no aggregations present in this sample because they are shattered into tiny particles with a more exposed and discrete surface, which is favorable for the strong interaction between the substance and the active acidic sites. The ordered stacking of the CN layers was enhanced after H2SO4 treatment because H2SO4 treatment could exfoliate the g-C3N4 layers and generate more exposed g-C3N4 units.24
 | ||
| Fig. 3 SEM images of Sg-C3N4 and g-C3N4. | ||
Elemental analysis of simple graphitic carbon nitride achieved by EDAX analysis verifies the existence of C and N in g-C3N4 (Fig. 4 & 5). After acid treatment, the value of the carbon–nitrogen ratio reduces. Additionally, elemental analysis of the acid-treated sample (Sg-C3N4) verifies the existence of S N, and C in the acid-treated sample. This proves that S was incorporated after the acid treatment. Similar results were also found in the elemental analysis (CNOS) of the samples (Table S1†).
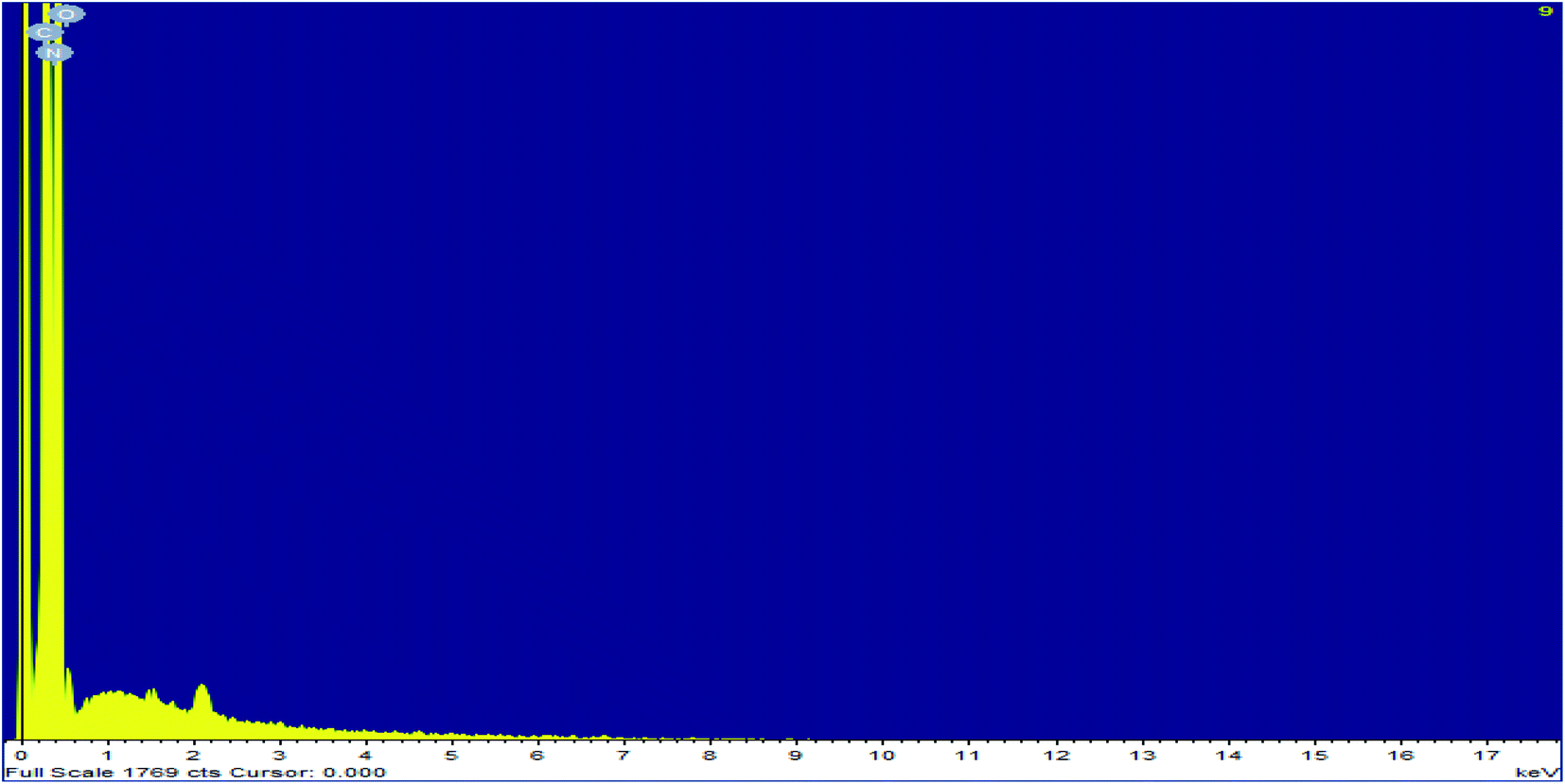 | ||
| Fig. 4 EDAX analysis of g-C3N4. | ||
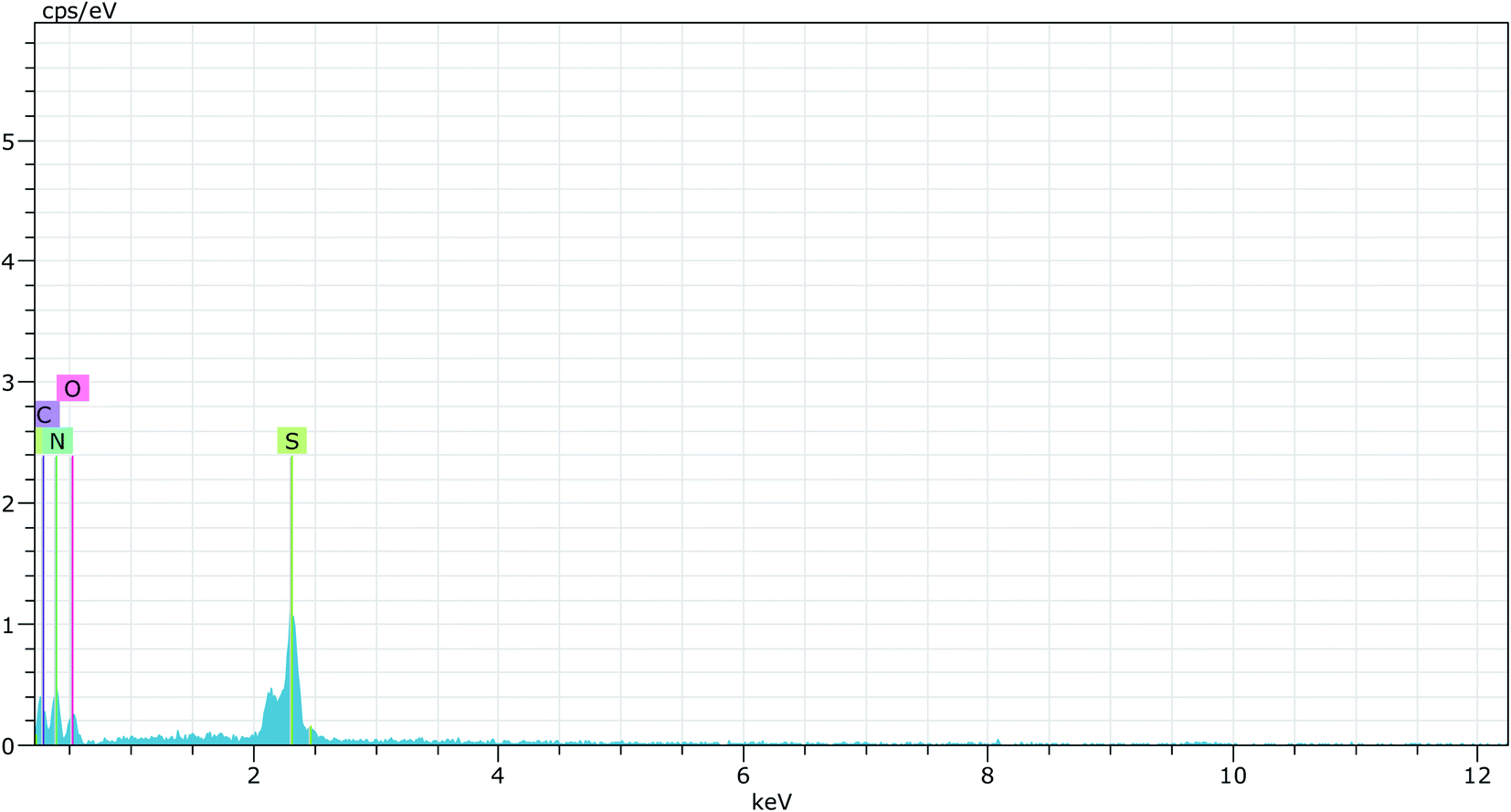 | ||
| Fig. 5 EDAX analysis of Sg-C3N4. | ||
FT-IR spectra of the prepared carbon nitride materials g-C3N4 and Sg-C3N4 are presented in Fig. 6. The sharp band at 808 cm−1 is assigned to the tris-s-triazine layers of the synthesized samples. Broad peaks at around 3040–3380 cm−1 are instigated by the N–H vibration modes for –NH2 groups. An absorption band at 1635 cm−1 corresponds to the typical stretching modes of CN heterocycles. A range of bands in the region of 1462–1570 cm−1 can be related to the tris-s-triazine of the melamine constituent. A signal at 1407 cm−1 is equivalent to the stretching vibrations of C–N bonds in tertiary N-atoms in the units of the prepared catalyst. Various bands in the range of 1209–1314 cm−1 correspond to sp2 hybridized C–(NH) entities. After the acid treatment, a sharp band at 649 cm−1 is attributed to the presence of –SO3H functionality in the Sg-C3N4 catalyst. The appearance of two strong peaks at 1352 and 1148 cm−1 (because of asymmetric and symmetric stretching vibrations) also confirms the occurrence of an –SO3H group. The broad peaks at around 3000–3400 cm−1 are instigated by free amino and hydroxyl functionalities. These results confirm that acid-treated samples initiate several new functional groups (NH2 & SO3H) in the carbon nitride nanosheet.
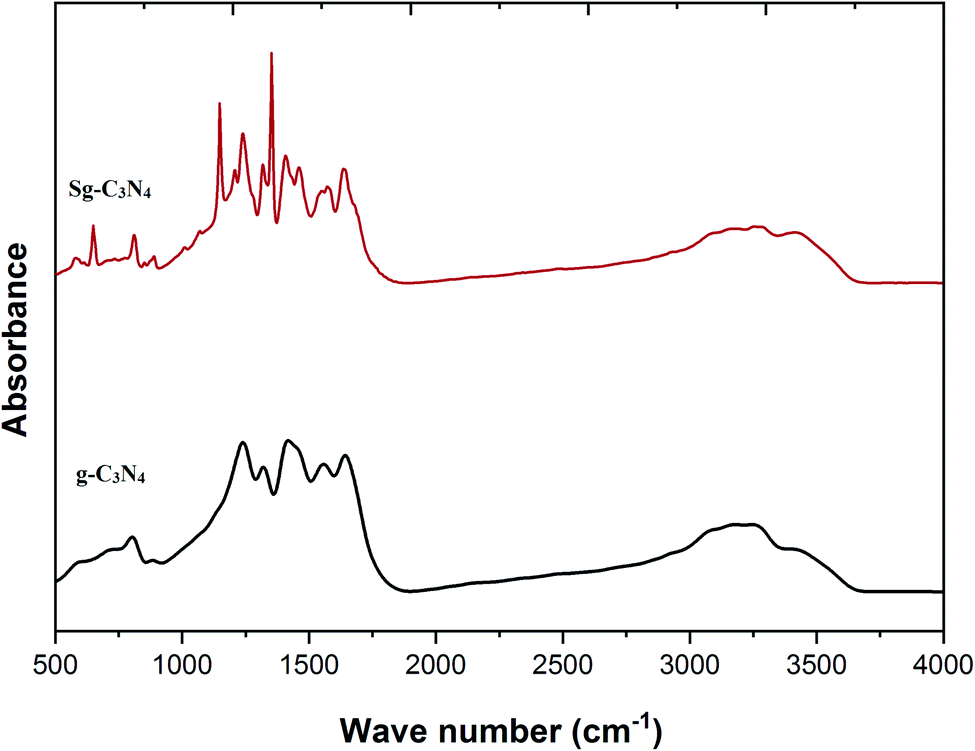 | ||
| Fig. 6 FT-IR spectra of g-C3N4 and Sg-C3N4. | ||
The occurrence of different surface atoms (chemical environment) and oxidation states in the prepared g-C3N4 and Sg-C3N4 catalysts were examined using the X-ray photoelectron spectroscopic (XPS) method (Fig. 7, and S3†). The XPS deconvoluted C 1s spectra of the samples show two characteristic peaks with binding energies of 288.2 and 284.4 eV owing to the occurrence of sp2-hybridized C elements in the prepared samples. The three peaks at 397.7, 399.2 and 400.3 eV in the N 1s spectrum of the samples belong to N element (sp2-bonded), tertiary N element and primary N element, respectively. The two peaks in the O 1s spectra of the samples with binding energies of 530.6 eV and 528.7 eV correspond to –OH functional groups. On the other hand, a study of the XPS survey spectrum of g-C3N4 with Sg-C3N4 reveals that intensity of the C 1s spectrum is reduced while the intensity of the N 1s and O 1s spectra is enhanced. A peak at around 232 eV in both survey spectra may arise due to the Auger spectra of C KLL.25
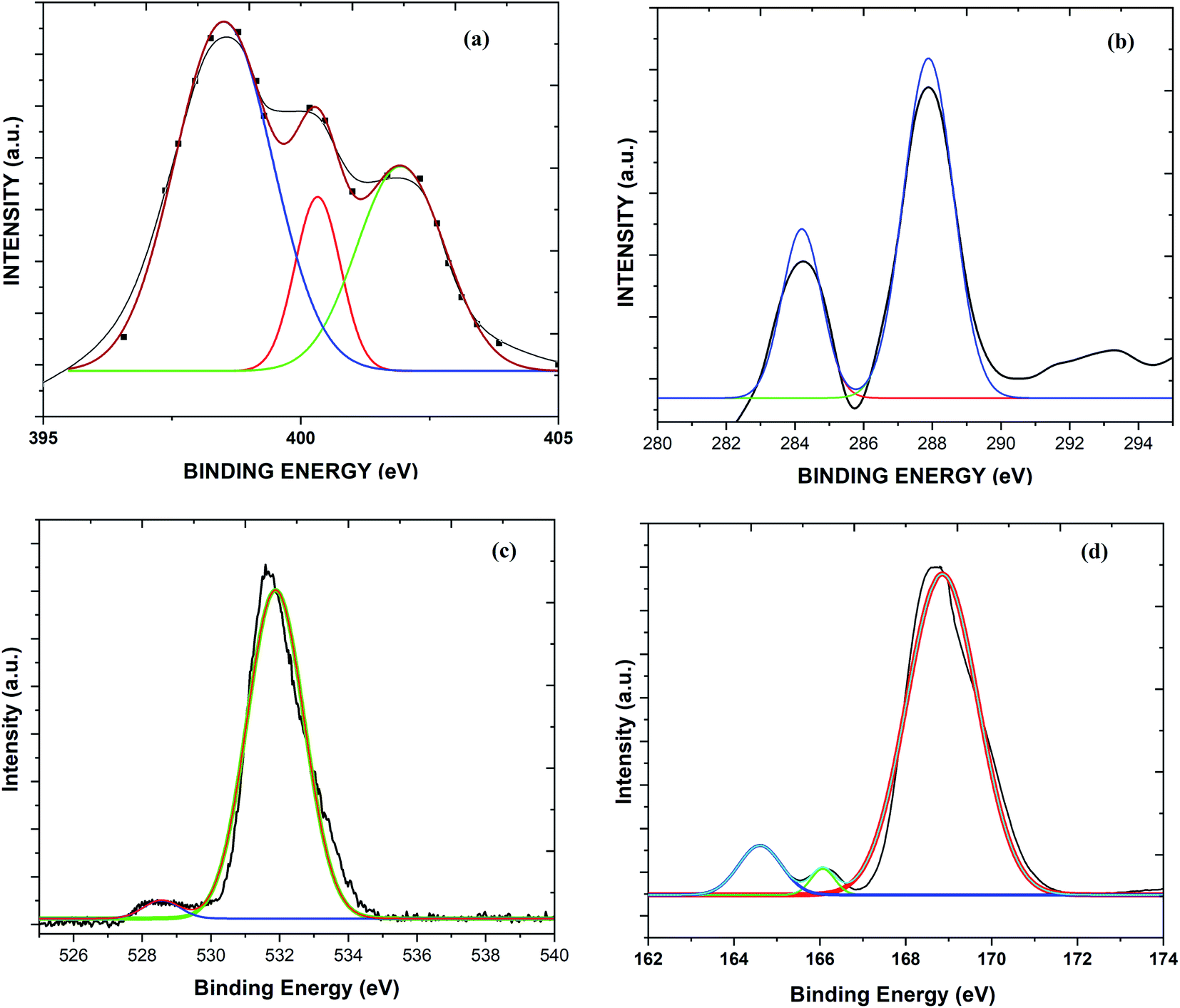 | ||
| Fig. 7 XPS spectra: (a) the N 1s spectrum of Sg-C3N4, (b) the C 1s spectrum of Sg-C3N4, (c) the O 1s spectrum of Sg-C3N4, and the (d) S 2p spectrum of Sg-C3N4. | ||
The XPS survey spectrum in Fig. 8 demonstrates the occurrence of C, N, O atoms in a simple carbon nitride sample, whereas the XPS spectrum of the Sg-C3N4 sample has C, N, S and O species without other impurities. XPS analysis of the elemental S 2p spectrum of Sg-C3N4 allocates three peaks with binding energies of 164.0 eV (C–S bond), 165.8 eV (–N–S bond) and 169.1 (–N–S bond). Furthermore, the characteristic peak at 169.1 eV is ascribed to the –S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group in Sg-C3N4, which further verifies the existence of –SO3H functionalities in the prepared Sg-C3N4 sample. FT-IR and X-ray photoelectron spectroscopy examinations verify the healthy incorporation of SO3H and NH2 functionalities into g-C3N4 after acid treatment.
O group in Sg-C3N4, which further verifies the existence of –SO3H functionalities in the prepared Sg-C3N4 sample. FT-IR and X-ray photoelectron spectroscopy examinations verify the healthy incorporation of SO3H and NH2 functionalities into g-C3N4 after acid treatment.
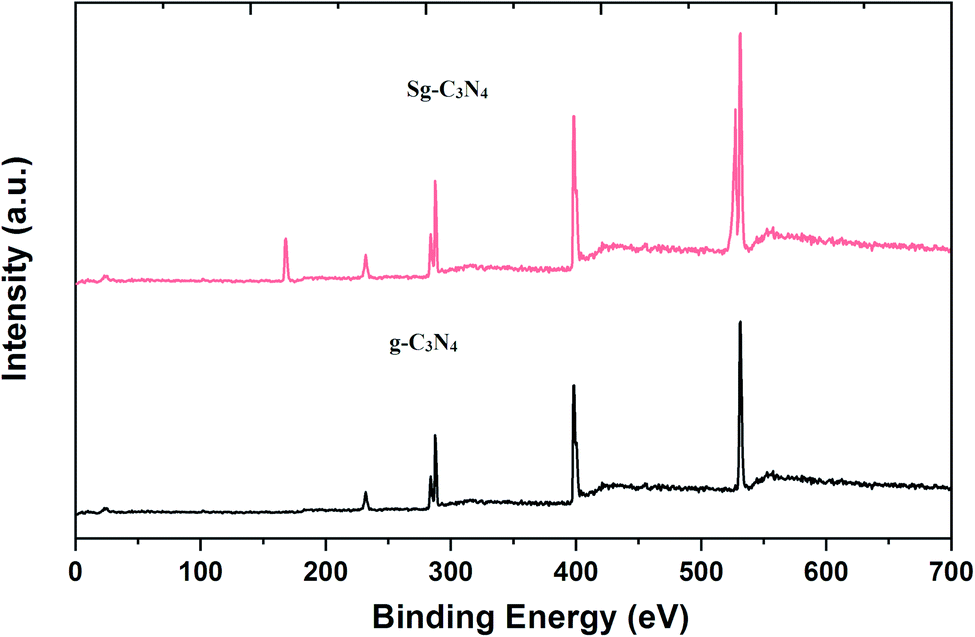 | ||
| Fig. 8 XPS survey scans of g-C3N4 and Sg-C3N4. | ||
After the identification of the fundamental nature of the samples, we assessed the catalytic activity of theses sample towards the synthesis of spiro-oxindoles. Our preliminary attempts concentrated on optimization of the production of spiro-pyrano chromen derivatives. The reaction of isatin 1 (2.0 mmol), malononitrile 2 (2.0 mmol) and 4-hydroxycoumarin 3 (2.0 mmol) was selected as a model reaction. Without any catalyst a lower yield of the product was formed. To enhance the efficacy of the protocol, different metal-free materials were screened as catalysts. As mentioned in Table 1, the most appealing outcome was obtained with Sg-C3N4 as the catalyst in terms of turnover frequency (TOF) compared to other catalysts. It was found that 20 wt% Sg-C3N4 in H2O is enough to drive the reaction forward. Any excess of Sg-C3N4 beyond this amount did not show any further increase in yield. g-C3N did not show any significant impact on the reaction, which demonstrates that acid treatment generated functionality plays a vital role in the reaction.
| S. No. | Catalyst | Solvent | Time (min) | Yieldb (%) | TOFc |
|---|---|---|---|---|---|
| a All reactions were performed with isatin (2.0 mmol), malononitrile (2.0 mmol), and 4-hydroxycoumarin (2.0 mmol) under reflux.b Isolated yield.c TOF (×10−3 mol g−1 min−1).d Multi-walled carbon nanotubes. | |||||
| 1 | — | H2O | 5 h | 42 | n.c. |
| 2 | I2 (20 wt%) | H2O | 60 | 52 | 0.092 |
| 3 | PTSA (20 wt%) | H2O | 60 | 59 | 0.105 |
| 4 | MWCNTs (20 wt%) | H2O | 60 | 28 | 0.050 |
| 5 | Graphite (20 wt%)d | H2O | 60 | — | — |
| 6 | Activated carbon (20 wt%) | H2O | 60 | 14 | 0.025 |
| 7 | GO (20 wt%) | H2O | 60 | 43 | 0.076 |
| 8 | rGO (20 wt%) | H2O | 60 | 20 | 0.035 |
| 9 | g-C3N4 (20 wt%) | H2O | 10 | 34 | 0.363 |
| 10 | Sg-C3N4 (20 wt%) | H2O | 10 | 96 | 1.024 |
| 11 | Sg-C3N4 (10 wt%) | H2O | 20 | 57 | 0.686 |
| 12 | Sg-C3N4 (30 wt%) | H2O | 10 | 96 | 0.598 |
| 13 | Sg-C3N4 (20 wt%) | CH3CN | 30 | 28 | 0.099 |
| 14 | Sg-C3N4 (20 wt%) | EtOH | 30 | 17 | 0.060 |
| 15 | Sg-C3N4 (20 wt%) | THF | 30 | 39 | 0.139 |
| 16 | Sg-C3N4 (20 wt%) | CH2Cl2 | 30 | 36 | 0.128 |
To confirm which functionality in Sg-C3N4 plays a vital role in the reaction, Sg-C3N4 was employed in basic conditions (refluxed with NaOH and NaCl) to acquire base-treated Sg-C3N4 (BSg-C3N4). The FT-IR intensity of the –SO3H group in BSg-C3N4 was relatively weak because of the deprotonation of the –SO3H group (Fig. S1; see ESI†). The intensity of the FT-IR band due to the sp2-C mode was improved because of the additional room in the π–π* network caused by the dehydration reaction forced by the base. The FT-IR band of –NH2 groups is stable in basic conditions so this intensity remains unchanged. This BSg-C3N4 was further converted into BaSg-C3N4 by acidic treatment with 0.1 M HCl. The FT-IR intensity of the –SO3H group can be restored because of reprotonation of the –SO3H group (Fig. S2; see ESI†). But the intensity of the FT-IR band due to –NH2 groups could not be restored due to the neutralization of basic groups by the acid treatment.
To investigate the mechanism for the catalytic action of the catalyst, some control trials were executed using BSg-C3N4 and BaSg-C3N4 catalysts (Scheme 2). When the model reaction was tried with BSg-C3N4, isatylidene malononitrile (X) was isolated as a major product instead of spiro-pyrano chromen. While BaSg-C3N4 confirmed substantial recovery in catalytic activity for the selective synthesis of spiro-pyrano chromen. However an excellent yield of spiro-pyrano chromen was observed with Sg-C3N4. No side product was formed under these conditions (Scheme 2a).
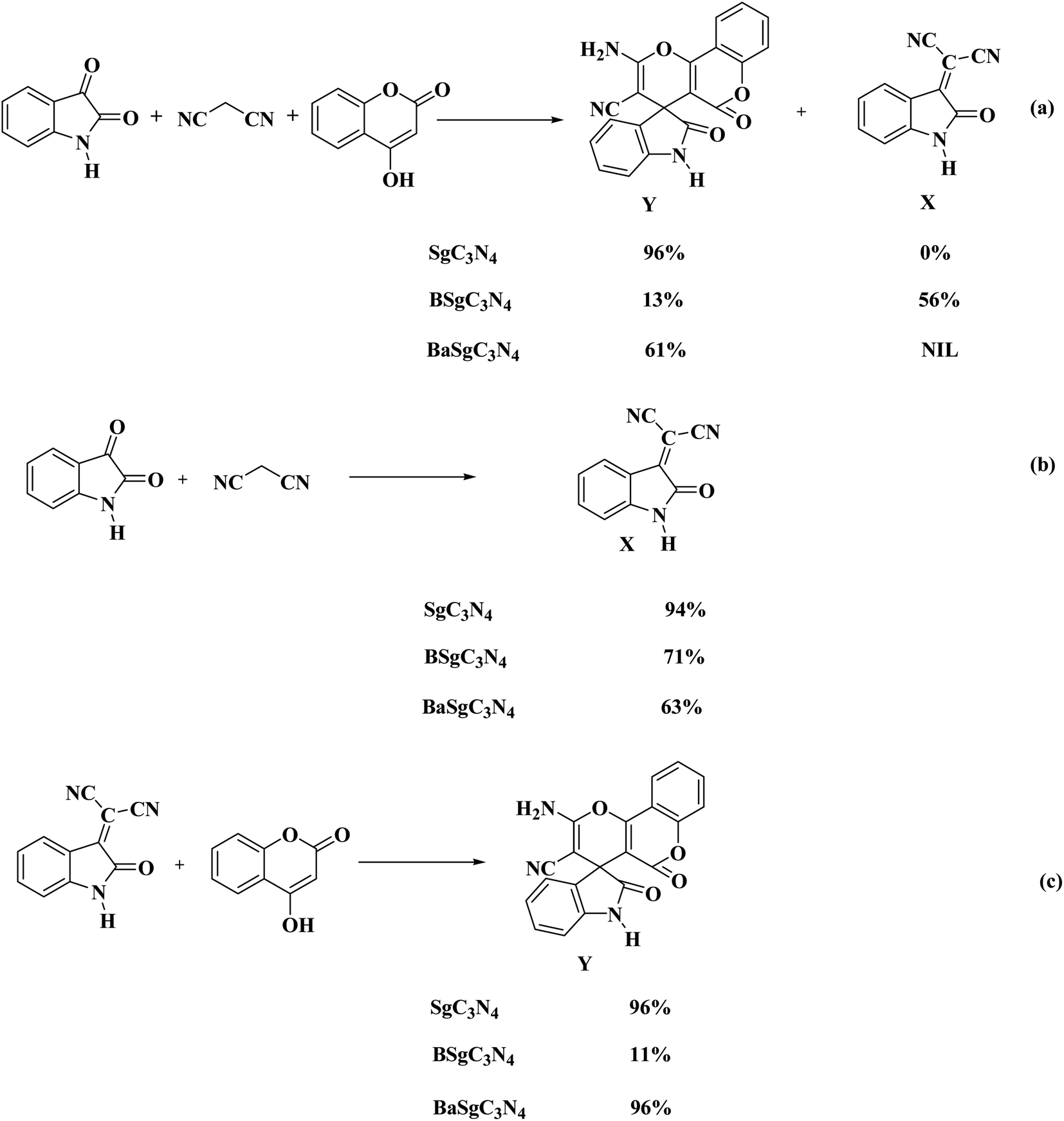 | ||
| Scheme 2 Various control experiments for the synthesis of spiro-pyrano chromen. | ||
To further confirm the site-specific role of this bifunctional catalyst, we carried out the stepwise synthesis of the desired product under control conditions. When the reaction of isatins with malononitrile was run with Sg-C3N4, corresponding Knoevenagel adducts were produced in excellent yield. While moderate yields were observed with BSg-C3N4 and BaSg-C3N4 catalysts (Scheme 2b). This outcome confirms the vital role of the –SO3H group and –NH2 groups for the formation of an Knoevenagel adduct. Moreover, we prepared the Knoevenagel adduct and subjected this adduct with 4-hydroxycoumarin to the control conditions. Sg-C3N4 was found to be the best catalyst for the synthesis of the desired product. BSg-C3N4 gave an inferior yield of the product. Significant improvement in catalytic activity was detected with BaSg-C3N4 (Scheme 2c). These experiments illustrate that –SO3H groups are the sites in Sg-C3N4 responsible for the above transformation.
Thus, these reaction profile indicates that the sites responsible for the Knoevenagel reaction are the –SO3H group and –NH2 groups and a further attack on 4-hydroxycoumarin was facilitated by the –SO3H groups of the Sg-C3N4 catalyst in a one-pot process.
Various solvents (like ethanol, CH3CN, THF, DCM and H2O) were also tested on the model reaction. The outcomes suggested that the solvents had a remarkable impact on the product yield. The best transformation was noticed when the conversion was carried out in water. g-C3N4 treated with 30% aqueous H2SO4 demonstrates the highest yield of the product among the different examined concentrations (10%, 20%, 30% and 40%).
Based on these outcomes, a probable mechanistic pathway for the synthesis of spiro-pyrano chromen is presented in Scheme 3. Firstly, Sg-C3N4 acts as a bifunctional catalyst and activates isatins and malononitrile for the Knoevenagel reaction and produces isatylidene malononitrile (X). Afterwards, the –SO3H group of Sg-C3N4 facilitates Michael addition and forms intermediate Y. Subsequent cyclization of intermediate Y followed by tautomerization produces desired product 3a.
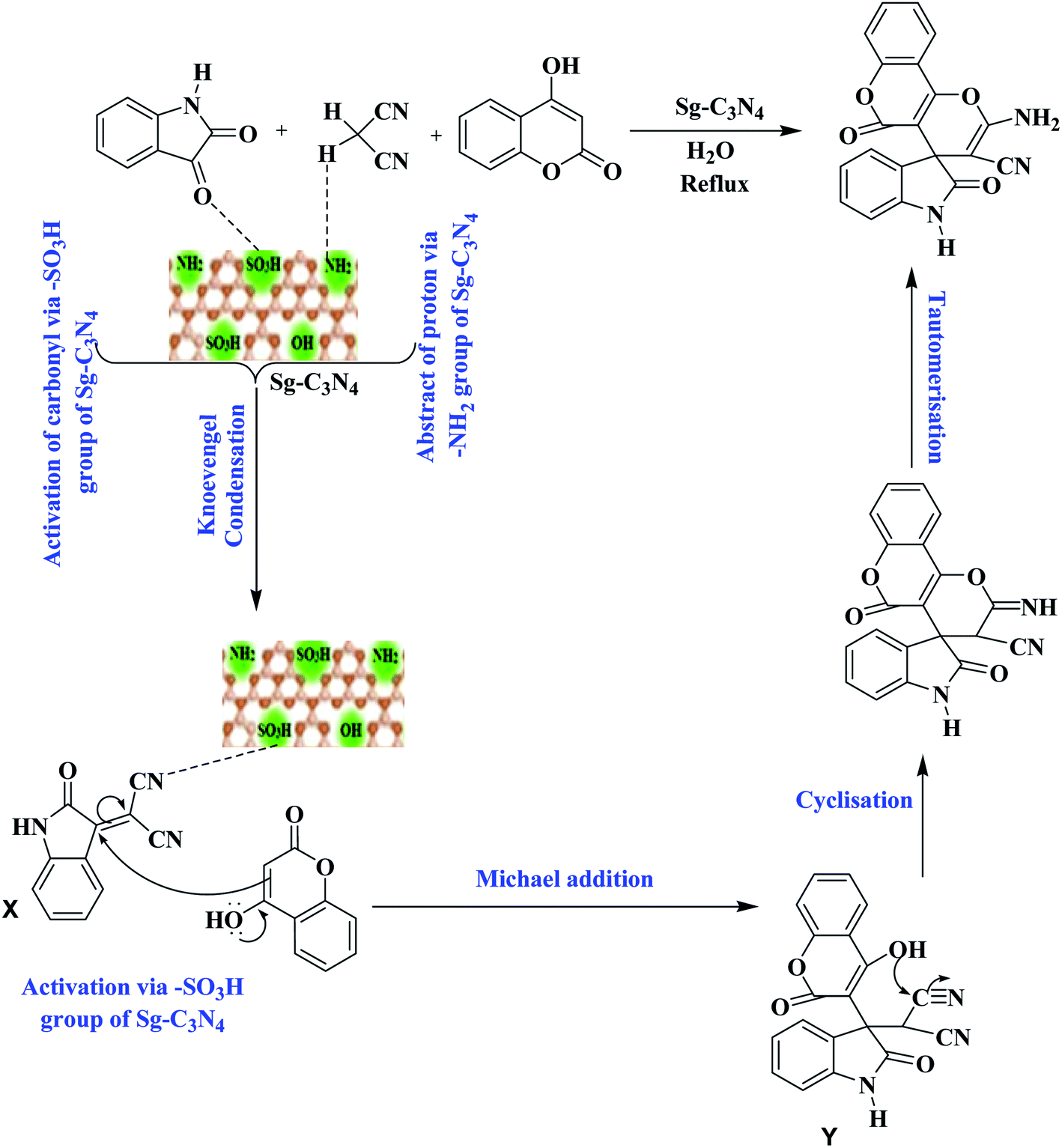 | ||
| Scheme 3 The probable mechanistic pathway for the production of spiro-pyrano chromens. | ||
To demonstrate the scope and versatility of this methodology under identical reaction conditions, a library of substituted medicinally relevant spiro-pyrano chromens was produced (Table 2). Isatins with different electron donating and withdrawing groups on the aromatic ring do not influence the reaction profile that much and give the desire product in excellent yield. N-substituted isatins also tolerate the reaction conditions and produce the desire product in good to excellent yield. The same one-pot method also proceeded efficiently with ethyl cyanoacetate in place of malononitrile. No noteworthy deviation in yields of the products was detected in the case of ethyl cyanoacetate. The conversion completes well and the desired compounds are isolated in good yields.
| a All reactions were performed with substituted isatins (2.0 mmol), malononitrile/ethyl cyanoacetate (2.0 mmol), and 4-hydroxycoumarin (2.0 mmol) using 20 wt% Sg-C3N4 in water and were completed in 30 to 45 min. |
|---|
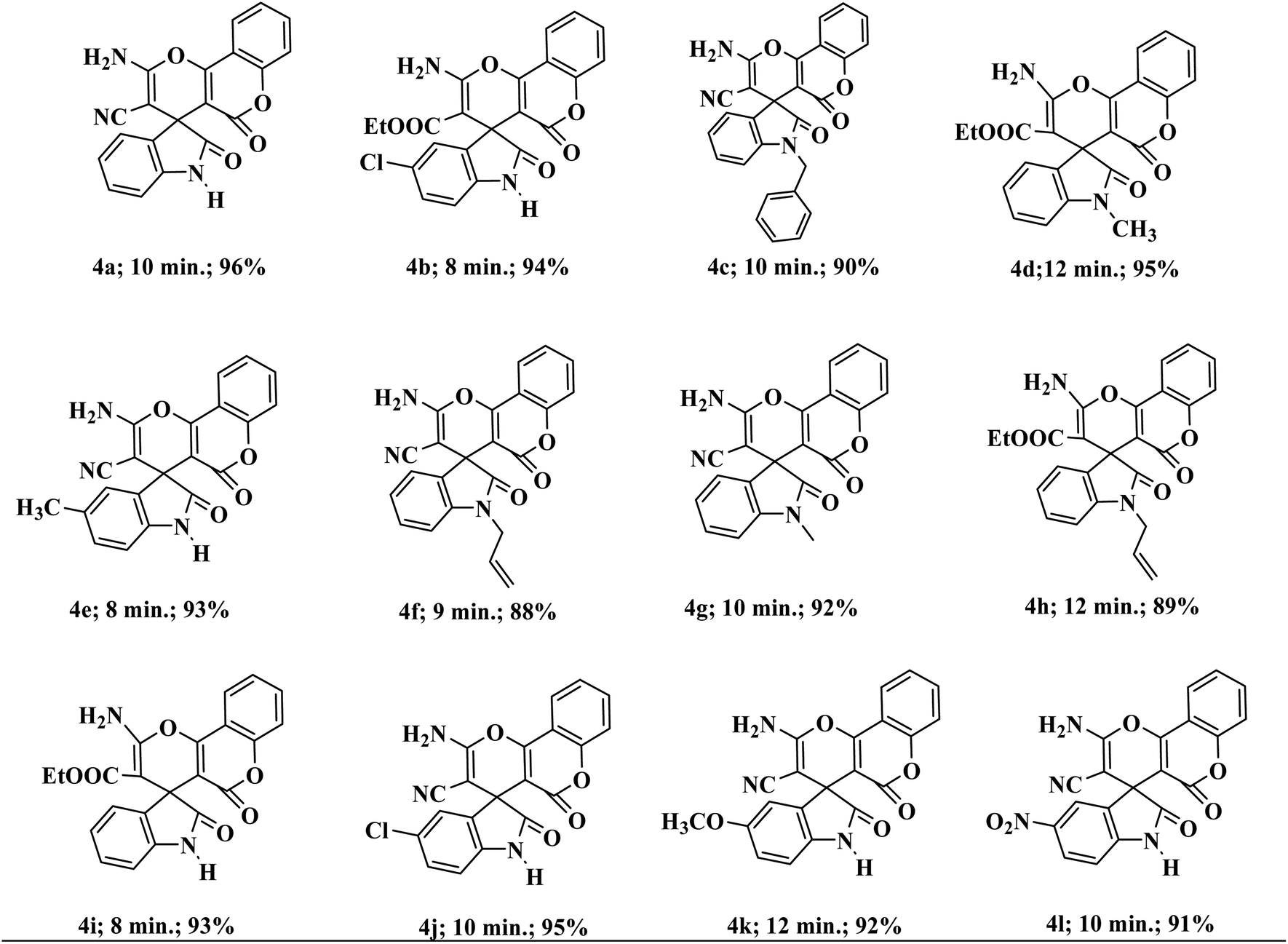 |
Encouraged by these results, we broadened the scope of the above approach for the production of other types of pharmaceutically imperative spiro indole-3,1′-naphthalene tetracyclic systems.26 According to the literature, synthesis of these compounds requires the previous production of Knoevenagel adducts, which involves two extra steps in the synthesis of target compounds.27 We have effectively applied the above optimized conditions for the production of these systems by a one-pot process using isatins, malononitrile and cyclohexanone in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 ratio. Various substituted isatins tolerated the reaction conditions and formed the corresponding functionalized spiro-oxindoles in excellent yield. The reaction also proceeded proficiently under similar experimental conditions with other types of cyclic ketones (Table 3).
:2:1 ratio. Various substituted isatins tolerated the reaction conditions and formed the corresponding functionalized spiro-oxindoles in excellent yield. The reaction also proceeded proficiently under similar experimental conditions with other types of cyclic ketones (Table 3).
| a All reactions were performed with substituted isatins (2.0 mmol), malononitrile (4.0 mmol), and cyclic ketones (2.0 mmol) using 20 wt% Sg-C3N4 in water and were completed in 30 to 45 min. |
|---|
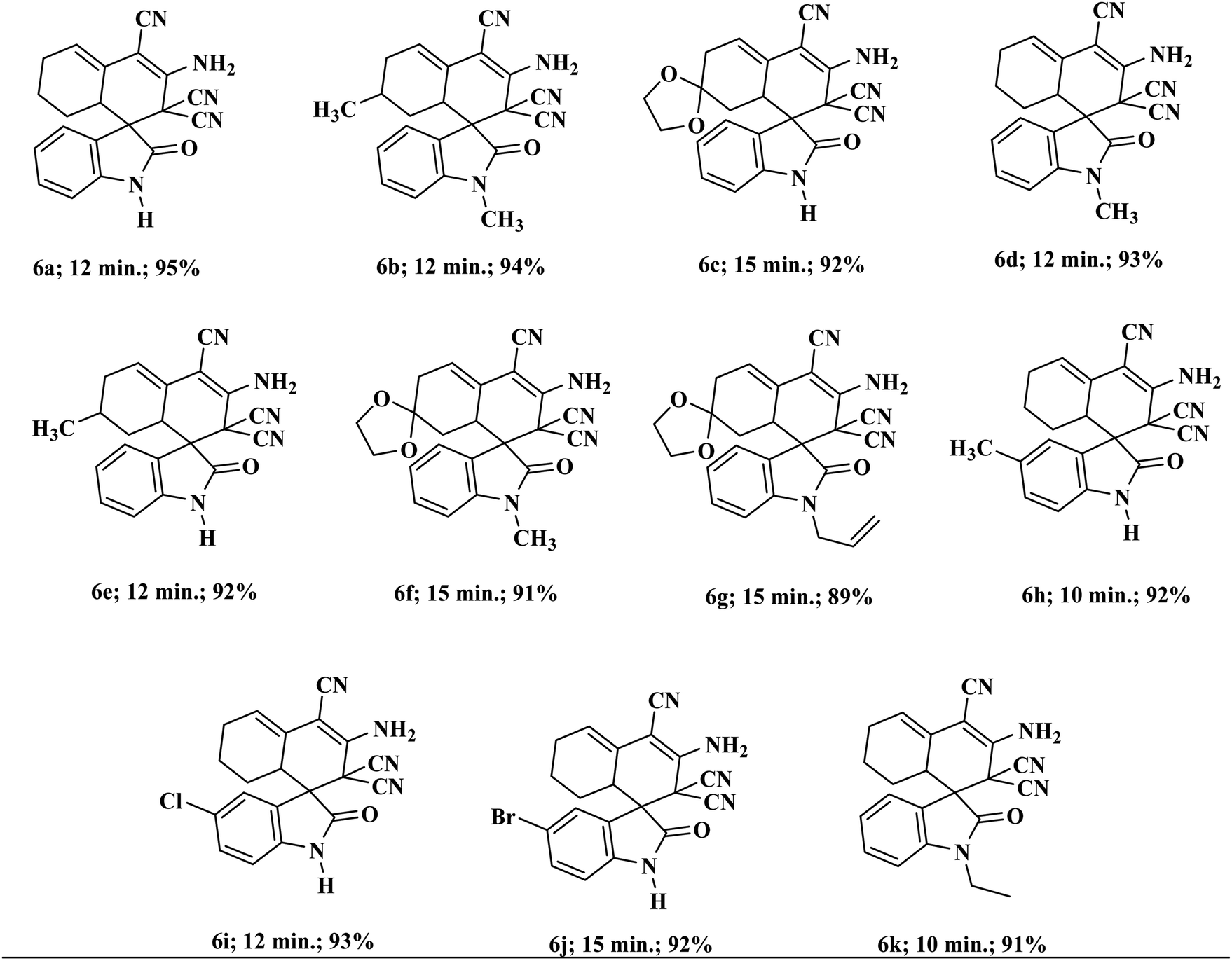 |
We have also done some control experiments to confirm which catalytic site in Sg-C3N4 was responsible for the above conversion (Scheme 4). To accomplish the appropriate conditions to afford the spiro indole-3,1′-naphthalene tetracyclic system, a modal reaction was performed between isatin, malononitrile and cyclohexanone under control conditions using, Sg-C3N4, BSg-C3N4 and BaSg-C3N4 catalysts (Scheme 4a). An admirable yield of the product was viewed with Sg-C3N4. No side product was produced under these conditions. Selective synthesis of a spiro indole-3,1′-naphthalene tetracyclic system was also observed with BSg-C3N4, but the yield of the product was lower compared to Sg-C3N4. While with BaSg-C3N4, Knoevenagel adducts were formed instead of a spiro indole-3,1′-naphthalene tetracyclic system. We have already established the bifunctional role of Sg-C3N4 for the synthesis of isatylidene malononitrile (Scheme 2b). Analogous results were found for the formation of cyclohexanone malononitrile (Scheme 4b). Moreover, we also carried out the reaction of isatylidene malononitrile and cyclohexanone malononitrile with Sg-C3N4, BSg-C3N4 and BaSg-C3N4 catalysts (Scheme 4c). Sg-C3N4 produced the desired product in excellent yield. BSg-C3N4 also acts as an active catalyst for the above transformation, but the yield of the product was lower compared to Sg-C3N4. A substantial loss in catalytic activity was observed when the reaction was tried with BaSg-C3N4. The desired product was not formed and the reactants remained unchanged under these conditions.
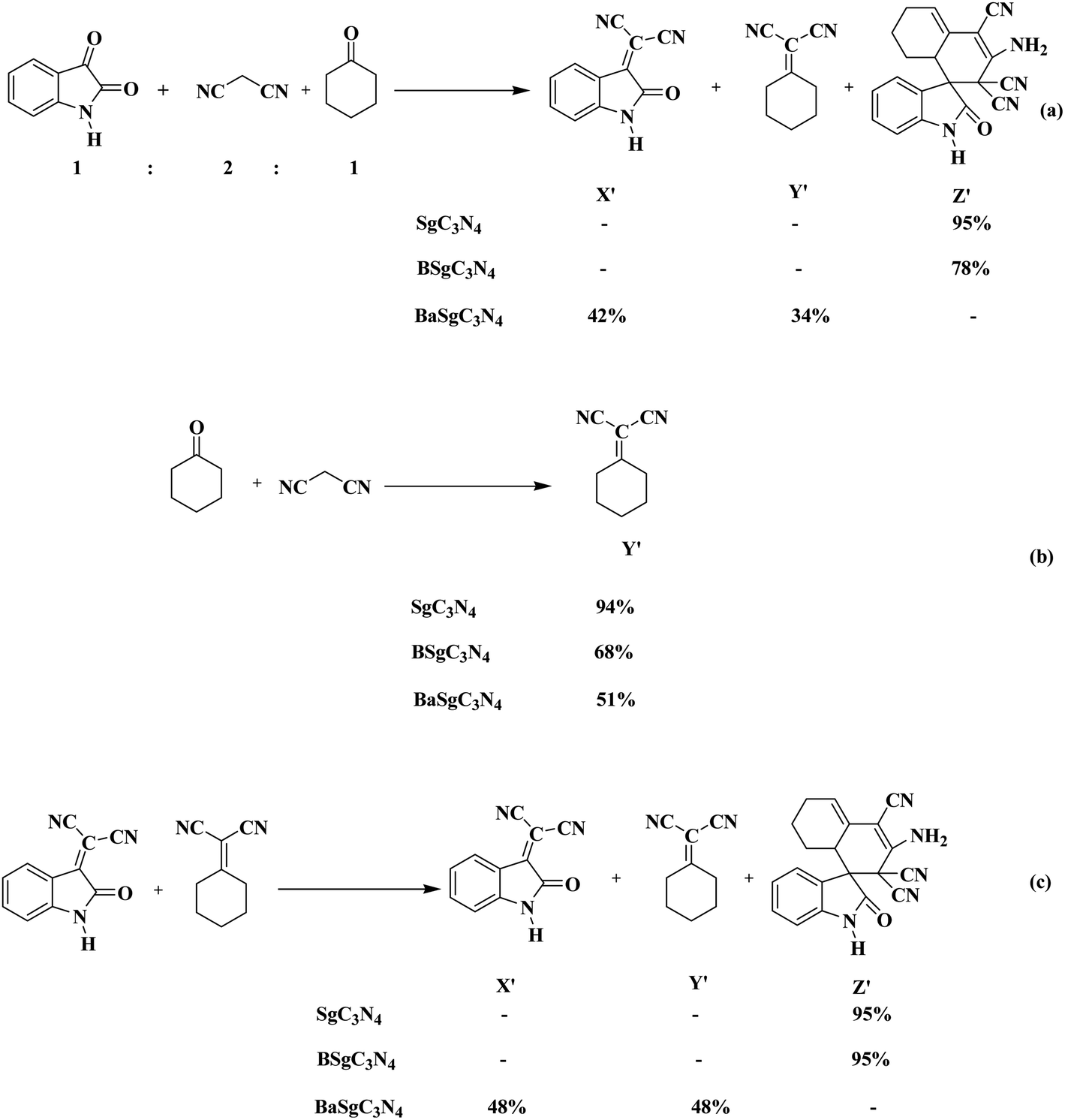 | ||
| Scheme 4 Various control experiments for the synthesis of a spiro indole-3,1′-naphthalene tetracyclic system (a–c). | ||
Hence, these results illustrate that the sites responsible for the Knoevenagel reaction are the –SO3H group and –NH2 groups, and furthermore the reaction of isatylidene malononitrile and cyclohexanone malononitrile was encouraged by the –NH2 groups of the Sg-C3N4 catalyst in a one-pot process.
Based on these outcomes, a probable mechanistic pathway for the production of a spiro indole-3,1′-naphthalene tetracyclic system is presented in Scheme 5. The –SO3H group and –NH2 groups of Sg-C3N4 facilitate the Knoevenagel reaction and produce isatylidene malononitrile (X) and cyclohexanone malononitrile (A). The –NH2 group of Sg-C3N4 activates the cyclohexanone malononitrile and forms anion B, which attacks the activated double bond of X with further cyclization into anion C. Subsequent intramolecular nucleophilic addition on the CN group forms an imine D. Isomerization of imine D yields the corresponding product 6.
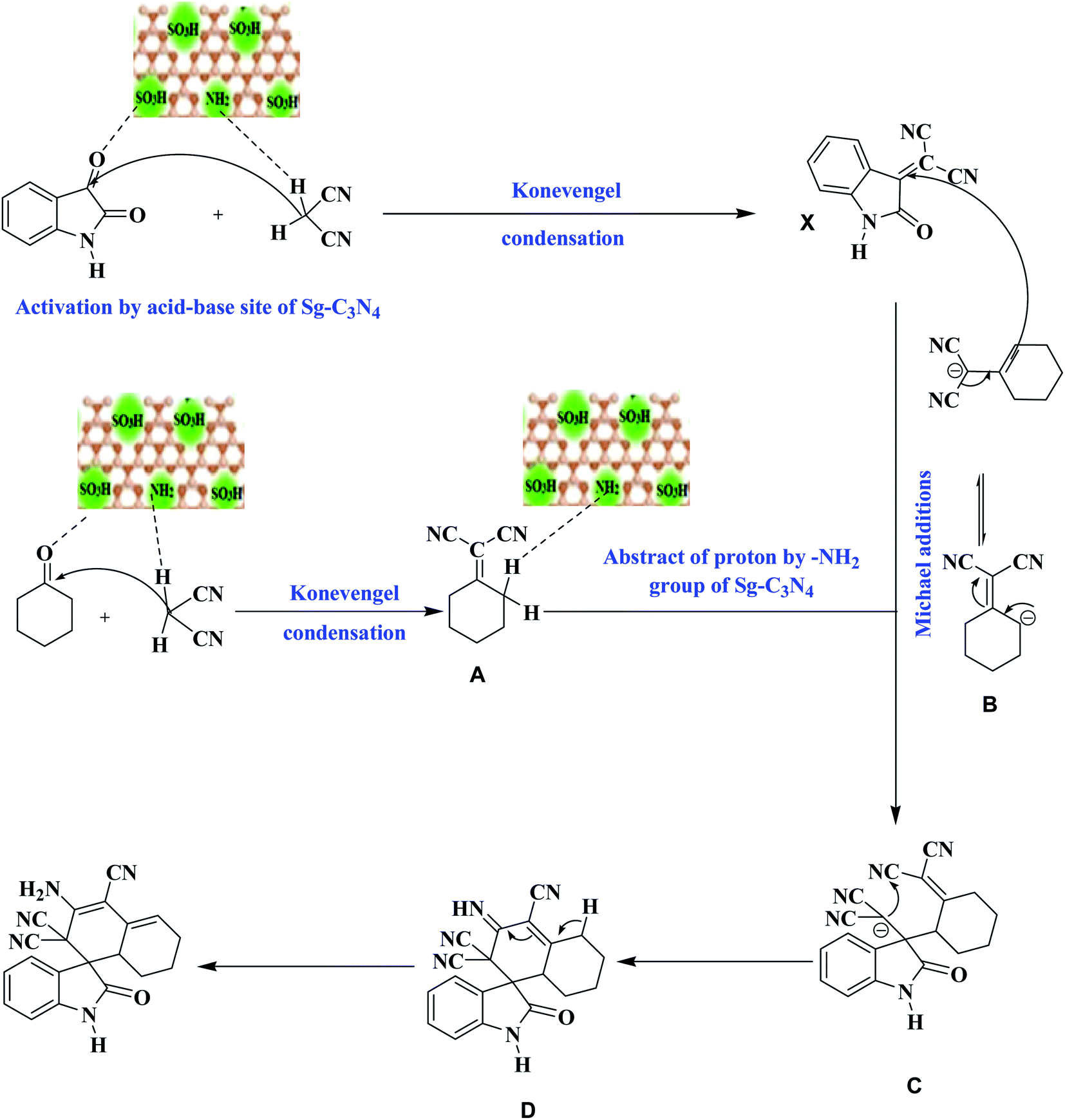 | ||
| Scheme 5 A probable mechanistic pathway for the synthesis of a spiro indole-3,1′-naphthalene tetracyclic system. | ||
The high recyclability and the heterogeneous nature of the used Sg-C3N4 catalyst were also established (see ESI†). The results show that the Sg-C3N4 sheets are heterogenous in nature and can be successfully reused for six cycles (Table 4).
 |
XRD, SEM and FT-IR of the reused catalyst confirmed the analogous characteristic nature of the catalyst compared to fresh catalyst. These outcomes suggest that the morphology and structure of Sg-C3N4 sheets remain unchanged during the reaction.
3 Experimental section
A general part including instruments used is provided in the ESI.†3.1. Synthesis of g-C3N4
Melamine (5 g) was heated at 550 °C (at a rate of 4 °C min−1) using a closed crucible with a lid in a muffle furnace for 3 h. After that, the obtained material was ground into a fine powder using a mortar and pestle and referred to as g-C3N4.3.2. Synthesis of Sg-C3N4
The as prepared g-C3N4 and 30% aqueous H2SO4 were placed in flask. And then it was fixed to a 12 mm tip diameter probe and the solution was sonicated at 50% power of the processor and 230 W output in a 4 s pulse mode for 4 h. The resulting mixture was poured into ice-cold water and centrifuged (12000 rpm for 10 min twice). The obtained solid material was dried under vacuum for 24 h and referred to as Sg-C3N4.
3.3. Synthesis of spiro-pyrano chromene derivatives
A mixture of isatins (2.0 mmol), ethyl cyanoacetate/malononitrile (2.0 mmol), and 4-hydroxycoumarin (2.0 mmol) with 20 wt% Sg-C3N4 in 20 ml of water were mixed. The solution was heated under reflux conditions for an appropriate time. The progress of the conversion was monitored with TLC. After completion of the conversion the Sg-C3N4 and solid product were filtered from the solution. The precipitate was dissolved in ethyl acetate and the catalyst was recovered by centrifugation (12000 rpm). The above residual solution was removed under reduced pressure. The final (crude) product was subjected to purification by recrystallization using ethanol.
3.4. Synthesis of a spiro indole-3,1′-naphthalene tetracyclic system
A mixture of malononitrile (4.0 mmol), isatins (2.0 mmol), and cyclic ketones (2.0 mmol) with 20 wt% Sg-C3N4 in 20 ml of water were mixed. The mixed solution was heated under reflux conditions for an appropriate time. The progress of the transformation was monitored with TLC. After completion of the transformation, Sg-C3N4 and the solid product were filtered from the solution. The precipitate was dissolved in ethyl acetate and Sg-C3N4 was recovered by centrifugation (12000 rpm). The solvent was evaporated from the crude. And then the obtained crude product was subjected to purification by recrystallization using ethanol.
4 Conclusions
In summary, a functionalized graphitic carbon nitride (Sg-C3N4) catalyst was prepared using an ultrasound-assisted technique and it was employed for the one-pot production of various spiro-pyrano chromenes and a spiro indole-3,1′-naphthalene tetracyclic system in aqueous media. Characterizations using different analytical tools showed that bifunctional acidic (–SO3H) and basic (–NH2) sites were created after functionalization. Additionally, the bifunctional nature (acidity/basicity) of the used catalyst Sg-C3N4 was confirmed via numerous control tests in one-pot reaction sequences. The uniqueness of this study is the simultaneous activation of the first reaction sequence by the synergistic participation of the bifunctional acidic and basic sites present in Sg-C3N4. The acidic sites present in Sg-C3N4 activated the second reaction sequence for the one-pot production of various spiro-pyrano chromenes, whereas the basic sites present in Sg-C3N4 activated the second reaction sequence for the one-pot production of a spiro indole-3,1′-naphthalene tetracyclic system. Sg-C3N4 showed no loss in activity or catalytic functionality over several runs. Diverse C–C, C–O, and C–N bonds, six-membered cycles, stereogenic centers, and spiro frameworks were designed in a single reaction.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial assistance from the CSIR, UGC and DST-SERB, New Delhi is gratefully acknowledged. We are thankful to the Malaviya National Institute of Technology, Jaipur, SICART, GUJARAT and Cochin University of Science & Technology, Kochi for spectral analyses.References
- O. C. Compton and S. T. Nguyen, Small, 2010, 6, 711–723 CrossRef CAS PubMed.
- K. P. Gopinath, D. V. N. Vo, D. G. Prakash, A. A. Joseph, S. Viswanathan and J. Arun, Environ. Chem. Lett., 2020, 1–26 CAS.
- Y. Liu, Q. Q. Li, H. Zhang, S. P. Yu, L. Zhang and Y. Z. Yang, N. Carbon Mater., 2020, 35, 323–335 CrossRef.
- (a) S. S. Dang, P. Siglinda and C. Gabriele, Chem. Rev., 2013, 113, 5782–5816 CrossRef PubMed; (b) C. Cha, S. R. Shin, N. Annabi, M. R. Dokmeci and A. Khademhosseini, ACS Nano, 2013, 7, 2891–2897 CrossRef CAS PubMed; (c) A. K. Wanekaya, Analyst, 2011, 136, 4383–4391 RSC; (d) K. Scida, P. W. Stege, G. Haby, G. A. Messina and C. D. García, Anal. Chim. Acta, 2011, 691, 6–17 CrossRef CAS PubMed; (e) L. L. Zhang and X. S. Zhao, Chem. Soc. Rev., 2009, 38, 2520–2531 RSC.
- A. K. Wanekaya, Analyst, 2011, 136, 4383–4391 RSC.
- J. Zhu, P. Xiao, H. Li and S. A. Carabineiro, ACS Appl. Mater. Interfaces, 2014, 6, 16449–16465 CrossRef CAS PubMed.
- C. Jia, L. Yang, Y. Zhang, X. Zhang, K. Xiao, J. Xu and J. Liu, ACS Appl. Mater. Interfaces, 2020, 12, 53571–53591 CrossRef CAS PubMed.
- A. Dandia, S. L. Gupta, P. Saini, R. Sharma, S. Meena and V. Parewa, Curr. Opin. Green Sustain. Chem., 2020, 3, 100039 CrossRef.
- (a) H. Yan, Y. Chen and S. Xu, Int. J. Hydrogen Energy, 2012, 37, 125–133 CrossRef CAS; (b) S. Patnaik, S. Martha, G. Madras and K. Parida, Phys. Chem. Chem. Phys., 2016, 18, 28502–28514 RSC; (c) F. Cheng, H. Wang and X. Dong, Chem. Commun., 2015, 51, 7176–7179 RSC.
- G. S. Singh and Z. Y. Desta, Chem. Rev., 2012, 112, 6104–6155 CrossRef CAS PubMed.
- (a) J. J. Badillo, A. Silva-Garcia, B. H. Shupe, J. C. Fettinger and A. K. Franz, Tetrahedron Lett., 2011, 52, 5550 CrossRef CAS PubMed; (b) S. Duce, F. Pesciaioli, L. Gramigna, L. Bernardi, A. Mazzanti, A. Ricci, G. Bartoli and G. Bencivenni, Adv. Synth. Catal., 2011, 353, 860 CrossRef CAS; (c) X.-N. Wang, Y.-Y. Zhang and S. Ye, Adv. Synth. Catal., 2010, 352, 1892 CrossRef CAS; (d) L.-T. Shen, P.-L. Shao and S. Ye, Adv. Synth. Catal., 2011, 353, 1943 CrossRef CAS; (e) F. Zhong, X. Han, Y. Wang and Y. Lu, Angew. Chem., Int. Ed., 2011, 50, 7837 CrossRef CAS PubMed; (f) N. V. Hanhan, N. R. Ball-Jones, N. T. Tran and A. K. Franz, Angew. Chem., Int. Ed., 2012, 51, 989 CrossRef CAS PubMed; (g) W.-B. Chen, Z.-J. Wu, Q.-L. Pei, L.-F. Cun, X.-M. Zhang and W.-C. Yuan, Org. Lett., 2010, 12, 3132 CrossRef CAS PubMed.
- D. Uemura, T. Chou, T. Haino, A. Nagatsu, S. Fukuzawa, S. Z. Zheng and H. S. Chen, J. Am. Chem. Soc., 1995, 117, 1155–1156 CrossRef CAS.
- (a) T. H. Kang, K. Matsumoto, M. Todha, Y. Murakami, H. Takayama, M. Kitajima, N. Aimi and H. Watanabe, Plant. Med. Phytother., 2002, 444, 39 CAS; (b) O. Dideberg, J. Lamotte-Brasseur, L. Dupont, H. Campsteyn, M. Vermeire and L. Angenot, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1977, 33, 1796 CrossRef; (c) L. Angenot, Plantes médicinales et phytothérapie, 1978, 12, 123 CAS; (d) C. H. Chou, C. L. Gong, C. C. Chao, C. H. Lin, C. Y. Kwan, C. L. Hsieh and Y. M. Leung, J. Nat. Prod., 2009, 72, 830 CrossRef CAS PubMed.
- A. Fensome, W. R. Adams, A. L. Adams, T. J. Berrodin, J. Cohen, C. Huselton, A. Illenberger, J. C. Karen, M. A. Hudak, A. G. Marella, E. G. Melenski, C. C. McComas, C. A. Mugford, O. D. Slayeden, M. Y. udt, J. Zhang, P. Zhang, Y. Zhu, R. C. Winneker and J. E. Wrobel, J. Med. Chem., 2008, 51, 1861 CrossRef CAS PubMed.
- K. Ding, Y. Lu, Z. Nikolovska-Coleska, G. Wang, S. Qiu, S. Shangary, W. Gao, D. Qin, J. Stukey, K. Krajewski, P. P. Roller and S. Wang, J. Med. Chem., 2006, 49, 3432 CrossRef CAS PubMed.
- V. V. Vintonyak, K. Warburg, H. Kruse, S. Grimme, K. Hubel, D. Rauth and H. Aldmann, Angew. Chem., Int. Ed., 2010, 49, 5902 CrossRef CAS PubMed.
- G. Kumari, M. Modi, S. K. Gupta and R. K. Singh, Eur. J. Med. Chem., 2011, 46, 1181 CrossRef CAS PubMed.
- (a) M. M.-C. Lo, C. S. Newmann, S. Nagayams, E. O. Perlstein and S. L. Schreiber, J. Am. Chem. Soc., 2005, 127, 10130 CrossRef PubMed; (b) K. Ding, Y. Lu, Z. Nikolovska-Coleska, S. Qui, Y. Ding, W. Gao, J. Stuckey, K. Krajewski, P. P. Roller, Y. Tomita, D. A. Parrish, J. R. Deschamps and S. Wang, J. Am. Chem. Soc., 2004, 126, 16077 CrossRef PubMed.
- (a) B. K. S. Yeung, B. Zou, M. Rottmann, S. B. Lakshminarayana, S. H. Ang, S. Y. Leong, J. Tan, J. Wong, S. Keller-Maerki, C. Fischli, A. Goh, E. K. Schmitt, P. Krastel, E. Francotte, K. Kuhen, D. Plouffe, K. Henson, T. Wagner, E. A. Winzeler, F. Petersen, R. Brun, V. Dartois, T. T. Diagana and T. H. Keller, J. Med. Chem., 2010, 53, 5155 CrossRef CAS PubMed; (b) M. Rottmann, C. McNamara, B. K. S. Yeung, M. C. S. Lee, B. Zhou, B. Russell, P. Seitz, D. M. Plouffe, N. V. Dharia, J. Tan, S. B. Cohen, K. R. Spencer, G. E. Gonzalez-Paez, S. B. Lakshminarayana, A. Goh, R. Suwanarusk, T. Jegla, E. K. Schmitt, H.-P. R. Beck, F. Nosten, L. Renia, V. Dartois, T. H. Keller, D. A. Fidock, E. A. Winzeler and T. T. Diagana, Science, 2010, 329, 1175 CrossRef CAS PubMed; (c) S. H. Ang, P. Crastel, S. Y. Leong, L. J. Tan, W. L. J. Wong, B. K. S. Yeung and B. Zou, US Pat., 2009/0275560 A1, Novartis AG, 2009 Search PubMed; (d) Z. Zhang, Hoffmann La Roche AG spiroindolinone derivatives, PCT Int. Appl. WO 2008/055812, 2008.
- T.-H. Kang, K. Matsumoto, Y. Murakami, H. Takayama, M. Kitajima, N. Aimi and H. Watanabe, Eur. J. Pharmacol., 2002, 444, 39 CrossRef CAS PubMed.
- (a) G. Shanthi, G. Subbulakshmi and P. T. Perumal, Tetrahedron, 2007, 63, 2057–2063 CrossRef CAS; (b) S. Nagaraju, B. Paplal, K. Sathish, S. Giri and D. Kashinath, Tetrahedron Lett., 2017, 58, 4200–4204 CrossRef CAS; (c) S. L. Zhu, S. J. Ji and Y. Zhang, Tetrahedron, 2007, 63, 9365–9372 CrossRef CAS; (d) K. Jadidi, R. Ghahremanzadeh and A. Bazgir, J. Comb. Chem., 2009, 11, 341–344 CrossRef CAS PubMed; (e) R. Sridhar, B. Srinivas, B. Madhav, V. P. Reddy, Y. V. D. Nageswar and K. R. Rao, Can. J. Chem., 2009, 87, 1704–1707 CrossRef CAS; (f) J. Devi, S. J. Kalita and D. C. Deka, Chemistryselect, 2018, 3, 1512–1516 CrossRef CAS; (g) P. Das, A. Dutta, A. Bhaumik and C. Mukhopadhyay, Green Chem., 2014, 16, 1426–1435 RSC; (h) K. R. Moghadam and L. Y. T. Miri, Tetrahedron, 2011, 67, 5693–5699 CrossRef; (i) F. Tufail, M. Saquib, S. Singh, J. Tiwari, P. Dixit, J. Singh and J. Singh, New J. Chem., 2018, 42, 17279–17290 RSC; (j) B. Karmakar, A. Nayak and J. Banerji, Tetrahedron Lett., 2012, 53, 5004–5007 CrossRef CAS; (k) F. Kamali and F. Shirini, J. Mol. Struct., 2021, 1227, 129654 CrossRef CAS.
- (a) A. Dandia, V. Parewa, A. K. Jain and K. S. Rathore, Green Chem., 2011, 13, 2135–2145 RSC; (b) A. Dandia, V. Parewa, S. Kumari, S. Bansal and A. Sharma, Green Chem., 2016, 18, 2488–2499 RSC; (c) A. Dandia, S. L. Gupta, A. Indora, P. Saini, V. Parewa and K. S. Rathore, Tetrahedron Lett., 2017, 58, 1170–1175 CrossRef CAS; (d) A. Dandia, S. Bansal, R. Sharma, D. K. Mahawar, K. S. Rathore, M. L. Meena and V. Parewa, J. Photochem. Photobiol., A, 2020, 389, 112242 CrossRef CAS; (e) A. Dandia, S. Khan, R. Sharma, S. Parihar and V. Parewa, ChemistrySelect, 2017, 2, 9684–9690 CrossRef CAS; (f) A. Dandia, S. Parihar, R. Sharma, K. S. Rathore and V. Parewa, Nanocatalysis in green organic synthesis, Elsevier, 2020, pp. 71–103 Search PubMed; (g) A. Dandia, P. Saini, R. Sharma and V. Parewa, Green organic synthesis by photochemical protocol, Elsevier, 2020, pp. 155–198 Search PubMed; (h) A. Dandia, R. Sharma, A. Indora and V. Parewa, ChemistrySelect, 2018, 3, 8285–8290 CrossRef CAS.
- (a) A. Dandia, S. Parihar, P. Saini, K. S. Rathore and V. Parewa, Catal. Lett., 2019, 149, 3169–3175 CrossRef CAS; (b) A. Dandia, D. K. Mahawar, R. Sharma, R. S. Badgoti, K. S. Rathore and V. Parewa, Appl. Organomet. Chem., 2019, 33, 5232 CrossRef; (c) A. Dandia, S. Bansal, R. Sharma, K. S. Rathore and V. Parewa, RSC Adv., 2018, 8, 30280–30288 RSC; (d) A. Dandia, A. Sharma, A. Indora, K. S. Rathore, A. Jain and V. Parewa, Mol. Catal., 2018, 459, 97–105 CrossRef CAS.
- S. Subhajyoti and R. Srivastava, Sustainable Energy Fuels, 2017, 1, 1390–1404 RSC.
- J. Chastain and R. C. King Jr, Handbook of X-ray photoelectron spectroscopy. Perkin-Elmer, USA, vol. 261, 1992 Search PubMed.
- K. Tatsuhiko, T. Toshiya, K. Takeshi, W. Yoichiro, S. Akira and F. Yoshifumi, WO2011/010715A1, 2011.
- (a) T. H. Babu, A. A. Joseph, D. Muralidharan and P. T. Perumal, Tetrahedron Lett., 2012, 51, 994 CrossRef; (b) X.-F. Huang, Y.-F. Zhang, Z.-H. Qi, N.-K. Li, Z.-C. Geng, K. Li and X.-W. Wang, Org. Biomol. Chem., 2014, 12, 4372 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra03881h |
| This journal is © The Royal Society of Chemistry 2021 |