
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective extraction of trivalent actinides using CyMe4BTPhen in the ionic liquid Aliquat-336 nitrate
Péter Zsabka a,
Karen Van Heckea,
Lesley Adriaensena,
Andreas Wildenb,
Giuseppe Modolob,
Marc Verwerfta,
Koen Binnemansc and
Thomas Cardinaels*ac
a,
Karen Van Heckea,
Lesley Adriaensena,
Andreas Wildenb,
Giuseppe Modolob,
Marc Verwerfta,
Koen Binnemansc and
Thomas Cardinaels*ac
aBelgian Nuclear Research Center (SCK CEN), Institute for Nuclear Materials Science, Boeretang 200, 2400 Mol, Belgium. E-mail: tcardina@sckcen.be
bForschungszentrum Jülich GmbH, Institute of Energy and Climate Research – IEK-6, Nuclear Waste Management and Reactor Safety, Wilhelm-Johnen-Str. 1, 52428 Jülich, Germany
cKU Leuven, Department of Chemistry, Celestijnenlaan 200 F, P.O. Box 2404, 3001 Heverlee, Belgium
First published on 3rd February 2021
Abstract
The extraction of Am(III), Cm(III) and Eu(III) by 2,9-bis(5,5,8,8-tetramethyl-5,6,7,8-tetrahydro-1,2,4-benzotriazin-3-yl)-1,10-phenanthroline (CyMe4BTPhen) from nitric acid solution was studied using the ionic liquid Aliquat-336 nitrate ([A336][NO3]) as diluent. Results show a high selectivity of the solvent for Am(III) and Cm(III) over Eu(III), but rather slow extraction kinetics. The kinetics of CyMe4BTPhen were largely improved by the addition of 0.005 mol L−1 N,N,N′,N′-tetra-n-octyl-diglycolamide (TODGA) as a phase transfer reagent and by the use of 1-octanol as co-diluent. The addition of the phase transfer catalyst and co-diluent did not compromise the selectivity towards the actinide/lanthanide separation and thus this four-component system can be successfully applied to separate Am(III) and Cm(III) from the lanthanides.
Introduction
Extraction of uranium, plutonium and neptunium from spent nuclear fuels is a mature industrial technology and is being applied in the Plutonium Uranium Reduction Extraction (PUREX) process.1 The minor actinides (MA: Am, Cm, Np) are mainly responsible for the residual radiotoxicity and heat production of (vitrified) nuclear waste after the decay of the short-lived fission products. The concept of Partitioning and Transmutation (P&T) aims at recovering MA elements for later transmutation by fast neutrons to reduce the radiotoxicity and heat production of the final waste. A complete removal of MA would allow a reduction of the residual heat load of the final waste form on the underground repository by a factor of 100 and a reduction on the repository footprint by a factor of 5, compared to the open nuclear fuel cycle scenario.2,3European Commission funded research projects (NEWPART,4 PARTNEW,5 EUROPART,6 ACSEPT,7,8 SACCESS,9 GENIORS10) resulted in the synthesis and testing of a large number of heterocyclic N-donor extractant molecules.8,11–14 As a result of this hydrometallurgical process development work, Selective ActiNide EXtraction (SANEX) processes were developed making use of the highly selective soft-donor ligands 6,6′-bis(5,5,8,8-tetramethyl-5,6,7,8-tetrahydro-1,2,4-benzotriazin-3-yl)-2,2′-bipyridine (CyMe4BTBP)15,16 and 2,9-bis(5,5,8,8-tetramethyl-5,6,7,8-tetrahydro-1,2,4-benzotriazin-3-yl)-1,10-phenanthroline (CyMe4BTPhen)17,18 in 1-octanol diluent and n-dodecane/1-octanol mixtures. These highly selective N-donor ligands are often poorly soluble in commonly used aliphatic diluents. The use of more polar diluents or diluent mixtures could increase the solubility of these types of ligands.
The CyMe4BTPhen ligand has a pre-organized cis-locked conformation with a cavity for chelation of the metal ion, and was also found to have a higher surface activity (and consequently a higher local concentration) at the organic/aqueous interphase than CyMe4BTBP (Fig. 1). Nuclear magnetic relaxation dispersion titration and Time-Resolved Laser Fluorescence Spectroscopy (TRLFS) studies proved that the prevalent complex structure is characterized by an 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 M(III):ligand stoichiometry.17
:2 M(III):ligand stoichiometry.17
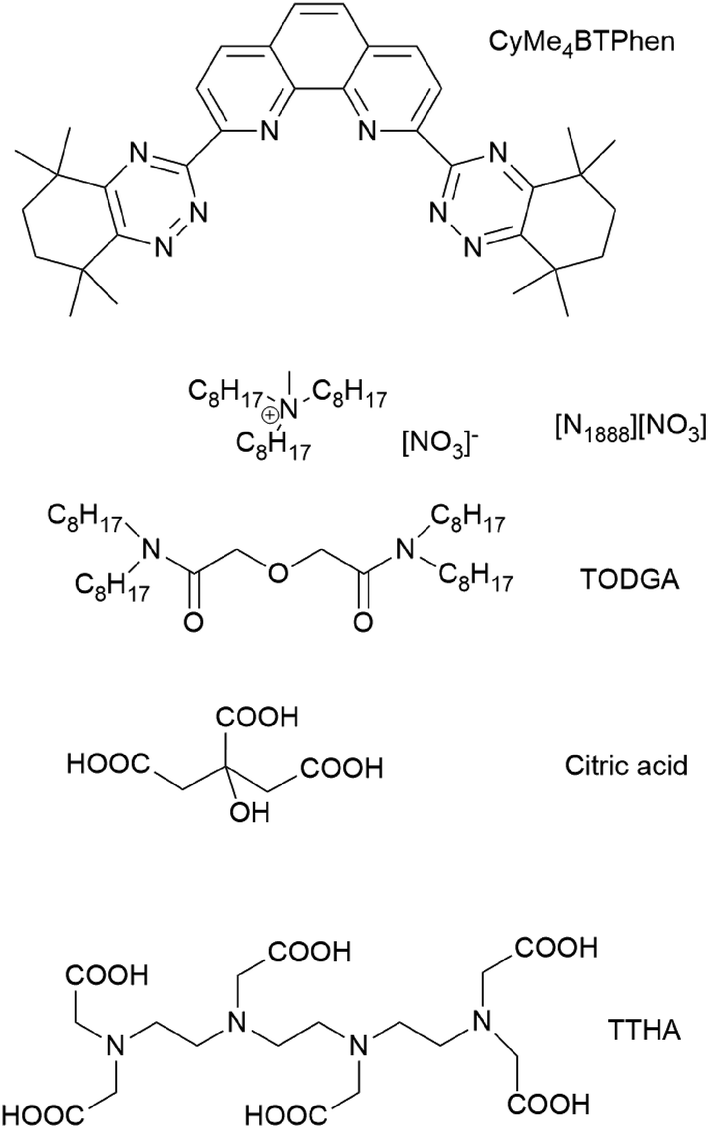 | ||
| Fig. 1 Chemical structures of the organic compounds used in this study. | ||
In organic diluents such as 1-octanol or 1-octanol/toluene mixtures both extractants showed slow extraction kinetics. Extraction equilibrium of Am(III) was reached after 60 min with 0.02 mol L−1 CyMe4BTBP in 1-octanol and after 15 min with 0.01 mol L−1 CyMe4BTPhen in 1-octanol, respectively.17,19 In 1-octanol or 1-octanol/toluene (40/60 vol%) mixtures containing 1 mmol L−1 CyMe4BTPhen, extraction equilibrium was reached beyond 2 h.20 The slow extraction kinetics is a disadvantage in continuous process applications. Modified diluent properties can significantly mitigate this issue, as shown recently by Distler et al.21 The authors used a fluorinated carbonate diluent (BK-1) for both 1 mmol L−1 CyMe4BTBP and 1 mmol L−1 CyMe4BTPhen. These solvents reached equilibrium for Am(III) extraction within 45 min and 15 min, respectively.21
Besides desired rapid kinetics, the organic solvent intended for use in the solvent extraction of minor actinides needs to be highly resistant against radiation-induced degradation. According to calculations, in a continuous minor actinide partitioning process the solvent would receive a total absorbed dose during one year of operation between 100 kGy and 1 MGy.22 Other sources suggest 320 kGy absorbed total dose during 2 years of operation.23 The diluent is present at significantly higher concentrations than the ligand, therefore the primary interaction of radiation occurs between the diluent molecules and the incident gamma rays or charged particles. Radiation stability of CyMe4BTBP and CyMe4BTPhen in 1-octanol both in the presence and absence of 1 mol L−1 HNO3 was studied by Schmidt et al.24,25 These studies revealed that the ligands undergo radiolysis as a function of absorbed gamma dose. The main radiolysis products identified are addition products of the N-heterocyclic ligands and the α-hydroxyoctyl radicals formed by diluent radiolysis. This type of adduct formation mechanism was previously discovered in irradiated solutions of (2,6-bis(5-(2,2-dimethylpropyl)-1H-pyrazol-3-yl)pyridine) (C5-BPP) in 1-octanol.26 Improvement of the radiation stability of the solvent could be achieved by the complete or at least partial replacement of 1-octanol by another diluent that is more resistant to radiation-induced degradation. Such diluent mixtures of 1-octanol/toluene or 1-octanol/hydrogenated tetrapropylene (TPH) were tested by Lewis et al.17
Ionic liquids (ILs) are a relatively new class of compounds that can provide several advantages over molecular diluents in solvent extraction processes at the back-end of the nuclear fuel cycle.27–30 ILs can be used as extractants, diluents or both, depending on the chosen conditions and metal ions to be separated. A main reason and driving force to consider ILs instead of classical organic diluents is their radiation stability. Indeed, some types of ionic liquids proved to show a much higher stability towards ionizing radiation.31,32 The low vapour pressure of ionic liquids renders them safer to use than the more common aliphatic diluents. In addition, the high intrinsic electrical conductivity of ionic liquids can prevent the build-up of static charge in the organic solvent, a phenomenon sometimes encountered when using molecular diluents for solvent extraction processes, which can result in fire.33,34 In the dry state, most ionic liquids have a broad electrochemical window.35 However, major drawbacks of ionic liquids are their significantly high viscosity and slower phase separation36 as well as their often higher ecotoxicity37 compared to aliphatic organic diluents. In former studies, the room-temperature ionic liquid Aliquat-336 ([A336][NO3]), was identified as a promising candidate that could be used as an alternative for aliphatic diluents in trivalent MA partitioning processes.38–43 This compound has a higher flash point (TFp ≥ 110 °C) than other diluents often used in the nuclear fuel cycle: kerosene (TFp ≥ 37–65 °C), n-dodecane (TFp ≥ 71 °C) or 1-octanol (TFp ≥ 81 °C).34,44 The polar nature of the ionic liquid diluent tri-n-octylmethylammonium nitrate, [N1888][NO3] (Fig. 1), being the main component of [A336][NO3], can tolerate high Nd(III) or nitric acid loadings without third phase formation.40 [A336][NO3] is composed of only the four elements C, H, O and N (so-called CHON-compatible), which is considered beneficial compared to the more often studied ionic liquids containing perfluorinated anions. CHON-compatible compounds are completely incinerable without leaving any solid residues. Fluorine containing compounds are to be avoided in nuclear fuel cycle applications since the preferred treatment of secondary waste is incineration.
We previously showed that the kinetics of An(III) and Ln(III) extraction by CyMe4BTPhen in [A336][NO3] is slow at 22 °C, but can be improved to some extent by an increase to 40 °C.41 We hypothesized that the decreased diluent viscosity at higher temperature contributed to increasing the phase transfer rate of metal ions. In that former study, the solvent was composed of CyMe4BTPhen in [A336][NO3] and TEDGA was added to the aqueous phase, with the aim to separate Am(III) from Cm(III) and the lanthanides (thus a selective Am(III) separation process).
The objective of the present study was to investigate the feasibility of using CyMe4BTPhen in the ionic liquid Aliquat-336 nitrate to separate efficiently the actinides Am(III) + Cm(III) from the lanthanides (thus an An/Ln separation process). In addition, efforts were made to enhance extraction kinetics of the system so that the solvent suits operational requirements of continuous counter-current solvent extraction equipment. Several solvent compositions were tested where TODGA (Fig. 1) was used as a phase transfer reagent and 1-octanol was added to [A336][NO3] as a co-diluent. The use of TODGA as a phase transfer reagent was established within the SANEX process development using CyMe4BTBP.15,45–48 The extraction behavior of an organic solvent containing various volume fractions of 1-octanol was also subject of the present investigation. The polarity of the diluent (or diluent mixture) is increased by the co-diluent 1-octanol, as opposed to former investigations,17 where the added co-diluents were of less polar nature, i.e. toluene or hydrogenated tetrapropylene (TPH).
Experimental
Chemicals
Aliquat® 336 chloride ([A336][Cl], quaternary amine content: 88.2–90.6%) of which the main component is tri-n-octylmethylammonium chloride ([N1888][Cl]), 1-octanol (purity >99%), citric acid (purity >99%, Fig. 1), and oxalic acid dihydrate (purity: 99.9%) were obtained from Sigma-Aldrich (Steinheim, Germany).N,N,N′,N′-Tetra-n-octyldiglycolamide (TODGA), (purity: 99%) and 2,9-bis(5,5,8,8-tetramethyl-5,6,7,8-tetrahydro-1,2,4-benzotriazin-3-yl)-1,10-phenanthroline (CyMe4BTPhen, purity >98%) were purchased from Technocomm Limited (Edinburgh, UK). Triethylene-tetramine-N,N,N′,N′′,N′′′,N′′′-hexaacetic acid (TTHA, Fig. 1) (purity >98.0%) was obtained from Tokyo Chemical Industries Co. Inc. (Tokyo, Japan).
AgNO3 (purity: 99.9%), Dy(NO3)3·6H2O (purity: 99.9%), and Yb(NO3)3·5H2O (purity: 99.9%) were obtained from Sigma-Aldrich (Steinheim, Germany). KNO3 (purity >97.0%), NaNO3 (purity >97.0%), and standardised NaOH solution (Titrisol) were obtained from Merck KGaA (Darmstadt, Germany). Gd(NO3)3·6H2O (purity: 99.9%) and Eu(NO3)3·6H2O (purity: 99.99%) were obtained from Alfa Aesar GmbH (Karlsruhe, Germany). La(NO3)3·6H2O (purity: 99.0%) was obtained from Fluka (Seelze, Germany). Y(NO3)3·6H2O (purity: 99.9%), Ce(NO3)3·6H2O (purity: 99.9%), Pr(NO3)3·6H2O (purity: 99.9%), Nd(NO3)3·6H2O (purity: 99.9%), and Sm(NO3)3·6H2O (purity: 99.9%) were obtained from Strem Chemicals (Kehl, Germany). Trace metal grade nitric acid was obtained from Fischer Chemicals (Seastar Chemicals Inc, Canada). Milli-Q water was used for all dilutions (resistivity: minimum 18 MΩ cm). For the purpose of ICP-MS calibration, a multi-element standard solution, obtained from Claritäs ppt Spex CertiPrep (Spex CertiPrep, USA) was used.
241Am tracer in 1 mol L−1 HNO3 solution (radionuclidic purity >99%) was available from legacy stocks of SCK CEN. The solution was purified from Pu traces using Dowex 1X4 100–200 mesh resin (Eichrom Technologies, USA) in chloride form. 244Cm (radionuclidic purity >99.9%) and 152Eu (radionuclidic purity >99%) radiotracers in 1 mol L−1 HNO3 solutions were obtained from Eckert and Ziegler Nuclitec GmbH (Braunschweig, Germany). The ionic liquid [A336][Cl] was converted into its nitrate form via a metathesis reaction, according to a method as described elsewhere.39 All other chemicals were used as received, without any further purification.
A stock solution for the lanthanide(III) nitrate containing aqueous phases was prepared by dissolving weighed amounts of the nitrate salts of La, Ce, Pr, Nd, Sm, Eu, Gd, Dy, Yb and Y in 0.1 mol L−1 HNO3 to obtain a 10−3 mol L−1 initial concentration for each lanthanide. This stock solution was diluted with the required nitric acid concentrations to obtain a 10−5 mol L−1 lanthanide concentration. The exact nitric acid concentration of the stock solution was determined by titration using an autotitrator (716 MPT Titrino, Metrohm, Switzerland).
Solvent extraction and analytical procedures
An organic solvent composed of [A336][NO3] or a mixture of [A336][NO3] and 1-octanol and the selected CyMe4BTPhen/TODGA concentration was used in all extraction experiments in an acid pre-equilibrated form. The organic phase was pre-equilibrated with an equal volume of an acidic solution at the same acid concentration as used for the feed solution of the extraction step by shaking at 30 °C and 2400 rpm using a TMS-200 Thermoshaker (Nemus Life, Sweden) equipped with an in-house fabricated metal block. The aqueous phases used as feed solutions were previously spiked with 10 μL of a tracer stock solution (0.01 mol L−1 HNO3 containing 0.3 kBq/μL ≈ 9.79 × 10−8 mol L−1 241Am, 0.3 kBq/μL ≈ 4.11 × 10−9 mol L−1 244Cm and 0.3 kBq/μL ≈ 2.80 × 10−9 mol L−1 152Eu) and homogenized before the addition of the organic phase.In a typical extraction experiment, an equal volume of the spiked aqueous phase was added to a weighed organic phase and mixed at 30 °C at 2400 rpm. The volumes of each phases were typically 0.5 mL or 1 mL (depending on the chosen vial size of 1.5 or 4 mL, respectively). Following equilibration, the phase disengagement was enhanced by centrifugation of the vials (5 min at 4000 rpm), and weighed 300 μL aliquots of the aqueous and organic phases were collected in 1.5 mL glass vials for gamma activity measurement.
Gamma spectrometric analysis of 241Am (using the 59.5 keV γ-peak) and 152Eu (using the 121.8 keV) was conducted using an HPGe detector (model: GC2520) with DSA-1000 Multi-Channel Analyzer and Genie2000 software (Canberra Semiconductors N. V., Olen, Belgium). The α-particle emitter radionuclide activities were determined for each separated phase using α spectrometry. Weighed aliquots of the organic or aqueous phase were pipetted on a C-1S cupped stainless steel α planchet (GA-MA and Associates, Inc., Florida, USA), heated under an infrared lamp (Theratherm 150 W, Osram, Germany) and subsequently burned with a gas torch. The 241Am (Eα = 5.485 MeV) and 244Cm (Eα = 5.805 MeV) α peaks were measured with an α spectrometer (Alpha Analyst, Canberra) equipped with Passivated Implanted Planar Silicon (PIPS) α detectors (Canberra Olen N.V., Olen, Belgium). The spectra were analyzed using Apex Alpha software (Canberra). Organic phase samples showed a baseline separation (i.e. no overlap/tailing of the higher energy 244Cm peaks with 241Am peaks). Samples prepared from the aqueous phases required an area correction due to tailing, typically less than 10% of the total peak area. Whenever the peak overlap was found to be greater than that, a new, diluted sample was prepared to mitigate the self-absorption in the alpha samples.
The concentration of the stable elements was determined by ICP-MS method using an X2Series II ICP-MS instrument (ThermoFischer Scientific, Bremen, Germany). A 1:100 dilution was prepared from the separated aqueous phases. All ICP-MS samples, calibration standards, and quality control standards were made using the same diluted nitric acid solution to avoid matrix effects. Calibration of the instrument was performed using a multi-element calibration standard covering the expected concentration range in the final dilutions (0–50 μg L−1). As internal standards, In and Tl (Spex CertiPrep Ltd, UK) were used. Nd (CPI International, California, USA) was used as the analyte for the quality control standard at a 10 μg L−1 concentration.
The distribution ratio (D) of a given element was calculated as the ratio of the equilibrium activity or concentration in the organic phase over the equilibrium activity or concentration in the aqueous phase (eqn (1)). In the case of ICP-MS samples, the organic phase concentration was determined by calculation from the initial and final equilibrium aqueous phase concentrations, as given by eqn (2).
| D = [Morg.eq.]/[Maq.eq.] | (1) |
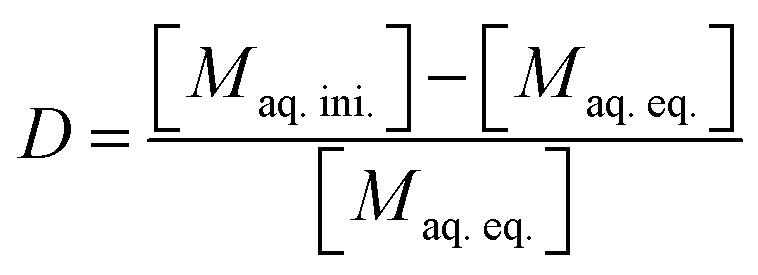 | (2) |
The separation factor (SF) of two elements was calculated from the distribution ratios of the respective elements in accordance with eqn (3):
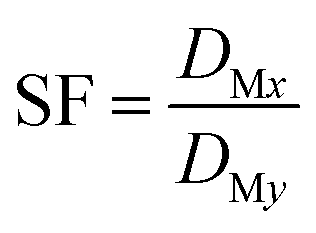 | (3) |
The distribution ratios carry a combined overall uncertainty of 10% between 0.01 < D < 100. Outside of this range, where the activity or the concentration of an analyte in one of the phases is close to the minimum detectable activity or to the limit of quantification, the overall uncertainty can reach 25%.
Results and discussion
Extraction of Am(III), Cm(III) and Eu(III) using CyMe4BTPhen in [A336][NO3]
The kinetics of extraction of An(III) and Ln(III) from acidic feed solution was studied in batch extractions using tracer concentrations of 241Am(III), 244Cm(III) and 152Eu(III). The distribution ratios of the radiotracers as a function of the equilibration time are shown in Fig. 2.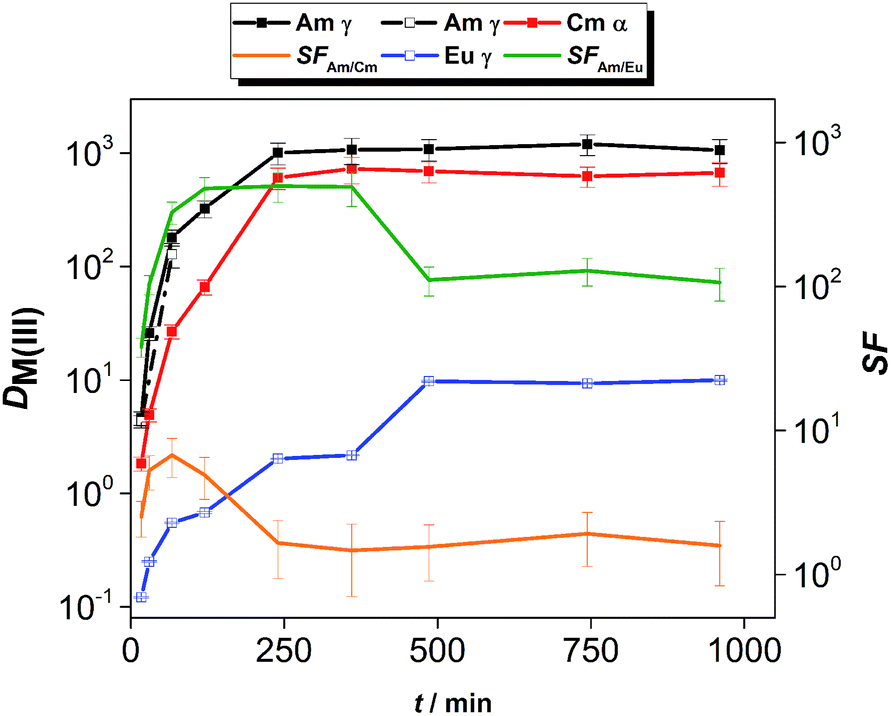 | ||
| Fig. 2 Distribution ratios of Am(III), Cm(III) and Eu(III) and the corresponding separation factors as a function of the equilibration time. Org.: 0.01 mol L−1 CyMe4BTPhen in [A336][NO3], Aq.: 1 mol L−1 HNO3, Ln(III) = 10−5 mol L−1; T = 30 °C, shaking at 2400 rpm. | ||
The distribution ratios of Am(III) and Cm(III) reached equilibrium after 4 h of contact time, while equilibrium of extraction for Eu(III) was reached after 8 h of contact time. The equilibrium distribution ratio was also reached faster for Am(III) than for Cm(III). This resulted in higher than equilibrium separation factors for Am(III) over Cm(III): the SFAm/Cm increased steeply (reaching values between 5.2 and 6.7) until 1 h contact time. For longer contact times, the SFAm/Cm decreased and reached an equilibrium value of 1.5–1.9, comparable to the values reported from diluents such as 1-octanol or 1-octanol/toluene mixtures.17,18
The kinetics of extraction of Am(III), Cm(III) and Eu(III) using 0.01 mol L−1 CyMe4BTPhen in [A336][NO3] is significantly slower than what was observed by Lewis et al. in 1-octanol diluent.17 In that study with 1-octanol diluent, the equilibrium distribution ratios were obtained after 60 min of contact time. Similarly, higher than equilibrium SFAm/Cm values (reaching 7.9) were reported in a solvent composed of 0.005 mol L−1 CyMe4BTPhen in 1-octanol/toluene (40/60).20 The authors proposed that the difference in extraction kinetics of the adjacent Am(III) and Cm(III) ions is caused by the presumed faster water exchange rates in the aqua complexes of Am(III) ions, compared to the aqua complexes of Cm(III) ions. Similarly, even higher SFAm/Cm values (reaching 18) outside equilibrium conditions were observed in our already mentioned previous study, at T = 22 °C in a solvent composed of 0.01 mol L−1 CyMe4BTPhen in [A336][NO3].41 Our previous studies showed that the [A336][NO3] diluent itself shows negligible extraction of An(III) and Ln(III) from 0.01 mol L−1 up to 6 mol L−1 HNO3.39
The slow extraction kinetics of ionic liquid-based solvents are often considered as a disadvantage in continuous solvent extraction processes, since not all apparatus can provide sufficient residence time needed to reach equilibrium. On the other hand, one could try to work with shorter contact times and operate outside equilibrium conditions to make advantage of the higher SFAm/Cm under such conditions as also proposed by Distler et al.49 When working within equilibrium conditions, the slow kinetics would result in prolonged exposure of the solvent to the highly radioactive aqueous feed solution and increase significantly the footprint of the process. The use of CyMe4BTPhen in [A336][NO3] diluent can be nonetheless justified, knowing its high stability against radiation-induced degradation.31 In addition, distribution ratios exceeding 10 for Am(III) and Cm(III) with sufficient separation from Ln(III) can already be achieved within contact times no longer than 30 min (however further studies will be required to verify if such solvent can be of practical use in continuous processes). With respect to the selectivity of the system, the solvent composed of 0.01 mol L−1 CyMe4BTPhen in [A336][NO3] clearly favors the extraction of An(III) over Eu(III). However, the equilibrium distribution ratio of Eu(III) reached 10, so most of the Eu(III) is extracted, but this can be avoided by working at lower acidity since distribution ratios increase for both extractants with the increase of feed acidity (see here below).
The effect of aqueous feed acidity on the distribution ratios of Am(III), Cm(III) and Eu(III) was studied with an equilibration time of 12 h (Fig. 3). Over the entire range of 0.01–6 mol L−1 aqueous feed acidity, the solvent had a higher affinity for Am(III) over both Cm(III) and Eu(III). The distribution ratios for An(III) were very high; DAm > 100 when extracting from 0.1 mol L−1 HNO3 feed solution and DAm ≫ 100 when extracting from feed solutions ≥1 mol L−1 HNO3. The DAm, DCm and DEu followed the same trend, and the Am/Cm separation factor at equilibrium was rather insensitive to the variation of feed acidity (SFAm/Cm = 1.3–2.1). The Am/Eu separation factor was globally almost always above 100. The observed trend of increasing DAm and DCm values with increasing nitric acid concentration above 1 mol L−1 HNO3 is different from the results of acid dependency studies performed with 0.01 mol L−1 CyMe4BTPhen in 1-octanol or 1-octanol/toluene mixtures by Lewis et al.20 The decrease of the distribution ratios at higher acidities when using various molecular diluent mixtures was attributed by the authors to the protonation of the N-donor moieties on the ligand, being competitive with the metal ion extraction, which was observed here only for the highest tested nitric acid concentration (6 mol L−1).
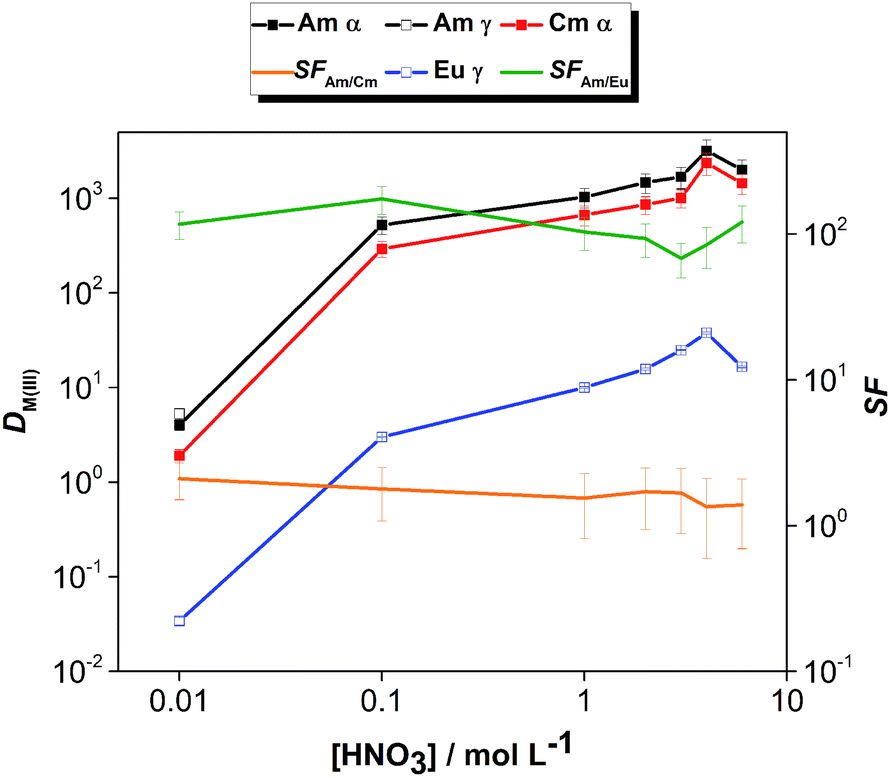 | ||
| Fig. 3 Distribution ratios of Am(III), Cm(III) and Eu(III) as a function of the aqueous feed acidity. Org.: 0.01 mol L−1 CyMe4BTPhen in [A336][NO3], Aq.: 0.01–6 mol L−1 HNO3, Ln(III) = 10−5 mol L−1, T = 30 °C, shaking at 2400 rpm for 12 h. | ||
Extraction of Am(III), Cm(III) and Eu(III) using CyMe4BTPhen in [A336][NO3]/1-octanol and TODGA as phase transfer catalyst
The solvent composed of 0.01 M CyMe4BTPhen in [A336][NO3] ionic liquid showed slow extraction kinetics (Fig. 2). With the aim of improving the extraction kinetics, both TODGA (as phase transfer catalyst) and 1-octanol (as co-diluent) were added to the CyMe4BTPhen in [A336][NO3] solvent.A concentration of 0.005 mol L−1 TODGA was chosen because this concentration was successful to enhance the kinetics in former studies.15
The distribution ratios of the three radionuclide tracers Am(III), Cm(III) and Eu(III), and the stable Ln(III) ions from La(III) – Dy(III), Yb(III) and Y(III) are shown in Fig. 4 for solvents with 0–30 vol% 1-octanol co-diluent. At each composition, An(III) were selectively extracted (DAm ≈ 4–11, DCm ≈ 2–5) without the co-extraction of Ln(III) (DLn ≪ 1). Separation factors for Am(III)/Eu(III) ranged between 156 and 493; separation factors for Am(III)/Cm(III) were ca. 2. Thus, the four solvent compositions (with 0–10–20–30 vol% 1-octanol co-diluent) proved to provide comparable selectivity for the Am/Ln and Am/Cm couples, showing that such diluent modification does not impair the An(III)/Ln(III) selectivity of the process.
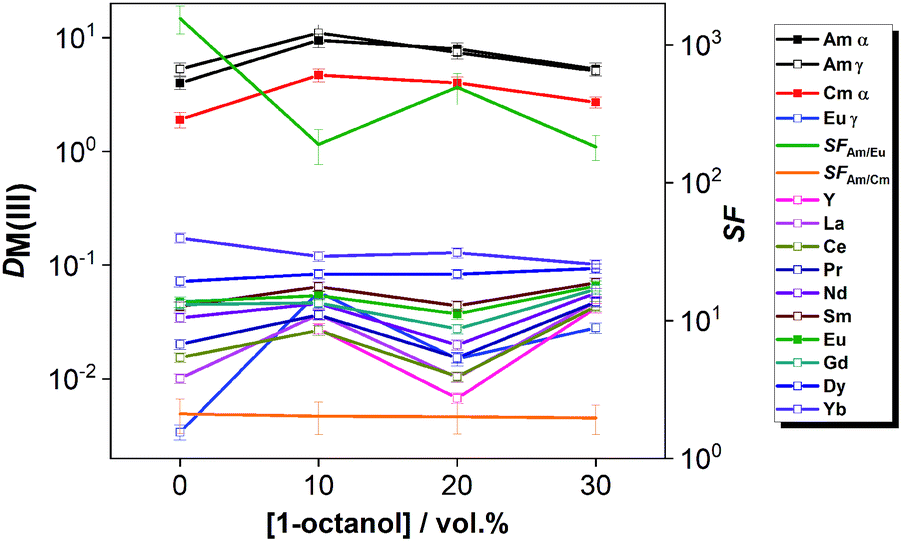 | ||
| Fig. 4 Distribution ratios of An(III) and Ln(III) with various concentrations of 1-octanol. Org.: 0.01 mol L−1 CyMe4BTPhen + 0.005 mol L−1 TODGA in diluent mixtures composed of 0–30 vol% 1-octanol in [A336][NO3]; extraction, Aq.: 0.01 mol L−1 HNO3, [Ln(III)] = 10−5 mol L−1, tracers of 241Am(III), 244Cm(III) and 152Eu(III). All equilibrations were performed for t = 12 h at T = 30 °C and shaking at 2400 rpm. | ||
A 70/30 vol% [A336][NO3]/1-octanol diluent mixture was chosen for the further studies as its hydrodynamic behavior seemed promising in continuous counter-current scoping tests.
Fig. 5 shows the results of the kinetics studies conducted on the solvent composed of 0.01 mol L−1 CyMe4BTPhen + 0.005 mol L−1 TODGA in 70 vol% [A336][NO3] and 30 vol% 1-octanol. For the actinides, Am(III) and Cm(III), equilibrium was reached after ca. 40 min (in comparison to ca. 4 h when using pure [A336][NO3] as diluent, see Fig. 2). Ln(III) ions reached equilibrium within 30 min contact time (in comparison to 8 h when using pure [A336][NO3] as diluent, see Fig. 2). The distribution ratios of the An(III) were much higher than those of the Ln(III), i.e. ca. 1000 for Am(III) and ca. 300 for Cm(III) at equilibrium. The distribution ratios increased along the lanthanide series.
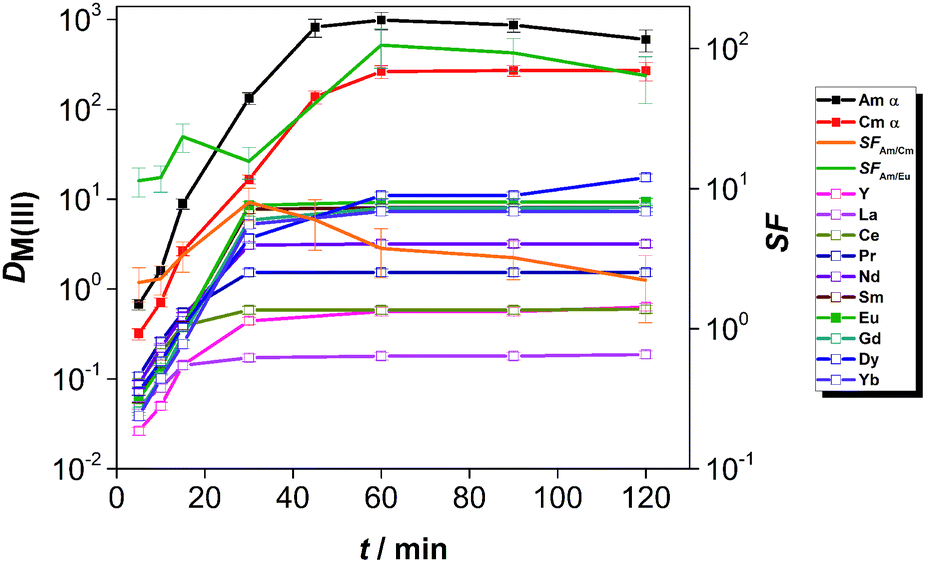 | ||
| Fig. 5 Distribution ratios of An(III) and Ln(III) as a function of equilibration time. Org.: 0.01 mol L−1 CyMe4BTPhen + 0.005 mol L−1 TODGA in a diluent mixture composed of 70 vol% [A336][NO3] and 30 vol% 1-octanol; Aq.: 1 mol L−1 HNO3, [Ln(III)] (La–Dy, Yb, Y) = 10−5 mol L−1; spiked with tracers of 241Am, 244Cm and 152Eu. T = 30 °C, shaking at 2400 rpm. | ||
The effect of various additives on the kinetics of Eu(III) extraction by either CyMe4BTBP or CyMe4BTPhen in 1-octanol was also studied by Lewis et al. with the rotating membrane cell technique.17 TODGA, added at a 0.005 mol L−1 concentration, resulted in a significant increase (by a factor of 100) of the extraction rate constant of the CyMe4BTBP solvent. However, less improvement was observed when TODGA was added to the CyMe4BTPhen solvent as a phase transfer reagent (kinetics improved by a factor of 4).
The distribution ratios of Am(III), Cm(III) and Ln(III) were studied as a function of the aqueous feed acidity using the solvent composed of 0.01 mol L−1 CyMe4BTPhen + 0.005 mol L−1 TODGA in 70/30 vol% [A336][NO3]/1-octanol (Fig. 6). The distribution ratios of the Ln(III) increased with aqueous feed acidity up to 4 mol L−1 HNO3. The heavy lanthanide Yb(III) and Y(III) (which typically behaves similarly to the heavy lanthanides), followed a different trend, the distribution ratios increased more steeply with the increase of feed acidity. At higher acidities, the distribution ratios showed a slight decrease up to 6 mol L−1 HNO3 for most of the elements, except for Yb and Y, where further increase of D values was observed.
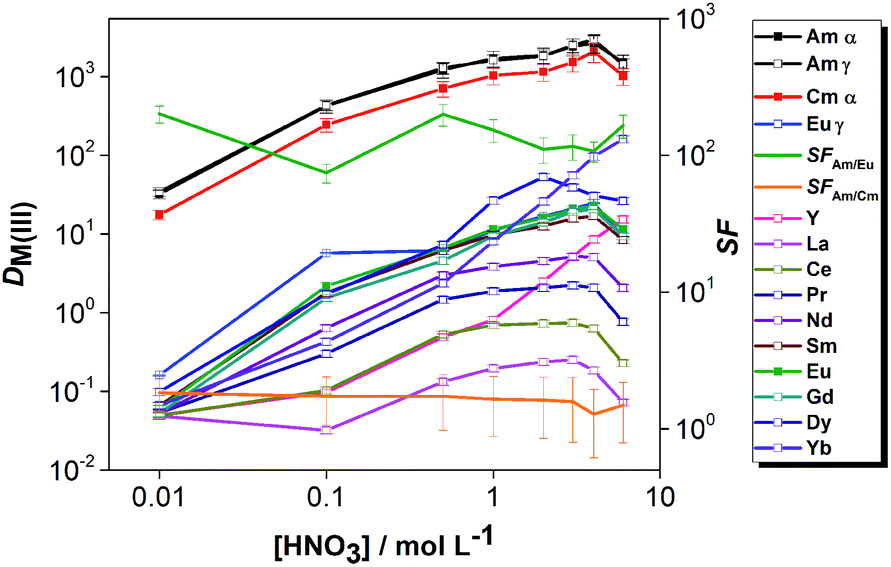 | ||
| Fig. 6 Distribution ratios of An(III) and Ln(III) as a function of the nitric acid concentration of the feed solution. Org.: 0.01 mol L−1 CyMe4BTPhen + 0.005 mol L−1 TODGA in a diluent mixture composed of 70 vol% [A336][NO3] and 30 vol% 1-octanol; Aq.: 0.1–6 mol L−1 HNO3, [Ln(III)] (La–Dy, Yb, Y) = 10−5 mol L−1; spiked with tracers of 241Am, 244Cm and 152Eu. T = 30 °C, shaking at 2400 rpm, t = 4 h. | ||
The acid dependency of distribution ratios for Am(III) and Cm(III) were similar to the two-component system. The SFAm/Eu is higher at low feed acidities in the case of both the two- and four-component solvents. This can be expected due to the extraction of Ln(III) by TODGA at higher acidities and the strong competition of protons with An(III)-ions for the electron-donating N-atoms of CyMe4BTPhen molecules.
The acid dependency studies (Fig. 3 and 6) showed that DAm(III) and DCm(III) are high enough even at 0.01 mol L−1 aqueous feed acidity to allow quantitative extraction of the actinides (if sufficient extraction stages are applied), while DEu(III) ≪ 1 showing that the lanthanides can be retained in the aqueous phase.
Back-extraction studies
The feasibility of stripping the An(III) from a loaded organic phase containing both CyMe4BTPhen and TODGA in [A336][NO3]/1-octanol is shown in Table 1. A hydrophilic complexant is necessary to allow An(III) stripping, given the high DAn(III) values even in the case of dilute nitric acid solutions (Fig. 6). For this purpose and in order to increase the An(III)/Ln(III) selectivity in the stripping section, 0.05 mol L−1 TTHA was used in a 0.5 mol L−1 citrate solution buffered to pH 4.| Process step | 1-Octanol (vol%) | DAm γ | DAm α | DCm α | DEu γ |
|---|---|---|---|---|---|
| a DEu was determined additionally by ICP-MS and it ranges between 1.3 ± 0.8 (low accuracy due to the low concentration of Eu in the aqueous phase). | |||||
| Extraction | 0 | 5.3 ± 0.7 | 4.0 ± 0.5 | 1.9 ± 0.3 | 0.034 ± 0.003 |
| Back-extraction | <0.001 | <0.001 | <0.001 | <Q. L.a | |
| Extraction | 30 | 5.1 ± 0.5 | 5.3 ± 0.7 | 2.7 ± 0.3 | 0.028 ± 0.003 |
| Back-extraction | <0.001 | <0.001 | <0.001 | <Q. L.a | |
Under these conditions, Am(III) and Cm(III) were quantitatively back-extracted into the aqueous phase, while Eu(III), as representative element for the Ln(III), was retained in the organic phase. In a previous study it was shown that the hydrophilic complexant TEDGA can also be used to either co-strip An(III) + Ln(III) or (by careful selection of the TEDGA concentration) to perform a selective Cm(III) + Ln(III) stripping.41 Depending on the desired aqueous product composition (acidity, Am/Cm ratio) the stripping conditions can be varied.
From the outcome of this study, combined with the results of our previous work, we can state that a full Aliquat-336 nitrate based process could be designed (see Fig. 7). This further opens the opportunity to exploit the benefits of ionic liquids in partitioning processes, such as their higher stability towards ionizing radiation. In the first step, the lanthanides and actinides can be coextracted from a PUREX raffinate using a [A336][NO3]/TODGA solvent.39 In the second step, the actinides can be separated from the lanthanides using [A336][NO3]/CyMe4BTPhen (this study), either with or without the use of a phase transfer catalyst and co-diluent (also a temperature increase could resolve the slow kinetics of the system). The final product of this process would be a combined Am(III) + Cm(III) product. If one desires an Am(III)-only product, an alternative second step using a [A336][NO3]/CyMe4BTPhen solvent with addition of TEDGA to the aqueous phase can be applied.41
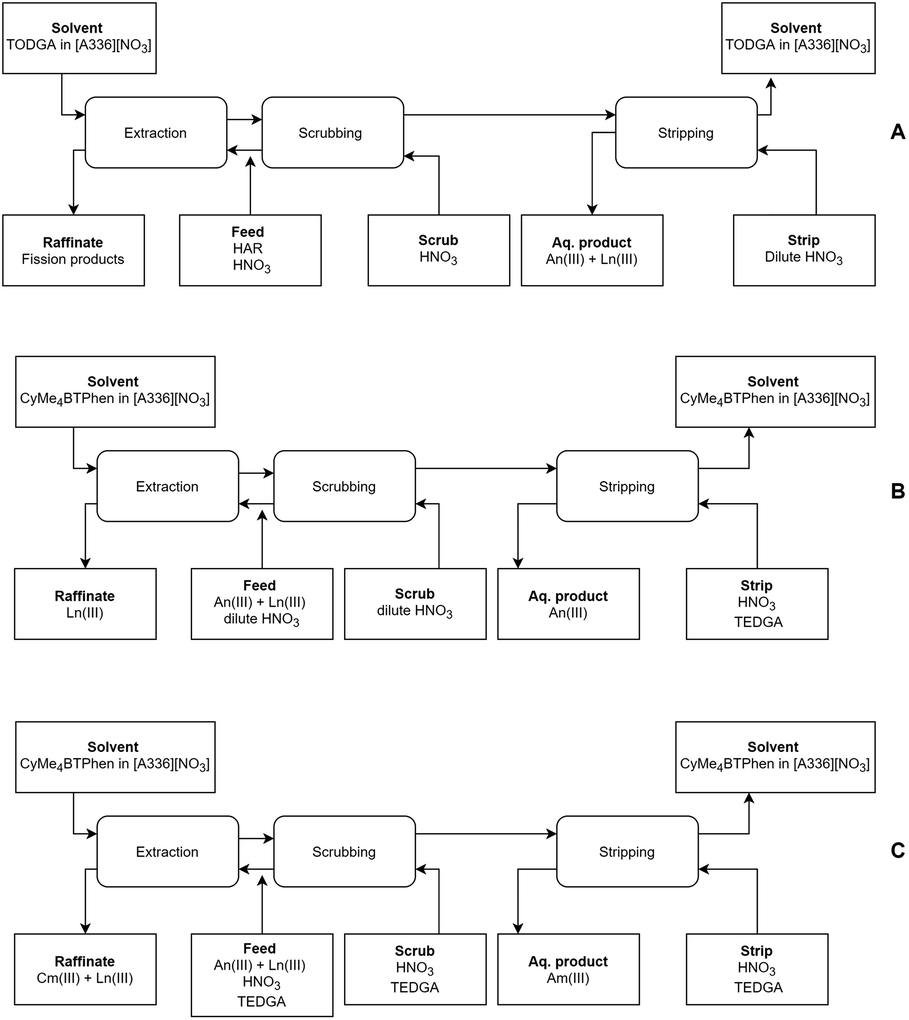 | ||
| Fig. 7 Preliminary flow sheets for the separation of Am(III) + Cm(III) (A + B), or separation of Am(III)-only (A + C) from a highly active raffinate feed solution using a [A336][NO3]-based process. For flow sheet B, TODGA (as phase transfer catalyst) and 1-octanol (as co-diluent) can be added to the solvent to improve kinetics. For flow sheet C, 1-octanol (as co-diluent) can be added to the solvent to improve kinetics. | ||
Conclusions
The extraction of trivalent actinides Am(III) and Cm(III) by the soft donor extractant CyMe4BTPhen alone in undiluted [A336][NO3] and in a diluent mixture of [A336][NO3]/1-octanol with TODGA added as phase transfer catalyst was studied. The slow extraction kinetics by the solvent composed of 0.01 mol L−1 CyMe4BTPhen in undiluted [A336][NO3] could be improved by the addition of TODGA as a phase transfer catalyst and 1-octanol as co-diluent. Since the improvement of the kinetics did not significantly impair the An(III)/Ln(III) selectivity, this system is promising for further development towards continuous processes. Short residence time annular centrifugal contactors will be used in the future to implement the concept obtained from these batch extraction experiments.Author contributions
Péter Zsabka: solvent extraction experiments, sample preparation, radioanalytical measurements, data treatment, conceptualization, investigation, writing – original draft. Karen Van Hecke: supervision, writing – review & editing. Lesley Adriaensen: supervision. Andreas Wilden: conceptualization, writing – review & editing. Giuseppe Modolo: conceptualization, writing – review & editing. Marc Verwerft: funding acquisition, supervision. Koen Binnemans: conceptualization, supervision, writing – review & editing. Thomas Cardinaels: conceptualization, funding acquisition, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Magda Ooms for the help with gamma and alpha spectrometry measurements and Karen Van Hoecke for the ICP-MS measurements. P. Z. Acknowledges the SCK CEN Academy for providing funding for a PhD fellowship and the FOD Economy for providing additional support via the Energietransitiefonds.References
- C. V. Ellison and T. C. Runion, US Pat., 2,990,240, 1961 Search PubMed.
- C. Poinssot, S. Bourg and B. Boullis, Prog. Nucl. Energy, 2016, 92, 234–241 CrossRef CAS.
- J. Serp, C. Poinssot and S. Bourg, Energies, 2017, 10, 1445–1464 CrossRef.
- C. Madic, M. J. Hudson, J. O. Liljenzin, J. P. Glatz, R. Nannicini, A. Facchini, Z. Kolarik and R. Odoj, New partitioning techniques for minor actinides – final report, NEWPART European Union project FI4I-CT-96-0010, European Commission Brussels, 2000 Search PubMed.
- C. Madic, F. Testard, M. J. Hudson, J. O. Liljenzin, B. Christiansen, M. Ferrando, A. Facchini, A. Geist, G. Modolo, A. Gonzalez-Espartero and J. d. Mendoza, Partnew – New solvent extraction processes for minor actinides – final report, France, 2004 Search PubMed.
- C. Madic, M. J. Hudson, P. Baron, N. Ouvrier, C. Hill, S. Bourg, F. Arnaud, A. G. Espartero, J. F. Desreux, G. Modolo, R. Malmbeck, G. d. Angelis and J. Uhlir, presented in part at the Information Exchange Meeting, Nimes (France); 25–29 Sep. 2006, Nuclear Energy Agency of the OECD (NEA), 2007 Search PubMed.
- S. Bourg, C. Hill, C. Caravaca, C. Ekberg and C. Rhodes, presented in part at the Proceedings of the GLOBAL 2009 congress – The Nuclear Fuel Cycle: Sustainable Options and Industrial Perspectives, France, 2009 Search PubMed.
- S. Bourg, C. Hill, C. Caravaca, C. Rhodes, C. Ekberg, R. Taylor, A. Geist, G. Modolo, L. Cassayre, R. Malmbeck, M. Harrison, G. de Angelis, A. Espartero, S. Bouvet and N. Ouvrier, Nucl. Eng. Des., 2011, 241, 3427–3435 CrossRef CAS.
- A. Geist, R. Taylor, C. Ekberg, P. Guilbaud, G. Modolo and S. Bourg, Procedia Chem., 2016, 21, 218–222 CrossRef.
- www.geniors.eu, accessed 2019/07/11.
- Z. Kolarik, Chem. Rev., 2008, 108, 4208–4252 CrossRef CAS.
- P. J. Panak and A. Geist, Chem. Rev., 2013, 113, 1199–1236 CrossRef CAS.
- F. W. Lewis, M. J. Hudson and L. M. Harwood, Synlett, 2011, 18, 2609–2632 Search PubMed.
- M. J. Hudson, L. M. Harwood, D. M. Laventine and F. W. Lewis, Inorg. Chem., 2013, 52, 3414–3428 CrossRef CAS.
- A. Wilden, C. Schreinemachers, M. Sypula and G. Modolo, Solvent Extr. Ion Exch., 2011, 29, 190–212 CrossRef CAS.
- D. Magnusson, B. Christiansen, M. R. S. Foreman, A. Geist, J. P. Glatz, R. Malmbeck, G. Modolo, D. Serrano-Purroy and C. Sorel, Solvent Extr. Ion Exch., 2009, 27, 97–106 CrossRef CAS.
- F. W. Lewis, L. M. Harwood, M. J. Hudson, M. G. Drew, J. F. Desreux, G. Vidick, N. Bouslimani, G. Modolo, A. Wilden, M. Sypula, T. H. Vu and J. P. Simonin, J. Am. Chem. Soc., 2011, 133, 13093–13102 CrossRef CAS.
- S. Lange, A. Wilden, G. Modolo, F. Sadowski, M. Gerdes and D. Bosbach, Solvent Extr. Ion Exch., 2017, 35, 161–173 CrossRef CAS.
- A. Geist, C. Hill, G. Modolo, M. R. S. J. Foreman, M. Weigl, K. Gompper and M. J. Hudson, Solvent Extr. Ion Exch., 2006, 24, 463–483 CrossRef CAS.
- F. W. Lewis, L. M. Harwood, M. J. Hudson, A. Afsar, D. M. Laventine, K. Šťastná, J. John and P. Distler, Solvent Extr. Ion Exch., 2018, 36, 115–135 CrossRef CAS.
- P. Distler, M. Mindová, J. John, V. A. Babain, M. Y. Alyapyshev, L. I. Tkachenko, E. V. Kenf, L. M. Harwood and A. Afsar, Solvent Extr. Ion Exch., 2020, 38, 180–193 CrossRef CAS.
- M. Nilsson, S. Andersson, C. Ekberg, M. Foreman, M. Hudson and G. Skarnemark, Radiochim. Acta, 2006, 94, 103–106 CAS.
- J.-F. Parisot, Treatment and recycling of spent nuclear fuel Actinide partitioning – Application to waste management, N. E. Division, CEA/CEN, Direction Scientifique, Paris, 2008 Search PubMed.
- H. Schmidt, A. Wilden, G. Modolo, J. Švehla, B. Grüner and C. Ekberg, Nukleonika, 2015, 60, 879–884 Search PubMed.
- H. Schmidt, A. Wilden, G. Modolo, D. Bosbach, B. Santiago-Schübel, M. Hupert, J. Švehla, B. Grüner and C. Ekberg, Procedia Chem., 2016, 21, 32–37 CrossRef.
- A. Wilden, G. Modolo, M. Hupert, B. Santiago-Schübel, E. Löfström-Engdahl, J. Halleröd, C. Ekberg, B. J. Mincher and S. P. Mezyk, Solvent Extr. Ion Exch., 2016, 34, 1–12 CrossRef CAS.
- P. K. Mohapatra, Dalton Trans., 2017, 46, 1730–1747 RSC.
- A. Rout, S. Karmakar, K. A. Venkatesan, T. G. Srinivasan and P. R. Vasudeva Rao, Sep. Purif. Technol., 2011, 81, 109–115 CrossRef CAS.
- M. Sivaramakrishna, D. R. Raut, S. Nayak, S. K. Nayak and P. K. Mohapatra, Sep. Purif. Technol., 2017, 181, 69–75 CrossRef CAS.
- A. Bhattacharyya, S. A. Ansari, T. Gadly, S. K. Ghosh, M. Mohapatra and P. K. Mohapatra, Dalton Trans., 2015, 44, 6193–6201 RSC.
- P. Zsabka, K. Van Hecke, A. Wilden, G. Modolo, M. Hupert, V. Jespers, S. Voorspoels, M. Verwerft, K. Binnemans and T. Cardinaels, Solvent Extr. Ion Exch., 2020, 38, 212–235 CrossRef CAS.
- A. Sengupta, P. K. Mohapatra, M. Iqbal, J. Huskens and W. Verboom, Sep. Purif. Technol., 2013, 118, 264–270 CrossRef CAS.
- J. N. Chubb, P. Lagos and J. Lienlaf, J. Electrost., 2004, 63, 119–127 CrossRef.
- I. M. Smallwood, Handbook of organic solvent properties, John Wiley & Sons Inc., London, 1996 Search PubMed.
- O. R. Luca, J. L. Gustafson, S. M. Maddox, A. Q. Fenwick and D. C. Smith, Org. Chem. Front., 2015, 2, 823–848 RSC.
- P. Giridhar, K. A. Venkatesan, T. G. Srinivasan and P. R. V. Rao, J. Nucl. Radiochem. Sci., 2004, 5, 17–20 CrossRef CAS.
- D. Zhao, Y. Liao and Z. Zhang, Clean, 2007, 35, 42–48 CAS.
- A. Rout, K. A. Venkatesan, T. G. Srinivasan and P. R. Vasudeva Rao, Sep. Sci. Technol., 2013, 48, 2576–2581 CrossRef CAS.
- P. Zsabka, K. Van Hecke, L. Adriaensen, A. Wilden, G. Modolo, M. Verwerft, K. Binnemans and T. Cardinaels, Solvent Extr. Ion Exch., 2018, 36, 519–541 CrossRef CAS.
- C. Venkateswara Rao, A. Rout and K. A. Venkatesan, New J. Chem., 2019, 43, 5099–5108 RSC.
- P. Zsabka, K. Van Hecke, A. Wilden, G. Modolo, M. Verwerft, K. Binnemans and T. Cardinaels, Solvent Extr. Ion Exch., 2020, 38, 194–211 CrossRef CAS.
- P. Zsabka, T. Opsomer, K. Van Hecke, W. Dehaen, A. Wilden, G. Modolo, M. Verwerft, K. Binnemans and T. Cardinaels, Solvent Extr. Ion Exch., 2020, 38, 719–734 CrossRef CAS.
- A. Rout, K. A. Venkatesan, M. P. Antony and P. R. Vasudeva Rao, Sep. Purif. Technol., 2016, 158, 137–143 CrossRef CAS.
- E. A. Mezhov, V. A. Kuchumov and V. V. Druzhenkov, Radiokhimiya, 2002, 44, 126–130 Search PubMed.
- G. Modolo, M. Sypula, A. Geist, C. Hill, C. Sorel, R. Malmbeck, D. Magnusson and M. R. S. J. Foreman, presented in part at the 10th Information Exchange Meeting on Actinide and Fission Product Partitioning and Transmutation, Mito, Japan, 6-10, October 2008 Search PubMed.
- D. Magnusson, A. Geist, A. Wilden and G. Modolo, Solvent Extr. Ion Exch., 2013, 31, 1–11 CrossRef CAS.
- A. Wilden, G. Modolo, C. Schreinemachers, F. Sadowski, S. Lange, M. Sypula, D. Magnusson, A. Geist, F. W. Lewis, L. M. Harwood and M. J. Hudson, Solvent Extr. Ion Exch., 2013, 31, 519–537 CrossRef CAS.
- G. Modolo, A. Wilden, H. Daniels, A. Geist, D. Magnusson and R. Malmbeck, Radiochim. Acta, 2013, 101, 155–162 CrossRef CAS.
- P. Distler, K. Stamberg, J. John, L. M. Harwood and F. W. Lewis, Sep. Sci. Technol., 2018, 53, 277–285 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |