
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of counter ions of ammonium for nitrogen doping and carbon properties in hydrothermal carbonization: characterization and supercapacitor performance†
Kenneth G.
Latham
 *ab,
Marveh
Forghani
c,
Wesley M.
Dose
de,
Jessica A.
Allen
f and
Scott W.
Donne
a
*ab,
Marveh
Forghani
c,
Wesley M.
Dose
de,
Jessica A.
Allen
f and
Scott W.
Donne
a
aDiscipline of Chemistry, University of Newcastle, Callaghan, NSW 2308, Australia. E-mail: kenneth.latham@umu.se
bUmeå University, Department of Chemistry, SE-90 187, Umeå, Sweden
cInstitute of Future Transport and Cities, Coventry University, Priory Street, Coventry CV1 5FB, UK
dDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, Cambridge, UK
eDepartment of Engineering, University of Cambridge, 17 Charles Babbage Road, CB3 0FS, Cambridge, UK
fSchool of Engineering, University of Newcastle, Callaghan, NSW 2308, Australia
First published on 7th December 2020
Abstract
Ammonium has been successfully utilized to nitrogen dope carbon structures via hydrothermal carbonization, although the influence of different attached counter ions (anions) on the resultant carbon physicochemical properties and electrochemical performance has not been examined before. Four different counter ions (SO42−, PO43−, Cl−, and Fe(SO4)2) attached to ammonium were seen to influence the hydrothermal reaction, nitrogen incorporation levels, physicochemical properties, activation ability and supercapacitor performance. For instance, nitrogen K-edge NEXAFS found differences in the levels of pyridinic and pyrrolic groups with PO43− incorporating predominately pyridinic nitrogen groups. PO43− also achieved the highest surface area (2132.6 m2 g−1), however this material was unstable as a supercapacitor, losing almost 50% of its performance over 500 cycles. SO42− resulted in the highest level of nitrogen incorporation (5.53 at%) and hydrothermal yield (45.5%), while Fe(SO4)22− resulted in the lowest (2.92 at%). However, Fe(SO4)22− produced unique flower like structures not seen in any of the other anions. Cl− produced the highest performing material, achieving 190 F g−1 at 10 mV s−1 in 1 M KOH and had moderate nitrogen incorporation (3.42 at%). Overall, this study indicates that the anion has substantial influence on the physicochemical properties of the material, allowing an additional level of tailoring.
1. Introduction
With their high charge–discharge rate (<1–3 s), large power density (>10 kW kg−1), long cycle life (>105 cycles), low cost and natural abundance of electrode precursor materials (e.g., biomass), carbon based supercapacitors (CSC) are a fundamental technology for addressing future energy needs. Despite these advantages, CSC still suffer from lower energy density and cell voltage than lithium-ion batteries. To address this issue, a range of strategies have been investigated to enhance the energy density and working voltage of supercapacitors without sacrificing power density or cycle life. These are, (i) structural improvements, such as pore size tailoring in hierarchical porous carbons,1 (ii) incorporation of transition metal oxides2 or (iii) physicochemical enhancements via heteroatom doping.3,4 Tailoring the porosity enhances electrolyte accessibility,1 while transition metal oxides increase capacitance via their pseudocapacitive characteristics. Heteroatom doping with N, B, O, P and S has been shown to increase capacitance through providing sites for pseudocapacitance,4 increasing wettability,5,6 creating lattice defects for charge storage,7 and enhancing electrical conductivity.4,8Among the heteroatom doped carbon materials, nitrogen is by far the most prominent dopant for its enhancement of electronic conductivity,7 improved surface wettability through hydrophilic groups and increased surface roughness,5 as well as ease of incorporation into carbon materials through a wide range of methods.3 Incorporating pyridinic, pyridonic, pyrrolic and oxidized –NOx, groups has been suggested to create sites where reversible faradaic redox reactions (pseudocapacitance) can occur, providing additional charge storage sites.3,9 Quaternary or graphitic nitrogen is also possible and suggested to promote the transport of electrons through the carbon lattice, reducing resistance.3
Nitrogen doped carbon materials have also shown promise in enlarging the cell voltage window, with Chi et al. showing excellent stability and capacitance retention of a nitrogen doped carbon surface after a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-cycle floating-like test between 3 and 4 V using an organic electrolyte (1 M TEABF4/PC).10 For comparison, the upper cut off voltage for CSC using organic electrolytes is generally between 2.5–2.7 V.11 As a result, nitrogen doped materials have consistently outperformed their non-doped counterparts when used in supercapacitors.
000-cycle floating-like test between 3 and 4 V using an organic electrolyte (1 M TEABF4/PC).10 For comparison, the upper cut off voltage for CSC using organic electrolytes is generally between 2.5–2.7 V.11 As a result, nitrogen doped materials have consistently outperformed their non-doped counterparts when used in supercapacitors.
Several approaches exist for synthesizing nitrogen doped carbon materials, such as chemical vapor deposition,12,13 plasma5 and arc discharge,14 pyrolysis of nitrogen containing precursors15,16 and hydrothermal carbonization.17,18 However, it is important to ensure that the production method is sustainable and environmentally friendly. For this reason, hydrothermal carbonization, a low temperature wet carbonization method,19 is preferred and excels in producing highly nitrogen doped carbon materials with a range of nitrogen functionalities.17,18,20,21
Using hydrothermal carbonization, nitrogen doped carbon materials can be synthesized by directly carbonizing nitrogen containing precursor (e.g., tannin,22 microalgae,23,24 chitosan21,25) or reacting a carbonaceous precursor (e.g., glucose, biomass) in the presence of a nitrogen containing agent (e.g., ammonium salt,17,26 ammonia,18 urea,27 glycine28). The first approach is simpler, although this provides limited control over the degree of nitrogen incorporation, precursor materials or the types of nitrogen groups formed. The second approach provides a greater level of flexibility with the nitrogen level and type being independent of the main carbon source. For instance, Park et al. demonstrated that the type and amount of nitrogen functional groups could be easily adjusted through modifying ratio between the nitrogen and non-nitrogen containing precursors as well as the reaction temperature.29 Additionally, nitrogenation agents containing multiple elements (e.g., (NH4)2HPO326,30) can further enhance the material beyond the influence of nitrogen via synergistic co-doping effects. For instance, phosphorus has a higher n-type behavior and stronger electron expansion ability than nitrogen, which can generate additional charge storage areas and improve electrical conductivity.31 Phosphorus doped carbons have also been shown to have larger stable voltage windows.32 Chlorine functionalization was shown by Pinkert et al. to increase the specific capacitance by 27% over a non-doped carbon at a cycle rate of 10 mV s−1 in EMIBF4 from −3.4 V to 3.4 V.33 Thus, co-doping is a way to further enhance the carbon surface beyond nitrogen doping. It is for this reason that ammonium salts represent an interesting group of compounds for enhanced hydrothermal doping, which have already been shown to co-dope carbon materials via hydrothermal carbonization.34 However, former studies examining the hydrothermal carbonization of glucose, fructose and sucrose with (NH4)2SO4 and (NH4)2HPO4 indicated that the ammonium counter-ion (anion), has an influence on the hydrothermal reaction.30 This study found vast differences in yield and pH between the (NH4)2SO4, (NH4)2HPO4 and non-doped hydrothermally carbonized materials.30
In this study we have examined the influence of four ammonium salts, (NH4)2SO4, (NH4)2HPO4, NH4Cl and (NH4)2Fe(SO4)2·6H2O on the hydrothermal carbonization of sucrose and supercapacitor performance. Extensive surface studies were conducted via XPS and synchrotron based NEXAFS to understand the differences in surface functionalities on the hydrothermal materials and activated hydrothermal materials. These surface studies were correlated with in-depth electrochemical studies using step potential electrochemical spectroscopy (SPECS), which can identify the different charge storage mechanisms occurring on these materials.35,36
2. Experimental
2.1 Hydrothermal carbon preparation
A series of five 350 mL deionized water solutions were prepared via the following method. Each solution contained 23.94 g of sucrose (Sigma Aldrich; 99%) and a single ammonium salt dissolved in the solution, with the exception of the control solution that contained no salt. The ammonium salts were selected for their stability in the hydrothermal system. Although relatively common, ammonium carbonate and ammonium nitrate were not selected as they could produce large amounts of CO, CO2 and NOx in the reactor, which could be a safety hazard. The amount of ammonium salt was 9.25 g for ammonium sulfate ((NH4)2SO4, Sigma Aldrich; 99%), 9.25 g for ammonium phosphate dibasic ((NH4)2HPO4, Sigma Aldrich 99%), 27.45 g for ammonium iron(II) sulfate hexahydrate ((NH4)2Fe(SO4)2·6H2O, Sigma Aldrich 99%) and 7.50 g for ammonium chloride (NH4Cl, Sigma Aldrich 99.5%). All solutions contained 0.2 M of ammonium salt, other than ammonium chloride which contained 0.4 M. This ensured an equivalent nitrogen concentration for each salt. The solutions were sealed in separate poly(tetrafluoroethylene)-lined reactors for hydrothermal synthesis at 200 °C over 4 hours. After reaction, the reactors were left to cool naturally in the oven before filtering the resultant suspension. The obtained hydrothermal carbon was first washed with Milli-Q water before being washed using acetone in a Soxhlet extractor at a cycle rate of one cycle per 30 min for 24 h. The washing procedure removed any non-chemically bound residual compounds that had dried on the surface from the liquid solution. The solid product remaining after washing was denoted here as H-Con for the pure sucrose control sample, and H-SO, H-PO, H-Cl, H-Fe for the ammonium sulfate, phosphate, chloride, and ferrous sulfate respectively.2.2 Activation of hydrothermal carbons
The dried hydrothermal carbons were activated by adding 1 g of hydrothermal carbon to 4 mL of 85% H3PO4 (Sigma Aldrich; 85%) and annealing at 600 °C for one hour under a constant 50 mL min−1 flow of N2. After heating, the activated carbons were washed with water to remove any acid residues until a neutral pH was achieved. The activated samples are denoted with an A-, such as A-SO for H-SO that has been activated.2.3 Yield, morphology, surface area and porosity
Yield was calculated based on the following formula:The morphology of the hydrothermal carbons was examined with a Philips XL30 scanning electron microscope (SEM) at three different magnifications, 500×, 3000× and 24
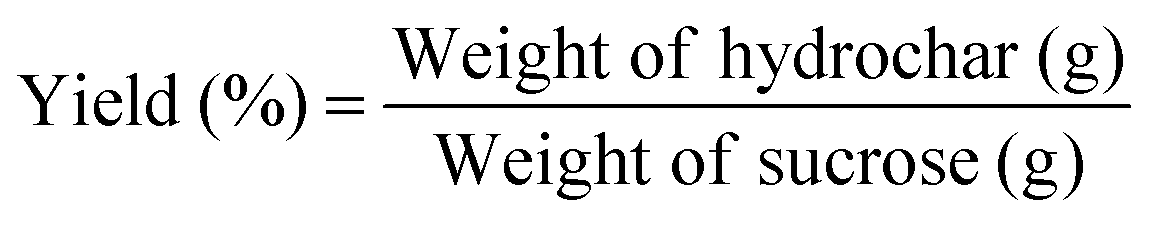 000×. Hydrothermal carbon samples were mounted on aluminium stubs with double sided carbon tape and gold coated at four different angles to ensure complete coverage of gold.
000×. Hydrothermal carbon samples were mounted on aluminium stubs with double sided carbon tape and gold coated at four different angles to ensure complete coverage of gold.
Surface area and porosity was examined by gas adsorption using a Micromeritics ASAP 2020 Surface Area and Porosity Analyzer. A representative 0.10 g sample of the carbon material was degassed under vacuum at 110 °C for 2 hours prior to analysis. An adsorption isotherm was then determined over the partial pressure range of 10−7–1 using N2 gas as the adsorbate at 77 K. The specific surface area was extracted from the gas adsorption data using the linearized BET isotherm in the partial pressure range 0.05 < P/P0 < 0.3.37
2.4 Surface characterization
000) were focused to a beam size of ∼100 μm2 onto samples within the UHV chamber. Photon flux at the carbon edge was ∼5 × 1010 photons per s per 200 mA of ring current. The UHV chamber, which has a base pressure lower than 2 × 10−10 mbar, was equipped with a channeltron detector that measured X-ray adsorption via partial electron yield at the magic angle of 54.7°, which is measured between the surface plane of the sample and the direction vector of the incident linearly polarized light beam.
The recorded signal was normalized to the incident photon flux using the stable monitor method, in which the sample signal is compared consecutively to clean reference samples and the time variation in flux is measured via a gold mesh.40 The normalized spectra were scaled by setting the pre-edge signal (280 eV for carbon and 380 eV for nitrogen) to zero and the post edge (320 eV for carbon, 415 eV for nitrogen) to 1. The photon energy was calibrated by measuring the NEXAFS spectrum of HOPG simultaneously to the sample signal and normalizing to the excitation peak at 291.65 eV.
To understand the effects of synchrotron radiation beam damage on the hydrothermal carbons, a sequence of scans was initially measured on the same spot until significant changes were observed in the carbon and nitrogen K-edge spectra. The scan time was subsequently restricted to an appropriate interval below this (∼0.5 s per 0.1 eV step) in which beam damage during a single scan was negligible.
2.5 Electrochemical testing
:10:5 using a mortar and pestle. To this mixture, 5 mL of N-methyl-2-pyrrolidone (NMP, anhydrous, 99.5%, Sigma Aldrich) was added to form an ink. The working electrode was prepared by dropping two 40 μL aliquots of ink onto the end of a separate clean 13 mm diameter stainless steel rod and left to dry at ambient temperature.
The counter electrode was prepared using a similar method except the activated hydrothermal carbon was replaced with carbon black conductive agent (Cabot, Vulcan XC72R) giving a weight ratio of 95:5 carbon black to PTFE. Additionally, 160 μL of the counter electrode ink was drop cast onto the stainless steel rod to ensure a greater mass of electrode material than the working electrode. The working electrodes weighed ∼4 mg cm−1, whereas the counter electrodes weighted ∼11.8 mg cm−1. This ensured that any limitations in the system were the result of the working electrode. Both electrodes were dried under air at 60 °C for 8 hours, the average mass of active material contained in the working electrodes was 3.3 ± 0.2 mg cm−1.
The specific capacitance (Cs, F g−1) was calculated from the CV curves according to the following equation:
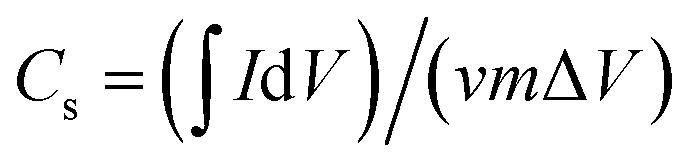
3. Results and discussion
3.1 Morphology, yield, pH, surface area and porosity
Before discussing the experimental results, it is important to indicate how ammonium salts can influence the hydrothermal reaction. These are through: (i) shifting the solution pH, (ii) the presence of reactive NH4+ and, (iii) the catalytic behaviour of the ammonium counter-ion. Each of these factors have been separately shown to influence the reaction. For instance, at low pH the breakdown of the precursor into soluble compounds is promoted, however the formation of solid carbon is suppressed.34,45 This also occurs at high pH,34 with the ideal range for hydrothermal reaction sitting between pH 3 to 7. Introducing NH4+ results in the formation of ammonia and subsequently amines.17 These amines catalyse some of the reaction pathways in hydrothermal carbonization, forming pyrrole and pyridines.17,28,38 Lastly, Ming et al. demonstrated that the presence of Na2SO4 increased the conversion efficiency for glucose from 3.8% to 32.9%, but was only 5% in the presence of NaCl,46 indicating that the anion in salts also has an important role in hydrothermal reactions.The influence of the aforementioned factors can be observed in Table 1, where the yield of hydrochar and solution pH change with different counter-ions. The differences in initial pH are due to how each ammonium salt disassociates in solution, for instance, the ammonium ion disassociates in ammonium chloride via the following equilibrium:
| Cl− + NH4+ + H2O ⇌ NH3 + H3O+ + Cl− |
| 2NH4+ + HPO4 ⇌ NH3 + NH4+ + H2PO4 |
| NH4+ + H2PO4 ⇌ NH3 + H3PO4 |
| Sample | Yield (%) | Yield – AWa (%) | pH initial | pH final | H+ produced (mM) | Average particle diameter (μm) |
|---|---|---|---|---|---|---|
| a Yield remaining after washing with acetone. b Particle size was not calculated as individual particles were not present. | ||||||
| H-Con | 29.6 ± 1.0 | 22.1 ± 0.02 | 5.72 ± 0.02 | 2.35 ± 0.02 | 4.4 ± 0.2 | 4.57 ± 1.15 |
| H-SO | 45.5 ± 0.5 | 44.7 ± 0.02 | 5.36 ± 0.02 | 0.92 ± 0.02 | 121.6 ± 5.6 | 4.12 ± 0.95 |
| H-Cl | 28.8 ± 1.1 | 27.8 ± 0.01 | 5.15 ± 0.01 | 1.12 ± 0.01 | 75.4 ± 1.7 | 4.01 ± 1.77 |
| H-Fe | 42.6 ± 0.7 | 41.2 ± 0.01 | 3.02 ± 0.01 | 1.08 ± 0.03 | 83.2 ± 5.8 | 3.46 ± 1.34 |
| H-PO | 35.2 ± 0.2 | 34.7 ± 0.02 | 8.58 ± 0.02 | 2.44 ± 0.04 | 3.7 ± 0.3 | NAb |
The creation of H2PO4 and H3PO4 has a buffering effect within the solution, increasing the pH. The pH decreased for every sample during hydrothermal carbonization, indicating the formation of acidic species. For saccharides, this tends to be formic, lactic, acetic, propanoic and levulinic acid.30 The addition of (NH4)2SO4, NH4Cl and (NH4)2Fe(SO4)2 lowered the pH to around 1, however this is unlikely to be the result of increased organic acid production as the pKa of the above-mentioned organic acids are from 3.75 to 4.87, indicating that these compounds are likely to be in their protonated form. This is more likely to be due to the formation of H2SO4 and HCl in the liquid as NH4+ is consumed by the carbonization process, leaving free SO42− and Cl− ions.
The observed differences in hydrochar yield are most likely from the ammonium counter ion, and not the influence of NH4+ or the influence of pH. NH4+ concentration was the same for all of the hydrothermal solutions and the pH is similar in H-Cl and H-Fe, but the yield of H-Cl is 28.8% whereas H-Fe is 42.6%. The reason for the increase in yield for H-SO and H-Fe has been ascribed to the presence of SO42− in the solution, which has been shown to accelerate the carbonization process from the saccharide to intermediates.46 Additionally, Ming et al. found that the addition of Na+ and SO42− to the hydrothermal solution in the form of Na2SO4 created aromatic species in the solution that were not observed without salt addition.46 This suggested that Na2SO4 could play a catalytic role in accelerating the conversion of intermediates to carbon materials. However, they did not examine whether these aromatics were present with NaCl or NaNO3 added, thus it is unclear if the effect is from the anion (SO42−) or the cation (Na+). Considering that we have compared the anions in this study and seen that SO42− increases the yield of over Cl and PO43−, this indicates that SO42− is the reason for the increased yield. Additionally, Lu et al., found that the presence of NaCl increased the conversion to intermediates, but not the final yield of carbon material,47 further demonstrating that it is the presence of SO42− and not Na+ that promotes the formation of carbon.
Changing the counter-ion also resulted in morphological differences (Fig. 1). H-Con, H-Cl, H-Fe and H-SO all displayed spherical particles with an average diameter of 3.46 ± 1.34 μm for H-Fe to 4.57 ± 1.15 μm for H-Con, whereas H-PO consisted of a conjoined structure. Interestingly, the incorporation of (NH4)2FeSO4 in H-Fe also resulted in the formation of ‘flower-like’ structures appearing alongside the commonly observed spherical particles.
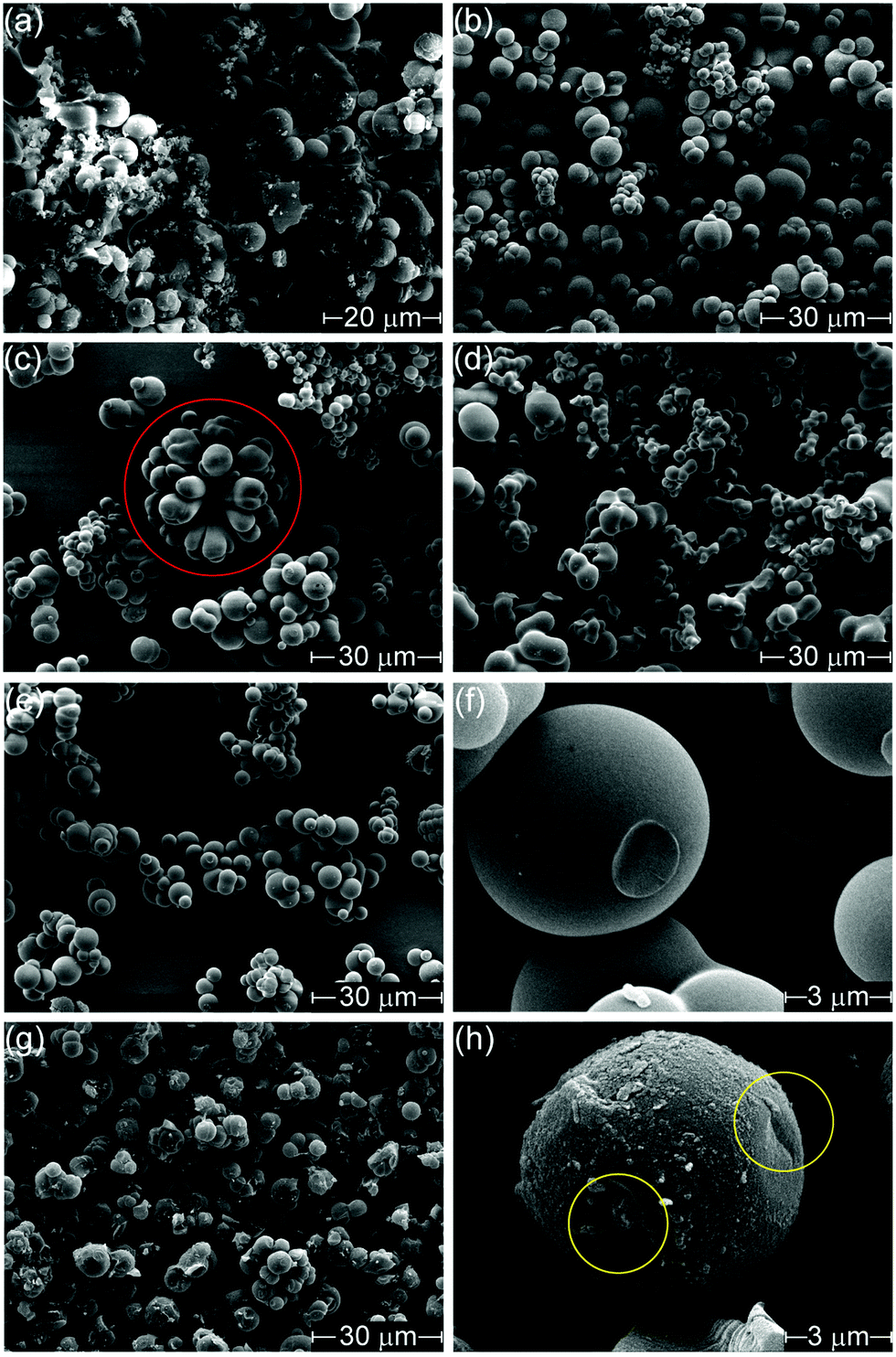 | ||
| Fig. 1 SEM images of (a) H-Con at 4000× (b) H-Cl (c) H-Fe with flower shape outlined in red, (d) H-PO, (e) H-SO, (f) H-SO at 24000×, (g) A-SO and, (h) A-SO at 24000× with removed sections outlined in yellow. Images were taken at 3000× magnification, unless otherwise specified. | ||
The surface area (Table 2) for the non-activated materials was <21 m2 g−1 for all samples, a typical result for hydrothermally derived carbon from saccharides as they contain almost no porosity.
| Sample | BET surface area (m2 g−1) | Micropore surface area (m2 g−1) |
|---|---|---|
| H-Con | 21 | 2.5 |
| H-SO | 3 | 0.5 |
| H-Cl | 16 | 2.7 |
| H-Fe | 3 | 0.6 |
| H-PO | 10 | 3.6 |
| A-Con | 1517 | 320 |
| A-SO | 738 | 172 |
| A-Cl | 1968 | 384.8 |
| A-Fe | 667 | 160.5 |
| A-PO | 2133 | 419.3 |
Activation with H3PO4 consists of two equally important stages, impregnation and carbonization. Increasing the impregnation or carbonization time increases the surface area of the materials, as impregnation time allows H3PO4 to penetrate further into the material and carbonization time allows for micro porosity development.48 The activation process (Fig. 1(g) and (h)) roughened the surface, chipping off sections of the spherical particles and forming small cracks. This can easily be observed by comparing the spherical particle of H-SO in Fig. 1(f) with the activated spherical particle of A-SO in Fig. 1(h). Specific surface area increased in the order of A-Fe (667 m2 g−1), A-SO (738 m2 g−1), A-Con (1517 m2 g−1), A-Cl (1968 m2 g−1) and A-PO (2133 m2 g−1). The differences in surface area suggest that the influence of the anion on the chemical and morphological structure of the hydrothermal carbon has an impact on the activation process. As the impregnation time and carbonization time was kept the same between the samples, it suggests that physicochemical properties that the counter-ion changed had an influence on the ability of H3PO4 to be impregnated into the structure prior to carbonization. Both of the samples that had SO42− ions (H-SO and H-Fe) reported low surface areas, suggesting that although SO42− increases the yield, it is detrimental for H3PO4 activation.
These differences are further highlighted in the pore size distribution date (Fig. 2), where A-Con contains a small distribution of pores between 10–22 Å, A-Cl and A-PO have broader distributions and A-Fe and A-SO have limited porosity development.
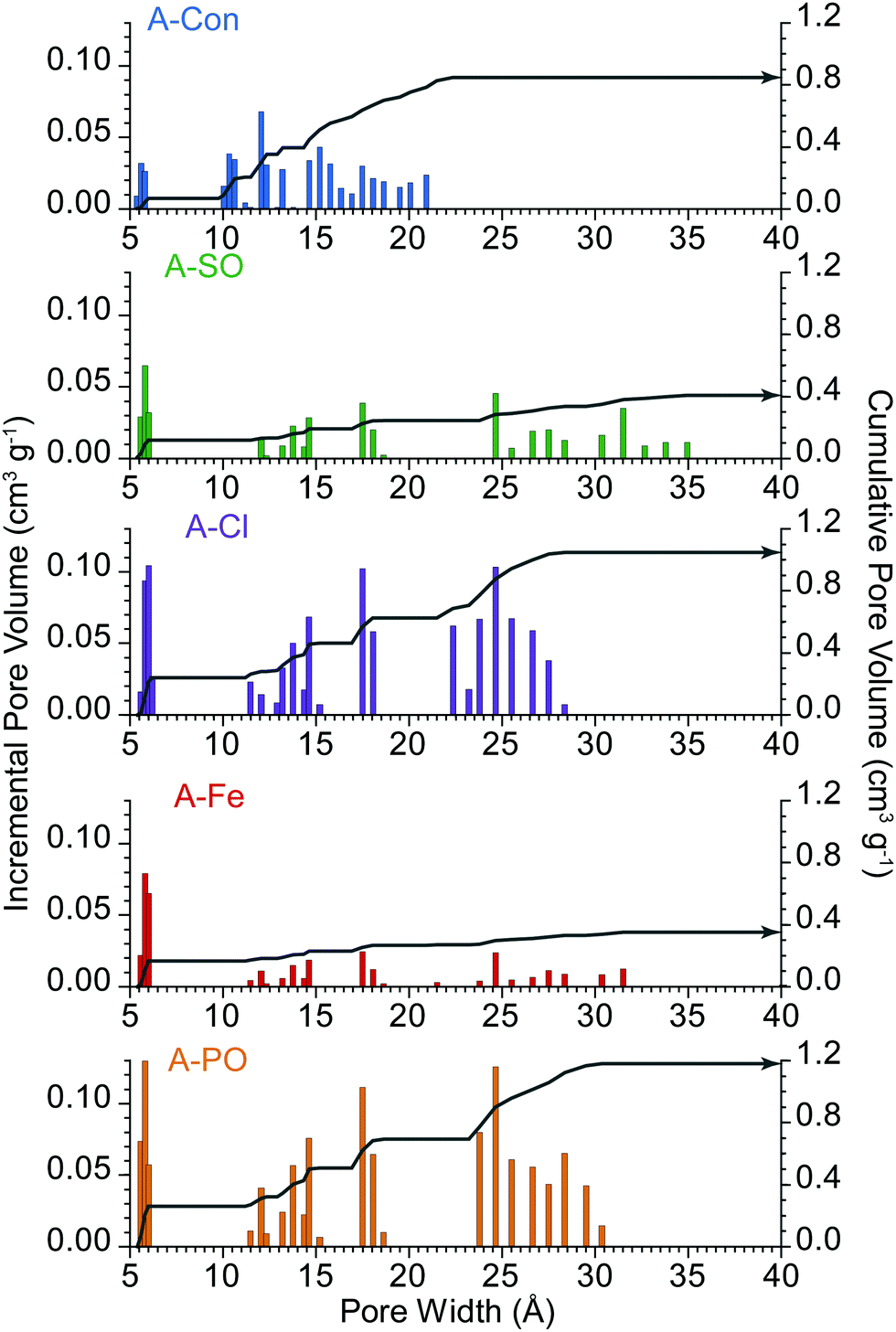 | ||
| Fig. 2 Pore size distribution of the activated materials. Incremental pore volume is displayed via the columns and cumulative pore volume represented by the grey line. | ||
Overall, the differences in surface area and morphology clearly indicate that the counter ion influences the physicochemical properties of the hydrothermal carbon, which subsequently influences the impact that the activation agent has on the carbon structure. The variation here also indicates that not all ammonium salts are beneficial for creating the high surface area carbon materials required in supercapacitors.
3.2 Surface functionality Identified by XPS and NEXAFS
Surface functionality plays a critical role in supercapacitors, with functional groups providing positive and negative benefits depending on their chemistry and electrolyte used.49,50Tables 3 and 4 display the XPS results for the hydrothermal and activated materials, with C1s, O1s and N1s peak deconvolutions. The presence of NH4+ was not detected at 406 eV in the N1s spectra, indicating that the nitrogen groups on the surface are incorporated into the carbon structure.| Carbon | Other | |||||||
|---|---|---|---|---|---|---|---|---|
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C C |
C–C, C–Hx | C–OH | –CO |
–COOH | –CO3 | Total | ||
| H-Con | 23.75 | 28.75 | 17.76 | 7.43 | 2.63 | 1.42 | 81.75 | |
| H-SO | 30.67 | 27.35 | 11.61 | 4.66 | 2.15 | 1.18 | 77.62 | 1.11 S |
| H-Cl | 27.82 | 31.59 | 8.76 | 4.60 | 2.99 | 1.10 | 76.87 | 0.63 Cl |
| H-Fe | 17.35 | 29.05 | 15.77 | 6.88 | 4.36 | 1.44 | 74.83 | 1.17 S |
| H-PO | 24.03 | 35.49 | 11.71 | 3.63 | 2.07 | 0.75 | 77.68 | 0.7 P |
| Nitrogen | Oxygen | |||||||
|---|---|---|---|---|---|---|---|---|
| Pyridinic | Amine | Pyrroles | Quaternary | Total | OC |
O–C | Total | |
| H-Con | 0.00 | 0.00 | 0.00 | 0.00 | 9.92 | 8.36 | 18.28 | |
| H-SO | 1.47 | 2.03 | 1.49 | 0.54 | 5.53 | 9.43 | 6.31 | 15.74 |
| H-Cl | 0.53 | 0.82 | 1.92 | 0.15 | 3.42 | 12.36 | 6.72 | 19.08 |
| H-Fe | 0.46 | 1.24 | 0.70 | 0.52 | 2.92 | 10.74 | 10.33 | 21.07 |
| H-PO | 1.47 | 1.86 | 1.45 | 0.38 | 5.16 | 7.21 | 9.24 | 16.45 |
| Carbon | Phosphorus | |||||||
|---|---|---|---|---|---|---|---|---|
| CC |
C–C, C–Hx | C–OH | –CO |
–COOH | –CO3 | Total | ||
| A-Con | 40.35 | 19.52 | 8.09 | 4.64 | 2.77 | 2.96 | 78.35 | 4.15 |
| A-SO | 31.68 | 22.20 | 8.07 | 5.49 | 3.39 | 3.37 | 74.2 | 2.7 |
| A-Cl | 35.11 | 19.49 | 8.58 | 4.56 | 3.45 | 2.96 | 74.15 | 4.06 |
| A-Fe | 36.13 | 17.79 | 9.46 | 3.90 | 3.04 | 2.71 | 73.02 | 4.85 |
| A-PO | 42.01 | 12.80 | 9.71 | 3.95 | 3.18 | 3.07 | 74.72 | 3.69 |
| Nitrogen | Oxygen | |||||||
|---|---|---|---|---|---|---|---|---|
| Pyridinic | Amine | Pyrroles | Quaternary | Total | OC |
O–C | Total | |
| A-Con | 7.32 | 10.19 | 17.5 | |||||
| A-SO | 1.45 | 0.85 | 2.24 | 0.77 | 5.31 | 6.38 | 11.23 | 17.79 |
| A-Cl | 0.74 | 0.22 | 1.30 | 0.77 | 3.02 | 10.09 | 7.43 | 17.52 |
| A-Fe | 1.28 | 0.53 | 1.92 | 0.45 | 4.18 | 8.67 | 9.29 | 17.96 |
| A-PO | 0.61 | 0.28 | 1.10 | 0.39 | 2.37 | 12.32 | 6.90 | 19.22 |
The N-doped carbons contained less oxygen surface functionalities than H-Con, a trend that is attributed to the occurrence of Maillard reactions forming various nitrogen groups instead of furans in the hydrothermal reactor.17,28 The type of counter ion also influenced the amount of nitrogen present on the surface. For instance, (NH4)2HPO4 achieved the highest incorporation at 5.16 at% (H-PO), while (NH4)2FeSO4 produced the lowest at 2.92 at% (H-Fe). Additionally, the ratio of nitrogen groups varies based on the counter ion suggesting a nitrogen functionality can be tuned via changing the counter ion. This can be seen by looking at the concentrations of each group between the samples, but is more easily demonstrated if the concentrations are converted into a ratio based on one of the groups. For instance, normalizing to pyridinic groups, the ratio between functionalities in H-SO is 1pyridinic:1.37amine:1.01pyrrolic:0.36quaternary, whereas H-Cl is 1pyridinic:0.67amine:1.60pyrrolic:0.13quaternary. Thus, Cl− produces significantly less amine groups on the surface but a higher number of pyrroles than SO42−. The normalized results for each sample is presented in the ESI† for reference. Sample H-Cl is particularly promising as the formation of amine groups is suppressed compared to pyrroles and pyridinic groups. Amine groups are undesirable for supercapacitors, as these groups are potentially more likely (less energy required) to be cleaved from the surface compared to ring nitrogen, leading to cell degradation under cycling.
Oxygen group distribution was also impacted by the ammonium anion. Normalizing to C–O– groups, H-Con has a ratio of 1C–O–:0.41CO:0.14COO−:0.08CO3 in the C1s peak, whereas H-Cl was 1C–O–:0.52CO:0.32COO−:0.12CO3 and H-PO was 1C–O–:0.31CO:0.17COO−:0.06CO3. Again, Cl− has produced a more theoretically ideal surface, with a lower ratio of cleavable C–O– groups relative to potentially (electrochemically) reversible CO functionalities.50
Duel heteroatom doping has also occurred, with the counter ion being incorporated into the structure in H-SO (1.11 at% S), H-Cl (0.63 at% Cl), H-Fe (1.17 at% S) and H-PO (0.7 at% P). Despite (NH4)2HFeSO4 containing three potential dopants (N, S and Fe), only N and S were detected on the surface of H-Fe. This was unexpected, as Fe has been previously incorporated hydrothermally using Fe(NO3)3·9H2O.51 However, that study incorporated a series of pre-treatment steps that were not performed here, such as soaking in a Fe(NO3)3·9H2O solution and pH adjustment, which might be critical for Fe incorporation into hydrothermal carbon.
The XPS results indicate that changing the counter ion has a substantial influence on the surface functionality and can provide another method to tune the surface of hydrothermal carbon for various applications. However, the tunability provided from the counter ion may be less important if the tuning is not carried out after activation. Fortunately, activation with H3PO4 actually enhances the benefits already provided from using different anions. For instance, the ratio of nitrogen groups in A-Cl is 1:0.29:1.75:1.03 (H-Cl is 1:0.67:1.59:0.13) for pyridinic, amine, pyrrolic and quaternarty, indicating an increase in the ratio of benificial pyrrolic, pyridinic and quaternary nitrogen groups during activation. Amine groups are reduced in all of the samples during activation, leaving a distribution of quaternary, pyrrolic and pyridinic functionalities. The activation step also incorporated between 2.7 to 4.85 at% phosphorus onto the surface, leaving the surface with oxygen, nitrogen and phosphorus functionalities.
While XPS is a robust method for quantifying elemental concentration on carbon surfaces, deconvolution is necessary to obtain quantitative information about the functional groups associated with each elemental peak. This can be problematic when a single broad peak is obtained that overlaps several functional group regions,38 making it unclear if the peak represents a single functionality or multiple functional groups. Thus, a secondary technique is required to confirm the presence of these functional groups. FTIR is commonly utilized for this reason, however nitrogen and oxygen functional groups can be difficult to differentiate with FTIR. Therefore we have used carbon K-edge and nitrogen K-edge NEXAFS to confirm the functionalities indicated by XPS and probe the surface of these materials further.
The carbon K-edge contains two easily observable differences between H-Con and the nitrogenated hydrothermal carbons (Fig. 3(a)) at 286.5 eV and 288.5 eV. The peak at 286.5 eV is associated with the furanic structure of hydrothermal carbon and its intensity decreases in the nitrogenated materials. Previous work established that nitrogen reduces the number of furan structures in the hydrothermal carbon structure, reducing the intensity of the peak at 286.5 eV.17 The same effect is clearly being replicated here with each ammonium salt.
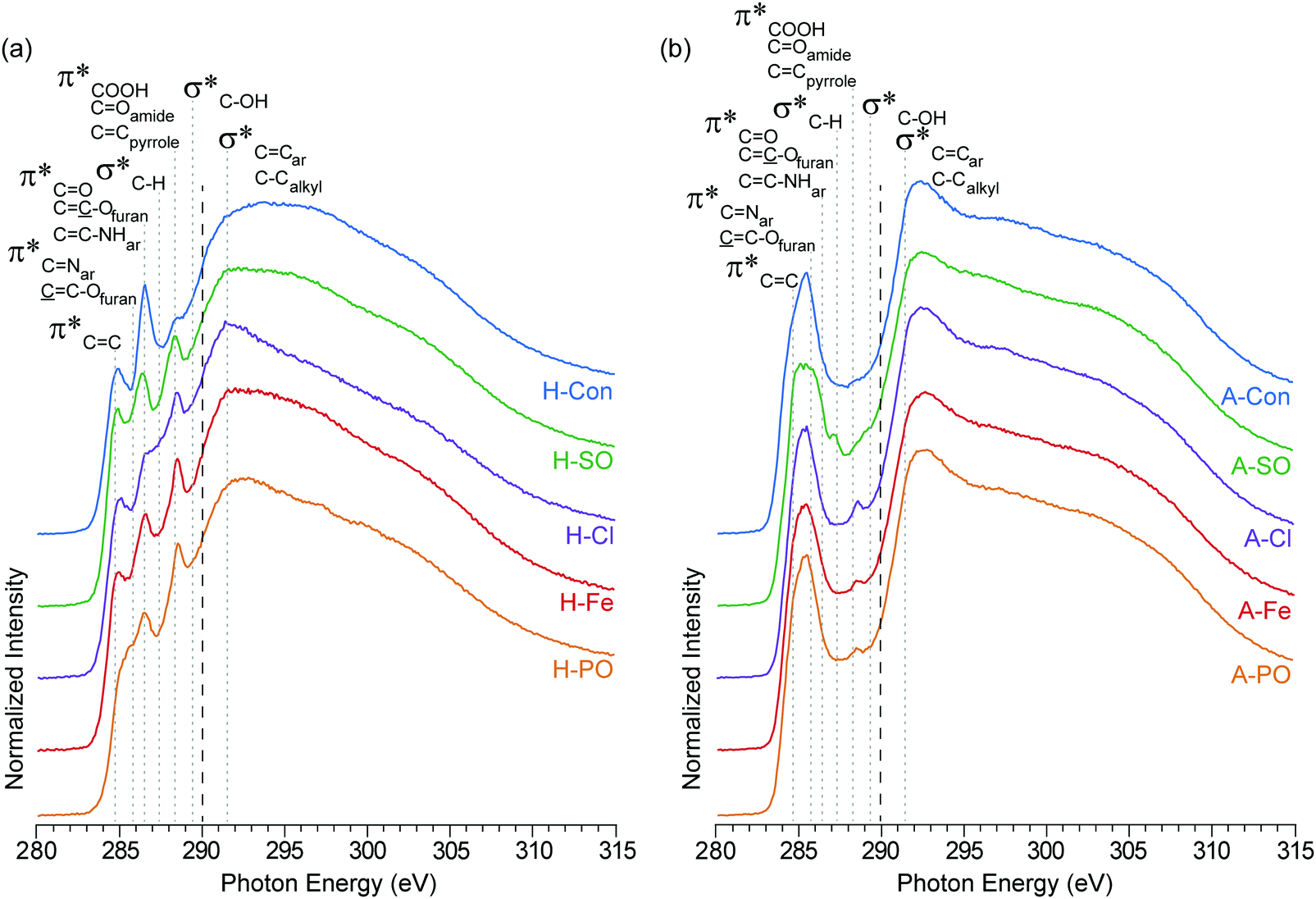 | ||
| Fig. 3 NEXAFS carbon K-edge spectra of (a) hydrothermal carbons (H-) and (b) activated hydrothermal carbons (A-). Functional group locations for the main C1s → π* and C1s → σ* are added for reference and based on previous NEXAFS work on hydrothermal carbons.17 The long dashed lines represent the position of the arctangent step function at 290.0 eV. | ||
The second difference at 288.5 eV, associated with C1s → π* transitions on COOH, COamides, and CCpyrroles,52 displays an observable increase in intensity for all of the nitrogenated carbons compared to H-Con. As the concentration of COOH groups does not vary significantly in the XPS, the increased peak height at 288.5 eV is attributed to the presence of amides and pyrroles. These groups can also be observed in the nitrogen K-edge spectra (Fig. 4) at 401.3 eV, confirming their presence. Overall, the carbon K-edge indicates that the hydrothermal N-doped materials have a similar carbon structure with variations in the concentration of functional groups, further demonstrating that tuning the surface functionality is possible through changing the anion.
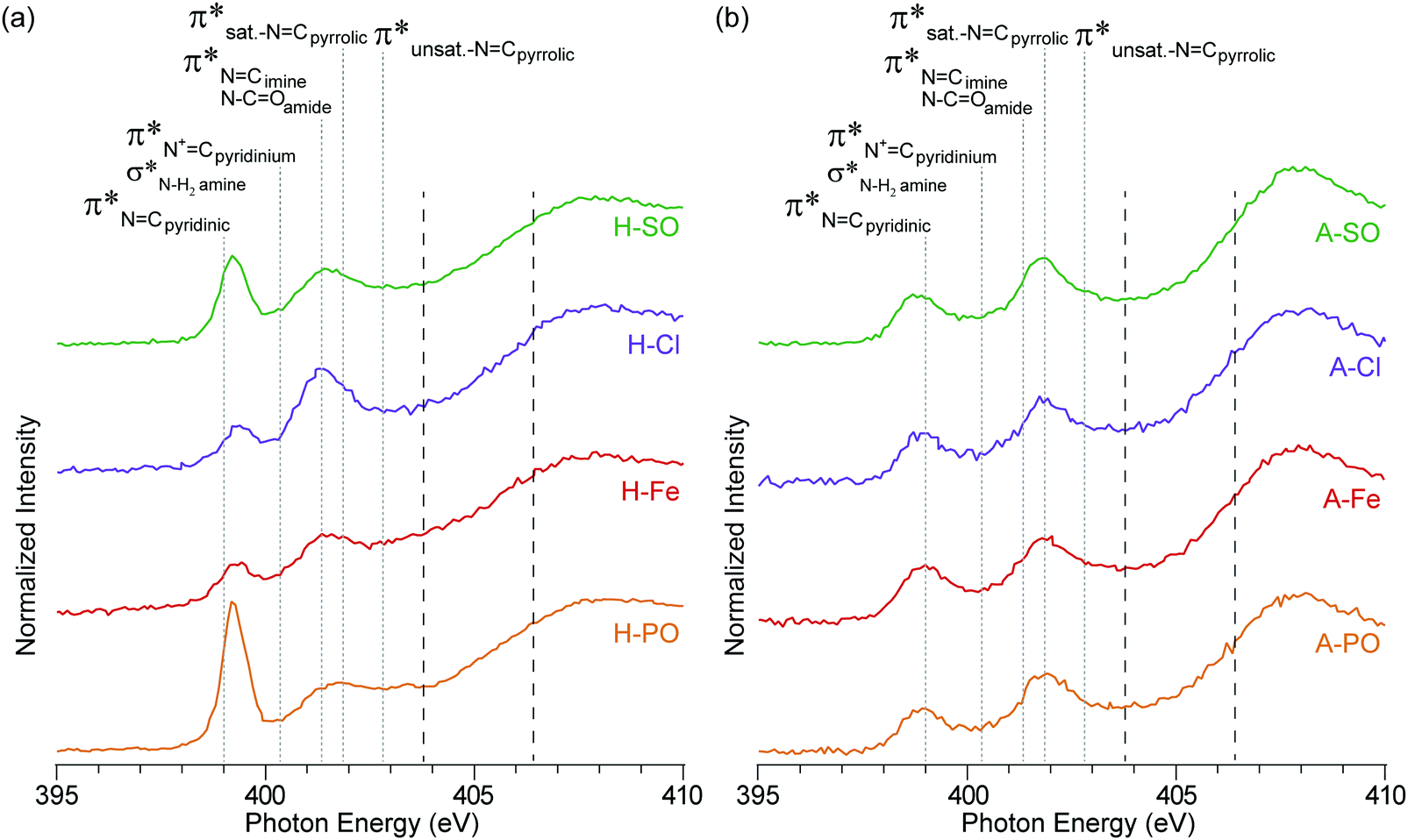 | ||
| Fig. 4 NEXAFS nitrogen K-edge spectra of (a) hydrothermal carbons and (b) activated hydrothermal carbons. Functional group locations for the main N1s → π* and N1s → σ* are added for reference and based on previous NEXAFS work on hydrothermal carbons.17 The long dashed lines represent the position of the arctangent step function at 403.9 eV and 406.5 eV. | ||
After activation with H3PO4, the sharp peaks associated with oxygen/nitrogen functionalities between 286.5 to 290 eV are almost completely removed, while peaks associated with aromatic groups increase. The loss of oxygen functionality and increased aromatic concentration after activation is a common occurrence, which is escalated with increasing temperature.42 What is interesting is the peak at 288.5 eV (COOH, COamides, and CCpyrroles52) is still present in the N-doped carbons, but not in A-Con. This suggests that pyrroles have survived the activation process, as they are also observed in the nitrogen K-edge spectra (Fig. 4) at 401.3 eV.
In addition to pyrrolic groups (401.3 eV), the nitrogen K-edge spectra also indicates the presence of pyridinic groups (399 eV). The first peak at 399 eV exclusively occurs from CN N1s → π* transitions on pyridinic groups, whereas the second peak is related to a combination of N1s → π* transitions on imine (401.3 eV), amide (401.3 eV) and pyrrolic groups (401.9 eV). In H-PO a third peak can be also observed at 402.9 eV, related to unsaturated NCpyrrolic structures.
The influence of the counter ion is again displayed here, with a clear difference in peak heights for the pyridinic groups at 399 eV (PO43− > SO42− > Cl− > Fe2+ + SO42−) and pyrrolic groups at 401.3 eV (Cl− > SO42− > PO43− > Fe2+ + SO42−). The differences in intensity here between the samples match the concentration differences in the XPS results.
The intensity of the nitrogen peaks decreased after activation, indicative of nitrogen loss, although the degree of nitrogen loss was not equally shared across all nitrogen functionalities. Pyridinic groups, especially in A-PO and A-SO, lost considerable intensity after activation, compared to pyrroles. This difference could stem from the weakly basic nature of pyridine, which would have accepted a proton from H3PO4 and increased its reactivity. From a previous study pyridinic groups appeared to also be impacted more by temperature than pyrrolic.42 This loss was further escalated when H3PO4 was used as an activation agent.
3.3 Electrochemical analysis
To understand if the counter ion influences electrochemical performance, the activated samples underwent cyclic voltammetry and SPECS analysis. KOH was selected as the electrolyte due to it being utilized commonly in the literature allowing comparison and its theoretical ability to allow nitrogen based pseudocapacitance reactions.38O and then quinone with an overall loss of oxygen from the surface from 26.77 at% to 14.85 at%.53 The surface of A-PO has the highest oxygen content (19.22 at%), with the highest number of CO groups (12.32 at%) and the lowest nitrogen content. However, the CV's in He et al.'s work did not show the shifting redox peaks that are observed in the CV of A-PO over 1000 cycles (Fig. 6). Unfortunately, it is unclear what functional groups is associated with these redox peaks or if these peaks have been observed previously as studies on supercapacitors typically don’t report failures. However, it does further demonstrate that the ammonium counter ion appears to have an impact, although negative, on the electrochemical performance of this material.
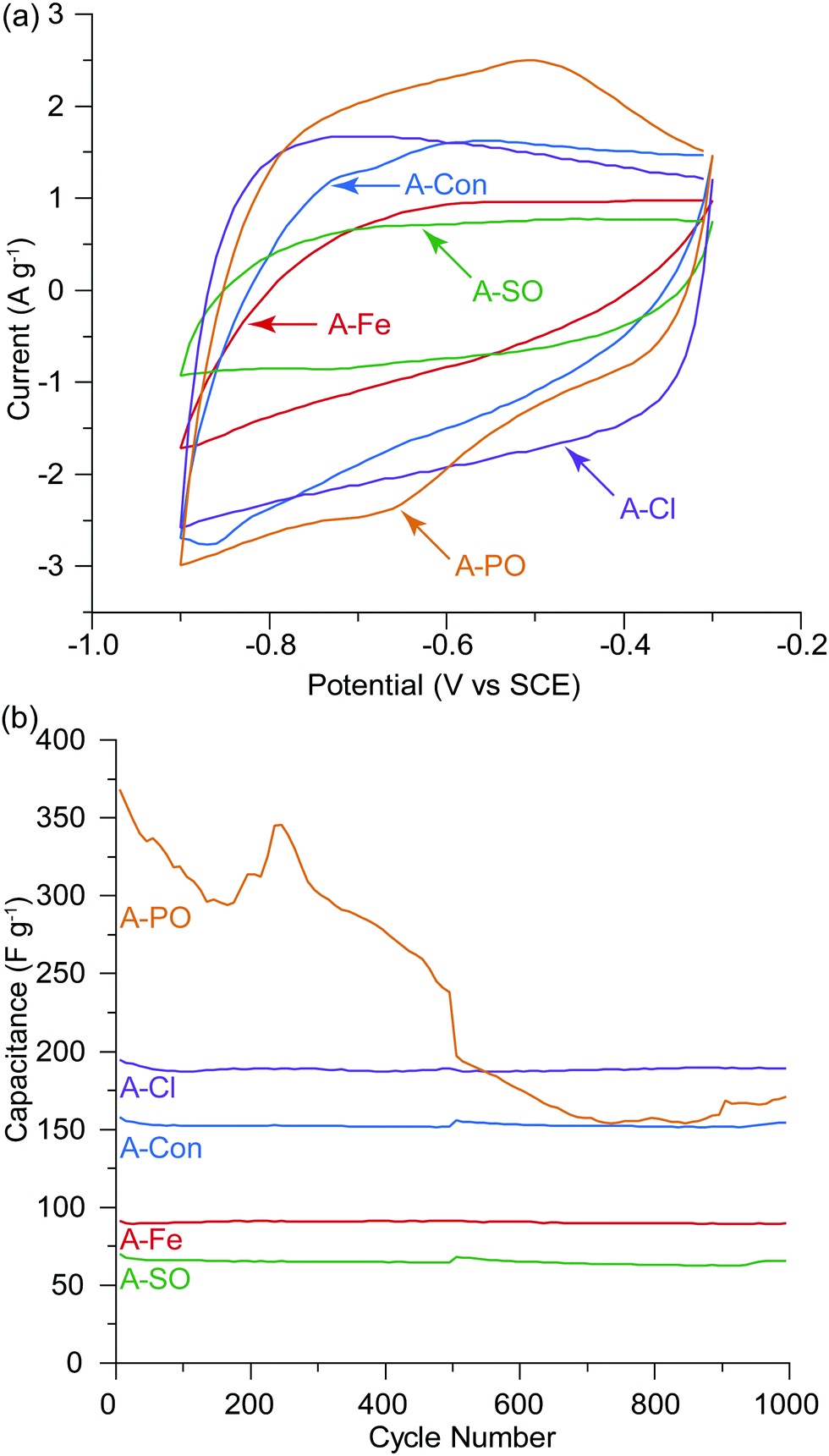 | ||
| Fig. 5 (a) Cyclic voltammograms at the 500th cycle before the SPECS experiment, (b) capacitance per cycle over 1000 cycles, 10 mV s−1 in 1 M KOH. The SPECS experiment was conducted at the 500th cycle, hence the slight discontinuity in the capacitance at cycle 500. Decomposition of the electrode/electrolyte was observed for A-PO. | ||
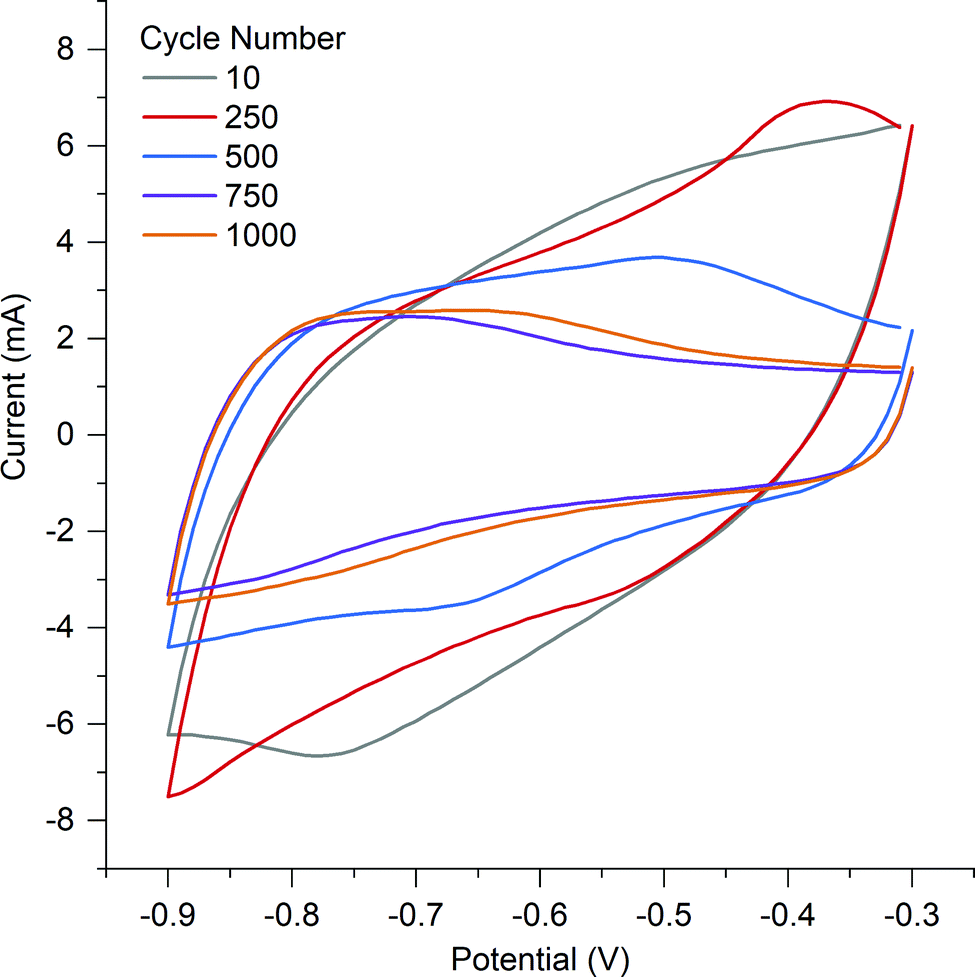 | ||
| Fig. 6 Cyclic voltammetry of A-PO at different cycles. | ||
A-Cl provided the highest capacitance and cycle stability (194.7 F g−1), followed by A-Con (155.4 F g−1), A-Fe (89.8 F g−1) and A-SO (67.7 F g−1). The results for A-Cl and A-Con are in the same range of a study by Misnon et al. using 1 M KOH, where they achieved from 110 to 210 F g−1 for their activated carbon from oil palm kernel.54 Apart from A-PO which has a clearly unstable surface characteristics, it is difficult to see the influence of the counter ion or nitrogen in a CV as the capacitance stored in the electrical double later is so large. Thus, SPECS was employed to break down the different capacitance contributions.
surface), as well as the surface of the pores in the bulk of the electrode (DL2pores). This method has been used to differentiate between different charge storage mechanisms and to characterize the kinetic behavior of the material or electrode under study over a full range of sweep rates. Further information on this method can be found in these studies.43,44
The advantage of the SPECS method is that a synthetic voltammogram for each charge storage mechanism can be generated, since the current response for each potential step has been deconvoluted into its individual components.43,44Fig. 7(a) and (b) show the synthetic voltammograms for A-Con sample at the cycle rates of 5 and 50 mV s−1 respectively. As expected, the average current increases with an increasing sweep rate. It can be seen that the synthetic voltammogram for the capacitive current at the porous surface area is greater than the capacitive current at the geometric surface area at the low sweep rate of 5 mV s−1, as at lower sweep rate there is more time for ions to cover the surface of inner pores. While at higher sweep rate the contribution of geometric capacitive process (DL1surface) is greater than the capacitive process at the porous surface area (DL2pores). Fig. 7(a) and (b) also indicate that overall the capacitive processes are the dominant charge storage mechanisms and diffusion limited processes have a slight contribution in charge storage mechanisms. This is due to the nature of carbon-based electrode materials, which mainly store charge through the capacitive processes. The origin of the diffusion limited processes in carbon-based electrodes is from diffusion into restrictive pores.
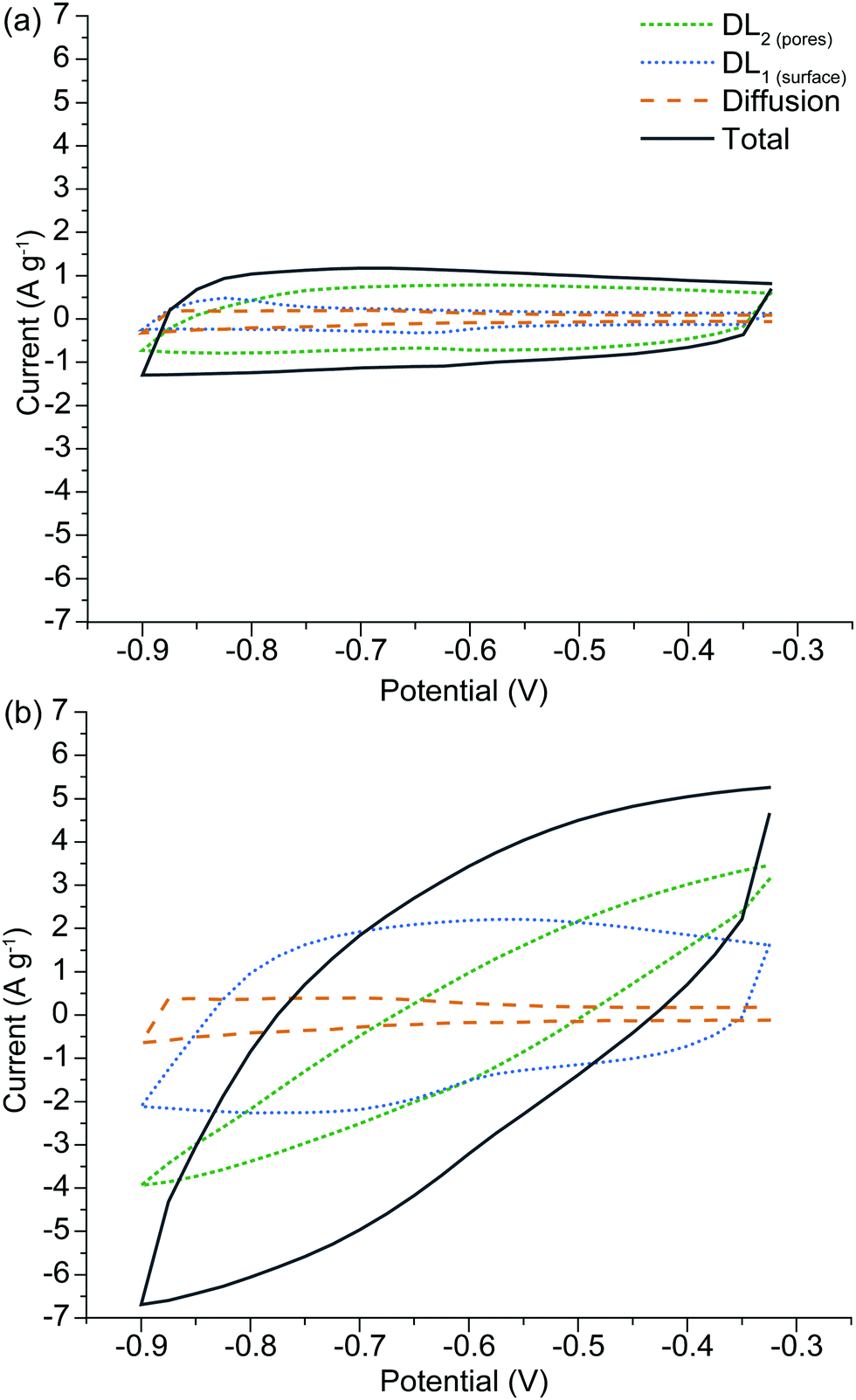 | ||
| Fig. 7 Synthetic voltammogram obtained from the SPECS data for A-H, comparing behavior at different sweep rates of (a) 5 mV s−1 and (b) 50 mV s−1. | ||
The synthetic voltammograms generated from the SPECS analysis can also be used to determine the specific capacitance as a function of cycle rate. The added benefit here is that because the SPECS data was deconvoluted into individual processes, from which the contributions made by each process to the overall voltammogram have been calculated, the capacitance of the individual processes can be calculated vs cycle rate. Fig. 8 shows the total specific capacitance and the specific capacitance of the geometric (DL1surface), porous (DL2pores) surfaces, the diffusion limited capacitance, and the residual capacitance, obtained from the SPECS data as a function of the sweep rate. A-PO has not been included in the SPECS analysis as the material was unstable under cycling.
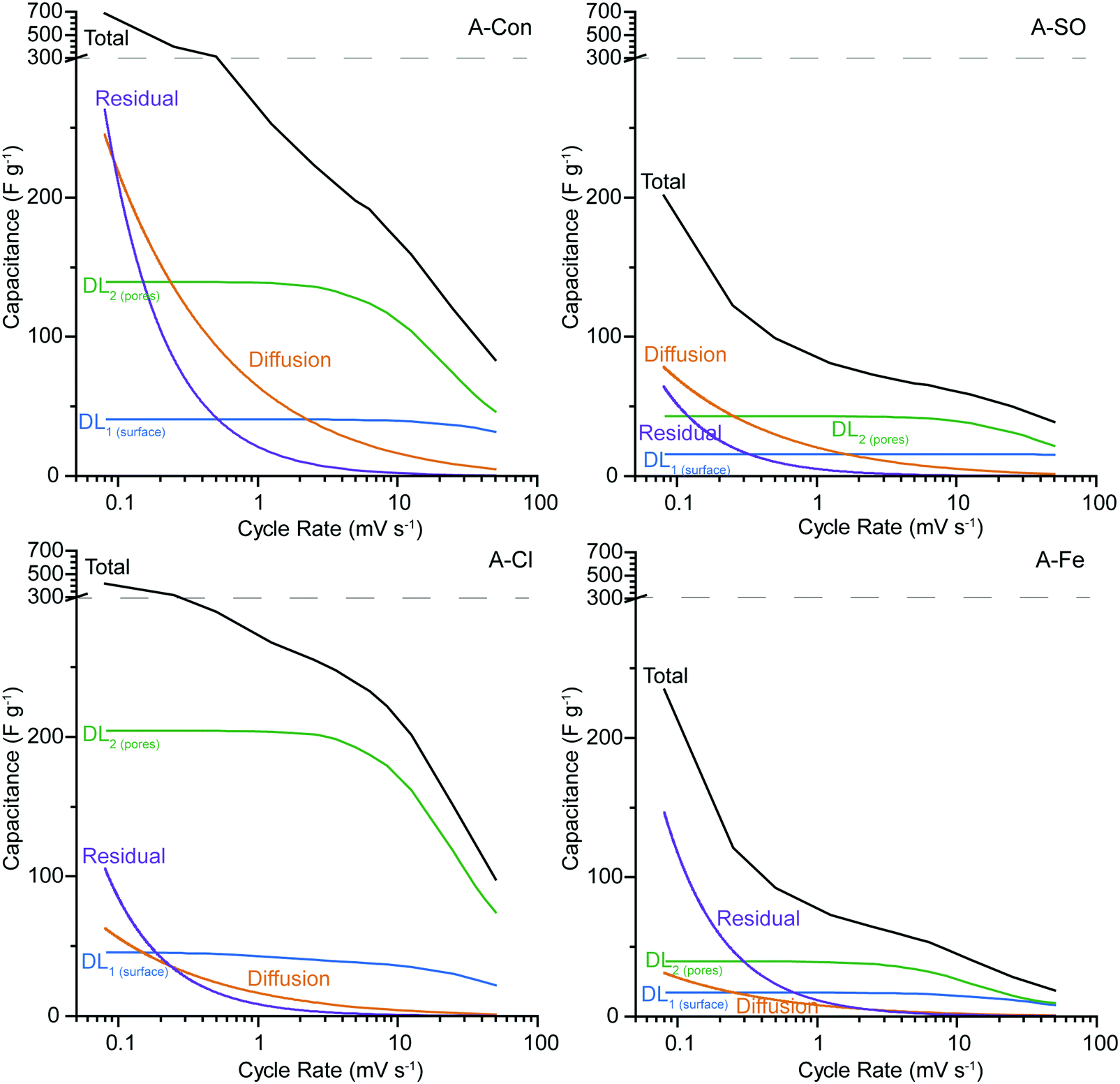 | ||
| Fig. 8 SPECS analysis for (a) A-Con, (b) A-SO, (c) A-Cl, and (d) A-Fe. The y-axis has been split at 300 F g−1 (dashed line) to allow easier comparison between samples. A-PO was not included as it was unstable under cycling. | ||
In all four samples, DL1surface displays limited change at different cycle rates, due to being associated with the geometric surface area and thus has no ion transport limitations. The higher capacitance for DL1surface in A-Con and A-Cl is likely due to having a higher geometric surface area for the material. The contribution from DL2pores follows the specific surface area, with higher surface areas producing a larger contribution from DL2pores, simply due to having higher total pore volume. However, it is important to consider the pore size distribution (Fig. 2) and how that impacts on the capacitance contribution from DL2pores and the diffusion limited capacitance.
The bare ion radii of K+ is 1.33 Å (d = 2.66 Å) and the solvated ion radii is 3.31 Å (d = 6.62 Å),55 therefore only a single solvated ion will fit in the pores between 3 to 6 Å. Considering that the SPECS experiments were performed in an aqueous electrolyte, it is likely that all of the ions are in their solvated state, unlike in organic and ionic liquids where desolvated ions are more common because of their weaker polarity (solvent) and charge density (ions). Despite the solvated ion fitting into these smaller pores, this restrictive environment reduces the likelihood that an ion will be pushed into these pores. Thus, only at lower cycle rates when there is sufficient time for an ion to travel down these pores do they become active. This is the reason why DL2pores starts to plateau at lower sweep rates, as the double layer is accessing the maximum surface area available to it at this point. It is also at this point that diffusion limited capacitance starts to increase significantly, as it represents the mass transport of ions into increasingly smaller pores. The higher level of diffusion and lower DL2pores in A-Con compared to A-Cl is directly related to its porosity, with almost 65% of the porosity in A-Con located in pores smaller than 15 Å, compared to A-Cl where only 44% is located in this region. This is further demonstrated when examining the ratio between the capacitance contributions of DL1surface and DL2pores for A-Con and A-Cl. As DL1surface is almost identical in A-Con and A-Cl, we can consider that the geometric surface is similar in both materials, however in A-Con the capacitance from DL2pores is about 1.5 times higher at 50 mV s−1, whereas it is 3.3 times higher in A-Cl at 50 mV s−1. Even at lower sweep rates, where smaller pores will become accessible, A-Cl still has a higher ratio (5.0 at 5 mV s−1) than A-Con (3.2 at 5 mV s−1). Thus, the surface of A-Cl is far more accessible than A-Con.
The residual current and diffusion are higher in A-Con compared to the nitrogen doped materials, suggesting that this surface is a lot more electrochemically active at slower scan rates. The residual represents kinetically slow redox processes that are ongoing beyond the duration of each potential step. These processes or electrode instabilities become apparent after the electrode is left to rest at a fixed potential for an extended period of time. Thus, the residual can be considered to be similar to a floating test across the full potential window. Thus, the combination of a more electrochemically active surface (higher residual) and restricted pores (higher diffusion) results in the total performance of A-Con being higher at cycle rates <1 mV s−1 than the nitrogen doped materials. However, if the residual current is not zero, then there are potential stability issues with the system. Thus, A-Con is likely to fail earlier than A-Cl due to these instabilities in the electrode surface.
Examining whether the incorporation of nitrogen into these materials has enhanced their performance is difficult, as the surface area and porosity is vastly different between each of the ammonium salt added materials and the control. What is observed here is that the ammonium counter ion has an influence on the hydrothermal reaction, which subsequently influences the ability for the hydrothermal carbon to increase its surface area under activation and these impacts are displayed in the electrochemistry. However, it is interesting that again we have a nitrogen doped material that does not display clear redox peaks in the CV, which seemingly contradicts the literature which continuously states that nitrogen introduces pseudocapacitance through redox active sites. In fact studies examining nitrogen doped carbon materials don’t appear to display any clear redox behaviour in their cyclic voltammetry data,16,20,56 unlike the electrochemical redox behaviour of the quinone group which is easily observable.57 What might be occurring here is that nitrogen is providing a localised charge storage site on the surface of the material, which is electrically connected to the carbon and the electrical double layer. Thus, a specific redox peak will not be observable. Additionally, the benefit of nitrogen may come from its other suggested properties, such as wettability,5 electrode stability and lower resistance.7
To examine if nitrogen doping does indeed have an impact on the resistance, electrochemical impedance spectroscopy (EIS) was used to examine A-S, A-SO, A-Cl and A-Fe with the Nyquist plot presented in Fig. 9. Each of the samples displayed the typical shape of a semicircle followed by a tail. Instead of fitting each of the EIS to RC circuits, a physical interpretation of the plot was performed based on the examination of EIS for supercapacitors by Mei et al.58 The electrode resistance is represented as the distance between 0 and the first data point on the Re(Z) axis and the bulk electrolyte resistance is represented as the diameter of the semicircle. The sum of these two properties represents the internal resistance of the electrode. Calculating the internal resistance resulted in the nitrogenated materials having a slightly lower resistance (A-SO 13.77 Ω, A-Cl 15.72 Ω, A-Fe 14.01 Ω) than A-Con (16.82 Ω), indicating that the suggestion that nitrogen improves conductivity is likely to be valid here as well.
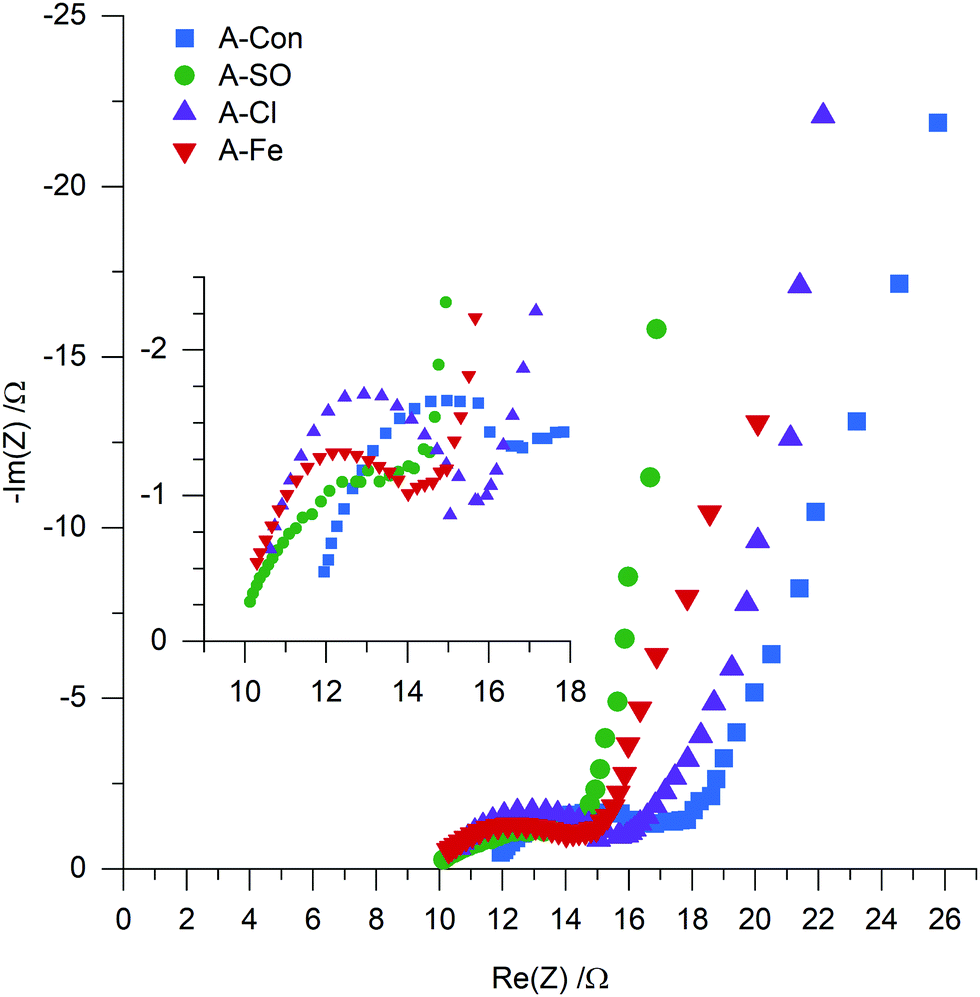 | ||
| Fig. 9 EIS of A-Con, A-SO, A-Cl and A-Fe. | ||
These benefits could be the reason why the residual current is far lower in the nitrogen doped materials compared to A-Con, as the incorporation of nitrogen creates a more stable electrochemical surface. Overall, further examination of how nitrogen contributes to capacitance through the examination of pseudocapacitance, conductivity, wettability and electrochemical testing is needed to understand its impact.
4. Conclusions
In this work we examined how the anion in ammonia influences the hydrothermal reaction, nitrogen incorporation levels, physicochemical properties, ability to be activated and supercapacitor performance. Morphologically, SO42− and Cl− created spherical particles between 1–5 mm, while PO43− consisted of conjoined spherical particles and Fe((SO4)2)2+ consisted of spherical and ‘flower like’ structures. Chemically, the degree of nitrogen incorporation varied between 2.92 at% for Fe((SO4)2)2− to 5.53 at% for SO42−, as well as the ratio between the pyridinic, pyrrolic, amine and quaternary nitrogen groups that are formed when introducing ammonia into the hydrothermal reaction. The nitrogen K-edge NEXAFS results indicated that PO43− and SO42− created a higher level of pyridinic groups, which were subsequently removed upon activation with H3PO4. This was stipulated to be caused by the weakly basic nature of pyridinic groups compared to pyrrolic groups, making them more susceptible for attack with H3PO4. The surface area after activation also varied dramatically from 667 m2 g−1 for Fe((SO4)2)2− to 2133 m2 g−1 for PO43− with the pore size distribution widening in the carbon that were synthesized in the presence of ammonium salts. Supercapacitor performance followed surface area, however, the highest surface area material produced from PO43− was unstable under cycling, losing over half of its capacitance over the first 500 cycles. The SPECS analysis revealed that the addition of ammonium chloride to the hydrothermal solution created a hydrothermal carbon that produced a beneficial pore distribution after activation, with a higher degree of the surface accessible to the electrolyte than the material without salt addition. SPECS also revealed that although the ammonium salt added materials displayed lower pseudocapacitance contributions, the surface was considerably more stable than the material without salt addition. Overall, this study demonstrates that the ammonium counter ion influences the pH, hydrothermal yield, particle size and degree of nitrogen incorporation into the hydrothermal carbon. These impacts flow through into the activated carbon, with the most prominent being the difference in surface area after activation. As surface area is one of the major factors determining supercapacitor performance, selecting the correct counter-ion is crucial to enhancing the electrochemical performance of nitrogen doped hydrothermal carbons for supercapacitors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
KGL acknowledges the University of Newcastle for a PhD scholarship. This research was undertaken on the Soft X-ray beamline at the Australian Synchrotron, part of ANSTO. The XPS work was performed in part at the Materials Node of the Australian National Fabrication Facility, a company established under the National Collaborative Research Infrastructure Strategy to provide nano and microfabrication facilities for Australia's researchers.References
- T. Liu, F. Zhang, Y. Song and Y. Li, Revitalizing carbon supercapacitor electrodes with hierarchical porous structures, J. Mater. Chem. A, 2017, 5(34), 17705–17733 RSC.
- Y. Ji, Y. Deng, F. Chen, Z. Wang, Y. Lin and Z. Guan, Ultrathin Co3O4 nanosheets anchored on multi-heteroatom doped porous carbon derived from biowaste for high performance solid-state supercapacitors, Carbon, 2020, 156, 359–369 CrossRef CAS.
- Y. Deng, Y. Xie, K. Zou and X. Ji, Review on recent advances in nitrogen-doped carbons: preparations and applications in supercapacitors, J. Mater. Chem. A, 2016, 4(4), 1144–1173 RSC.
- J. P. Paraknowitsch and A. Thomas, Doping carbons beyond nitrogen: an overview of advanced heteroatom doped carbons with boron, sulphur and phosphorus for energy applications, Energy Environ. Sci., 2013, 6(10), 2839 RSC.
- K. Terpiłowski, A. E. Wiącek and M. Jurak, Influence of nitrogen plasma treatment on the wettability of polyetheretherketone and deposited chitosan layers, Adv. Polym. Technol., 2018, 37(6), 1557–1569 CrossRef.
- S. C. H. Kwok, J. Wang and P. K. Chu, Surface energy, wettability, and blood compatibility phosphorus doped diamond-like carbon films, Diamond Relat. Mater., 2005, 14(1), 78–85 CrossRef CAS.
- Y. Taluja, B. SanthiBhushan, S. Yadav and A. Srivastava, Defect and functionalized graphene for supercapacitor electrodes, Superlattices Microstruct., 2016, 98, 306–315 CrossRef CAS.
- Y. Zhang and M. Antonietti, Photocurrent generation by polymeric carbon nitride solids: an initial step towards a novel photovoltaic system, Chem. – Asian J., 2010, 5(6), 1307–1311 CrossRef CAS.
- J. Biemolt, I. M. Denekamp, T. K. Slot, G. Rothenberg and D. Eisenberg, Boosting the Supercapacitance of Nitrogen-Doped Carbon by Tuning Surface Functionalities, ChemSusChem, 2017, 10(20), 4018–4024 CrossRef CAS.
- Y. W. Chi, C. C. Hu, H. H. Shen and K. P. Huang, New Approach for High-Voltage Electrical Double-Layer Capacitors Using Vertical Graphene Nanowalls with and without Nitrogen Doping, Nano Lett., 2016, 16(9), 5719–5727 CrossRef CAS.
- C.-F. Liu, Y.-C. Liu, T.-Y. Yi and C.-C. Hu, Carbon materials for high-voltage supercapacitors, Carbon, 2019, 145, 529–548 CrossRef CAS.
- Y. Xia and R. Mokaya, Synthesis of Ordered Mesoporous Carbon and Nitrogen-Doped Carbon Materials with Graphitic Pore Walls via a Simple Chemical Vapor Deposition Method, Adv. Mater., 2004, 16(17), 1553–1558 CrossRef CAS.
- F. Wu, C. Wang, H.-Y. Hu, M. Pan, H.-F. Li, N. Xie, Z. Zeng, S. Deng, M. H. Wu, K. Vinodgopal and G.-P. Dai, Controlled synthesis of N-doped carbon microspheres from melamine-based carbon by chemical vapor deposition, Chem. Phys. Lett., 2019, 730, 124–129 CrossRef CAS.
- T. V. Pham, J. G. Kim, J. Y. Jung, J. H. Kim, H. Cho, T. H. Seo, H. Lee, N. D. Kim and M. J. Kim, High Areal Capacitance of N-Doped Graphene Synthesized by Arc Discharge, Adv. Funct. Mater., 2019, 29(48), 1905511 CrossRef CAS.
- F. Liu, Z. Wang, H. Zhang, L. Jin, X. Chu, B. Gu, H. Huang and W. Yang, Nitrogen, oxygen and sulfur co-doped hierarchical porous carbons toward high-performance supercapacitors by direct pyrolysis of kraft lignin, Carbon, 2019, 149, 105–116 CrossRef CAS.
- S. Li and Z. Fan, Nitrogen-doped carbon mesh from pyrolysis of cotton in ammonia as binder-free electrodes of supercapacitors, Microporous Mesoporous Mater., 2019, 274, 313–317 CrossRef CAS.
- K. G. Latham, M. I. Simone, W. M. Dose, J. A. Allen and S. W. Donne, Synchrotron based NEXAFS study on nitrogen doped hydrothermal carbon: Insights into surface functionalities and formation mechanisms, Carbon, 2017, 114, 566–578 CrossRef CAS.
- F. Schipper, S. Kubo and T.-P. Fellinger, Nitrogen-doped porous carbon via ammonothermal carbonization for supercapacitors, J. Sol-Gel Sci. Technol., 2018, 89(1), 101–110 CrossRef.
- M. M. Titirici and M. Antonietti, Chemistry and materials options of sustainable carbon materials made by hydrothermal carbonization, Chem. Soc. Rev., 2010, 39(1), 103–116 RSC.
- Z. Xu, J. Chen, X. Zhang, Q. Song, J. Wu, L. Ding, C. Zhang, H. Zhu and H. Cui, Template-free preparation of nitrogen-doped activated carbon with porous architecture for high-performance supercapacitors, Microporous Mesoporous Mater., 2019, 276, 280–291 CrossRef CAS.
- X. Tong, Z. Chen, H. Zhuo, Y. Hu, S. Jing, J. Liu and L. Zhong, Tailoring the physicochemical properties of chitosan-derived N-doped carbon by controlling hydrothermal carbonization time for high-performance supercapacitor application, Carbohydr. Polym., 2019, 207, 764–774 CrossRef CAS.
- F. L. Braghiroli, V. Fierro, M. T. Izquierdo, J. Parmentier, A. Pizzi and A. Celzard, Nitrogen-doped carbon materials produced from hydrothermally treated tannin, Carbon, 2012, 50(15), 5411–5420 CrossRef CAS.
- M. Sevilla, W. Gu, C. Falco, M. M. Titirici, A. B. Fuertes and G. Yushin, Hydrothermal synthesis of microalgae-derived microporous carbons for electrochemical capacitors, J. Power Sources, 2014, 267, 26–32 CrossRef CAS.
- M. Ren, T. Zhang, Y. Wang, Z. Jia and J. Cai, A highly pyridinic N-doped carbon from macroalgae with multifunctional use toward CO2 capture and electrochemical applications, J. Mater. Sci., 2018, 54(2), 1606–1615 CrossRef.
- Q. Wu, G. Zhang, M. Gao, L. Huang, L. Li, S. Liu, C. Xie, Y. Zhang and S. Yu, N-doped porous carbon from different nitrogen sources for high-performance supercapacitors and CO2 adsorption, J. Alloys Compd., 2019, 786, 826–838 CrossRef CAS.
- C. Wang, Y. Zhou, L. Sun, P. Wan, X. Zhang and J. Qiu, Sustainable synthesis of phosphorus- and nitrogen-co-doped porous carbons with tunable surface properties for supercapacitors, J. Power Sources, 2013, 239, 81–88 CrossRef CAS.
- S. Guo, Y. Gao, Y. Wang, Z. Liu, X. Wei, P. Peng, B. Xiao and Y. Yang, Urea/ZnCl2 in situ hydrothermal carbonization of Camellia sinensis waste to prepare N-doped biochar for heavy metal removal, Environ. Sci. Pollut. Res. Int., 2019, 26(29), 30365–30373 CrossRef CAS.
- N. Baccile, G. Laurent, C. Coelho, F. Babonneau, L. Zhao and M.-M. Titirici, Structural Insights on Nitrogen-Containing Hydrothermal Carbon Using Solid-State Magic Angle Spinning 13C and 15N Nuclear Magnetic Resonance. The, J. Phys. Chem. C, 2011, 115(18), 8976–8982 CrossRef CAS.
- H. Park, P. P. M. Schleker, Z. Liu, N. Kowalew, T. Stamm, R. Schlögl, R.-A. Eichel, S. Heumann and J. Granwehr, Insights into Water Interaction at the Interface of Nitrogen-Functionalized Hydrothermal Carbons. The, J. Phys. Chem. C, 2019, 123(41), 25146–25156 CrossRef CAS.
- K. G. Latham, A. Ferguson and S. W. Donne, Influence of ammonium salts and temperature on the yield, morphology and chemical structure of hydrothermally carbonized saccharides, SN Appl. Sci., 2019, 1(1), 54 CrossRef.
- H. Liu, M. Wang, D. D. Zhai, X. Y. Chen and Z. J. Zhang, Design and theoretical study of carbon-based supercapacitors especially exhibiting superior rate capability by the synergistic effect of nitrogen and phosphor dopants, Carbon, 2019, 155, 223–232 CrossRef CAS.
- D. Hulicova-Jurcakova, A. M. Puziy, O. I. Poddubnaya, F. Suarez-Garcia, J. M. Tascon and G. Q. Lu, Highly stable performance of supercapacitors from phosphorus-enriched carbons, J. Am. Chem. Soc., 2009, 131(14), 5026–5027 CrossRef CAS.
- K. Pinkert, M. Oschatz, L. Borchardt, M. Klose, M. Zier, W. Nickel, L. Giebeler, S. Oswald, S. Kaskel and J. Eckert, Role of surface functional groups in ordered mesoporous carbide-derived carbon/ionic liquid electrolyte double-layer capacitor interfaces, ACS Appl. Mater. Interfaces, 2014, 6(4), 2922–2928 CrossRef CAS.
- K. G. Latham, G. Jambu, S. D. Joseph and S. W. Donne, Nitrogen Doping of Hydrochars Produced Hydrothermal Treatment of Sucrose in H2O, H2SO4, and NaOH, ACS Sustainable Chem. Eng., 2014, 2(4), 755–764 CrossRef CAS.
- K. G. Latham and S. W. Donne, Nitrogen Doped Heat-Treated and Activated Hydrothermal Carbon: Examination of Electrochemical Performance Using Step Potential Electrochemical Spectroscopy, J. Electrochem. Soc., 2018, 165(11), A2840–A2848 CrossRef CAS.
- M. F. Dupont and S. W. Donne, Separating faradaic and non-faradaic charge storage contributions in activated carbon electrochemical capacitors using electrochemical methods, J. Electrochem. Soc., 2015, 162(7), A1246–A1254 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, Adsorption of Gases in Multimolecular Layers, J. Am. Chem. Soc., 1938, 60(2), 309–319 CrossRef CAS.
- K. G. Latham, A. Rawal, J. M. Hook and S. W. Donne, Molecular structures driving pseudo-capacitance in hydrothermal nanostructured carbons, RSC Adv., 2016, 6(16), 12964–12976 RSC.
- E. Gann, A. T. Young, B. A. Collins, H. Yan, J. Nasiatka, H. A. Padmore, H. Ade, A. Hexemer and C. Wang, Soft X-ray scattering facility at the Advanced Light Source with real-time data processing and analysis, Rev. Sci. Instrum., 2012, 83(4), 045110 CrossRef CAS.
- B. Watts, L. Thomsen and P. C. Dastoor, Methods in carbon K-edge NEXAFS: Experiment and analysis, J. Electron Spectrosc. Relat. Phenom., 2006, 151(2), 105–120 CrossRef CAS.
- B. Ravel and M. Newville, ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT, J. Synchrotron Radiat., 2005, 12(Pt 4), 537–541 CrossRef CAS.
- K. G. Latham, W. M. Dose, J. A. Allen and S. W. Donne, Nitrogen doped heat treated and activated hydrothermal carbon: NEXAFS examination of the carbon surface at different temperatures, Carbon, 2018, 128, 179–190 CrossRef CAS.
- M. Forghani and S. W. Donne, Modification of the Step Potential Electrochemical Spectroscopy Analysis Protocol to Improve Outcomes, J. Electrochem. Soc., 2019, 166(13), A2727–A2735 CrossRef CAS.
- M. Forghani, H. Mavroudis, J. McCarthy and S. W. Donne, Electroanalytical characterization of electrochemical capacitor systems using step potential electrochemical spectroscopy, Electrochim. Acta, 2020, 332 Search PubMed.
- J. Liang, Y. Liu and J. Zhang, Effect of Solution pH on the Carbon Microsphere Synthesized by Hydrothermal Carbonization, Procedia Environ. Sci., 2011, 11, 1322–1327 CrossRef CAS.
- J. Ming, Y. Wu, G. Liang, J.-B. Park, F. Zhao and Y.-K. Sun, Sodium salt effect on hydrothermal carbonization of biomass: a catalyst for carbon-based nanostructured materials for lithium-ion battery applications, Green Chem., 2013, 15(10), 2722–2726 RSC.
- X. Lu, J. R. Flora and N. D. Berge, Influence of process water quality on hydrothermal carbonization of cellulose, Bioresour. Technol., 2014, 154, 229–239 CrossRef CAS.
- H. Teng, T.-S. Yeh and L.-Y. Hsu, Preparation of activated carbon from bituminous coal with phosphoric acid activation, Carbon, 1998, 36(9), 1387–1395 CrossRef CAS.
- L.-Z. Fan, S. Qiao, W. Song, M. Wu, X. He and X. Qu, Effects of the functional groups on the electrochemical properties of ordered porous carbon for supercapacitors, Electrochim. Acta, 2013, 105, 299–304 CrossRef CAS.
- H. Liu, H. Song, X. Chen, S. Zhang, J. Zhou and Z. Ma, Effects of nitrogen- and oxygen-containing functional groups of activated carbon nanotubes on the electrochemical performance in supercapacitors, J. Power Sources, 2015, 285, 303–309 CrossRef CAS.
- H. Sun, G. Zhou, S. Liu, H. M. Ang, M. O. Tade and S. Wang, Nano-Fe(0) encapsulated in microcarbon spheres: synthesis, characterization, and environmental applications, ACS Appl. Mater. Interfaces, 2012, 4(11), 6235–6241 CrossRef CAS.
- X. Q. Yang, J. Chen, P. D. Hale, T. Inagaki, T. A. Skotheim, Y. Okamoto, L. Samuelson, S. Tripathy, K. Hong, M. F. Rubner and M. L. denBoer, A FTIR and NEXAFS study of polypyrrole Langmuir-Blodgett films, Synth. Met., 1989, 28(1–2), 251–256 CrossRef.
- Y. He, Y. Zhang, X. Li, Z. Lv, X. Wang, Z. Liu and X. Huang, Capacitive mechanism of oxygen functional groups on carbon surface in supercapacitors, Electrochim. Acta, 2018, 282, 618–625 CrossRef CAS.
- I. I. Misnon, N. K. M. Zain, R. A. Aziz, B. Vidyadharan and R. Jose, Electrochemical properties of carbon from oil palm kernel shell for high performance supercapacitors, Electrochim. Acta, 2015, 174, 78–86 CrossRef CAS.
- E. R. Nightingale, Phenomenological Theory of Ion Solvation. Effective Radii of Hydrated Ions. The, J. Phys. Chem., 1959, 63(9), 1381–1387 CrossRef CAS.
- T. Wei, X. Wei, Y. Gao and H. Li, Large scale production of biomass-derived nitrogen-doped porous carbon materials for supercapacitors, Electrochim. Acta, 2015, 169, 186–194 CrossRef CAS.
- C. Han, H. Li, R. Shi, T. Zhang, J. Tong, J. Li and B. Li, Organic quinones towards advanced electrochemical energy storage: recent advances and challenges, J. Mater. Chem. A, 2019, 7(41), 23378–23415 RSC.
- B.-A. Mei, O. Munteshari, J. Lau, B. Dunn and L. Pilon, Physical Interpretations of Nyquist Plots for EDLC Electrodes and Devices. The, J. Phys. Chem. C, 2017, 122(1), 194–206 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00601g |
| This journal is © The Royal Society of Chemistry 2021 |