
Photothermal CO2 hydrogenation to hydrocarbons over trimetallic Co–Cu–Mn catalysts†
Zhen-Hong
He
 *a,
Zhu-Hui
Li
a,
Zhong-Yu
Wang
a,
Kuan
Wang
a,
Yong-Chang
Sun
a,
Sen-Wang
Wang
b,
Wei-Tao
Wang
a,
Yang
Yang
a and
Zhao-Tie
Liu
*ab
*a,
Zhu-Hui
Li
a,
Zhong-Yu
Wang
a,
Kuan
Wang
a,
Yong-Chang
Sun
a,
Sen-Wang
Wang
b,
Wei-Tao
Wang
a,
Yang
Yang
a and
Zhao-Tie
Liu
*ab
aShaanxi Key Laboratory of Chemical Additives for Industry, College of Chemistry and Chemical Engineering, Shaanxi University of Science & Technology, 710021 Xi'an, China. E-mail: hezhenhong@sust.edu.cn; ztliu@snnu.edu.cn
bSchool of Chemistry & Chemical Engineering, Shaanxi Normal University, 710119 Xi'an, China
First published on 23rd June 2021
Abstract
Photocatalytic CO2 reduction is a highly vital process for converting CO2 into valuable chemicals. However, the reaction always proceeds less efficaciously at low temperature. A combination of optical and thermal conditions is one of the feasible approaches to achieve the reaction with high efficiency and has gained much attention recently. In the present work, we prepared several Co–Cu–Mn trimetallic catalysts via a simple co-precipitation method, which were used in catalyzing photothermal CO2 reduction to hydrocarbons. The metal composition and reduction temperature of the catalysts had important effects on their structural and photoelectrical characteristics and adsorption behaviors, further resulting in diverse catalytic performances. Among the prepared trimetallic catalysts, Co7Cu1Mn1Ox(200), with a Co/Cu/Mn molar ratio of 7/1/1 and reduced at 200 °C in H2 for 2 h, could produce CH4 with an activity of 14.5 mmol gcat−1 h−1 in 10% CO2/30% H2/60% N2, and CH4 and C2+ hydrocarbons with the activities of 15.9 and 7.5 mmol gcat−1 h−1 in 25% CO2/75% H2, respectively. The present strategy for constructing trimetallic oxide catalysts for the photothermal reaction not only provides a highly active catalyst for CO2 utilization, but also offers a potential possibility for reducing the high temperature of conventional thermal reactions.
Introduction
CO2 is the primary greenhouse gas, whose concentration in the atmosphere continues to rise.1 It is also an important C1 source and can be converted into many valuable chemicals such as CO,2 hydrocarbons,3–5 olefins,6 acids,7–9 alcohols,10–12etc. Although many efforts have been invested in this area, it is still very difficult to convert CO2 into chemicals due to its thermodynamic stability and chemical inertness.1 CO2 hydrogenation to hydrocarbons represents an essential and green way for its utilization.13 However, the conventional thermal process is always conducted under harsh conditions, leading to the formation of carbon deposit, the increase of energy consumption, etc. The photocatalytic method is an ideal way for the reaction, which could be carried out under mild conditions and using solar energy. However, this route suffers from low activity especially at room temperature, impeding its further applications. Recently, the photothermal catalytic method has gained increasing attention, especially in CO2 reduction.14,15The photothermal routes always contain four types, including thermal-assisted photocatalysis, photo-assisted thermocatalysis, photothermal co-catalysis, and photo-driven thermocatalysis.16 Generally, these routes combine the advantages of both photocatalysis and thermal catalysis including high activity, proceeding under mild conditions, efficient energy utilization, etc. To date, many catalysts such as Ru/silicon nanowire,17 RuO2/SrTiO3,18 RuO2/3D silicon photonic crystals,19 Ru@FL-LDHs (Ru/Mg–Al LDHs matrix),20 Ru/Al2O3,21 Ru/TiO2,22 Pd@Nb2O5,23 CoFeAl-LDH nanosheet,13 Ni/SiO2·Al2O3,14 Cu2O/Zn-MOF,24 Cu2O/graphene,25etc. have been used for photothermal CO2 hydrogenation, and their catalytic performances are given in Table S1.† Significant advances have been made in recent years; however, most of the above catalysts suffer from drawbacks such as using expensive noble metals, working at high temperature, and exhibiting low activity. Besides, the obtained products mainly focused on C1 chemicals such as CO and CH4. The synthesis of C2+ products via a photothermal route is a more difficult but significant issue, which is rarely involved in the above developed catalytic systems.26 Considering the above aspects, to develop a cost-efficient, highly active and reusable catalyst for photothermal CO2 reduction into CH4 and C2+ hydrocarbons is particularly important.
On the other hand, it has been well known that pure metals used in CO2 hydrogenation always showed a low activity due to their low CO2 binding capacity.27 Oxides could not only anchor metal atoms but also provide vacancies for the reaction, and in this way, they could participate in the reaction and enhance the catalytic performances.28 Trimetallic oxide catalysts show unique electronic and structural features different from their individual components. The complex composition of trimetallic catalysts could form multiple interfaces conducive to catalysis.29 In this aspect, trimetallic catalysts have been widely used in many reactions such as CO and CO2 hydrogenation,30–32 ethanol steam reforming,33 ammonia oxidation,34etc. with good performances. In particular, complicated metal–trimetallic oxides can provide many vacancies and active sites for catalysis, and have great potential for application in CO2 reduction. Herein, a series of Co–Cu–Mn trimetallic oxides were prepared and used for photothermal CO2 reduction, and the screened Co7Cu1Mn1Ox(200) catalyst, with a Co/Cu/Mn ratio of 7/1/1 and reduced at 200 °C in H2, showed a high CH4 formation activity (14.5 mmol gcat−1 h−1) even at low CO2 (10%) and H2 (30%) concentrations, which lies in the highest activity range in Table S1.† Interestingly, after increasing the CO2 and H2 concentrations to 25% and 75%, 15.9 and 7.5 mmol gcat−1 h−1 of CH4 and C2+ hydrocarbons were produced with selectivities of 65.4% and 30.8%, respectively. Moreover, the catalyst has a good temporal stability under continuous-flow conditions. The metal/oxides were confirmed to be very important for the catalytic performance, in which the Co0 species were the active sites, which together with the oxides acted as supports and semiconductors for providing a local heating environment via the photothermal effects. The Cu species could promote the reduction of Co and Mn oxides, and also promote the coupling of C–C bonds to produce C2+ products especially at a high CO2 concentration. The Mn species could on one hand enhance the adsorption of CO2 and H2, and also provide the photothermal effect to produce a local heating effect to promote the reaction. The present strategy for constructing trimetallic catalysts for the photothermal reaction does not only provide a simple but efficient way to convert CO2 into valuable hydrocarbons, but also offer a general method for reducing the reaction temperature of conventional high-temperature reactions.
Results and discussion
Preparation and characterization of catalysts
Trimetallic Co–Cu–Mn catalysts were prepared by a simple co-precipitation method, and the details are given in the ESI.† The prepared catalysts were denoted as CoaCubMncOx(T), in which a/b/c and T represent the Co/Cu/Mn molar ratio and the reduction temperature, respectively. CoaCubMncOx means unreduced catalysts. The morphologies of the catalysts were examined by TEM tests. Initially, the Co7Cu1Mn1Ox catalyst mainly showed the morphology of nanoparticles with the size range of 6–15 nm (Fig. 1a and b). The lattice spacings of 0.186, 0.246, 0.219, 0.152, 0.197, and 0.135 nm in the HRTEM image could be successively indexed to the (331) and (311) planes of Co3O4, the (411) and (611) planes of Mn2O3, and the (−112) and (−221) planes of CuO (Fig. 1c).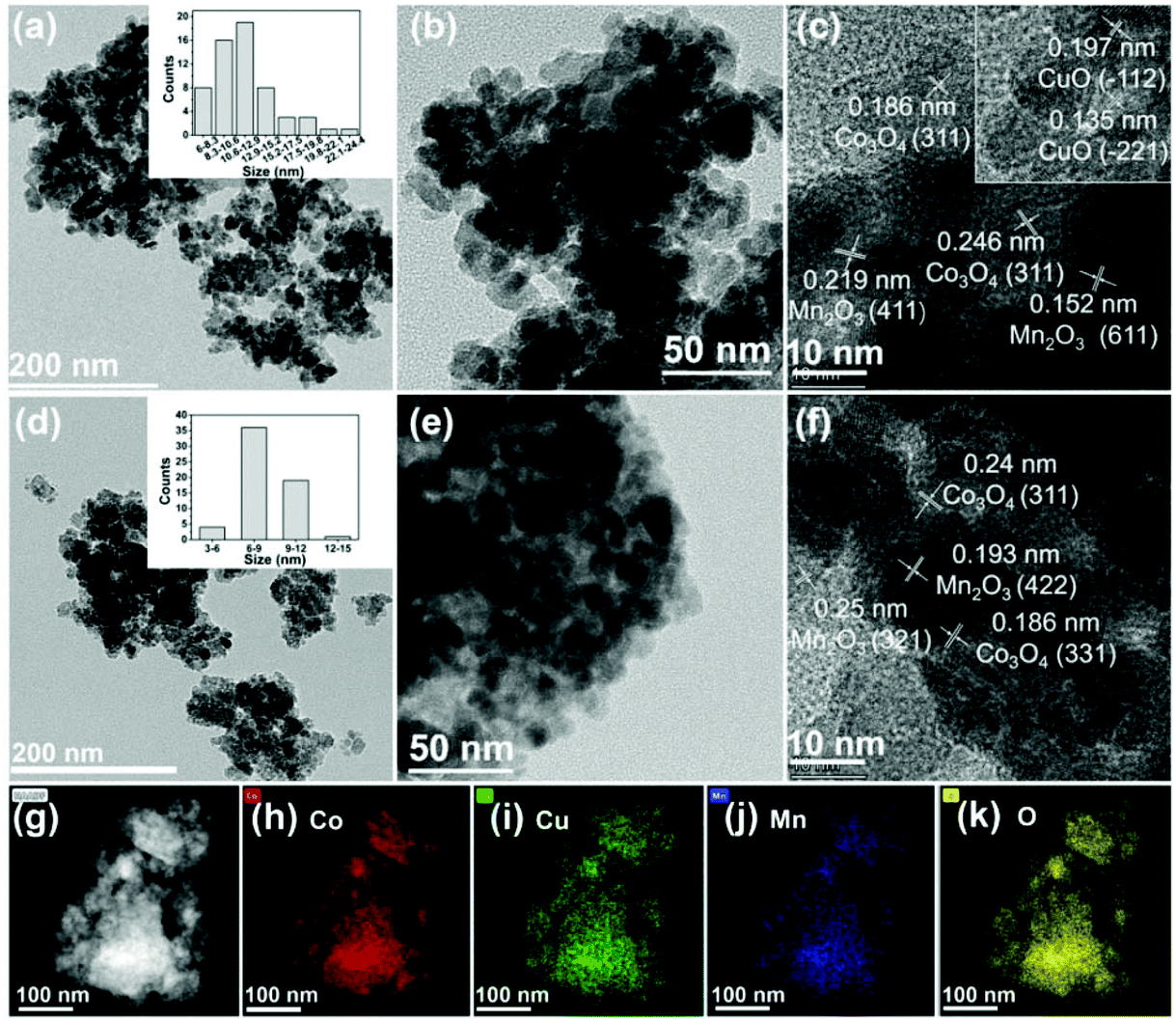 | ||
| Fig. 1 TEM images (a, b, d, and e) and HRTEM images (c and f) for the Co7Cu1Mn1Ox (a–c) and Co7Cu1Mn1Ox(200) (d–f) catalysts, HAADF-STEM image for the Co7Cu1Mn1Ox(200) catalyst (g) and the corresponding EDS elemental mapping of Co, Cu, Mn, and O (h–k). The insets in (a) and (d) are the size distribution histograms for the Co7Cu1Mn1Ox and Co7Cu1Mn1Ox(200) catalysts. | ||
After reduction, the Co7Cu1Mn1Ox(200) catalyst maintained the nanoparticle morphology in the size range of 6–12 nm (Fig. 1d and e). Co3O4 and Mn2O3 were also detected, as shown in Fig. 1f. The HAADF-STEM and EDX elemental mappings of the Co7Cu1Mn1Ox(200) catalyst showed that Co, Cu, Mn, and O elements are homogeneously dispersed over the whole grain (Fig. 1g to k).
For comparison, the TEM images of catalysts with different Co/Cu/Mn ratios were also studied, and the results are shown in Fig. S1.† With the increasing Co content, the morphology changed from nanosheets to nanoparticles. For example, the Co3Cu1Mn1Ox(200) and Co5Cu1Mn1Ox(200) catalysts were nanosheets, while the Co7Cu1Mn1Ox(200) and Co9Cu1Mn1Ox(200) catalysts were mainly nanoparticles, which were beneficial for exposing more active sites.
The molar ratios of the bimetallic and trimetallic catalysts were tested by ICP-OES, and the results are given in Table S2.† The molar ratios of metals were consistent with the feed amounts.
XRD examinations showed that all the tested unreduced catalysts such as Co7Cu1Mn1Ox, Co7Mn1Ox, and Co7Cu1Ox showed the XRD patterns of Co3O4 (JCPDS file no. 73-1701). No obvious CuOx or MnOx peaks were found, implying that they were easily incorporated into the Co3O4 crystals and were well dispersed (Fig. 2a). The Co7Cu1Mn1Ox(200) catalyst showed the same peaks as that of the Co7Cu1Mn1Ox catalyst, but with lower intensities, indicating the crystallinity decrease of the Co3O4 component after reduction.
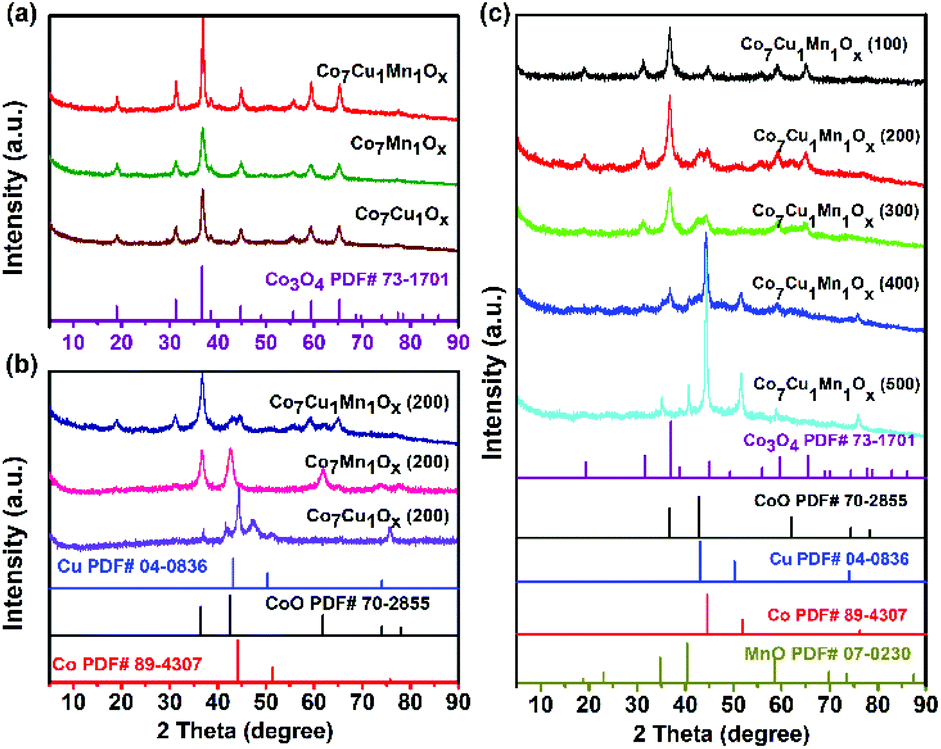 | ||
| Fig. 2 XRD patterns of (a) the unreduced Co7Mn1Ox, Co7Cu1Ox, and Co7Cu1Mn1Ox catalysts; (b) Co7Mn1Ox(200), Co7Cu1Ox(200), and Co7Cu1Mn1Ox(200); and (c) Co7Cu1Mn1Ox catalysts reduced at different temperatures. | ||
The XRD pattern of the Co7Cu1Mn1Ox(200) catalyst was almost the same as that of the unreduced catalyst, and it is mainly attributed to the Co3O4 phase (Fig. 2b). However, two small peaks at 43.0° and 61.8° were carefully detected, which could be assigned to the CoO (200) and (220) planes (JCPDS file no. 70-2855). No Co0, Cu0, Cu2O, or Mn2O3 species were detected due to their low amounts. The Co7Mn1Ox(200) catalyst showed three peaks at 36.8°, 42.7°, and 61.8°, which could be assigned to the CoO (111), (200), and (220) planes (JCPDS file no. 78-0431), respectively. Two phases of Co0 fcc and hcp were identified in the Co7Cu1Ox(200) catalyst, and the formation of two phases is commonly observed in the synthesis of cobalt nanostructures.35 The peaks at 41.5°, 44.4°, 47.4°, and 75.9° could be indexed to Co0 hcp (100), (002), (101), and (110) facets (JCPDS file no. 01-071-4239), respectively. The peaks at 44.4° and 51.5° could be contributed to the Co0 fcc (111) and (200) planes (JCPDS file no. 01-071-4651).35 The results indicated that Co3O4 could be reduced to Co0 by the promotion of Cu species, and this will be further verified by the following H2-TPR tests. No MnOx or CuOx species were detected due to their good dispersion. It is worth noting that although Cu could promote the reduction of CoOx species to Co0, no Co0 was detected in the bulk phase of the Co7Cu1Mn1Ox(200) catalyst.
The XRD patterns of the catalysts with different Co/Cu/Mn ratios also showed the main peaks of Co3O4 and CoO (Fig. S2†); however, the Co3Cu1Mn1Ox(200) and Co5Cu1Mn1Ox(200) catalysts showed a wide peak at around 20°, indicating that they possess amorphous structures.
The results of the N2 adsorption/desorption tests are shown in Fig. S3 and Table S3.† The reduction temperature affects the catalyst structure remarkably, and the BET surface areas of the tested trimetallic catalysts declined gradually with the increase of the reduction temperature, which is probably because the high reduction temperature leads to catalyst sintering. The bimetallic Co–Cu catalyst shows the lowest BET surface area, while the Co–Mn catalyst presents the highest. Indeed, the surface areas of the CoOx and Co7Cu1Mn1Ox(100, 200, and 300) catalysts did not differ greatly from one another, thus their diverse performances did not stem from the difference in their surface areas.
To distinguish the valences of the Co, Cu, and Mn species on the surface of the catalysts, the XPS of Co 2p, Cu 2p, Mn 2p and 3s, and AES of Cu L3M4,5M4,5 were analyzed (Fig. 3).5,36,37 In the XPS of the Co7Cu1Mn1Ox catalyst, the Co element was composed of Co3+ and Co2+ species, which was confirmed by the peaks at 779.5 eV and 780.9 eV, which can be attributed to the Co3+ and Co2+ 2p3/2 peaks (Fig. 3a).38 The Cu2+ 2p XPS showed two peaks at 934.5 eV and 954.2 eV, which could be assigned to Cu2+ 2p3/2 and 2p1/2, respectively (Fig. 3b and c). The XPS peaks of Co species in the Co7Cu1Mn1Ox(200) catalyst were the same as those in the unreduced catalyst; however, the Cu species was found to be Cu+. The Mn XPS indicated that the catalyst contains Mn3+ oxidation state (Fig. 3d and e). The XPS results indicated that the surface of the Co7Cu1Mn1Ox(200) catalyst mainly contained Co2+, Co3+, Cu+, a small amount of Cu2+, and Mn3+, respectively, but no Co0 or Cu0 species were detected for air oxidation.
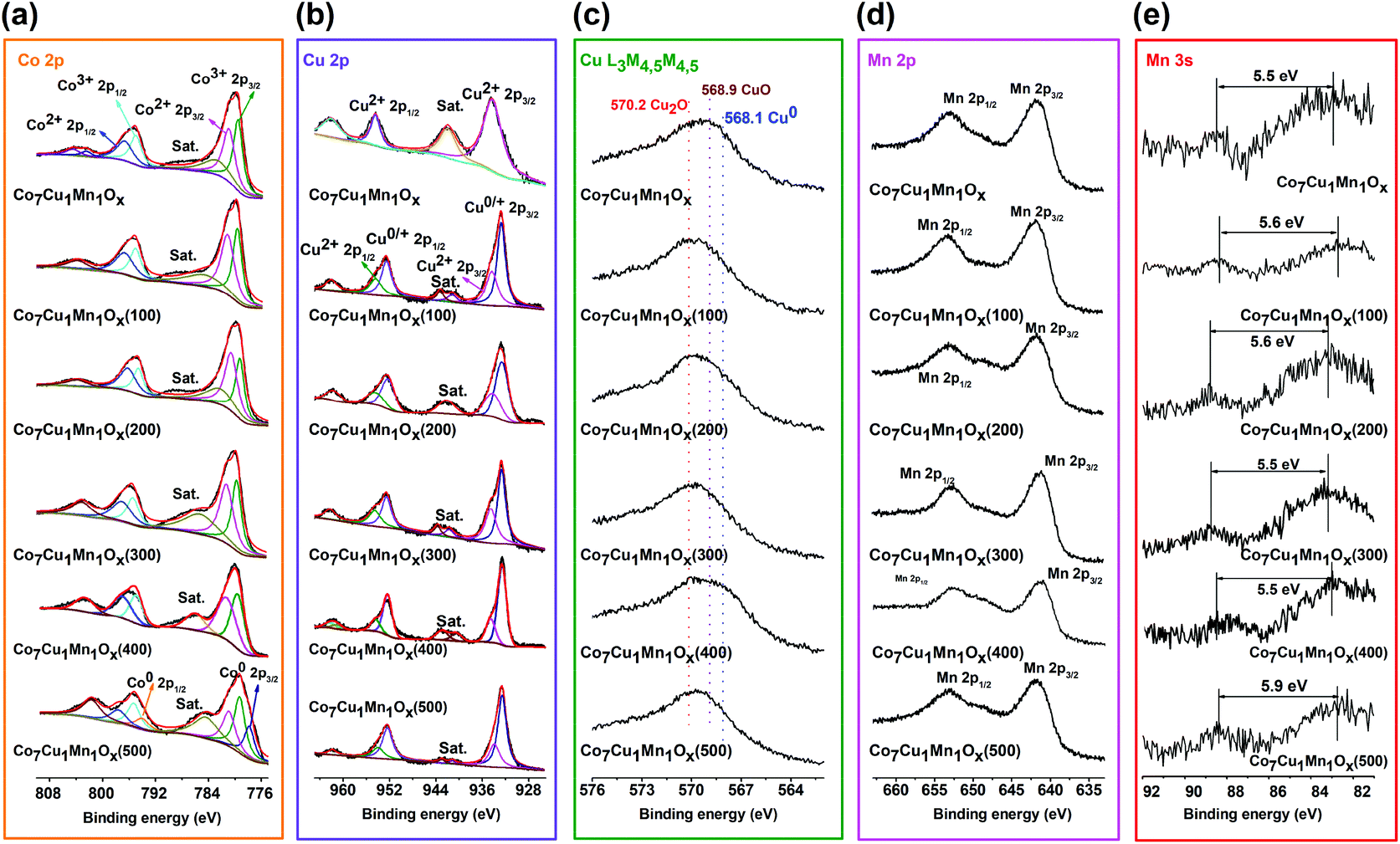 | ||
| Fig. 3 XPS spectra of the unreduced Co7Cu1Mn1Ox catalyst and the Co7Cu1Mn1Ox(100, 200, 300, and 500) catalysts. (a) Co 2p XPS, (b) Cu 2p XPS, (c) Cu L3M4,5M4,5 AES spectra, (d) Mn 2p XPS, and (e) Mn 3s XPS, respectively. | ||
For comparison, the XPS spectra of the catalysts reduced at different temperatures were also tested. The Co element in these catalysts was mainly composed of Co2+ and Co3+ species but with different contents. The Co2+ component increased with the increase of the reduction temperature. No obvious Co0 species was found until the reduction temperature reached 500 °C. As for the Cu XPS, the area of the satellite peak around 942 eV decreased with increasing reduction temperature, indicating that Cu0/+ 2p3/2 increased. Besides, all the reduced catalysts possess the dominant Cu+ component. The Mn 3s XPS showed that the Co7Cu1Mn1Ox(500) catalyst is composed of Mn2+ species, i.e., MnO, while the others showed Mn3+ species.
The XPS spectra of the Co–Cu–Mn trimetallic catalysts with different molar ratios were also investigated, and the results are given in Fig. S4.† All the elements had the same valences but contained different contents, such as Co2+, Co3+, Mn3+, Cu+, and a small amount of Cu2+, which are similar to the Co7Cu1Mn1Ox(200) catalyst, indicating that they are probably active in the reaction. This was confirmed by the following catalytic activity studies (Table 1).
| Entry | Catalyst | Conv. (%) | Activityb (mmol gcat−1 h−1) | CH4b Sele. (%) | ||
|---|---|---|---|---|---|---|
| CH4 | C2+ | CO | ||||
| a Reaction conditions: catalyst 50 mg, CO2/H2/N2 10%/30%/60%, full irradiation, 200 °C, 3 h. b Detected by GC and obtained from three successive runs. c N2 was replaced by Ar. | ||||||
| 1 | None | 0 | 0 | 0 | 0 | — |
| 2 | CoOx(200) | 5.6 | 3.1 ± 0.1 | 0.4 ± 0.1 | 0 | 88.6 |
| 3 | CuOx(200) | 0 | 0 | 0 | 0 | — |
| 4 | MnOx(200) | 0 | 0 | 0 | 0 | — |
| 5 | Cu1Mn1Ox(200) | 0 | 0 | 0 | 0 | — |
| 6 | Co7Mn1Ox(200) | 0.8 | 0.2 ± 0.1 | 0 | 0.3 ± 0.1 | 40.0 |
| 7 | Co7Cu1Ox(200) | 14.0 | 5.9 ± 0.2 | 2.4 ± 0.1 | 0.4 ± 0.1 | 67.8 |
| 8 | Co7Cu1Mn1Ox(200) | 27.4 | 14.5 ± 0.2 | 1.4 ± 0.1 | 1.1 ± 0.1 | 85.3 |
| 9 | Co3Cu1Mn1Ox(200) | 12.4 | 7.2 ± 0.2 | 0.5 ± 0.2 | 0 | 93.5 |
| 10 | Co5Cu1Mn1Ox(200) | 16.8 | 9.4 ± 0.3 | 0.8 ± 0.2 | 0.2 ± 0.2 | 84.7 |
| 11 | Co9Cu1Mn1Ox(200) | 18.9 | 10.1 ± 0.2 | 0.9 ± 0.2 | 0.7 ± 0.1 | 90.4 |
| 12 | Co1Cu3Mn1Ox(200) | 3.1 | 1.6 ± 0.2 | 0.3 ± 0.1 | 0 | 84.2 |
| 13 | Co1Cu1Mn3Ox(200) | 2.1 | 1.1 ± 0.2 | 0.2 ± 0.1 | 0 | 84.6 |
| 14c | Co7Cu1Mn1Ox(200) | 28.4 | 15.1 ± 0.2 | 1.3 ± 0.1 | 1.2 ± 0.1 | 85.8 |
| 15 | Co7Cu0.25Mn1.75Ox(200) | 13.6 | 7.7 ± 0.1 | 0.4 ± 0.2 | 0.4 ± 0.1 | 90.6 |
| 16 | Co7Cu0.75Mn1.25Ox(200) | 22.9 | 12.7 ± 0.2 | 0.7 ± 0.1 | 0.9 ± 0.1 | 88.8 |
| 17 | Co7Cu1.25Mn0.75Ox(200) | 24.0 | 11.5 ± 0.2 | 2.6 ± 0.1 | 0.9 ± 0.1 | 76.7 |
| 18 | Co7Cu1.75Mn0.25Ox(200) | 14.1 | 5.0 ± 0.1 | 2.5 ± 0.1 | 1.3 ± 0.1 | 56.8 |
The Raman spectrum of the Co7Cu1Mn1Ox(200) catalyst showed a peak at 680 cm−1, which could be assigned to the A1 g mode of Co3O4 (Fig. S5†).39 This profile is similar to those of the Co7Cu1Mn1Ox and Co7Cu1Mn1Ox (100, 300, and 400) catalysts, indicating that Co3O4 exists in these catalysts. Interestingly, the Co7Cu1Mn1Ox(500) catalyst showed a wide peak at 540 cm−1, assigned to the one-phonon LO mode of CoO,40 which was formed by the partial oxidation of Co0 by local laser heating.45 No obvious Cu or Mn species were detected, indicating that they have permeated into the Co3O4 crystal lattice.
The above characterization studies showed that the Co7Cu1Mn1Ox(200) catalyst mainly possesses Co3O4 in the bulk phase, while the surface of the catalyst was composed of Co3+, Co2+, Mn3+, Cu+, and a small amount of Cu2+ species, which are in the forms of Co3O4, Mn2O3, Cu2O, and CuO. The catalysts reduced at 100, 200, and 300 °C, and the catalysts with different Co/Cu/Mn molar ratios contained similar components but of different concentrations, indicating that they possess similar structures and properties.
Catalytic performances
Photothermal CO2 hydrogenation was carried out in a tubular reactor (Fig. S6),† and the procedure details are given in the ESI.† The products of photothermal CO2 reduction include CO, CH4, and C2+ hydrocarbons (mainly C2 to C6) in the present work. Initially, the catalytic performances over different catalysts are given in Table 1. No products were detected in the absence of a catalyst, and the sole metal catalysts or the bimetallic catalysts showed a very low activity in the reaction. The Co7Cu1Mn1Ox(200) catalyst showed the best catalytic performances with a CH4 formation activity of 14.5 mmol gcat−1 h−1 and a selectivity of 85.3% (entries 1–7 vs. 8). These activities were achieved under the irradiation of a 300 W Xe lamp (234 mW cm−2, 300–1100 nm) and heat generated by an additional electric heater (Fig. S6†). Unlike the CuOx(200) and MnOx(200) catalysts, the CoOx(200) catalyst could show a very low CH4 formation activity (only 3.1 mmol gcat−1 h−1) (entries 2–4), demonstrating that the Co species is probably the active species, while the CuOx and MnOx species are the promoters for the Co species. Indeed, the Cu and Mn species are always used as promoters or active sites for the Co-catalyzed CO2 reduction.41 The activities of the bimetallic Cu1Mn1Ox or Co7Mn1Ox catalysts confirmed that Cu or Mn are not the active sites (entries 5–7). Interestingly, the Co7Cu1Ox(200) catalyst showed higher activities for the formation of CH4 and C2+ hydrocarbons than those of the pure CoOx(200) and CuOx(200) catalysts, indicating that the Cu species could not only enhance the activity of the Co-based catalyst in CO2 reduction to CH4, but also promote the formation of C2+ products. For comparison, other trimetallic catalysts with different ratios were also assessed; however, all of them possessed lower activities than that of the Co7Cu1Mn1Ox(200) catalyst (entries 8 vs. 9–13).The activities achieved in CO2/H2/N2 and CO2/H2/Ar were very similar (entries 8 vs. 14), indicating that the catalytic performances were not affected by the type of diluent gas. In addition, the C2+ activities of the Co9Cu1Mn1Ox(200), Co7Cu1Mn1Ox(200), Co5Cu1Mn1Ox(200), and Co3Cu1Mn1Ox(200) catalysts were only 0.9, 1.4, 0.8, and 0.5 mmol gcat−1 h−1, respectively, and inconsistent with the change rule of the catalyst composition. Because the formation of C2+ products is closely related to the Cu species, the uneven distribution of Cu species on the catalyst surface is probably the reason for the discrepancy between the C2+ activity and the Co/Cu/Mn molar ratio of the catalyst.
The Co species are the active sites; however, the promoters of Cu and Mn species are also very important for the reaction. To confirm this, the catalysts with the same component of Co element were investigated, and the results indicated that the Cu and Mn concentrations affected the catalytic performances remarkably (entries 15–18). The Co7Cu0.75Mn1.25Ox(200) and Co7Cu1.25Mn0.75Ox(200) catalysts showed high CH4 formation activities, while the Co7Cu0.25Mn1.75Ox(200) and Co7Cu1.75Mn0.25Ox(200) catalysts offered relatively low values, indicating that the low concentrations of Cu and Mn are not beneficial for catalysis.
The catalytic performance studies showed that the coexistence of Co, Cu, and Mn in the Co/Cu/Mn trimetal is very important for the reaction, and their molar ratios and the synergistic effects of these multicomponents are closely related to the catalytic performances, and a similar phenomenon could be found in the previous report.42
Effect of the reduction temperature of the Co7Cu1Mn1Ox catalysts
The above characterization results indicated that the reduction temperature affected the metal valence compositions of the catalysts, and it was proved to be able to affect their catalytic performances.43,44 To verify this, the catalysts reduced at different temperatures were studied, and the results are shown in Fig. 4. Indeed, the Co7Cu1Mn1Ox and Co7Cu1Mn1Ox(100, 200, and 300) catalysts showed good catalytic performances during the reaction, indicating that they have similar catalyst compositions and properties. However, the Co7Cu1Mn1Ox(400 and 500) catalysts showed decreased catalytic activities, which has a great relationship with the high concentration of Co0 in the bulk phase but less Co3O4 and Mn2O3.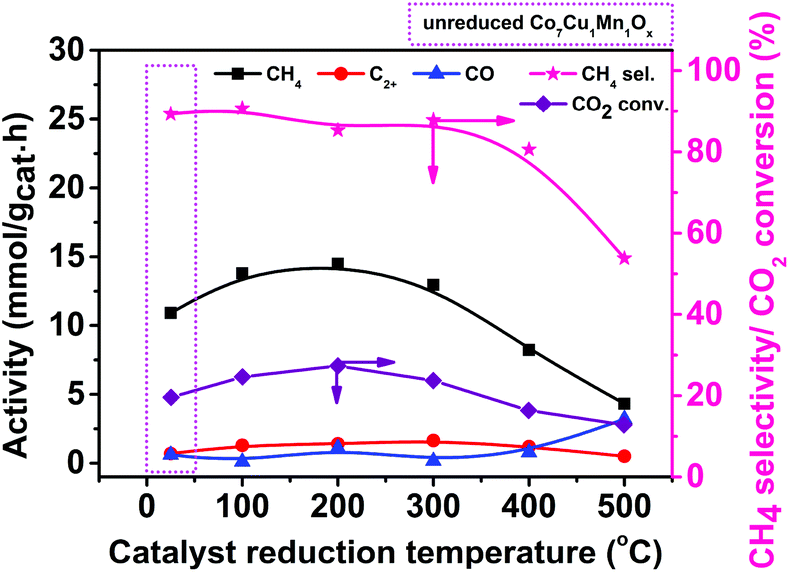 | ||
| Fig. 4 Effect of the reduction temperature of the Co7Cu1Mn1Ox(T) catalysts on the catalytic performances of photothermal CO2 reduction. Reaction conditions: catalyst 50 mg, CO2/H2/N2 10%/30%/60%, 200 °C, 3 h, and 300 W Xe lamp full irradiation. | ||
The catalysts after being used for 9 h and 21 h were characterized by XPS and XRD in Fig. 5, and the results showed that Co0 and Cu0 species could be found during the reaction. Co0 was widely recognized as the active site for CO2 hydrogenation. The XRD patterns of the catalysts used for 9 h showed similar peaks to those of Co3O4 and CoO, revealing that Co3O4 still exists in the bulk phase. Compared with the Co7Cu1Mn1Ox(200) catalyst in Fig. 2, the peak at 36.6° decreased remarkably. The catalyst used for 21 h showed similar results but with more Co0 and Cu0 species. The XPS and XRD characterization studies showed that the catalyst compositions were stable during the reaction.
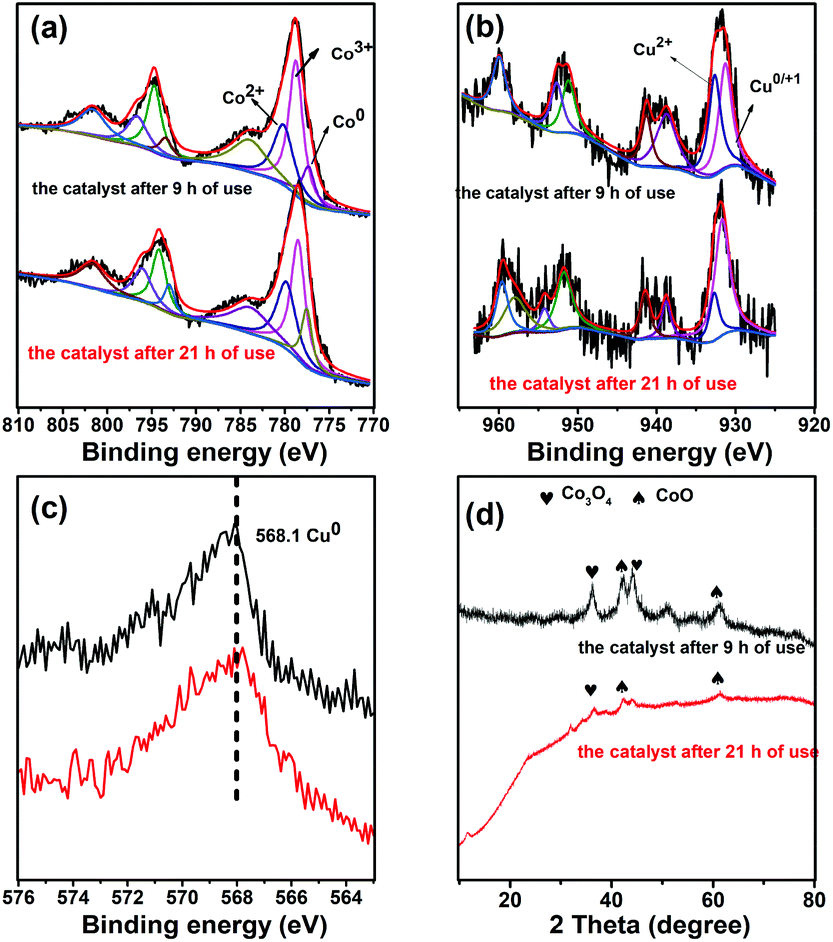 | ||
| Fig. 5 XPS tests (a, Co 2p; b, Cu 2p; c, Cu 3s LMM AES) and XRD pattern (d) for the catalysts after 9 h and 21 h of uses, respectively. | ||
The highest CH4 activity was achieved over the Co7Cu1Mn1Ox(200) catalyst. Besides, the Co7Cu1Mn1Ox(100 and 300) catalysts also showed acceptable CH4 formation activities. Generally, in thermal catalysis, Co0 species are recognized as the active components. However, the Co7Cu1Mn1Ox(500) catalyst showed the lowest activity in the reaction, which mainly contained the Co0 species but no Co3O4 or Mn2O3. It should be noted that in the present photothermal catalysis, the oxides including Co3O4, Mn2O3, etc. could promote the reaction via the interaction effect between the Co species and other oxides.
The H2-TPR tests were carried out to study the interaction effect of different metal oxides, and the results are shown in Fig. 6. Pure Co3O4 showed two peaks at 273 °C and 336 °C, which could be assigned to the reduction peaks of Co3O4 to CoO and CoO to Co0, respectively.46 Mn2O3 showed two peaks located at 292 °C and 388 °C, which could be assigned to the reduction of Mn2O3 to Mn3O4 and Mn3O4 to MnO, respectively.47,48 The Co7Cu1Ox catalyst had two peaks at lower temperatures of 156 °C and 210 °C indicating that Cu could obviously promote the reduction of Co3O4 and CoO, which is consistent with previous reports.45 However, the Co7Mn1Ox catalyst showed two main peaks at 280 °C and 428 °C, which could be assigned to the reduction of (Co,Mn)3O4 to (Co,Mn)O, and (Co,Mn)O to Co0 and MnO,5,49,50 respectively, revealing that Mn species impedes the reduction of CoOx. The Co7Cu1Mn1Ox catalyst had three peaks at 128, 183, and 284 °C, which could be attributed to the reduction peaks of CuO to Cu0, Co3O4 to CoO, and CoO to Co0 and MnO, respectively.50 The H2-TPR results indicated that the Co, Cu, and Mn species show strong interaction effects, and the previous report showed that the presence of Cu could alter the electronic interactions with Co and Ni in the Co–Cu–Ni trimetallic catalysts.30
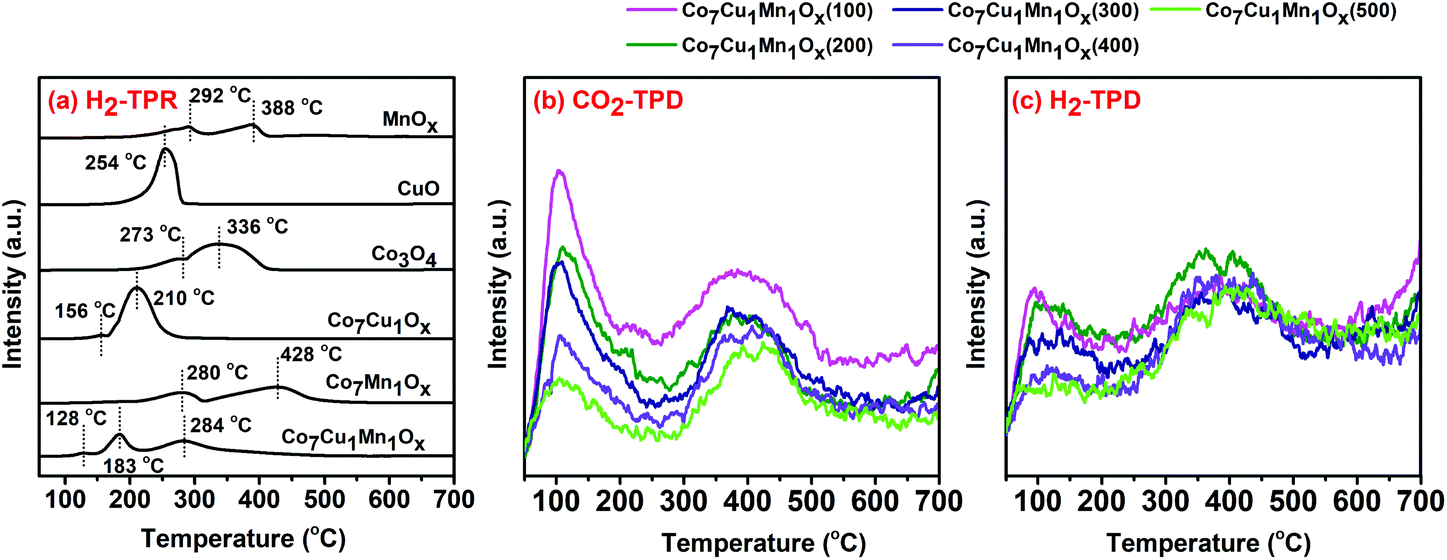 | ||
| Fig. 6 H2-TPR(a), CO2-TPD (b), and H2-TPD (c) tests for the different catalysts. | ||
The surface adsorption capacities could significantly affect the catalytic activity. Thus, we conducted the CO2-TPD and H2-TPD tests for the diverse trimetallic catalysts and the results indicated that with the increase of the reduction temperature, the adsorption amounts of CO2 and H2 on the catalysts decreased. The Co7Cu1Mn1Ox(100 and 200) catalysts contained higher CO2 and H2 adsorption amounts than the others, which is beneficial for them to catalyze the CO2 photothermal reduction.
To investigate the effects of the Mn and Cu species on the Co-based catalysts, the CO2-TPD tests for Co–Mn and Co–Cu bimetallic catalysts were performed (Fig. S7†). The Mn species was found to be able to improve CO2 and H2 adsorption for the Co-based catalysts, while the Cu species reduced their adsorption since the Co–Cu bimetallic catalyst could be deeply reduced at 200 °C. Even so, the Co7Cu1Ox(200) catalyst exhibited a strong CO2 adsorption peak at around 350 °C, showing that the Cu species could promote strong adsorption of CO2 on the catalyst.
We investigated the catalytic performances of the Co–Cu and Co–Mn bimetallic catalysts reduced at different temperatures, and the results are given in Table S4.† With the increase of the reduction temperature, the activities of the Co–Cu and Co–Mn catalysts increased initially but were then reduced at high reduction temperatures of 400 and/or 500 °C. The highest activities were lower than that of the Co7Cu1Mn1Ox(200) catalyst (Table 1, entry 8), indicating that the Cu and Mn species were both needed for the photothermal CO2 reduction.
Effect of the reaction conditions
The above investigation showed the composition and pretreatment of the catalyst affected the activity remarkably. In addition, the reaction conditions also had a great influence on the catalytic performance. The effects of the reaction conditions on the catalytic performance were also investigated, and the results are shown in Fig. 7. Almost no product was detected in the absence of CO2 or H2, indicating that the products are generated from CO2. This was further confirmed by a control experiment of adding 13CO2 in the reaction, in which 13CH4 was detected with a mass spectrometer (Fig. S8†).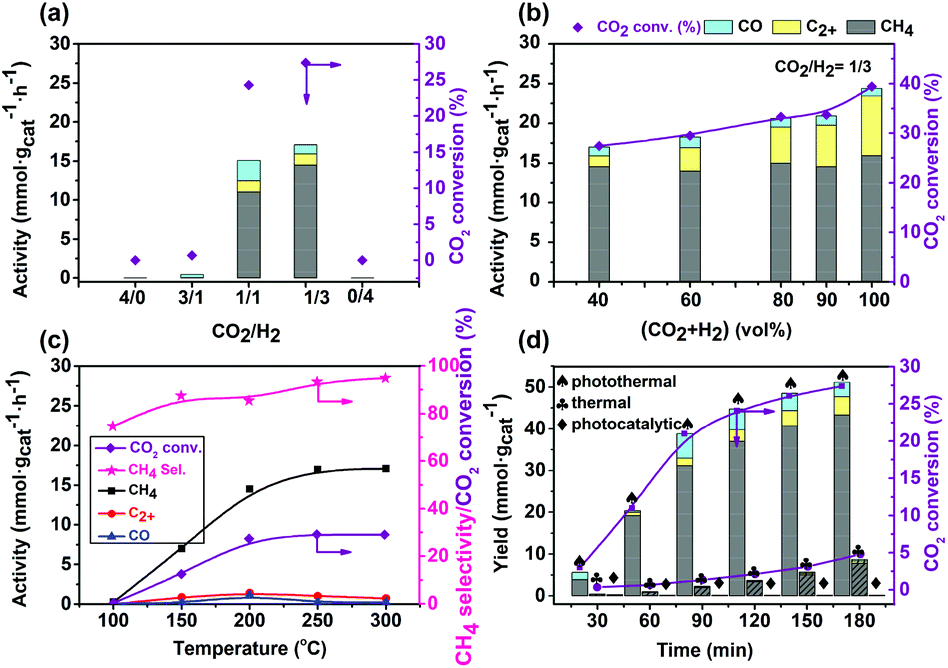 | ||
| Fig. 7 Effects of the reaction conditions on the catalytic performances of photothermal CO2 reduction over the Co7Cu1Mn1Ox(200) catalyst; (a) effect of the CO2/H2 ratio, (b) effect of the total concentration of CO2 and H2, (c) effect of the reaction temperature, and (d) effect of the reaction time. Reaction conditions: catalyst 50 mg, 300 W Xe lamp full irradiation and (a) (CO2 + H2)/N2 40%/60%, 200 °C, 3 h; (b) CO2/H2 1/3, 200 °C, 3 h; (c) CO2/H2/N2 10%/30%/60%, 3 h; and (d) CO2/H2/N2 10%/30%/60%, 200 °C, spades (/) photothermal conditions, clubs (♣) thermal conditions, and diamonds (◆) photocatalytic conditions respectively. | ||
The best performances for CH4 formation were achieved in CO2/H2 with a molar ratio of 1/3 (Fig. 6a). Surprisingly, changing the CO2 and H2 concentrations slightly affected the activity of CH4 formation (around 15 mmol gcat−1 h−1), but had an influence on C2+ hydrocarbon formation. In the absence of a dilute gas, the highest C2+ activity reached 7.5 mmol gcat−1 h−1 with a 30.8% selectivity. Generally, C2+ hydrocarbon formation involves CO2 reduction and C–C coupling steps, and is much more difficult to achieve under photothermal conditions.51 Thus, the present Cu7Cu1Mn1Ox(200) catalyst could catalyze the C2+ hydrocarbon formation, but only the CO2 and H2 concentrations need to be enhanced. The C2+ formation is strongly linked to the amount of surface adsorbed carbon species such as CO, –CH2–, etc. A high amount of carbon species can be coupled to generate C2+ hydrocarbons over a suitable catalyst, thus an increase of the CO2 concentration could enhance the C2+ selectivity. However, generally, to obtain C2+ products, high pressure and/or CO2/H2 molar ratio are always needed.42 The present Co7Cu1Mn1Ox(200) catalyst has a good CO2 adsorption capacity, which was confirmed by the CO2-TPD tests. The high C2+ selectivity indicates that the Co7Cu1Mn1Ox(200) catalyst possesses good CO2 reduction and C–C coupling abilities even under ambient pressure and a low CO2/H2 molar ratio (1/3). These multi-functional applications of synthesis of CH4 and C2+ hydrocarbons stemmed from the multiple interfaces created by the complicated compositions of Co0, Co3O4, Mn2O3, CuO and Cu2O species, etc.
The reaction temperature has a significant influence on the catalytic performance, which was monitored using a thermocouple positioned above the catalyst level (Fig. S6†). As shown in Fig. 7c, high temperature promoted the reaction remarkably. With the increase of the reaction temperature, the activity of CH4 formation increased. The C2+ selectivity increased initially and then slightly decreased at a temperature higher than 200 °C. This is probably because at high temperature, the catalyst was sintered and reduced, which affected the product distribution especially with the CH4 selectivity increased and C2+ products decreased.
The evolution of activity on reaction time was also studied and the results are shown in Fig. 7d. In the first 30 min of reaction, CH4 was formed with a very low activity, indicating that the catalyst had an induction period. During this period, the surface Co species, especially the surface CoO was reduced to the active Co0 species. With prolonged time, the CH4 formation activity increased remarkably. During our experiments, we found that the pressure of the reaction gas decreased remarkably after 3 h, but further prolonging the time did not show any remarkable decrease, indicating that the reaction has reached equilibrium. Thus, we chose 3 h as the optimum reaction time. In addition, control experiments were conducted to investigate the effect of optical and thermal conditions. The reaction could hardly occur under light irradiation but without extra heating, and the sole thermal conditions offered a very low CH4 formation activity (only one-sixth that of the photothermal reaction). However, their combination is beneficial for the CO2 reduction, indicating that the excellent catalytic performances stemmed from their synergistic promotion effect.
As mentioned above, the present catalytic system could achieve the photothermal CO2 reduction to C2+ hydrocarbons, but only needs to enhance the concentrations of CO2 and H2 to 25% and 75%. Fig. S9† shows the catalytic performances of the reaction. Under the conditions of 200 °C and 3 h, the highest C2+ selectivity and activity reached 30.8% and 7.5 mmol gcat−1 h−1 obtained after 3 h of reaction.
The results indicated that the multifunctional applications of the trimetallic Co–Cu–Mn catalysts derive from not only the multicomponents of the catalysts, but also the interaction effects between the components.
Photoelectric properties of the Co–Cu–Mn catalysts
The study of the catalytic performances confirmed that Co3O4 and Mn2O3 oxides are very important for the reaction, and this differs from the fully reduced Co-based catalysts in H2 such as Co/CoOx29 and Co6/MnOx5 catalysts in CO2 thermal hydrogenation. This diversification is probably because photothermal catalysis needs more light-sensitive components such as Co3O4 and Mn2O3 due to their semiconductor nature,52,53 while the thermal catalyst favours low-valent metals such as Co0.To further investigate the light-sensitive properties of the Co–Cu–Mn trimetallic catalysts, the UV-vis DRS spectra were investigated, and the results are shown in Fig. S10a.† The unreduced catalysts including Co7Mn1Ox, Co7Cu1Ox, and Co7Cu1Mn1Ox showed higher absorption than the corresponding reduced catalysts throughout the UV-vis region. This was because the former mainly contain Co3O4, which has strong absorption of d–d transitions.54 Upon reduction, the Co3O4 amount decreased, leading to the decrease of absorption accordingly. As for the Co7Cu1Mn1Ox(200, 300, 400, and 500) catalysts, with the increase of the reduction temperature, the absorption strength decreased gradually for the deep reduction of Co3O4 to CoO and Co0 species. Both the Co7Cu1Ox(200) and Co7Cu1Mn1Ox(500) catalysts showed a lower absorbance than the other samples because they did not contain the Co3O4 species, which was confirmed by the XRD tests.
The band gap energies (Eg) for the tested catalysts were calculated using the Kubelka–Munk (K–M) model.55 The Co7Cu1Mn1Ox(200) catalyst had a lower Eg value (1.86 eV) than those of the catalysts reduced at higher temperatures (Fig. S10b†). The Co7Cu1Mn1Ox(500) catalyst, however, had the highest Eg (3.26 eV) value and showed a very low activity in the reaction. The low Eg value of the Co7Cu1Mn1Ox(200) catalyst led to the feature that the electrons in the semiconductor could be easily excited under light irradiation. The lower band gap is conducive to visible light excitation, which was confirmed by the control experiments using different types of light including full irradiation (300–1100 nm), UV (300–420 nm), and visible light (420–800 nm). The catalytic performances obtained under visible light illumination are almost similar to those under UV-vis and full irradiation, as shown in Table S5.†
The photocurrent–time curves of the Co7Cu1Mn1Ox(100, 200, 300, and 400) catalysts are shown in Fig. S10c.† All of these catalysts showed obvious photocurrent response behavior, indicating that they could be excited to generate electron–hole pairs.
Co3O4, Mn2O3, Cu2O, and CoO are p-type semiconductors; however, after reduction, oxygen vacancies were created and regulated the p-type to a n-type semiconductor, which is confirmed by the Mott–Schottky curves shown in Fig. S10d.† All the catalysts showed dipolar (p-type and n-type) semiconductor properties, resulting from the multiple interfaces of the complex trimetallic catalyst.
The EIS Nyquist plots of the tested samples including Co7Cu1Mn1Ox(100, 200, 300, and 500) showed similar arc radii, revealing that they have similar impedance values and electron transfer rates (Fig. S10e†).56
The photothermal effect was investigated via testing the stable temperature of the samples under vacuum (Fig. S11†). Under similar conditions, the catalyst temperatures were monitored using an infrared camera. Among the tested catalysts, the MnOx catalyst showed the highest temperature (104.6 °C) under the full irradiation, while the CuOx catalyst offered the lowest (88.3 °C). The Co7Cu1Mn1Ox(200) catalyst showed a higher temperature (102.2 °C) than that of the Co7Cu1Mn1Ox(500) (95.8 °C), indicating that the Co7Cu1Mn1Ox(200) catalyst could provide a higher local heating environment. The results revealed that the presence of Mn could provide a higher local temperature for the catalyst.
In summary, the results obtained due to the photoelectrical characteristics indicated that the Co7Cu1Mn1Ox(200) catalyst is a p-type and n-type semiconductor, showing a low band gap. All of these properties are closely related to the excellent catalytic activity in photothermal CO2 reduction. However, it should be noted that the unique catalytic performances of the Co7Cu1Mn1Ox(200) catalyst were derived from the synergistic effect of the adsorption effect, light-response properties, and photothermal effect.
Light effect and the proposed mechanism for photothermal CO2 reduction
In the present catalytic system, the synergistic effect between light and heat had a great influence on the catalytic activity. Under light irradiation, the electrons of the Co7Cu1Mn1Ox(200) catalyst could be excited to generate the electron–hole pairs. Generally, in photocatalysis, the excited electrons and holes can participate in reactions.57 However, under photothermal conditions, the Eg value of the semiconductor is normally found to be lower, and the excited electrons recombine more easily with the photogenerated holes than under the traditional photocatalysis without heating.To investigate the effects of the generated electron–hole pairs, we carried out two control experiments by adding sacrificial electron donors including p-xylene (∼2.18 V vs. Ag/AgClsat) and anisole (1.92 V vs. Ag/AgClsat) in the absence of H2.18,25 As shown in Fig. S12,† both the reactions gave a very low CH4 yield despite the fact that p-xylene and anisole could quench the photogenerated holes more efficiently than H2. These results indicated that photocatalysis plays a minor role in the present photothermal CO2 reduction. In this aspect, similar to the Ru-,18 Rh-,58 Au-,58 and Ag-based59 catalysts, the charge separation and recombination under light irradiation resulted in a local thermal effect at the surface of the catalyst, offering more energy to promote CO2 reduction.
The mechanism for the photothermal CO2 reduction to hydrocarbons is still unclear, and two reaction routes are generally accepted.60 The first involves a CO intermediate, in which CO2 is initially converted to CO, and CO could be further hydrogenated to hydrocarbons.61 The other route is the direct CO2 conversion. In this route, CO2 is hydrogenated to carbonate, formate, and methoxy intermediates, and then hydrogenated to hydrocarbons. In this work, the CO formation activity was very low. To study the reaction pathway, we conducted the control experiments of replacing CO2 by CO under the conditions given in Table 1, entry 8. Interestingly, the activities of CH4 and C2+ were 4.9 and 7.3 mmol gcat−1 h−1, and their selectivities were 40.2% and 59.8%, respectively (Table 1, entry 8, Fig. S13, S14 vs. Table S6, entry 2, Fig. S15†). To exclude that the high CO concentration may occupy the active sites on the catalyst surface, a mixed gas of 2%CO/8%CO2 (total 10%) was also reacted (Table S6, entry 3 and Fig. S16†). However, the activities of CH4 and C2+ were 7.2 and 2.8 mmol gcat−1 h−1 with the selectivities of 72.0% and 28.0%, respectively. The results implied that the presence of CO could not improve but reduce the CH4 activity, meaning that in the photothermal CO2 reduction, CH4 is probably not formed via the CO intermediate route. However, CO was found to be beneficial for the C2+ synthesis, revealing that it is probably an intermediate for the formation of the C2+ hydrocarbon product.
The characterization results confirmed that the trimetallic Co7Cu1Mn1Ox(200) catalyst contains CoO, Co3O4, Cu2O, Mn2O3, etc. During the reaction, the CoO species could be reduced to Co0, which was recognized as the active site. The CoO and Cu2O species could be reduced by H2 to Co0 and Cu0 during the reaction, which was confirmed by the XPS results (Fig. 5). In the present photothermal CO2 hydrogenation, metal oxides such as Co3O4 and Mn2O3 were very important for the reaction because they are semiconductors, which could generate electrons and holes under light illumination. A quick recombination of electrons and holes provides a local heating environment for the catalysts, and further promotes the reaction.5 In addition, Mn2O3 could offer Lewis acidic sites to enhance CO2 and H2 adsorption capacity of the catalyst.
Cu species could promote the partial reduction of cobalt oxides to CoO and Co0 and maintain the presence of CoO and Co3O4 prior to and during the reaction, and reduce the temperature of catalyst reduction. Besides, the CO2-TPD tests and the control experiments confirmed that the Cu species or the Cu–Co interfaces could provide strong CO2 adsorption sites to convert CO2 to CO, and further form C2+ hydrocarbons.
It should be mentioned that, although CH4 was probably not formed via the CO route, CO could be produced with an activity of 1.1 mmol gcat−1 h−1 (entry 8, Table 1). Importantly, the higher concentration of CO could make it easily coupled into the C2+ products over the same Co7Cu1Mn1Ox(200) catalyst. Thus, C2+ hydrocarbons could be formed by increasing the CO2 concentration.
In addition, the present protocol could be extended to photothermal CO hydrogenation. We conducted the reaction over the Co7Cu1Mn1Ox(200) catalyst under the similar conditions given in Table 1, entry 8, and the results are shown in Fig. S17.† The photothermal activity is about 7 times higher than that of the thermal activity, and the selectivity of the C2+ products is higher than that of CH4, which is similar to CO2 hydrogenation. The results indicated that the present trimetallic Co–Cu–Mn catalyst has multifunctional applications as shown in photothermal CO hydrogenation.
The reusability and stability studies of the Co7Cu1Mn1Ox(200) catalyst
The reusability of the Co7Cu1Mn1Ox(200) catalyst was investigated, and the results are shown in Fig. 8a. For comparison, the CO2 reduction reactions were alternatively conducted under photothermal and thermal conditions. The results revealed that the present catalyst possesses good reusability in the reaction, and after using for 18 times, it still offered an activity of higher than 10 mmolCH4 gcat−1 h−1. Besides, the results also confirmed that the activities under photothermal conditions were much higher than those under thermal conditions. The used catalysts were characterized by XRD and XPS, and the results are shown in Fig. 5, respectively.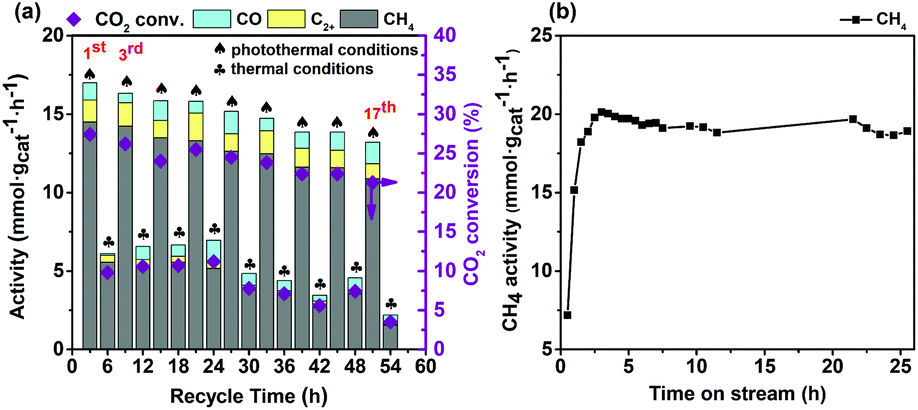 | ||
| Fig. 8 Reusability (a) and temporal stability (b) studies of Co7Cu1Mn1Ox(200) in photothermal CO2 reduction. Reaction conditions: (a) catalyst 50 mg, 200 °C, CO2/H2/N2 10%/30%/60%, full irradiation for photothermal conditions; (b) catalyst 50 mg, 200 °C, CO2/H2/N2 10%/30%/60% 20 mL min−1, full irradiation. | ||
The activity loss is probably caused by catalyst agglomeration, which was confirmed by the TEM images of the catalyst after being used 18 times (Fig. S18).† The size of the used catalyst increased in the range 40–90 nm.
Fig. 8b shows the temporal stability of the Co7Cu1Mn1Ox(200) catalyst in a continuous flow operation (details are given in the ESI†). The catalyst offered approximately 20 mmol gcat−1 h−1 of CH4 after a short induction period of 3 h, and maintained higher than 18 mmol gcat−1 h−1 even after 25 hours of reaction. The result showed that the present Co7Cu1Mn1Ox(200) catalyst has a long-term temporal stability, confirming that it has a potential industrial application.
Conclusions
In summary, a trimetallic Co–Cu–Mn catalytic system for photothermal CO2 reduction was constructed by a simple co-precipitation method. The metal compositions and reduction temperatures of the catalysts affected their activities remarkably, and the screened Co7Cu1Mn1Ox(200) catalyst could offer 14.5 mmol gcat−1 h−1 of CH4 at low CO2 (10%) and H2 (30%) concentrations. Importantly, the catalyst system could produce C2+ hydrocarbons with an activity of 7.5 mmol gcat−1 h−1 and a selectivity of 30.8% by only improving the CO2 and H2 concentrations to 25% and 75%, respectively. High activities stemmed from the multicomponents of the catalyst, especially the metallic Co0, Cu0, and CoO, Co3O4, Cu2O, and Mn2O3 semiconductors. The Cu species could tune the reduction characteristics, and further tune the balance of Co2+ and Co3+. The Mn species could enhance the CO2 and H2 adsorption, and also offer a high local temperature for the nanocatalyst. The present strategy of partial reduction of the Co–Cu–Mn trimetallic catalyst not only highlights the route for using solar energy to produce valuable chemicals under mild conditions especially low CO2 concentration, but also constructs the highly efficient trimetallic catalysts with multifunctional applications in photothermal catalysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial supports from the National Natural Science Foundation of China (21978160, 21776170, 22078182, 21706152, and 21908139), the Natural Science Basic Research Plan in Shaanxi Province of China (Program No. 2019JLM-16, 2019JQ-772, and 2019JQ-782) and the Key Industrial Innovation Project of Shaanxi Provincial Science and Technology Department (2019ZDLGY06-04).Notes and references
- M. He, Y. Sun and B. Han, Angew. Chem., Int. Ed., 2013, 52, 9620–9633 CrossRef CAS PubMed.
- L. Wang, Y. Dong, T. Yan, Z. Hu, A. A. Jelle, D. M. Meira, P. N. Duchesne, J. Y. Y. Qiu, E. E. Storey, Y. Xu, W. Sun, M. Ghoussoub, N. P. Kherani, A. S. Helmy and G. A. Ozin, Nat. Commun., 2020, 11, 2432 CrossRef CAS PubMed.
- B. Zhou, P. Ou, N. Pant, S. Cheng, S. Vanka, S. Chu, P. T. Rashid, G. Botton, J. Song and Z. Mi, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 1330–1338 CrossRef CAS PubMed.
- M. Cui, Q. Qian, J. Zhang, Y. Wang, B. B. A. Bediako, H. Liu and B. Han, Chem, 2021, 7, 726–737 CAS.
- Z. He, M. Cui, Q. Qian, J. Zhang, H. Liu and B. Han, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 1254–12659 Search PubMed.
- Z. Ma and M. D. Porosoff, ACS Catal., 2019, 9, 2639–2656 CrossRef CAS.
- R. De, S. Gonglach, S. Paul, M. Haas, S. S. Sreejith, P. Gerschel, U.-P. Apfel, T. H. Vuong, J. Rabeah, S. Roy and W. Schöfberger, Angew. Chem., Int. Ed., 2020, 59, 10527–10534 CrossRef CAS PubMed.
- Y. Wang, X. Shang, J. Shen, Z. Zhang, D. Wang, J. Lin, J. C. S. Wu, X. Fu, X. Wang and C. Li, Nat. Commun., 2020, 11, 3043 CrossRef CAS PubMed.
- A. Weihard, S. P. Argent and V. Sans, Nat. Commun., 2021, 12, 231 CrossRef PubMed.
- Z. He, Q. Qian, J. Ma, Q. Meng, H. Zhou, J. Song, Z. Liu and B. Han, Angew. Chem., Int. Ed., 2016, 55, 737–741 CrossRef CAS PubMed.
- L. Wang, E. Guan, Y. Wang, L. Wang, Z. Gong, Y. Cui, X. Meng, B. C. Gates and F.-S. Xiao, Nat. Commun., 2020, 11, 1033 CrossRef CAS PubMed.
- M. Rahaman, K. Kiran, I. Z. Montiel, V. Grozovski, A. Dutta and P. Broekmann, Green Chem., 2020, 22, 6497–6509 RSC.
- G. Chen, R. Gao, Y. Zhao, Z. Li, G. I. N. Waterhouse, R. Shi, J. Zhao, M. Zhang, L. Shang, G. Sheng, X. Zhang, X. Wen, L.-Z. Wu, C.-H. Tung and T. Zhang, Adv. Mater., 2017, 1734663 Search PubMed.
- F. Sastre, A. V. Puga, L. Liu, A. Corma and H. García, J. Am. Chem. Soc., 2014, 136, 6798–6801 CrossRef CAS PubMed.
- J. Albero, E. Dominguez, A. Corma and H. Garcia, Sustain. Energy Fuels, 2017, 7, 1303–1307 RSC.
- F. Zhang, Y.-H. Li, M.-Y. Qi, Y. M. A. Yamada, M. Anpo, Z.-R. Tang and Y.-J. Xu, Chem. Catal., 2021, 1, 1–26 CrossRef.
- P. G. O'Brien, A. Sandhel, T. E. Wood, A. A. Jelle, L. B. Hoch, D. D. Perovic, C. A. Mims and G. A. Ozin, Adv. Sci., 2014, 1, 1400001 CrossRef PubMed.
- D. Mateo, J. Albero and H. García, Joule, 2019, 3, 1–14 CrossRef.
- A. A. Jelle, K. K. Ghuman, P. G. O'Brien, M. Hmadeh, A. Sandhel, D. D. Perovic, C. V. Singh, C. A. Mims and G. A. Ozin, Adv. Energy Mater., 2018, 8, 1702277 CrossRef.
- J. Ren, S. Ouyang, H. Xu, X. Meng, T. Wang, D. Wang and J. Ye, Adv. Energy Mater., 2016, 1601657 Search PubMed.
- X. Meng, T. Wang, L. Liu, S. Ouyang, P. Li, H. Hu, T. Kako, H. Iwai, A. Tanaka and J. Ye, Angew. Chem., Int. Ed., 2014, 53, 11478–11482 CrossRef CAS PubMed.
- C. Wang, S. Fang, S. Xie, Y. Zheng and Y. H. Hu, J. Mater. Chem. A, 2020, 8, 7990–7394 Search PubMed.
- J. Jia, H. Wang, Z. Lu, P. G. O'Brien, M. Ghoussoub, P. Duchesne, Z. Zheng, P. Li, Q. Qiao, L. Wang, A. Gu, A. A. Jelle, Y. Dong, Q. Wang, K. K. Ghuman, T. Wood, C. Qian, Y. Shao, C. Qiu, M. Ye, Y. Zhu, Z.-H. Lu, P. Zhang, A. S. Helmy, C. V. Singh, N. P. Kherani, D. D. Perovic and G. A. Ozin, Adv. Sci., 2017, 1700252 CrossRef PubMed.
- M. Cabrero-Antonino, S. Remiro-Buenamañana, M. Souto, A. A. García-Valdivia, D. Choquesillo-Lazarte, S. Navalón, A. Rodríguez-Diéguez, G. M. Espallargas and H. García, Chem. Commun., 2019, 55, 10932–10935 RSC.
- D. Mateo, J. Albero and H. García, Energy Enviorn. Sci., 2017, 10, 2392–2400 RSC.
- M.-P. Jiang, K.-K. Huang, J.-H. Liu, D. Wang, Y. Wang, X. Wang, Z.-D. Li, X.-Y. Wang, Z.-B. Geng, X.-Y. Hou and S.-H. Feng, Chem, 2020, 6, 2335–2346 CAS.
- J. A. Rodriguez, P. Liu, D. J. Stacchiola, S. D. Senanayake, M. G. White and J. G. Chen, ACS Catal., 2015, 5, 6696–6706 CrossRef CAS.
- R. Lang, X. Du, Y. Huang, X. Jiang, Q. Zhang, Y. Guo, K. Liu, B. Qiao, A. Wang and T. Zhang, Chem. Rev., 2020, 120, 11986–12043 CrossRef CAS PubMed.
- K. Zhao, M. Calizzi, E. Moioli, M. Li, A. Borsay, L. Lombardo, R. Mutschler, W. Luo and A. Zuttel, J. Energy Chem., 2021, 53, 241–250 CrossRef.
- M. Ao, G. H. Pham, V. Sage, V. Pareek and S. Liu, Fuel Process. Technol., 2019, 193, 141–148 CrossRef CAS.
- Y. Xiang, R. Barbosa, N. Kruse and X. Li, ACS Catal., 2015, 5, 2929–2934 CrossRef CAS.
- Y. Wang, S. Kattel, W. Gao, K. Li, P. Liu, J. G. Chen and H. Chen, Nat. Commun., 2019, 10, 1166 CrossRef PubMed.
- A. A. S. Goncalves, P. B. Faustino, J. M. Assaf and M. Jaroniec, ACS Appl. Mater. Interfaces, 2017, 9, 6079–6092 CrossRef CAS PubMed.
- Y. Li, H. S. Pillai, J. Lattimer, N. M. Adli, S. Karakalos, M. Chen, L. Guo, H. Xu, J. Yang, D. Su, H. Xin and G. Wu, ACS Catal., 2020, 10, 3945–3957 CrossRef CAS.
- M. Sharma, J.-H. Jang, D. Y. Shin, J. A. Kwon, D.-H. Lim, D. Choi, H. Sung, J. Jang, S.-Y. Lee, K. Y. Lee, H.-Y. Park, N. Jung and S. J. Yoo, Energy Environ. Sci., 2019, 12, 2200–2211 RSC.
- L. Martin, H. Martinezz, D. Poinot, B. Pecquenard and F. L. Cras, J. Phys. Chem. C, 2013, 117, 4421–4430 CrossRef CAS.
- Z. Geng, Y. Wang, J. Liu, G. Li, L. Li, K. Huang, L. Yuan and S. Feng, ACS Appl. Mater. Interfaces, 2016, 8, 27825–27831 CrossRef CAS PubMed.
- L. Lukaskuk, N. Yigit, R. Rameshan, E. Kolar, D. Teschner, M. Havecker, A. Knop-Gericke, R. Schlogl, K. Fottinger and G. Rupprechter, ACS Catal., 2018, 8, 8630–8641 CrossRef PubMed.
- J. Su, Z. Zhang, D. Fu, X. Xu, B. Shi, X. Wang, R. Si, Z. Jiang, J. Xu and Y. Han, J. Catal., 2016, 336, 94–106 CrossRef CAS.
- B. Rivas-Murias and V. Salgueiriño, J. Raman Spectrosc., 2017, 48, 837–841 CrossRef CAS.
- X. Jiang, X. Nie, X. Guo, C. Song and J. G. Chen, Chem. Rev., 2020, 120, 7984–8034 CrossRef CAS PubMed.
- Y. Xiang, V. Chitry, P. Liddicoat, P. Felfer, J. Cairney, S. Ringer and N. Kruse, J. Am. Chem. Soc., 2013, 135, 7114–7117 CrossRef CAS PubMed.
- Z.-H. He, C.-S. Jiang, K. Wang, Z.-Y. Wang, N. Li, W.-T. Wang and Z.-T. Liu, Catal. Today, 2020, 356, 579–588 CrossRef CAS.
- M. Tahir, B. Tahir, N. A. S. Amin and A. Muhammad, Energy Convers. Manag., 2016, 119, 368–378 CrossRef CAS.
- Z.-H. He, N. Li, K. Wang, W.-T. Wang and Z.-T. Liu, Mol. Catal., 2019, 470, 120–126 CrossRef.
- O. A. Bulavchenko, E. Y. Gerasimov and T. N. Afonasenko, Dalton Trans., 2018, 47, 17153–17159 RSC.
- W. Yang, Y. Peng, Y. Wang, Y. Wang, H. Liu, Z. Su, W. Yang, J. Chen, W. Si and J. Li, Appl. Catal., B, 2020, 278, 119279 CrossRef CAS.
- W. Yang, S. Wang, K. Li, S. Liu, L. Gan, Y. Peng and J. Li, Chem. Eng. J., 2019, 364, 448–451 CrossRef CAS.
- C.-I. Ahn, D.-W. Jeong, J. M. Cho, H.-S. Na, W.-J. Jang, H.-S. Roh, J.-H. Choi, S.-H. Um and J.-W. Bae, Microporous Mesoporous Mater., 2016, 221, 204–211 CrossRef CAS.
- O. A. Bulavchenko, E. Y. Gerasimov and T. N. Afonasenko, Dalton Trans., 2018, 47, 17153–17159 RSC.
- G. Chen, R. Gao, Y. Zhao, Z. Li, G. I. N. Waterhouse, R. Shi, J. Zhao, M. Zhang, L. Shang, G. Sheng, X. Zhang, X. Wen, L. Wu, T. Tung and T. Zhang, Adv. Mater., 2018, 30, 1704663 CrossRef PubMed.
- J. Zhao, Z. Zhao, N. Li, J. Nan, R. Yu and J. Du, Chem. Eng. J., 2018, 353, 805–813 CrossRef CAS.
- Y. Zeng, H. Li, Y. Xia, L. Wang, K. Yin, Y. Wei, X. Liu and S. Luo, ACS Appl. Mater. Interfaces, 2020, 12, 44608–44616 CrossRef CAS PubMed.
- Z. Li, J. Liu, Y. Zhao, R. Shi, G. I. N. Waterhouse, Y. Wang, L.-Z. Wu, C.-H. Tung and T. Zhang, Nano Energy, 2019, 60, 467–475 CrossRef CAS.
- A. Umasadharshini, M. Bououdina, M. Venkateshwarlu, C. Manoharan and P. Dhamodharan, Surf. Interfaces, 2020, 19, 100535 CrossRef.
- Q. Wan, J. Zhang, B. Zhang, D. Tan, L. Yao, L. Zheng, F. Zhang, L. Lin, X. Cheng and B. Han, Green Chem., 2020, 22, 2750–2754 RSC.
- N. Li, M. Liu, B. Yang, W. Shu, Q. Shen, M. Liu and J. Zhou, J. Phys. Chem. C, 2017, 121, 2923–2932 CrossRef CAS.
- X. Zhang, X. Li, D. Zhuang, N. Q. Su, W. Yang, H. O. Everitt and J. Liu, Nat. Commun., 2017, 8, 14542 CrossRef CAS PubMed.
- P. Christopher, H. Xin and S. Linic, Nat. Chem., 2011, 3, 467–472 CrossRef CAS PubMed.
- B. Miao, S. S. K. Ma, X. Wang, H. Su and S. H. Chan, Catal. Sci. Technol., 2016, 6, 4048–4058 RSC.
- B. Tahir, M. Tahir and N. S. Amin, Energy Convers. Manage., 2015, 90, 272–281 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc01152a |
| This journal is © The Royal Society of Chemistry 2021 |