
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect synthesis of polycarbonate diols from atmospheric flow CO2 and diols without using dehydrating agents†
Yu
Gu
 a,
Masazumi
Tamura
*b,
Yoshinao
Nakagawa
a,
Kenji
Nakao
c,
Kimihito
Suzuki
c and
Keiichi
Tomishige
*a
a,
Masazumi
Tamura
*b,
Yoshinao
Nakagawa
a,
Kenji
Nakao
c,
Kimihito
Suzuki
c and
Keiichi
Tomishige
*a
aDepartment of Applied Chemistry, School of Engineering, Tohoku University, 6-6-07 Aoba, Aramaki, Aoba-ku, Sendai, 980-8579, Japan. E-mail: tomi@erec.che.tohoku.ac.jp
bResearch Center for Artificial Photosynthesis, The Advanced Research Institute for Natural Science and Technology, Osaka City University, 3-3-138 Sugimoto, Sumiyoshi, Osaka, 558-8585, Japan. E-mail: mtamura@osaka-cu.ac.jp
cNippon Steel Corporation, 20-1 Shintomi, Futtsu, Chiba, 293-8511, Japan
First published on 26th July 2021
Abstract
Polymer synthesis with CO2 as a C1 chemical has attracted much attention from the viewpoint of green chemistry. The direct transformation of CO2 and diols into polycarbonate diols is promising as an alternative method to the hazardous phosgene process, however, challenging due to the inert characteristic of CO2 and thermodynamic limitation. Herein, we present the direct synthesis of polycarbonate diols from atmospheric pressure CO2 and α,ω-diols using a heterogeneous CeO2 catalyst and a CO2 flow semi-batch reactor. The target alternating polycarbonate diol from CO2 and 1,6-hexanediol was obtained with high yield (92%) and selectivity (97%) without using any dehydrating agents. Activation of atmospheric pressure CO2 by a CeO2 catalyst and the shift of equilibrium towards the product by removing the coproduced water (gas stripping) are responsible for the high yield. The flow reaction system with a CeO2 catalyst was applicable to the reactions of CO2 and primary mono-alcohols or 1,2-diols, giving the target organic carbonates in high selectivity (>99%).
Introduction
The reduction of CO2 in the atmosphere is an urgent issue for mankind to achieve a sustainable society because of global warming and related problems such as abnormal weather, sea level rise, ecological system change, and so on. CO2 is renewable, abundant, cheap, non-flammable and non-toxic, and the transformation of CO2 as a C1 source into valuable chemicals is one of the promising strategies.1–5 Various effective catalyst systems have been developed for the reductive transformation of CO2 into methanol, CO, hydrocarbons, formic acid, etc.6–20 and for the non-reductive transformation into organic carbonates, carbamates, ureas, and their polymers.20–47 Organic carbonates such as dialkyl carbonates, cyclic carbonates, and polycarbonates are valuable chemicals and conventionally made by the phosgene process with problems such as the use of hazardous phosgene and dichloromethane, and the production of waste salt from the reaction with sodium hydroxide.28–31 The direct conversion of CO2 with alcohols to the carbonates can be an ideal substitute because water is the only by-product, and the substrates are easily available and safe. However, the high yield synthesis of organic carbonates, especially polycarbonates, is difficult because of the low reactivity of CO2 and low yield restricted by the equilibrium (Fig. 1).28–31,48 CeO2-based catalysts have been reported to be effective heterogeneous ones for the reaction owing to the high potential for CO2 activation and the equilibrium yields of the corresponding dialkyl carbonates were lower than 2% even with pressurized CO2 up to 20 MPa.28–31 It is reported that high CO2 pressure is required for the spontaneous progress of the reaction (e.g., ΔG < 0, PCO2 > 2.41 × 104 MPa in the dimethyl carbonate synthesis at 353 K, theoretically).49 Such enormous pressure is technically difficult and consequently, the coproduced water should be removed to shift the equilibrium to the product side. Various dehydration systems equipped with membranes,50,51 physical adsorbents,41,52–55 and chemical dehydrating agents56–68 have been developed (Fig. 1). Membranes such as inorganic ceramic membranes and organic polymer membranes50,51 and physical adsorbents such as molecular sieves52–55 provided low yields of organic carbonates despite the high CO2 pressure (0.4–30 MPa), and the low durability, regeneration treatment, and/or complex reaction system are also problematic. The synthesis combined with a reactive dehydration reaction using chemical dehydrating agents such as nitriles, acetals, epoxides, ionic liquids, and so on has been regarded as effective systems.56–69 A handful of dehydrating agents including 2-cyanopyridine,62–64 cyanopyrazine,62,64 and 2-furonitrile60 with a CeO2 catalyst enabled sufficient removal of water to shift the equilibrium to the product side and provided over 90% yield of dialkyl carbonates. Various problems for industrialization, however, remain unsolved, including the regeneration of the dehydrating agents, high CO2 pressure, formation of dehydrating agent-derived by-products, and decomposition of dehydrating agents.56,59,60,64 On the other hand, the synthesis of dialkyl carbonates from CO2 and alcohols without using dehydrating agents has also been reported. Ti-Based and Cu–Ni-based MOFs and graphene oxide immobilized Cs2Mo6Br14 provided 6–20% yield of the dialkyl carbonates,70–73 and the catalysts themselves may play a role in H2O removal from the reaction system.31,72 | ||
| Fig. 1 Direct synthesis of organic carbonates from CO2 and alcohols with/without dehydration systems and the strategy of this work. | ||
Polymers are essential materials in our life, and the development of greener synthesis methods for them is indispensable to attain a sustainable society. In this context, the direct synthesis of polymers from CO2 is a hot topic, and various effective reaction systems with suitable chemicals and/or catalysts have been developed: CO2 can polymerize with cyclic ethers (epoxides and oxetanes) to polycarbonates,21–25,40 alkenes or alkynes to polyesters,74–76 dihalide and diols to polycarbonates,77,78 diols to polycarbonates,59,60,79,80 and diamines to polyureas,81–83 and so on.84 Polycarbonates are useful chemicals with high demand (4.3 million tons in 2015, and expected to reach 7.7 million tons by 202485) as engineering plastics and intermediates for the production of polyurethanes. Polycarbonates have been conventionally produced by using phosgene, which has many drawbacks such as the high toxicity of phosgene, use of a large amount of solvents and caustic sodium hydroxide, and formation of a large amount of waste salt by neutralization.25,59,85 As an alternative method, the Asahi Kasei process was industrialized in 2012 using CO2 as the carbonyl source, and the process is composed of four steps including the carbonate synthesis from ethylene oxide and CO2, and transesterification of carbonates and alcohols.86,87 Considering the versatility, availability and handling ability of alcohols, the direct synthesis of polycarbonates from CO2 and diols is a promising process. The atom efficiency of the reaction is higher than those of the reactions applied in other processes (direct process: 94%, phosgene process: 63%, Asahi Kasei transesterification process: 81% in the case of bis(6-hydroxyhexyl)carbonate synthesis from CO2 and 1,6-hexanediol. Formulas are shown in Fig. S1†). Despite the theoretical advantages, the reaction of CO2 and diols has only been reported with additional reagents: alkyl α,ω-dihalides were used as a co-reactant with CO2 and diols under 1 MPa CO2, affording polycarbonates with an Mn of 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1.77 A CeO2 catalyst with nitrile dehydrating agents such as 2-cyanopyridine and 2-furonitrile was also reported to be effective for the polymerization of CO2 and α,ω-diols under 5 MPa CO2, producing the polycarbonates with Mn values lower than 6000 g mol−1 due to the nitrile-related side reactions (a detailed comparison of these works is shown in Table S1†).59,60,79,80 The high pressure of CO2 and use of dihalides and nitriles are not preferable and the development of a practical and hazardous reagent-free process for the synthesis of polycarbonate diols from low-pressure CO2 and diols is highly required.
000 g mol−1.77 A CeO2 catalyst with nitrile dehydrating agents such as 2-cyanopyridine and 2-furonitrile was also reported to be effective for the polymerization of CO2 and α,ω-diols under 5 MPa CO2, producing the polycarbonates with Mn values lower than 6000 g mol−1 due to the nitrile-related side reactions (a detailed comparison of these works is shown in Table S1†).59,60,79,80 The high pressure of CO2 and use of dihalides and nitriles are not preferable and the development of a practical and hazardous reagent-free process for the synthesis of polycarbonate diols from low-pressure CO2 and diols is highly required.
Gas stripping is a separation process, wherein a gas passes through a liquid mixture to selectively remove some components with lower boiling points from the liquid phase.88,89 As for the synthesis of polycarbonate diols from CO2 and diols, the boiling points of diols and the target polycarbonate diols can be adequately higher than that of H2O, and flowing CO2 will serve as a reactant as well as a stripper for the removal of the coproduced water. Therefore, we envisioned that a high yield of target polycarbonate diols can be obtained by selectively removing the coproduced water from the reaction media by gas stripping with CO2 flow and shifting the equilibrium to the product side if diols can react with atmospheric pressure CO2 by using an appropriate catalyst. It is necessary to precisely control the reaction conditions such as reaction temperature, solvents, CO2 flow rate, and so on to decrease the water concentration of the reaction media to a low level.
Herein, we found that the combination of a CeO2 catalyst and CO2 flow reaction setup with gas stripping was an effective reaction system for the direct synthesis of polycarbonate diols from atmospheric CO2 and diols.
Results and discussion
Application of a CO2 flow semi-batch reactor in the reaction of 1,6-hexanediol and CO2 over CeO2
We commenced with the reaction of 1,6-hexanediol and flow CO2 at atmospheric pressure over CeO2, which is a well-known catalyst for this type of reaction as mentioned above, in triethylene glycol dimethyl ether (triglyme) solvent at 473 K by using a CO2 flow semi-batch reactor (Fig. 2a, the details of the reactor are shown in Fig. S2†). The boiling points of 1,6-hexanediol (522 K) and triglyme solvent (489 K) are higher than the reaction temperature of 473 K, however, their vaporization under the gas flow conditions leads to material loss. A condenser was connected to the outlet of the flask and the temperature was set to 297 K to trap the vaporized substrate and solvent in the gas flow. Triglyme was selected as a solvent because it can dissolve 1,6-hexanediol, and the solidified 1,6-hexanediol (melting point: 315 K) at the condenser was rinsed away with the trapped triglyme. The reaction of flow CO2 and 1,6-hexanediol proceeded smoothly to reach 50% yield in 24 h with a high diol balance (>99%) (Fig. 2a). The corresponding dimer, bis(6-hydroxyhexyl)carbonate, was formed as the sole product until 4 h, and the formation of the polymer, which is defined as a trimer and further polymerized products in this paper, appeared at a longer reaction time. The water content in the reaction media measured by coulometric Karl-Fischer titration (Fig. S3†) was below the detection limit (<4 ppm), suggesting that the coproduced water was efficiently removed from the reaction media. The amount of the coproduced water at the initial 4 h is calculated to be 1.0 mmol (0.018 g) from the conversion. Assuming that all the coproduced water was diffused to the gas phase at a constant rate, the water concentration at a gas flow rate of 200 mL min−1 can be estimated to be 0.38 g m−3, which is far below the water concentration of saturated water vapor at 297 K (condenser temperature), 22 g m−3. Therefore, the coproduced water in the gas flow cannot be trapped in the condenser.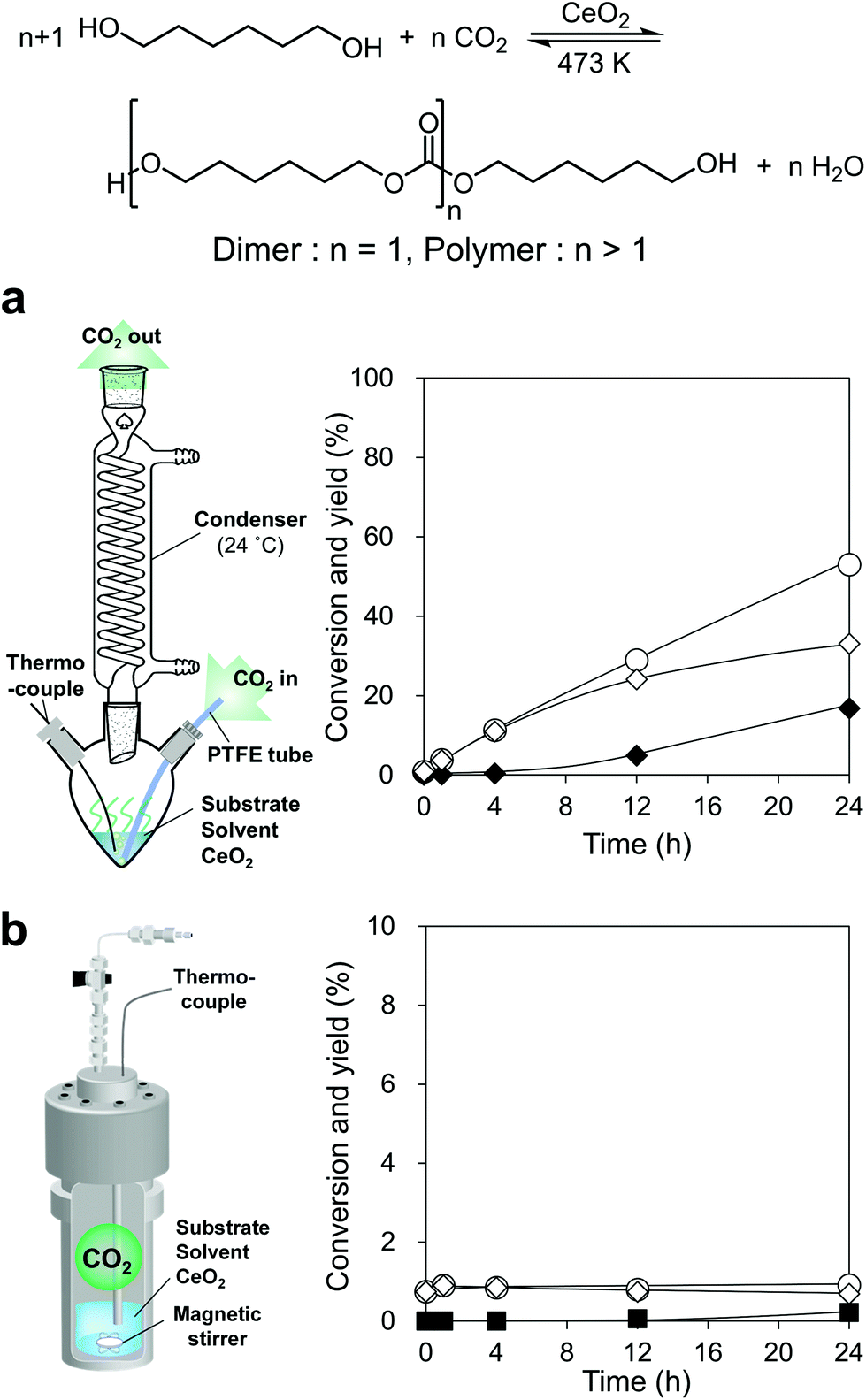 | ||
| Fig. 2 Reaction of CO2 and 1,6-hexanediol over CeO2 with two different reactors: (a) CO2 flow semi-batch reactor and time-course under atmospheric CO2 flow and (b) sealed batch reactor and time-course under 7.5 MPa CO2. ○, Conversion; ◊, dimer yield; ◆, polymer yield; and ■, Others yield (Others include ester and ether). Reaction conditions: (a) CeO2 0.10 g, 1,6-hexanediol 2.0 g (17 mmol), triglyme 3 g, 473 K, CO2 flow rate 200 mL min−1. (b) CeO2 0.10 g, 1,6-hexanediol 2.0 g (17 mmol), triglyme 3 g, 473 K, CO2 7.5 MPa, the detailed data are shown in Table S2.† | ||
To check the equilibrium level in a sealed batch reactor under atmospheric pressure CO2, 1,6-hexanediol was reacted with 0.1 MPa of CO2 in an autoclave reactor, giving the result that the formation amount of the target carbonate was below the detection limit (yield < 0.01%, Table S3†). Even in the reaction with high-pressure CO2 (7.5 MPa) in the sealed batch reactor (Fig. 2b), the conversion was low and became constant at 0.9% (4–24 h) with a dimer produced as the main product. These results indicate that the equilibrium yield at high CO2 pressure (7.5 MPa) is about 1%, and the equilibrium yield with atmospheric pressure CO2 at 473 K in the sealed batch reactor can be estimated to be below 0.01%. The water content of the reaction mixture in the sealed batch reactor under 7.5 MPa was 600–700 ppm (Fig. S3†), which was mainly from the water impurity of the reagents and the coproduced H2O from the carbonate formation. The water amount was clearly higher than that in the CO2 flow semi-batch reactor. As a result, the conversion in the CO2 flow semi-batch reactor largely exceeded the equilibrium level, which is hardly achievable in a sealed-batch reactor without dehydrating agents. As above, the coproduced water in the CO2 flow semi-batch reactor can be removed from the reaction media, leading to high conversion and yields by shifting the chemical equilibrium to the product side.
Effect of reaction parameters on the reaction of 1,6-hexanediol and CO2 in a CO2 flow semi-batch reactor
To confirm the specificity of CeO2 for the reaction in a CO2 flow semi-batch reactor, the reactions without a catalyst and with various solid oxides were carried out (Table S4†). No products were detected without using a catalyst (yield < 0.01%). CeO2 exhibited excellent catalytic activity towards the reaction of flow CO2 and 1,6-hexanediol under atmospheric pressure, giving 17% conversion of the diol and >99% selectivity to organic carbonate species including the dimer and the polymer. In the cases of γ-Al2O3 and SiO2-Al2O3, there were no carbonate products, while linear and cyclic ethers of 1,6-hexanediol were produced as major products with 5-hexen-1-ol and its isomers as minor products. The strong acidity of these catalysts is reported to be responsible for their catalytic activity.90–92 Other metal oxides used in the experiments were inactive, and no products were obtained. The initial formation rate of the dimer with CeO2 was found to be 15 mmol gcat−1 h−1 (Fig. S4†), which is adequately high compared with the previous reports (0.1–11 mmol gcat−1 h−1) using organic dehydrating agents except for the cases with 2-cyanopyridine (75 mmol gcat−1 h−1),28 CCl3COOMe (70 mmol gcat−1 h−1),28 and 2,2-diethoxypropane (44 mmol gcat−1 h−1).61 Therefore, the catalyst system is quite meaningful from the practical viewpoint.Various reaction parameters were investigated in the same reaction (Fig. 3). The conversion at 1 h was plotted as a function of the CeO2 amount (Fig. 3a), showing that the conversion increased with the increase of the CeO2 amount in the range of 0–0.08 g and reached 3.8% in this range. The reaction progress was controlled by the catalyst amount in the range of 0–0.08 g, indicating that the rate-determining factor is the catalytic activity of CeO2 in the reaction of 1,6-hexanediol with CO2. With larger amounts of CeO2 from 0.08 to 0.20 g, the conversion became constant at 3.8%, indicating that the rate-determining factor here is the removal of water from the reaction media.
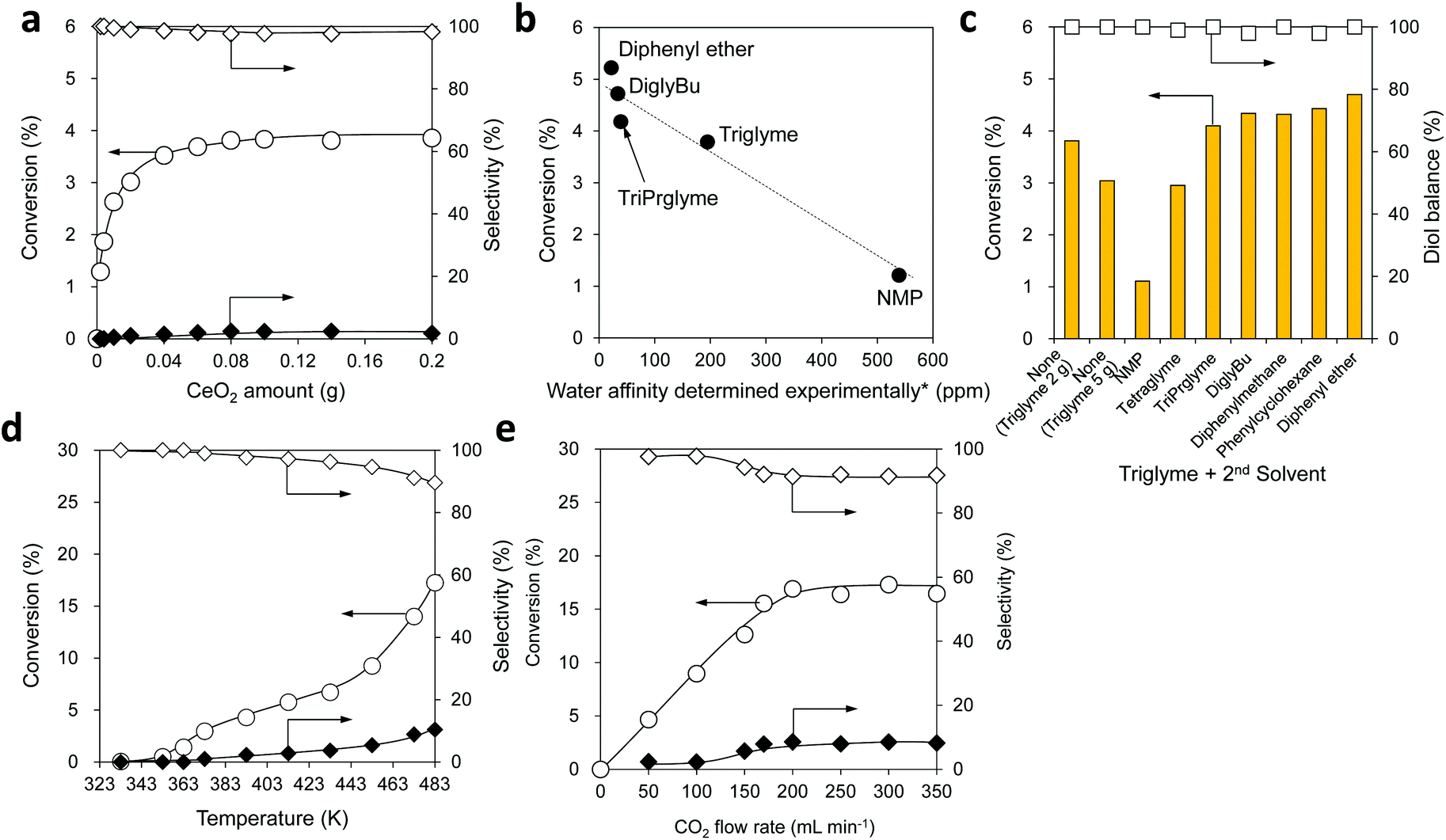 | ||
| Fig. 3 Effect of reaction parameters: (a) CeO2 amount, (b) relationship between the water affinity of solvents and the conversion, (c) solvent effect, (d) reaction temperature, and (e) CO2 flow rate. Marks in (a), (d) and (e): ○, Conversion; ◊, dimer selectivity; and ◆, polymer selectivity. Mark and bar in (c):□, Diol balance; bar, conversion. *The ability of solvents to retain water under the reaction conditions. Reaction conditions: (a) CeO2 0–0.20 g, 1,6-hexanediol 2.0 g (17 mmol), triglyme 3 g, CO2 flow rate 200 mL min−1, 473 K, 1 h. (b) CeO2 0.10 g, 1,6-hexanediol 2.0 g (17 mmol), solvent 3 g, CO2 flow rate 200 mL min−1, 473 K, 1 h. (c) CeO2 0.10 g, 1,6-hexanediol 2.0 g (17 mmol), triglyme 2 g, 2nd solvent 0 or 3 g, CO2 flow rate 200 mL min−1, 473 K, 1 h. (d) CeO2 0.10 g, 1,6-hexanediol 2.0 g (17 mmol), triglyme 2 g, diphenyl ether 3 g, CO2 flow rate 200 mL min−1, 333–483 K, 4 h. (e) CeO2 0.10 g, 1,6-hexanediol 2.0 g (17 mmol), triglyme 2 g, diphenyl ether 3 g, CO2 flow rate 0–350 mL min−1, 483 K, 4 h. | ||
Next, the solvent effect on the reaction was investigated with an adequate amount of the CeO2 catalyst (0.10 g). The reaction under the neat conditions provided a similar conversion to the previous case, however, the diol balance decreased to 87%, and the solidified 1,6-hexanediol was observed in the condenser (Fig. S5 and S6†), leading to a block of the flow line at a longer reaction time. Other solvents with high boiling points including N-methyl-2-pyrrolidone (NMP), tetraethylene glycol dimethyl ether (tetraglyme), tripropylene glycol dimethyl ether (isomer mixture, triPrglyme), diethylene glycol dibutyl ether (diglyBu), diphenylmethane, phenylcyclohexane, and diphenyl ether were applied to the reaction. The solvents with low solubility in water (≤2 g L−1-H2O) including diglyBu, diphenylmethane, phenylcyclohexane, and diphenyl ether, provided higher conversion (4.7–5.2%) to the carbonates than triglyme (Fig. S5†). However, the solidification of 1,6-hexanediol occurred in the condenser due to their poor ability to dissolve 1,6-hexanediol (Table S5†), resulting in a lower diol balance (90–97%) and a difficulty in the prolonged reaction similar to the case of the neat conditions. On the other hand, the reactions with triglyme or tetraglyme that are miscible with water provided a higher diol balance (>99%) and adequate conversion (3.8%).
We estimated the ability of the solvents to retain water under the same reaction conditions by addition of water (0.10 g) in NMP, triglyme, triPrglyme, diglyBu, and diphenyl ether solvents, and the retained water amount in the solvent is called water affinity determined experimentally (Fig. 3b, x-axis, the detailed method is described in the ESI†). The solvents with lower solubility in water (diphenyl ether, <1 g L−1-H2O; diglyBu, 2 g L−1-H2O; triPrglyme, 280 g L−1-H2O) exhibited lower water affinity, and NMP, which is miscible with water and a highly polar aprotic solvent, showed higher water affinity. The conversion of the reaction of 1,6-hexanediol and CO2 at 1 h was plotted as a function of the water affinity (Fig. 3b), showing a good relationship between the water affinity and conversion; the conversion decreased with higher water affinity. These results indicate that solvents with lower water affinity can promote the removal of water from the reaction media.
To achieve both high conversion and good diol balance, we studied a dual-solvent reaction system with a 2nd solvent in combination with triglyme (1st solvent) (Fig. 3c). The addition of NMP, which has higher water affinity, gave much lower conversion (1.1%) than that in the case of using 2 g of triglyme alone (3.8%), and the increase of the triglyme amount or combination with tetraglyme as a 2nd solvent also showed lower conversions (3.0%). The addition of solvents with lower water affinity such as triPrglyme, diglyBu, diphenylmethane, phenylcyclohexane, and diphenyl ether as a 2nd solvent gave higher conversion (4.1–4.7%) than that with triglyme alone (3.8%). Since diphenyl ether as a 2nd solvent provided higher conversion (4.7%) than the other solvents, diphenyl ether was selected as a 2nd solvent in combination with triglyme in an optimized ratio (2 g of triglyme and 3 g of diphenyl ether, Fig. S7 and S8†) and used in the subsequent experiments.
The effect of reaction temperature was investigated with an adequate amount of CeO2 (0.10 g) in the range between 333 and 483 K, which is below the boiling point of the 1st solvent, triglyme (489 K) (Fig. 3d). The conversion increased with an increase in the reaction temperature from 333 K to 483 K, and production of the polymer was observed when the conversion exceeded 3% at temperatures above 373 K. The results suggest that a higher reaction temperature facilitates the removal of water from the reaction media, which shifts the equilibrium to the product side to result in higher conversion.
The effect of the CO2 flow rate was investigated in the presence of an adequate CeO2 amount (0.10 g) (Fig. 3e). The conversion increased almost linearly with the increase of the CO2 flow rate from 0 to 200 mL min−1 and became constant at 17% when the flow rate was above 200 mL min−1. From the results, a CO2 flow rate of 200 mL min−1 was selected for the subsequent synthesis of polycarbonate diol.
The effect of the mixed gas flow of CO2 and N2 was studied in the presence of an adequate amount of CeO2 (0.10 g) (Fig. 4, left side, the detailed results are shown in Table S6†). The flow rate of CO2 was fixed to 70 mL min−1 and the total flow rate was changed from 70 to 250 mL min−1 by N2 flow. The conversions were not changed at any flow rates (70–250 mL min−1). The behaviour is different from that with pure CO2 flow (Fig. 4, right side, the detailed results are shown in Table S7†), where the higher CO2 flow rate provided higher conversion in the range of 70–200 mL min−1. These results indicate that the gas flow rate does not contribute to the removal of water, which can be explained by the low water concentration in the gas flow (0.38 g m−3). A possible interpretation of the results is that a higher CO2 flow rate increased the concentration of CO2 in the reaction media, helping the equilibrium shift to the product side and afford higher conversion.
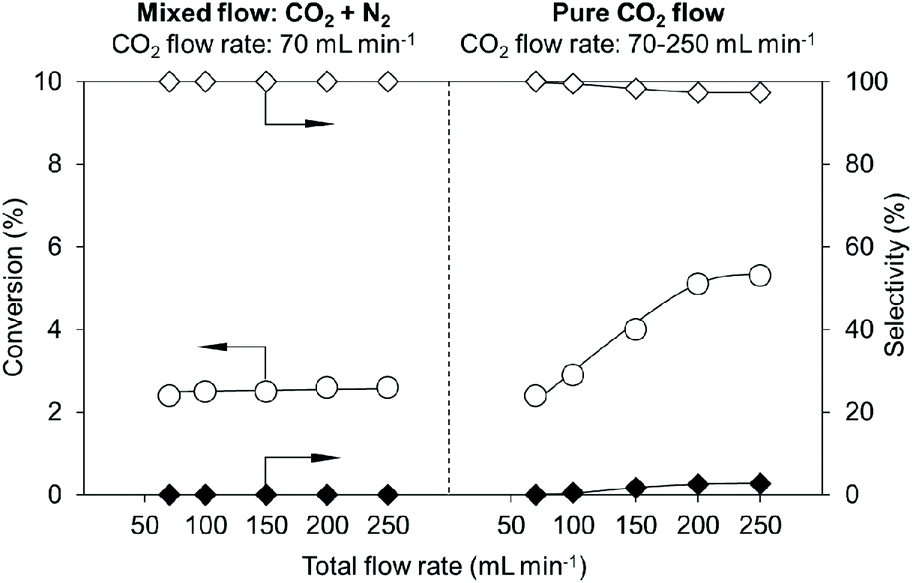 | ||
| Fig. 4 Effect of mixed gas flow of CO2 and N2 in the reaction of flow CO2 and 1,6-hexanediol with a CeO2 catalyst. Reaction conditions: CeO2 0.10 g, 1,6-hexanediol 2.0 g (17 mmol), triglyme 2 g, diphenyl ether 3 g, CO2 flow rate 70 mL min−1 [compensated with N2 (0–180 mL min−1), left graph]; CO2 flow rate 70–250 mL min−1 (right graph), 483 K, 1 h. ○, Conversion; ◊, dimer selectivity; and ◆, polymer selectivity. | ||
Fig. 5 shows the effect of the 1,6-hexanediol concentration on the reaction at 483 K. The CeO2 amount was decreased to 0.03 g, where the rate-determining step is the reaction over CeO2, not the removal of water to shift the equilibrium (Fig. 3a). The conversion increased with an increase in the 1,6-hexanediol concentration at lower concentrations from 0.9 to 2.4 M (Fig. 5), and the selectivity to the dimer slightly decreased as it was polymerized into the polymer. At higher concentrations from 2.4 to 4.9 M, the conversion and selectivities were almost the same, suggesting that the adsorption of 1,6-hexanediol was saturated over CeO2. The standard reaction conditions were set at a concentration of 2.4 M, which was in this range.
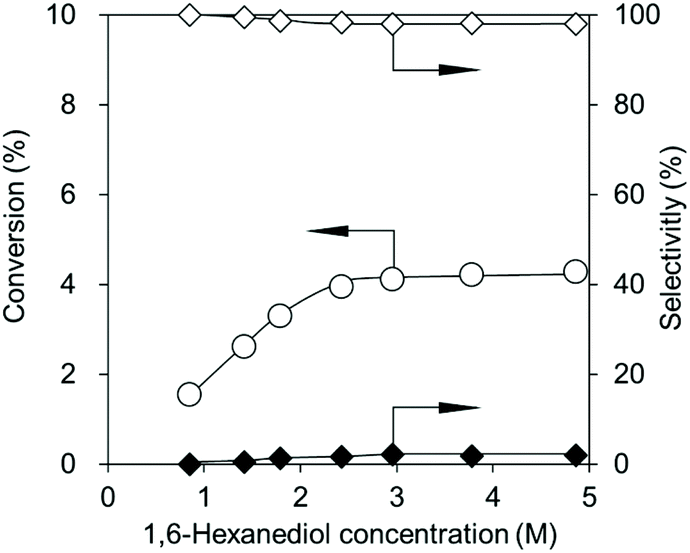 | ||
| Fig. 5 Effect of the 1,6-hexanediol concentration in the reaction of flow CO2 and 1,6-hexanediol with a CeO2 catalyst. Reaction conditions: CeO2 0.03 g, 1,6-hexanediol 2.0 g (17 mmol), solvent volume 1.5–18 mL (triglyme:diphenyl ether = 2:3 volume ratio), CO2 flow rate 200 mL min−1, 483 K, 1 h. ○, Conversion; ◊, dimer selectivity; and ◆, polymer selectivity. | ||
Time-course of the reaction of 1,6-hexanediol and CO2 over CeO2 in the CO2 flow semi-batch reactor and analyses of the produced polymer
Fig. 6a exhibits the time-course of the reaction of flow CO2 and 1,6-hexanediol at 483 K with a CeO2 catalyst and a mixture solvent of triglyme and diphenyl ether. The reaction proceeded smoothly to reach 95% conversion at 96 h. The selectivity to the dimer was high at a low conversion level and decreased with a longer reaction time, and conversely, the selectivity to the polymer kept increasing and reached 97% at 96 h. No non-polymeric by-product was observed during the reaction. The water content in the reaction media drastically decreased from 900 ppm to <4 ppm (below detection limit) during the heating-up to the reaction temperature of 483 K within several minutes and was unmeasurable at a longer reaction time. The conversion of CO2 was also calculated using the data of the 4 h reaction, in which the dimer and the trimer were the only products. 2000 mmol of CO2 were introduced into the reactor in 4 hours and the formation amount of the carbonates was 2 mmol, meaning that the conversion of CO2 was 0.1%.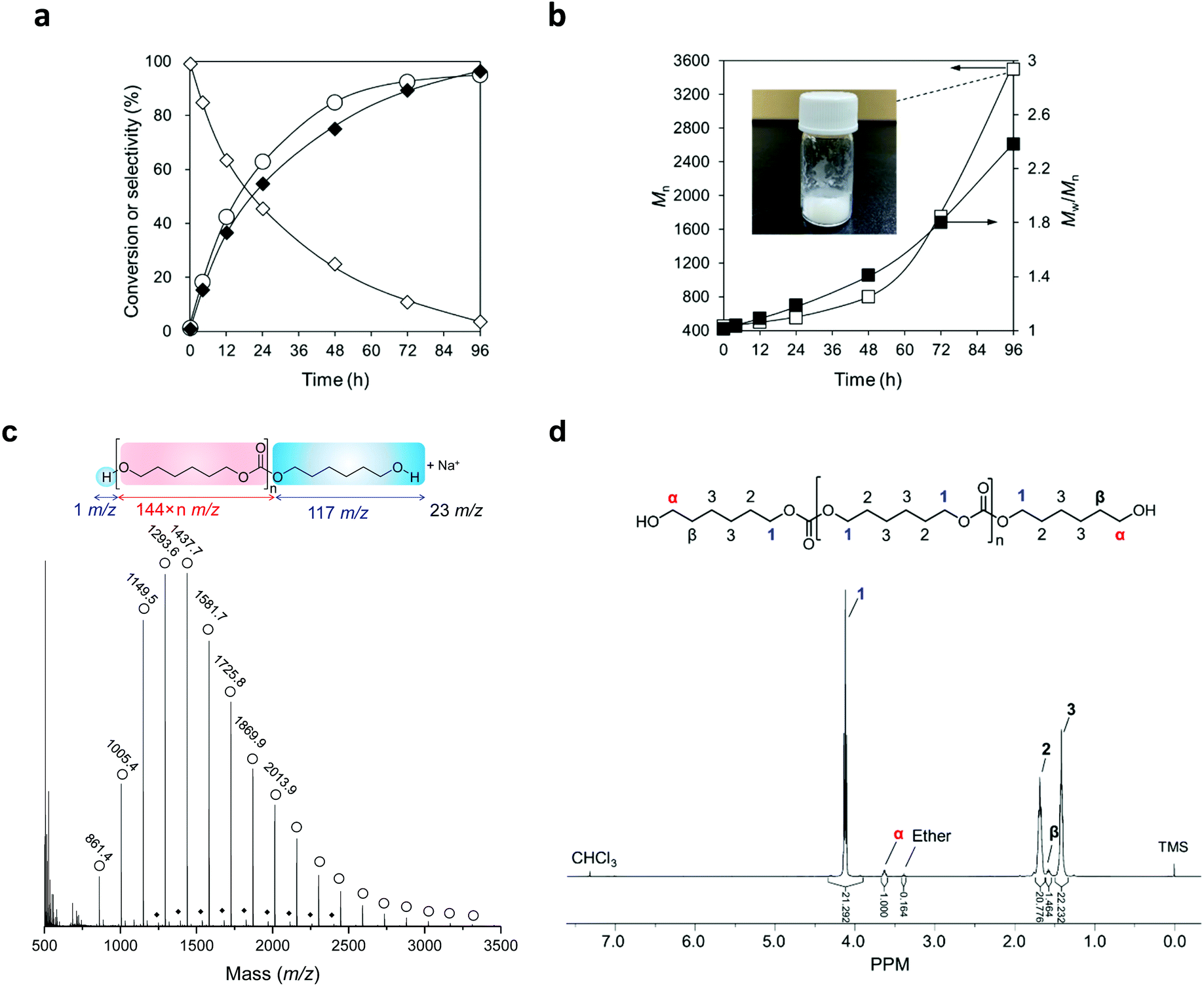 | ||
| Fig. 6 Synthesis and characterization of polycarbonate diol from CO2 and 1,6-hexanediol over CeO2 in a CO2-flow reaction system: (a) Time-course and (b) molecular weight (Mn) and dispersity (Mw/Mn) profile. (c) MALDI-TOF/MS analysis. (d) 1H-NMR analysis with CDCl3 solvent. Marks in (a): ○, Conversion; ◊, dimer selectivity; and ◆, polymer selectivity; marks in (b) □, Mn; ■, Mw/Mn. Reaction conditions of (a) and (b): CeO2 0.10 g, 1,6-hexanediol 2.0 g (17 mmol), triglyme 2 g, diphenyl ether 3 g, CO2 flow rate 200 mL min−1, 483 K, 0–96 h. | ||
The average molecular weight (Mn) and dispersity (Mw/Mn) of the produced products were analysed by size-exclusion chromatography (SEC) (Fig. 6b). Both the Mn and Mw/Mn of the products kept increasing during the reaction, and polymer with an Mn of 3500 (in the case of polycarbonate diols, 24 repeating units) was obtained at 96 h. The increase of Mn became faster when higher conversion was achieved with a longer reaction time, which is a characteristic of step-growth polymerization.
The polymer produced at 96 h was isolated by precipitation (the detailed method is described in the ESI†), and the isolated polymer was a pure white wax-like solid (Fig. 6b). The isolated polymer was analysed by MALDI-TOF mass (Fig. 6c) and 1H-NMR (Fig. 6d) spectroscopy (the detailed method is described in the ESI†). A series of strong signals with a mass increase of 144 was observed in MALDI-TOF mass spectrum and they were assigned to poly(hexamethylene carbonate) diol (Fig. 6c). The other series of minor signals with a lower mass number of 44 than the strong signals was identified as poly(hexamethylene carbonate) diol with one ether linkage. The 1H NMR chart of the obtained polymer (Fig. 6d) showed the characteristic signals at 3.6 ppm (marked as “α” in the chart) and 4.0–4.2 ppm (marked as “1”), which can be assigned to the protons of the methylene group adjacent to the OH group and carbonate group, respectively. The average molecular weight of the isolated polycarbonate diol is estimated to be 3200 g mol−1 by calculating the ratio of the proton numbers on the two positions (“α” and “1”). The value was similar to that determined by SEC (3500 g mol−1 based on polystyrene standards). A triplet signal at 3.4 ppm is assigned to the protons of the methylene group adjacent to the ether bond, which is in agreement with the result of MALDI-TOF mass spectra, and the ratio of the ether linkage to the carbonate linkage is 0.8%. There were no signals at 4.9 and 5.8 ppm, where the vinylic proton appears (an enlarged chart is shown in Fig. S9†).
The proposed reaction mechanism of the polycarbonate diols from CO2 and 1,6-hexanediol over CeO2 is shown in Fig. 7 based on the previous reports on the synthesis of dimethyl carbonate from CO2 and methanol over CeO2, where the reaction mechanism was proposed by the kinetic studies and analyses such as DRIFTS and isotopic labelling studies.64,79,93 The present reaction proceeded via the formation of dialkyl carbonate (dimer) from CO2 and 1,6-hexanediol over CeO2 (Fig. 6a), and hence the reaction mechanism of the present catalyst system would be similar to the previous one: (i) The dissociative adsorption of 1,6-hexanediol on the CeO2 surface gives the alkoxide adspecies. (ii) The insertion of CO2 into the alkoxide adspecies provides Ce-hydroxyhexyl carbonate adspecies. (iii) The nucleophilic attack of the oxygen anion in the alkoxide adspecies to the carbonate group in the Ce-hydroxyhexyl carbonate adspecies affords the corresponding linear carbonate (dimer). (iv) Diffusion and purge of the coproduced water from the reaction media to the gas phase by the gas flow. (v) Further reaction of the produced dimer with CO2 and 1,6-hexanediol or the produced copolymer results in a step-growth polymerization with the production of poly(hexamethylene carbonate) diol. The water removal step in (iv) played a vital role in the reaction progress, which shifts the equilibrium and enables the dimer yield high enough to start the polymerization. Without this step, no products will be achieved under atmospheric pressure as shown in Table S3.† The high activity of CeO2 for the reaction is due to the acid–base bifunctionality: CeO2 has both weak acidic sites and medium basic sites, which can be used for the activation of CO2 and alcohols.31,64,90 Side reactions catalysed by strong acid catalysts, which afford by-products such as ethers and alkenes, are also suppressed because of the weak acidity of CeO2.
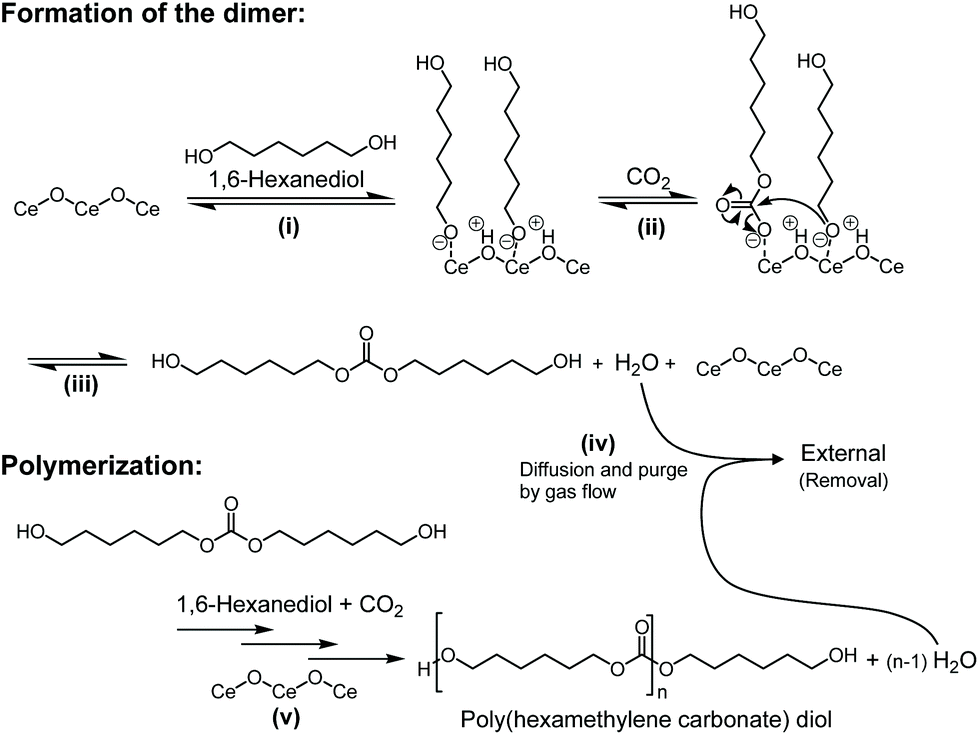 | ||
| Fig. 7 Proposed reaction mechanism of the copolymerization of flow CO2 and 1,6-hexanediol over a CeO2 catalyst. | ||
The high durability of CeO2 was confirmed by the reusability test (Fig. 8). 0.8 gram of the CeO2 catalyst was used, and a CeO2 amount below 0.8 g would lead to a decrease in the conversion. The CeO2 was used 4 times, providing similar conversions and selectivities. No distinct difference in the XRD patterns, crystallite size, and specific surface area was observed between fresh CeO2 catalyst and the spent CeO2 catalyst which has been used 4 times (Fig. 9).
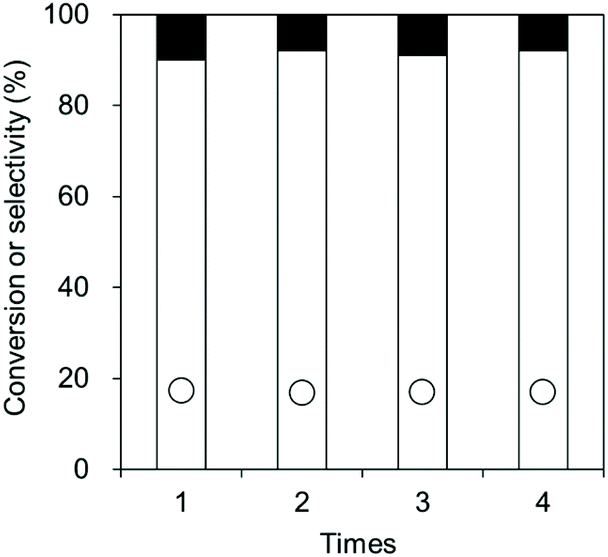 | ||
| Fig. 8 Reusability test of the CeO2 catalyst in the reaction of flow CO2 and 1,6-hexanediol. ○: Conversion, white bar: dimer selectivity, and black bar: polymer selectivity. Reaction conditions: CeO2 0.08 g, 1,6-hexanediol 2.0 g (17 mmol), triglyme 2.0 g, diphenyl ether 3.0 g, CO2 flow rate 200 mL min−1, 483 K, 4 h. | ||
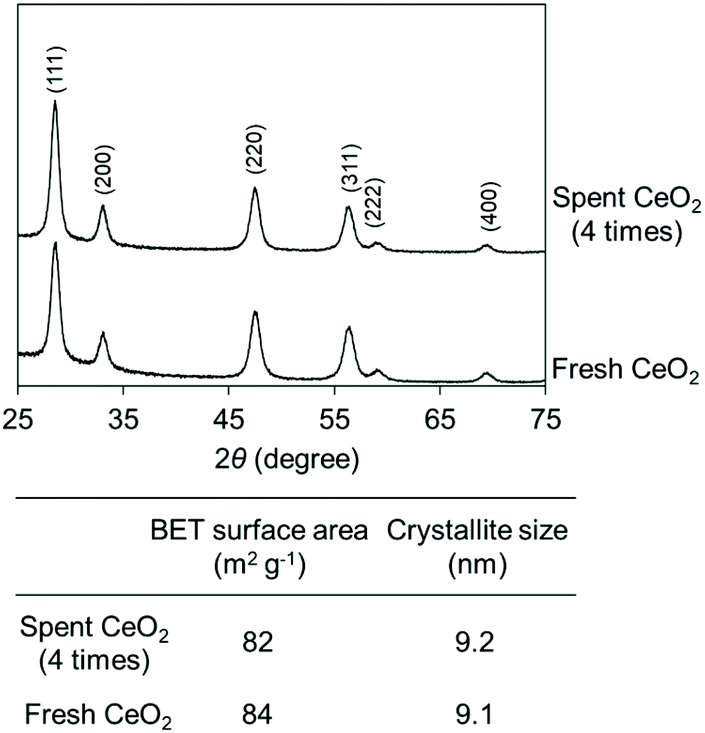 | ||
| Fig. 9 XRD patterns and BET surface area before and after the reuse test. The reaction results are shown in Fig. 8. The crystallite size of CeO2 was estimated using the Scherrer equation [crystalline plane (111) was used for the estimation]. | ||
Substrate scope of the CO2 flow reaction system with a CeO2 catalyst
The scope of the reaction system was investigated with various alcohols (Table 1). C4–C10 α,ω-diols (entries 1–4) including trans-1,4-cyclohexanedimethanol, an α,ω-diol with a rigid structure, provided the target dimer and polycarbonate diols in high selectivity (>99%). Moreover, 1-decanol (entry 6), a primary alcohol, also reacted to give the corresponding linear carbonate in high selectivity. In the case of 1,2-propanediol (entry 5) which has primary and secondary OH groups, the propylene carbonate, a cyclic organic carbonate, was obtained without the production of linear carbonates.| Entry | Alcohol | B.P.a (K) | T (K) | Conv. (%) | Selectivity (%) | ||
|---|---|---|---|---|---|---|---|
| Reaction conditions: CeO2 0.10 g, alcohol 17 mmol, triglyme 2 g, diphenyl ether 3 g, CO2 flow rate 200 mL min−1, 393–483 K, 48 h.a Boiling point of the alcohols.b Polymers are defined as trimers and further polymerized into polycarbonate diols. | |||||||
| 1 |

|
523 | 483 | 85 |
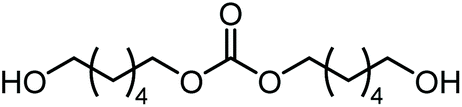
|
Polymerb | Others |
| 25 | 75 | <1 | |||||
| 2 |

|
578 | 473 | 58 |
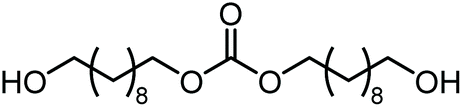
|
Polymerb | Others |
| 52 | 48 | <1 | |||||
| 3 |
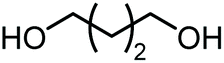
|
508 | 453 | 30 |
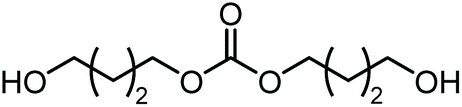
|
Polymerb | Others |
| 73 | 27 | <1 | |||||
| 4 |
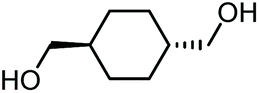
|
558 | 473 | 55 |
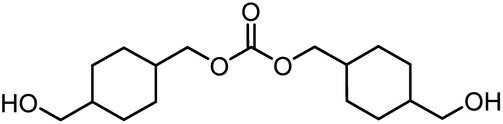
|
Polymerb | Others |
| 71 | 29 | <1 | |||||
| 5 |
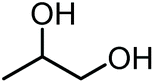
|
458 | 393 | 13 |
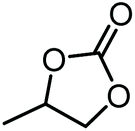
|
Polymerb | Others |
| >99 | <1 | <1 | |||||
| 6 |
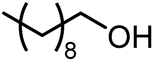
|
503 | 483 | 53 |

|
Others | |
| >99 | <1 | ||||||
Conclusions
We demonstrated the direct transformation of atmospheric pressure flowing CO2 with 1,6-hexanediol into the corresponding polycarbonate diol without any dehydration agents, achieving high yield and selectivity to the target polycarbonate diol. CeO2 worked as the most effective and reusable heterogeneous catalyst and the CO2 flow system enabled the removal of water from the reaction media to overcome the reaction equilibrium. The wide applicability of the reaction system to other alcohols was confirmed and the corresponding organic (poly)carbonates were obtained in high selectivity (>99%). The developed catalyst system is expected to substitute for hazardous processes such as the phosgene process for the synthesis of polycarbonate diols and also contribute to the synthesis of other CO2-based organic carbonates with large molecular weights. Although the reaction conditions are comparatively severe at present, the catalyst system has high potential for the green process by the rationalization of the catalytic process.Experimental
Materials
CeO2 was prepared by calcining commercial CeO2 (Daiichi Kigenso Kagaku Kogyo, purity of CeO2 = 99.97%) in air at 873 K for 3 h, and the specific surface area (BET method) of the calcined CeO2 is 84 m2 g−1. Other commercial metal oxides including ZrO2 (Daiichi Kigenso Kagaku Kogyo), SiO2 (Fuji Silysia Chemical), γ-Al2O3 (Sumitomo Chemical), MgO (Ube Industries), and TiO2 (Nippon Aerosil) were also calcined under the same conditions before use. La2O3, Dy2O3, Gd2O3, Eu2O3, Sm2O3, Y2O3, Pr6O11, and ZnO purchased from Kanto Chemical, and SiO2–Al2O3 (product code: JRC-SAL-3) received from the Catalysis Society of Japan were used without any treatment. All the reagents and gases used in the experiments were used without further purification, which are as follows: 1,6-hexanediol [>97.0%, TCI (Tokyo Chemical Industry)], 1,10-decanediol (>97.0%, TCI), 1,4-butanediol (>98.0%, FUJIFILM Wako Pure Chemical), trans-1,4-cyclohexanedimethanol (>98.0%, TCI), 1,2-propanediol (>99.0%, TCI), 1-decanol (>98.0%, TCI), diphenyl ether (>99.0%, TCI), triethylene glycol dimethyl ether (>99.0%, stabilized with BHT, TCI), and diethylene glycol dimethyl ether (>99.0%, TCI), 1-methyl-2-pyrrolidone (>99.0%, TCI), tetraethylene glycol dimethyl ether (>98.0%, TCI), dipropylene glycol dimethyl ether (mixture of isomers) (>94.0%, TCI), diethylene glycol dibutyl ether (>98.0%, TCI), diphenylmethane (>99.0%, TCI), phenylcyclohexane (>97.0%, TCI), CO2 (G1 grade, purity ≥ 99.995 vol%, H2O ≤ 2.6 ppm, Taiyo Nippon Sanso), and N2 (G1 grade, purity ≥ 99.99995 vol%, H2O ≤ 0.5 ppm).Methods
Author contributions
Yu Gu: Investigation, methodology, formal analysis, and writing – original draft. Masazumi Tamura: Conceptualization, funding acquisition, formal analysis, and writing – original draft. Yoshinao Nakagawa: Writing – review & editing. Kenji Nakao: Formal analysis. Kimihito Suzuki: Formal analysis. Keiichi Tomishige: Formal analysis, supervision, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This paper is based on the results obtained from a “NEDO Feasibility Study Program” commissioned by the New Energy and Industrial Technology Development Organization (NEDO).References
- Carbon Dioxide as Chemical Feedstock, ed. M. Aresta, Wiley-VCH, Weinheim, Germany, 2010 Search PubMed.
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed.
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS PubMed.
- D. U. Nielsen, X.-M. Hu, K. Daasbjerg and T. Skrydstrup, Nat. Catal., 2018, 1, 244–254 CrossRef CAS.
- M. Aresta and A. Dibenedetto, Front. Energy Res., 2020, 8, 159 CrossRef.
- C. S. Diercks, Y. Liu, K. E. Cordova and O. M. Yaghi, Nat. Mater., 2018, 17, 301–307 CrossRef CAS PubMed.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- W. Li, H. Wang, X. Jiang, J. Zhu, Z. Liu, X. Guo and C. Song, RSC Adv., 2018, 8, 7651–7669 RSC.
- L. Guo, J. Sun, Q. Ge and N. Tsubaki, J. Mater. Chem. A, 2018, 6, 23244–23262 RSC.
- W.-H. Wang, Y. Himeda, J. T. Muckerman, G. F. Manbeck and E. Fujita, Chem. Rev., 2015, 115, 12936–12973 CrossRef CAS PubMed.
- X. Jiang, X. Nie, X. Guo, C. Song and J. G. Chen, Chem. Rev., 2020, 120, 7984–8034 CrossRef CAS PubMed.
- J. R. Cabrero-Antonino, R. Adam and M. Beller, Angew. Chem., Int. Ed., 2019, 58, 12820–12838 CrossRef CAS PubMed.
- A. Álvarez, A. Bansode, A. Urakawa, A. V. Bavykina, T. A. Wezendonk, M. Makkee, J. Gascon and F. Kapteijn, Chem. Rev., 2017, 117, 9804–9838 CrossRef PubMed.
- H. Yang, C. Zhang, P. Gao, H. Wang, X. Li, L. Zhong, W. Wei and Y. Sun, Catal. Sci. Technol., 2017, 7, 4580–4598 RSC.
- X. Wang, H. Wang and Y. Sun, Chem, 2017, 3, 211–228 CAS.
- C. Chen, J. F. Khosrowabadi Kotyk and S. W. Sheehan, Chem, 2018, 4, 2571–2586 CAS.
- A. Modak, P. Bhanja, S. Dutta, B. Chowdhury and A. Bhaumik, Green Chem., 2020, 22, 4002–4033 RSC.
- M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kühn, Angew. Chem., Int. Ed., 2011, 50, 8510–8537 CrossRef CAS PubMed.
- B. Yu and L.-N. He, ChemSusChem, 2015, 8, 52–62 CrossRef CAS PubMed.
- Z. Chen, S. Du, J. Zhang and X.-F. Wu, Green Chem., 2020, 22, 8169–8182 RSC.
- A. J. Kamphuis, F. Picchionia and P. P. Pescarmona, Green Chem., 2019, 21, 406–448 RSC.
- D. J. Darensbourg, Chem. Rev., 2007, 107, 2388–2410 CrossRef CAS PubMed.
- X.-B. Lu and D. J. Darensbourg, Chem. Soc. Rev., 2012, 41, 1462–1484 RSC.
- G. Fiorani, W. S. Guo and A. W. Kleij, Green Chem., 2015, 17, 1375–1389 RSC.
- B. Grignard, S. Gennen, C. Jérôme, A. W. Kleij and C. Detrembleur, Chem. Soc. Rev., 2019, 48, 4466–4514 RSC.
- C. Martín, G. Fiorani and A. W. Kleij, ACS Catal., 2015, 5, 1353–1370 CrossRef.
- H. Sugimoto and S. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5561–5573 CrossRef CAS.
- K. Tomishige, Y. Gu, T. Chang, M. Tamura and Y. Nakagawa, Mater. Today Sustainability, 2020, 9, 100035 CrossRef.
- K. Tomishige, Y. Nakagawa and M. Tamura, Chem. Rec., 2019, 19, 1354–1379 CrossRef CAS PubMed.
- M. Tamura, M. Honda, Y. Nakagawa and K. Tomishige, J. Chem. Technol. Biotechnol., 2014, 89, 19–33 CrossRef CAS.
- K. Tomishige, Y. Gu, Y. Nakagawa and M. Tamura, Front. Energy Res., 2020, 8, 117 CrossRef.
- Y. Gu, A. Miura, M. Tamura, Y. Nakagawa and K. Tomishige, ACS Sustainable Chem. Eng., 2019, 7, 16795–16802 CrossRef CAS.
- M. Tamura, A. Miura, M. Honda, Y. Gu, Y. Nakagawa and K. Tomishige, ChemCatChem, 2018, 10, 4821–4825 CrossRef.
- H.-Y. Yuan, J.-C. Choi, S. Onozawa, N. Fukaya, S. J. Choi, H. Yasuda and T. Sakakura, J. CO2 Util., 2016, 16, 282–286 CrossRef CAS.
- Q. Zhang, H.-Y. Yuan, N. Fukaya, H. Yasuda and J.-C. Choi, ChemSusChem, 2017, 10, 1501–1508 CrossRef CAS PubMed.
- J.-C. Choi, H.-Y. Yuan, N. Fukaya, S. Onozawa, Q. Zhang, S. J. Choi and H. Yasuda, Chem. – Asian J., 2017, 12, 1297–1300 CrossRef CAS PubMed.
- Q. Zhang, H.-Y. Yuan, N. Fukaya, H. Yasuda and J.-C. Choi, Green Chem., 2017, 19, 5614–5624 RSC.
- Q. Zhang, H.-Y. Yuan, N. Fuakaya and J.-C. Choi, ACS Sustainable Chem. Eng., 2018, 6, 6675–6681 CrossRef CAS.
- C. Claver, M. B. Yeamin, M. Reguero and A. M. Masdeu-Bultó, Green Chem., 2020, 22, 7665–7706 RSC.
- G. A. Bhat, M. Luo and D. J. Darensbourg, Green Chem., 2020, 22, 7707–7724 RSC.
- W. S. Putro, A. Ikeda, S. Shigeyasu, S. Hamura, S. Matsumoto, V. Y. Lee, J.-C. Choi and N. Fukaya, ChemSusChem, 2021, 14, 842–846 CrossRef CAS PubMed.
- L. Guo, K. J. Lamb and M. North, Green Chem., 2021, 23, 77–118 RSC.
- H. Chen, P. Chauhanb and N. Yan, Green Chem., 2020, 22, 6874–6888 RSC.
- M. Tamura, K. Noro, M. Honda, Y. Nakagawa and K. Tomishige, Green Chem., 2013, 15, 1567–1577 RSC.
- M. Tamura, K. Ito, Y. Nakagawa and K. Tomishige, J. Catal., 2016, 343, 75–85 CrossRef CAS.
- M. Tamura, M. Honda, K. Noro, Y. Nakagawa and K. Tomishige, J. Catal., 2013, 305, 191–203 CrossRef CAS.
- M. Honda, S. Sonehara, H. Yasuda, Y. Nakagawa and K. Tomishige, Green Chem., 2011, 13, 3406–3413 RSC.
- H. Ohno, M. Ikhlayel, M. Tamura, K. Nakao, K. Suzuki, K. Morita, Y. Kato, K. Tomishige and Y. Fukushima, Green Chem., 2021, 23, 457–469 RSC.
- E. Leino, P. Mäki-Arvela, V. Eta, D. Yu. Murzin, T. Salmi and J.-P. Mikkola, Appl. Catal., A, 2010, 383, 1–13 CrossRef CAS.
- C.-F. Li and S.-H. Zhong, Catal. Today, 2003, 82, 83–90 CrossRef CAS.
- A. Dibenedetto, M. Aresta, A. Angelini, J. Ethiraj and M. Aresta, Chem. – Eur. J., 2012, 18, 10324–10334 CrossRef CAS PubMed.
- P. Unnikrishnan, P. Varhadi and D. Srinivas, RSC Adv., 2013, 3, 23993–23996 RSC.
- J. C. Choi, L. N. He, H. Yasuda and T. Sakakura, Green Chem., 2002, 4, 230–234 RSC.
- K. Iwakabe, M. Nakaiwa, T. Sakakura, J.-C. Choi, H. Yasuda, T. Takahashi and Y. Ooshima, J. Chem. Eng. Jpn., 2005, 38, 1020–1024 CrossRef CAS.
- D. C. Stoian, E. Taboada, J. Llorca, E. Molins, F. Medina and A. M. Segarra, Chem. Commun., 2013, 49, 5489–5491 RSC.
- M. Honda, M. Tamura, Y. Nakagawa and K. Tomishige, Catal. Sci. Technol., 2014, 4, 2830–2845 RSC.
- A. Bansode and A. Urakawa, ACS Catal., 2014, 4, 3877–3880 CrossRef CAS.
- Q. Zhang, H.-Y. Yuan, X.-T. Lin, N. Fukaya, T. Fujitani, K. Sato and J.-C. Choi, Green Chem., 2020, 22, 4231–4239 RSC.
- M. Tamura, K. Ito, M. Honda, Y. Nakagawa, H. Sugimoto and K. Tomishige, Sci. Rep., 2016, 6, 24038 CrossRef PubMed.
- Y. Gu, K. Matsuda, A. Nakayama, M. Tamura, Y. Nakagawa and K. Tomishige, ACS Sustainable Chem. Eng., 2019, 7, 6304–6315 CrossRef CAS.
- T. Chang, M. Tamura, Y. Nakagawa, N. Fukaya, J.-C. Choi, T. Mishima, S. Matsumoto, S. Hamura and K. Tomishige, Green Chem., 2020, 22, 7321–7327 RSC.
- M. Honda, M. Tamura, Y. Nakagawa, S. Sonehara, K. Suzuki, K.-i. Fujimoto and K. Tomishige, ChemSusChem, 2013, 6, 1341–1344 CrossRef CAS PubMed.
- M. Honda, M. Tamura, K. Nakao, K. Suzuki, Y. Nakagawa and K. Tomishige, ACS Catal., 2014, 4, 1893–1896 CrossRef CAS.
- M. Honda, M. Tamura, Y. Nakagawa, K. Nakao, K. Suzuki and K. Tomishige, J. Catal., 2014, 318, 95–107 CrossRef CAS.
- K. Tomishige and K. Kunimori, Appl. Catal., A, 2002, 237, 103–109 CrossRef CAS.
- M. Honda, A. Suzuki, B. Noorjahan, K.-i. Fujimoto, K. Suzuki and K. Tomishige, Chem. Commun., 2009, 4596–4598 RSC.
- M. Honda, S. Kuno, B. Noorjahan, K.-i. Fujimoto, K. Suzuki, Y. Nakagawa and K. Tomishige, Appl. Catal., A, 2010, 384, 165–170 CrossRef CAS.
- M. Honda, S. Kuno, S. Sonehara, K.-i. Fujimoto, K. Suzuki, Y. Nakagawa and K. Tomishige, ChemCatChem, 2011, 3, 365–370 CrossRef CAS.
- T. Zhao, X. Hu, D. Wu, R. Li, G. Yang and Y. Wu, ChemSusChem, 2017, 10, 2046–2052 CrossRef CAS PubMed.
- S. Kumar, P. Kumar and S. L. Jain, J. Mater. Chem. A, 2014, 2, 18861–18866 RSC.
- S. Verma, R. B. N. Baig, M. N. Nadagouda and R. S. Varma, Sci. Rep., 2017, 7, 655 CrossRef PubMed.
- S. Kumar, M. B. Gawande, I. Medřík, M. Petr, O. Tomanec, V. Kupka, R. S. Varma and R. Zbořil, Green Chem., 2020, 22, 5619–5627 RSC.
- A. Poungsombate, T. Imyen, P. Dittanet, B. Embley and P. Kongkachuichay, J. Taiwan Inst. Chem. Eng., 2017, 80, 16–24 CrossRef CAS.
- R. Nakano, S. Ito and K. Nozaki, Nat. Chem., 2014, 6, 325–331 CrossRef CAS PubMed.
- Y.-C. Xu, H. Zhou, X.-Y. Sun, W.-M. Ren and X.-B. Lu, Macromolecules, 2016, 49, 5782–5787 CrossRef CAS.
- Y. Zhang, J. Xia, J. Song, J. Zhang, X. Ni and Z. Jian, Macromolecules, 2019, 52, 2504–2512 CrossRef CAS.
- Z. Chen, N. Hadjichristidis, X. Feng and Y. Gnanou, Polym. Chem., 2016, 7, 4944–4952 RSC.
- S. Oi, K. Nemoto, S. Matsuno and Y. Inoue, Macromol. Rapid Commun., 1994, 15, 133–137 CrossRef CAS.
- Z.-J. Gong, Y.-R. Li, H.-L. Wu, S. D. Lin and W.-Y. Yu, Appl. Catal., B, 2020, 265, 118524 CrossRef.
- Y.-C. Yu, T.-Y. Wang, L.-H. Chang, P.-J. Wu, B.-Y. Yu and W.-Y. Yu, J. Taiwan Inst. Chem. Eng., 2020, 116, 36–42 CrossRef CAS.
- J. Shang, S. Liu, X. Ma, L. Lu and Y. Deng, Green Chem., 2012, 14, 2899–2906 RSC.
- Z. Ying, Y. Dong, J. Wang, Y. Yu, Y. Zhou, Y. Sun, C. Zhang, H. Cheng and F. Zhao, Green Chem., 2016, 18, 2528–2533 RSC.
- S. Jiang, H.-Y. Cheng, R.-H. Shi, P.-X. Wu, W.-W. Lin, C. Zhang, M. Arai and F.-Y. Zhao, ACS Appl. Mater. Interfaces, 2019, 11, 47413–47421 CrossRef CAS PubMed.
- R. Liu, X. Liu, K. Ouyang and Q. Yan, ACS Macro Lett., 2019, 8, 200–204 CrossRef CAS.
- Polycarbonate market size worth $25.37 Billion by 2024, https://www.grandviewresearch.com/press-release/global-polycarbonate-market, (accessed June 2021).
- S. Fukuoka, I. Fukawa, T. Adachi, H. Fujita, N. Sugiyama and T. Sawa, Org. Process Res. Dev., 2019, 23, 145–169 CrossRef CAS.
- S. Fukuoka, M. Kawamura, K. Komiya, M. Tojo, H. Hachiya, K. Hasegawa, M. Aminaka, H. Okamoto, I. Fukawa and S. Konno, Green Chem., 2003, 5, 497–507 RSC.
- Separation Process Principles, ed. J. D. Seader and E. J. Henley, John Wiley & Sons Inc., Hoboken NJ, 2006, ch. 6, pp. 193–242 Search PubMed.
- Y.-L. Hwang, G. E. Keller II and J. D. Olson, Ind. Eng. Chem. Res., 1992, 31, 1753–1759 CrossRef CAS.
- K. Tomishige, T. Sakaihori, Y. Ikeda and K. Fujimoto, Catal. Lett., 1999, 58, 225–229 CrossRef CAS.
- S. Roy, G. Mpourmpakis, D.-Y. Hong, D. G. Vlachos, A. Bhan and R. J. Gorte, ACS Catal., 2012, 2, 1846–1853 CrossRef CAS.
- M. A. Ardagh, Z. Bo, S. L. Nauert and J. M. Notestein, ACS Catal., 2016, 6, 6156–6164 CrossRef CAS.
- Y. Yoshida, Y. Arai, S. Kado, K. Kunimori and K. Tomishige, Catal. Today, 2006, 115, 95–101 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc01172c |
| This journal is © The Royal Society of Chemistry 2021 |