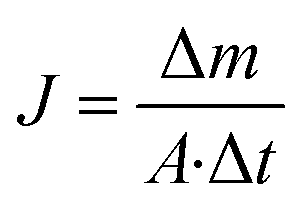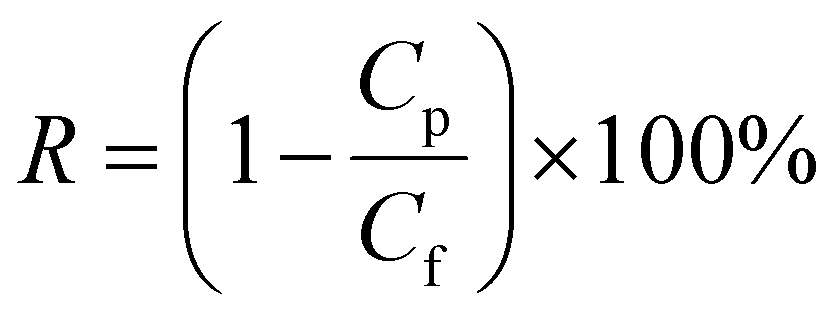DOI:
10.1039/D0RA07629E
(Paper)
RSC Adv., 2020,
10, 40562-40568
Large-pore-size membranes tuned by chemically vapor deposited nanocoatings for rapid and controlled desalination†
Received
6th September 2020
, Accepted 22nd October 2020
First published on 6th November 2020
Abstract
Though membranes with pore size larger than 1 μm are much desired to increase the permeate flux of membrane distillation (MD), the vulnerability of large-pore-size membranes to pore wetting results in the penetration of saline water and consequent failure of MD operation. We report modification of large-pore-size membranes by chemically vapor deposited nanocoatings to achieve both high salt rejection and high permeate flux. The chemical vapor modification not only led to enhanced surface hydrophobicity and increased liquid entry pressure in membranes, but also significantly improved membrane wetting resistance at high temperature. Membranes with 1.0 and 2.0 μm pore size were successfully used for MD desalination with salt rejection higher than 99.99% achieved. Enlarging the pore size from 0.2 μm to 2.0 μm contributed to 48–73% enhancement in the permeate flux of the modified membranes. The modified large-pore-size membranes maintained the high permeate flux at elevated saline concentration and extended the operation time.
1. Introduction
Freshwater shortage is one of the most critical global challenges of our time.1 Driven by population growth and climate change, 1.0–1.3 billion people are forecasted to live with overly exploited water conditions by 2050.2,3 There is tremendous interest in desalination to produce freshwater for human use. Though reverse osmosis accounts for over 60% of global desalination capacity,4 the process requires significant energy input when the osmotic pressure of the feed increases.5 Membrane distillation (MD), a combination of membrane separation and thermal distillation processes, has the advantage of desalinating high-salinity water using low-grade or waste heat,6 making it a promising technology to increase sustainable water production and alleviate the global water crisis.5,7,8
During the MD process, the water vapor is thermally transported through hydrophobic membrane pores that are not wetted by saline water. The driving force is the vapor pressure differentials generated by the temperature differentials across the membrane.5,9 The vapor flux is linearly related to the vapor pressure differentials by the permeability coefficient C,10–13 which can be associated with membrane properties:5,11,14–16
| |
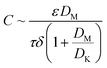 | (1) |
where
ε,
τ, and
δ are the membrane porosity, tortuosity, and thickness, respectively, and
DK and
DM are the Knudsen diffusion coefficient and molecular diffusion efficient of water vapor, respectively. Since the molecule-wall collision of Knudsen diffusion decreases in large pores, increasing pore size is expected to improve
DK and thus the vapor flux.
17–19 Large-pore-size membranes also have benefits in reducing pore plugging by particulate matters.
20 However, the pore wetting in large-pore-size membranes results in the penetration of saline water and the consequent failure of MD operation.
21–23 As a result, the pore size of most MD membranes reported in literatures was limited to 0.2 and 0.45 μm.
12,24,25 Membrane orientation was reported to significantly impact the water flux and salt rejection of asymmetric MD membranes due to the wetting of relatively large pores.
26 A detailed study about the effects of pore size on MD permeate flux and salt rejection demonstrated that water flux increased as pore size increased, but when membrane pore size reached 1 μm, salt rejection deteriorated rapidly, especially at high salt concentration in the feed.
27 Significant compromise of salt rejection rate was also observed in MD using 1 μm polytetrafluoroethylene membranes.
28
Membrane modification has been explored to alleviate pore wetting by increasing the membrane hydrophobicity and the liquid entry pressure, the minimum transmembrane pressure at which water starts penetrating the pores.29 Hierarchical structures with multilevel roughness have been created using nanoparticle deposition to improve wetting resistance.30–34 However, the decrease in membrane porosity after direct nanoparticle modification usually resulted in the increase in the mass transfer resistance. For example, polyvinylidene difluoride membranes demonstrated superhydrophobicity after the TiO2 nanoparticle deposition, but the permeate flux declined by 13.5%.30 On the other hand, efforts in enhancing permeate flux using nanoparticles usually compromised the liquid entry pressure (LEP) when compared with the pristine MD membranes.32,35 The observed alternating up-and-down between permeate flux and LEP calls for the need to study modification schemes to increase both the permeate flux and LEP of MD membranes.
This paper reports a novel chemical vapor method to modify large-pore-size MD membranes for improving permeate flux and LEP simultaneously. The vapor-based modification provides a facile route for tailoring porous structures and has been reported to enhance the wetting resistance of polysulfone,36,37 polyamide,38 and nylon membranes.39 In this study, we investigated the desalination performance of chemically tuned MD membranes with various pore sizes, aiming to understand the effect of enlarging pore size on improving permeate flux and desalting efficiency. The changes in membrane characteristics, wetting dynamics, and MD performance after the vapor modification were systematically studied. The permeate flux of modified membranes under varying operating conditions such as feed concentration and temperature was also investigated.
2. Materials and methods
2.1. Materials
1H,1H,2H,2H-Heptadecafluorodecyl acrylate (HDFA, 97%) was purchased from TCI America. tert-Butyl peroxide (TBP, 98%) and sodium chloride (NaCl, ≥99%) were purchased from Sigma-Aldrich. Isopropyl alcohol (99.9%) was purchased from Pharmco-Aaper. Polypropylene (PP) membranes with nominal pore size of 0.2, 0.45, 1, and 2 μm were purchased from TISCH Scientific (Table 1). Silicon wafer (P/boron 〈100〉) was purchased from WRS Materials.
Table 1 Characteristics of the pristine PP nanofiber membranes
| Pore diametera (μm) |
Thicknessa (μm) |
Porosityb (%) |
LEPb (inHg) |
Contact angleb (°) |
Gas permeabilitya (mL min−1 at 10 psi) |
| Supplier provided. Lab measured. |
| 0.2 |
180–250 |
45.7 ± 0.8 |
42.9 ± 1.0 |
128.6 ± 2.1 |
60 |
| 0.45 |
180–250 |
60.0 ± 1.1 |
41.0 ± 0.9 |
131.9 ± 2.5 |
140 |
| 1 |
180–250 |
62.3 ± 0.9 |
37.0 ± 0.4 |
131.2 ± 1.6 |
200 |
| 2 |
180–250 |
60.2 ± 0.6 |
35.8 ± 0.3 |
133.0 ± 2.2 |
250 |
2.2. Membrane preparation
PP membranes were coated as received with poly(1H,1H,2H,2H-heptadecafluorodecyl acrylate) (PHDFA) synthesized using the vapor deposition method described in previous studies.40,41 The initiator of TBP was vaporized at room temperature and fed into the reactor using a mass flow controller (MKS, model 1479A). The monomer of HDFA was vaporized at 80 °C and fed into the reactor through a needle valve. The flow rate of TBP and HDFA was maintained constant at 0.48 sccm and 0.20 sccm, respectively. Inside the reactor, the Nichrome filament (Ni80/Cr20, Goodfellow) was resistively heated to 290 °C, while the stage substrate was maintained at 32 °C by water cooling. These temperatures were measured using directly attached thermocouples (Omega, Type K). The pressure inside the reactor was maintained at 0.2 torr using a butterfly valve (MKS, model 253B). The PHDFA vapor deposition proceeded 3–30 min on PP membranes. A silicon wafer was placed beside the membranes to record the reference coating thickness using an interferometry system with a 633 nm He–Ne laser (JDS Uniphase). The reference coating thickness increased with deposition time. For the 3, 8, 15, and 30 min deposition, the reference coating thickness on the silicon wafer was measured to be 30, 75, 150, and 300 nm, respectively.
2.3. Membrane characterization
The morphology of PP membranes before and after coating was observed using the FEI Quanta 600 field-emission scanning electron microscope (SEM) at an acceleration voltage of 20 kV. Fourier transform infrared (FTIR) spectra were collected using a Nicolet 6700 spectrometer with a DTGS detector under the transmission mode at 4 cm−1 resolution. Surface hydrophobicity was analyzed using a goniometer (ramé-hart, model 250-F1). The static contact angle was measured by the standard sessile drop method using an 8 μL droplet of deionized water. The dynamics of contact angle change was measured every two min using a 15 μL droplet of deionized water at 65 °C on membranes placed on a metal plate heated to 65 °C. Four measurements were taken on each sample. For comparison, the dynamics of contact angle change on PHDFA-coated silicon wafer was recorded under the same condition, and the contact angle change was within measurement error, indicating that the size of the water droplet did not change within the 10 min test.
Membrane porosity εm was determined using the gravimetric method.29,42 The membrane was immersed in isopropyl alcohol under ultrasonication for 30 min, allowing the membrane pores to be completely filled with the liquid. The mass of the wet membrane was measured after removing excess isopropyl alcohol on the membrane surface. The porosity εm (%) was calculated using the following equation:
| |
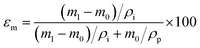 | (2) |
where
m1 (g) is the mass of the wet membrane,
m0 (g) is the mass of the dry membrane,
ρi (g cm
−3) is the isopropyl alcohol density,
ρp (g cm
−3) is the polymer density. Liquid entry pressure (LEP) was measured by placing the membrane in a 7 mm membrane holder. A 10 mm layer of deionized water was placed on the feed side of the membrane. Vacuum was applied on the permeate side to incrementally increase the pressure against the membrane.
43 The vacuum was recorded using a vacuum gauge (WIKA). LEP was detected when the first drop of water appearing at the permeate side. Each membrane was tested three times.
2.4. MD desalination performance
The performance of MD desalination was evaluated using the Franz cell (PermeGear) shown in Fig. S1.† The effective distillation area was 7 mm in diameter. Saline water with 30 or 100 g L−1 NaCl was used as the feed solution. The feed temperature was controlled at 50 or 65 °C with an error of 0.5 °C. A variable gear pump (Cole-Parmer) was used to circulate the feed solution at a constant flow rate in the range of 0.81 to 1.04 L min−1. The permeate was collected and weighed using an analytical balance (Mettler Toledo). The permeate flux 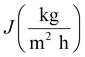 was calculated according to the following equation:
was calculated according to the following equation:| |
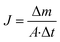 | (3) |
where Δm (kg) is the mass of distillates collected over a predetermined time, A (m2) is the effective surface area of membrane, and Δt (h) is the time of the MD test. Salt concentration was analyzed using an inductively coupled plasma spectrometer (Spectro Arcos II). The salt rejection R (%) was calculated using equation:| |
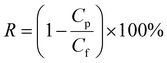 | (4) |
where Cp and Cf represent the sodium concentration in the permeate and feed, respectively.
3. Results and discussion
3.1. Chemical vapor modification of membranes
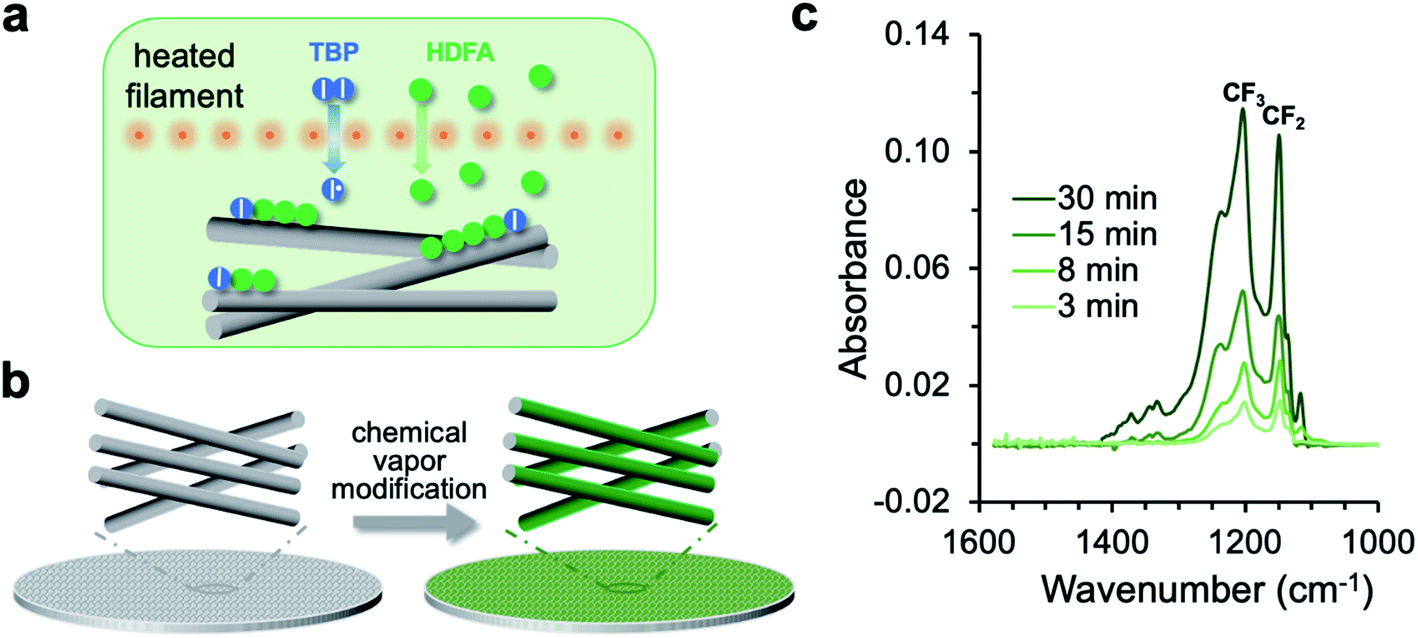
The chemical vapor modification functionalized the membrane using a one-step, solvent-free process. As shown in Fig. 1a, the process involved thermal decomposition of the initiator TBP into free radicals and subsequent addition polymerization of the HDFA monomer,41 which formed PHDFA nanocoatings right on the fiber surface. The high diffusivity of vapor molecules allowed for the functionalization of each individual fiber inside the membrane without clogging the inter-fiber space (Fig. 1b). The chemical composition of PHDFA coating was confirmed using FTIR spectroscopy. The absorption peaks at 1741, 1206 and 1149 cm−1 (Fig. S2†) are assigned to the stretching of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, –CF3, and –CF2 functional groups in PHDFA, respectively.44 The intensities of the absorption peaks were observed to proportionally increase with the deposition time of PHDFA coatings (Fig. 1c).
O, –CF3, and –CF2 functional groups in PHDFA, respectively.44 The intensities of the absorption peaks were observed to proportionally increase with the deposition time of PHDFA coatings (Fig. 1c).
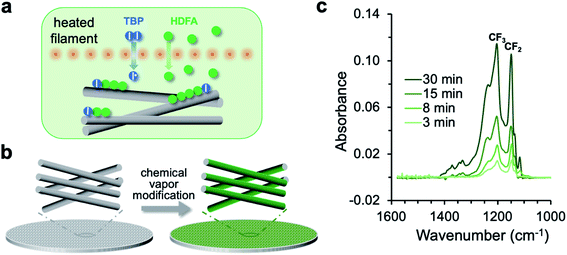 |
| | Fig. 1 (a) Schematic for the vapor-phase decomposition of the initiator TBP into free radicals and subsequent polymerization of HDFA on fiber surfaces. I–I and I˙ stand for TBP and free radicals, respectively. (b) Chemical vapor modification of individual fibers inside the membrane. (c) FTIR absorption of the CF2 and CF3 moieties in PHDFA coatings with varying deposition time. | |
The pristine PP membrane was consisted of overlapped microfibers (Fig. 2a and b), forming interconnected pore structure with high vapor permeability and thermal efficiency for the MD process.45,46 Because of the conformal nature of chemical vapor modification, which was confirmed in our previous studies,47 the PP membranes after PHDFA coating retained the porous microstructure. The microfibers had minimal change in the dimension, as shown in Fig. 2c and d. This result was attributed to the ultrathin thickness of deposited PHDFA coating. For 8 min chemical vapor deposition, the actual thickness of PHDFA coating on fibers was lower than the recorded reference thickness of 80 nm, due to much higher surface area inside the porous structure than that of the flat reference surface.38
 |
| | Fig. 2 SEM images of 2 μm PP membranes before (a and b) and after (c and d) 8 min chemical vapor deposition of PHDFA. | |
3.2. Characteristics of chemically tuned membranes
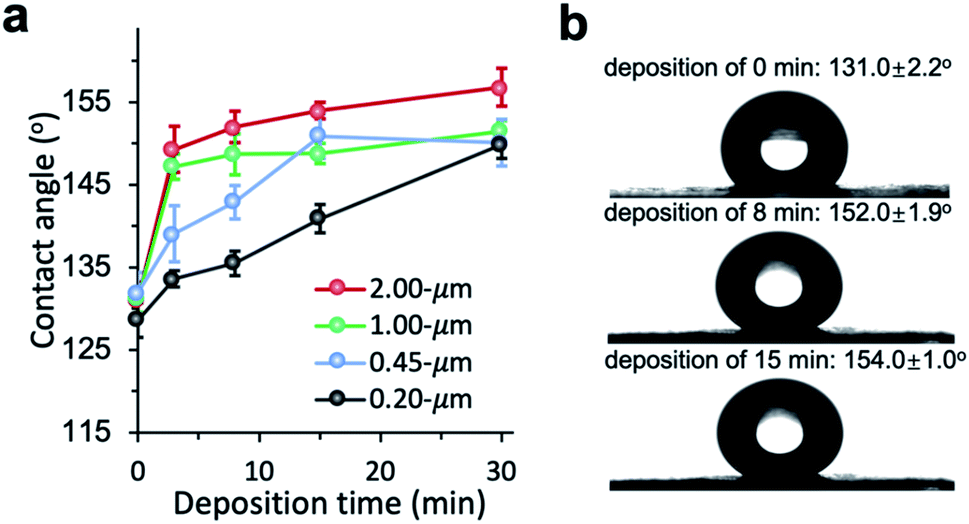
The chemical vapor modification significantly improved the surface hydrophobicity of PP membranes. The effect of deposition time on the membrane wettability was revealed in the water contact angle measurement. Since the difference in the contact angle between the top and the bottom side is negligible within the measurement error, Fig. 3 only shows the contact angle of the top side. The contact angle of 0.2 μm membrane increased with deposition time almost linearly, reaching 149.9° at 30 min deposition time. The contact angle of 0.45 μm membrane was also observed to increase with deposition time up to 15 min in a close-to-linear manner. Interestingly, with only 3 min deposition, the contact angle of 1 and 2 μm membranes was the same as the contact angle of 0.2 μm membrane at 30 min deposition time. Overall, the contact angle of larger-pore-size membranes was higher than that of smaller-pore-size membranes at the same deposition time. Possibly PHDFA was coated more completely around the microfibers inside larger-pore-size membranes due to their higher accessibility to vapor reactants, enabling water droplets to be more readily suspended above the air pockets between microfibers and form Cassie–Baxter state.36,48
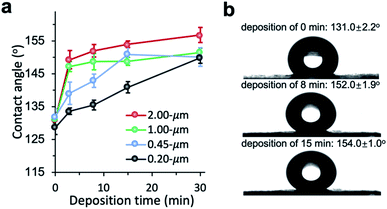 |
| | Fig. 3 (a) Effect of vapor deposition time on the wettability of PP membranes with varying pore sizes. (b) Optical images of water droplet on 2 μm PP membranes with different deposition time. | |
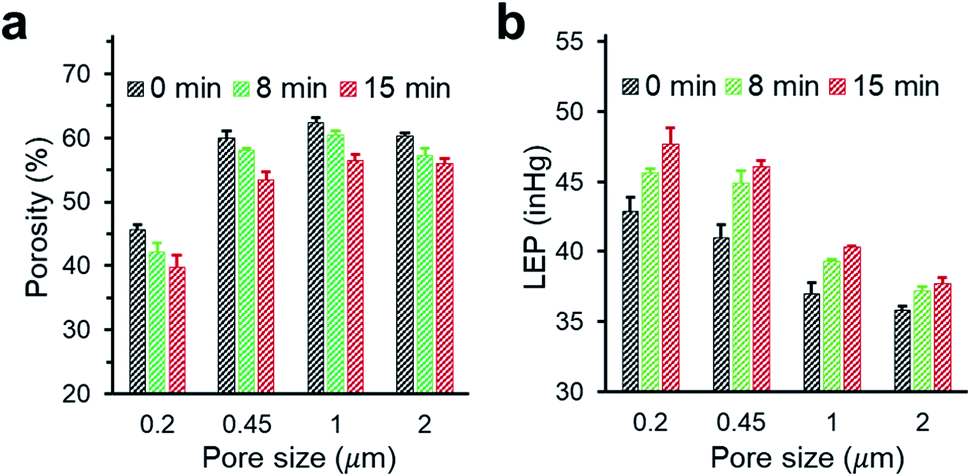
We investigated the changes in membrane porosity and LEP as a function of the deposition time of PHDFA nanocoatings (Fig. 4). LEP was observed to increase with deposition time for all the membrane pore sizes, indicating the improvement in wetting resistance with increasing deposition time. This phenomenon was consistent with the increase of CA with deposition time (Fig. 3a). It is noted that LEP also depends on the membrane geometrical structure.43 At the same deposition time with PHDFA coatings, despite of higher CA for larger-pore-size membranes, LEP was observed to decrease with pore size. The membrane porosity was measured to decrease with deposition time due to the negative correlation between porosity and the amount of polymer deposited into membranes. Membrane porosity increased as pore size increased from 0.2 μm to 0.45 μm; however, there was no significant difference in porosity between membranes with pore size of 0.45, 1.0, and 2.0 μm.
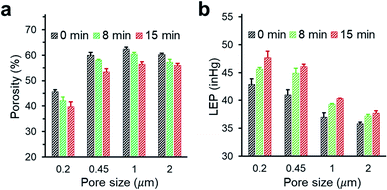 |
| | Fig. 4 The porosity (a) and LEP (b) of PP membranes with different pore size after PHDFA coating with varying deposition time. | |
3.3. MD performance
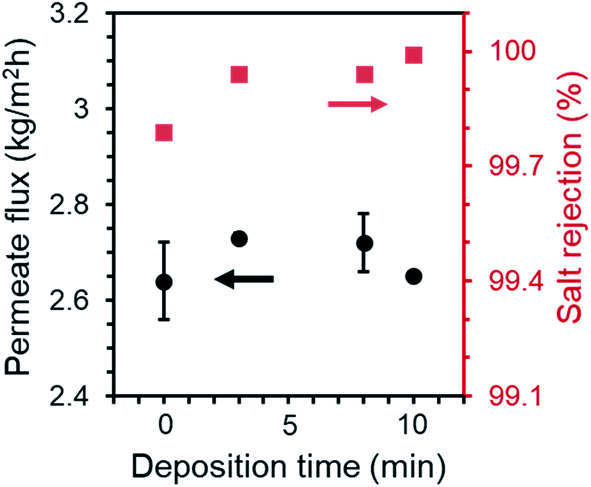
The MD performance of PP membranes was significantly improved after the chemical vapor modification. For the 0.45 μm PP membrane, the pristine membrane exhibited a salt rejection of 99.7%, suggesting that a tiny amount of saline water penetrated through the membrane possibly due to pores that are over-sized.27 The salt rejection of PHDFA-modified membranes was improved to above 99.9% (Fig. 5), owing to the enhancement in the membrane wetting resistance. The permeate flux first increased with the deposition time then decreased, because on one hand the low-surface-energy coating reduced the liquid–solid interface enabling the enhancement of the liquid–vapor interface where water evaporation occurs,49 yet on the other hand the coating reduced the membrane porosity resulting in counteracting effect on permeate flux (Fig. 4a). Nevertheless, compared with the pristine membrane, both the salt rejection and the permeate flux of the modified 0.45 μm membranes were improved with deposition time less than 10 min.
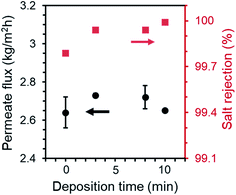 |
| | Fig. 5 Permeate flux and salt rejection of 0.45 μm PP membranes after PHDFA coating with varying deposition time. Test condition: feed temperature at 50 °C, flow rate at 0.81 L min−1, NaCl concentration at 30 g L−1. | |
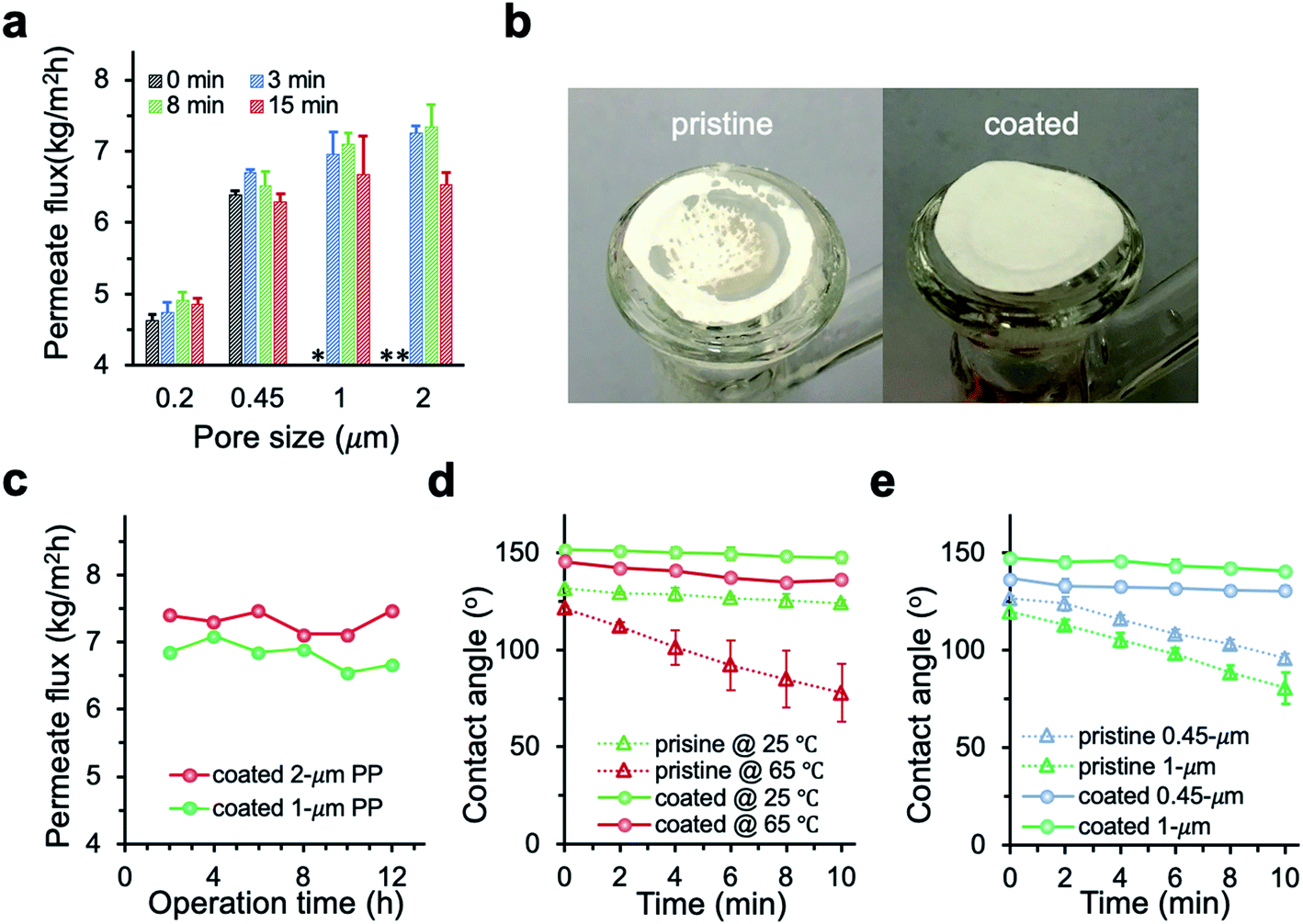
The permeate flux of membranes with different pore size was systematically investigated (Fig. 6a). We observed a huge improvement in the permeate flux of pristine membranes when the pore size increased from 0.2 to 0.45 μm. However, further increase of the pore size to 1 and 2 μm resulted in intermittent or frequent leaking in pristine membranes (Fig. 6b), which is consistent with the pore wetting reported for membranes with pore size larger than 1 μm.11,12,24,28 With 3 min PHDFA coating, both 1 and 2 μm membranes overcame the wetting problem, and the permeate flux further increased when the deposition time increased to 8 min. In addition, the desalination performance of the modified large-pore-size membranes was stable. During the 12 h test, the coated 1 and 2 μm membranes maintained stable flux of 6.7 ± 0.2 and 7.5 ± 0.2 kg m−2 h−1, respectively (Fig. 6c), and the salt rejection exceeded 99.99%. It is worth noting that though the porosity of modified 2 μm membrane was slightly lower than that of the modified 1 μm membrane (Fig. 4a), the permeate flux of modified 2 μm membrane was improved 12% compared with that of modified 1 μm membrane. The flux increase was attributed to enhanced Knudsen diffusion at larger pore size, which resulted in the increase of vapor permeability through the pores.
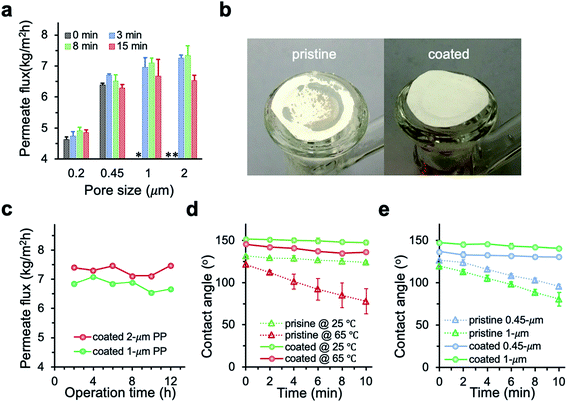 |
| | Fig. 6 (a) Permeate flux of PP membranes after varying deposition time. Coated 1 and 2 μm membranes demonstrated enhanced permeate flux compared with smaller-pore-size membranes, while the un-coated 1 and 2 μm membranes leaked during MD. * and ** indicate intermittent and frequent leaking, respectively. Test condition: feed temperature at 65 °C, flow rate at 1.04 L min−1, NaCl concentration at 30 g L−1. (b) Pictures of the pristine and coated 2 μm PP membranes after MD desalination. The dark area on the pristine PP indicated membrane wetting. (c) Time traces of the permeate flux for 1 and 2 μm membranes with 8 min deposition of PHDFA. (d) Contact angle change for 2 μm membranes before and after 8 min deposition of PHDFA. (e) Contact angle change at 65 °C for 0.45 and 1 μm membranes before and after 8 min deposition of PHDFA. | |
To further understand how the PHDFA modification prevented water from penetrating through large-pore-size membranes, we measured the dynamics of water contact angle change on the 2 μm membrane surface at different temperatures (Fig. 6d). For pristine 2 μm membrane, the contact angle dropped 7.4° and 43.2° in 10 min at 25 °C and 65 °C, respectively. The pronounced wetting with the increase of temperature could be attributed to the decrease of water surface tension and the membrane property change at high temperature.50 For the 2 μm coated membrane, the profile of contact angle change remained similar as temperature increased from 25 °C to 65 °C. The contact angle changes of coated membranes with 0.45 and 1 μm pores at 65 °C were similarly small as that of the 2 μm modified membrane, while the contact angle of pristine 0.45 and 1 μm membranes decreased much quickly with time (Fig. 6e). Since water surface tension decreased the same on pristine and coated membranes as temperature increased, the dramatic difference in the contact angle change between pristine and coated membranes suggested that PHDFA-coated membranes had relatively small heat-induced property change and thus excellent wetting resistance at high temperature.
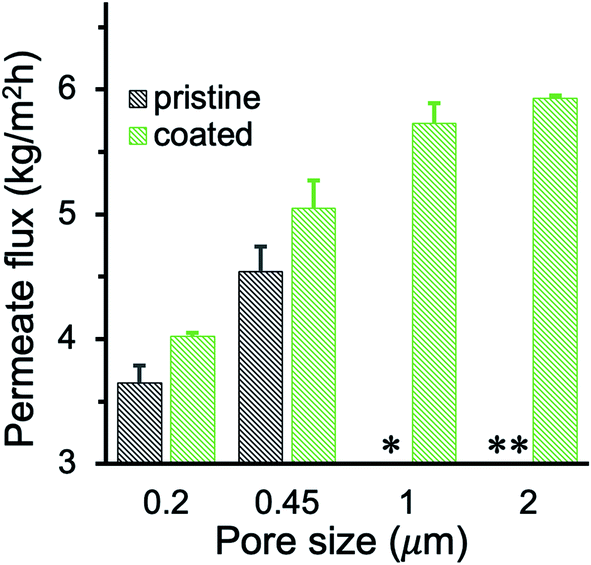
Membrane wetting usually becomes more significant with the increase of the feed salinity.51 To investigate the anti-wetting capacity of membranes, MD was also conducted using high salinity feed with 100 g L−1 NaCl. The PHDFA-coated membranes demonstrated excellent wetting resistance with salt rejection above 99.99%. The permeate flux using 100 g L−1 NaCl as feed was shown in Fig. 7. The value was lower than the corresponding permeate flux using 30 g L−1 NaCl as the feed (Fig. 6a) due to the reduction of vapor pressure with the increase of salt concentration.27 Nonetheless, compared with the permeate flux obtained using un-coated membranes, the permeate flux of coated large-pore-size membranes improved 33–67%, indicating the potential of their application in desalinating high-salinity water.
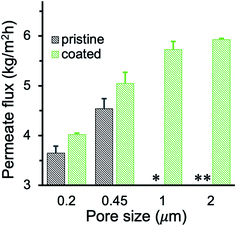 |
| | Fig. 7 Permeate flux of PP membranes before and after 8 min of PHDFA coating using high-salinity feed with 100 g L−1 NaCl. The pristine 1 and 2 μm membranes leaked during MD while the coated membranes remained nonwetted. * and ** indicate intermittent and frequent leaking, respectively. | |
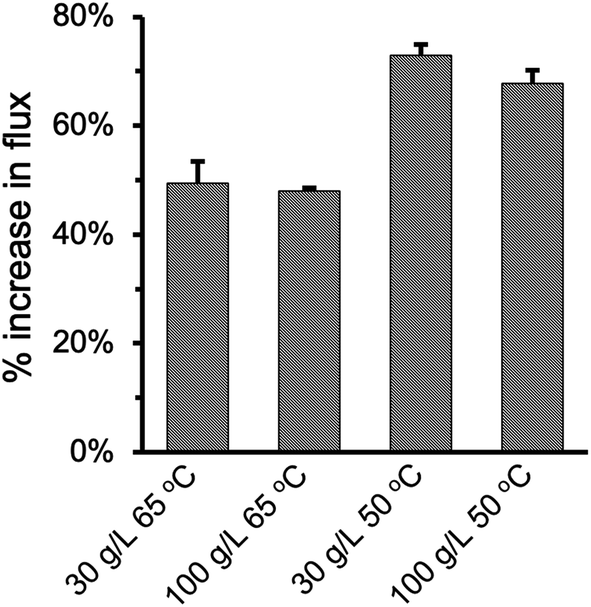
Fig. 8 summarized the effect of enlarging pore size on improving permeate flux at varying feed temperature and NaCl concentration. As the pore size increased from 0.2 μm to 2.0 μm in the PHDFA-coated membranes, the permeate flux increased by 48.0–73.0%. Such enhancement in flux with pore size increase is notable compared with reported strategies such as surface modification52 and MD module design.53 The percentage increase was 20% higher at 50 °C than they were at 65 °C, which suggests that increasing pore size has a more significant effect on Knudsen diffusion at lower temperature.19,54 On the other hand, saline concentration does not affect the permeate flux enhancement that much. The percentage increase in flux only decreased slightly as NaCl concentration increased from 30 g L−1 to 100 g L−1.
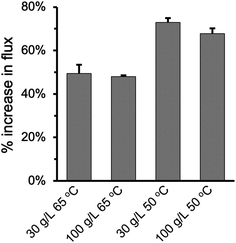 |
| | Fig. 8 Percentage increase in the permeate flux under different operation conditions when the nominal pore size increases from 0.2 μm to 2 μm for PP membranes after 8 min of PHDFA coating. | |
4. Conclusions
A novel vapor modification method was used to chemically tune the pores inside MD membranes with PHDFA nanocoatings. The membrane surface hydrophobicity and liquid entry pressure were significantly improved after the PHDFA modification. The modified membranes maintained high wetting resistance at high feed temperature. The modification improved both the permeate flux and the salt rejection of 0.2 and 0.45 μm MD membranes. In addition, the PHDFA modification enabled the use of 1.0 and 2.0 μm membranes for MD desalination with salt rejection exceeding 99.99%. The permeate flux of modified membranes improved 48.0–73.0% as the pore size increased from 0.2 μm to 2.0 μm. The modified large-pore-size membranes maintained the high permeate flux at high saline concentration and extended operation time.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This study was financially supported by the Oklahoma Center for the Advancement of Science and Technology under AR19-018. We thank the Oklahoma State University (OSU) Microscopy Laboratory for SEM experiments and the OSU Soil, Water and Forage Analytical Laboratory for the analysis of salt concentration.
Reference
- A. Y. Hoekstra, Nat. Clim. Change, 2014, 4, 161–163 CrossRef.
- C. A. Schlosser, K. Strzepek, X. Gao, C. Fant, É. Blanc, S. Paltsev, H. Jacoby, J. Reilly and A. Gueneau, Earth's Future, 2014, 2, 341–361 CrossRef.
- M. E. a. W. A. Phillip, Science, 2011, 333, 712–717 CrossRef.
- M. Safavi and T. Mohammadi, Chem. Eng. J., 2009, 149, 191–195 CrossRef CAS.
- A. Deshmukh, C. Boo, V. Karanikola, S. Lin, A. P. Straub, T. Tong, D. M. Warsinger and M. Elimelech, Energy Environ. Sci., 2018, 11, 1177–1196 RSC.
- J. A. Bush, J. Vanneste and T. Y. Cath, Sep. Purif. Technol., 2016, 170, 78–91 CrossRef CAS.
- D. L. Shaffer, L. H. Arias Chavez, M. Ben-Sasson, S. Romero-Vargas Castrillon, N. Y. Yip and M. Elimelech, Environ. Sci. Technol., 2013, 47, 9569–9583 CrossRef CAS.
- M. A. Shannon, P. W. Bohn, M. Elimelech, J. G. Georgiadis, B. J. Marinas and A. M. Mayes, Nature, 2008, 452, 301–310 CrossRef CAS.
- D. R. L. Kevin and W. Lawson, J. Membr. Sci., 1997, 124, 1–25 CrossRef.
- L. Li and K. K. Sirkar, J. Membr. Sci., 2016, 513, 280–293 CrossRef CAS.
- L. Eykens, K. De Sitter, C. Dotremont, L. Pinoy and B. Van der Bruggen, Ind. Eng. Chem. Res., 2016, 55, 9333–9343 CrossRef CAS.
- A. Alkhudhiri, N. Darwish and N. Hilal, Desalination, 2012, 287, 2–18 CrossRef CAS.
- R. W. SchofieldA, G. FaneC and J. D. Fell, J. Membr. Sci., 1987, 33, 299–313 CrossRef.
- S. K. Bhatia, M. R. Bonilla and D. Nicholson, Phys. Chem. Chem. Phys., 2011, 13, 15350–15383 RSC.
- L. M. Pant, S. K. Mitra and M. Secanell, Int. J. Heat Mass Transfer, 2013, 58, 70–79 CrossRef CAS.
- J. Lee and R. Karnik, J. Appl. Phys., 2010, 108, 044315 CrossRef.
- J. Cai and F. Guo, Desalination, 2017, 407, 46–51 CrossRef CAS.
- J. Phattaranawik, J. Membr. Sci., 2003, 215, 75–85 CrossRef CAS.
- D. Winter, J. Koschikowski, D. Düver, P. Hertel and U. Beuscher, Desalination, 2013, 323, 120–133 CrossRef CAS.
- M. E. XIaohua Zhu, Environ. Sci. Technol., 1997, 31, 3654–3662 CrossRef.
- Z. Wang, Y. Chen, X. Sun, R. Duddu and S. Lin, J. Membr. Sci., 2018, 559, 183–195 CrossRef CAS.
- D. M. Warsinger, A. Servi, G. B. Connors, M. O. Mavukkandy, H. A. Arafat, K. K. Gleason and J. H. Lienhard V, Environ. Sci.: Water Res. Technol., 2017, 3, 930–939 RSC.
- A. Sadeghzadeh, S. Bazgir and M. M. A. Shirazi, Sep. Purif. Technol., 2020, 239, 116498 CrossRef CAS.
- E. Drioli, A. Ali and F. Macedonio, Desalination, 2015, 356, 56–84 CrossRef CAS.
- D. H. Seo, S. Pineda, Y. C. Woo, M. Xie, A. T. Murdock, E. Y. M. Ang, Y. Jiao, M. J. Park, S. I. Lim, M. Lawn, F. F. Borghi, Z. J. Han, S. Gray, G. Millar, A. Du, H. K. Shon, T. Y. Ng and K. Ostrikov, Nat. Commun., 2018, 9, 683 CrossRef.
- S. Nejati, C. Boo, C. O. Osuji and M. Elimelech, J. Membr. Sci., 2015, 492, 355–363 CrossRef CAS.
- A. Alkhudhiri and N. Hilal, Desalination, 2017, 403, 179–186 CrossRef CAS.
- J. Xu, Y. B. Singh, G. L. Amy and N. Ghaffour, J. Membr. Sci., 2016, 512, 73–82 CrossRef CAS.
- A. C. M. F. K. Smolders, Desalination, 1989, 72, 249–262 CrossRef.
- A. Razmjou, E. Arifin, G. Dong, J. Mansouri and V. Chen, J. Membr. Sci., 2012, 415–416, 850–863 CrossRef CAS.
- D. Hou, J. Wang, X. Sun, Z. Ji and Z. Luan, J. Membr. Sci., 2012, 405–406, 185–200 CrossRef CAS.
- J. E. Efome, M. Baghbanzadeh, D. Rana, T. Matsuura and C. Q. Lan, Desalination, 2015, 373, 47–57 CrossRef CAS.
- J. A. Prince, G. Singh, D. Rana, T. Matsuura, V. Anbharasi and T. S. Shanmugasundaram, J. Membr. Sci., 2012, 397–398, 80–86 CrossRef CAS.
- M. M. Ghaleni, S. Kaviani, K. Rajwade, M. Bavarian, F. Perreault and S. Nejati, Adv. Mater. Interfaces, 2020, 7, 1902159 CrossRef CAS.
- M. Bhadra, S. Roy and S. Mitra, Desalination, 2016, 378, 37–43 CrossRef CAS.
- C. Yang, X.-M. Li, J. Gilron, D.-f. Kong, Y. Yin, Y. Oren, C. Linder and T. He, J. Membr. Sci., 2014, 456, 155–161 CrossRef CAS.
- M. Tian, Y. Yin, C. Yang, B. Zhao, J. Song, J. Liu, X.-M. Li and T. He, Desalination, 2015, 369, 105–114 CrossRef CAS.
- F. Guo, A. Servi, A. Liu, K. K. Gleason and G. C. Rutledge, ACS Appl. Mater. Interfaces, 2015, 7, 8225–8232 CrossRef CAS.
- A. T. Servi, J. Kharraz, D. Klee, K. Notarangelo, B. Eyob, E. Guillen-Burrieza, A. Liu, H. A. Arafat and K. K. Gleason, J. Membr. Sci., 2016, 520, 850–859 CrossRef CAS.
- B. Zhi, Q. Song and Y. Mao, RSC Adv., 2018, 8, 4779–4785 RSC.
- Y. Ye and Y. Mao, RSC Adv., 2017, 7, 24569–24575 RSC.
- Y. Liao, R. Wang, M. Tian, C. Qiu and A. G. Fane, J. Membr. Sci., 2013, 425–426, 30–39 CrossRef CAS.
- P. Yazgan-Birgi, M. I. Hassan Ali and H. A. Arafat, J. Membr. Sci., 2018, 552, 68–76 CrossRef CAS.
- D. Soto, A. Ugur, T. A. Farnham, K. K. Gleason and K. K. Varanasi, Adv. Funct. Mater., 2018, 28, 1707355 CrossRef.
- L. Eykens, K. De Sitter, C. Dotremont, L. Pinoy and B. Van der Bruggen, Sep. Purif. Technol., 2017, 182, 36–51 CrossRef CAS.
- I. Hitsov, T. Maere, K. De Sitter, C. Dotremont and I. Nopens, Sep. Purif. Technol., 2015, 142, 48–64 CrossRef CAS.
- Y. Ye, Y. Mao, H. Wang and Z. Ren, J. Mater. Chem., 2012, 22, 2449–2455 RSC.
- J. Li, S. Guo, Z. Xu, J. Li, Z. Pan, Z. Du and F. Cheng, J. Membr. Sci., 2019, 574, 349–357 CrossRef CAS.
- L.-H. Chen, A. Huang, Y.-R. Chen, C.-H. Chen, C.-C. Hsu, F.-Y. Tsai and K.-L. Tung, Desalination, 2018, 428, 255–263 CrossRef CAS.
- A. M. Barbe, P. A. Hogan and R. A. Johnson, J. Membr. Sci., 2000, 172, 149–156 CrossRef CAS.
- J. Ge, Y. Peng, Z. Li, P. Chen and S. Wang, Desalination, 2014, 344, 97–107 CrossRef CAS.
- Z. Xu, X. Yan, Z. Du, J. Li and F. Cheng, Sep. Purif. Technol., 2020, 251, 117304 CrossRef CAS.
- Q. Li, B. Lian, W. Zhong, A. Omar, A. Razmjou, P. Dai, J. Guan, G. Leslie and R. A. Taylor, Sep. Purif. Technol., 2020, 248, 117072 CrossRef CAS.
- A. P. Straub and M. Elimelech, Environ. Sci. Technol., 2017, 51, 12925–12937 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra07629e |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence and
Yu Mao
and
Yu Mao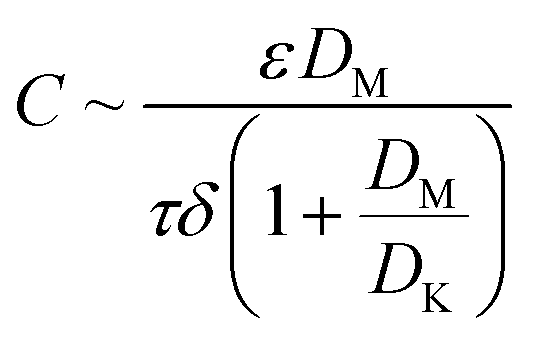
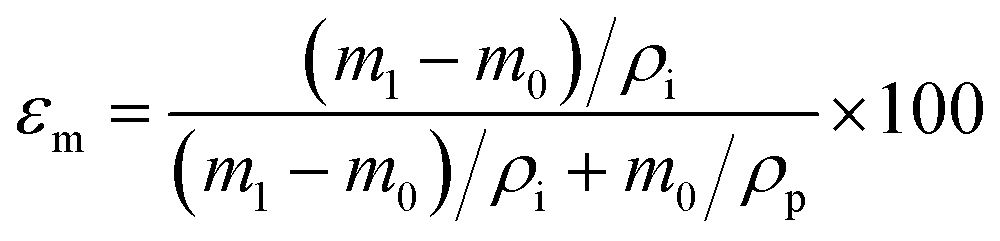
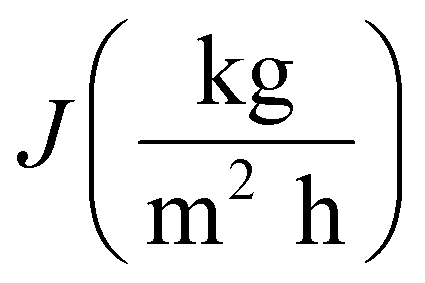 was calculated according to the following equation:
was calculated according to the following equation: