
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTin(II) chloride dihydrate/choline chloride deep eutectic solvent: redox properties in the fast synthesis of N-arylacetamides and indolo(pyrrolo)[1,2-a]quinoxalines†
Sergio Alfonso Trujilloa,
Diana Peña-Solórzanoa,
Oscar Rodríguez Bejaranob and
Cristian Ochoa-Puentes *a
*a
aLaboratorio de Síntesis Orgánica Sostenible, Departamento de Química, Universidad Nacional de Colombia–Sede Bogotá, Carrera 45 # 26–85, A.A. 5997, Bogotá, Colombia. E-mail: cochoapu@unal.edu.co
bLaboratorio de Electroquímica y Termodinámica Computacional, Departamento de Química, Universidad Nacional de Colombia–Sede Bogotá, Carrera 45 # 26–85, A.A. 5997, Bogotá, Colombia
First published on 6th November 2020
Abstract
In this contribution a physicochemical, IR and Raman characterization for the tin(II) chloride dihydrate/choline chloride eutectic mixture is reported. The redox properties of this solvent were also studied by cyclic voltammetry finding that it can be successfully used as an electrochemical solvent for electrosynthesis and electroanalytical processes and does not require negative potentials as verified by the reduction of nitrobenzene. The potential use of this eutectic mixture as a redox solvent was further explored in obtaining aromatic amines and N-arylacetamides starting from a wide variety of nitroaromatic compounds. In addition, a fast synthetic strategy for the construction of a series of indolo(pyrrolo)[1,2-a]quinoxalines was developed by reacting 1-(2-nitrophenyl)-1H-indole(pyrrole) with aldehydes. This simple protocol offers a straightforward method for the construction of the target quinoxalines in short reaction times and high yields where the key step involves a tandem one-pot reductive cyclization-oxidation.
Introduction
Nitro compounds are versatile nitrogenated molecules found in natural products and synthetic derivatives. Its isolation from plants, bacteria, fungi, and mammals has allowed the discovery of several bioactive compounds which exhibited a high structural diversity together with antibiotic,1,2 antitumor3 and immunosuppressive4 activities. Different synthetic derivatives have been also employed as solvents, agrochemicals,5 drugs, dyes6 and fine chemicals.Due to their reactivity, nitro compounds are considered as versatile synthetic tools and, in particular, aliphatic nitro compounds are suitable starting materials to perform carbon–carbon bond forming reactions such as nitro-Mannich, Henry, and nitro-Michael reactions.7–9 They are also fundamental building blocks employed in the synthesis of amines which are obtained by the reduction of the nitro group through metal–acid combinations, catalytic hydrogenation, hydride transfer reductions, catalytic transfer hydrogenation and metal-free reduction methods.10 The synthetic utility of this transformation is highlighted in several one pot procedures for the obtention of diverse structures including imines, amides11 and α-aminophosphonates12 together with heterocyclic compounds such as benzimidazoles,13–15 quinazolinones,16,17 pyrrolines,18 isoindolinone derivatives,19 tetrahydroquinolines,20–23 and quinoxalines,24,25 where a tandem one-pot reduction and intramolecular cyclization reaction is achieved.
Among the different nitrogenated heterocycles, quinoxalines are considered privileged structures with broad medicinal and industrial applications. This bioactive motif is found in marketed drugs with antibiotic, antibacterial, antitumor, antiviral, antifungal, anti-inflammatory, antimicrobial activities,26 while many more are under study for potential biomedical applications (Fig. 1).27
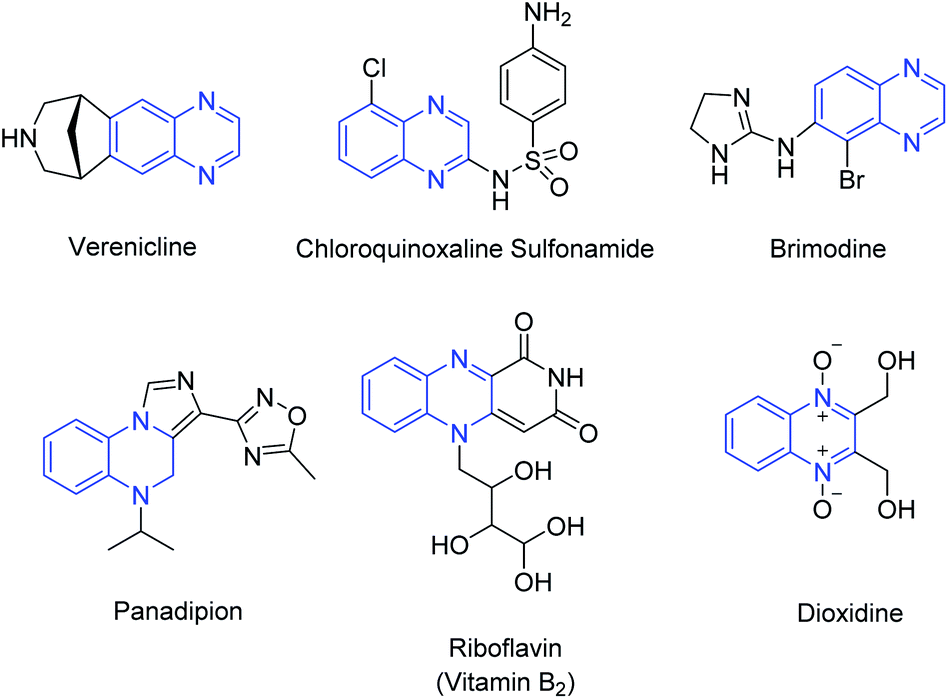 | ||
| Fig. 1 Representative bioactive and naturally occurring quinoxalines. | ||
Besides the pharmaceutical interest, various synthetic derivatives have also found applications in organic electronics,28–30 dyes,31 chemically controllable switches,32 cavitands and macrocyclic receptors.33–35
Within the diverse structural derivatives reported in literature for this heterocyclic system, the indolo(pyrrolo)[1,2-a]quinoxalines have attracted much interest from the synthetic and biological community. Considering the integrity of the benzene ring, the construction of this polycyclic skeleton follows different strategies depending on the number of atoms contained in each fragment. Thus, synthetic equivalents with several combinations of atoms might be used. On the other hand, a more convenient strategy based on pyrrole or indole derivatives can be developed for the formation of the pyrazine ring;36 and for this 1-(2-aminophenyl)pyrrole/indole has been widely used as starting material in combination with a broad scope of substrates including aldehydes,37–40 alcohols,41 ketones,42,43 α-aminoacids,44 carboxylic acids,45 2-methylpyridine/quinoline46 and benzylamines47 (Scheme 1). Analogously, other synthetic approaches have employed arylhalides,48 arylisocyanides,49 and nitrobenzene24 having ortho-substituted pyrrole or indole rings (Scheme 1).
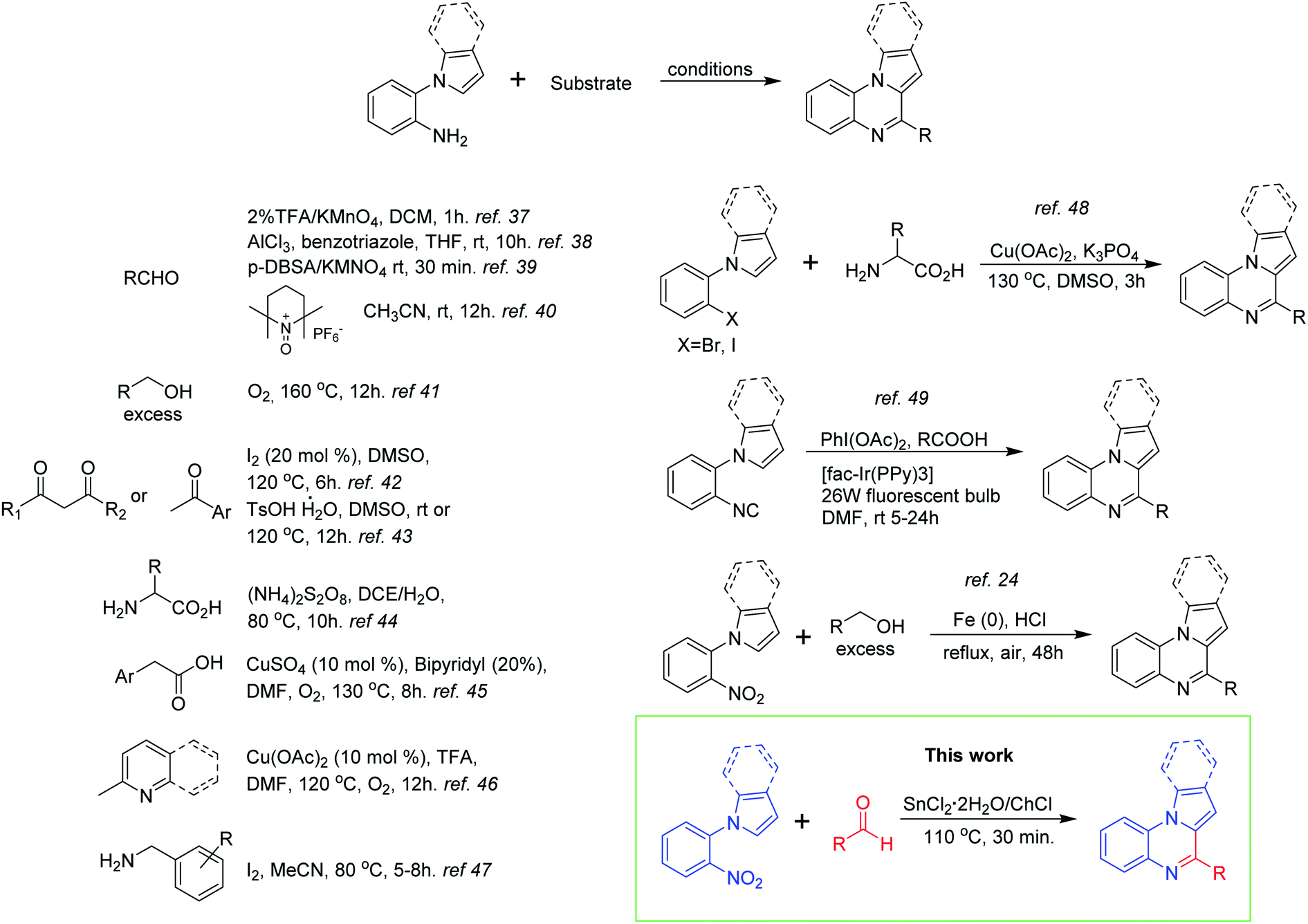 | ||
| Scheme 1 Synthetic strategies for the synthesis of indolo(pyrrolo)[1,2-a]quinoxalines. | ||
Although the methods depicted before are ingeniously developed and represent new alternatives for the obtention of the indolo(pyrrolo)[1,2-a]quinoxaline skeleton, some of them suffered from several disadvantages such as the use of strong oxidizing agents, ligands and costly catalysts, require an elaborated set up, and have long reaction times with moderated yields. Keeping this in mind, we envisioned that the tin(II) chloride dihydrate/choline chloride deep eutectic solvent (DES) might be a suitable reductive reaction medium to obtain the target compounds starting from 1-(2-nitrophenyl)-indole/pyrrole and aldehydes via a one-pot reductive cyclization strategy (Scheme 1). The use of tin(II) chloride/choline chloride DES in organic synthesis has been previously introduced by Azizi50 and Tavakol.51,52 The first author found that this eutectic mixture effectively catalyses the ring opening of epoxides by a range of nucleophiles (amines, thiols, alcohols, cyanide and azide) providing the target products with good chemo, regio, and stereoselectivity.50 On the other hand, Tavakol developed an environmentally friendly synthesis of quinoline derivatives via a one-pot multi-component reaction between aryl aldehydes, aniline derivatives and enolizable aldehydes employing the ChCl/SnCl2 DES as a green catalyst and solvent.51 The same author also reported the synthesis of xanthene derivatives by the reaction between 2-naphthol derivatives and aromatic or aliphatic aldehydes in the presence of the DES.52 Other applications of this eutectic mixture include the extractive desulfurization of liquid fuels53 and its use as catalysts for epoxy resin crosslinking.54
Results and discussion
Before starting the synthetic studies of this work, we first performed a physicochemical characterization and investigated the redox properties of the tin(II) chloride dihydrate/choline chloride DES in order to gain more insight into the properties and potential applications for this solvent.The FTIR spectra of pure choline chloride (a), SnCl2·2H2O (b) and the DES (c) are presented in Fig. 2. For choline chloride, the FTIR spectrum shows the characteristic bands associated to the OH, CH2, CH3 and C–N+ groups at 3200 cm−1 (O–H stretch), 1470–1440 cm−1 (CH3 and CH2 bend), 1050 cm−1 (C–O stretch) and 882 cm−1 (C–N+ symmetric stretching). Similarly, the FTIR spectrum of pure SnCl2·2H2O (Fig. 2b) showed the usual broad band of stretching vibrations of the OH bond between 3100 and 3500 cm−1 together with the bending H–O–H vibrations in the region between 1610–1620 cm−1, while absorption bands below 900 cm−1 belong to Sn–Cl and Sn–OH bonds. These spectra agree with others previously published.55,56 On the other hand, it is observed that several bands corresponding to choline chloride co-exist after the DES formation (Fig. 2c). However, a vibrational broad band appears at 3500 cm−1 which is assigned to the O–H stretching. This band broadens because of the presence of water molecules indicating the formation of hydrogen bonds. The bending H–O–H vibration appears in the region between 1615–1624 cm−1, and the absorption corresponding to C–N+ symmetric stretching is observed at 869 cm−1.
 | ||
| Fig. 2 FTIR spectrum for (a) choline chloride, (b) tin(II) chloride dihydrate, and (c) the eutectic mixture SnCl2·2H2O/ChCl. | ||
The Raman spectra for choline chloride (a), SnCl2·2H2O (b) and the DES (c) are depicted in Fig. 3. It could be seen that Raman spectra of ChCl presents bands in the range of 2800–3100 cm−1 which are assigned to anti-symmetric and symmetric CH2 and CH3 stretching vibrations. Bands assigned to CH3 and COH deformations are at 1410–1430 cm−1 and the Raman band at 720 cm−1 can be assigned to the symmetric stretching vibration of the four C–N bonds.57,58 The Raman spectra of tin(II) chloride dihydrate shows peaks in frequencies below 300 cm−1. It is known that the unit cell of SnCl2·2H2O contains four molecules of the pyramidal dichloroaquatin(II) complexes, SnCl2(H2O), and this coordinated water molecule at an apex of each pyramid forms a two-dimensional hydrogen bonded network with another noncoordinating water molecule. Based on this, Raman frequencies in the range from 52 to 222 cm−1 has been attributed to the deformation and stretching modes of the Cl–Sn–Cl, Cl–Sn–O and Sn–Cl groups, while the band at 247 cm−1 corresponds to the O–H⋯O hydrogen bonded network.59 It could be seen that Raman spectra of SnCl2·2H2O/ChCl DES present a combined spectrum of the two components where the broad band at 254 cm−1 suggest the formation of hydrogen bonds between ChCl and SnCl2·2H2O through water molecules from SnCl2·2H2O and Cl− and OH groups from ChCl.
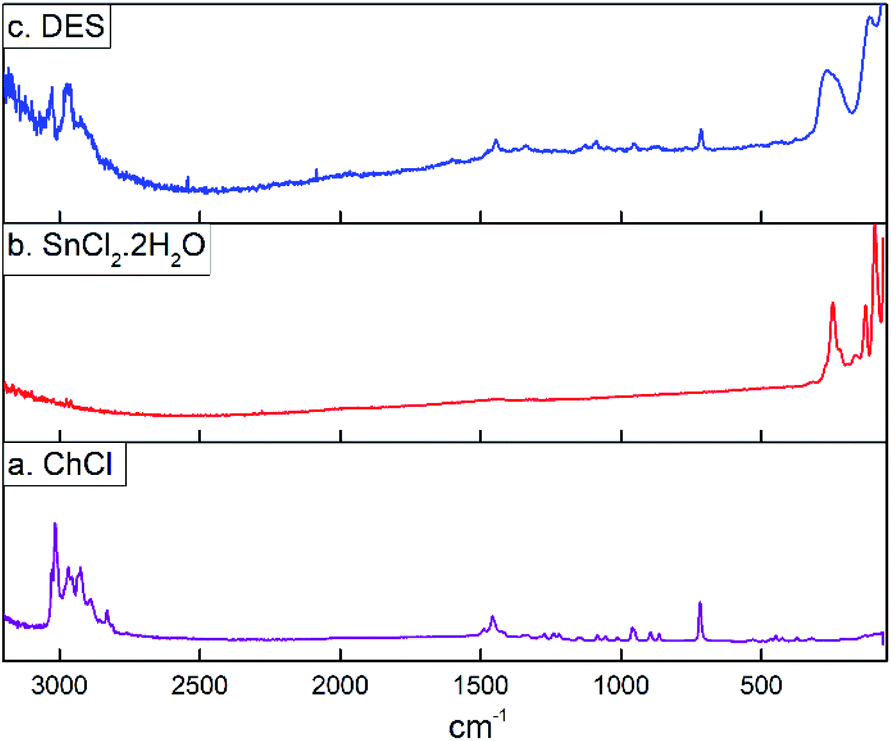 | ||
| Fig. 3 Raman spectrum for (a) choline chloride, (b) tin(II) chloride dihydrate, and (c) the eutectic mixture SnCl2·2H2O/ChCl. | ||
Next, the refractive index (nD), conductivity (k) and density (ρ) were studied as function of temperature in the range from 35 to 80 °C. The temperature dependence of the refractive index is shown in Fig. 4. The refractive index shows a decrease when the temperature is increased lying in the range from 1.6420 to 1.6304 and it has higher values compared to common polar protic and aprotic organic solvents such as hexamethylphosphoramide (HMPT), N-methylacetamide (NMA), N,N-dimethylacetamide (DMA), dioxane, acetonitrile, N-methylformamide (NMF), and methanol.60
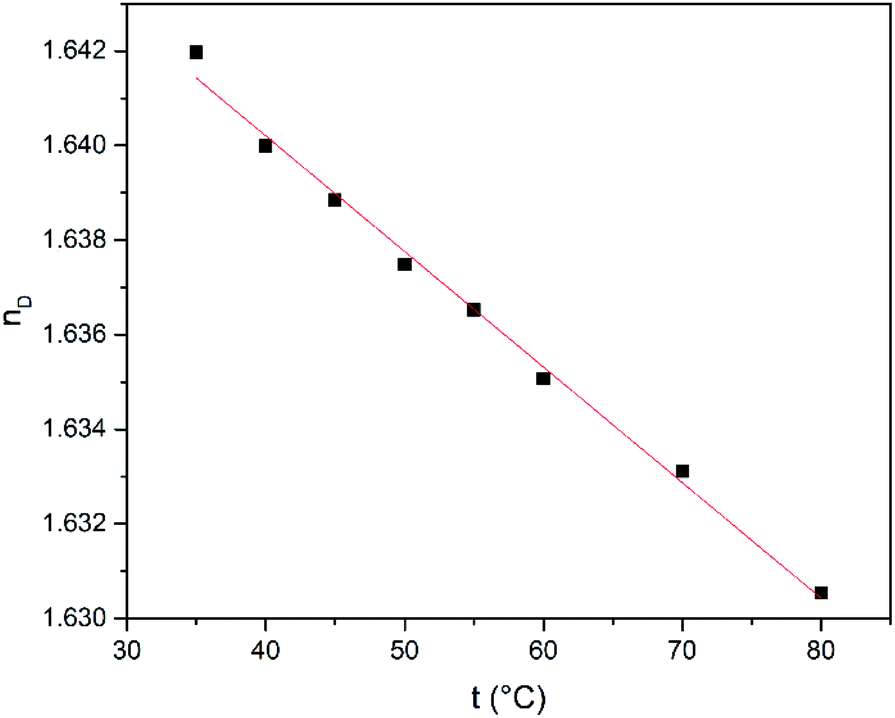 | ||
| Fig. 4 Refractive index (nD) of SnCl2·2H2O/ChCl as a function of temperature. | ||
The temperature-dependent behaviour of refractive index was fitted linearly according with the eqn (1) where nD is the refractive index, t is the temperature, a and b are unitless terms given in Table 1.
| nD = a(t/°C) + b | (1) |
| Parameters and correlation coefficient | Physical properties | ||
|---|---|---|---|
| nD | k | ρ | |
| a | −3 × 10−4 | −1.1 × 10−3 | |
| b | 1.651 | 2.0808 | |
| k0 | 2.42 × 10−4 | ||
| Ek/R | 645.8 | ||
| R2 | 0.9902 | 0.9995 | 0.9993 |
In contrast to refractive index, higher values for conductivity are observed at elevated temperatures (Fig. 5) which shows an increase in the kinetic energy of all species that coexist in the DES.
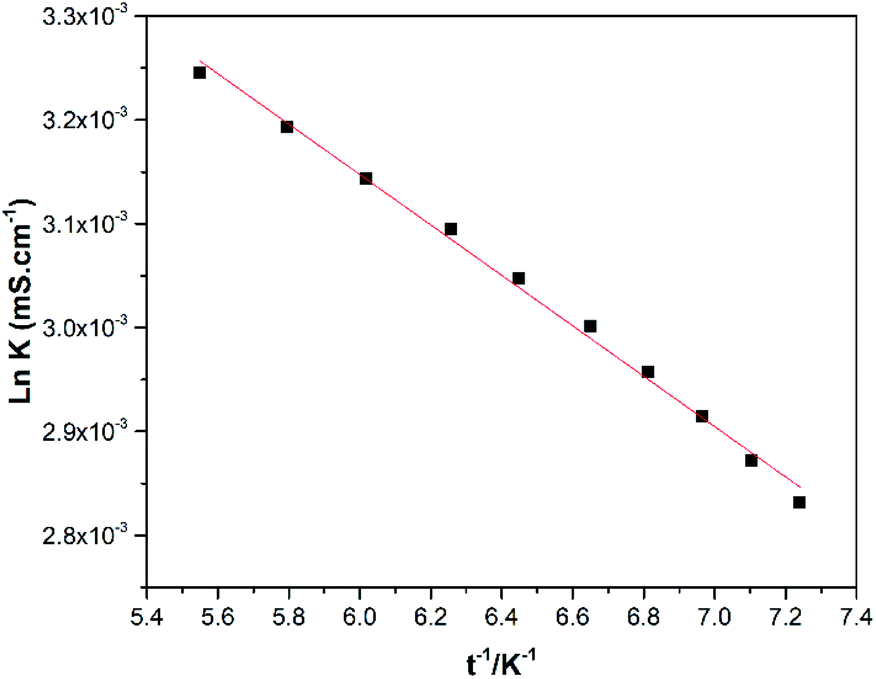 | ||
| Fig. 5 Conductivity as a function of temperature for the SnCl2·2H2O/ChCl DES. | ||
The dependence between conductivity and temperature is modelled with the Arrhenius-like eqn (2), where the values of k0 and (Ek/R) are shown in Table 1.
ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) k = lnk0 − (Ek/RT) k = lnk0 − (Ek/RT)
| (2) |
Similarly to refractive index, the density shows an inverse correlation where it decreases when the temperature increases (Fig. 6). In addition, density values measured for this DES are close two or three-fold higher of that found for the aforementioned organic solvents.60
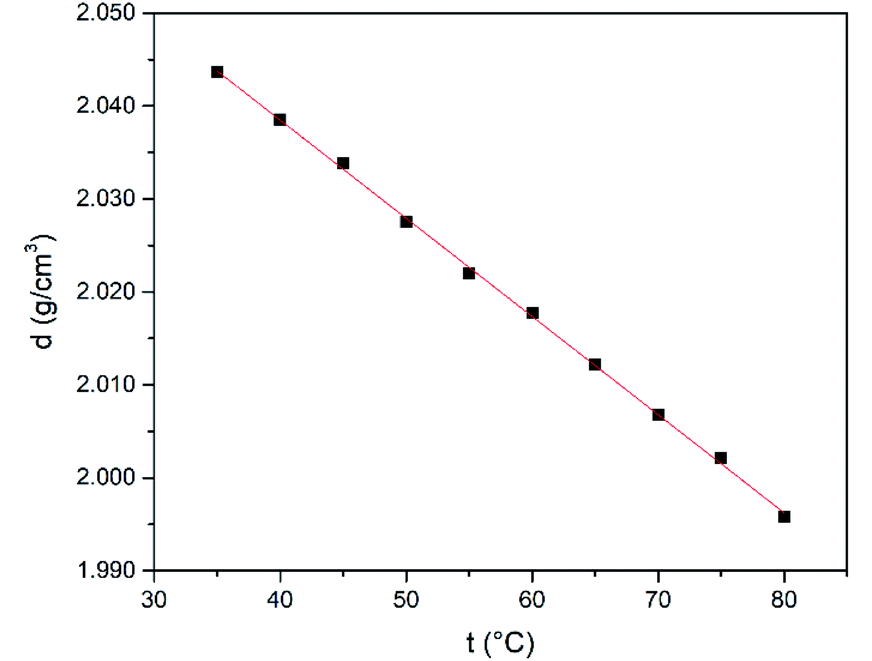 | ||
| Fig. 6 Density as a function of temperature for the SnCl2·2H2O/ChCl DES. | ||
The variation of density with temperature was modelled according to eqn (3), where ρ is the density, t is the temperature, a and b are constants (Table 1).
| ρ (g cm−3) = a(t/°C) + b | (3) |
Gano et al.53 have also studied the effect of temperature on some basic physical properties of the synthesized DES finding that the viscosity, density and refractive index decrease with temperature, while the conductivity increased with temperature. Our observations are in agreement with the pervious study and allow to predict the behaviour of the DES at different temperatures within the studied range (30 to 80 °C).
The redox properties of the SnCl2·2H2O/ChCl DES were also studied by cyclic voltammetry. Solvents and electrolytes for electrochemical cells should resist a possible oxidation and reduction. The electrochemical potential window (EPW) is an indicator of electrochemical stability of DES and the potential redox of a given analyte in a reaction process involving DES is expected to fall within the EPW. The EPW is also useful in the selection of a DES as an electrochemical solvent for specific electrochemical applications. In this work, we determine the potential window of the DES employing four working electrodes. Results are shown in Table 2.
| Working electrode | Anodic limit (V) | Cathodic limit (V) | Potential window (V) |
|---|---|---|---|
| Glassy carbon | 2 | −0.4 | 2.4 |
| Pt | 2 | −0.3 | 2.3 |
| Au | 0.5 | −0.3 | 0.8 |
| Sn | 1.9 | −0.4 | 2.3 |
Anodic and cathodic limits of the EPW were determined with the oxidation current onset potential for chlorine gas evolution and the reduction current onset potential for tin deposition respectively. Although all systems show relatively narrow potential ranges, these results are consistent with the behaviours of similar systems previously reported.61,62 All this indicates that the SnCl2·2H2O/ChCl DES can be a good electrochemical solvent for applications in electroanalytical chemistry and electrosynthesis.
The use of DES as a novel solvent in reduction reactions is another application field that we explored. In order to demonstrate the reduction ability of the DES, the electrochemical properties for a model reaction were examined by cyclic voltammetry (CV) mixing 10 mM of nitrobenzene in SnCl2·2H2O/ChCl mixture and glassy carbon (GC) as working electrode (Fig. 7).
 | ||
| Fig. 7 Cyclic voltammograms of nitrobenzene (10 mM); in SnCl2·2H2O/ChCl DES at scan rate 100 mV s−1 using GC disc versus Ag/AgCl as reference electrode and Pt wire counter electrode at 60 °C. (a) Recorded after adding nitrobenzene; (b) ten cycles, recorded one minute after nitrobenzene was added. | ||
Fig. 7a displays the cyclic voltammogram recorded immediately after adding nitrobenzene to the mixture. Here, only one irreversible cathodic peak at −0.35 V is observed, which most likely corresponds to the reduction of nitrobenzene to aniline. Fig. 7b shows the voltammograms recorded one minute after the addition of nitrobenzene to the mixture, and the absence of reduction peak of nitrobenzene to aniline suggests that the reduction in homogeneous phase by the action of the DES components occurs very fast. The kinetic studies of this process will be presented in a future work.
With the results of the electrochemical characterization in our hands, we study the reduction of several aromatic nitrocompounds employing the eutectic mixture as solvent and reducing agent (Table 3). As seen in Table 3, all mono-nitroaromatic compounds are effectively reduced in very short reaction times. Electron-donating and electron-withdrawing groups does not influence the outcome of the reaction and the corresponding anilines 1a–6a, 10a–16a are obtained in excellent yields. The tolerance of ketone and ester functionalities is demonstrated in the reduction of p-nitroacetophenone 15 and methyl 4-nitrobenzoate 16 which yields p-aminoacetophenone 15a and methyl 4-aminobenzoate 16a in 93 and 90% yield respectively. In addition, dinitrobenzene isomers 7–9 also afforded the target diaminobenzenes (entries 7 to 9) in yields comparable to that of the mononitro analogues (entries 4 to 6).
|
|||||
|---|---|---|---|---|---|
| Entry | Compd | R (nitrocompd) | Compd | R (aniline) | Yieldb |
| a Reaction conditions: nitroaromatic compound (1 mmol), 1.6 g of SnCl2·2H2O/ChCl (2:1 molar ratio) DES, 80 °C, 1–10 min.b Isolated yield.c 3.2 g of SnCl2·2H2O/ChCl DES was used. |
|||||
| 1 | 1 | H | 1a | H | 96 |
| 2 | 2 | o-OH | 2a | o-OH | 94 |
| 3 | 3 | p-OH | 3a | p-OH | 93 |
| 4 | 4 | o-NH2 | 4a | o-NH2 | 96 |
| 5 | 5 | m-NH2 | 5a | m-NH2 | 95 |
| 6 | 6 | p-NH2 | 6a | p-NH2 | 96 |
| 7 | 7 | o-NO2c | 4a | o-NH2 | 94 |
| 8 | 8 | m-NO2c | 5a | m-NH2 | 93 |
| 9 | 9 | p-NO2c | 6a | p-NH2 | 94 |
| 10 | 10 | o-Me | 10a | o-Me | 97 |
| 11 | 11 | m-Me | 11a | m-Me | 96 |
| 12 | 12 | p-Me | 12a | p-Me | 94 |
| 13 | 13 | p-Cl | 13a | p-Cl | 95 |
| 14 | 14 | p-Br | 14a | p-Br | 98 |
| 15 | 15 | p-(CO)CH3 | 15a | p-(CO)CH3 | 93 |
| 16 | 16 | p-CO2CH3 | 16a | p-CO2CH3 | 90 |
Reactions performed at low temperatures (40 and 60 °C) also afford the desired products in longer reaction times (between 35 minutes to one hour) due to the increased viscosity of the DES and lower solubility of nitroarenes. The reaction conditions here established allow the reduction of a wide variety of nitroarenes employing a simple experimental setup with shorter reaction times in comparison to traditional methods reported with stannous chloride.63,64 In light of these interesting results, we decided to explore the synthesis of some N-arylacetamides through a one-pot reductive-acetylation of nitroarenes employing the DES. To perform this synthetic transformation starting from nitroaromatic compounds, several combination of reagents such as red phosphorus, iodine and carboxylic acids,65 platinum complexes/carboxylic acids,66 platinum nanoparticles,67 magnetic copper68 and nickel69 catalyst with acetic anhydride have been reported. In our model reaction, nitrobenzene reacted with acetic anhydride in presence of the DES affording acetanilide in 92% yield (Table 4 entry 1). Other nitroarenes also reacted smoothly yielding the corresponding N-arylacetamides in 70–94% (Table 4).
|
|||||
|---|---|---|---|---|---|
| Entry | Compd | R (nitrocompd) | compd | R (acetaniline) | Yieldb |
| a Reaction conditions: nitroaromatic compound (1 mmol), 1.6 g of SnCl2·2H2O/ChCl (2:1 molar ratio) DES, 80 °C, 1–10 min, then Ac2O (1.2 mmol), AcONa·3H2O (2 mmol), H2O (1 mL) 10 min.b Isolated yield.c 2.4 mmol of Ac2O, and 4 mmol AcONa·3H2O were used.d 3.2 g of SnCl2·2H2O/ChCl DES, 2.4 mmol of Ac2O, and 4 mmol AcONa·3H2O were used. |
|||||
| 1 | 1 | H | 1b | H | 92 |
| 2 | 2 | o-OHc | 2b | o-OAc | 70 |
| 3 | 3 | p-OHc | 3b | p-OAc | 89 |
| 4 | 8 | m-NO2d | 8b | m-NHAc | 88 |
| 5 | 9 | p-NO2d | 9b | p-NHAc | 91 |
| 6 | 10 | o-Me | 10b | o-Me | 87 |
| 7 | 11 | m-Me | 11b | m-Me | 89 |
| 8 | 12 | p-Me | 12b | p-Me | 93 |
| 9 | 13 | p-Cl | 13b | p-Cl | 94 |
| 10 | 14 | p-Br | 14b | p-Br | 92 |
As seen in Table 4, nitroaromatic compounds 10–14 bearing methyl and halogen substituents afforded the corresponding acetanilides 10b–14b in good yields. On the other hand, bis-acetylated compounds 2b, 3b, 8b and 9b were obtained with nitroarenes bearing amino and hydroxy groups (Table 4, entries 2–5). Any attempts to selectively achieve the acetylation on the amino over the hydroxy group in the reaction of compounds 2 and 3 were unsuccessful.
To further expand the scope of applications for the SnCl2·2H2O/ChCl DES, our synthetic efforts were next directed toward the obtention of indolo(pyrrolo)[1,2-a]quinoxalines. Different synthetic strategies for the construction of this heterocyclic skeleton are depicted in Scheme 1, however the development of a simple and efficient synthetic methodology involving readily available starting materials, few synthetic steps and high yields of products is highly desirable. Therefore, we envisaged that the reaction of aldehydes with the nitroarene 17 or 18, which are easily obtained in one step from o-nitroaniline70 and o-fluoronitrobenzene42 respectively, might be a suitable synthetic proposal to obtain the target quinoxalines (Scheme 2). To develop our proposal, we first synthesized the nitroarene 17 starting from o-nitroaniline and dimethoxytetrahydrofurane following the methods reported in literature70 (Scheme 2), and the reaction between compound 17 and benzaldehyde in SnCl2·2H2O/ChCl DES was taken as model reaction.
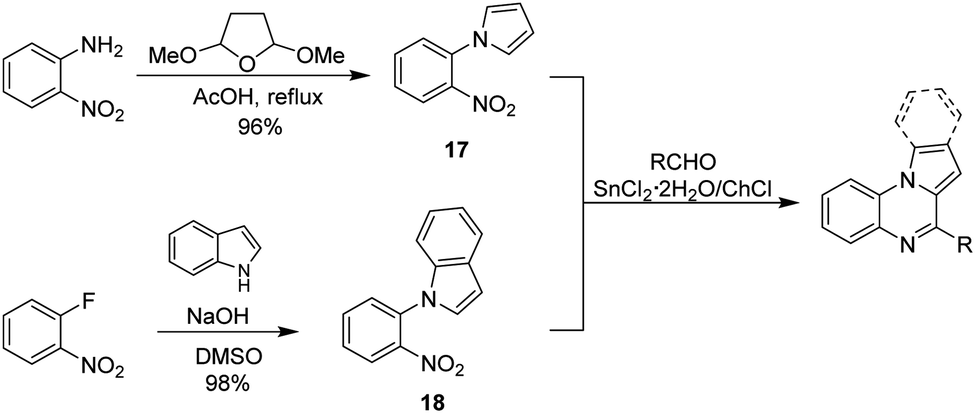 | ||
| Scheme 2 Synthetic proposal for the obtention of indolo(pyrrolo)[1,2-a]quinoxalines. | ||
Initially, equimolar amounts of 17 and benzaldehyde (1 mmol each) were poured into the DES (2.0 g, 1.53 g SnCl2·2H2O and 0.47 g ChCl) and the reaction was conducted at 80 and 110 °C obtaining the pyrrolo[1,2-a]quinoxaline 17a in 75 and 92% yield after 1 hour and 30 minutes respectively. Reactions performed with tin(II) chloride (1.53 g, 6.8 mmol) under solvent free conditions (110 °C, 30 min) and employing ethyl acetate as solvent (3 h, reflux) showed that the heterocyclic compound was not formed in the first case, and a 45% yield was obtained in the second experiment. In addition, a reduction on the amount of the DES (1.0 g of DES containing 763 mg, 3.4 mmol of tin(II) chloride) decreases the yield to 38%. These results show that under the optimal reaction conditions (2 g of DES, 110 °C, 30 minutes) the target compound can be obtained in a good yield, and to identify the benefits and limitations of our method, we began exploring the substrate scope by reacting different aliphatic and aromatic aldehydes. Results are summarized in Table 5.
|
||||
|---|---|---|---|---|
| Entry | Compd | R | Yieldb | Overall yieldc |
| a Reaction conditions: compound 17 or 18 (1 mmol), aldehyde (1 mmol), 2.0 g of SnCl2·2H2O/ChCl (2:1 molar ratio) DES, 110 °C, 30 min.b Isolated yield.c Calculated considering the yield for compounds 17 and 18. |
||||
| 1 | 17a | C6H5 | 92 | 88 |
| 2 | 17b | 4-CH3O-C6H4 | 85 | 82 |
| 3 | 17c | 4-(CH3)2CH-OC6H4 | 86 | 83 |
| 4 | 17d | 4-CH3-C6H4 | 89 | 85 |
| 5 | 17e | 3,4-(OCH2O)-OC6H3 | 84 | 81 |
| 6 | 17f | 2-Cl-C6H4 | 98 | 94 |
| 7 | 17g | 4-Cl-C6H4 | 75 | 72 |
| 8 | 17h | 4-Br-C6H4 | 87 | 84 |
| 9 | 17i | 2-OH-C6H4 | 89 | 85 |
| 10 | 17j | 2-Py | 88 | 84 |
| 11 | 17k | n-C9H19 | 83 | 80 |
| 12 | 18a | C6H5 | 94 | 92 |
| 13 | 18b | 4-CH3O–C6H4 | 94 | 92 |
| 14 | 18c | 4-(CH3)2CH-OC6H4 | 91 | 89 |
| 15 | 18d | 4-CH3-C6H4 | 94 | 92 |
| 16 | 18e | 3,4-(OCH2O)-OC6H3 | 92 | 90 |
| 17 | 18f | 2-Cl-C6H4 | 92 | 90 |
| 18 | 18g | 4-Cl-C6H4 | 89 | 87 |
| 19 | 18h | 4-Br-C6H4 | 95 | 93 |
| 20 | 18i | 2-Py | 89 | 87 |
| 21 | 18j | n-C9H19 | 90 | 88 |
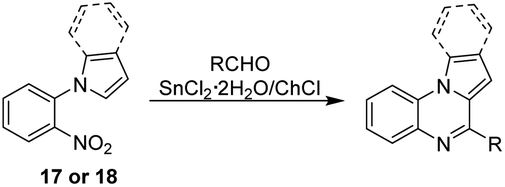
According to Table 5, all aromatic aldehydes bearing electron-donating and electron-withdrawing groups reacted easily with the nitroarene 17 affording the corresponding pyrrolo[1,2-a]quinoxalines 17a–17k in 75–98% yield. No clear tendency was observed regarding the influence of the substitution pattern or electronic character of the groups on the aromatic ring (Table 5, entries 1–9). Heteroaromatic and aliphatic aldehydes such as 2-pyridinecarboxaldehyde and n-decanal also provide the corresponding derivatives 17j and 17k in satisfactory yields (Table 5, entries 10 and 11).
Once the series of pyrrolo[1,2-a]quinoxalines were obtained, we focused on the synthesis of the indolo[1,2-a]quinoxalines, and for this, the nitroarene 18 was successfully prepared from o-fluoronitrobenzene and indole.42 Next, compound 18 reacted with different aldehydes affording the desired compounds 18a–18j in 89–95% yield (Table 5). As expected, similar results for the reactivity were observed in comparison to the pyrroloquinoxaline series: the reaction of aromatic and heteroaromatic aldehydes with 18 in the presence of the DES leads to the formation of compounds 18a–18i in good to excellent yields (Table 5, entries 12 to 20), and n-decanal yield the corresponding product 18j in 90% yield (Table 5, entry 21).
To evaluate the synthetic efficiency of the method here developed, the overall yield for each synthesized compound was calculated. As shown in Table 5, the first group of derivatives corresponding to pyrrolo[1,2-a]quinoxalines 17a–17j, was obtained in 72–94% overall yields, while for the second group of indolo[1,2-a]quinoxalines 18a–18j a 87–92% overall yield was achieved. These remarkable results are attributed to two factors: the few synthetic steps involved (two for both series of compounds starting from inexpensive and commercially available reagents) and the good to excellent yields obtained in each step.
To evaluate if the DES also plays a role as catalyst in the reaction, and to propose a plausible reaction mechanism two control experiments were performed (Scheme 3).
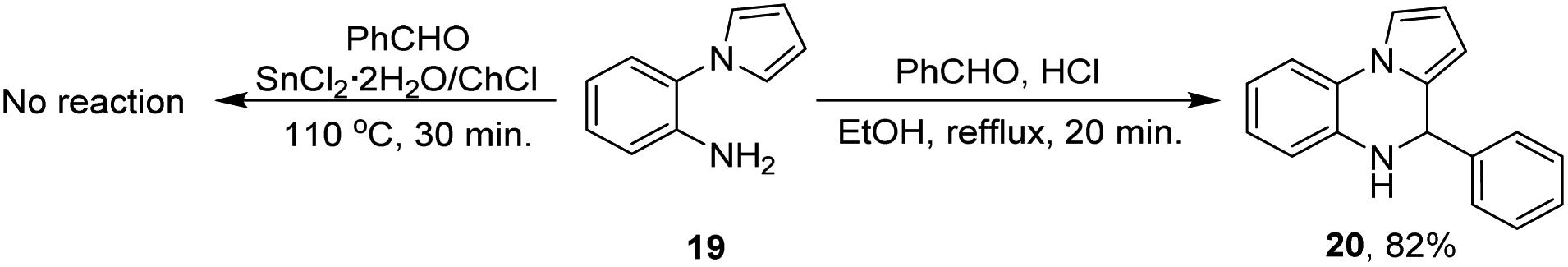 | ||
| Scheme 3 Control experiments performed to propose a plausible reaction mechanism. | ||
For the first experiment, the amine 19 and benzaldehyde were poured into the DES and the reaction was run under the optimal reaction conditions already established, however the compound 17a was not obtained. Taking into account that HCl might be formed during the reaction of compounds 17 and 18 with the DES, the second experiment was run with the amine 19, benzaldehyde and HCl (drops) in ethanol. After 30 minutes, the dihydroquinoxaline 20 was obtained in 82% yield.
Control experiments demonstrate that although the deep eutectic solvent promotes a fast reduction of the nitro group, it does not catalyze the cyclization process, and therefore the DES plays a dual role as solvent and reducing agent. On the other hand, it is hypothesized that the cyclization step might be promoted by hydrochloric acid generated in situ in the reaction medium in which the dihydroquinoxaline 20 undergo an aerobic oxidation.
With the experimental results previously obtained, a plausible reaction mechanism for the obtention of compound 17a is proposed in Scheme 4. As depicted in Scheme 4, the reaction begins with the reduction of the nitroarene 17 mediated by the SnCl2·2H2O present in the DES affording the corresponding amino derivative 19. It is known that the reduction of nitro groups might involve the formation of several intermediates, however the precise mechanism still remains unclear.71 Next, the reaction of the intermediate amine 19 with benzaldehyde (activated by HCl) leads to the formation of the imine 21 which follows an intramolecular cyclization promoted by the nucleophilic attack of the carbon C-1 of the pyrrole core over the azomethine carbon of 21 affording the dihydroquinoxaline 20 which after an aerobic oxidation yield the corresponding pyrrolo[1,2-a]quinoxaline 17a.
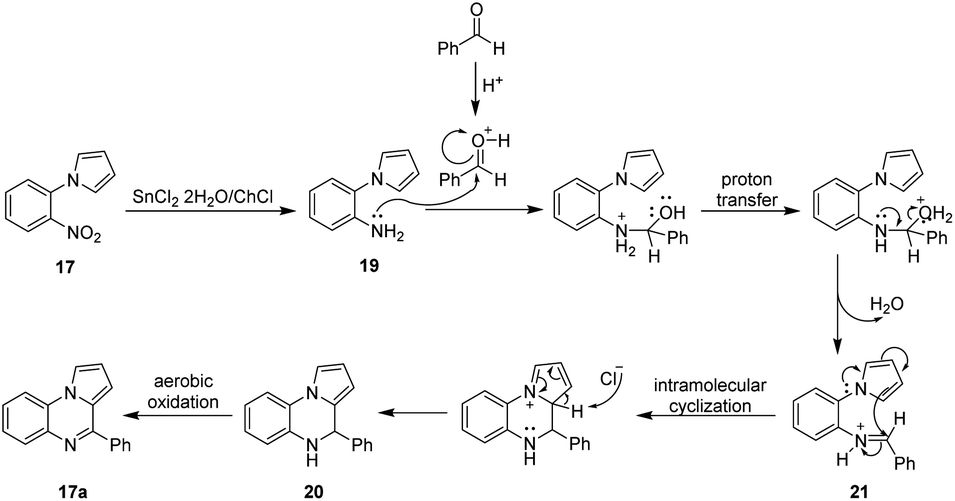 | ||
| Scheme 4 Plausible reaction mechanism for the obtention of compound 17a. | ||
Finally, gram scale experiments (10 mmol) for the obtention of compounds 1a, 1b and 17a were conducted in order to demonstrate the versatility of this synthetic method. As result of these experiments, the target compounds were obtained in 90, 87 and 85% yield respectively.
Experimental
Materials and methods
All the chemicals and solvents were purchased from commercial suppliers (Aldrich, Merck). Melting points, reported without correction, were measured using a Stuart SMP10 apparatus. The FT-IR spectra were obtained with a Shimadzu IR prestige 21 spectrophotometer (Columbia, MD, USA). 1H and 13C NMR spectra were recorded with a Bruker AVANCE III system operating at 400 MHz, using residual and deuterated solvent peaks of CDCl3 (δH 7.26; δC 77.0) and DMSO (δH 2.50; δC 39.5) as reference standards. Elemental analyses were performed on a Thermo Scientific Flash 2000 CHNS/O analyser and their results agreed with the calculated values. Raman spectra was acquired with Thermo Scientific DXR Raman Microscope, excitation was accomplished by using a single line of 780 nm wavelength from a frequency-stabilized single mode diode laser. The incident power was about 20 mW at the sample point. The density of the DES was measured using a liquid densitometer (Anton Paar DMA4500M). An Abbe type refractometer (model 2WAJ equipped with a sodium D1 line) was used to measure the DES refractive. The conductivity and its temperature dependence were determined using a Jenway 470 portable conductivity/TDS meter calibrated by measuring the conductivities of aqueous solutions of KCl at different concentrations. The variation of the temperature for the determination of the physical properties was done by using a Lauda Alpha water circulator. Cyclic voltammetry was carried out using a Gamry Interface 1000E potentiostat from Gamry Instruments. The electrochemical measurements were performed in a conventional three-electrode cell, with the 0.071 cm2 Glassy Carbon, the 0.0314 cm2 Au, the 0.0314 cm2 Pt, and 0.0314 cm2 Sn disks as the working electrodes, Ag/AgCl as the reference electrode, and Pt wire as the counter electrode. The working electrodes were polished with 1 and 0.3 μm γ-alumina paste, rinsed and sonicated for two minutes with deionized water, and dried prior to all measurements. The experiments were performed at 60 °C using a scan rate of 100 mV s−1.General experimental procedure for the synthesis of compounds 1a–6a, 10a–16a
To an open headspace vial equipped with a magnetic stir bar, the nitroarene (1.0 mmol) and 1.6 g of SnCl2·2H2O/ChCl DES were added, and the resulting solution was then stirred and heated at 80 °C. After running the reaction to an appropriate time (TLC analysis), the mixture was cooled to room temperature, neutralized with NaHCO3 and extracted with AcOEt (3 × 10 mL). After drying over anhydrous Na2SO4 and evaporating the solvent under reduced pressure, the desired product was obtained in high purity without further purification.General experimental procedure for the synthesis of N-arylacetamides 1b–3b, 8b–14b
To an open headspace vial equipped with a magnetic stir bar, the nitroarene (1.0 mmol) and 1.6 g of SnCl2·2H2O/ChCl (2:1 molar ratio) DES were added, and the resulting solution was then stirred and heated at 80 °C. After complete reduction of the nitroarene (TLC), Ac2O (1.2 mmol) and AcONa·3H2O (2.0 mmol) in 2 mL water were added and heating was continued until the reaction was complete (TLC analysis). Next, the reaction mixture was cooled to room temperature, neutralized with NaHCO3 and extracted with AcOEt (3 × 10 mL). After drying over anhydrous Na2SO4 and evaporating the solvent under reduced pressure, the crude material was purified by column chromatography on silica gel to afford the desired product.
General experimental procedure for the synthesis of indolo(pyrrolo)[1,2-a]quinoxalines 17a–k and 18a–j
To an open headspace vial equipped with a magnetic stir bar, the nitroarene 17 or 18 (1.0 mmol), the corresponding aldehyde (1 mmol) and 2.0 g of SnCl2·2H2O/ChCl (2:1 molar ratio) DES were added, and the resulting solution was then stirred and heated at 110 °C. After running the reaction to an appropriate time (TLC analysis), the mixture was allowed to cool to room temperature, neutralized with NaHCO3 and extracted with AcOEt (3 × 10 mL). After drying over anhydrous Na2SO4 and evaporating the solvent under reduced pressure, the crude material was purified by column chromatography on silica gel to afford the desired product.
Conclusions
In conclusion, we have studied the IR and Raman spectrum, and different physicochemical properties as function of temperature for the SnCl2·2H2O/ChCl DES. The FTIR and Raman spectrum suggest the formation of hydrogen bonds between ChCl and SnCl2·2H2O through Cl− and OH groups from ChCl and water molecules from SnCl2·2H2O. The refractive index and density show an inverse correlation with temperature while the conductivity increases with temperature. The electrochemical characterization the eutectic mixture presents a good potential window, so it could be used as a solvent for electrochemical cells in several types of applications. The eutectic mixture proved to be an effective reducing agent in which several nitroaromatic compounds afforded the corresponding anilines in short reaction times and high yields. In addition, this DES also promoted the reductive-acetylation of nitroarenes yielding acetanilides in 70–94%. Finally, a simple, fast and high yield method for the synthesis of indolo(pyrrolo)[1,2-a]quinoxalines via one-pot reductive cyclization–oxidation reaction was developed. According with this protocol, the target quinoxalines are obtained in moderate to excellent overall yields in two steps starting form readily available starting materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support from Universidad Nacional de Colombia. C. O.-P. sincerely thanks Prof. Humberto Zamora for providing some infrastructure facilities. Dedicated to Prof. Dr Burkhard König on the occasion of his 57th birthday.Notes and references
- K. Masuda, K. Suzuki, A. Ishida-Okawara, S. Mizuno, K. Hotta, S. Miyadoh, O. Hara and M. Koyama, J. Antibiot., 1991, 44, 533–540 CrossRef CAS.
- R. D. Charan, G. Schlingmann, V. S. Bernan, X. Feng and G. T. Carter, J. Nat. Prod., 2005, 68, 277–279 CrossRef CAS.
- J.-y. Ueda, J. Hashimoto, A. Nagai, T. Nakashima, H. Komaki, K. Anzai, S. Harayama, T. Doi, T. Takahashi, K. Nagasawa, T. Natsume, M. Takagi and K. Shin-ya, J. Antibiot., 2007, 60, 321–324 CrossRef CAS.
- K. Kurosawa, K. Takahashi and E. Tsuda, J. Antibiot., 2001, 45, 541–547 CrossRef.
- D. J. Cole, Pestic. Sci., 1999, 55, 756 CAS.
- J. Cong, X. Yang, J. Liu, J. Zhao, Y. Hao, Y. Wang and L. Sun, Chem. Commun., 2012, 48, 6663–6665 RSC.
- A. M. Faisca Phillips, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Front. Chem., 2020, 8, 30 CrossRef.
- X. Gu, Y. Dai, T. Guo, A. Franchino, D. J. Dixon and J. Ye, Org. Lett., 2015, 17, 1505–1508 CrossRef CAS.
- T. Marcelli, R. N. S. van der Haas, J. H. van Maarseveen and H. Hiemstra, Angew. Chem., Int. Ed., 2006, 45, 929–931 CrossRef CAS.
- M. Orlandi, D. Brenna, R. Harms, S. Jost and M. Benaglia, Org. Process Res. Dev., 2018, 22, 430–445 CrossRef CAS.
- S. Sobhani, F. O. Chahkamali and J. M. Sansano, RSC Adv., 2019, 9, 1362–1372 RSC.
- M. K. Kolli, P. Elamathi, G. Chandrasekar, V. R. Katta and G. Balvantsinh Raolji, Synth. Commun., 2018, 48, 638–649 CrossRef CAS.
- D. Yang, D. Fokas, J. Li, L. Yu and C. M. Baldino, Synthesis, 2005, 2005, 47–56 CrossRef.
- Z. Wu, P. Rea and G. Wickham, Tetrahedron Lett., 2000, 41, 9871–9874 CrossRef CAS.
- E. J. Hanan, B. K. Chan, A. A. Estrada, D. G. Shore and J. P. Lyssikatos, Synlett, 2010, 2010, 2759–2764 CrossRef.
- A. H. Romero, J. Salazar and S. E. López, Synthesis, 2013, 45, 2043–2050 CrossRef CAS.
- A. D. Roy, A. Subramanian and R. Roy, J. Org. Chem., 2006, 71, 382–385 CrossRef CAS.
- D. I. S. P. Resende, S. Guieu, C. G. Oliva and A. M. S. Silva, Synlett, 2015, 26, 846–850 CrossRef CAS.
- A. Cho, S. Byun and B. M. Kim, Adv. Synth. Catal., 2018, 360, 1253–1261 CrossRef CAS.
- A. Patti and S. Pedotti, Tetrahedron, 2010, 66, 5607–5611 CrossRef CAS.
- R. A. Bunce, B. Nammalwar and L. M. Slaughter, J. Heterocycl. Chem., 2009, 46, 854–860 CrossRef CAS.
- R. A. Bunce, D. M. Herron, L. B. Johnson and S. V. Kotturi, J. Org. Chem., 2001, 66, 2822–2827 CrossRef CAS.
- E. Merişor, J. Conrad, I. Klaiber, S. Mika and U. Beifuss, Angew. Chem., Int. Ed., 2007, 46, 3353–3355 CrossRef.
- M. d. F. Pereira and V. Thiéry, Org. Lett., 2012, 14, 4754–4757 CrossRef CAS.
- E. Merisor, J. Conrad, S. Mika and U. Beifuss, Synlett, 2007, 2007, 2033–2036 CrossRef.
- T. Kaushal, G. Srivastava, A. Sharma and A. Singh Negi, Bioorg. Med. Chem., 2019, 27, 16–35 CrossRef CAS.
- J. A. Pereira, A. M. Pessoa, M. N. D. S. Cordeiro, R. Fernandes, C. Prudêncio, J. P. Noronha and M. Vieira, Eur. J. Med. Chem., 2015, 97, 664–672 CrossRef CAS.
- K. R. Justin Thomas, M. Velusamy, J. T. Lin, C.-H. Chuen and Y.-T. Tao, Chem. Mater., 2005, 17, 1860–1866 CrossRef.
- A. P. Kulkarni, Y. Zhu and S. A. Jenekhe, Macromolecules, 2005, 38, 1553–1563 CrossRef CAS.
- A. Tsami, T. W. Bünnagel, T. Farrell, M. Scharber, S. A. Choulis, C. J. Brabec and U. Scherf, J. Mater. Chem., 2007, 17, 1353–1355 RSC.
- J.-Y. Jaung, Dyes Pigm., 2006, 71, 245–250 CrossRef CAS.
- M. J. Crossley and L. A. Johnston, Chem. Commun., 2002, 10, 1122–1123 RSC.
- P. P. Castro, G. Zhao, G. A. Masangkay, C. Hernandez and L. M. Gutierrez-Tunstad, Org. Lett., 2004, 6, 333–336 CrossRef CAS.
- J. L. Sessler, H. Maeda, T. Mizuno, V. M. Lynch and H. Furuta, Chem. Commun., 2002, 8, 862–863 RSC.
- S. K. Dey, M. Al Kobaisi and S. V. Bhosale, ChemistryOpen, 2018, 7, 934–952 CrossRef CAS.
- A. A. Kalinin and V. A. Mamedov, Chem. Heterocycl. Compd., 2011, 46, 1423 CrossRef CAS.
- P. K. Agarwal, D. Sawant, S. Sharma and B. Kundu, Eur. J. Org. Chem., 2009, 2009, 292–303 CrossRef.
- A. K. Verma, R. R. Jha, V. K. Sankar, T. Aggarwal, R. P. Singh and R. Chandra, Eur. J. Org. Chem., 2011, 2011, 6998–7010 CrossRef CAS.
- A. Preetam and M. Nath, RSC Adv., 2015, 5, 21843–21853 RSC.
- H.-r. Huo, X.-Y. Tang and Y.-f. Gong, Synthesis, 2018, 50, 2727–2740 CrossRef CAS.
- J. Li, J. Zhang, H. Yang, Z. Gao and G. Jiang, J. Org. Chem., 2017, 82, 765–769 CrossRef CAS.
- Z. Zhang, C. Xie, X. Tan, G. Song, L. Wen, H. Gao and C. Ma, Org. Chem. Front., 2015, 2, 942–946 RSC.
- C. Xie, L. Feng, W. Li, X. Ma, X. Ma, Y. Liu and C. Ma, Org. Biomol. Chem., 2016, 14, 8529–8535 RSC.
- H. Liu, F. Zhou, W. Luo, Y. Chen, C. Zhang and C. Ma, Org. Biomol. Chem., 2017, 15, 7157–7164 RSC.
- J. J. Lade, B. N. Patil, M. V. Vhatkar, K. S. Vadagaonkar and A. C. Chaskar, Asian J. Org. Chem., 2017, 6, 1579–1583 CrossRef CAS.
- C. Dai, S. Deng, Q. Zhu and X. Tang, RSC Adv., 2017, 7, 44132–44135 RSC.
- M. Ramamohan, R. Sridhar, K. Raghavendrarao, N. Paradesi, K. B. Chandrasekhar and S. Jayaprakash, Synlett, 2015, 26, 1096–1100 CrossRef CAS.
- H. Liu, T. Duan, Z. Zhang, C. Xie and C. Ma, Org. Lett., 2015, 17, 2932–2935 CrossRef CAS.
- Z. He, M. Bae, J. Wu and T. F. Jamison, Angew. Chem., Int. Ed., 2014, 53, 14451–14455 CrossRef CAS.
- N. Azizi and E. Batebi, Catal. Sci. Technol., 2012, 2, 2445–2448 RSC.
- D. Shahabi and H. Tavakol, J. Mol. Liq., 2016, 220, 324–328 CrossRef CAS.
- D. Shahabi and H. Tavakol, J. Iran. Chem. Soc., 2017, 14, 135–142 CrossRef CAS.
- Z. S. Gano, F. S. Mjalli, T. Al-Wahaibi and Y. Al-Wahaibi, Int. J. Chem. Eng. Appl., 2015, 6, 367–371 CAS.
- H. Mąka, T. Spychaj and J. Adamus, RSC Adv., 2015, 5, 82813–82821 RSC.
- N. Delgado-Mellado, M. Larriba, P. Navarro, V. Rigual, M. Ayuso, J. García and F. Rodríguez, J. Mol. Liq., 2018, 260, 37–43 CrossRef CAS.
- C. A. Ibarguen, A. Mosquera, R. Parra, M. S. Castro and J. E. Rodríguez-Páez, Mater. Chem. Phys., 2007, 101, 433–440 CrossRef CAS.
- C. F. Araujo, J. A. P. Coutinho, M. M. Nolasco, S. F. Parker, P. J. A. Ribeiro-Claro, S. Rudić, B. I. G. Soares and P. D. Vaz, Phys. Chem. Chem. Phys., 2017, 19, 17998–18009 RSC.
- C. Yuan, K. Chu, H. Li, L. Su, K. Yang, Y. Wang and X. Li, Chem. Phys. Lett., 2016, 661, 240–245 CrossRef CAS.
- M. Hiroshi, K. Hideko and K. Ryôiti, Chem. Lett., 1973, 2, 1061–1066 CrossRef.
- P. Pacák, Chem. Pap., 1991, 45, 227–232 Search PubMed.
- A. P. Abbott, G. Capper, D. L. Davies and R. Rasheed, Inorg. Chem., 2004, 43, 3447–3452 CrossRef CAS.
- A. Cojocaru, S. Costovici, L. Anicai and T. Visan, Metal. Int., 2008, 14, 38–46 Search PubMed.
- F. D. Bellamy and K. Ou, Tetrahedron Lett., 1984, 25, 839–842 CrossRef CAS.
- A. B. Gamble, J. Garner, C. P. Gordon, S. M. J. O'Conner and P. A. Keller, Synth. Commun., 2007, 37, 2777–2786 CrossRef CAS.
- X. Du, M. Zheng, S. Chen and Z. Xu, Synlett, 2006, 2006, 1953–1955 CrossRef.
- Y. Watanabe, Y. Tsuji, T. Kondo and R. Takeuchi, J. Org. Chem., 1984, 49, 4451–4455 CrossRef CAS.
- M. L. Kantam, R. S. Reddy, K. Srinivas, R. Chakravarti, B. Sreedhar, F. Figueras and C. Venkat Reddy, J. Mol. Catal. A: Chem., 2012, 355, 96–101 CrossRef CAS.
- Z. Shokri, B. Zeynizadeh and S. A. Hosseini, J. Colloid Interface Sci., 2017, 485, 99–105 CrossRef CAS.
- B. Zeynizadeh, Z. Shokri and M. Hasanpour Galehban, Appl. Organomet. Chem., 2019, 33, e4771 CrossRef.
- G. D. Ho, D. Tulshian, A. Bercovici, Z. Tan, J. Hanisak, S. Brumfield, J. Matasi, C. R. Heap, W. G. Earley, B. Courneya, R. Jason Herr, X. Zhou, T. Bridal, D. Rindgen, S. Sorota and S.-W. Yang, Bioorg. Med. Chem. Lett., 2014, 24, 4110–4113 CrossRef CAS.
- S. Yamabe and S. Yamazaki, J. Phys. Org. Chem., 2016, 29, 361–367 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization data for all compounds and selected 1H and 13C-NMR spectra. See DOI: 10.1039/d0ra06871c |
| This journal is © The Royal Society of Chemistry 2020 |