
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of bodinieric acids A and B, both C-18 and C-19-functionalized abietane diterpenoids: DFT study of the key aldol reaction†‡
Ramón J. Zaragozá *a and
Miguel A. González-Cardenete*b
*a and
Miguel A. González-Cardenete*b
aDepartamento de Química Orgánica, Universidad de Valencia, Dr. Moliner 50, 46100 Burjassot, Valencia, Spain
bInstituto de Tecnología Química (UPV-CSIC), Universitat Politècnica de Valencia-Consejo Superior de Investigaciones Científicas, Avda de los Naranjos s/n, 46022 Valencia, Spain. E-mail: migoncar@itq.upv.es
First published on 16th April 2020
Abstract
The first synthesis of C-18- and C-19-bifunctionalized abietane diterpenoids, bodinieric (or callicapoic) acids, via an aldol reaction has been developed. This key aldol reaction was very sensitive to steric hindrance. This fact has been studied by deuterium exchange experiments and DFT methods. Optimization of this reaction led to the synthesis of anti-inflammatory bodinieric acids A and B, starting from abietic acid.
Introduction
Diterpenoids of the abietane class have been known for almost 200 years since the discovery of abietic acid (1, Fig. 1) but new members are still being discovered and useful pharmacological applications are under study.1 From the organic synthesis point of view, the preparation of abietanes has long been a matter of interesting research.2 The abietane carbon framework characterized by a tricyclic ring system (Fig. 1) with several stereocenters and a wide grade of oxygenation pattern on the skeleton invites synthetic chemists to be creative and efficient. The recent report, in 2018, on isolation of related abietane congeners of dehydroabietic acid (2, DHA) with both C-18- and C-19-oxygenated carbons, such as bodinieric acid A (3, deacetylcallicapoic acid M5), and B (4, also known as callicapoic acid M4) attracted our attention.3 Their important biological activities as a selective spleen tyrosine kinase (SYK) inhibitors have led to a patent.4 These natural products reminded us an old synthetic work on another diterpenoid, possessing exactly the same stereochemistry at C4 and rare constitution, that is, both C18 and C19 oxidized and with the same oxidation pattern at this carbons in ring A, (−)-scopadulcic acid A (5, Fig. 1), as we had worked in the field 20 years ago.5 We became interested in developing a scalable synthetic route towards those acids for further study, including the control of the stereoselectivity at the quaternary stereocenter at C-4. Herein, we describe the first synthesis of bodinieric acids A (3) and B (4) through a key aldol reaction of which we give further insight based on deuterium exchange experiments and DFT computational studies. | ||
| Fig. 1 Abietane numbering system and some examples. | ||
Results and discussion
The original retrosynthetic analysis is outlined in Scheme 1. In a similar approach to the work of Ziegler and Wallace to complete the synthesis of (±)-scopadulcic acid A in 1995,6 we envisaged that bodinieric acid B (4) could be readily obtained from the aldol 6 (Scheme 1) by Pinnick oxidation. Conversion of the isopropyl moiety into a methyl ketone moiety would then afford bodinieric acid A (3). Aldol 6 would be prepared by treatment with formalin and base of a mixture of known aldehydes 7a,b used in the synthesis of callitrisic acid (4-epidehydroabietic acid) from dehydroabietic acid (2) by Pelletier and Herald.7 The necessary aldehydes for the key aldol reaction could be obtained after opening of the epoxide 8, which in turn is synthesized from a mixture of olefins 9 coming from an oxidative decarboxylation of DHA (2), obtainable from (−)-abietic acid (1). | ||
| Scheme 1 Retrosynthetic Analysis. | ||
With this straightforward synthetic plan in mind, we prepared DHA (2) from commercial (−)-abietic acid (1) following our method by methylation, aromatization with Pd/C catalyst and hydrolysis (Scheme 2).1c,8 We thought that the preparation of the exo-olefin 4(18), though as a mixture of regioisomers, would be easy and fast by treatment of DHA (2) with lead tetraacetate. But like other researchers found, the reaction was capricious and low yielding in the desired exo-olefin.7,9 We sought an alternative and decided to use the methodology of Barrero and co-workers based on the elimination of a formate moiety (Scheme 2).10 Thus, methyl dehydroabietate1c (10) obtained from (−)-abietic acid (1) was converted into dehydroabietinal (11) in almost quantitative yield, which was oxidized with m-CPBA in the presence of disodium phosphate to furnish formate 12 in 73% yield. Heating in collidine at reflux of 12 led to a mixture of olefin regioisomers 9, ca. 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 (4,18-; 3,4-; 4,5-), in 87% yield where the major component contained the exocyclic 4,18-double bond. Continuing with our synthetic plan, epoxidation of that mixture of olefins 9 (ca. 69% purity in exo-olefin) with an excess of m-CPBA afforded epoxide 8, which was treated with BF3 etherate in toluene to give a 1:1 mixture of epimeric aldehydes 7a and 7b (88%, two steps). The introduction of the remaining carbon at C-4 and manipulation of the functionality at this site were the remaining steps to be accomplished. To our surprise, our first attempt of the planned aldol reaction6 (aq. HCHO/Na2CO3) with aldehydes 7a,b (ca. 1:1 mixture) did not work as expected since only β-aldehyde 7a reacted, recovering unaltered α-aldehyde 7b. At this point, we hypothesized that using a stronger base such as NaOH might help but little progress in the aldol reaction was observed when using recovered α-aldehyde 7b and NaOH as a base, even with slow addition of aq. HCHO.
:1:1 (4,18-; 3,4-; 4,5-), in 87% yield where the major component contained the exocyclic 4,18-double bond. Continuing with our synthetic plan, epoxidation of that mixture of olefins 9 (ca. 69% purity in exo-olefin) with an excess of m-CPBA afforded epoxide 8, which was treated with BF3 etherate in toluene to give a 1:1 mixture of epimeric aldehydes 7a and 7b (88%, two steps). The introduction of the remaining carbon at C-4 and manipulation of the functionality at this site were the remaining steps to be accomplished. To our surprise, our first attempt of the planned aldol reaction6 (aq. HCHO/Na2CO3) with aldehydes 7a,b (ca. 1:1 mixture) did not work as expected since only β-aldehyde 7a reacted, recovering unaltered α-aldehyde 7b. At this point, we hypothesized that using a stronger base such as NaOH might help but little progress in the aldol reaction was observed when using recovered α-aldehyde 7b and NaOH as a base, even with slow addition of aq. HCHO.
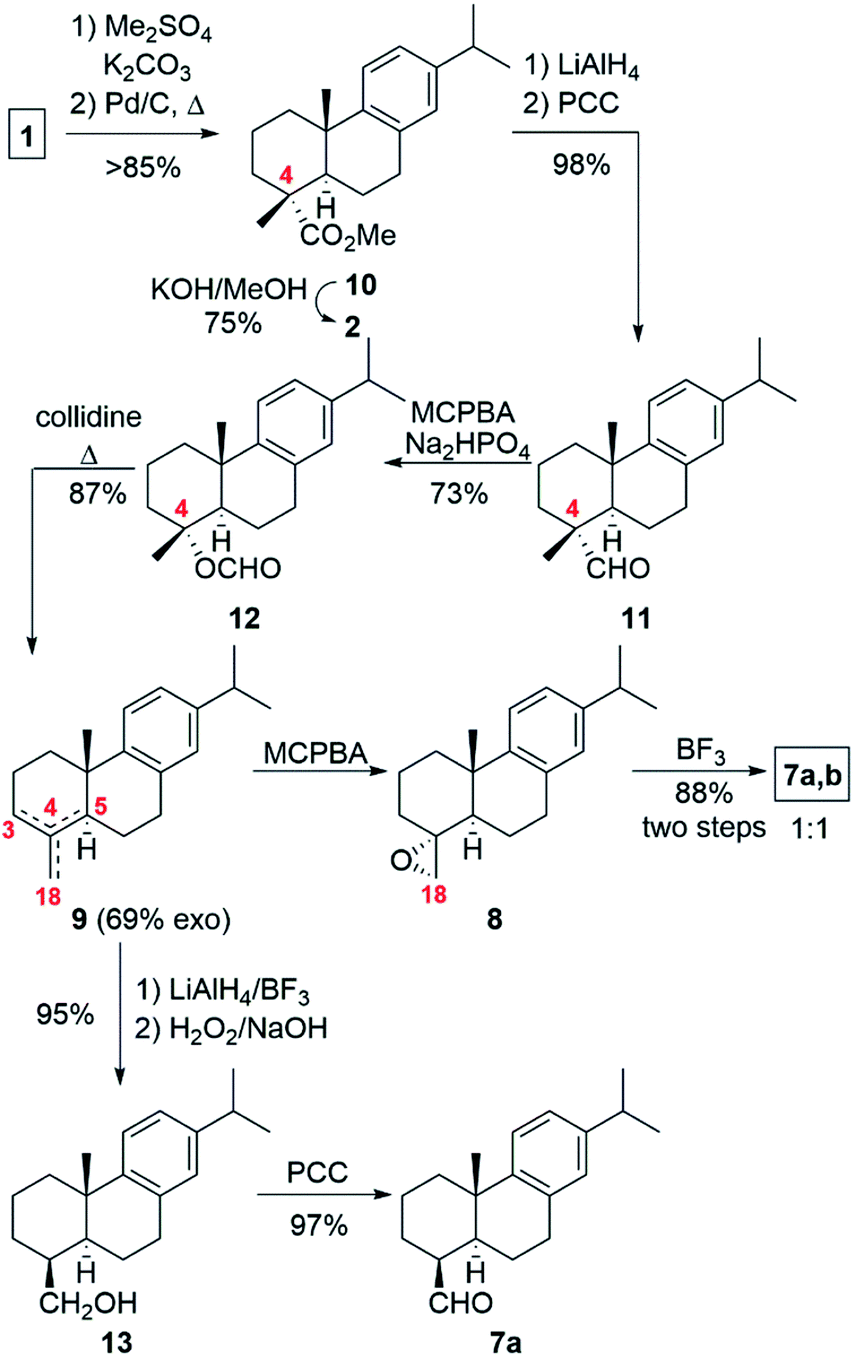 | ||
| Scheme 2 Synthesis of the key intermediate 7a. | ||
Initially, it was attributed this high stereoselectivity to the presence of the axial C-20 methyl moiety, which probably hinders the approach of the base to the most hindered β-face.
This fact was investigated by computational methods and deuterium exchange experiments (see below) in order to determine the causes of the different behavior in the enolization step and hydroxymethylation of both aldehyde epimers at C-4, 7a and 7b.
In view of those results and other failed attempts to perform the aldol reaction with the epimer 7b, including organocatalysis, we modified slightly the planned synthetic sequence. Thus, the reactive aldehyde 7a (CHO-18β) was synthesized stereoselectively. To this end, 18-norabieta-8,11,13-trien-19-ol (13) was prepared by hydroboration/oxidation generating in situ BH3, treating BF3 etherate with LiAlH4, followed by addition of H2O2 in basic media.11 Oxidation of 13 with pyridinium chlorochromate (PCC) afforded the intermediate aldehyde 7a in high yield (92%, two steps).
The completion of the syntheses of both bodinieric acids A and B is shown in Scheme 3. At this stage, the key aldol reaction of aldehyde 7a with aq. HCHO/Na2CO3 rapidly gave the desired α stereoselectivity as a single isomer, hydroxy-aldehyde 6 in high yield (70%). Selective oxidation of the aldehyde group of 6 with NaClO2 provided bodinieric acid B (4, callicapoic acid M4) in 82% yield, whose 1H and 13C NMR spectra (in CDCl3 and acetone-d6) and HRMS (calcd for C20H29O3 [M + H]+: 317.2117; found: 317.2111) were in complete agreement with the reported data for the natural product 4.3,12 Interestingly, Zhang and co-workers and Wang and co-workers defined wrongly this natural product as 18-hydroxydehydroabietic acid instead of 18-hydroxycallitrisic acid which contains the C-19 carboxylic acid, and 18-hydroxy-8,11,13-abietatetraen-19-oic acid instead of 18-hydroxy-8,11,13-abietatrien-19-oic acid, respectively.3 Our next synthetic target was bodinieric acid A (Scheme 3). Starting from bodinieric acid B (4), selective dehydrogenation at C-15 with dichloro dicyano quinone (DDQ) provided the isopropenyl moiety in 14 (bodinieric acid C, as a ca. 3:2 mixture with unreacted starting material). Subsequent oxidative cleavage of 14 with catalytic OsO4 and oxone as co-oxidant13 furnished bodinieric acid A (3) in moderate yield (53% brsm, two steps) which was not further optimized (10.2% overall yield from abietic acid, 13 steps, see Scheme S1‡). The data for synthetic 3 were in excellent agreement with the isolation data (1H NMR, 13C NMR, [α]D).3a A conformational study of 3 was also carried out to estimate the 13C data with the GIAO (gauge including atomic orbital) method, implemented in the Gaussian package (see Fig. S1 and S2 and Tables, S1 and S2‡).14 Sixteen different conformations were obtained in acetone (solvent used in NMR experiments), the most stable being that indicated in Fig. 2. This conformation contains a hydrogen bond between the carbonyl of the acid group and the hydroxyl proton. A good correlation is observed between the experimental and theoretical 13C NMR data. In particular, our experimental values for 3 gave a mean deviation of 1.82 (Table S2‡). Only some sensitive carbons to the rotation of the carboxylic group such as C5 give a high deviation.
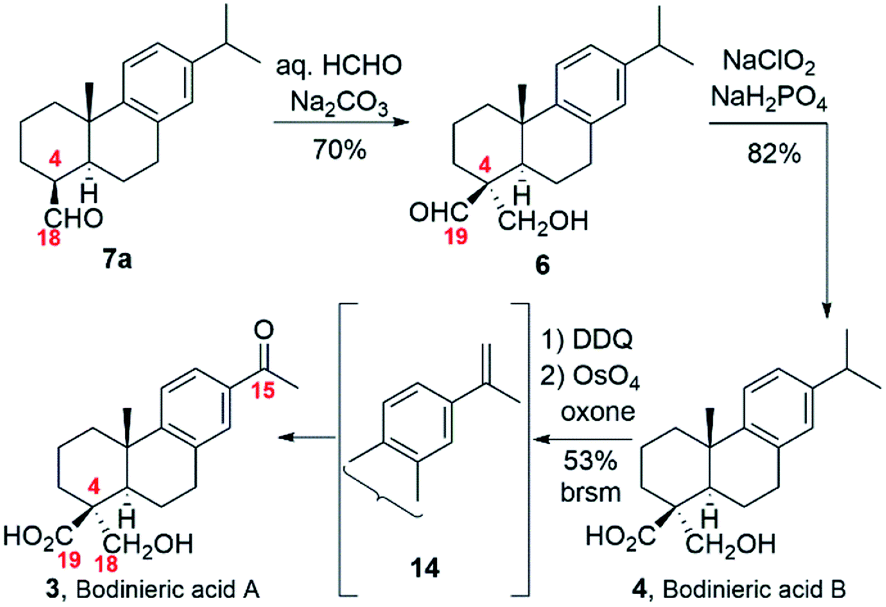 | ||
| Scheme 3 Synthesis of bodinieric acids A and B. | ||
 | ||
| Fig. 2 Geometry at B3LYP/6-31G** level of the most stable conformation of the bodinieric acid A (3), in acetone. | ||
Deuterium exchange experiments and computational study
To further get insight on the mechanism of the enolization, an experiment of deuterium exchange of hydrogens H-4 was designed and executed successfully. Thus, a ca. 1:1 mixture of aldehydes 7a/7b was subjected to equivalent conditions of the aldol reaction, without adding formaldehyde, with the corresponding solvents capable of releasing protons as deuterated versions (MeOD and D2O). The results (see ESI, Fig. S9–S12‡) indicate that after 60 minutes of reaction the proton H-4 of isomer 7a (β-aldehyde) was exchanged by deuterium, while that of isomer 7b (α-aldehyde) apparently remained unaltered. It is believed that with longer reaction times the isomer 7b should also react but in the real system with formaldehyde this reaction will be disfavored since formaldehyde will decompose in basic media.
The enolate formation of both aldehyde epimers 7 using as base HO− and CO32− was studied by DFT methods. In Scheme 4, there is a representation of the mechanism of enolate generation, starting from 7a and 7b. Initially, both epimers form a molecular complex (MC1) with the base, which evolves through the corresponding transition state (TS) to another molecular complex (MC2). MC2 is composed by the enolate and the conjugated acid of the used base. We have considered that ion HO− is solvated by three H2O molecules surrounding the oxygen with negative charge (see Fig. S7‡).15 In a similar way, the anion CO32− is also surrounded by three H2O molecules (see Fig. S8‡). All calculations have been made with those explicit H2O molecules. The energy results are presented in Table S4‡ (see also Fig. S3–S6‡) and the geometries of all the involved species are in Fig. S7 and S8.‡
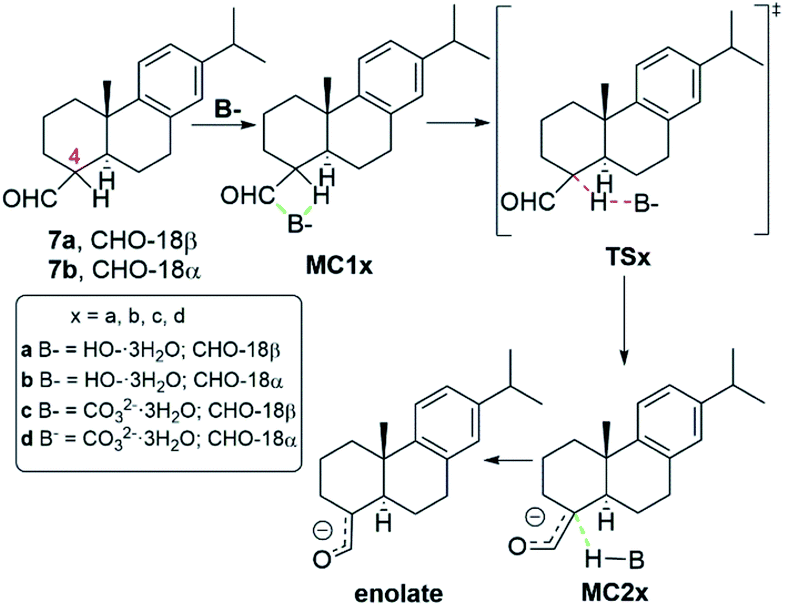 | ||
| Scheme 4 Mechanism of enolate formation starting from the alpha and beta aldehyde epimers 7. | ||
As it can be seen in Table S4‡ and in Fig. 3, the enolate formation of the epimer 7a (CHO-18β) with HO− occurs through the TSa with a Gibbs free energy difference ΔG = 6.2 kcal mol−1 from MC1a. The enolate generation of the epimer 7b (CHO-18α) is clearly disfavored from the kinetic point of view (TSb, ΔG = 12.5 kcal mol−1 from MC1b). Nevertheless, both enolate formations are very endergonic (MC2a, ΔG = 3.5 kcal mol−1 and MC2b, ΔG = 7.5 kcal mol−1). As a result, a very low concentration of complex MC2 is obtained and therefore, low concentration of enolate.
 | ||
| Fig. 3 Free energy profile (ΔG in kcal mol−1) at B3LYP/6-31G** level of species involved in the enolate formation of 7a and 7b with HO− (left) and CO32− (right), in water. | ||
It must be considered that formaldehyde under the conditions of reaction is an aqueous solution stabilized by methanol. Those conditions provide a low concentration of formaldehyde, since there are several equilibria on reacting formaldehyde with water to give methylene glycol (HOCH2OH) and poly(oxymethylene)glycols and with methanol to form hemiformal (CH3OCH2OH) and poly(oxymethylene)hemiformals.16 Thus, the low concentration of enolate and free formaldehyde makes the alkylation a slow reaction. This fact could be also accompanied by a side reaction, the Cannizzaro reaction,17 that normally occurs under basic conditions. As a final result formaldehyde decomposes without reacting with the enolate.
On the other side, the use of CO32− as base changes the scenario notably. As it is shown in Table S4,‡ Fig. 3 and 4, the reaction of 7b (CHO-18α) with CO32− leads to MC2d through TSd. The Gibbs free energy difference is 8.1 kcal mol−1 but the process is still very endergonic (MC2d, ΔG = 3.7 kcal mol−1) and therefore, disfavored. However, the enolate formation of 7a (CHO-18β) is fast (TSc, ΔG = 6.9 kcal mol−1), leading to MC2c with only 0.1 kcal mol−1 above the energy of MC1c. This allows a high concentration of the complex MC2c and therefore a high concentration of the corresponding enolate. In the soft basic media of CO32− vs. OH−, the Cannizzaro reaction would be less important and, therefore, less decomposition of formaldehyde will occur. These two circumstances explain the experimental finding that only the epimer 7a reacts in a carbonate media (CO32−). As it can be seen in Fig. 4, both the transition state and the molecular complex resulting from the enolate formation of aldehyde 7b with CO32− (TSd and MC2d, respectively), have a high steric hindrance with H-2β, H-6β and methyl 20. This is the main cause of the increase in energy of these species. This circumstance is even worse in the real situation since in the simulation only three water molecules have been considered and, in fact, there are more water molecules solvating the base.
 | ||
| Fig. 4 Geometries at B3LYP/6-31G** level of TSc, TSd, MC2c and MC2d involved in the enolate formation of 7a and 7b with CO32−, in water. | ||
The use of a more real model would have several consequences: (a) on considering a higher number of explicit water molecules would imply the stabilization of the reactant base and the MC1 complex due to H-bonding as compared to the transition state, which raises the barrier height and hinders reaction.18
(b) It is known that steric hindrance, i.e. SN2 reactions, increases the energy of the transition state.19 In our case, this effect would be similar especially for the steric characteristics of TSd, which would hinder the removal of the proton by the base.
(c) If we consider the effect of the solvated cation the steric effects would be even higher.20
As an overall result, it can be deduced that in a more real computational model the reactants will be stabilized in comparison with the TS. Therefore, including more explicit water molecules would probably lead to slower reactions,18a which is in agreement with the deuterium exchange experiment.
Conclusions
In summary, the first stereoselective synthesis of both C-18 and C-19 oxygenated abietane diterpenoids, in particular, bodinieric acids A and B, has been completed using an aldol reaction as key step enabling the synthesis of additional congeners of this new group of natural products. Those metabolites were isolated very recently and had not been previously prepared synthetically. Our synthesized materials established and confirmed the originally proposed absolute stereochemistry of each natural product since the structure of the starting material (−)-abietic acid is already known by single-crystal X-ray determination.21 The key aldol reaction was very sensitive to steric hindrance. This fact has been studied by deuterium exchange experiments and DFT methods. Further application of the intermediates obtained and biological evaluation of these synthetic natural products and related analogues are underway and will be reported in due course.Experimental section
General experimental procedures
The melting points were measured with a Büchi M-560 apparatus. Optical rotations were measured using a 10 cm cell in a Jasco P-2000 polarimeter in DCM unless otherwise stated. NMR spectra were recorded on a 300 MHz spectrometer (1H: 300 MHz, 13C: 75 MHz) and referenced to the solvent peak at 7.26 ppm (1H) and 77.00 ppm (13C) for CDCl3 and 2.05 ppm (1H) and 29.84 ppm (13C) for acetone-d6. All spectra were recorded in CDCl3 as solvent unless otherwise stated. Complete assignments of 13C NMR multiplicities were made on the basis of DEPT experiments and further assignment of signals with the help of COSY and HSQC experiments. J values are given in Hz. MS data were acquired on a QTOF spectrometer. Reactions were monitored by TLC using Merck silica gel 60 F254 (0.25 mm-thick) plates. Compounds on TLC plates were detected under UV light at 254 nm and visualized by immersion in a 10% sulfuric acid solution and heating with a heat gun. Purifications were performed by flash chromatography on Merck silica gel (230–400 mesh). Commercial reagent grade solvents and chemicals were used as purchased unless otherwise noted. Combined organic extracts were washed with brine, dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The starting material, methyl dehydroabietate (10), was obtained following our reported protocol8 with the following modifications: abietic acid (1, >70%, TCI) was methylated under standard conditions with dimethyl sulfate (1.2 equiv.) and K2CO3 (1.3 equiv.) in acetone (0.33 M in abietic acid, 30 g scale). The resulting methyl abietate was aromatized with 10% Pd/C catalyst (1% of ester mass), heating at 240 °C for 3 h. Next, compound 10 was reduced with LiAlH4 (5 equiv.) in THF (0.23 M in methyl dehydroabietate, 8 g scale) and subsequently the crude product was oxidized with PCC (1.5 equiv.) in DCM (0.24 M in dehydroabietinol, 5 g scale) to give dehydroabietinal 11 (quantitative) which was used in the next step without further purification. The carbon numbering of all synthetic compounds corresponds to that of natural products.Computational methods
All calculations were carried out with the Gaussian 09 suite of programs.14 Initially, density functional theory22 calculations (DFT) have carried out using the B3LYP23 exchange-correlation functionals, together with the standard 6-31G** basis set, in gas phase.24 Subsequently, the inclusion of solvent effects have been considered by using a relatively simple self-consistent reaction field (SCRF) method25 based on the polarizable continuum model (PCM).26 Geometries have been fully optimized with PCM. As solvents we have used H2O. In the calculations, three water molecules have been explicitly included. The values of enthalpies, entropies and free energies in H2O were calculated with the standard statistical thermodynamics at 298.15 K.24 The stationary points were characterized by frequency computations in order to verify that TSs have one and only one imaginary frequency. The intrinsic reaction coordinate (IRC) paths27 were traced in order to check the energy profiles connecting each TS to the two associated minima of the proposed mechanisms using the second order González–Schlegel integration method.28:1) to give 3.7 g (73%) of formate 12 as a yellowish oil: [α]25D +9.5 (c 1.0, DCM) (lit.,29 +27.9 (c 1.0, CHCl3); 1H NMR (300 MHz) δ 8.08 (1H, s, OCHO), 7.17 (1H, d, J = 8.1), 7.01 (1H, dd, J = 8.1, 2.1), 6.91 (1H, s), 2.93 (2H, m), 2.84 (1H, m), 2.64 (1H, m), 2.25 (1H, m), 1.59 (3H, s), 1.23 (3H, d, J = 6.9), 1.23 (3H, d, J = 6.9)), 1.20 (3H, s), 0.89 (2H, m); 13C NMR (75 MHz) δC 160.5 (d), 145.9 (s), 134.6 (s), 127.0 (d), 124.5 (d), 124.1 (d), 87.1 (s), 49.7 (d), 38.4 (s), 38.0 (t), 37.5 (t), 33.5 (d), 30.1 (t), 24.8 (q), 24.0 (q), 24.0 (q), 20.0 (t), 19.9 (q), 18.4 (t); HRMS (ESI) m/z 323.1975 [M + Na]+, calcd for C20H28O2Na: 323.1987.:1:1 (4,18-; 3,4-; 4,5-). The 1H and 13C NMR data for the major exocyclic isomer are:9a 1H NMR (300 MHz) δ 7.22 (1H, d, J = 8.1), 7.01 (1H, dd, J = 8.1, 2.2), 6.93 (1H, s), 4.85 (1H, d, J = 1.8), 4.60 (1H, d, J = 1.8), 2.90–2.77 (3H, m), 1.24 (6H, d, J = 7.2), 1.00 (3H, s); 13C NMR (75 MHz) δC 150.7 (s), 145.7 (s), 144.6 (s), 134.8 (s), 127.1 (d), 125.3 (d), 123.9 (d), 106.3 (t), 47.9 (d), 39.2 (s), 38.4 (t), 36.4 (t), 33.5 (d), 30.0 (t), 24.0 (q), 24.0 (q), 23.8 (t), 22.8 (q), 21.4 (t).:1) to give 245 mg (88% based on the Δ4(18)-isomer in the alkenes 9 mixture) of epimeric aldehydes 7a,b (NMR data vide infra) as a colorless oil which were used in the next step directly (aldol reaction).:4) to give 2.0 g (>95%) of 13 as a colorless oil: [α]25D +110.9 (c 1.0, DCM). The 1H NMR data were in agreement with those reported in the literature:30 1H NMR (300 MHz) δ 7.18 (1H, d, J = 8.1), 7.01 (1H, dd, J = 8.1, 2.1), 6.90 (1H, d, J = 2.1), 3.75 (2H, m), 2.93–2.84 (3H, m), 2.27 (1H, br d, J = 11.5), 1.24 (6H, d, J = 6.9), 1.06 (3H, s); 13C NMR (75 MHz) δC 146.1 (s), 145.6 (s), 134.7 (s), 127.0 (d), 124.5 (d), 123.9 (d), 61.7 (t), 44.5 (d), 44.0 (d), 38.6 (t), 36.9 (s), 33.4 (d), 30.6 (t), 27.6 (t), 25.3 (t), 24.3 (q), 24.0 (q), 24.0 (q), 18.2 (t); HRMS (ESI) m/z 336.2343 [M + CH3CN + Na]+, calcd for C21H31NONa: 336.2303.:1 MeOH–DCM mixture (120 mL) and treated with excess 37% aqueous HCHO (25 mL, 0.336 mol, 48 equiv.) and Na2CO3 (400 mg, 3.8 mmol, 0.54 equiv.) under an Ar atmosphere. After being stirred for 5 h, the mixture was diluted with DCM (120 mL) and washed with a mixture of 30 mL of water and 30 mL of brine, dried and concentrated. The crude oily residue was chromatographed on silica eluting with n-hexane–EtOAc (7:3) to give 1.5 g (70%) of aldol 6 as a colorless solid which solidified (white solid) upon standing at 5 °C: mp 113–115 °C; [α]24D +76.3 (c 1.0, DCM). 1H NMR (300 MHz) δ 9.97 (1H, d, J = 1.2), 7.18 (1H, d, J = 8.4), 7.02 (1H, dd, J = 8.4, 2.1), 6.91 (1H, d, J = 2.1), 3.96 (1H, d, J = 10.8), 3.56 (1H, d, J = 10.8), 3.00–2.90 (2H, m), 2.83 (1H, sept., J = 6.9), 2.34 (2H, m), 2.40–1.60 (6H, m), 1.42 (1H, ddd, J = 12.6, 12.1, 5.1), 1.23 (6H, d, J = 6.9), 1.09 (3H, s); 13C NMR (75 MHz) δC 205.9 (d), 146.1 (s), 144.8 (s), 134.2 (s), 126.9 (d), 124.8 (d), 124.1 (d), 67.2 (t), 54.0 (s), 47.2 (d), 38.0 (t), 37.5 (s), 33.5 (d), 30.7 (t), 29.0 (t), 24.7 (q), 23.9 (q), 23.9 (q), 19.1 (t), 18.6 (t); HRMS (ESI) m/z 301.2170 [M + Na]+, calcd for C20H29O2: 301.2168.When this procedure was first carried out with the ca. 1:1 mixture of aldehydes 7a,b, as starting material, we recovered unreacted aldehyde 7b (19-norabieta-8,11,13-trien-18-al, Rf = 0.50 in n-hexane–EtOAc (7:3)), which eluted prior to the aldol product 6, as a semisolid: 1H NMR (300 MHz) δ 9.54 (1H, d, J = 4.5), 7.21 (1H, d, J = 8.1), 7.01 (1H, br d, J = 8.4), 6.91 (1H, d, J = 2.1), 2.90–2.75 (3H, m), 1.23 (6H, d, J = 6.9), 1.12 (3H, s); 13C NMR (75 MHz) δC 205.2 (d), 146.1 (s), 144.4 (s), 134.6 (s), 127.1 (d), 124.4 (d), 123.9 (d), 51.2 (d), 41.6 (d), 37.1 (t), 36.0 (s), 33.5 (d), 29.0 (t), 26.4 (t), 24.0 (q), 24.0 (q), 23.0 (t), 22.7 (q), 20.6 (t).
:1) to give 432 mg (82%) of acid 4 as an amorphous white solid: [α]22D +104.9 (c 0.10, MeOH) (lit.,3a +57.3 (c 0.10, MeOH)) (lit.,3b +22.2 (c 0.10, MeOH)). The 1H and 13C NMR data were in agreement with those of the natural product in both acetone-d6 (ref. 3a) and CDCl3 (ref. 3b): 1H NMR (300 MHz, CD3COCD3) δ 7.21 (1H, d, J = 8.1), 6.97 (1H, dd, J = 8.1, 2.1), 6.86 (1H, d, J = 2.1), 3.87 (1H, d, J = 10.2), 3.66 (1H, d, J = 10.2), 2.90–2.70 (3H, m), 2.31 (2H, m), 1.78 (1H, m), 1.65 (1H, m), 1.19 (6H, d, J = 6.9), 1.15 (3H, s); 13C NMR (75 MHz, CD3COCD3) δC 177.4 (s), 146.7 (s), 146.3 (s), 135.7 (s), 127.5 (d), 126.2 (d), 124.6 (d), 70.2 (t), 50.6 (s), 47.5 (d), 40.1 (t), 38.8 (s), 34.3 (d), 32.5 (t), 32.3 (t), 24.4 (q), 24.3 (q), 24.0 (q), 21.7 (t), 20.4 (t); 1H NMR (300 MHz, CDCl3) δ 7.18 (1H, d, J = 8.1), 6.99 (1H, dd, J = 8.4, 2.1), 6.89 (1H, br s), 4.17 (1H, d, J = 10.2), 3.50 (1H, d, J = 10.8), 2.90–2.70 (3H, m), 2.45 (1H, br d, J = 12.9), 2.29 (1H, br d, J = 12.9), 2.10–2.00 (3H, m), 1.72 (2H, m), 1.22 (6H, d, J = 6.9), 1.15 (3H, s); 13C NMR (75 MHz, CDCl3) δC 181.3 (s), 145.8 (s), 145.3 (s), 134.6 (s), 126.8 (d), 125.3 (d), 124.0 (d), 71.4 (t), 49.9 (s), 47.7 (d), 38.9 (t), 38.1 (s), 33.4 (d), 31.9 (t), 31.5 (t), 23.9 (q), 23.9 (q), 23.4 (q), 20.8 (t), 19.3 (t); HRMS (ESI) m/z 317.2111 [M + H]+, calcd for C20H29O3: 317.2117.:2 mixture with starting material based on 1H NMR integration) as a pale-brown semisolid which was used in the next step without further purification (same Rf). The 1H and 13C NMR data (from the mixture) were in agreement with those of the natural product (14, 18-hydroxy-8,11,13,15-abietatetraen-19-oic acid, callicapoic acid M3) in CDCl3,3b with the exception for signal reported at 8.25 ppm that we believe contains a typrographical error and it should be 7.25 ppm: 1H NMR (300 MHz, CDCl3) δ 7.25 (1H, d, J = 8.1), 7.18 (1H, d, J = 8.1), 7.13 (1H, br s), 5.32 (1H, s), 5.02 (1H, s), 4.13 (1H, d, J = 10.5), 3.55 (1H, d, J = 10.5), 2.90–2.70 (3H, m), 2.43 (1H, br d, J = 12.3), 2.30 (1H, m), 2.11 (3H, s), 1.73 (2H, m), 1.16 (3H, s); 13C NMR (75 MHz, CDCl3) δC 181.0 (s), 147.2 (s), 142.9 (s), 138.4 (s), 134.7 (s), 126.0 (d), 125.3 (d), 123.1 (d), 111.7 (t), 71.2 (t), 49.9 (s), 47.5 (d), 38.8 (t), 38.2 (s), 31.8 (t), 31.5 (t), 23.3 (q), 21.7 (q), 20.8 (t), 19.3 (t).The crude alkene-acid 14 (70 mg, 0.142 mmol, ca. 3:2 mixture) obtained above was dissolved in DMF (720 μL, 0.2 M) and OsO4 (20 μL, 2.5% in tert-BuOH, 0.01 equiv.) was added and stirred for 5 min. Oxone (178 mg, 0.58 mmol, 4 equiv.) was added (the solution darkened) in one portion and the reaction was stirred at rt for 3 h 30 min. Then, 10% aqueous Na2SO3 (1 mL) and Et2O (7 mL) were added and stirred for 1 h. After separation of layers, the aqueous phase was extracted with 2 mL of Et2O and the combined organic extracts were washed with 1 N HCl, brine, dried and concentrated. The resulting pale oily residue was chromatographed on silica eluting with n-hexane–acetone (5:5) to give 13 mg of unreacted impurity (isopropyl moiety) contained in the starting alkene-acid (isopropenyl moiety), followed by 17.0 mg (38% for the two steps, 53% brsm) of bodinieric acid A (3) as an amorphous white solid: [α]22D +125 (c 0.5, MeOH) (lit.,3a +121 (c 0.52, MeOH)). The 1H and 13C NMR data were in agreement with those of the natural product in acetone-d6:3a 1H NMR (300 MHz, CD3COCD3) δ 7.71 (1H, dd, J = 8.1, 2.1), 7.66 (1H, d, J = 2.1), 7.45 (1H, d, J = 8.4), 3.85 (1H, d, J = 10.2), 3.69 (1H, d, J = 10.2), 2.52 (3H, s), 2.35 (2H, m), 1.87 (2H, m), 1.66 (1H, m), 1.19 (3H, s); 13C NMR (75 MHz, CD3COCD3) δC 197.7 (s), 177.3 (s), 154.6 (s), 136.5 (s), 135.6 (s), 130.0 (d), 126.7 (d), 126.3 (d), 69.9 (t), 50.6 (s), 46.9 (d), 39.7 (t), 39.5 (s), 32.3 (t), 32.1 (t), 26.6 (q), 23.7 (q), 21.4 (t), 20.3 (t); HRMS (ESI) m/z 315.1599 [M − H]+, calcd for C19H23O4: 315.1596.
Deuterium exchange experiment of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :1 mixture of aldehydes 7a,b with base in deuterated solvent
:1 mixture of aldehydes 7a,b with base in deuterated solvent
A 1:1 mixture of aldehydes 7a/7b (64 mg, 0.237 mmol) was dissolved in 1:1 DCM:MeOD (5 mL) and 1 mL of D2O was added followed by Na2CO3 (15 mg). After being stirred for 3 minutes an aliquot (2 mL) was taken from the reaction mixture and diluted with 4 mL of DCM in a separation funnel. Then, 2 mL of D2O with a bit of sodium chloride was added, agitated and the resulting phases were separated. The organic layer was dried on MgSO4 and concentrated. Another aliquot (2 mL) was taken after 15 minutes of reaction time and processed similarly. The remaining 2 mL of reaction mixture after 60 minutes of reaction time were processed accordingly. Thus, we obtained three residues of 20, 21, and 21 mg, respectively, as colorless oils which were then studied spectroscopically by NMR (see Fig. S9–S11‡).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support has been provided by the Spanish Government [CSIC (201680I008)] and the Universidad de Valencia, Spain.Notes and references
- (a) M. A. González, Nat. Prod. Rep., 2015, 32, 684 RSC; (b) M. A. González, Eur. J. Med. Chem., 2014, 87, 834 CrossRef PubMed; (c) D. Hamulić, M. Stadler, S. Hering, J. M. Padrón, R. Bassett, F. Rivas, M. A. Loza-Mejía, M. A. Dea-Ayuela and M. A. González-Cardenete, J. Nat. Prod., 2019, 82, 823 CrossRef PubMed.
- M. A. González, Tetrahedron, 2015, 71, 1883 CrossRef.
- Two research groups discovered these natural products from two different species of Callicarpa naming them as bodinieric acids and callicapoic acids, see: (a) J.-B. Gao, S.-J. Yang, Z.-R. Yan, X.-J. Zhang, D.-B. Pu, L.-X. Wang, X.-L. Li, R.-H. Zhang and W.-L. Xiao, J. Nat. Prod., 2018, 81, 998 CrossRef CAS PubMed; (b) Z.-H. Wang, C. Niu, D.-J. Zhou, J.-C. Kong and W.-K. Zhang, Molecules, 2017, 22, 842 CrossRef PubMed.
- W. Xiao, R. Zhang, J. Gao, S. Yang, X. Zhang, D. Pu and X. Li, patent number CN 108129295, application number CN 2018-10028568, 2018.
- M. Arnó, M. A. González, M. L. Marín and R. J. Zaragozá, J. Org. Chem., 2000, 65, 840 CrossRef PubMed.
- F. E. Ziegler and O. B. Wallace, J. Org. Chem., 1995, 60, 3626 CrossRef CAS.
- S. W. Pelletier and D. L. Herald, J. Chem. Soc. D, 1971, 10b–11 RSC.
- M. A. González, D. Perez-Guaita, J. Correa-Royero, B. Zapata, L. Agudelo, A. Mesa-Arango and L. Betancur-Galvis, Eur. J. Med. Chem., 2010, 45, 811 CrossRef PubMed.
- (a) J. W. Huffman, J. Org. Chem., 1970, 35, 478 CrossRef CAS; (b) R. H. Burnell, C. Côté and N. Théberge, J. Nat. Prod., 1993, 56, 1459 CrossRef CAS.
- A. F. Barrero, E. J. Alvarez-Manzaneda, R. Alvarez-Manzaneda, R. Chahboun, R. Meneses and M. Aparicio, Synlett, 1999, 713 CrossRef CAS.
- A. W. Burghstahler and J. N. Marx, J. Org. Chem., 1969, 34, 1562 CrossRef.
- In the original paper of Wang and co-workers (ref. 3b) for natural callicapoic acid M4 (4), the observed optical rotation ([α]D = +22.2, c = 0.10, MeOH) is different from the reported value ([α]D = +57.3, c = 0.10, MeOH) for bodinieric acid B (4) by Zhang and co-workers (ref. 3a), and also different of the value for our synthetic 4 ([α]D= +104.9, c = 0.10, MeOH). Our synthesized material 4 was used for the synthesis of bodinieric acid A (3) and we obtained a value for our synthetic 3 ([α]D= +125.0, c = 0.5, MeOH) which is in excellent agreement with the optical rotation ([α]D = +121.0, c = 0.52, MeOH) reported by Zhang and co-workers (ref. 3a).
- B. R. Travis, R. S. Narayan and B. Borhan, J. Am. Chem. Soc., 2002, 124, 3824 CrossRef CAS PubMed.
- M. J. Frisch, et al., Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- J. M. Hermida-Ramón and G. Karlström, J. Phys. Chem. A, 2003, 107, 5217 CrossRef.
- (a) J. G. M. Winkelman, O. K. Voorwinde, M. Ottens, A. A. C. M. Beenackers and L. P. B. M. Janssen, Chem. Eng. Sci., 2002, 57, 4067 CrossRef CAS; (b) M. Detcheberry, P. Destrac, X.-M. Meyer and J.-S. Condoret, Fluid Phase Equilib., 2015, 392, 84 CrossRef CAS.
- M. B. Smith, and J. March, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 6th edn, Wiley, New Jersey, 2007, pp. 1863–1865 Search PubMed.
- (a) D. K. Bohme and G. I. Mackay, J. Am. Chem. Soc., 1981, 103, 979 CrossRef; (b) C. K. Regan, S. L. Craig and J. I. Brauman, Science, 2002, 295, 2245 CrossRef CAS PubMed.
- X. Liu, J. Zhang, L. Yang and W. L. Hase, J. Am. Chem. Soc., 2018, 140, 10995 CrossRef CAS PubMed.
- P. Raiteri, R. Demichelis and J. D. Gale, J. Phys. Chem. C, 2015, 119, 24447 CrossRef CAS.
- M. A. González, M. J. Gil-Gimeno and A. J. Blake, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, 3346 CrossRef.
- (a) R. G. Parr and W. Yang, Density Functional Theory of Atoms and Molecules, Oxford University Press, New York, 1989 Search PubMed; (b) T. Ziegler, Chem. Rev., 1991, 91, 651 CrossRef CAS.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed.
- W. J. Hehre, L. Radom, P. von R. Schleyer and J. A. Pople, Ab initio Molecular Orbital Theory, Wiley, New York, 1986 Search PubMed.
- K. Fukui, J. Phys. Chem., 1970, 74, 4161 CrossRef CAS.
- (a) C. González and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523 CrossRef; (b) C. González and H. B. Schlegel, J. Chem. Phys., 1991, 95, 5853 CrossRef.
- (a) J. Tomasi and M. Persico, Chem. Rev., 1994, 94, 2027 CrossRef CAS; (b) B. Y. Simkin, and I. Sheikhet, Quantum Chemical and Statistical Theory of Solutions-A Computational Approach, Ellis Horwood, London, 1995 Search PubMed.
- (a) E. Cances, B. Mennunci and J. A. Tomasi, J. Chem. Phys., 1997, 107, 3032 CrossRef CAS; (b) M. Cossi, V. Barone, R. Cammi and J. Tomasi, Chem. Phys. Lett., 1996, 255, 327 CrossRef CAS; (c) V. Barone, M. Cossi and J. Tomasi, J. Comput. Chem., 1998, 19, 404 CrossRef CAS.
- R. Caputo, L. Previtera, P. Monaco and L. Mangoni, Tetrahedron, 1974, 30, 963 CrossRef CAS.
- J. R. Tagat, D. V. Nazareno, M. S. Puar, S. W. McCombie and A. K. Ganguly, Bioorg. Med. Chem. Lett., 1994, 4, 1101 CrossRef CAS.
Footnotes |
| † Dedicated to Professor Gerald Pattenden on the occasion of his 80th birthday for his outstanding contributions in natural products synthesis. |
| ‡ Electronic supplementary information (ESI) available: Copies of 1H NMR and 13C NMR spectra for all relevant compounds, including COSY and HSQC spectra for 3 and 4, Table S1 (B3LYP/6-31G** conformational energies for 3), comparisons of 13C NMR data of natural and synthetic compounds (Tables S2 and S3), Table S4 (B3LYP/6-31G** energies of the species involved in the enolization of 7a and 7b), figures for different conformers of 3, and figures of the energy profiles (IRC, ΔE) corresponding to the transition structures TSa–d. Geometries of the species involved in the enolization and XYZ matrices computed at B3LYP/6-31G** level (PDF). Zoom of NMR spectra of the deuterium exchange experiment. See DOI: 10.1039/d0ra02711a |
| This journal is © The Royal Society of Chemistry 2020 |