
Recent developments in dehydration of primary amides to nitriles
Muthupandian
Ganesan
 *a and
Paramathevar
Nagaraaj
*b
*a and
Paramathevar
Nagaraaj
*b
aToxicology Division, Regional Forensic Science Laboratory, Forensic Sciences Department, Forensic House, Chennai-4, India. E-mail: ganchemiitd@gmail.com
bDepartment of Chemistry, Anna University, Chennai-25, India. E-mail: nagaiitd@gmail.com
First published on 21st September 2020
Abstract
Dehydration of amides is an efficient, clean and fundamental route for the syntheses of nitriles in organic chemistry. The two imperative functional groups viz., amide and nitrile groups have been extensively discussed in the literature. However the recent development in the century-old dehydration method for the conversion of amides to nitriles has hardly been reported in one place, except a lone review article which dealt with only metal catalysed conversions. The present review provides broad and rapid information on the different methods available for the nitrile synthesis through dehydration of amides. The review article has major focus on (i) non-catalyzed dehydrations using chemical reagents, and (ii) catalyzed dehydrations of amides using transition metal, non-transition metal, organo- and photo-catalysts to form the corresponding nitriles. Also, catalyzed dehydrations in the presence of acetonitrile and silyl compounds as dehydrating agents are highlighted.
 Muthupandian Ganesan | Dr M. Ganesan was born in 1983 in Madurai, Tamilnadu state, India. He received his B.Sc. and M.Sc. from Madurai Kamaraj University and then his Ph.D. from the Indian Institute of Technology Delhi (Prof. N. G. Ramesh Group). He did his postdoctoral research at the Nanyang Technological University of Singapore and Chungnam National University of South Korea. Since 2017, he has been working as a junior scientific officer at Regional Forensic Science Laboratory, Forensic Sciences Department (Govt. of Tamilnadu), Chennai, India. |
 Paramatheva Nagaraaj | Dr P. Nagaraaj graduated in chemistry from Gandhigram Rural Institute (deemed University), Dindigul, Tamil Nadu, India in the year 2003. He earned his Master's degree from Pondicherry Central University, Pondicherry in the year 2005. In 2010, he received his doctorate degree from the Indian Institute of Technology Delhi (IITD) under Professor N. G. Ramesh. Later in 2010–2015, he worked as a scientist at the Spices Board of India (Govt. of India). Currently (since 2015) he is working as an assistant professor in the Department of Chemistry, CEG Anna University, Chennai, India. His current research areas are organic synthesis, green chemistry & catalysis, sensors, polymers and nanochemistry. |
1. Introduction
Nitriles are naturally found in various bacteria, fungi, plants and animals and are extensively applied as raw materials for the production of a variety of pharmaceuticals, agrochemicals, polymers, materials, etc.1 Examples of pharmaceuticals containing nitrile groups include vildagliptin, an anti-diabetic drug2 and anastrazole, a drug used for treating breast cancer (Fig. 1).3 Moreover, nitrile functionalities have been utilized as versatile intermediates in organic synthesis and they can be readily converted to various other important functional groups viz., aldehydes,4 carboxylic acids,5 esters,6 primary amines,7 imines,8 substituted oximes,9 heterocycles,10 amides,11etc.12 Synthesis of such a highly valuable nitrile functionality has been one of the promising research fields in organic chemistry.13 | ||
Fig. 1 Examples of –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N containing drug molecules. N containing drug molecules. | ||
Various methods13,14 have been developed for the syntheses of nitriles using different synthetic strategies including the addition reaction of multiple bonds (I),15 oxidation of amines (II),16 reduction of nitroalkanes (II),17 reactions of alcohols (II),18 dehydration or rearrangement of oximes (III) and amidoximes (IV),19 dehydration of amides or water shuffling with amides (V),20 dehydrosulfurization of thioamides (V),21 substitution of aryl and alkyl-halides/azides (VI),22 and reaction of aldehydes (VI)23 (Scheme 1). The classical methods viz., nucleophilic substitution of alkyl halides, the Sandmeyer reaction of diazotized aniline, the Rosenmund–von Braun reaction of aryl halides, etc., have limited applications due to the usage of highly toxic cyanide sources.24 On the other side, synthetic procedures using non-metallic cyanide sources25 have also been available for the large-scale production of nitriles like acrylonitrile by ammoxidation of n-propene, adiponitrile by hydrocyanation of 1,3-butadiene, and so on.26 However, these methods are also limited by their poor functional group tolerance due to harsh reaction conditions.27 Alternatively, alkyl/aryl halides have also been catalytically converted to the corresponding amides using naturally abundant small molecules viz., CO2, NH3 and urea.28 In fact, syntheses of nitriles from nitrogen containing compounds such as amine, amides, etc., will be better alternatives than their non-nitrogen counterparts, as additional steps for the incorporation of the nitrogen functionality, involving the usage of stoichiometric reagents and generation of metal waste, can be avoided. Amides, apart from their easy availability, with a general formula of RCONR′R′′, are commonly found in numerous synthetic and natural products, besides their crucial role as basic building blocks of proteins in every form of life.29 Direct synthesis of nitriles by dehydration of amides with benign and low-cost catalysts and dehydrating agents under simple reaction conditions is particularly attractive from the green chemistry point of view. Furthermore, in the conversion of amides to nitriles, water molecules are the only value added side product which theoretically adds merit to the sustainable synthesis (Scheme 2). Thus, the dehydration of amides would be one of the fundamental and clean synthetic routes for the syntheses of nitriles.13 Simple heating at very high temperatures could lead to the dehydration of the amide groups to form the corresponding nitriles, whether catalysts and/or dehydrating agents are present or not. However, in order to overcome the problems associated with such harsh temperature, the dehydration methods were often carried out using stoichiometric amounts of dehydrating agents in the presence of catalysts, since the conversion of primary amides to nitriles by eliminating water is typically endergonic.13 Another interesting method for the preparation of nitriles from amides is “transfer hydration” which is based on the “water” acceptor–donor capacity of the substrate.16 Under this method, powerful water donors such as amides and aldoximes are dehydrated into nitriles in the presence of dehydrating agents such as acetonitrile. However, selectivity and excess use of the water donor are the major drawbacks of this method. Hence, it is a challenging goal to develop eco-friendly ways for the conversion of amides to the corresponding nitriles.
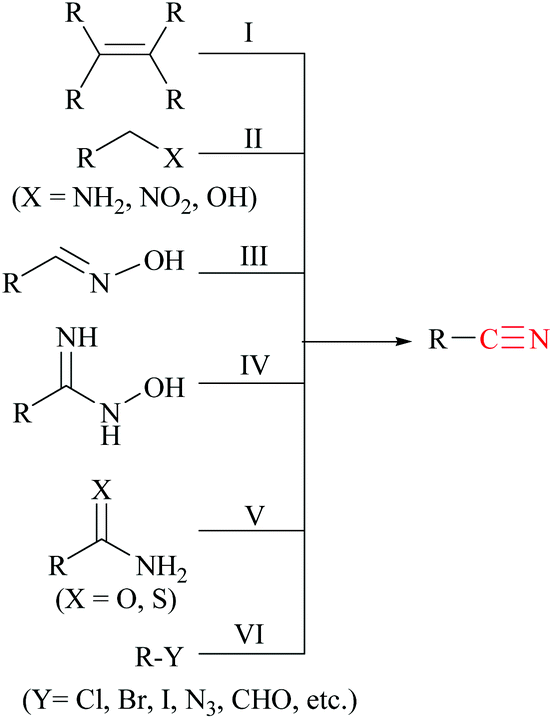 | ||
| Scheme 1 Various reported routes for nitrile synthesis. | ||
 | ||
| Scheme 2 Dehydration of amides leading to clean products. | ||
It is worth noting that recently, Al-Huniti and Croatt have reviewed the metal catalyzed conversion of amides to nitriles.13 However, our aim is to present broad and rapid information on the various available methods for the dehydration of amides to nitriles under non-catalyzed and catalyzed conditions. The catalytic dehydrations included for the discussion are based on transition metal, non-transition metal, organo- and photo-catalysts in the presence of acetonitrile and silyl compounds as dehydrating agents. It is hoped that by compiling the widely scattered information on the dehydration of amides, the present review will grasp the attention of a broad readership due to the potential applications of nitriles.
The present review shall describe the merits and demerits of the present methods and pertaining challenges for dehydration of amides by categorizing them into two parts: part I shall deal with the non-catalyzed dehydration methods using chemical reagents and in part II applications of catalysts in dehydration shall be discussed.
2. Non-catalyzed dehydration of amides using chemical reagents
The first dehydration of amides was performed by Wöhler and Liebig in 1832 who reported that pyrogenetic decomposition of benzamide over barium oxide yielded phenyl cyanide and water.30 The first use of phosphorus pentoxide to dehydrate ammonium salts or amides was introduced by Dumas in 1847.31 However, chemical reagents either as dehydrating agents or as catalysts are usually required for the mild conversion of amides to nitriles. Thus, the direct preparation of nitriles was generally achieved by the dehydration of the corresponding amides using classical dehydrating reagents such as Lewis acids, basic reducing agents and other efficient chemical reagents that are discussed in the following sections.Rickborn and Jensen reported the dehydration of 4-tert-butylcyclohexanecarboxamide to the corresponding nitrile using phosphorus pentoxide (P2O5), thionyl chloride and also phosphorus oxychloride as dehydrating agents.32 Kenyon and Ross used P2O5 for the dehydration of 2-methyl-3-phenylpropanamide to obtain the corresponding nitrile.33 Mahajan et al. reported the dehydration of 2-amino-5-chlorobenzamide using P2O5 to form 2-amino-5-chlorobenzonitrile and phosphoric acid (Scheme 3).34
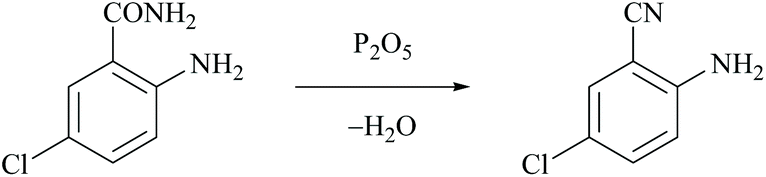 | ||
| Scheme 3 P2O5 as a dehydrating agent. | ||
Konwar and coworkers developed a method for the dehydration of oximes and amides in hydrated media to their corresponding nitriles in good yields using an AlCl3·6H2O/KI/H2O/CH3CN system (Scheme 4).35 The same group has also reported anhydrous AlCl3-KI mediated dehydration of amides. In the presence of water, aluminium is coordinated with amide-nitrogen and in anhydrous medium it is coordinated with amide-oxygen and facilitates the removal of water from the amide to form the corresponding nitrile (Scheme 4).
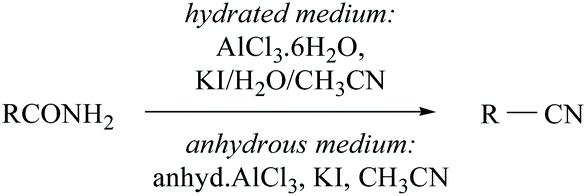 | ||
| Scheme 4 AlCl3 as a dehydrating agent. | ||
In the preparation of oxathioazol-2-one, an inhibitor of threonine protease, using chlorocarbonylsulfenyl chloride as a coupling reactant and sodium carbonate as a base, the starting carboxamide in the presence of dioxane as solvent afforded the expected piperidin-3-yl-oxathiazol-2-one compound in 68% yield.36 However, in the absence of sodium carbonate as a base, the reaction of carboxamide with chlorocarbonylsulfenyl chloride alone using pyridine as solvent initially involved coupling which is followed by a rapid elimination of sulphur oxide to form its corresponding nitrile. Thus the combination of chlorocarbonylsulfenyl chloride and pyridine constitutes a dehydrating system and facilitates the dehydration of the amide to give the corresponding nitrile by destabilizing the intermediate, piperidin-3-yl-oxathiazol-2-one (Scheme 5).36
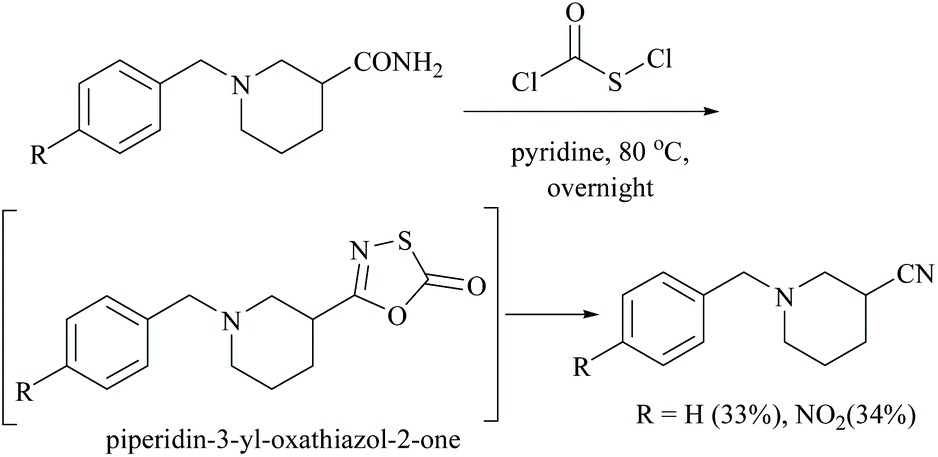 | ||
| Scheme 5 Chlorocarbonylsulfenyl chloride and pyridine mediated dehydration. | ||
Unnikrishnan and coworkers disclosed a one-pot synthesis of nitriles from amides and oximes using an in situ–generated Burgess-type reagent as the dehydrating agent (Scheme 6). The treatment of chlorosulfonyl isocyanate with β-aminoalcohol [1-(2-hydroxyethyl)-piperidine] yielded in a single step a cyclic Burgess-type reagent which without further purification was used for the dehydration of aromatic amides.37 Itabashi and coworkers reported the dehydration of benzamide, acetamide, thiobenzamide and nicotinamide using phosphorus tris(diethylamide) [P(NEt2)3] under reflux conditions in THF for 3 hours, to give the corresponding nitriles in 89.6%, 98.5%, 90.4% (in benzene) and 70.5% yields, respectively (Scheme 6).38
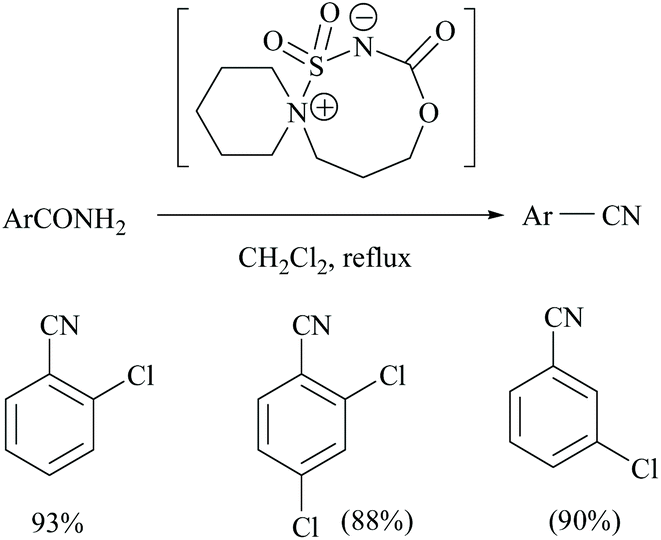 | ||
| Scheme 6 Burgess-type reagent as the dehydrating agent. | ||
Touil and coworkers developed three different methods for the conversion of primary amides into the corresponding nitriles, mediated by the highly electrophilic phosphorus-III reagents, tris-(dimethylamino)phosphine [P(NMe2)3], phosphorus trichloride (PCl3), or triphenylphosphite [P(OPh)3], respectively (Scheme 7).39 The primary amides coupled with P(NMe2)3, PCl3, or P(OPh)3 followed by rapid elimination with a base: diethylamine or DBU to form the corresponding nitriles under MW irradiation or reflux conditions. Among these three methods, method C with P(OPh)3 and DBU under MW irradiation exhibited the best result. Notably, aromatic amides bearing electron-donating groups on the aromatic rings produced higher yields and the reaction conditions were found to be compatible with cyanoamides, malonamides, α,β-unsaturated amides, an unprotected amine functionality, the existing nitrile functionality, and a thiophene ring.
 | ||
| Scheme 7 Use of electrophilic phosphorus-III reagents. | ||
Dennis reported the dehydration of amides to nitriles with the help of silazanes, aminosilanes, alkoxysilanes, and chlorosilanes as dehydrating agents at high temperatures.40 For example, hexamethyltricyclotrisilazane with acetamide or benzamide underwent dehydration at a temperature range of 180–200 °C, forming acetonitrile and benzonitrile in 95 and 85% yields, respectively (Scheme 8). Moreover, with benzamide, silazanes formed a siloxane polymer of molecular weight Mw 120![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 as the by-product. Under these conditions, N-methylbenzamide also gave the benzonitrile when heated to above 200 °C.
000 as the by-product. Under these conditions, N-methylbenzamide also gave the benzonitrile when heated to above 200 °C.
 | ||
| Scheme 8 Silyl compounds as dehydrating agents. | ||
The conversion of primary amides into nitriles has also been found to use oxalyl chloride as an activating agent only along with other reagents.41 In 1978, Swern used for the first time a combination of DMSO and oxalyl chloride for the oxidation of primary and secondary alcohols.42 The combination has been used for many other conversions.43 Sun et al. wished to use the Swern reaction conditions catalytically for the dehydration of amides which often involve the usage of stoichiometric amounts of DMSO and (COCl)2.41 The authors successfully carried out the catalytic dehydration of amides to obtain nitriles in good yields, using a catalytic amount of DMSO in the presence of (COCl)2 and Et3N (Scheme 9). The proposed mechanism for the catalytic Swern oxidation involved initial formation of chlorodimethylsulfonium chloride from the reaction of DMSO and (COCl)2, followed by nucleophilic substitution of amide-oxygen in the presence of the Et3N-base. In the last step, elimination of HCl and DMSO led to the formation of the corresponding nitrile. The replacement of the base Et3N with other bases such as DBU and NMM was not successful. Notably, the yields obtained were found to be comparable with those of the catalytic Appel type reaction (discussed in a later section). The Boc protected substrate was found to be compatible under catalytic Swern oxidation, whereas under catalytic Appel's reaction, Boc-protected amino acids did not undergo the conversion. Moreover, the present method was superior in terms of higher yield than the one reported by Nakajima and Ubukata41 using Swern oxidation with stoichiometric DMSO.
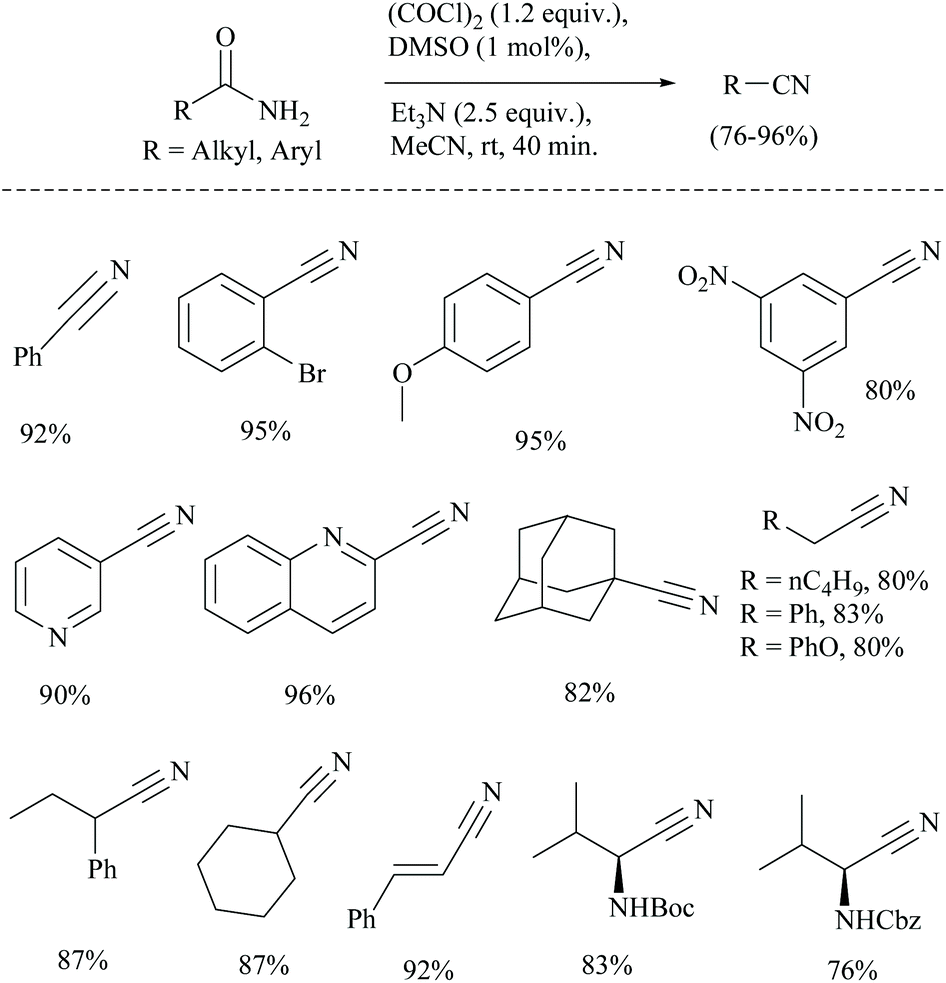 | ||
| Scheme 9 Oxalyl chloride with a DMSO catalyst. | ||
Monson and Priest reported the dehydration of aliphatic and aromatic carboxamides to their corresponding nitriles in hexamethylphosphoric triamide (HMPT) at 220–240 °C in good yields viz., propionamide (94%), n-butyramide (75%), hexanamide (78%), banzamide (67%), phenylacetamide (75%) and adipamide (49%) (Scheme 10).44 The plausible mechanism for this reaction involves the evolution of dimethylamine, the formation of a phosphorodiamidate derivative of the amide, and the elimination of the phosphorodiamidate leaving group to form the nitrile (Scheme 10).
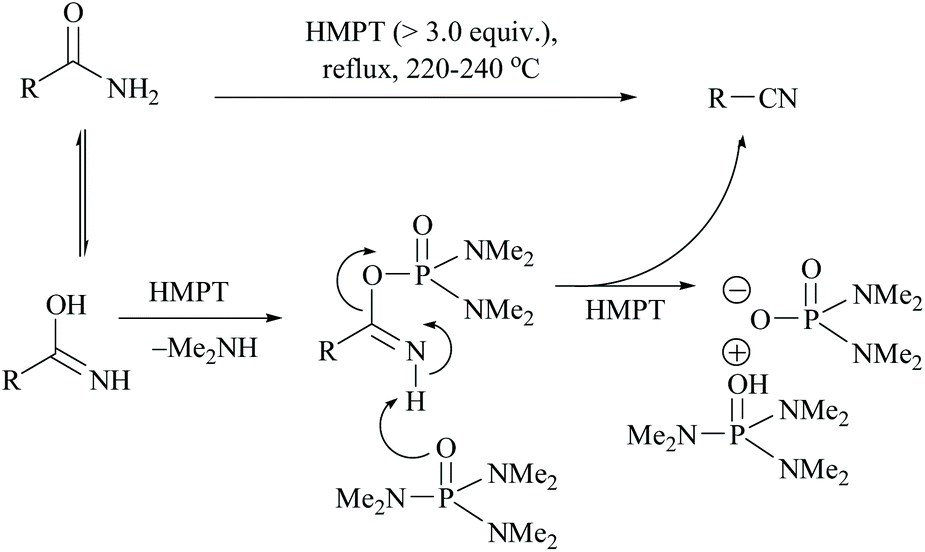 | ||
| Scheme 10 HMPT mediated dehydration. | ||
Cyanuric chloride (2,4,6-trichloro-s-triazine) has also been used as a dehydrating agent for carboxamide. Under these reaction conditions, only a one third molar equivalent of cyanuric chloride is needed to convert an amide group into its nitrile.45 The mechanism involves initially simple nucleophilic substitution of chloride in cyanuric chloride with formamide which is then substituted by carboxamide. For example, Rodriguez and coworkers reported the dehydration of N-protected α-amino-carboxamides to their corresponding nitriles in good yields using cyanuric chloride in DMF as the dehydrating agent (Scheme 11).46 Using similar conditions, Bonrath and coworkers reported the dehydration of heterocyclic carboxamides to the corresponding nitriles (Scheme 12). The oxazole amides underwent dehydration smoothly to form their corresponding nitriles.47 They also found that the nature of the formamide is not critical and any conventional N,N-disubstituted formamide can be used but the formamide must be water insoluble. However, nicotinic acid amide did not react under these conditions to 3-cyano-pyridine.
 | ||
| Scheme 11 Cyanuric chloride mediated dehydration. | ||
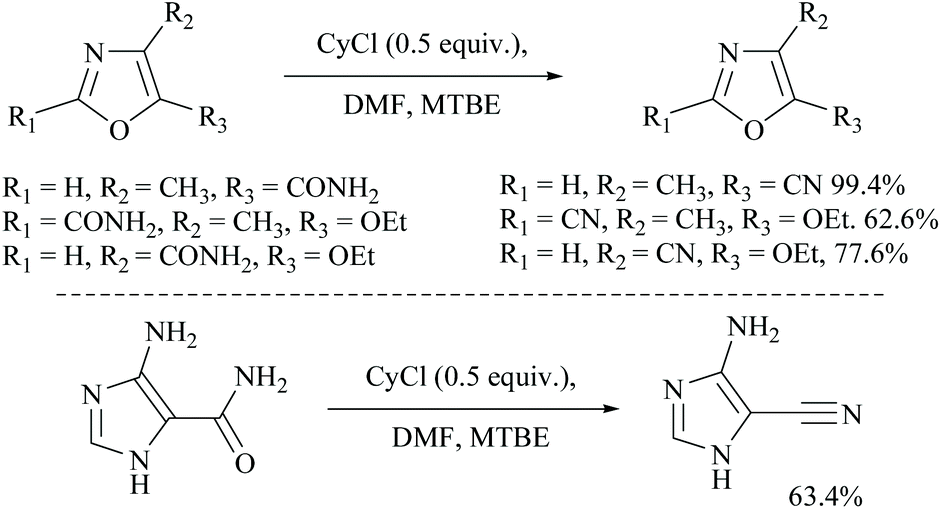 | ||
| Scheme 12 CyCl with MTBE for dehydration. | ||
Shia and coworkers disclosed a simple procedure for the conversion of primary amides to the corresponding nitriles, using ethyl dichlorophosphate as the dehydrating agent along with DBU (Scheme 13).48 Moreover, for comparison, the authors also carried out the dehydration of cyclohexanyl carboxamide distinctly using their own reagent system ethyl dichlorophosphate/DBU as well as with a trifluoroacetic anhydride (TFAA)/Et3N system which had already been reported for the dehydration of primary amides.49 (Scheme 14). They observed that their reagent afforded the nitrile in 92% yield; however with the TFAA system the nitrile was formed in only 64% yield along with trifluoro-acylated nitrile as a side product in 23% yield.
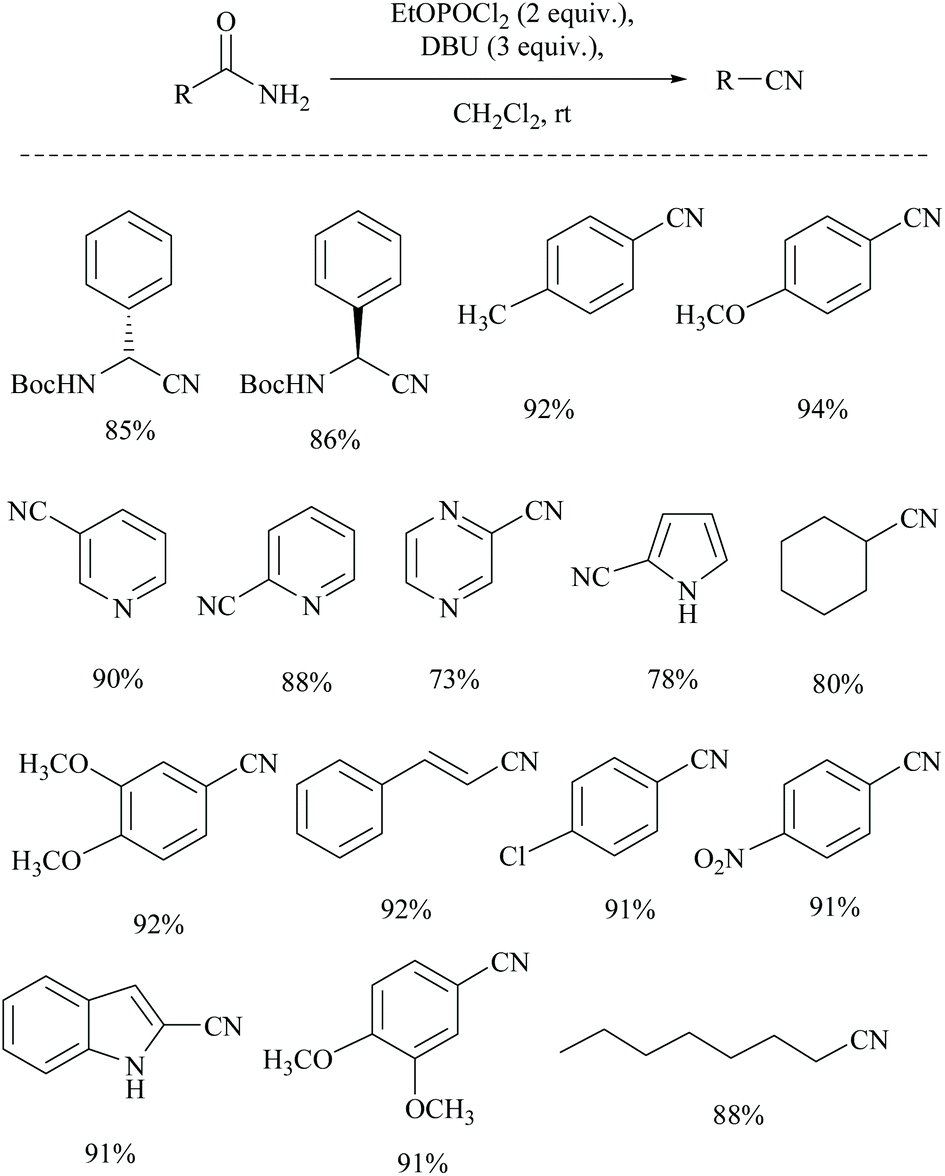 | ||
| Scheme 13 Ethyl dichlorophosphate as a dehydrating agent. | ||
 | ||
| Scheme 14 Ethyl dichlorophosphate vs. TFAA. | ||
In 2011, Sardarian and coworker50 reported a better method using “diethyl chlorophosphate” (EtO)2POCl as a dehydrating agent (Scheme 15), compared to the previous one with “ethyl dichlorophosphate” (Scheme 13).48,49 Moreover, the present method has an advantage that the dehydration could also be carried out in the absence of a solvent and base, without compromising the yield (Table 1).
 | ||
| Scheme 15 Diethyl chlorophosphate as a dehydrating agent. | ||
|
|
(EtO)2POCl | (EtO)POCl2 | ||||
|---|---|---|---|---|---|---|
| Toluene | Solvent free | |||||
| Yield, % | Time, min | Yield, % | Time, min | Yield, % | Time, min | |
| R = H | 83 | 20 | 89 | 15 | 98 | 60 |
| R = p-Me | 82 | 10 | 90 | 15 | 92 | 180 |
| R = p-OMe | 85 | 5 | 91 | 5 | 94 | 180 |
| R = p-NO2 | 89 | 15 | 91 | 25 | 93 | 120 |
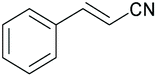
|
92 | 10 | 94 | 10 | 92 | 180 |
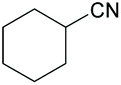
|
72 | 10 | 74 | 10 | 80 | 180 |

The proposed mechanism involves the reaction of the imidic form of primary amide with diethyl chlorophosphate to form the corresponding diethyl phosphate imide followed by the release of diethyl hydrogen phosphoric acid and nitriles. In order to study the effect of solvent, they carried out the dehydration in toluene as solvent under reflux conditions and the solvent method was found to produce relatively low yields. Notably, 2-cyanopyridine (2-CP) has been used as a dehydration agent in the synthesis of dimethylcarbonate (DMC) from CO2 and methanol.51 In order to improve the recyclability, the end product, 2-picolinamide (2-PA), was dehydrated back to 2-CP. In this context, many attempts have been made to achieve the dehydration of 2-PA to 2-CP.49,52,53 Some of them such as ethyldichlorophosphate,49 (N-methyl-N-trimethylsilyl) trifluoroacetamide,52 trifluoromethane sulfonic acid anhydride and trimethylamine53 are non-renewable and expensive too.
3. Catalyzed dehydration of amides
Recent years have seen an upsurge in the publications of catalytic dehydrations using a variety of metals and organic small molecules as catalysts in the presence of dehydrating agents. Transition metal catalyzed amide-dehydration reactions typically utilize acetonitrile41,54 or silane55,56 including N-methyl-N-(trimethylsilyl)trifluoroacetamide (MSTFA), even though a very high temperature can also cause dehydration but with poor yield. A catalyzed water shuffling method between amides and nitriles has also been gaining reasonable attention for converting amides to nitriles and vice versa.57 Metal based catalysts reported for the dehydration of primary amides to nitriles include complexes of transitions metals viz., Cu,41,58 In,59 Fe,56,60 Pd,61 Sn,19 Re,62 Rh,63 Ru,56,64 U,52 V,65 W,52,66 Zn,52,66,67 and Zr.68 The available literature reports are reviewed on the basis of the catalyst/dehydrating agent: (1) catalyzed dehydration at high temperature; (2) catalyzed dehydration using acetonitrile as a dehydrating agent, (3) transition metal catalyzed silylative dehydration, and (4) non-transition metal catalyzed, (5) organo-catalyzed, and (6) photocatalyzed dehydration.3.1. Metal catalysed dehydration at high temperature
Dehydration of amides in the absence of a dehydrating agent, at high temperature either with or without a catalyst, has been found to give the corresponding nitrile. However, in order to improve the yield, to make it suitable for thermally unstable amides possessing other functional groups, and to avoid the usage of stoichiometric reagents including dehydrating agents, it is wise to make use of catalysts. Catalyzed dehydrations by bringing down the actual required temperature and time are found to be compatible for a variety of amide substrates. One such condition was accomplished by Campbell and coworkers for non-catalyzed dehydration of primary amides under flash vacuum pyrolysis (FVP) conditions requiring heating to >800 °C; however the addition of WO3 to the furnace tube reduced the temperature by half to 400 °C for yielding benzonitrile in 97%.52,66Indeed, the first method in this context, using Wilkinson's heterogeneous catalyst, RhCl(PPh3)3, also required a very high temperature to remove the water molecules from the amide to form benzonitrile.63 Also, under such harsh conditions, dehalogenation was observed with p- and m-chlorobenzamides. Thus, Watanabe et al. replaced the catalyst with a ruthenium(II) complex for the dehydration of the amide using, notably, an equimolar amount of N,N′-dialkylurea which gets converted into isocyanate and finally into alkyl amines and CO2 (Scheme 16).64
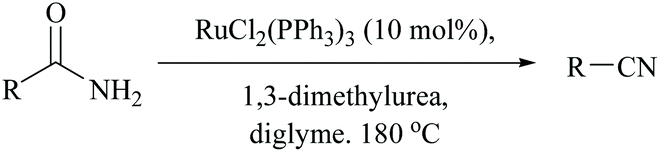 | ||
| Scheme 16 Ruthenium(II) complex for dehydration. | ||
Yamamoto and coworkers reported a functional group-tolerable and large-scalable method using ∼1 mol% of perrhenic acid under azeotropic reflux of mesitylene for the dehydration of amides including vinyl and aromatic amides (Scheme 17).58
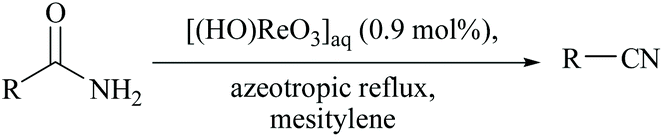 | ||
| Scheme 17 Perrhenic acid mediated dehydration. | ||
Kaneda and coworkers reported that monomeric vanadium oxide supported on hydrotalcite (V/HT) acted as a reusable solid heterogeneous catalyst for the dehydration of amides into the corresponding nitriles. Under these reaction conditions, 16 different nitriles were obtained in good yields (83–94%) (Scheme 18).65
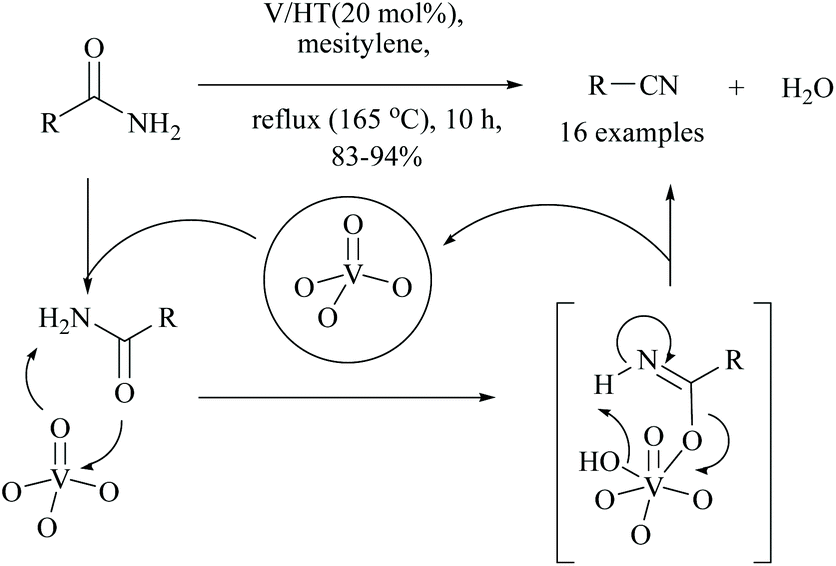 | ||
| Scheme 18 Vanadium catalyst for dehydration. | ||
In 2004, Bergman and Ruck68 reported a zirconium-mediated conversion of amides to the corresponding nitriles and thus joined the club consisting of the lone transition metal-mediated dehydration by TiCl469 (Scheme 19). In the present method, primary amides were converted into their corresponding nitriles through the formation of the corresponding N-acylimidozirconocene complex.
 | ||
| Scheme 19 Zirconium catalyst with LiCl for dehydration. | ||
Dimethylzirconocene reacted with amides to form a methyl-zirconium amide complex. This methyl-zirconium amide complex, with the help of a chloride source, underwent 1,2-hydro methyl elimination to form an N-acylimidozirconocene complex which reacted intra-molecularly to give the corresponding nitrile (Scheme 19). Mechanistic studies reveal that chloride coordination to zirconium is required for this transformation to occur which was proved by carrying out dehydration with two different methods. In method 1, [Cp2Zr(CH3)Cl] with an inbuilt chloride neatly formed the corresponding nitrile. However in method 2, with dimethylzirconocene, initially N-benzoylimidozirconocene was formed and left intact even after prolonged heating. The formed N-benzoylimidozirconocene continued to react only after the external addition of LiCl to form an N-benzoylimidozirconocene complex. In method 1, the residual LiCl by-product formed from the reaction between the lithium salt of benzamide and [Cp2Zr(CH3)Cl]. These observations indicated that the dehydration was dependent upon the chloride-anion additive effect.
3.2. Metal catalysed dehydration using acetonitrile as a dehydrating agent
Acetonitrile, the relatively inert solvent, gets activated in the presence of a metal catalyst and acts as a dehydrating agent for the conversion of primary amides to form the corresponding nitriles along with an equimolar amount of acetamide. However, dehydrations using nitriles as solvents are typically reversible, and the catalyst can promote the reaction in either direction depending on the type of solvent involved in that direction. Palladium-catalyzed dehydration of amides using acetonitrile as a dehydrating agent is known for its chemo-selectivity and compatibility over many other functionalities.54,61,70In 2005, Maffioli et al. reported for the first time PdCl2 catalysed reversible conversion of primary amides to nitriles in aqueous acetonitrile solvent (Scheme 20).61,70 The dehydration of primary amides involved “water transfer” from the amide to acetonitrile. They revealed three important findings: (i) the dehydration of amides using a palladium-catalyst requires the addition of water; no reaction occurs under anhydrous conditions, (ii) water was not generated during the reaction but transferred directly from the amide to acetonitrile, and (iii) PdCl2 and Pd(OAc)2 alter the pH of the reaction solutions to the ranges 2.8–3.5 and 4.7–5.0, respectively; thus Pd(OAc)2 can be a better choice of catalyst for the substrates which are sensitive to an acidic environment. The mechanism of palladium catalyzed dehydration involves the coordination of the palladium metal with nitrogen, and a H+-shift between the amide and nitrile (Scheme 20). The reversibility of this reaction i.e., conversion of the nitrile to amide by hydration under the same reaction conditions was validated. Benzonitrile (PhCN) was allowed to react with MeCONH2 under the same reaction conditions in which PhCN was used as a co-solvent instead of MeCN along with water. For making a homogeneous solution, a small amount of THF was also added and complete hydration of PhCN to benzamide was observed in 6 hours at room temperature.
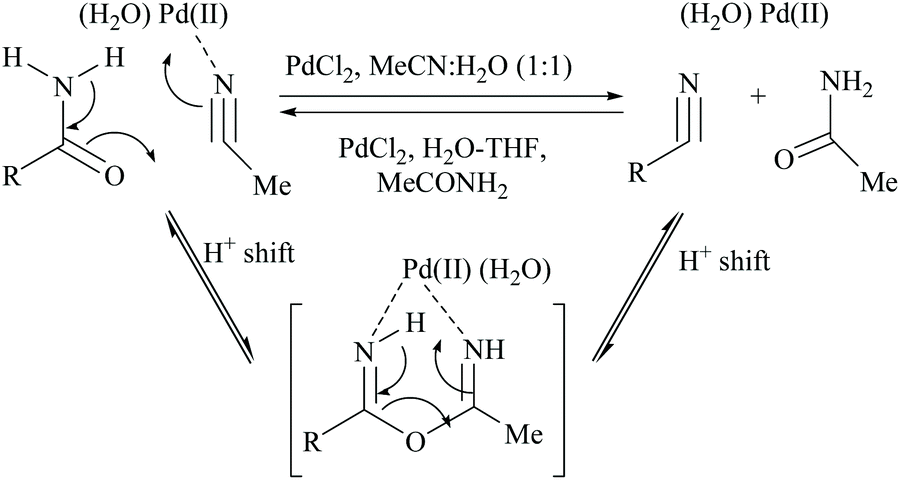 | ||
| Scheme 20 Palladium catalyst for dehydration. | ||
In order to drive the reversibility of this reaction towards the complete conversion of amides to nitriles, Okabe et al. very recently carried out the palladium-catalyzed dehydration of primary amides to nitriles using dichloroacetonitrile as a water acceptor (Scheme 21).71 The authors initially searched for a better water acceptor than MeCN in order to overcome the fact that the dehydration of electron-deficient amides with MeCN is energetically unfavourable. In this context, they focused on the use of electron-deficient nitriles such as dichloroacetonitrile as water acceptors to make the dehydration thermodynamically more favourable and also to obtain nitriles with the retention of the stereochemistry. The driving force to push the reaction towards nitrile formation is the energy required for hydration of the water acceptor. It was found that the hydration of dichloroacetonitrile was more exergonic than that of acetonitrile.
 | ||
| Scheme 21 Dichloroacetonitrile as a water acceptor. | ||
Using dichloroacetonitrile, a palladium trifluoroacetate-catalyzed dehydration method was effectively carried out for various α-aminoamides derived from natural amino acids in which many protecting groups were found to be intact. Notably, only the amide group with free –CONH2 gets dehydrated under these conditions (Scheme 22).
 | ||
| Scheme 22 Dehydration of α-aminoamides. | ||
Singh and coworkers used a very low catalyst loading (0.5 mol%) of a trinuclear palladium(II) complex in aqueous acetonitrile solvent for the dehydration of amides to the corresponding nitriles in good yields (72–96%) (Scheme 23).70 They used two NHC-Pd complexes with NHC containing soft donors viz., sulphur and selenium and found that Pd catalyst A (with S) was more efficient than Pd catalyst B. They found that the reaction conditions were compatible with aliphatic amide, valeramide, pyridyl and aryl groups with various halogen substituents. During the optimization the authors screened two other catalysts viz., Pd(CH3CN)2Cl2 and Pd(PhS(CH2)2SPh)Cl2, and even with 1 mol% loading these failed to achieve conversion, whereas in contrast, Maffioli and coworkers succeeded with 0.5 mol% for PdCl2. Since direct comparison is not available, we shall assume that this discrepancy in the observation of two groups is due to the different sources of Pd(II), PdCl2versus Pd(CH3CN)2Cl2.
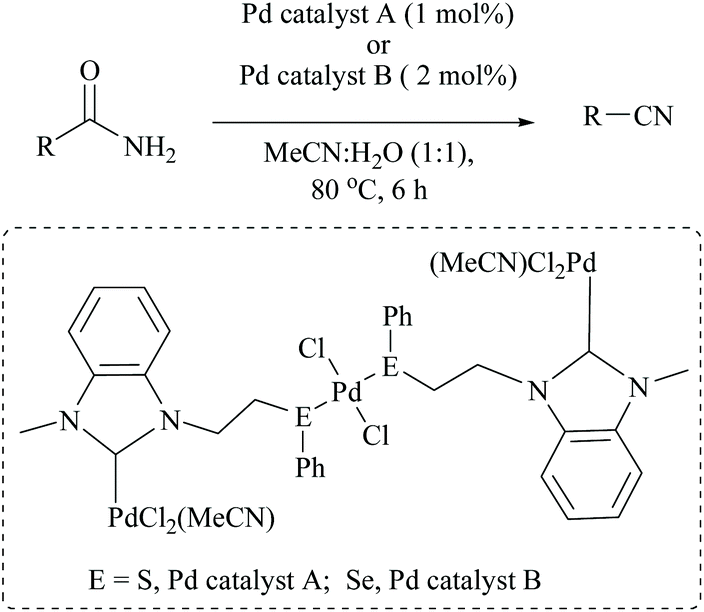 | ||
| Scheme 23 Palladium catalyst for dehydration. | ||
Croatt and coworkers, while carrying out their research on improving the biological activities of Alamethicin F50, a 20-mer peptaibol, by fluorination using Pd(OAc)2 and Selectfluor®. However, instead of fluorination, only dehydration of amide functionalities to form the corresponding tricyano product, along with semi-dehydrated analogues viz., mono- and di-cyano products, was detected (Scheme 24).70 This observation led them to explore further the application of this catalytic system to alkyl and aryl primary amides to obtain their corresponding nitriles in good yields. The role of the additive (20 mol% Selectfluor® in acetonitrile) was to act as an oxidant and to avoid the requirement of water for palladium catalysed dehydrations. The added Selectfluor® modified the Pd-catalytic pathway that generally uses either Pd(0) or Pd(II), by the formation of Pd(IV) which no longer requires the addition of water. Several non-, mono-, and disubstituted benzamides and aliphatic amides were converted to their corresponding nitriles in good yields. Notably, under these catalytic conditions, the cyclopropyl moieties were found to be intact.
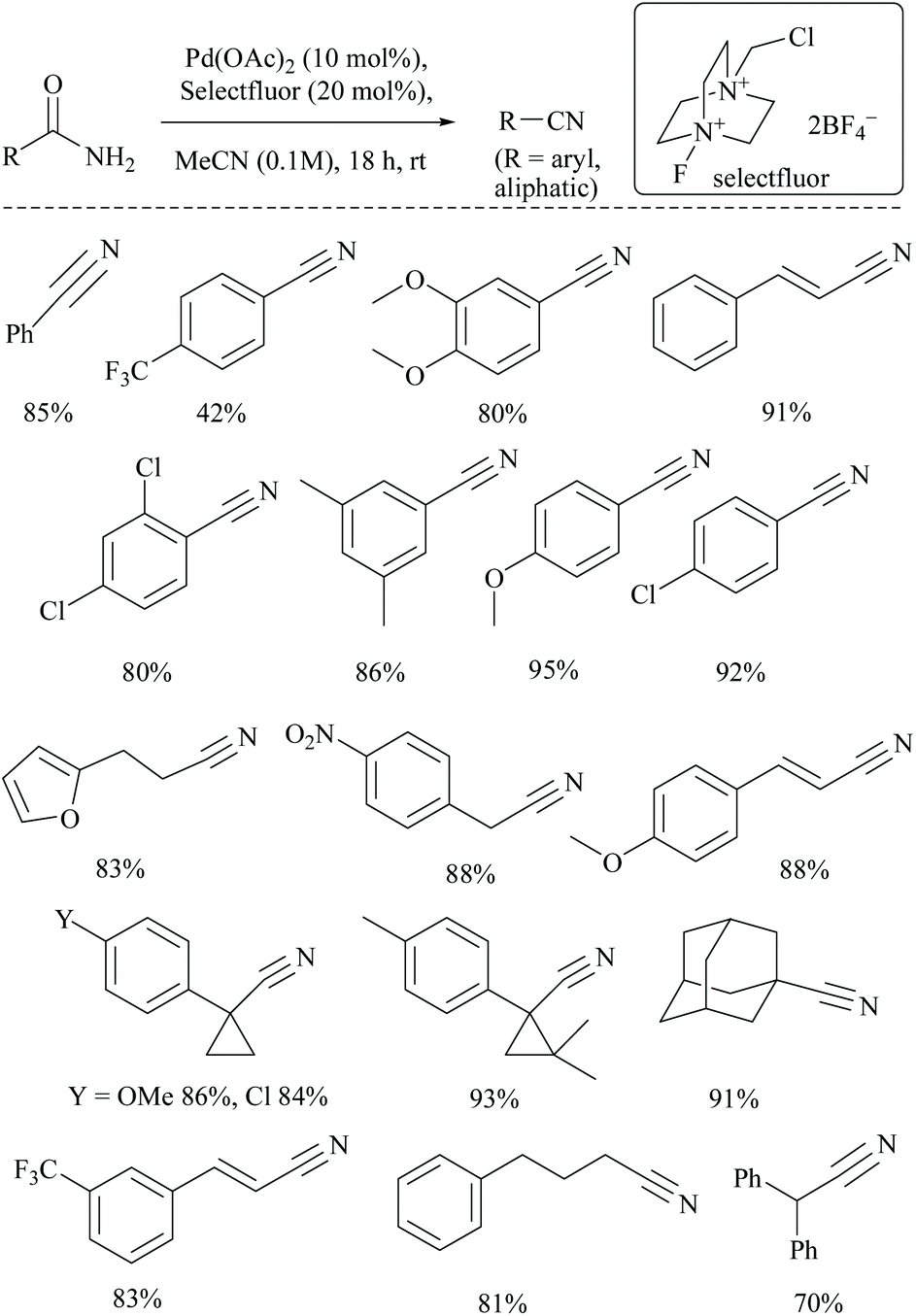 | ||
| Scheme 24 Palladium catalyst with Selectfluor®. | ||
Dai and coworkers reported two different palladium-catalyzed reaction conditions using non-aqueous acetonitrile for the conversion of α,β,γ,δ-unsaturated amides (Scheme 25) and aryl-alkyl amides (Scheme 26) into the corresponding nitriles.54 The general requirement of water for palladium-catalysed dehydration of amides was exempted by the addition of metal acetate, which takes different pathways to assist the palladium catalyst. However, metal acetate alone without the palladium catalyst did not result in the conversion. The optimized reaction conditions for the conversion of unsaturated amides were achieved with PdCl2, LiCl, and AgOAc in MeCN, whereas for the alkyl and aryl amides, the conditions with PdCl2, (PPh3)2, and Cu(OAc)2 in MeCN were used.
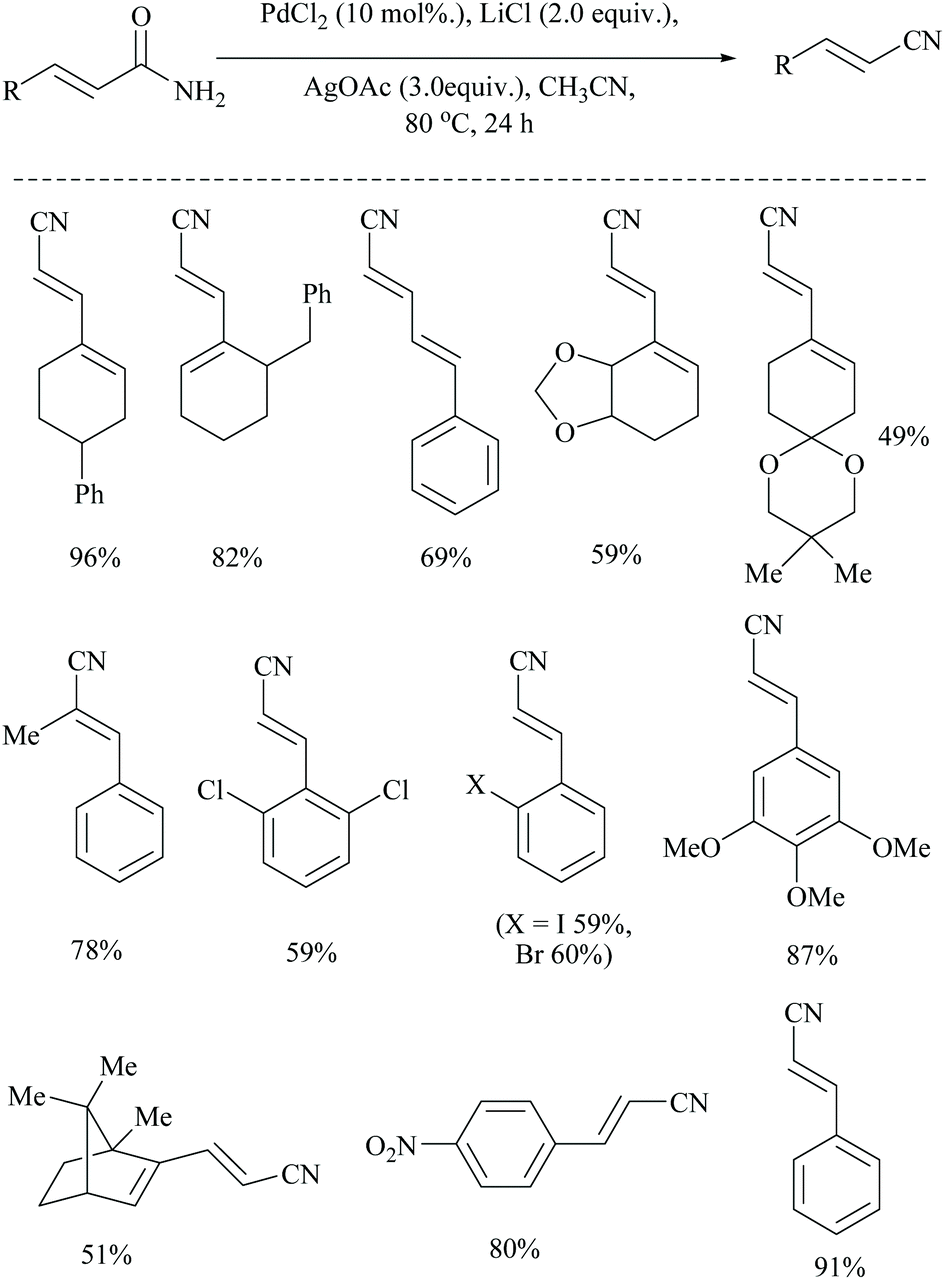 | ||
| Scheme 25 Palladium catalyst with Ag(OAc) for dehydration. | ||
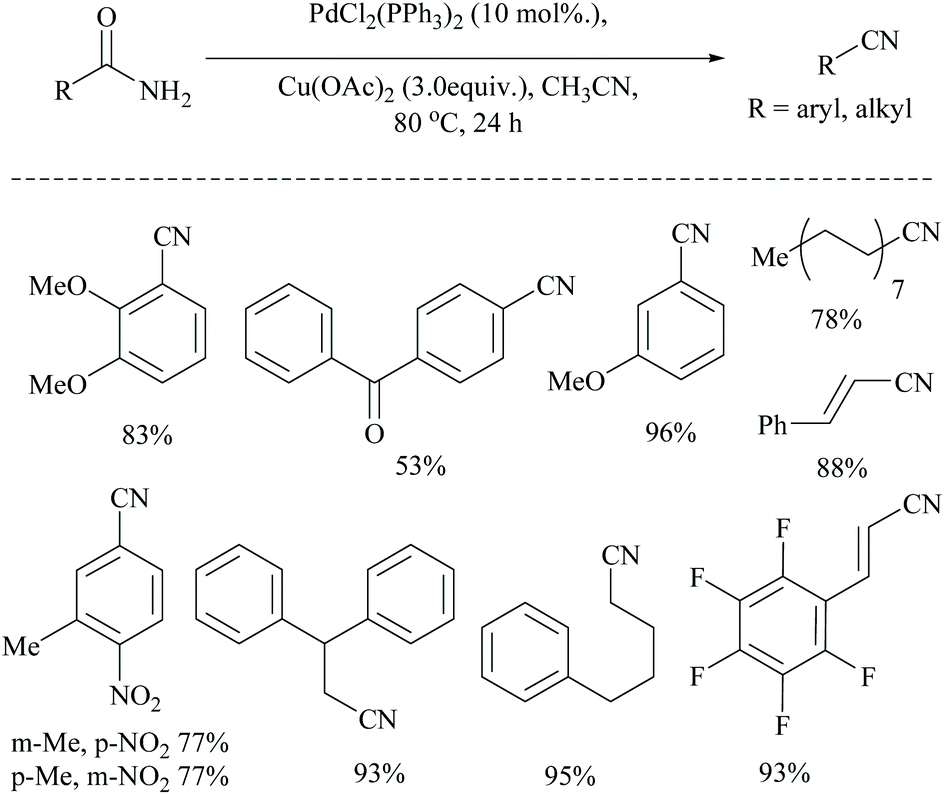 | ||
| Scheme 26 Palladium catalyst with Cu(OAc)2 for dehydration. | ||
Notably, both reaction conditions were found to be successful at moderately high temperatures (80 °C). In general, the role of AgOAc is to regenerate Pd(II) from Pd(0) in the catalytic cycle. However, in the present method, AgOAc and Cu(OAc)2 are merely the source of acetate counter anions which can be realized from the observation that the replacement of AgOAc with NaOAc did not affect the yield very much. But, the presence of acetate is highly required for the conversion, as it acts as a base throughout the mechanism. Being a dehydrating agent, when MeCN was replaced with DMF or toluene, it reduced the yield to less than 15%. Moreover, during the dehydration, MeCN is present as solvent in excess so as to drive the equilibrium towards the nitrile product formation by getting converted itself into acetamide.
A rapid method for the conversion of primary amides to nitriles and vice versa using ZnCl2 and a ZnCl2–H2O–THF system in the presence of aqueous acetonitrile and acetamide, respectively, under microwave irradiation was reported by Manjula and Pasha (Scheme 27).67 Zn(II) facilitates the transfer of water from the amide to acetonitrile and vice versa.
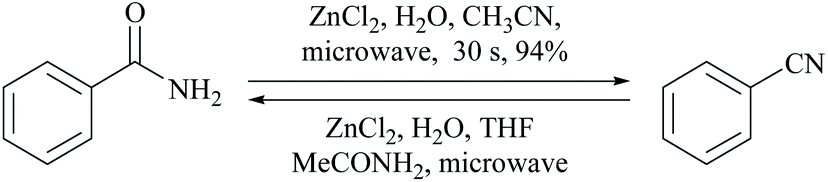 | ||
| Scheme 27 Zinc catalyst for dehydration. | ||
3.3. Catalyzed silylative dehydration – theoretical aspects
In silylative dehydrations of primary amides, transition metals get inserted into the Si–H bond and form hydrogen gas and silyl ether (Scheme 28). Indeed, silicon compounds have been used as silylating reagents, and they actively induce dehydration of the amides only in the presence of a catalyst.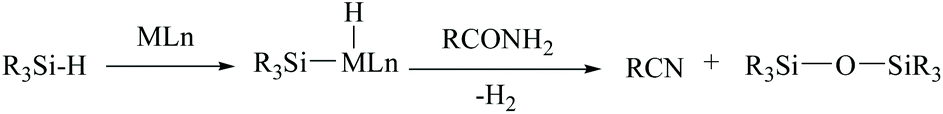 | ||
| Scheme 28 Silicon-based dehydration of amides. | ||
However, there have been recent reports that either the high energy barrier syn-elimination or low energy barrier β-elimination in the presence of catalysts was the responsible pathway for the silylative dehydration to occur. Before reviewing the literature available for the metal catalyzed silylative dehydration of carboxamide to form the corresponding nitrile, we believe that knowing the mechanism of amide-dehydration will be very useful in designing the reaction conditions. In addition, Density Functional Theory (DFT) studies are found to be very helpful in determining the structure–reactivity relationships for the catalytic dehydration of primary amides using silanes. Mandal and coworkers72 reported the DFT calculations for amide to nitrile conversion using dehydrating agents such as phenyl silane. They compared the energy profile of the catalyzed reaction with that of the non-catalyzed one by taking p-methoxy benzamide as an example. Uncatalyzed silylative dehydration undergoes N-silylation, N- to O-silyl migration, a second N-silylation to form N,O-disilylimidate, and finally elimination of silylether to form the nitrile (Scheme 29). Each silylation is followed by dehydrogenation to give relatively low energy silylated imidate intermediates. Notably, both the silylation and elimination of silylether were accompanied by large energy barriers (Scheme 29). Hence, the addition of a catalyst can greatly help in the silylative dehydration process. For example, aNHC catalyzed mono-silylation and N- to O-silyl migration have energy barriers of 27.0 (T1, Scheme 32) and 15.7 kcal mol−1 (calculated), respectively, which were found to be lower than those for uncatalyzed conditions. Buchwald's group58 also obtained similar results using a copper catalyst that N- to O-silyl migration needs only 13.5 kcal mol−1; hence O-silyl and N-silyl imidate forms equilibrate rapidly at room temperature (Scheme 30).
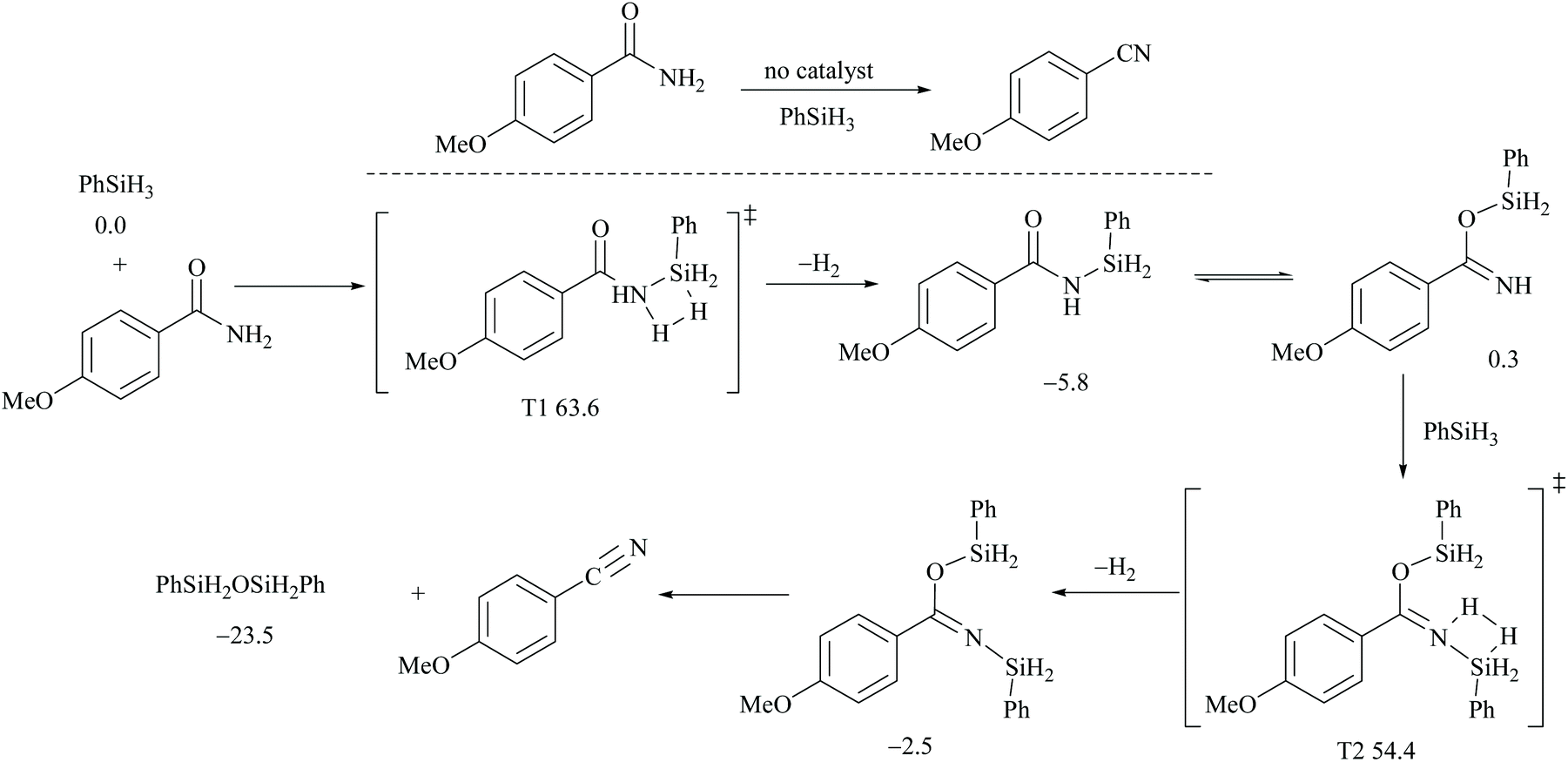 | ||
| Scheme 29 DFT calculations and Gibbs free energy ΔG (kcal mol−1): uncatalyzed dehydration. | ||
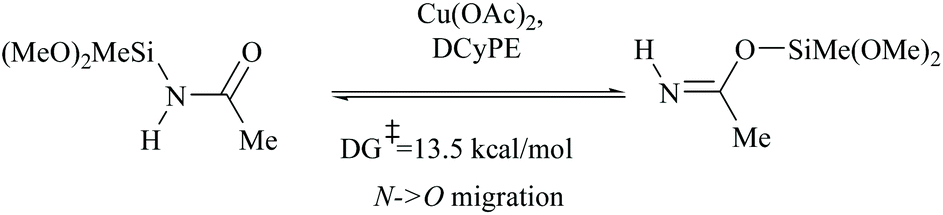 | ||
| Scheme 30 DFT calculations: Cu catalysed interconversion of N-silyl and O-silyl imidate forms. | ||
The catalyzed silylative dehydration follows either syn(1,2)-elimination or β-elimination. The syn-elimination pathway involves mono-N-silylation, N- to O-silyl migration, a second N-silylation to form N,O-disilylimidate, and finally syn-elimination of silylether to form the nitrile with the regeneration of the catalyst (Scheme 32). In the case of the β-elimination pathway, differently, the amide substrate underwent mono-N-silylation, N- to O-silyl migration, and finally β-elimination of the catalyst along with silyl oxide as an ion-pair to form the nitrile. In order to regenerate the catalyst, a second silylation is required to dissociate the ion-pair into silyl ether (Scheme 32). It is interesting to note that in the catalyzed dehydrations, the addition of a second mole of silane is entirely utilized for regenerating the catalyst and eliminating the silylether. Thus, the role of a catalyst can be observed until the second silylation which substitutes the catalyst.
DFT calculations for the copper catalyzed elimination process in disilyl imidate derived from the reaction of acetamide with DMMS showed that the barrier for syn-elimination was too high to be surmountable at ambient temperature (+37.0 kcal mol−1, Scheme 31). Also, the formation of N,O-disilylimidate itself requires a high energy barrier to be surmounted (37.7 kcal mol−1), making syn-elimination energetically least viable and therefore not feasible.
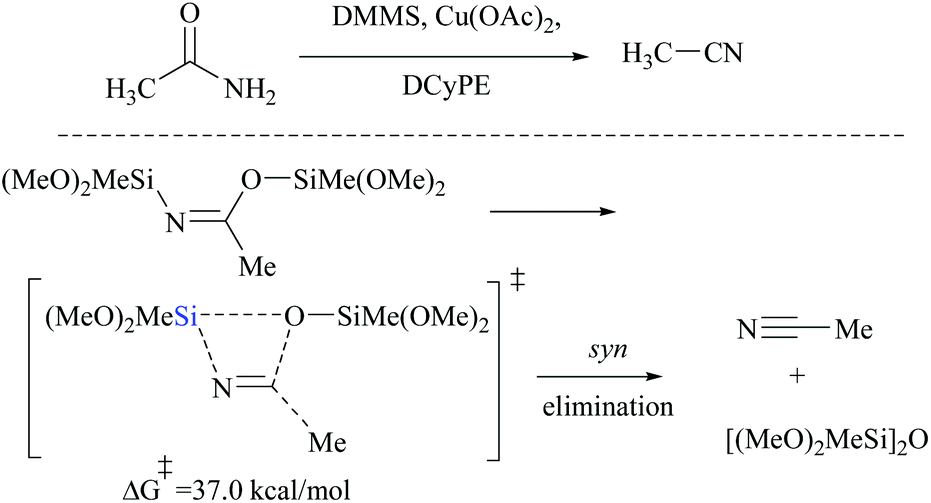 | ||
| Scheme 31 DFT calculations: Cu-catalyzed syn-elimination. | ||
Appreciably, for the β-elimination, aNHC and copper/DCyPE catalysts have low energy barriers of 6.6 and 4.8 kcal mol−1, respectively (Schemes 32 and 33). In copper catalysed dehydration, β-elimination of the silyl group was facilitated by reduced strain in the four-membered transition state (Scheme 33). This can be explained with the help of the difference in bond distances in their respective transition states. The β-elimination has a transition state with bond lengths, viz. Cu–O and Cu–N bond lengths (251 and 199 pm), longer than those of the syn-elimination transition state (Si–O, Si–N: 187, 200 pm). Due to the larger size of Cu compared to Si, the reduced strain and the thermodynamically favourable –CN bond make the β-elimination comparatively easy and rapid.
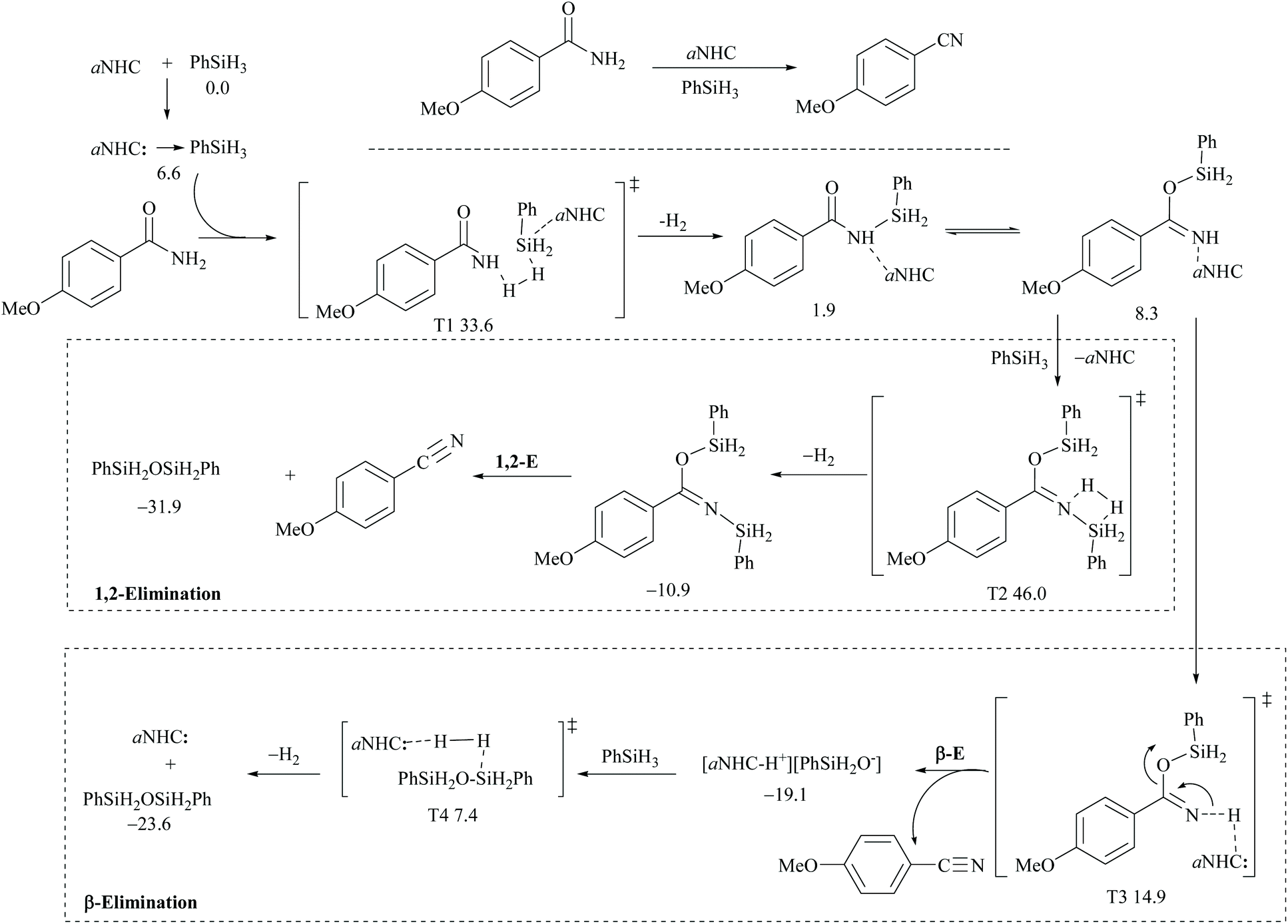 | ||
| Scheme 32 DFT calculations and Gibbs free energy ΔG (kcal mol−1): aNHC catalyzed 1,2-elimination and β-elimination. | ||
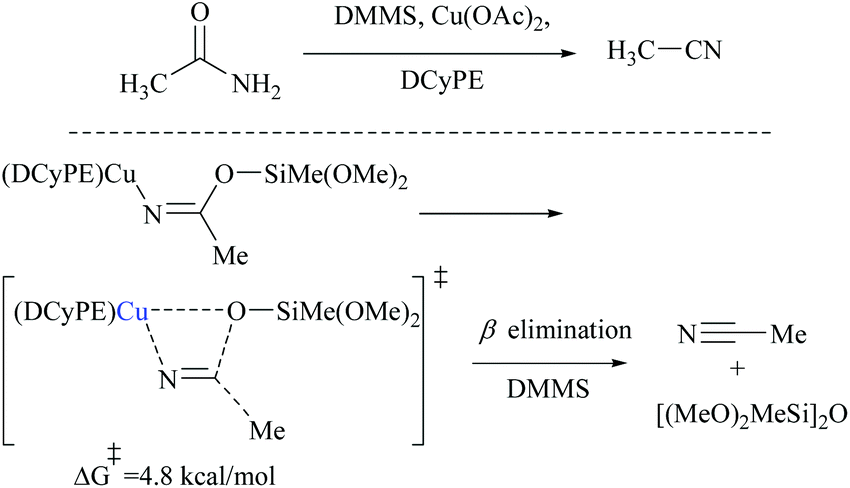 | ||
| Scheme 33 DFT calculations: Cu-catalyzed β-elimination. | ||
3.4. Metal catalysed silylative dehydration
In 2008, Nagashima and coworkers reported the first silicon-based dehydration of primary amides using a triruthenium carbonyl cluster (Scheme 34).56 They showed that the yield of nitriles depended on the type of silane mediating the dehydrations and that poly(methylhydroxysiloxane) (PMHS), triethyl and dimethoxymethyl silane were obtained in poor yields (Scheme 34). | ||
| Scheme 34 Ruthenium catalyzed silylative dehydration. | ||
The highest yield was observed with a bifunctional organosilane, 1,2-bis(dimethylsilyl)ethane, in the presence of 2.5 mol% of triruthenium catalyst. The reaction conditions were found to tolerate several functional groups including an alkene but not α,β-unsaturated amides. The authors also deduced the mechanism for the ruthenium catalyzed dehydration that the initially formed N,N-bis(trimethylsilyl)amide, at high temperatures, underwent N- to O-silyl migration followed by syn-elimination of silyl groups as its ether to give the corresponding nitrile and to regenerate the catalyst.
Silylative dehydrations of amides to nitriles catalyzed by iron hydrides have also been reported. For example, Beller and coworkers reported the first iron-catalyzed dehydration of primary amides to nitriles using silanes as the dehydrating agents (Scheme 35).56 In the presence of silane, (EtO)2MeSiH, the dehydration did not proceed without a catalyst and also even with 5 mol% of Fe(CO)5, FeCl2 or FeCl3. However, on the addition of various iron carbonyl clusters [CpFe(CO)2]2, Fe3(CO)12 and [Et3NH] [HFe3(CO)11], Fe(acac)3 and Fe2(CO)9, a good conversion of amides to nitriles (83–97%) was observed. The optimization of the reaction for dehydration of benzamide resulted in conditions with the iron catalyst [Et3NH] [HFe3(CO)11] which gave a better result (90%) in a short time (3 h). Also the silane, (EtO)2MeSiH, along with toluene solvent showed better conversion in comparison with other silanes such as PMHS, PhSiH3, Ph2SiH2, Et3SiH, TMSOSiMeH, and (EtO)3SiH as dehydrating agents. The result that PMHS gave poor yield was similar to that of Nagashima's method56 reported earlier. Notably, the dehydration of cinnamamide using four equivalents of silane in 1,4-dioxane as solvent gave the corresponding cinnamonitrile along with a reduced product 3-phenylpropionitrile in a 1:2 ratio. Also, no dehydration was observed with nicotinamide under these reaction conditions. They proposed a mechanism for the iron-catalyzed dehydration and found it to be similar to that reported earlier for the ruthenium catalyst (trirutheniumcarbonyl cluster, (ACE)Ru3(CO)7) by Nagashima and co-workers that involved metal catalysed N-silylation followed by its elimination as silyl ether.
 | ||
| Scheme 35 Iron-hydride catalyzed silylative dehydration. | ||
Li and coworkers reported an air-stable N-heterocyclic PSiP pincer iron hydride and an analogous nitrogen iron hydride for the dehydration of primary amides to nitriles (Scheme 36).73 They optimized the reaction conditions using three hydride catalysts and found that iron hydride III was better than other hydrides in terms of its air-stability and amide to nitrile conversion. Among different silanes, viz. (EtO)2SiMeH, Ph3SiH, Et3SiH, Ph2SiH2 and (EtO)3SiH that were tested, Ph2SiH2 was found to be a promising dehydrating agent due to its excellent conversion and lower toxicity. The proposed mechanism for the conversion of amides to nitriles involved the iron-hydride complex assisted O-silylation followed by its elimination in ether form. They noted that the presence of an electron withdrawing group on the phenyl ring had raised the yields of the nitrile product. Moreover, the catalytic system did not affect the double bonds in acrylonitrile and cinnamamide. The dehydration of acetoacetamide, however, yielded 3-hydroxybutyramide, and the carbonyl group of the acetyl group was selectively reduced but not that of the amide group. The authors also reported the use of silyl iron hydride bearing [PSiP]-pincer ligands as catalysts along with a Lewis acid as the promoter for dehydration of primary amides to nitriles (Scheme 37).73 Lewis acids accompanied by higher temperature, longer reaction time, and less yield have already been utilized for the dehydration of amides to nitriles.52,67,74,75 Thus, the authors wanted to study the influence of Lewis acids on the metal-hydride catalytic activity. The significance of a Lewis acid as an additive in the iron-hydride catalytic reaction was extensively studied and it was found that ZnBr2 was the best under these catalytic conditions. However, the Lewis acid ZnBr2 alone without the catalyst gave very poor yield. The same group has also achieved the reductive dehydration of amides to nitriles in good yields at lower temperature (60 °C) using hydrido thiophenolato iron(II) complexes [cis-Fe(H)(SAr)(PMe3)4] as catalysts with (EtO)3SiH as the dehydrating agent (Scheme 38).56 Though the reaction conditions with low catalyst loading (5 mol%) can tolerate aliphatic, acrylic and aromatic amides possessing nitro, halo and methoxy substituents, the keto-group of the amide was found be reduced much faster than dehydration.
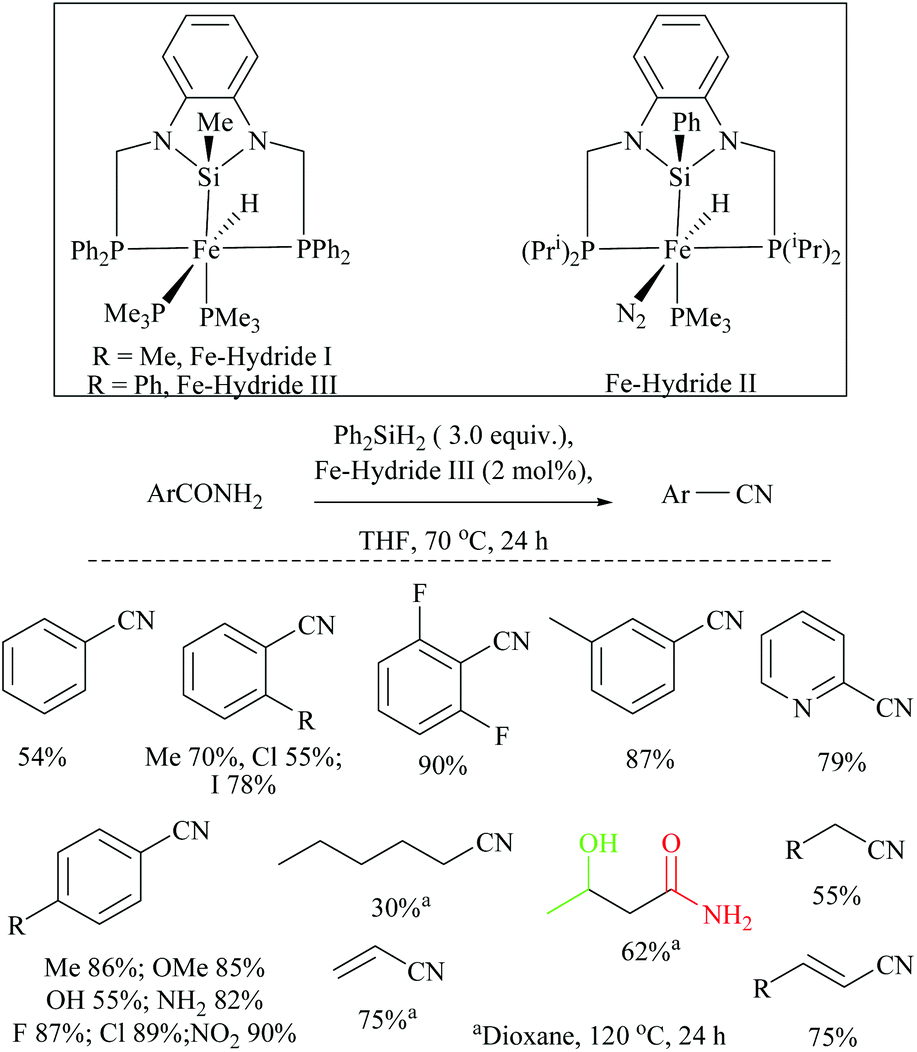 | ||
| Scheme 36 Iron-hydride catalyzed silylative dehydration. | ||
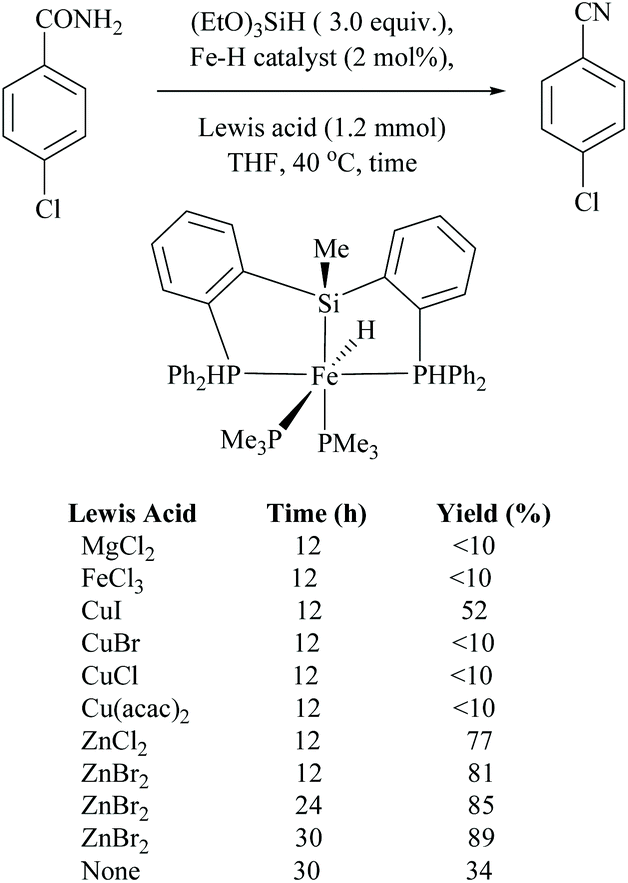 | ||
| Scheme 37 Iron-hydride catalyzed silylative dehydration. | ||
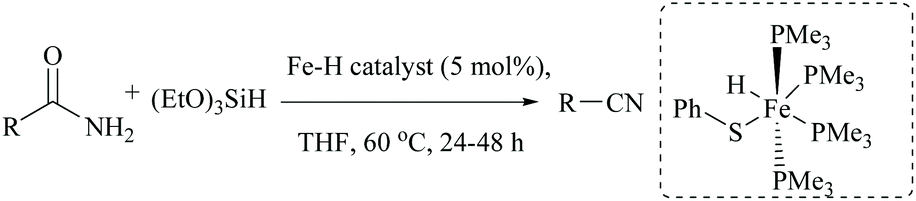 | ||
| Scheme 38 Iron-hydride catalyzed silylative dehydration. | ||
Similarly, the same group reported the applications of phosphorus-chalcogen chelated hydrido iron(II) complexes [(o-(R′2P)-p-R-C6H4Y)FeH(PMe3)3, Y = O, S, Se] and also cis-selenophenolato iron hydrides (cis-[(ArSe)FeH(PMe3)4]) as catalysts and (EtO)3SiH as a reducing agent for the dehydration of primary amides to nitriles.76 In 2011, Lemaire and coworkers reported the use of the inexpensive polymethylhydrosiloxane (PMHS) as a reducing agent for the conversion of primary amides to the corresponding amines along with nitriles as side products.77
Iron based N-heterocyclic carbene complexes as homogeneous catalysts have been used in many transformations including silylative dehydrations.78 Basic NHC with or without transition metal has also been used for silylative dehydrations.56,60,72 For example, when Darcel and coworkers56 carried out the dehydration at high temperatures (100 °C) using a light-activated iron Knolker-type complex bearing an NHC ligand, nitriles were formed in moderate yields (Scheme 39). However, like Beller's observation (Scheme 35),56 when they changed the catalytic system to [Et3NH] [HFe3(CO)11] and (EtO)2MeSiH, cinnamamide gave cinnamyl nitrile (65% conversion, 61% isolated yield) under these conditions, along with reduced product 3-phenylpropionitrile in very small amounts (∼2%). The group has also published the iron-catalyzed dehydration conditions but with different pairs of catalysts and silanes, viz. a cyclopentadienyl NHC-iron complex and phenylsilane (Scheme 40).56 Primary amides were smoothly dehydrated into nitriles in quantitative yields; however, under this system, secondary and tertiary amides were found to be reduced to the secondary and tertiary amines, respectively.
 | ||
| Scheme 39 Iron Knolker-type complex catalyzed silylative dehydration. | ||
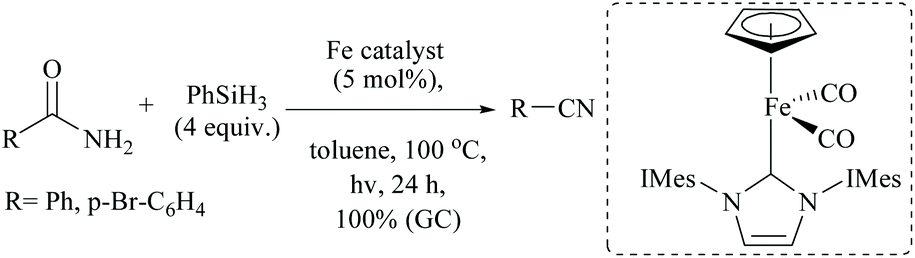 | ||
| Scheme 40 NHC-iron complex catalyzed silylative dehydration. | ||
In 2019, Li and coworkers, similar to their hydrido iron complexes along with (EtO)3SiH as the reducing agent,56,79 reported for the first time, the hydrido cobalt(III) catalyst to dehydrate aryl and aliphatic primary amides to the corresponding nitriles in 67–92% yields using (EtO)3SiH as the dehydrating agent at 60 °C (Scheme 41).60 The optimization of the reaction resulted in findings that as the catalyst [CNC]-pincer hydrido cobalt(III) had decomposed at very high temperature, the conversion of the amide to nitrile was found to be reduced. They also found that THF was the best solvent system over toluene, DMF, DMSO, dioxane and acetonitrile. Compared to other silanes such as PhSiH3, Ph3SiH, Et3SiH, TMDS, and PMHS, the highly flammable and toxic silane (EtO)3SiH as a reducing agent was found to give good conversion and was selective over double bonds. The electron withdrawing groups afforded nitriles in good yields; however electron donating groups showed moderate results.
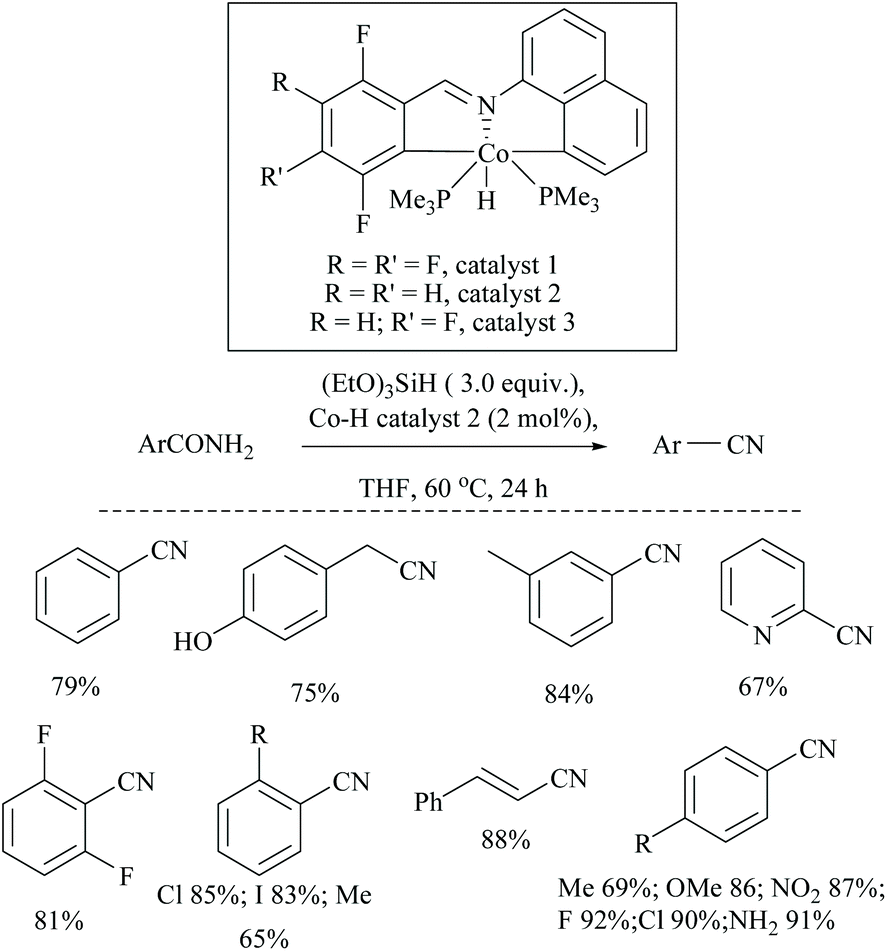 | ||
| Scheme 41 Cobalt-hydride catalyzed silylative dehydration. | ||
Mandal and coworkers reported the reduction of amides by a manganese catalyst with dual activity in transforming a primary amide into an amine or nitrile depending on the reaction conditions such as in the presence of a catalytic amount of a secondary amide (Scheme 42).80 They optimized the reaction conditions with KOtBu as a base, PhSiH3 as a silylating agent, N-methyl-benzamide as an inhibitor, and a Mn catalyst for the conversion of various aryl, hetero-aryl and aliphatic primary amides to the corresponding nitriles in good yields (72–94%). Interestingly, the catalyst exhibited excellent selectivity and only the primary amide was selectively reduced in the presence of a secondary amide. In the substrate 4-cyanobenzamide, only the amide group was selectively reduced into the nitrile, sparing the existing nitrile. The mechanism that they proposed was a non-radical, metal alkoxide transformation into a metal-hydride complex by σ-bond metathesis with the silane, followed by the formation of a primary amide coordinated complex with a vacant Mn(III) site. The activated amide can be converted into nitrile via N-silylation by tert-butoxide activated silane with the evolution of hydrogen gas followed by the elimination of the silyl group as its ether and regeneration of the catalyst. The same reaction conditions in the absence of PhCONHMe reduced the amide to the corresponding amine. Thus the single catalyst was either able to completely reduce the amide into amine or could give nitrile in the presence of the inhibitor PhCONHMe (Scheme 42).
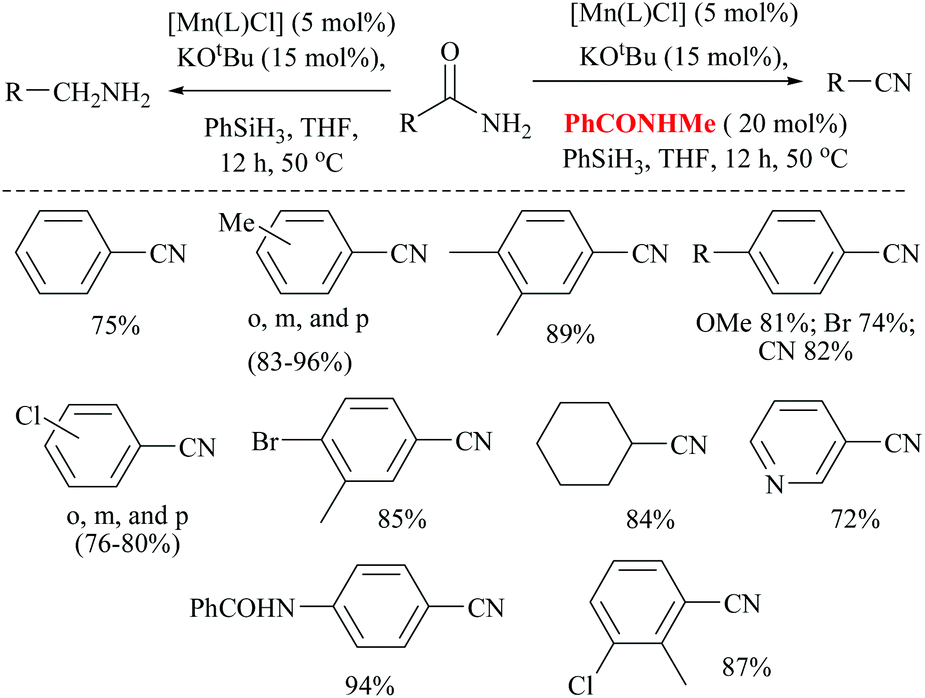 | ||
| Scheme 42 Manganese with PhCONHMe for dehydration. | ||
Enthaler's research group has extensively worked on catalyzed silylative dehydrations of amides by elegantly overcoming the problems associated with the selection of silanes and catalysts.58,60 Instead of simple silanes, an excellent silylating reagent MSTFA, in the presence of various metal salts, iron(II) chloride, copper(I) chloride, zinc(II) triflate, and uranyl nitrate, was utilized for the dehydration of amides.58 The addition of MSTFA meticulously ensured the safe handling of the reaction and elicited the scope of silylative dehydration by avoiding the release of hydrogen gas, but still followed through the 1,2-elimination of silyl groups. With copper(I) chloride as a metal catalyst in toluene, primary amides were dehydrated into their corresponding nitriles using MSTFA as a silylating reagent (Scheme 43).58 The mechanism involved first the copper metal coordination with oxygen, and then silylation of nitrogen followed by N- to O-silyl migration yielded nitrile upon elimination of silylether as the by-product (Scheme 44).
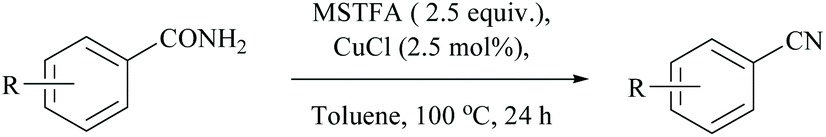 | ||
| Scheme 43 Copper catalyst for dehydration. | ||
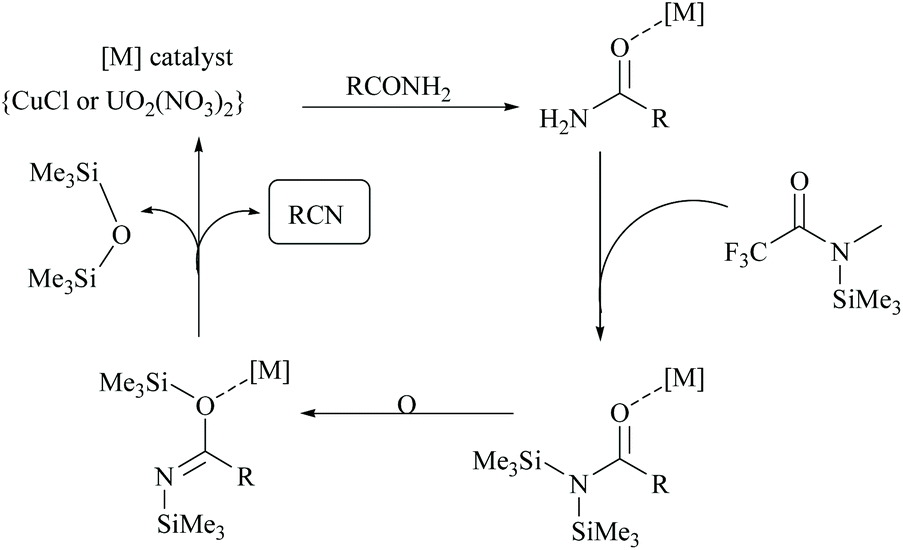 | ||
| Scheme 44 Mechanism of Cu and U-catalyzed dehydrations. | ||
The same group has also reported uranium catalyzed (Scheme 45) and zinc catalyzed dehydrations of primary amides using MSTFA as the silylating reagent.52 They found that the catalyst alone did not give the product without the help of MSTFA; however on increasing its concentration, the yield of the nitriles increased. The temperature of the reaction conditions below 70 °C did not give appreciable yields of the product and at least 100 °C was required (Scheme 45). Also, MSTFA in THF as a solvent with N-heterocyclic compounds did not give appreciable yields. The silylating agent MSTFA and hence the mechanism were expected to work well with other metals too.
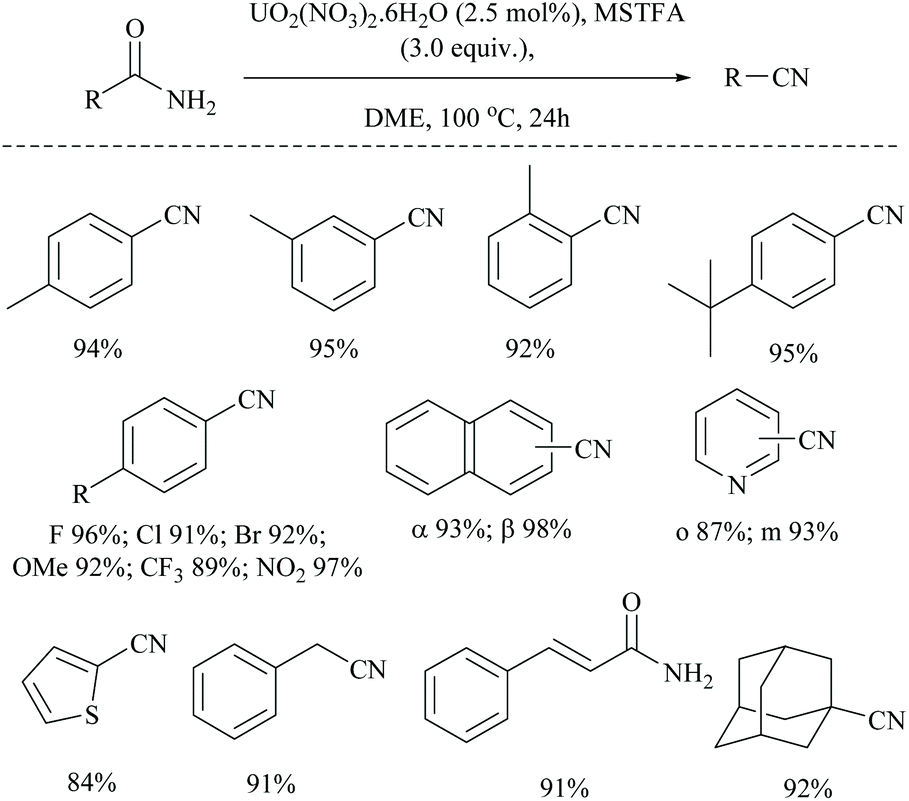 | ||
| Scheme 45 Uranium catalyzed silylative dehydration. | ||
In 2014, Mineno and coworkers reported an efficient MSTFA dehydration method using indium(II) triflate (5 mol%) (Scheme 46).59 The reaction with 3.5 equivalents of MSTFA in toluene under refluxing conditions was found to tolerate mono- and di-substituted aryl compounds, α,β-unsaturated amides and aliphatic amides. O-Silyl protected groups, viz. TBDMS and TBDPS groups, and also double bonds in oleamide were found to be resistant to the reaction conditions.
 | ||
| Scheme 46 Indium catalyzed silylative dehydration. | ||
Buchwald and coworkers reported a copper hydride (CuH)-catalyzed silylative dehydration of primary amides to nitriles using a DCyPE ligand (Scheme 47).58 Notably, in contrast to other silylative dehydrations, under these conditions, the reaction was carried out at room temperature with 3.0 equivalents of dimethoxymethylsilane (DMMS) as a dehydrating agent. Dehydrations were performed on a variety of amide structures viz., α-primary, secondary, and tertiary amides, aromatic amides with electron-withdrawing and -donating groups, and heterocyclic amides, including a thiophene, pyridine, pyrrolidine, and piperidine to give their corresponding nitriles in good yields. The optimization of the dehydration resulted in a copper(II) salt, a chelating diphosphine ligand (DCyPE), and monomeric DMMS. The addition of ligands was found to be necessary as in the absence of any ligand, the net dehydration reaction did not yield the nitrile product, even though it could catalyze the dehydrogenative silylation of acidic groups (Scheme 47). In addition, they chose four amide-substrates of different skeletons and subjected them to dehydration using six different classical and modern methods to obtain their corresponding nitriles using each of the methods described in Table 2. These six methods were followed as per the literature reported already and were compared with the one that the authors developed (copper-catalyst, DCyPE-ligand and DMMS – silylating agent). The yields of the corresponding nitrile products are shown in the following table. The results showed that the method developed by the authors performed better than other methods in terms of yield. In order to deduce a plausible mechanism, the authors have performed a computational study for the dehydration reaction of acetamide using DMMS as the silylating agent and copper(II) acetate as the catalyst (Schemes 30, 31, and 33). The DFT-optimized transition state structures followed the dehydration through β-elimination of monosilyl imidate in the presence of copper(II) acetate with a low energy barrier (+4.8 kcal mol−1) using a DCyPE supporting ligand and resulted in the formation of the nitrile (Scheme 33). They also proved that the dehydration did not follow a high energy barrier (+35 kcal mol−1) syn-elimination of N,O-disilyl imidate by carrying out the dehydration using the same reaction conditions but with a large excess (5 equiv.) of the silylating agent. The additional silanes under these reaction conditions resulted in only a trace amount of the nitrile product, which means that disilyl imidate did not lead to the formation of the nitrile product anyway.
 | ||
| Scheme 47 Copper catalyzed silylative dehydration. | ||
| Method |
|
|
|
|
|---|---|---|---|---|
| a Method 1: Cu(OAc)2, DCyPE, DMMS, temp, 12 h; method 2: POCl3, Et3N, rt, 12 h; method 3: propylphosphonic anhydride (T3P), toluene, 100 °C, 12 h; method 4: diiron nonacarbonyl, toluene, rt, 12 h; method 5: diiron nonacarbonyl, toluene, 100 °C, 12 h; method 6: phenylsilane, TBAF, rt, 30 min; method 7: phenylsilane, TBAF, 100 °C, 30 min. | ||||
| Method 1 | 97% | 85% | 80% | 93% |
| Method 2 | 98% | 27% | 38% | 42% |
| Method 3 | 86% | 48% | 13% | 68% |
| Method 4 | <5% | <5% | 24% | <5% |
| Method 5 | 85% | 23% | 75% | 49% |
| Method 6 | 7% | <5% | 16% | 5% |
| Method 7 | 87% | 8% | 62% | 47% |

4. Non-transition metal catalyzed dehydration
Wang and coworkers searched for other catalytic dehydration methods without any additives such as a dehydrating agent and an activator, which led them to the use of silica supported potassium oxide acting as a heterogeneous catalyst.81 The K2O/SiO2 catalyst on calcination at 300 °C for 3 h afforded 2-CP in 23.6% yield.81 Huang and coworkers reported selective reduction of primary amides to nitriles with hydrosilane or hydrosiloxane using a catalytic system triethylborane in combination with an alkali metal base (Scheme 48).74 They also studied the mechanism of BEt3/base catalyzed amide hydrosilylation reaction and found that a modified Piers’ silane Si–H⋯B activation mode facilitated the hydride abstraction by BEt3 by the coordination of the Si-center with a hydroxide or an alkoxide. The reaction proceeded with the hydrosilylation reaction of primary amides using either PhSiH3 at rt, or polymethylhydrosiloxane (PMHS) at 50 °C followed by reduction catalysed by BEt3/base. The role of the base was also examined and it was found that the base enhanced the catalytic activity and facilitated the hydride transfer from Si to B through modified Piers’ mode. | ||
| Scheme 48 BEt3/base reagent for dehydration. | ||
In 1971, the Appel reaction was applied for the synthesis of nitriles from amides in which the harmful CCl4 and stoichiometric Ph3P were utilized along with triethylamine.82 Shipilovskikh et al., in 2018, developed a catalytic Appel-type dehydration of amides to nitriles by modifying the original Appel reaction in which the combination of Ph3P, CCl4, and Et3N was replaced with a new system consisting of Ph3PO, oxalyl chloride, and Et3N (Scheme 49).82 The mechanisms of both the systems follow the same pathway through the initial formation of a common intermediate chlorophosphonium chloride.
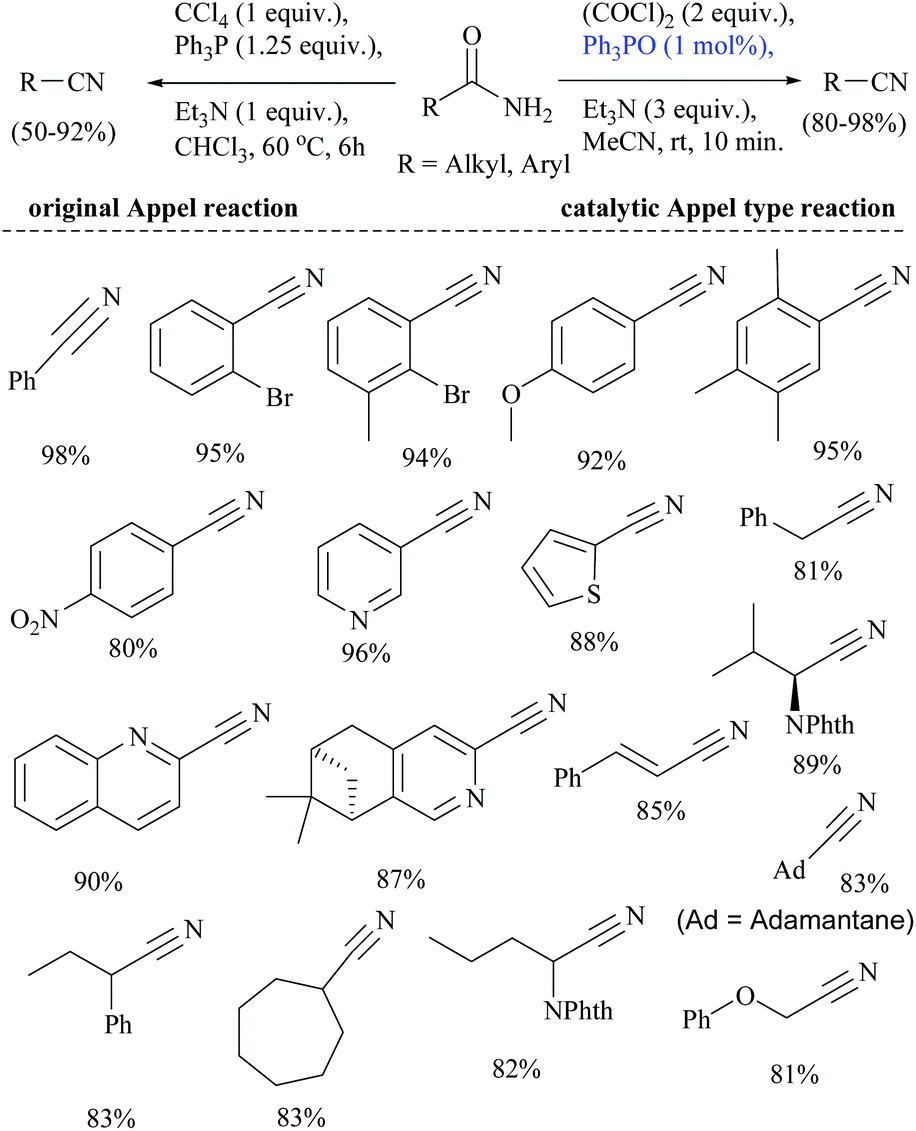 | ||
| Scheme 49 Catalytic Appel type reaction. | ||
Optimization of the reaction conditions resulted in finding that the oxalyl chloride alone and the presence of Et3N did not give the nitrile; however on the addition of Ph3PO as a catalyst, the trio (oxalyl chloride, Ph3PO and Et3N) produced the nitrile product in the presence of MeCN solvent. Under these new catalyzed Appel type reaction conditions, aromatic, heteroaromatic, and aliphatic amides, including derivatives of α-hydroxy and α-amino acids, were converted to the corresponding nitriles. However, the exception was N-Boc protected amino acids which did not yield the nitrile, due to their reaction with oxalyl chloride. Beller et al. reported the fluoride-catalyzed dehydration of many aromatic and aliphatic amides to nitriles in good yields, using phenylsilane as the a dehydrating agent in the presence of a catalytic amount of TBAF (Scheme 50).55
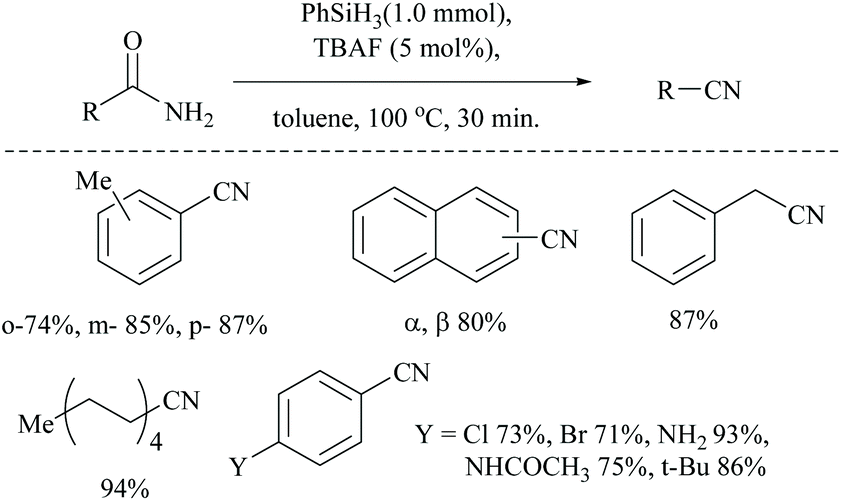 | ||
| Scheme 50 TBAF catalyzed silylative dehydration. | ||
5. Organocatalytic dehydration of amides
Mandal and coworkers reported N-heterocyclic carbene (NHC) catalyzed silylative dehydration of primary amides (Scheme 51).72 They found that this NHC catalyzed reaction bypasses the energy need for the usual 1,2-siloxane (syn)-elimination step in metal/silane coupled dehydrations of amides to nitriles. The dehydration followed low energy barrier β-elimination to give nitriles. Optimization of the dehydration has involved the use of various silanes (phenyl silane, DMMS, PMHS, HMTS, and butylsilane) along with different catalysts viz., NHCs, PPh3, Et3N, pyridine, etc. Under these conditions (phenylsilane and aNHC catalyst), various primary amides were converted into the corresponding nitriles.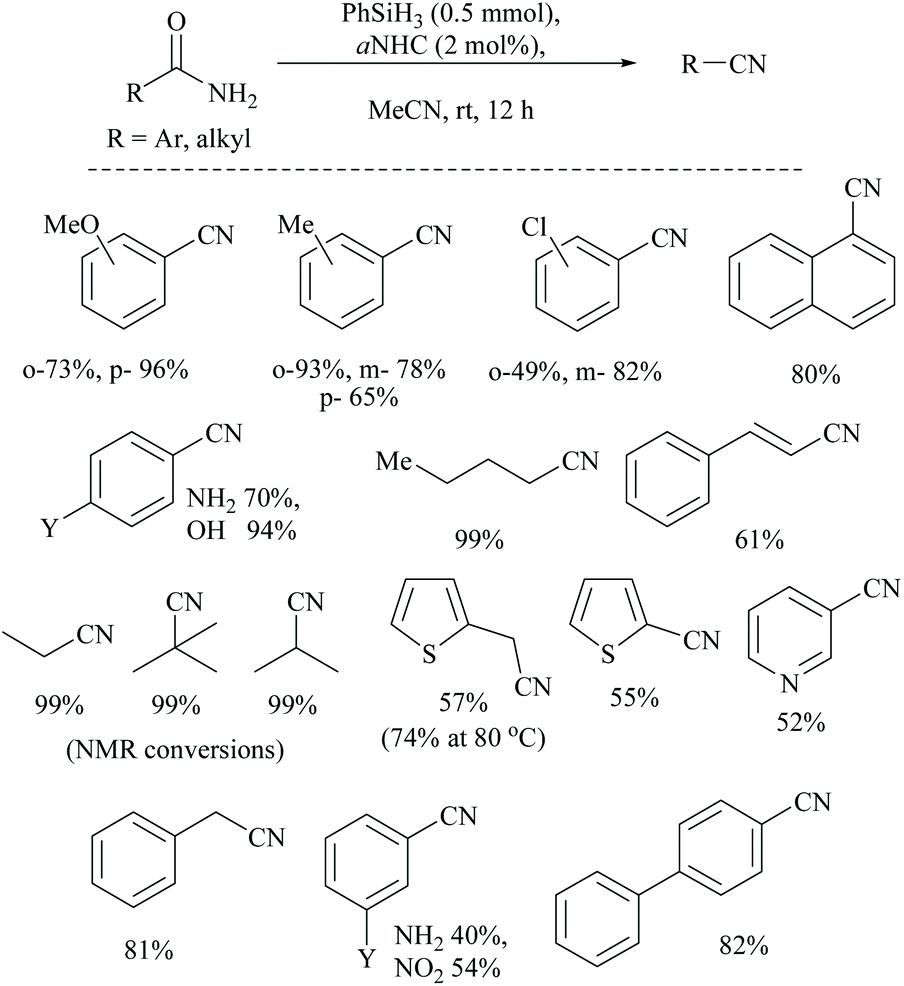 | ||
| Scheme 51 NHC catalyzed silylative dehydration. | ||
Alternatively, without silylation, simply using an oxalyl chloride/DBU base combination in the presence of cyclopropenone as an organo-catalyst has also been found to be successful for the conversion of the amide group into its corresponding nitrile. Yadav and coworkers reported the conversion of aldoximes and primary amides of aromatic, heterocyclic, and aliphatic compounds into nitriles by employing cyclopropenone as an organo-catalyst (Scheme 52).83 In the conversion of the amide to nitrile, oxalyl chloride along with the base DBU is used as an activating agent for the cyclopropenone to form the catalytically active species cyclopropenium cations (Scheme 52). Then, the cyclopropenium cation undergoes nucleophilic reaction with the amide to give the nitrile and regenerating the cyclopropenone catalyst. Under these reaction conditions, they found that the absence of either the cyclopropenone catalyst or the base DBU did not lead to the conversion even in the presence of oxalyl chloride.
 | ||
| Scheme 52 Organocatalytic dehydration. | ||
6. Organo-photocatalytic dehydration of amides
Photocatalyzed dehydration of an amide is a sustainable approach as it uses a clean light source in the presence of an organo catalyst. In this context, Yadav and coworkers reported a photosensitization based approach for the dehydration of primary amides into the corresponding nitriles using eosin-Y as an organophotoredox catalyst (Scheme 53).84 The plausible mechanism proposed for this conversion involved light absorption by eosin-Y to reach the triplet excited state which transfers a single electron to CBr4 to give a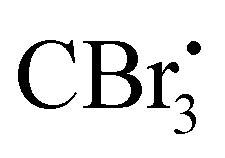 radical. Upon reaction with DMF,
radical. Upon reaction with DMF, 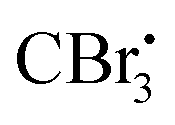 forms the radical species which in turn loses a single electron to the oxidatively quenched species EY+˙ and forms the corresponding iminium ion completing the redox catalytic cycle. Then, the iminium ion loses COBr2 to form a Vilsmeier–Haack reagent which reacts with the added amide in its imidol form to give the corresponding nitrile. A similar method has also been applied for the dehydrosulfurization of thioamides.85
forms the radical species which in turn loses a single electron to the oxidatively quenched species EY+˙ and forms the corresponding iminium ion completing the redox catalytic cycle. Then, the iminium ion loses COBr2 to form a Vilsmeier–Haack reagent which reacts with the added amide in its imidol form to give the corresponding nitrile. A similar method has also been applied for the dehydrosulfurization of thioamides.85
 | ||
| Scheme 53 Organo-photocatalytic dehydration. | ||
7. Conclusions
Nitriles are important precursors or fine products in academia and in industries. Therefore, an efficient and green synthesis of nitriles has always been one of the major topics of interest for organic synthetic chemists. Dehydration of amides, an efficient, clean and fundamental route for the syntheses of nitriles, was started with stoichiometric amounts of metal as well non-metal based dehydrating reagents. With the progress in this field the stoichiometric reagents were replaced with catalysts to afford high conversion and selectivity towards nitriles. The more recent advancements require less than one equivalent of the metal catalyst, but the reactions require either very high reaction temperatures or mild reaction conditions using a stoichiometric dehydrating agent. Overwhelmingly, numerous research studies are available with respect to acetonitrile and silyl compounds as dehydrating agents either with or without a metal catalyst. It is clear from the review that attempts have been made to develop an efficient catalytic system for dehydration of amides.In comparison with the metal catalyzed dehydrations, there are only a small number of publications available in the literature for non-transition metal and organo-catalyzed dehydrations. Moreover, a conclusion can be deduced based on the works by Buchwald and Mandal that the result outputs in terms of the reaction time and product yield of the organo-catalyzed silylative dehydrations are comparable with those of metal-catalyzed dehydrations. Hence, there is a better scope for the organo-catalyzed dehydration of amides. Indeed, the green and clean organo-photocatalyzed reaction has only a pair of research reports, each one on: the dehydration of carboxamides and the dehydrosulfurization of thioamides. Thus, in this direction, much more attention is needed in order to explore the sustainable dehydration methods.
Even though, in most of the cases, the dehydration of an amide gives a clean product, it can lead to multiple by-products depending on the other functionalities. Hence, in this context, new methods with the help of suitable reagents and catalysts for the selective dehydration of amides to nitriles are still needed to be explored. Moreover, most of the catalyzed dehydrations require inevitable use of non-renewable and expensive dehydrating agents. Additionally, these methods often require transition metals and/or stoichiometric reagents which might cause severe storage and handling problems as well as significant waste generation.
New strategies for promising methods, in order to address the problems associated with amide dehydration, viz. recyclability of reagents, selectivity, energy parameters and reaction mechanisms, are always in demand.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
One of the authors, Dr Ganesan, thanks the Directorate of Forensic Sciences, Tamilnadu State Government for granting permission to work on this review paper. The authors thank the respective heads of the organizations for support.References
- R. C. Larock, Comprehensive organic transformations, Wiley, New York, 2010 RSC; P. Pollak, G. Romeder, F. Hagedorn and H.-P. Gelbke, Nitriles, in Ullmann's encyclopedia of industrial chemistry, ed. M. Bohnet, C. G. Brinker and B. Cornils, Wiley-VCH, Weinheim, Germany, 2000 RSC; P. Hu, J. C. Chai, Y. L. Duan, Z. H. Liu, G. L. Cui and L. Q. Chen, Progress in nitrile-based polymer electrolytes for high performance lithium batteries, J. Mater. Chem. A, 2016, 4, 10070–10083 RSC; B. Ahrén, A. Schweizer, S. Dejager, E. B. Villhauer, B. E. Dunning and J. E. Foley, Mechanisms of action of the dipeptidyl peptidase-4 inhibitor vildagliptin in humans, Diabetes, Obes. Metab., 2011, 13, 775–783 CrossRef CAS; F. F. Fleming, L. Yao, P. C. Ravikumar, L. Funk and B. C. Shook, Nitrile-containing pharmaceuticals: efficacious roles of the nitrile pharmacophore, J. Med. Chem., 2010, 53, 7902–7917 CrossRef; F. F. Fleming and Q. Wang, Unsaturated nitriles: conjugate additions of carbon nucleophiles to a recalcitrant class of acceptors, Chem. Rev., 2003, 103, 2035–2078 CrossRef; A. Kleemann, J. Engel, B. Kutscher and D. Reichert, Pharmaceutical substance: synthesis patents, applications, Georg Thieme, Stuttgart, 4th edn, 2001 CrossRef; J. S. Miller and J. L. Manson, Designer magnets containing cyanides and nitriles, Acc. Chem. Res., 2001, 34, 563–570 CrossRef; R. J. H. Gregory, Cyanohydrins in nature and the laboratory: biology, preparations, and synthetic applications, Chem. Rev., 1999, 99, 3649–3682 CrossRef; D. Wöuhrle and G. Knothe, Polymers from nitriles. VII. Polymerization of fumaronitrile with triethylamine as initiator, J. Polym. Sci., Part A: Polym. Chem., 1988, 26, 2435–2447 CrossRef.
- B. Ahren, M. Landin-Olsson, P. A. Jansson, M. Svensson, D. Holmes and A. Schweizer, Inhibition of dipeptidyl peptidase-4 reduces glycemia, sustains insulin levels, and reduces glucagon levels in type 2 diabetes, J. Clin. Endocrinol. Metab., 2004, 89, 2078–2084 CrossRef CAS.
- R. Jakesz, W. Jonat, M. Gnant, M. Mittlboeck, R. Greil, C. Tausch, J. Hilfrich, W. Kwasny, C. Menzel, H. Samonigg, M. Seifert, G. Gademann, M. Kaufmann and J. Wolfgang, Switching of postmenopausal women with endocrine-responsive early breast cancer to anastrozole after 2 years’ adjuvant tamoxifen: combined results of ABCSG trial 8 and ARNO 95 trial, Lancet, 2005, 366, 455–462 CrossRef CAS.
- J. H. Ha and D. K. Ahn, Selective reduction of aromatic nitriles to aldehydes by lithium diisobutylpiperidinohydroaluminate (LDBPA), Bull. Korean Chem. Soc., 2006, 27, 121–122 CrossRef CAS.
- F. Chemat, A mild and convenient ‘dry’ hydrolysis of amides to carboxylic acids, Tetrahedron Lett., 2000, 41, 3855–3857 CrossRef CAS.
- M. Tamura, T. Tonomura, K. Shimizu and A. Satsuma, CeO2-catalysed one-pot selective synthesis of esters from nitriles and alcohols, Green Chem., 2012, 14, 984–991 RSC.
- D. Haddenham, L. Pasumansky, J. Desoto, S. Eagon and B. Singaram, Reductions of aliphatic and aromatic nitriles to primary amines with diisopropylaminoborane, J. Org. Chem., 2009, 74, 1964–1970 CrossRef CAS.
- D. Srimani, M. Feller, Y. Ben-David and D. Milstein, Catalytic coupling of nitriles with amines to selectively form imines under mild hydrogen pressure, Chem. Commun., 2012, 11853–11854 RSC.
- A. R. Burns, J. H. Kerr, W. J. Kerr, J. Passmore, L. C. Paterson and A. J. B. Watson, Tuned methods for conjugate addition to a vinyl oxadiazole; synthesis of pharmaceutically important motifs, Org. Biomol. Chem., 2010, 8, 2777–2783 RSC.
- K.-S. Yeung, M. E. Farkus, J. F. Kadow and N. A. Meanwell, A base-catalyzed, direct synthesis of 3,5-disubstituted 1,2,4-triazoles from nitriles and hydrazides, Tetrahedron Lett., 2005, 46, 3429–3432 CrossRef CAS; T. Horneff, S. Chuprakov, N. Chernyak, V. Gevorgyan and V. V. Fokin, Rhodium-catalyzed transannulation of 1,2,3-triazoles with nitriles, J. Am. Chem. Soc., 2008, 130, 14972–14974 CrossRef.
- K. Yamaguchi, M. Matsushita and N. Mizuno, Ruthenium nanocatalyst for hydration of nitriles in water, Angew. Chem., Int. Ed., 2004, 43, 1576–1580 CrossRef CAS.
- D. Enders and J. P. Shilvock, Some recent applications of a-amino nitrile chemistry, Chem. Soc. Rev., 2000, 29, 359 RSC; N. Otto and T. Opatz, Heterocycles from α–Aminonitriles, Chem. – Eur. J., 2014, 20, 13064 CrossRef CAS; D. B. Bagala and B. M. Bhanage, Recent advances in transition metal–catalyzed hydrogenation of nitriles, Adv. Synth. Catal., 2015, 357, 883 CrossRef; M.-X. Wang, Enantioselective biotransformations of nitriles in organic synthesis, Acc. Chem. Res., 2015, 48, 602 CrossRef; B. R. Pitta, O. W. Steward and F. F. Fleming, Electrophile-Dependent Alkylations of Lithiated 4-Alkoxyalk-4-enenitriles, J. Org. Chem., 2018, 83, 2753–2762 CrossRef; G. Barker, M. R. Alshawish, M. C. Skilbeck and I. Coldham, Remarkable configurational stability of magnesiated nitriles, Angew. Chem., Int. Ed., 2013, 52, 7700–7703 CrossRef; V. Y. Kukushkin and A. J. L. Pombeiro, Additions to metal-activated organonitriles, Chem. Rev., 2002, 102, 1771–1802 CrossRef; B. Gaspar and E. M. Carreira, Mild cobalt-catalyzed hydrocyanation of olefins with tosyl cyanide, Angew. Chem., Int. Ed., 2007, 46, 4519–4522 CrossRef.
- M. H. Al-Huniti and M. P. Croatt, Metal–catalyzed dehydration of primary amides to nitriles, Asian J. Org. Chem., 2019, 8, 1791–1799 CrossRef CAS; D. T. Mowry, The preparation of nitriles, Chem. Rev., 1948, 42, 189–283 CrossRef.
- L. R. Subramanian, Nitriles, in Science of Synthesis, ed. B. M. Trost and M. Lautens, Thieme, Stuttgart, Germany, 2011, vol. 19, p. 79 CrossRef; A. J. Fatiadi, Preparation and synthetic applications of cyano compounds. In triple–bonded functional groups. 1983 (eds S. Patai and Z. Rappoport). G. P. Ellis and I. L. Thomas. 6 Recent advances in the synthesis of nitriles, Prog. Med. Chem., 1974, 10, 245–285 CrossRef.
- T. V. Rajanbabu, Hydrocyanation of alkenes and alkynes, Org. React., 2011, 75, 1 Search PubMed; J. Podlech, Introduction of the cyanide group by conjugate addition, in Science of Synthesis, ed. S.-I. Murahashi, Thieme, Stuttgart, Germany, 2004, 19, 311–324 Search PubMed.
- K.-N. T. Tseng and N. K. Szymczak, Dehydrogenative oxidation of primary amines to nitriles, Synlett, 2014, 25, 2385–2389 CrossRef CAS.
- C. Czekelius and E. M. Carreira, Convenient transformation of optically active nitroalkanes into chiral aldoximes and nitriles, Angew. Chem., Int. Ed., 2005, 44, 612–615 CrossRef CAS.
- S. S. Deshmukh, S. N. Huddar, D. S. Bhalerao and K. G. Akamanchi, Oxidation of amidoximes with IBX and IBX/TEAB, ARKIVOC, 2010, 2, 118–126 Search PubMed.
- Y. Wang, S. Furukawa, Z. Zhang, L. Torrente-Murciano, S. A. Khan and N. Yan, Oxidant free conversion of alcohols to nitriles over Ni-based catalysts, Catal. Sci. Technol., 2019, 9, 86–96 RSC.
- H. Naka and A. Naraoka, Recent advances in transfer hydration of nitriles with amides or aldoximes, Tetrahedron Lett., 2020, 61, 151557 CrossRef CAS.
- M. I. Lim, W. Y. Ren and R. S. Klein, Facile conversion of primary thioamides into nitriles with butyltin oxides, J. Org. Chem., 1982, 47, 4594–4595 CrossRef CAS.
- W. Zhou, J. Xu, L. Zhang and N. Jiao, An efficient transformation from benzyl or allyl halides to aryl and alkenyl nitriles, Org. Lett., 2010, 12, 2888–2891 CrossRef CAS; J. He, K. Yamaguchi and N. Mizuno, Aerobic oxidative transformation of primary azides to nitriles by ruthenium hydroxide catalyst, J. Org. Chem., 2011, 76, 4552–4553 CrossRef; S. Iida and H. Togo, Direct oxidative conversion of alkyl halides into nitriles with molecular iodine in aqueous ammonia, Synlett, 2008, 1639–1642 Search PubMed; H. Yu, R. N. Richey, W. D. Miller, J. Xu and S. A. May, Development of Pd/C-catalyzed cyanation of aryl halides, J. Org. Chem., 2011, 76, 665–668 CrossRef.
- D. J. Quinn, G. J. Haun and G. Moura-Letts, Direct synthesis of nitriles from aldehydes with hydroxylamine-O-sulfonic acid in acidic water, Tetrahedron Lett., 2016, 57, 3844–3847 CrossRef CAS.
- K. W. Rosenmund and E. Struck, Das am Ringkohlenstoff gebundene Halogen und sein Ersatz durch andere Substituenten. I. Mitteilung: Ersatz des Halogens durch die Carboxylgruppe, Ber. Dtsch. Chem. Ges. A, 1919, 52, 1749–1756 CrossRef; T. Sandmeyer, Ber. Dtsch. Chem. Ges., 1884, 17, 2650–2653 CrossRef; A. J. Fatiadi, The Chemistry of Triple-Bonded Functional Group, Part 2, ed S. Patai and Z. Rappoport, John Wiley and Sons, New York, 1983 Search PubMed.
- A. W. Schuppe, G. M. Borrajo-Calleja and S. L. Buchwald, Enantioselective olefin hydrocyanation without cyanide, J. Am. Chem. Soc., 2019, 141, 18668–18672 CrossRef CAS; H. Chen, S. Sun, Y. A. Liu and X. Liao, Nickel-catalyzed cyanation of aryl halides and hydrocyanation of alkynes via C–CN bond cleavage and cyano transfer, ACS Catal., 2020, 10, 1397–1405 CrossRef; Y. Xing, R. Yu and X. Fang, Synthesis of tertiary benzylic nitriles via nickel-catalyzed markovnikov hydrocyanation of α-substituted styrenes, Org. Lett., 2020, 22, 1008–1012 CrossRef.
- P. Pollak, G. Romeder, F. Hagedorn and H.-P. Gelbke, Nitriles. In Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, Germany, 2002 Search PubMed.
- G. Y. Popava, T. V. Andrushkevich, Y. A. Chesalov, L. M. Plyasova, L. S. Dovlitova, E. V. Ischenko, G. I. Aleshina and M. I. Khramov, Formation of active phases in MoVTeNb oxide catalysts for ammoxidation of propane, Catal. Today, 2009, 144, 312–317 CrossRef.
- H. Wang, Y. Dong, C. Zheng, C. A. Sandoval, X. Wang, M. Makha and Y. Li, Catalytic cyanation using CO2 and NH3, Chem, 2018, 4, 2883–2893 Search PubMed; Y. Dong, H. Wang and Y. Li, Cu/Fe-catalyzed cyanation of aryl iodides using urea, Sci. Sin.: Chim., 2018, 48, 1603–1610 CrossRef CAS.
- E. Valeur and M. Bradley, Amide bond formation: beyond the myth of coupling reagents, Chem. Soc. Rev., 2009, 38, 606–631 RSC; The Amide Linkage: Structural Significance in Chemistry, Biochemistry, and Materials Science, ed. A. Greenberg, C. M. Breneman and J. F. Liebman, Wiley, New York, 2000 Search PubMed.
- F. Wöhler and J. Liebig, Researches on the radical of benzoic acid, Ann. Pharm., 1832, 3, 249–282 CrossRef.
- J. Dumas, F. Malaguti and LeBlanc, Déshydratation des amides avec de l'acide phosphorique anhydre, C. R. Seances Soc. Biol. Ses Fil., 1847, 26, 384 Search PubMed.
- B. Rickborn and F. R. Jensen, α-Carbon isomerization in amide dehydrations, J. Org. Chem., 1962, 27, 4608–4610 CrossRef CAS.
- J. Kenyon and W. A. Ross, The mechanism of the decarboxylation of substituted malonic acid derivatives, J. Chem. Soc., 1951, 3407 RSC.
- S. S. Mahajan and A. Mahalakshmi, Synthesis of 2-amino-5-chlorobenzonitrile, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2006, 45, 1299–1300 Search PubMed.
- D. Konwar, M. Boruah, G. K. Sarmah, N. K. Bhattacharyya, N. Borthakur, B. N. Goswami and K. R. Boruah, Aluminum chloride and sodium iodide (AlCl3—NaI): A versatile dehydrating agent, J. Chem. Res., 2001, 2001, 490–492 CrossRef CAS; M. Boruah and D. J. Konwar, AlCl3·6H2O/KI/H2O/CH3CN: A new alternate system for dehydration of oximes and amides in hydrated media, J. Org. Chem., 2002, 67, 7138–7139 CrossRef.
- M. Jukič, K. Grabrijan, S. Kadić, F. de Lera Garrido, I. Sosič, S. Gobec and A. Obreza, Chlorocarbonylsulfenyl chloride cyclizations towards piperidin-3-yl-oxathiazol-2-ones as potential covalent inhibitors of threonine proteases, Acta Chim. Slov., 2017, 64, 771–781 CrossRef.
- J. P. Rappai, J. Karthikeyan, S. Prathapan and P. A. Unnikrishnan, Simple and efficient one-pot synthesis of nitriles from amides and oximes using in situ–generated burgess-type reagent, Synth. Commun., 2011, 41, 2601–2606 CrossRef CAS.
- T. Sodeyania, M. Kodomari and K. Itabashi, Studies on preparations and physical properties of multivalent metal condensed phosphates. vii. The effect of water content on the formation of the types a and b of Al4(P4O12)3, Chem. Lett., 1973, 577–578 CrossRef.
- I. Talbi, M. L. Efrit and S. Touil, Efficient new protocols for converting primary amides into nitriles initiated by P(NMe2)3, PCl3, or P(OPh)3, ACS Omega, 2018, 3, 5078–5082 CrossRef CAS.
- W. E. Dennis, Nitrile synthesis. The dehydration of amides by silazanes, chlorosilanes, alkoxysilanes and aminosilanes, J. Org. Chem., 1970, 36, 3253–3255 CrossRef.
- R. Ding, Y. Liu, M. Han, W. Jiao, J. Li, H. Tian and B. Sun, Synthesis of nitriles from primary amides or aldoximes under conditions of a catalytic Swern oxidation, J. Org. Chem., 2018, 83, 12939–12944 CrossRef CAS; N. Nakajima and M. Ubukata, Preparation of nitriles from primary amides under Swern oxidation conditions, Tetrahedron Lett., 1997, 38, 2099–2102 CrossRef; N. Nakajima, M. Saito and M. Ubukata, Tetrahedron, 2002, 58, 3561 CrossRef; A. J. Speziale and L. R. Smith, The reaction of oxalyl chloride with amides. ii. oxazolidinediones and acyl isocyanates, J. Org. Chem., 1963, 28, 1805 CrossRef; A. J. Speziale, L. R. Smith and J. E. Fedder, The reaction of oxalyl chloride with amides. iv. Synthesis of acyl isocyanates, J. Org. Chem., 1965, 30, 430 Search PubMed.
- A. J. Mancuso, S. Huang and D. J. Swern, Oxidation of long-chain and related alcohols to carbonyls by dimethyl sulfoxide “activated” by oxalyl chloride, Org. Chem., 1978, 43, 2480–2482 CrossRef CAS.
- T. T. Tidwell, Oxidation of alcohols by activated dimethyl sulfoxide and related reactions: an update, Synthesis, 1990, 857–870 CrossRef CAS.
- R. Monson and D. Priest, Dehydration of amides to nitriles initiated by hexamethylphosphoric triamide, Can. J. Chem., 1971, 49, 2897–2898 CrossRef CAS.
- G. A. Olah, S. C. Narang, A. P. Fung and B. G. B. Gupta, Synthetic methods and reactions; 821. Cyanuric chloride, a mild dehydrating agent in the preparation of nitriles from amides, Synthesis, 1980, 657–658 Search PubMed.
- P. Maetz and M. Rodriguez, A simple preparation of N-protected chiral α-aminonitriles from N-protected α-amino acid amides, Tetrahedron Lett., 1997, 38, 4221–4222 CrossRef CAS.
- F. Aquino, R. Karge, H. Pauling and W. Bonrath, Dehydration of aromatic heterocyclic carboxamides to aromatic heterocyclic carbonitriles, Molecules, 1997, 2, 176–179 CrossRef CAS.
- C.-W. Kuo, J.-L. Zhu, J.-D. Wu, C.-M. Chu, C.-F. Yao and K.-S. Shia, A convenient new procedure for converting primary amides into nitriles, Chem. Commun., 2007, 301–303 RSC; K. Mai and G. Patil, Facile conversion of carboxamides to nitriles, Tetrahedron Lett., 1986, 27, 2203–2206 CrossRef CAS.
- F. Campagna, A. Carotti and G. Casini, A convenient synthesis of nitriles from primary amides under mild conditions, Tetrahedron Lett., 1977, 21, 1813–1815 CrossRef.
- Z. Shahsavari-Fard and A. R. Sardarian, Diethyl chlorophosphate: A new alternative reagent for dehydration of primary amides to nitriles in solvent and solvent-free conditions, J. Iran. Chem. Soc., 2011, 8, 204–208 CrossRef CAS.
- S. P. Wang, J. J. Zhou, S. Y. Zhao, Y. J. Zhao and X. B. Ma, Enhancements of dimethyl carbonate synthesis from methanol and carbon dioxide: The in situ hydrolysis of 2-cyanopyridine and crystal face effect of ceria, Chin. Chem. Lett., 2015, 26, 1096–1100 CrossRef CAS; M. Honda, M. Tamura, Y. Nakagawa, S. Sonehara, K. Suzuki, K. Fujimoto and K. Tomishige, Ceria–catalyzed conversion of carbon dioxide into dimethyl carbonate with 2−cyanopyridine, ChemSusChem, 2013, 6, 1341–1344 CrossRef.
- S. Enthaler and S. Inoue, An efficient zinc–catalyzed dehydration of primary amides to nitriles, Chem. – Asian J., 2012, 7, 169–175 CrossRef CAS; S. Enthaler, Straightforward uranium–catalyzed dehydration of primary amides to nitriles, Chem. – Eur. J., 2011, 17, 9316–9319 CrossRef.
- J. K. Rhee, J. I. Lim, W. B. Im and J. N. Yang, Facile conversion of carboxamides to nitriles, Bull. Korean Chem. Soc., 1993, 14, 301–302 CAS.
- W. Zhang, C. W. Haskins, Y. Yang and M. Dai, Synthesis of nitriles via palladium-catalyzed water shuffling from amides to acetonitrile, Org. Biomol. Chem., 2014, 12, 9109–9112 RSC.
- S. Zhou, K. Junge, D. Addis, S. Das and M. Beller, A general and convenient catalytic synthesis of nitriles from amides and silanes, Org. Lett., 2009, 11, 2461–2464 CrossRef CAS.
- B. Xue, H. Sun, Y. Wang, T. Zheng, X. Li, O. Fuhr and D. Fenske, Efficient reductive dehydration of primary amides to nitriles catalyzed by hydrido thiophenolato iron(II) complexes under hydrosilation conditions, Catal. Commun., 2016, 86, 148–150 CrossRef CAS; S. Elangovan, S. Quintero-Duque, V. Dorcet, T. Roisnel, L. Norel, C. Darcel and J.-B. Sortais, Knölker-type iron complexes bearing an n-heterocyclic carbene ligand: synthesis, characterization, and catalytic dehydration of primary amides, Organometallics, 2015, 34, 4521–4528 CrossRef; D. Bezier, G. T. Venkanna, J.-B. Sortais and C. Darcel, Well-defined cyclopentadienyl NHC iron complex as the catalyst for efficient hydrosilylation of amides to amines and nitriles, ChemCatChem, 2011, 3, 1747–1750 CrossRef; S. Zhou, D. Addis, S. Das, K. Junge and M. Beller, New catalytic properties of iron complexes: dehydration of amides to nitriles, Chem. Commun., 2009, 45, 4883–4885 RSC; S. Hanada, Y. Motoyama and H. Nagashima, Hydrosilanes are not always reducing agents for carbonyl compounds but can also induce dehydration: a ruthenium–catalyzed conversion of primary amides to nitriles, Eur. J. Org. Chem., 2008, 4097–4100 CrossRef; R. Calas, E. Frainnet and A. Bazouin, Action du triethylsilane sur les amides primaires monosubstitutes et sur les iminoethers; addition du triethylsilane a la fonction imine, Compt. Rend., 1963, 257, 1304 Search PubMed.
- M.-P. Heck, A. Wagner and C. J. Mioskowski, Conversion of primary amides to nitriles by aldehyde-catalyzed water transfer, Org. Chem., 1996, 61, 6486 CrossRef CAS.
- R. Y. Liu, M. Bae and S. L. Buchwald, Mechanistic insight facilitates discovery of a mild and efficient copper-catalyzed dehydration of primary amides to nitriles using hydrosilanes, J. Am. Chem. Soc., 2018, 140, 1627–1631 CrossRef CAS; Y. Furuya, K. Ishihara and H. Yamamoto, Perrhenic acid-catalyzed dehydration from primary amides, aldoximes, n-monoacylureas, and α-substituted ketoximes to nitrile compounds, Bull. Chem. Soc. Jpn., 2007, 80, 400–406 CrossRef; S. Enthaler and M. Weidauer, Copper-catalyzed dehydration of primary amides to nitriles, Catal. Lett., 2011, 141, 1079–1085 CrossRef.
- T. Mineno, M. Shinada, K. Watanabe, H. Yoshimitsu, H. Miyashita and H. Kansui, Highly-efficient conversion of primary amides to nitriles using indium(III) triflate as the catalyst, Int. J. Org. Chem., 2014, 4, 1–6 CrossRef.
- S. Enthaler, Straightforward iron–catalyzed synthesis of nitriles by dehydration of primary amides, Eur. J. Org. Chem., 2011, 4760–4763 Search PubMed; S. Ren, Y. Wang, F. Yang, H. Sun and X. Li, Dehydration of primary amides to nitriles catalyzed by [CNC]-pincer hydrido cobalt(III) complexes, Catal. Commun., 2019, 120, 72–75 CrossRef CAS; G. Chang, X. Li, P. Zhang, W. Yang, K. Li, Y. Wang, H. Sun, O. Fuhr and D. Fenske, Lewis acid promoted dehydration of amides to nitriles catalyzed by [PSiP]–pincer iron hydrides, Appl. Organomet. Chem., 2020, e5466 Search PubMed; D. Bézier, G. T. Venkanna, J. B. Sortais and C. Darcel, Well–defined cyclopentadienyl nhc iron complex as the catalyst for efficient hydrosilylation of amides to amines and nitriles, ChemCatChem, 2011, 3, 1747–1750 CrossRef; J. Hart-Davis, P. Battioni, J. L. Boucher and D. Mansuy, New catalytic properties of iron porphyrins: model systems for cytochrome p450-catalyzed dehydration of aldoximes, J. Am. Chem. Soc., 1998, 120, 12524 CrossRef.
- S. I. Maffioli, E. Marzorati and A. Marazzi, Mild and reversible dehydration of primary amides with PdCl2 in aqueous acetonitrile, Org. Lett., 2005, 7, 5237–5239 CrossRef CAS.
- K. Ishihara, Y. Furuya and H. Yamamoto, Rhenium(VII) oxo complexes as extremely active catalysts in the dehydration of primary amides and aldoximes to nitriles, Angew. Chem., Int. Ed., 2002, 41, 2983–2986 CrossRef CAS.
- J. Blum, A. Fisher and E. Greener, The catalytic decomposition of secondary carboxamides by transition-metal complexes, Tetrahedron, 1973, 29, 1073 CrossRef CAS; J. Blum and A. Fisher, A novel synthesis of nitriles from secondary amides, Tetrahedron Lett., 1970, 11, 1963–1966 CrossRef.
- Y. Watanabe, F. Okuda and Y. J. Tsuji, Ruthenium complex catalyzed dehydration of carboxamides to nitriles in the presence of urea derivatives, Mol. Catal., 1990, 58, 87 CrossRef CAS.
- S. Sueoka, T. Mitsudome, T. Mizugaki, K. Jitsukawa and K. Kaneda, Supported monomeric vanadium catalyst for dehydration of amides to form nitriles, Chem. Commun., 2010, 46, 8243–8245 RSC.
- S. Itagaki, K. Kamata, K. Yamaguchi and N. Mizuno, A monovacant lacunary silicotungstate as an efficient heterogeneous catalyst for dehydration of primary amides to nitriles, ChemCatChem, 2013, 5, 1725–1728 CrossRef CAS; J. A. Campbell, G. McDouglad, H. McNab, L. V. C. Rees and R. G. Tyas, Laboratory-scale synthesis of nitriles by catalysed dehydration of amides and oximes under flash vacuum pyrolysis (fvp) conditions, Synthesis, 2007, 3179–3184 Search PubMed.
- K. Manjula and M. A. Pasha, Rapid method of converting primary amides to nitriles and nitriles to primary amides by ZnCl2 using microwaves under different reaction conditions, Synth. Commun., 2007, 37, 1545–1550 CrossRef CAS.
- R. T. Ruck and R. G. Bergman, Zirconium-mediated conversion of amides to nitriles: a surprising additive effect, Angew. Chem., Int. Ed., 2004, 43, 5375–5377 CrossRef.
- W. Lehnert, Nitrile aus primären carbonsäureamiden mit TiCl4/base bei 0 °C, Tetrahedron Lett., 1971, 19, 1501 CrossRef.
- M. H. Al-Huniti, J. Rivera-Chávez, K. L. Colón, J. L. Stanley, J. E. Burdette, C. J. Pearce, N. H. Oberlies and M. P. Croatt, Development and utilization of a palladium-catalyzed dehydration of primary amides to form nitriles, Org. Lett., 2018, 20, 6046–6050 CrossRef CAS; S. I. Maffioli, Y. Zhang, D. Degen, T. Carzaniga, G. Del Gatto, S. Serina, P. Monciardini, C. Mazzetti, P. Guglierame, G. Candiani, A. I. Chiriac, G. Facchetti, P. Kaltofen, H.-G. Sahl, G. Dehò, S. Donadio and R. H. Ebright, Antibacterial nucleoside-analog inhibitor of bacterial RNA polymerase, Cell, 2017, 169, 1240–1248 CrossRef; P. Dubey, S. Gupta and A. K. Singh, Trinuclear complexes of palladium(II) with chalcogenated N-heterocyclic carbenes: catalysis of selective nitrile–primary amide interconversion and Sonogashira coupling, Dalton Trans., 2017, 46, 13065–13076 RSC.
- H. Okabe, A. Naraoka, T. Isogawa, S. Oishi and H. Naka, Acceptor-controlled transfer dehydration of amides to nitriles, Org. Lett., 2019, 21, 4767–4770 CrossRef CAS.
- P. K. Hota, S. Maji, J. Ahmed, N. M. Rajendran and S. K. Mandal, NHC-catalyzed silylative dehydration of primary amides to nitriles at room temperature, Chem. Commun., 2020, 56, 575–578 RSC.
- Y. Wang, H. Zhang, S. Xie, H. Sun, X. Li, O. Fuhr and D. Fenske, An air-stable N-heterocyclic [PSiP] pincer iron hydride and an analogous nitrogen iron hydride: synthesis and catalytic dehydration of primary amides to nitriles, Organometallics, 2020, 39, 824–833 CrossRef CAS.
- W. Yao, H. Fang, Q. He, D. Peng, G. Liu and Z. Huang, A BEt3-base catalyst for amide reduction with silane, J. Org. Chem., 2019, 84, 6084–6093 CrossRef CAS.
- Y. Y. Wang, L. Y. Fu, H. M. Qi, S. W. Chen and Y. H. Li, Bioinspired synthesis of nitriles from primary amides via zinc/anhydride catalysis, Asian J. Org. Chem., 2018, 7, 367–370 CrossRef CAS.
- K. Li, H. Sun, W. Yang, Y. Wang, S. Xie, X. Li, O. Fuhr and D. Fenske, Efficient dehydration of primary amides to nitriles catalyzed by phosphorus–chalcogen chelated iron hydrides, Appl. Organomet. Chem., 2020, 34, e5337 Search PubMed; T. Zheng, Y. Wang, Z. Yang, H. Sun and X. Li, Catalytic effect of iron hydrides on dehydration of primary amides to nitriles, Chin. J. Org. Chem., 2019, 39, 2941–2945 CrossRef CAS.
- S. Laval, W. Dayoub, L. Pehlivan, E. Métay, A. Favre-Réguillon, D. Delbrayelle, G. Mignani and M. Lemaire, Hydrosiloxane–Ti(OiPr)4: an efficient system for the reduction of primary amides into primary amines as their hydrochloride salts, Tetrahedron Lett., 2011, 52, 4072–4075 CrossRef CAS.
- Q. Liang and D. Song, Iron N-heterocyclic carbene complexes in homogeneous catalysis, Chem. Soc. Rev., 2020, 49, 1209–1232 RSC.
- S. Ren, S. Xie, T. Zheng, Y. Wang, S. Xu, B. Xue, X. Li, H. Sun, O. Fuhrd and D. Fenske, Synthesis of silyl iron hydride via Si–H activation and its dual catalytic application in the hydrosilylation of carbonyl compounds and dehydration of benzamides, Dalton Trans., 2018, 47, 4352–4359 RSC.
- S. K. Mandal, H. S. Das, S. Das, K. Dey, B. Singh, R. K. Haridasan, A. Das and J. Ahmed, Primary amides to amines or nitriles: a dual role by a single catalyst, Chem. Commun., 2019, 55, 11868–11871 RSC.
- Y. Li, Y. Zhao, S. Wang and X. Ma, Silica supported potassium oxide catalyst for dehydration of 2-picolinamide to form 2-cyanopyridine, Chin. Chem. Lett., 2019, 30, 494–498 CrossRef CAS.
- R. Appel, R. Kleinstuck and K. D. Ziehn, Über die gemeinsame Einwirkung von Phosphinen und Tetrachlorkohlenstoff auf Ammoniak (Derivate), II. Eine einfache nitril–synthese, Chem. Ber., 1971, 104, 1030–1034 CrossRef CAS; S. A. Shipilovskikh, V. Y. Vaganov, E. I. Denisova, A. E. Rubtsov and A. V. Malkov, Dehydration of amides to nitriles under conditions of a catalytic appel reaction, Org. Lett., 2018, 20, 728–731 CrossRef.
- A. Rai and L. D. S. Yadav, Cyclopropenone–catalyzed direct conversion of aldoximes and primary amides into nitriles, Eur. J. Org. Chem., 2013, 2013, 1889–1893 CrossRef CAS.
- A. K. Yadav, V. P. Srivastava and L. D. S. Yadav, Visible-light-mediated efficient conversion of aldoximes and primary amides into nitriles, RSC Adv., 2014, 4, 4181–4186 RSC.
- T. Xu, T. Cao, Q. Feng, S. Huang and S. Liao, Metal-free dehydrosulfurization of thioamides to nitriles under visible light, Chem. Commun., 2020, 56, 5151–5153 RSC.
| This journal is © the Partner Organisations 2020 |