
Recent developments in dehydration of primary amides to nitriles
 *a
and
Paramathevar
Nagaraaj
*b
*a
and
Paramathevar
Nagaraaj
*b
Abstract
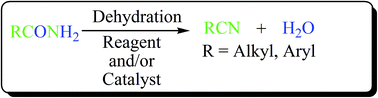
Dehydration of amides is an efficient, clean and fundamental route for the syntheses of nitriles in organic chemistry. The two imperative functional groups viz., amide and nitrile groups have been extensively discussed in the literature. However the recent development in the century-old dehydration method for the conversion of amides to nitriles has hardly been reported in one place, except a lone review article which dealt with only metal catalysed conversions. The present review provides broad and rapid information on the different methods available for the nitrile synthesis through dehydration of amides. The review article has major focus on (i) non-catalyzed dehydrations using chemical reagents, and (ii) catalyzed dehydrations of amides using transition metal, non-transition metal, organo- and photo-catalysts to form the corresponding nitriles. Also, catalyzed dehydrations in the presence of acetonitrile and silyl compounds as dehydrating agents are highlighted.
 Please wait while we load your content...
Please wait while we load your content...

