
Stimuli-responsive structural changes in metal–organic frameworks
Zhanning
Liu
 *,
Lu
Zhang
and
Daofeng
Sun
*
*,
Lu
Zhang
and
Daofeng
Sun
*
School of Materials Science and Engineering, China University of Petroleum (East China), Qingdao, Shandong 266580, China. E-mail: znliu@upc.edu.cn; dfsun@upc.edu.cn
First published on 8th July 2020
Abstract
In the past decades, metal–organic frameworks (MOFs) have attracted considerable interest not only for their intrinsic aesthetic appeal but also their wide potential applications. The most distinctive feature of MOFs is their flexibility, which makes them exhibit a much larger magnitude of responses toward external stimuli than traditional materials. In addition, the stimuli usually have a huge impact on the MOFs’ performances. A fundamental understanding of the structure evolution process under external stimuli is critical to reveal the structure–property relationships in MOFs. In this feature article, we tried to summarize how the crystal structure changes toward external stimuli, such as guest insertion, temperature, pressure, electric field, and light.
 Zhanning Liu | Zhanning Liu received his PhD degree in Metallurgical Physical Chemistry at the University of Science and Technology Beijing (USTB) under the supervision of Prof. Xianran Xing in 2019. Now he is working at China University of Petroleum (East China) as a postdoctoral fellow with Prof. Daofeng Sun. His current research interests include synthesis of responsive MOFs and exploring the structure property relationships. |
 Daofeng Sun | Daofeng Sun completed his PhD in physical chemistry under the supervision of Prof. Maochun Hong and Rong Cao from Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences (2003). He worked with Professor Hongcai Zhou at the University of Miami as a postdoctoral fellow during 2003–2006 before joining Shandong University in 2007. He moved to China University of Petroleum (East China) in 2013, and now is a full professor of Chemistry. His research interests focus on porous materials. |
Introduction
As a new class of porous materials, metal–organic frameworks (MOFs), also known as porous coordination polymers (PCPs), have attracted considerable interest owing to their wide potential applications. MOFs are usually assembled from metal ions or clusters that are connected by organic ligands. By virtue of the diversity of metal ions (or clusters) and organic ligands, numerous MOFs with kinds of topologies can be achieved. The last three decades have witnessed a blooming development of MOFs.1–7The inorganic–organic hybrid nature not only offers MOFs with combined or enhanced properties of individual components,8–10 but also endows them with novel functions. The pioneering works of Kitagawa and Ferey have led us to focus on the flexibility of MOFs.11–13 Generally, the flexibility means a material can alter its form easily according to the situation without breaking. MOFs are highly crystallized material and used to be regarded as “truly crystalline” structure.14 It may seem counterintuitive to associate the high degree order of MOFs with flexibility. While it should be noted that the MOFs are mainly constructed by coordination bonds which is much weaker than the covalent bonds in inorganic materials. The moderate coordination bonds can act as joints, triggering geometrical changes. For example, the metal–carboxylate interactions in MIL-53 enable the rotation of ligand around the carboxylate group, leading to the “breathing behavior” (transition among the large pore (LP), narrow pore (NP) and closed pore (CP) phases).15 Additionally, the incorporation of organic ligands can endow the framework with large intramolecular degrees of freedom,16 enabling a series of intramolecular distortions, which can hardly be realized in traditional inorganic materials. For instance, the [O2P(OH)2] group in γ-ZrPO4[O2P(OH)2]·2H2O can be partially replaced by the 1,10-decanediphosphonate ligand [O2POH–(CH2)10–HOPO2], which shows accordion-like transformation during the adsorption and desorption of the water molecules. This remarkable expansion and shrinkage mainly originate from the torsion of pillar by increasing the cis-position.17 It can be concluded that the combination of moderate metal–ligand interactions and the large intramolecular degrees of freedom can dramatically enhance the flexibility of MOFs, making them show much larger responses toward external stimuli than traditional porous materials. Generally, for inorganic materials, the atom movements are very small (<1 Å) and often occur during displacive phase transition.11,18 However, in some cases, the molecular displacements of MOFs can reach 5–10 Å, one order larger than those of inorganic compounds.15,19
Although the flexibility usually concerns solid-state rearrangement of atoms, some MOFs can maintain the single-crystallinity (single-crystal to single-crystal transition), enabling direct visualization of the structural changes. Additionally, the development of computational modeling and advanced structural determination methods, such as neutron diffraction and synchrotron X-ray total scattering techniques also helped to deep understand the structural evolution process.20–24
The large flexibility of MOFs made them show potential applications in gas storage/separation,25–29 sensing,30,31 catalysis,32,33 and drug delivery.34,35 For example, the flexible MOFs usually exhibit type V or VI (S shaped or multi-step) isotherms. Since this kind of isotherm corresponds to an abrupt gas uptake over a narrow range, its working capacity in the storage of gases is much higher than rigid MOFs.36 In addition, the catalytic activities of flexible MOFs can be switched on and off by changing the conformations of MOFs.32 While the flexibility also made them sensitive to the external stimuli. Considering that MOFs may work at different conditions, the environmental fluctuations may induce large structural changes of MOFs, which may further influence their performances. As the structure–property relationship commonly exists in materials, a small lattice parameter change can significantly influence material's performance. For example, a small lattice swelling of NiFe MOFs can induce partial electrons transfer from Ni to O, which resulting in about 50–100 times enhancement of the oxygen electrocatalytic properties.37 Based on this, in this feature article, our main focus is on the crystal structural evolution of MOFs including lattice expansion/contraction and symmetry changes under external stimuli such as guest insertion, temperature, pressure, electrical field, and light.
Guest insertion
The study of MOFs has a history of more than three decades,38 while the initially synthesized MOFs possess much lower stability. During the removal of guest molecules, the framework usually collapses. Since then, great efforts have been devoted to synthesize stable MOFs that can endure the reversible guest insertion and removal.39–41 Kitagawa et al. classified the MOFs into three categories:12,42 the first generation of MOFs don’t possess permanent porosity, which will collapse after the removal of guests; the second and third generations possess rigid and flexible frameworks, respectively. It should be noted that the pores of the third generation can be easily modified by external stimuli.The large inherent pores of MOFs can endure the insertion of kinds of guest molecules. In return, the conformations of MOFs will also be influenced by the size or shape of guests. During the guest insertion process, the lattice can not only expand or contract but also reversibly transform between different phases. Among them, the most commonly structural change is lattice expansion, while the magnitude of lattice expansion in MOFs is much higher than the inorganic compounds. Take the pure silica ITQ-12 zeolite for example, the as-synthesized ITQ-12 contains organic guests in the pores. After calcination, the volume decrease is only ∼1%. While Fujita et al. found that the MOF (ZnI2)3(TPT)2 (TPT = 2,4,6-tris(4-pyridyl)triazine) can reversibly shrink (or swell) with the volume changes up to ∼20% during the guest removal (or insertion).43
The most representative flexible MOF is MIL-88, which is composed of the Fe3(μ3-O)(RCOO)6 secondary building units (SBUs) and dicarboxylates. The MIL-88 series possess isoreticular framework structures with the acs topology. The insertion of guest solvents and the desolvation won’t cause structural symmetry changes or apparent bond breaking but can result in a very large volume change (Fig. 1a). Take MIL-88-D for example, the insertion of pyridine molecules will result in an expansion of lattice parameter a (b) (∼94%) and contraction of c (∼−16%). The magnitude of volume change is up to ∼233% in comparison with its desolvated form (from 2456 Å3 to 7758 Å3, Fig. 1b).19 Detailed structural analyses revealed that the O–O axis of the carboxylate functional groups can act as a “knee cap”, around which the trimer and the phenyl rings can change their relative angular orientations (Fig. 1c), so the framework can allow a reversible swelling. It should be noted that this swelling behavior is not only related to the coordination bonds, but also depends on the intramolecular flexibility of the ligands. For example, the introduction of functional groups on the organic ligand can effectively enhance the steric effect, which usually weakens the framework flexibility. The substitution of 1,4-benzenedicarboxylate (BDC) ligands with methyl functionalized ones can largely reduce the swelling magnitude to ∼25% in comparison with 136% for the pristine MIL-88B(Fe).44 While extending the length of ligands can endow them with various conformational changes, such as blending, flipping, etc. For example, the volume changes of 2,6-naphthalenedicarboxylate (NDC) bridged MIL-88C can also reach 170%, while the value for MIL-88A (connected by fumarate) is only 72%. Additionally, the (4,12) connected ftw type framework based on the Zr6–oxo cluster has always been regarded as rigid owing to their high symmetry and high connectivity.45–48 Farha et al. synthesized NU-1105 using an expanded pyrene-based tetracarboxylate ligand. During the adsorption/desorption of propane, the ligand can switch between a semi-planar and bowl-shaped conformation which lead to approximately 30% changes in pore volume and boost by ∼15% of its propane working capacity.49
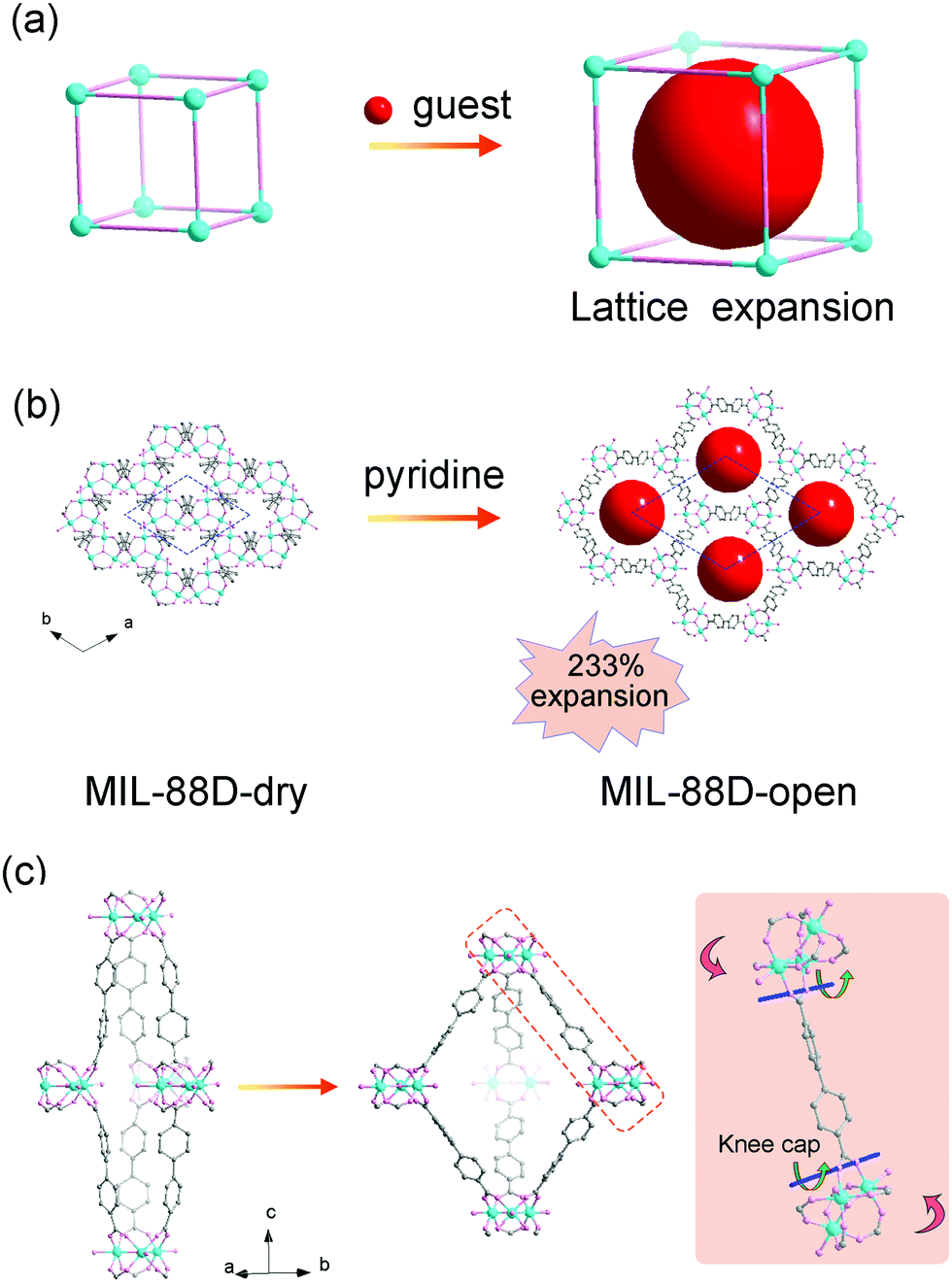 | ||
| Fig. 1 (a) Scheme of the guest insertion induced lattice expansion. (b) Crystal structure of the MIL-88D before and after pyridine loading. (c) The cage structure in MIL-88D before (left) and after (middle) guest loading. Right: Illustration of the framework displacements during swelling, which occur around the knee cap O–O axis (blue lines and green arrows). This allows the rotation around this axis of the whole trimeric units (red arrows). Partly taken from ref. 19 with permission. | ||
Intriguingly, the compound MCF-18, with similar architectures with MIL-88 but built from H2dcpb (2,6-di-p-carboxyphenyl-4,4′-bipyridine), exhibits reverse breathing mode with MIL-88. During the pyridine insertion, the lattice parameter c dramatically expands (∼121%) and the parameter a (b) contracts (∼3.7%).50 Further structural analyses revealed that the metal–carboxylate junctions can all bend toward the equatorial plane under thermal stimuli owing to the unique shape of dcpb2−, resulting in the different breathing direction with MIL-88.
Normally, the insertion of guest molecules will expand the framework, while in some cases, the unit cell volume can contract owing to specific host–guest interactions (Fig. 2a). Take MIL-53 for example, MIL-53 is composed of metal ions (Al3+, Fe3+, Cr3+, Ga3+ or In3+) and BDC ligand, with one-dimensional diamond shaped tunnel along the c axis. During the adsorption/desorption of guest water molecules, MIL-53(Cr) can switch between the LP phase and NP phase.15 The LP phase crystallizes in the orthorhombic phase with the space group Imcm, while the NP phase is a monoclinic phase with the space group C2/c. It should be noted that the phase transition usually results in re-definition of unit cell, making it difficult to unambiguously compare the lattice parameters of different phases. Although there is a symmetry difference between the two phases of MIL-53, the fragments in each unit are actually identical. Both phases possess four metal ions and four BDC ligands in each unit. So direct comparison of unit cell volume can be carried out. The adsorption of H2O molecules can cause a remarkable volume contraction, which is up to −32% (VLP = 1486.1 Å3 and VCP = 1012.8 Å3, Fig. 2b). This counterintuitive phenomenon may originate from the strong host–guest interactions and it is also known as “breathing effect”. The observed macroscopic flexibility is critically dependent on the composition of the framework. Since the ligands rotate around the metal–carboxylate bonds during the breathing transition, the strength of metal–ligand interactions can largely influence the magnitude of breathing behavior. For example, the aluminum analogue MIL-53(Al) shows similar breathing transition during the dehydration process, while the structural change of iron based MIL-53 (Fe) after dehydration is negligible (ΔV ≈ 87 Å3).51 Additionally, the introduction of functional groups on the ligands can modify the host–guest interactions, which can be utilized to finely control the breathing behavior.52 Some flexible MOFs with similar square or diamond shaped 1D channels have also been found to show adsorption induced contraction, such as [Zn2(1,4-bdc)2(dabco)] (dabco = 1,4-diazabicyclo[2.2.2]octane).53
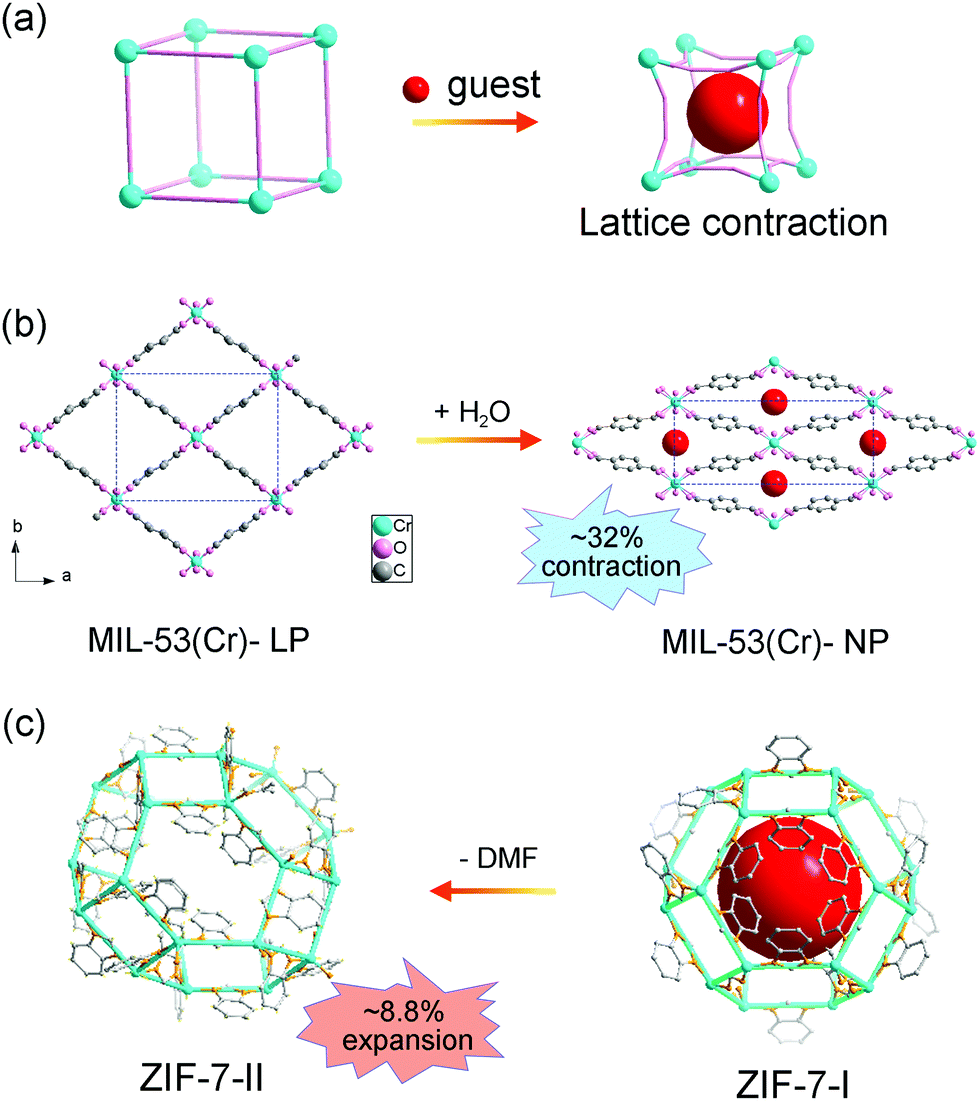 | ||
| Fig. 2 (a) Scheme of the guest insertion induced lattice contraction. Illustration of the crystal structure changes of MIL-53 (b) and ZIF-7 (c) during the guest insertion and desolvation. Partly taken from ref. 15 and 55 with permission. | ||
Zeolitic imidazolate frameworks (ZIFs), a subclass of MOFs, are usually constructed from metal ions and imidazole molecules, displaying zeolite type structures. ZIF-7 (Zn(bzIm)2, bzIm = benzimidazolate) is one of the earliest reported ZIFs with a prototypical structure related to the SOD topology.54 Similar with MIL-53, the adsorption/desorption of guest N,N-dimethylformamide (DMF) molecules can induce a lattice contraction. Redfern et al. found that ZIF-7 can reversibly desorption/adsorption of guest DMF molecules, which is mediated by the tilting of the tetrahedral unit.55 The two phases possess similar framework fragments (totally 18 metal ions and 36 imidazolate ligands in each unit). While the unit cell volume of DMF loaded ZIF-7-II (V = 7214 Å3) is much smaller than the pristine ZIF-7-I (V = 7917 Å3), ∼8.8% changes (Fig. 2c), which may be related with the strong host–guest interactions. Additionally, leaving ZIF-7-II to water for one week can result in an irreversible transition to dense planar ZIF-7-III. Du et al. investigated the adsorption property of C1 to C7 alcohol, and a continuous swelling of ZIF-7 was observed.56 The lattice parameter a can expand from 22.9 Å (methanol saturated) to 23.7 Å (1-pentanol saturated). Further increasing the size of probe molecules (1-hexanol or 1-heptanol) will decrease the number of adsorbents in the cage, so an obvious sudden shrinkage of lattice parameter was observed.
Although the gases are invisible and the interactions with the framework are usually much weaker than solvents, the adsorption of gases can also overcome the free energy difference between different states, inducing structural changes.20,57 The flexible MOFs usually exhibit S-shaped or multi-step adsorption isotherms.25–29 The Co(BDP) (H2BDP = 1,4-benzenedipyrazole), with a flexible “wine-rack” motif, can exhibit a rare five-step phase transition upon N2 adsorption (Fig. 3).58 The first four phases crystalize in the monoclinic phase with the same space group (C2/c) but dramatically different lattice parameters. The N2 adsorption results in a continuous volume expansion (from 1080 Å3 to 2338 Å3, ∼116%). Further increasing of N2 content will give rise to a tetragonal phase (P4122) with the V = 2618 Å3 (expands ∼142%). The breathing behavior is sensitive to the adsorbed gases. For a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 mixture of CO2 and CH4, the Co(BDP) can exhibit a large CO2 adsorption capacity and approaches complete exclusion of CH4, leading to an outstanding selectivity.
:50 mixture of CO2 and CH4, the Co(BDP) can exhibit a large CO2 adsorption capacity and approaches complete exclusion of CH4, leading to an outstanding selectivity.
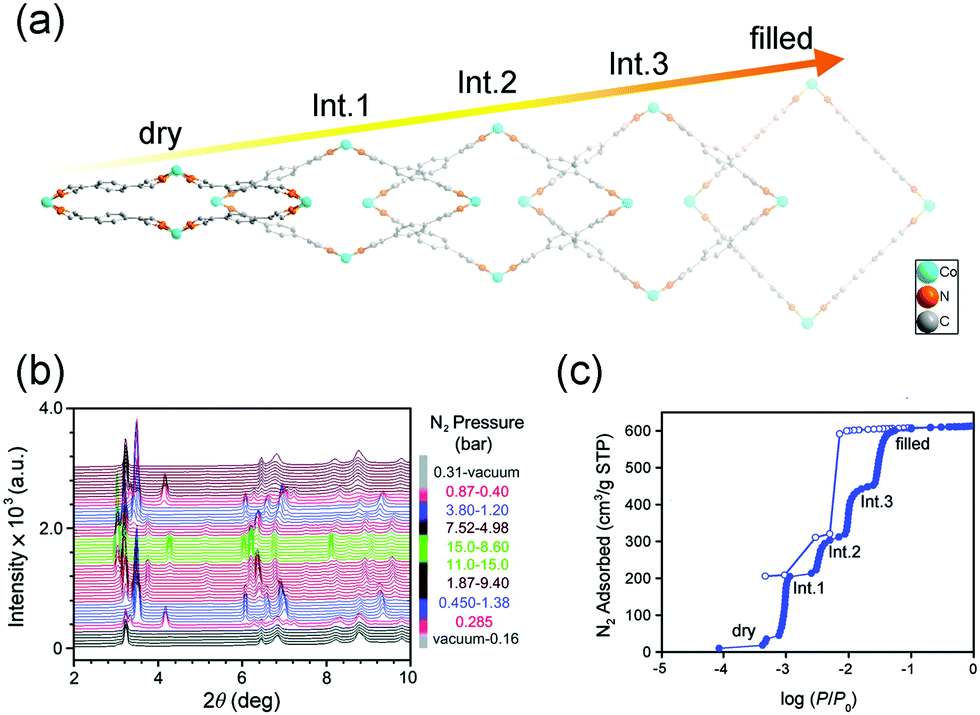 | ||
| Fig. 3 (a) Scheme of the continuous breathing behavior of the Co(BDP) during N2 adsorption. (b) In situ X-ray powder diffraction patterns of Co(BDP) measured under different gas pressure at 100 K. (c) N2 adsorption isotherm measured at 77 K, indicating the five pressure-dependent phases. Filled and empty symbols represent adsorption and desorption, respectively. Modified and reprinted with permission from ref. 58. | ||
The breathing behavior is related to not only the framework flexibility but the host–guest interactions, so it varies for different host–guest pairs. For example, Brammer et al. found that [Me2NH2][In(ABDC)2] (SHF-61) (Me = meythl, ABDC = 2-aminobenzene-1,4-dicarboxylate) can exhibit an intriguing solvent-switchable continuous breathing behavior (Fig. 4a).59 The SHF-61 is a doubly interpenetrated anionic 3D diamondoid framework with Me2NH2 as counter cations. The as-synthesized SHF-61 contains DMF solvents in the channels, and the desolvation will cause a continuous breathing effect accompanied by remarkable lattice changes (Δa ≈ 0.45 Å, Δb ≈ −6 Å, Δc ≈ 4 Å and ΔV ≈ −2000 Å3). The breathing mechanism involves the rotation of ABDC ligands and coordination geometry distortion of the In(III) centers. For the CHCl3 exchanged SHF-61, although it can also exhibit breathing phenomenon during the desolvation process, the magnitude is much smaller (Δb ≈ −0.6 Å, Δc ≈ 0.6 Å and ΔV ≈ −385 Å3) and the framework remains LP form after desolvation. The two different activation strategies can induce dramatically different CO2versus CH4 selectivities. What's more, partial desolvation of SHF-61-DMF can introduce a gating behavior and stepped isotherm for the adsorption of CO2, which is absent in the fully activated material.
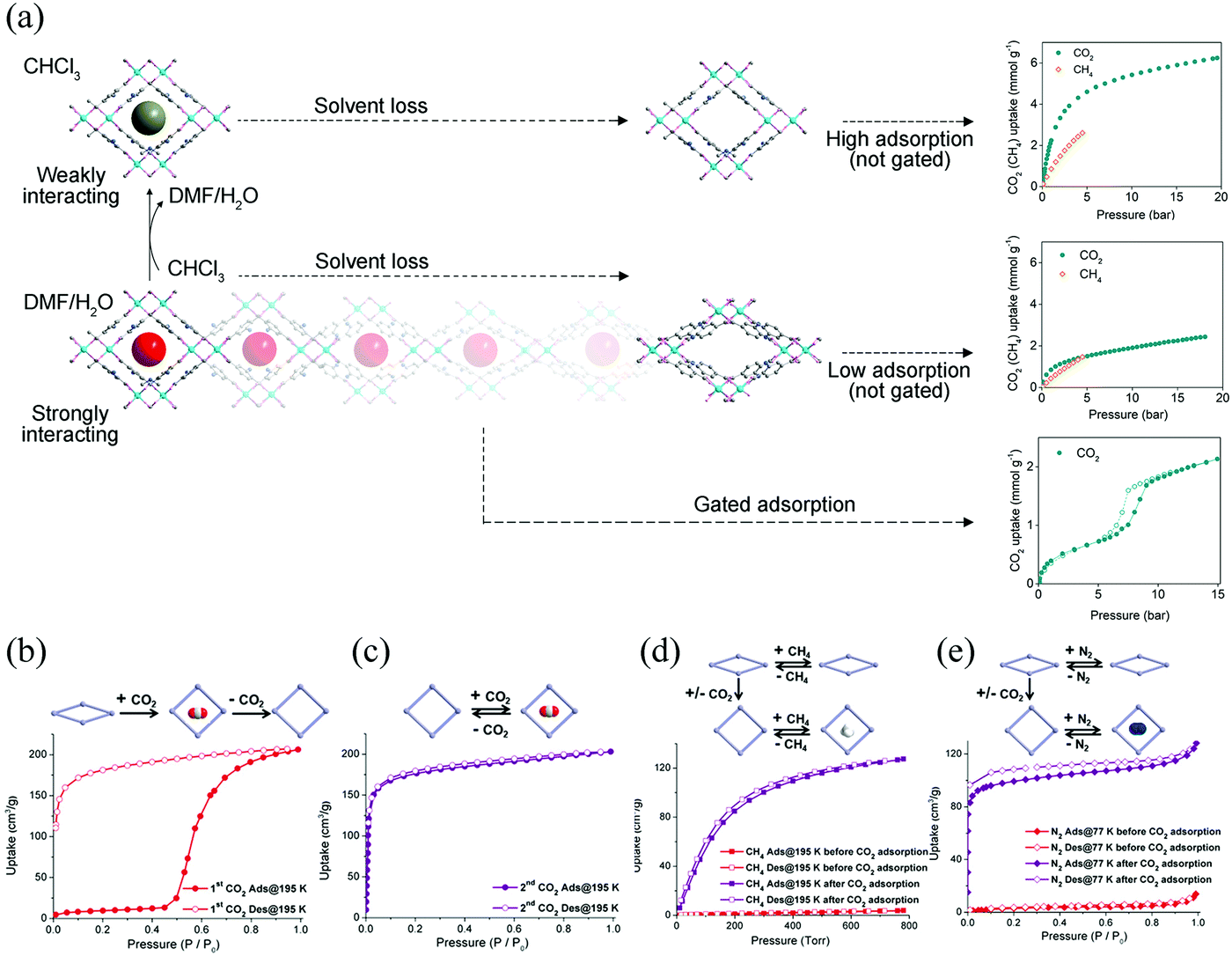 | ||
| Fig. 4 (a) Scheme of the guest–framework interactions in the solvent-dependent breathing properties of SHF-61. Left: Structural changes of the framework during the desolation process. Right: Gravimetric adsorption isotherms at 298 K for the activated SHF-61 toward CO2 and CH4. (b) The first cycle of CO2 adsorption/desorption isotherms of CPM-107 at 195 K. (c) The second cycle of CO2 adsorption/desorption isotherms of CPM-107 at 195 K. (d) CH4 adsorption/desorption isotherms of CPM-107 at 195 K before and after CO2 adsorption. (e) N2 adsorption/desorption isotherms at 77 K of CPM-107 before and after CO2 adsorption. Reprinted with permission from ref. 59 and 60. | ||
Similarly, the existence of gas–framework interactions before gas adsorption measurements can also influence the gate opening (or breathing) behavior. For instance, Bu et al. synthesized an anionic MOF CPM-107, which contains ordered cations and solvent molecules.60 The activated framework shows non-adsorption to common gases (CH4, H2, and N2) except CO2. The adsorption of CO2 can induce a transition from NP to LP phase. While, different from the other gate-opening MOFs, the LP phase can be retained after desorption (Fig. 4b and c) and can be accessed with CH4, N2, etc. indicating a “shape-memory” effect (Fig. 4d and e). The LP phase can also be re-locked after soaking in CH2Cl2 and then desolvated under vacuum.
Normally, the isothermal gas adsorption capacity of MOFs will increase with increasing pressure and decrease with increasing temperature, while the flexibility of MOFs can sometimes cause counterintuitive phenomena. For example, Bu et al. synthesized a flexible microporous MOF NKU-FlexMOF-1 using the Cu2(COO)4 paddle-wheel SBUs and H2FPBDC (5-(5-fluoropyridin-3-yl)-1,3-benzenedicarboxylic acid) ligands. The NKU-FlexMOF-1 exhibits enhanced gas sorption of C2H6, C3H6, and C3H8 with increasing temperature (Fig. 5).61In situ structural analyses revealed that this atypical behavior is a result of loading dependent conformational changes. The material can subtly adapt to different sorbents via an induced-fit mechanism., which can provide repulsion/dispersion interactions.
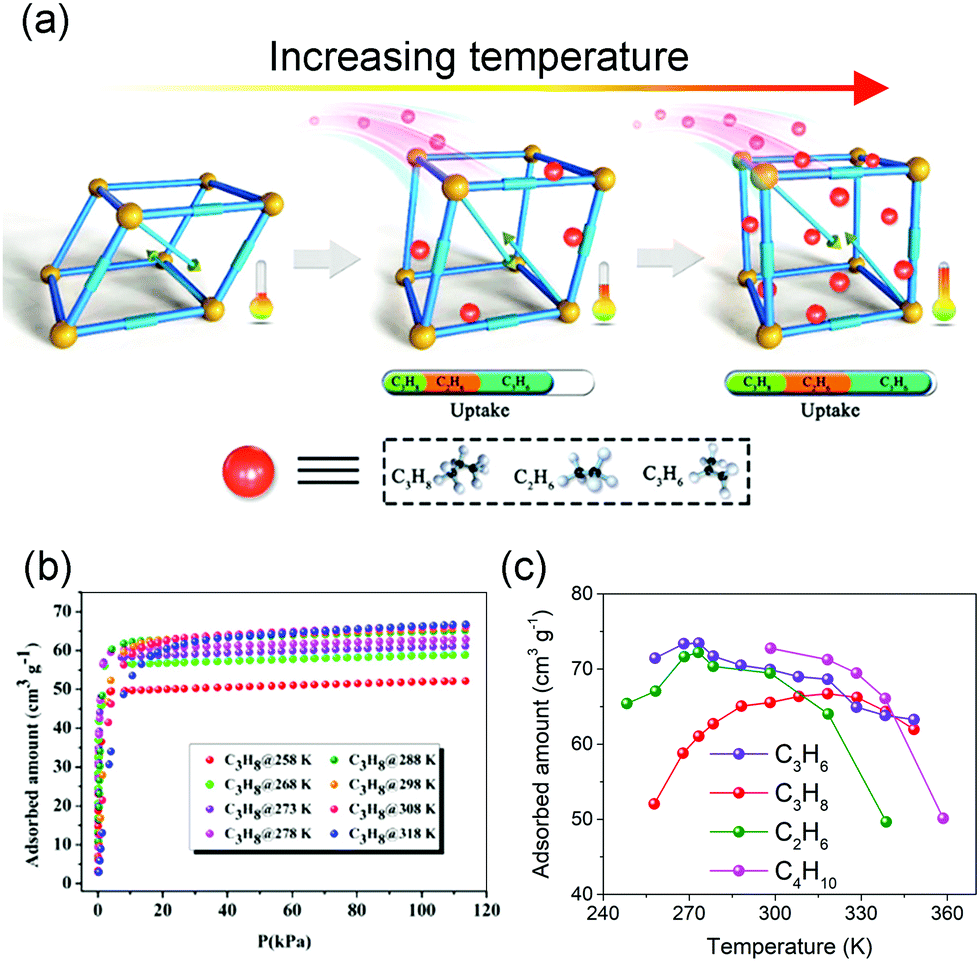 | ||
| Fig. 5 (a) Scheme of the enhanced gas adsorption in NKU-FlexMOF-1. (b) C3H8 adsorption isotherms from 258 to 318 K. (c) Temperature-dependent uptake amounts for C2H6 (green), C3H6 (purple), C3H8 (red) and C4H10 (pink). Reprinted with permission from ref. 61. | ||
Additionally, Krause et al. found that DUT-49 exhibits spontaneous desorption of gas during pressure increase, which was defined as “negative gas adsorption” (NGA).62 The structure of DUT-49 can be regarded as a face-centered cubic analogue arrangement of cuboctahedral metal–organic polyhedra (MOPs) with the fcu topology. The methane adsorption isotherm at 111 K showed an obvious decrease at 10 kPa (Fig. 6b), while the isotherm of N2 at 77 K show a typical slope. In situ synchrotron diffraction measurements revealed that the framework undergoes an enormous volumetric contraction of ∼53% (from 100072 Å3 to 47282 Å3, Fig. 6c) during the NGA, which is caused by the concerted rotation of the MOPs around the [111] axis (Fig. 6a). The structural deformation was also confirmed by the calculation of the elastic properties, in which the shear modulus is as low as 1.5 GPa. Density functional theory (DFT) calculations also revealed that the energy penalty of transition from LP phase to CP phase can be compensated by the adsorption of methane.
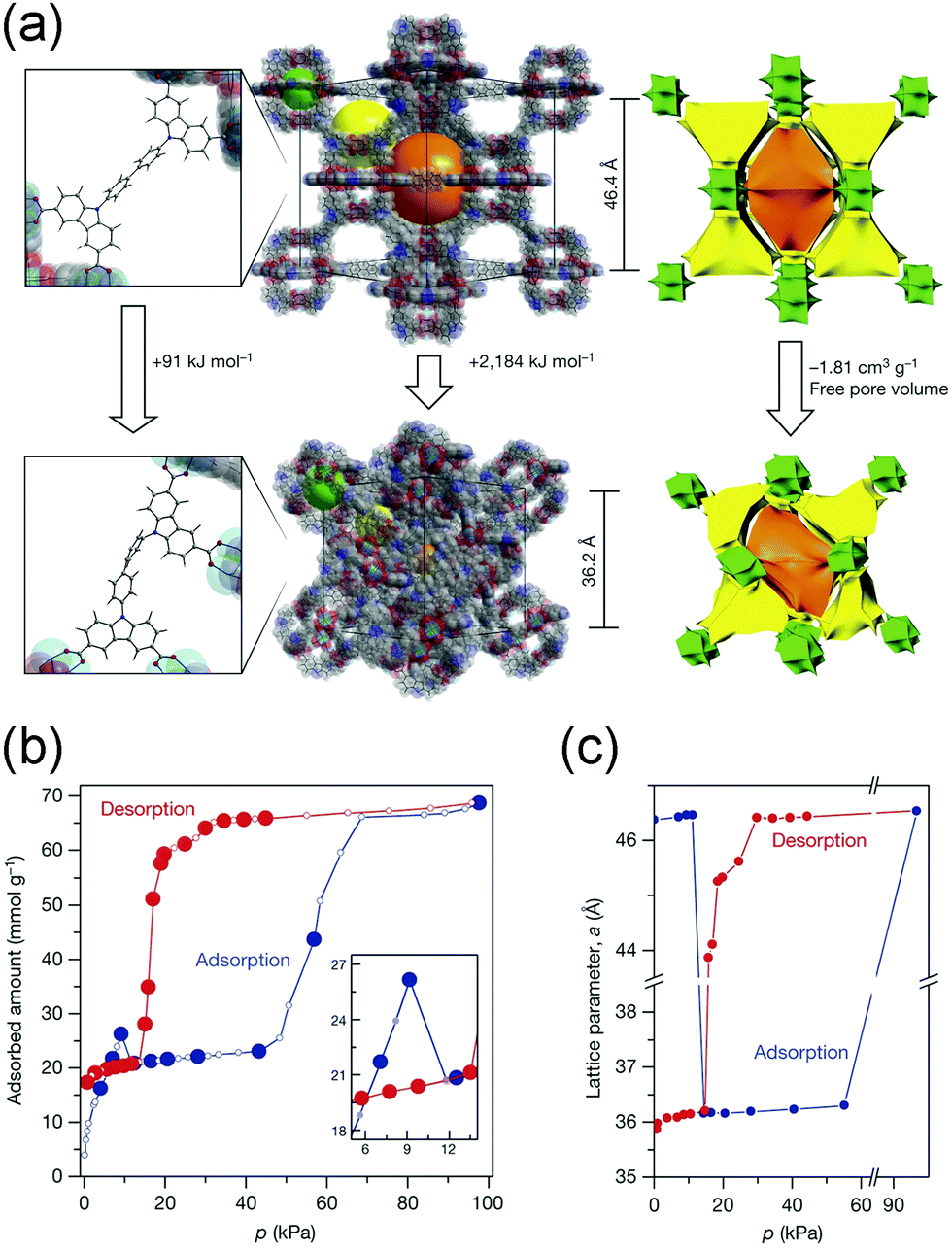 | ||
| Fig. 6 (a) Illustration of the structural changes from the DUT-49-LP to DUT-49-NP phase. (b) Methane adsorption and desorption isotherms at 111 K. (c) Lattice parameter change of DUT-49 during the adsorption (blue) and desorption (red) of the methane at 111 K. Reprinted with permission from ref. 62. | ||
In some cases, the adsorption induced structural changes is coupled with electronic structural changes, which can influence materials’ macroscopic colors and magnetism. Kepert et al. found that Fe2(azpy)4·(guest) (azpy = trans-4,4′-azopyridine) displayed guest-dependent spin crossover.63 The Fe2(azpy)4·(guest), an interpenetrated framework, exhibited a considerable flexibility during the solvent uptake and release. Take Fe2(azpy)4·(EtOH) (EtOH = ethanol) for example, the desorption of EtOH will induce a scissors-like motion, which can cause the pore volume decreasing from 12 to 2%. It should be noted that there are hydrogen-bonding interactions between the ethanol OH groups and sulfur atoms of the thiocyanate (Fig. 7a), which can induce a symmetry breaking, resulting in a phase transition (monoclinic system with the space group C2/c for Fe2(azpy)4·(EtOH); orthorhombic system with the space group Ibam for Fe2(azpy)4). It's intriguing that the Fe2(azpy)4·(EtOH) underwent “half-spin” crossovers, while this switching property is absent in the desorbed phase (Fig. 7b). It is speculated that the flexibility of the material may facilitate a low-energy pathway between the high-spin and low-spin structures. Similarly, the adsorption of gases can also influence materials’ electronic structures. Since the coordinatively unsaturated metal sites of MOFs tend to act as active sites to adsorb gases, the gas uptake will modify the local coordination modes of metal ions, which may concern the electronic structures. For example, the adsorption of CO for Fe2Cl2(bbta) (H2bbta = 1H,5H-benzol(1,2-d:4,5-d′′)bistriazole) can induce a transition from high-spin to low-spin state.64
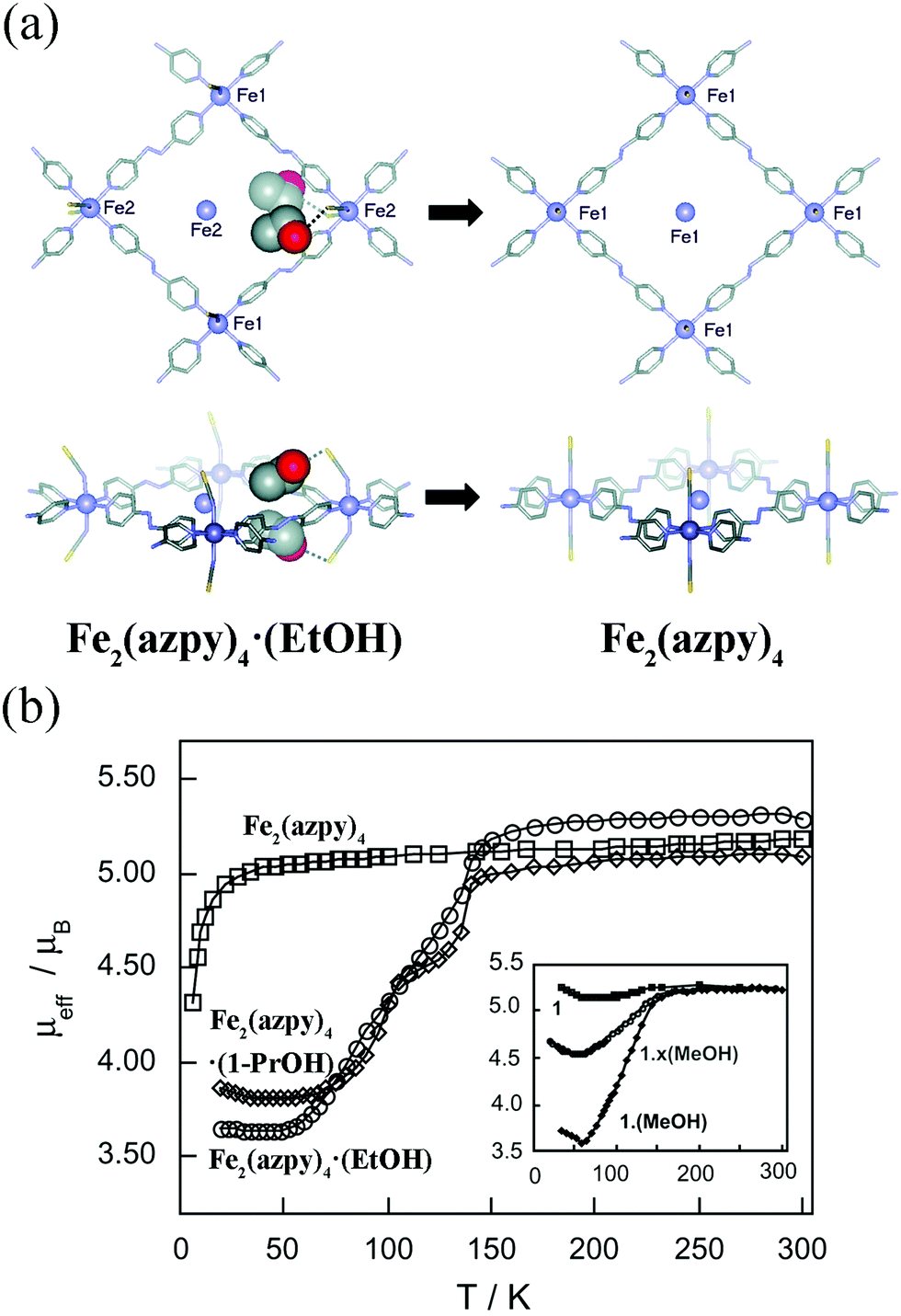 | ||
| Fig. 7 (a) The rhombic grid layers of Fe2(azpy)4·(EtOH) and Fe2(azpy)4. Removal of ethanol causes extensive modification to the structure, including a rotation of both the azpy andthiocyanate units, a translation of the interpenetrating layers by 1.0 Å (Fe atoms shown inside the rhomboids belong to the interpenetrating layers), and the removal of hydrogen-bonding interactions from the ethanol to the thiocyanate sulfur atoms (shown as dashed lines). (b) The temperature-dependent magnetic moment of Fe2(azpy)4·(guest). Reprinted with permission from ref. 63. | ||
Temperature
The most basic response of materials toward thermal stimuli is thermal expansion, which originates from the asymmetry potential energy well of paired atoms.65 The coefficients of thermal expansion (CTEs) can be defined as: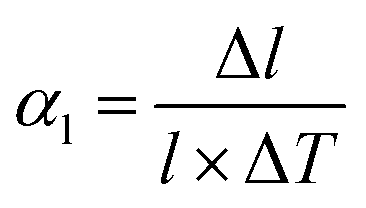
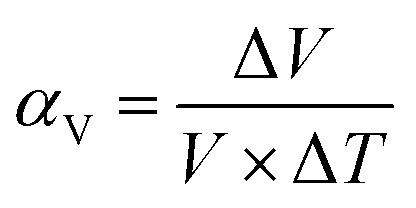
In the equations, αl and αV represent the linear and volumetric CTEs, respectively. Δl and ΔV stands for the relative changes in length and volume, l, V are the initial length and volume. The ΔT represents the change of temperature. For traditional inorganic materials, the linear CTE locates in the range 1–20 mK−1, while some MOFs can exhibit abnormal behavior benefitting from the large flexibility, such as negative thermal expansion (NTE),66 zero thermal expansion (ZTE) and colossal positive thermal expansion (PTE, |αl| > 100 mK−1) etc.67 According to the definition of CTE, herein we divided the NTE into two parts, linear (or areal) NTE and volumetric NTE.
Brown et al. found that the breathing behavior of MIL-53 can not only be triggered by guest insertion, but also thermal stimuli. The desolvated LP phase of MIL-53 can transform to the NP phase at 325–375 K, while the reversed transition occurs at 125–150 K, exhibiting a large temperature hysteresis.68 Owing to the existence of a phase transition and an abrupt lattice change, it's hard to ascertain the CTEs. While Attfield et al. found that the thermal induced breathing effect can be restrained by substituting the OH− in the 1D inorganic SBUs with F−, and the volumetric CTEs of the orthorhombic MIL-53 can switch from positive to negative.69
Burtch et al. studied the thermal expansion behavior of Zn2(BDC)(dabco) in the range 150–375 K.70 This compound crystallizes in the tetragonal phase (P4/nbm) and possesses a pillared-layered motif (Fig. 8a). The layers are constructed from Zn2(COO)2 paddle wheel SBUs and BDC ligands, which are further pillared by dabco. Upon heating, Zn2(BDC)(dabco) exhibits NTE in the ab plane with CTE ∼ −10 mK−1, and PTE along the c axis with the CTE + 15–21 mK−1. While the decoration of the BDC ligand with four methyl groups (TMBDC, 2,3,5,6-tetramethyl-1,4-benzenedicarboxylate) can switch the NTE to PTE without breaking the lattice symmetry (Fig. 8b), which may be caused by the increased steric effect. Additionally, partial substituting of BDC with TMBDC (2,3,5,6-tetramethyl-1,4-benzenedicarboxylate) can give rise to a series of solid solutions, and can continuously tune the thermal expansion behavior in ab plane from negative to positive. Intriguingly, decoration of the BDC ligand with 2-methoxyethoxy substituents at the 2,5 sites of the phenyl will result in a phase transition from the parent tetragonal system to the monoclinic system (C2/m, NP),71 which may be caused by the lower symmetry of the ligand and larger steric hindrance (Fig. 8c). The 1D channel along c axis will also change from the original square shape to the diamond-shape. Upon heating, the diamond-shaped channel showed win-rack distortion and gave rise to the LP phase (tetragonal phase) at 489 K. To avoid the influences of the difference between the NP and LP phase, re-defined lattice vectors were used to describe the thermal expansion behaviors. Before the breathing transition (303–463 K), the compound undergoes a continuous opening up, accompanied by a large anisotropic thermal expansion behavior. The CTEs along two vertical axes can reach −380 mK−1 and +1161 mK−1 at 463 K, respectively. Further increasing temperature will cause the NP to LP transition and a volume change up to 19% can be observed. Kim et al. found that the insertion of guest DMF molecules can effectively tune the NTE in ab plane to PTE, which may be caused by the host–guest interactions and the steric effects.72 Some similar flexible MOFs with the “wine-rack” motif have been reported to show linear or areal NTE (Table 1).
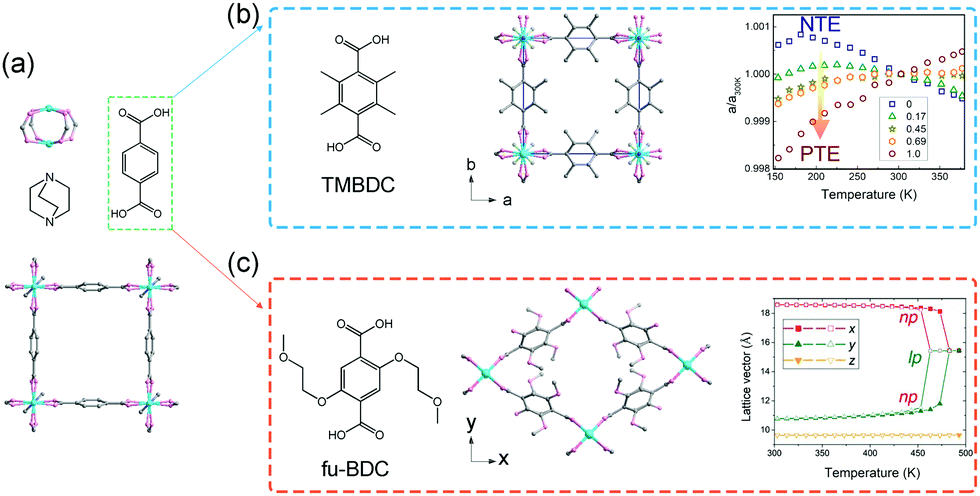 | ||
| Fig. 8 (a) Crystal structure of the Zn2(BDC)(dabco) viewing along the c axis and its building units. (b) Illustration of the structure after TMBDC decoration and the lattice parameter changes as a function of temperature and composition. (c) Illustration of the structure after fu-BDC decoration and the temperature-dependent lattice changes. Partly taken from ref. 70 and 71 with permission. | ||
| Name | CTE (mK−1) | Temperature range (K) | Ref. |
|---|---|---|---|
| HMOF-1 | α a = +177, αb = −21, αc = +3 | 160–320 | 73 |
| Ag(mim) | α x1 = +130, αx2 = +44, αx3 = −25 | 20–300 | 74 |
| MCF-34 | α a = +2, αb = +224, αc = −107 | 123–673 | 75 |
| MCF-34·DMA | α a = +25, αb = +213, αc = −101 | 156–271 | |
| MCF-34·DMF(LT) | α a = +16, αb = +152, αc = −56 | 127–208 | |
| MCF-34·DMF(HT) | α a = +36, αb = +237, αc = −116 | 233–299 | |
| Ag4(tpt)4{δ-[Mo8O26]}·1.5H2O | α x1 = −18, αx2 = +75, αx3 = −5 | 100–340 | 76 |
| Ag4(tpt)4{δ-[Mo8O26]} | α x1 = −22, αx2 = −7, αx3 = +77 | 100–500 | |
| FJI-H11-R | 100–300 | 77 | |
| R = Me | α a = −38, αc = +653 | ||
| R = Et | α a = −33, αc = +489 | ||
| R = iPr | α a = −25, αc = +404 | ||
| [Cd(HBTC)(BPP)]·1.5DMF·2H2O | α a = −43, αb = +2, αc = +133 | 100–260 | 78 |
| SION-2 | α x1 = −153, αx2 = +43, αx3 = +230 | 100–340 | 79 |
It should be noted that, the linear (or areal) NTE is not only restricted in the MOFs with the “wine-rack” motif. For instance, we found that some flexible MOFs with helical axes can exhibit 1D NTE along the helical direction owing to the spring-like thermal motion.80–82 Zhang et al. found that the MCF-82 can exhibit “jack-like” thermal motion, resulting in a colossal anisotropic thermal expansion with the CTE reach +482/−218 mK−1.83 Recently, we found that the PbC4O4 (H2C4O4 = square acid) with a pillar-layered structure can exhibit a rare 1D ZTE along the packing direction, the linear CTE is only −0.61 mK−1 (Fig. 9a and b).84 By the utilization of synchrotron X-ray total scattering and Raman spectra, we found that, in the inorganic Pb–O layer, the combination of out-plane rotation and geometric deformation of rhombic Pb2O2 plane results in a NTE behavior (Fig. 9c and d). While the organic squarate pillar exhibits normal PTE. So the observed uniaxial ZTE originates from the compensation effect between the NTE in the inorganic layer and PTE in the organic pillar (Fig. 9e).
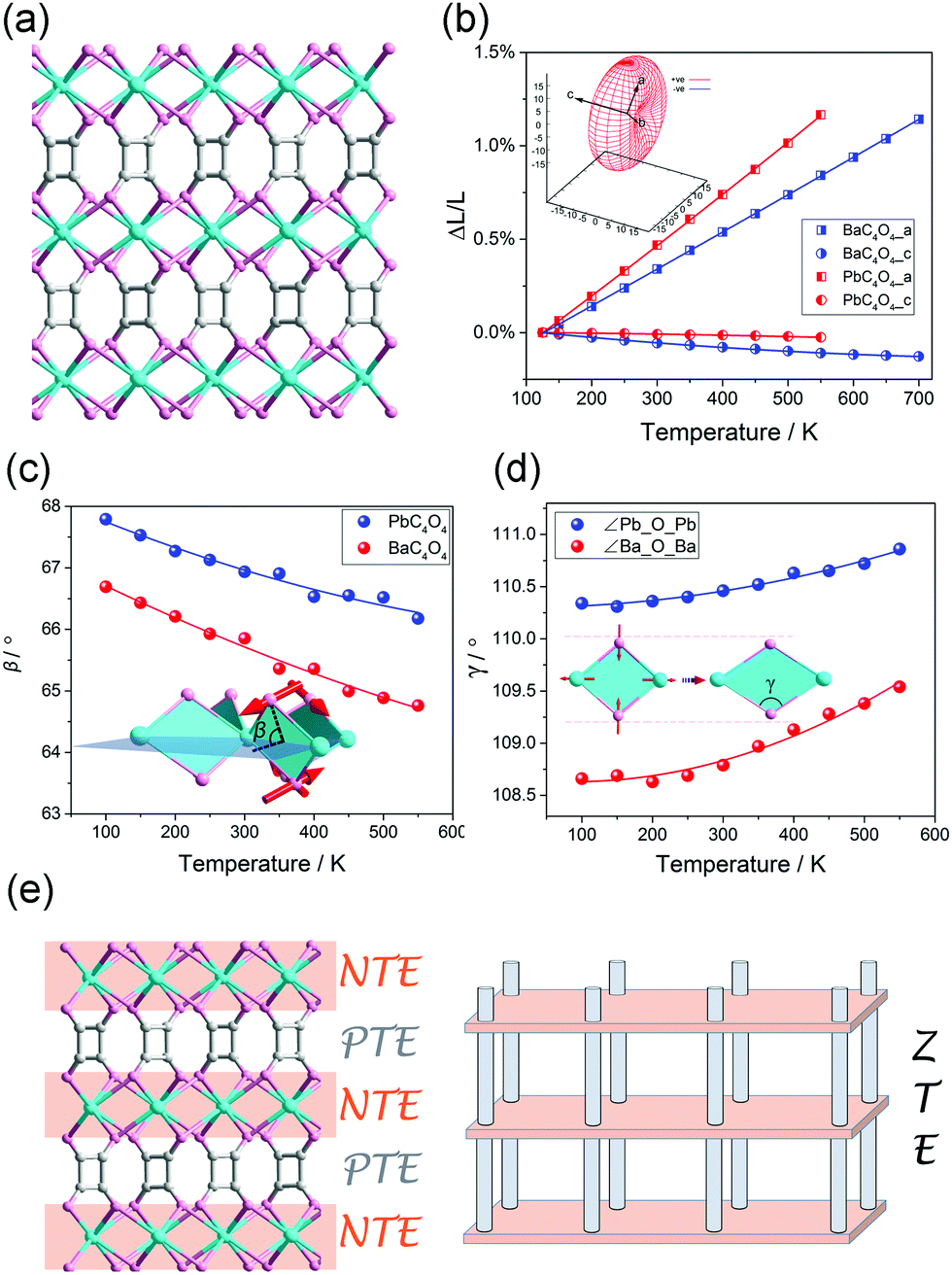 | ||
| Fig. 9 (a) Crystal structure of pillar-layered MC4O4 (M = Ba, Pb). (b) Lattice parameter changes of the MC4O4 (M = Ba, Pb) obtained from Le bail fitting. Illustration of the out-plane rotation (c) and geometric deformation (d) of rhombic Pb2O2 (or Ba2O2) plane. (e) Scheme of the mechanism of 1D ZTE which concerns of the NTE in the inorganic layers and PTE of the organic pillar. Reprinted with permission from ref. 84. | ||
Compared with the linear NTE, the volumetric NTE is much rarer. Yaghi et al. performed in situ diffraction measurements to study the gas adsorption sites in MOF-5. They found that the unit cell volume of gas loaded MOF-5 in low temperature is slightly larger than its RT value (∼200 Å3), so they proposed that MOF-5 may exhibit volumetric NTE.85 Later, Snurr et al. proved that MOF-5 can exhibit isotropic NTE by the mean of molecular simulation,66 and they also found that the magnitude of NTE can be enhanced by extending the length of the ligands. Among them, the IRMOF-16 shows the strongest NTE with the volumetric CTE −60 to −80 mK−1, about three times as that of ZrW2O8 (−8.9 mK−1). Kepert et al. found that Cu3(BTC)2 (BTC = 1,3,5-benzenetricarboxylate) exhibit isotropic NTE in the temperature range 100–500 K with the average linear CTE αa = −4.1(1) mK−1.86 Detailed structural analyses shed light on that the NTE is caused by the transverse vibration of the aromatic ring and the twisting vibration of the paddle-wheel unit.87 Extending the length of ligand will result in a two-interpenetrated Cu3(BTB)2 (BTB = 4,4′,4′′-benzene-1,3,5-triyl-tribenzoate).88 Normally, framework interpenetration will weaken the magnitude of NTE owing to the steric effect. While Kepert et al. found that the large thermal expansion of the π⋯π interactions can result in repulsion between the central phenyl of the BTB ligands, which will be help to enhance the magnitude of NTE (αa = −13 mK−1).89 Some other MOFs have also been reported to exhibit NTE, such as UiO-66 and its analogue.90,91
The volumetric NTE usually originates from thermal vibration of the subnetwork (ligand rotation, blending etc.), of which the magnitude is usually much smaller than the framework geometry changes (such as breathing behavior). For a flexible MOF, the contribution from the thermal vibration may be frustrated by the geometry changes of the framework. So the reported volumetric NTE materials were mainly restricted in isotropic MOFs (cubic phase). To enrich the kinds of volumetric NTE MOFs, we investigated the thermal expansion behavior of the “rigid” orthorhombic MIL-68(In) (Fig. 10a). The results show that MIL-68(In) can exhibit a rare 3D NTE along its all axes.92 The combination of single crystal X-ray diffraction and synchrotron based X-ray pair distribution function (PDF) analyses revealed that the NTE in ab plane derives from the transverse vibration of the phenyl group (Fig. 10b), while the NTE along c axis is a result of the rotation of the rigid InO6 octahedrons (Fig. 10c).
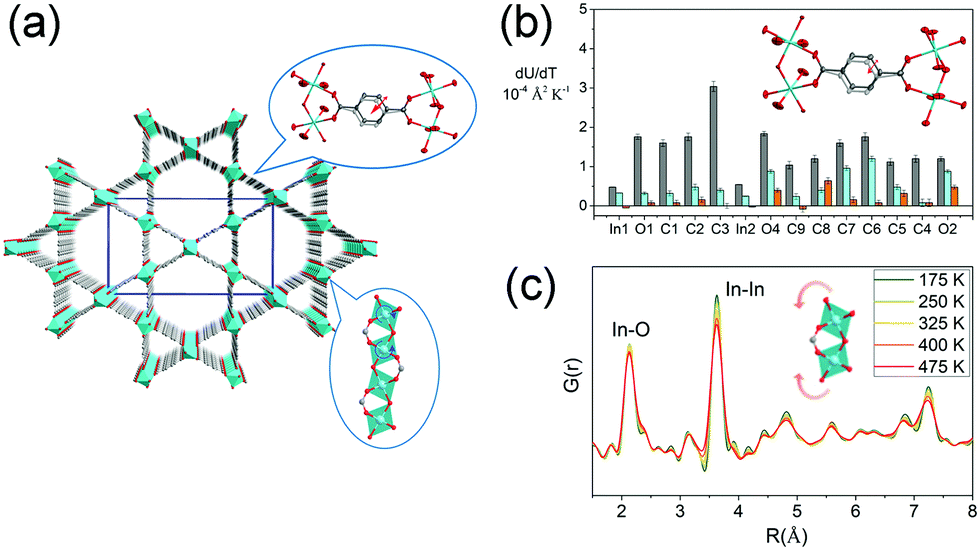 | ||
| Fig. 10 (a) Crystal structure of MIL-68(In) viewing along c axis. (b) Rates of change of anisotropic atomic displacement parameters (ADPs) with temperature. Rates are given for the three ADP axes. The major, intermediate, and minor axes are shown in gray, turquoise, and orange, respectively. The inset shows the illustration of the transverse vibration of the phenyl ring. (c) Partial atomic pair distribution function G(r) at different temperatures. The inset shows the illustration of the rotation of InO6 octahedrons. Partly taken from ref. 92 with permission. | ||
Recently, we found that the thermal expansion behavior is strongly dependent on the molecular packing mode. Firstly, we synthesized three kinds of metal squarate frameworks. Owing to the different packing mode of the squrate ligands (staggered packing and eclipsed packing), two kinds of polymorphs were achieved (Fig. 11a and b): cubic phase (termed as Zn_sq_C, Cd_sq_C) and trigonal phase (termed as Cd_sq_R).93 Without the high symmetry constraints, the trigonal phase Cd_sq_R can undergo a “wine-rack” distortion upon heating owing to the in-plane rotation of the squarate ligand (Fig. 11d). This distortion gave rise to an anisotropic thermal expansion behavior (PTE along c axis and NTE along a axis). The combination of single crystal X-ray diffraction and synchrotron based X-ray PDF shed light on that the different magnitude of the out-plane rotation of the squrate ligand caused the different thermal expansion behavior (Fig. 11c). The volumetric CTEs are +11.3(7) mK−1, +2.3(6) mK−1 and −13.9(6) mK−1 for Zn_sq_C, Cd_sq_C and Cd_sq_R respectively.
 | ||
| Fig. 11 Crystal structure of the cubic phase (a) and trigonal phase (b) of MC4O4·2H2O (M = Zn, Cd). (c) Temperature-dependent relative volume changes of the three compounds. (d) Scheme of the wine-rack distortion of the framework in the trigonal phase Cd_sq_R and illustration of the transverse vibration of the squarate ligand. Reprinted with permission from ref. 93. | ||
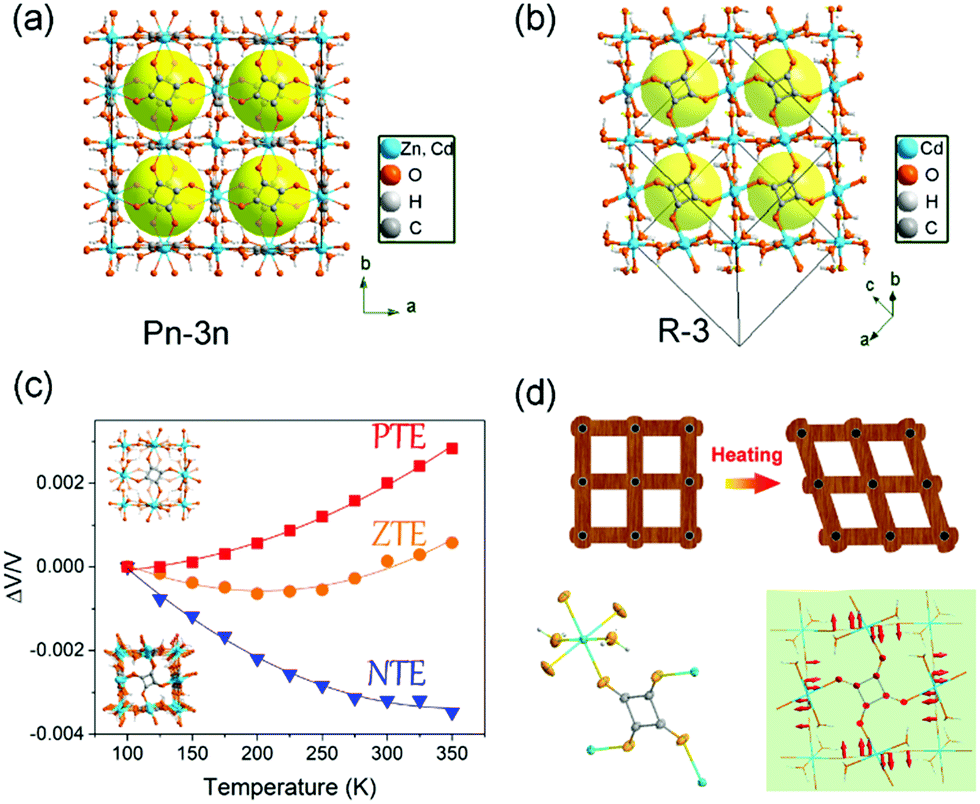
While in some cases, the thermal vibration won’t cause significant lattice changes. For example, under thermal stimuli, the benzene unit of the ligand in MOF-5 can rotate along its 1,4-axis,94 making it a candidate of molecular rotator.95 Although the lattice parameter won’t be influenced by the rotation behavior, the pore structure does change significantly. For example, the rotation of pyridyl rings in Mg(4-PyC)2 (4-PyC = 4-pyridine carboxylic acid) can cause a switching between a porous and nonporous state.96 Recently, Kitagawa et al. designed a Cu-based MOF with a butterfly-type ligand phenothiazine-5,5-dioxide (OPTZ) (Fig. 12a and b). Under thermal stimuli, the OPTZ moieties can exhibit a series of low energy motions including rotation and flipping.97 The thermal flipping of the OPTZ unit can gradually enlarge the size of the cage gate (Fig. 12c), which will facilitate the admission of gas molecules at high temperatures and block them at low temperatures. Since the optimal adsorption temperature (Tmax) is dependent on the gas type and gate-admission temperature, by finely control the temperature, this MOF can effectively separate O2/Ar and C2H4/C2H6 (the selectivity is up to ∼350 and ∼75, respectively). What's more, the temperature fluctuation can influence the thermal motion of counter ions in cationic MOFs, which can be utilized to control the gate opening and closing.98
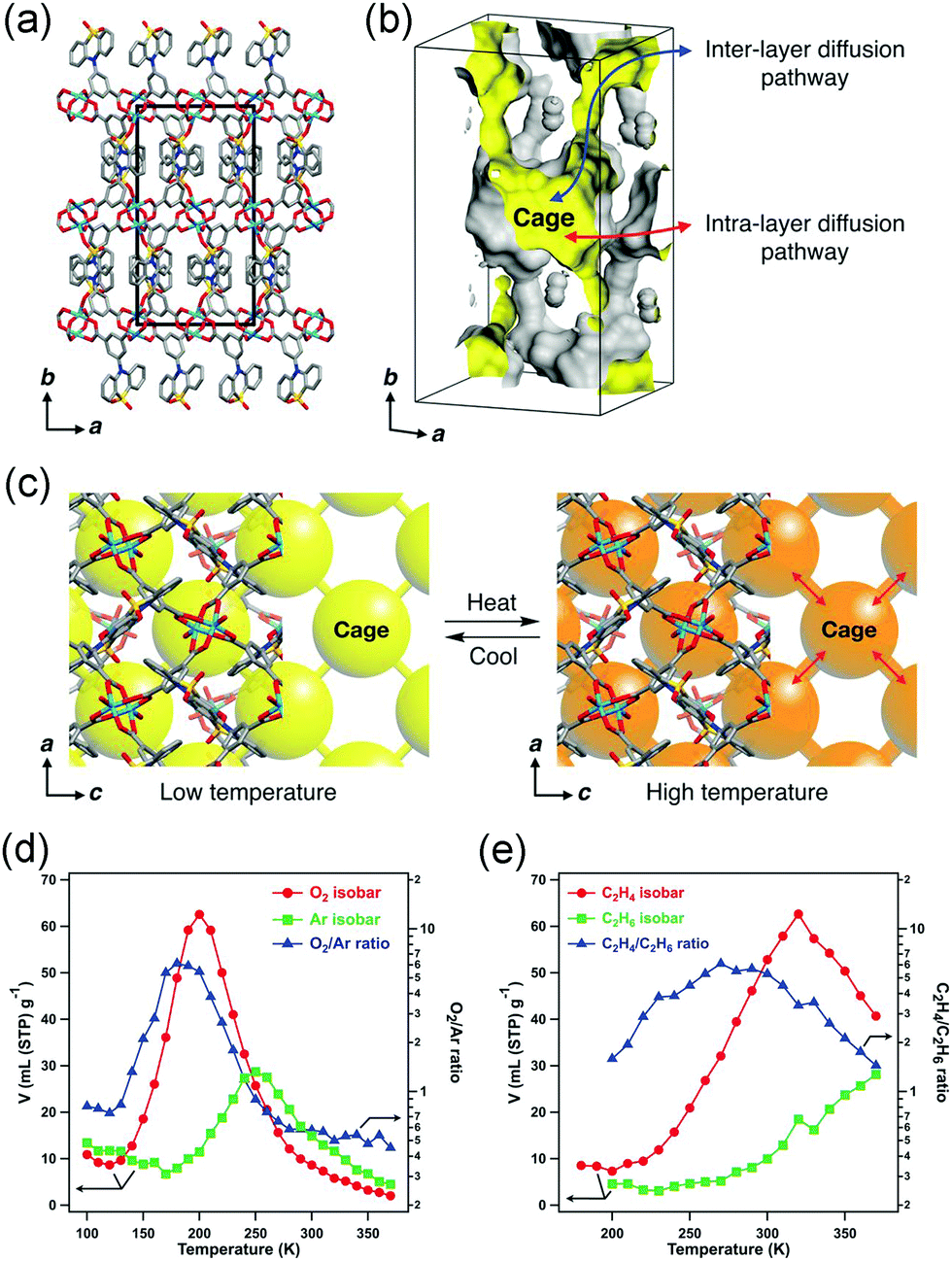 | ||
| Fig. 12 (a) Crystal structure of Cu(OPTZ) viewing along c axis. (b) Cross-sectional view of a Connolly surface of the void in Cu(OPTZ), where a small probe radius of 0.8 Å was used to visualize that the cage structure is interconnected with narrow channels. (c) Scheme of the voids structure changes at low and high temperatures. (d) O2 and Ar adsorption isobars at 1 bar and the O2/Ar uptake ratio. (e) C2H4 and C2H6 adsorption isobars at 1 bar and the C2H4/C2H6 uptake ratio. Reprinted with permission from ref. 97. | ||
Since the coordination bonds possess reversible nature, their breaking and re-formation can be realized by thermal stimuli. For example, [Yb2(C4H4O4)3] can reversibly transform between two polymorphs.99 At ambient temperature, the Yb2(C4H4O4)3 adopts α form, while when the temperature increases to 408 K, it will translate to the β form accompanied by ∼10% volume expansion (both phases two asymmetric units in the unit cell). During this transition, the coordination number of Yb atom will be reduced from eight to seven. The thermal induced breaking of the Yb–O3′ bond will cause the rupture of the chains along a axis to the dimeric units (Fig. 13a and b), resulting in the expansion of lattice parameter a and unit cell volume.
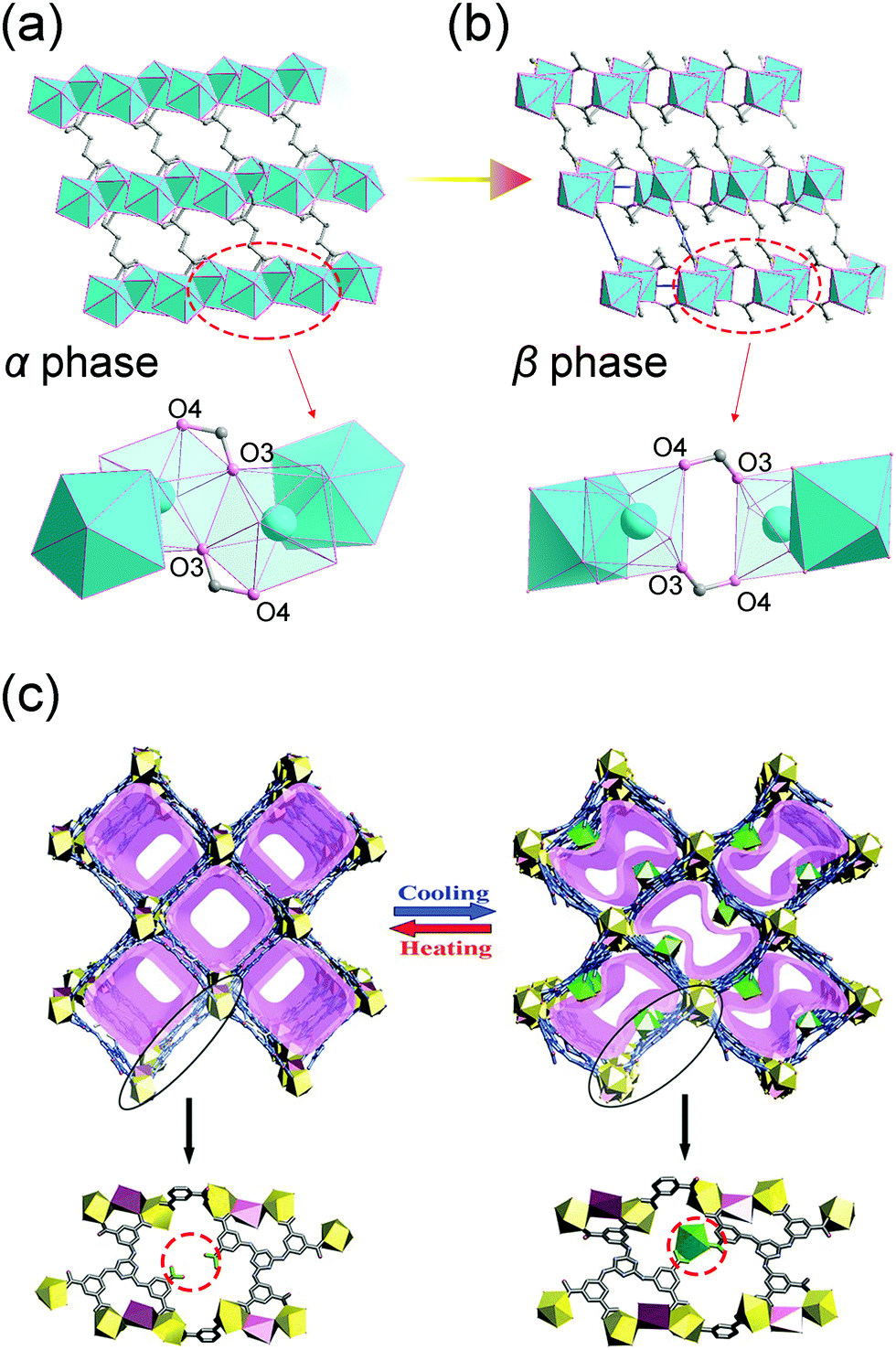 | ||
| Fig. 13 Illustration of the structural changes of [Yb2(C4H4O4)3] at low (a) and high (b) temperatures. Top: The overview of the framework. Bottom: Local SBU changes. (c) Scheme of the temperature induced structural changes. Top: The overview of the 3D framework. Bottom: The local enlarged view of the structural difference. Partly taken from ref. 99 and 100 with permission. | ||
Similarly, Zhang et al. synthesized a Eu based flexible MOF that shows a single-crystal to single-crystal (SCSC) phase transition (Fig. 13c).100 At ambient temperature, this compound possesses one-dimensional square channels, which are filled with intrinsic free Eu3+. For each ligand, there is one uncoordinated carboxylate group, which can act as a hook. At low temperature (193 K), the compound can quickly capture the intrinsic free Eu3+ by the carboxylate-hooks, while increasing the temperature will cause the captured Eu3+ to escape the hooks.
Cheetham et al. found that the compound ZIF-4, composed of Zn2+ and imidazole ligands, can undergo a series of phase transitions under thermal stimuli. For example, upon cooling, the activated porous ZIF-4 reversibly translates to a dense phase accompanied with ∼23% volume contraction (at 140 K) (Fig. 14a and b). The combination of synchrotron X-ray powder diffraction analyses and DFT calculations revealed that the isotropic framework contraction originates from the concerted rotation of the imidazolate linkers.101 While this transition can be blocked by the guest DMF molecules insertion. Heating the ZIF-4 to 573 K can induce an irreversible transition to a dense topologically disordered phase (amorphous) with the same composition as crystalline ZIF-4 (Fig. 14c). Further increasing the temperature to 673 K results in a recrystallization behavior and a more condensed phase with the zni topology (termed as ZIF-zni) can be achieved (Fig. 14d).102
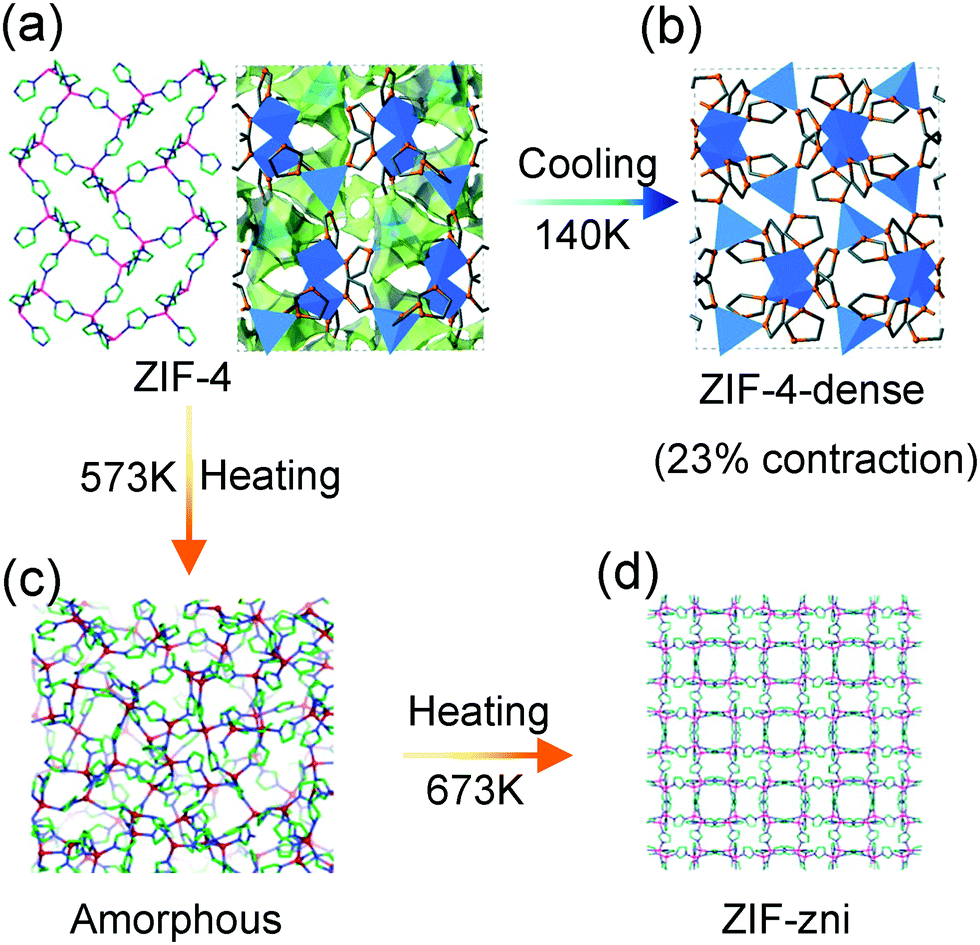 | ||
| Fig. 14 Scheme of the temperature induced structural changes of ZIF-4. Crystal structure of ZIF-4 at ambient temperature (a), 140 K (b), 573 K (c) and 673 K (d). Reprinted with permission from ref. 101 and 102. | ||
Pressure
Owing to the high surface areas of MOFs, the application of mechanical pressure can easily distort the framework or the pore structure. Normally, two kinds of pressure can be applied to a material, including hydrostatic pressure and uniaxial pressure (non-hydrostatic pressure).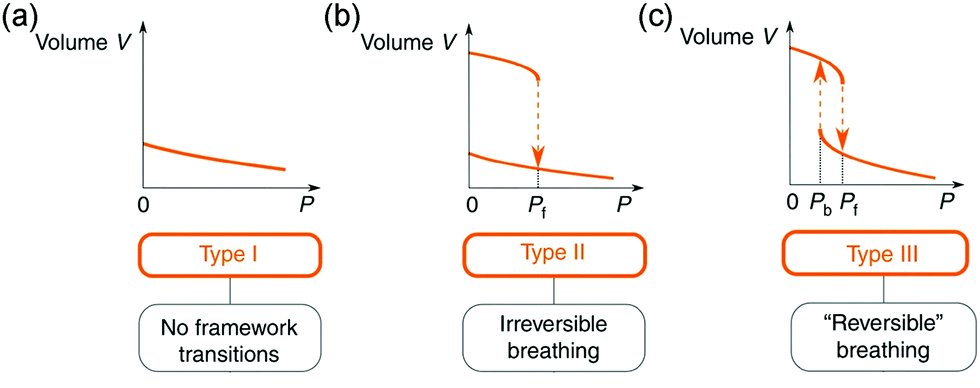 | ||
| Fig. 15 Illustration of the possible volume change of a material under hydrostatic pressure. (a) Type 1: continuous volume decrease without framework transition. (b) Type II, an irreversible abrupt volume decrease at a certain pressure. (c) Type III, reversible breathing. Reprinted with permission from ref. 103. | ||
Generally, materials contract in all directions when a hydrostatic pressure is applied. However, a few anisotropic materials can exhibit negative linear or area compressibility (NLC or NAC).104 It should be noted that the negative volumetric compressibility is forbidden by the laws of thermodynamics. Some experimentally observed volume expansions under hydrostatic compression are actually caused by the entrance of the pressure transmitting medium into the pores (ZIF-8, MOF-5, HKUST-1).105–107
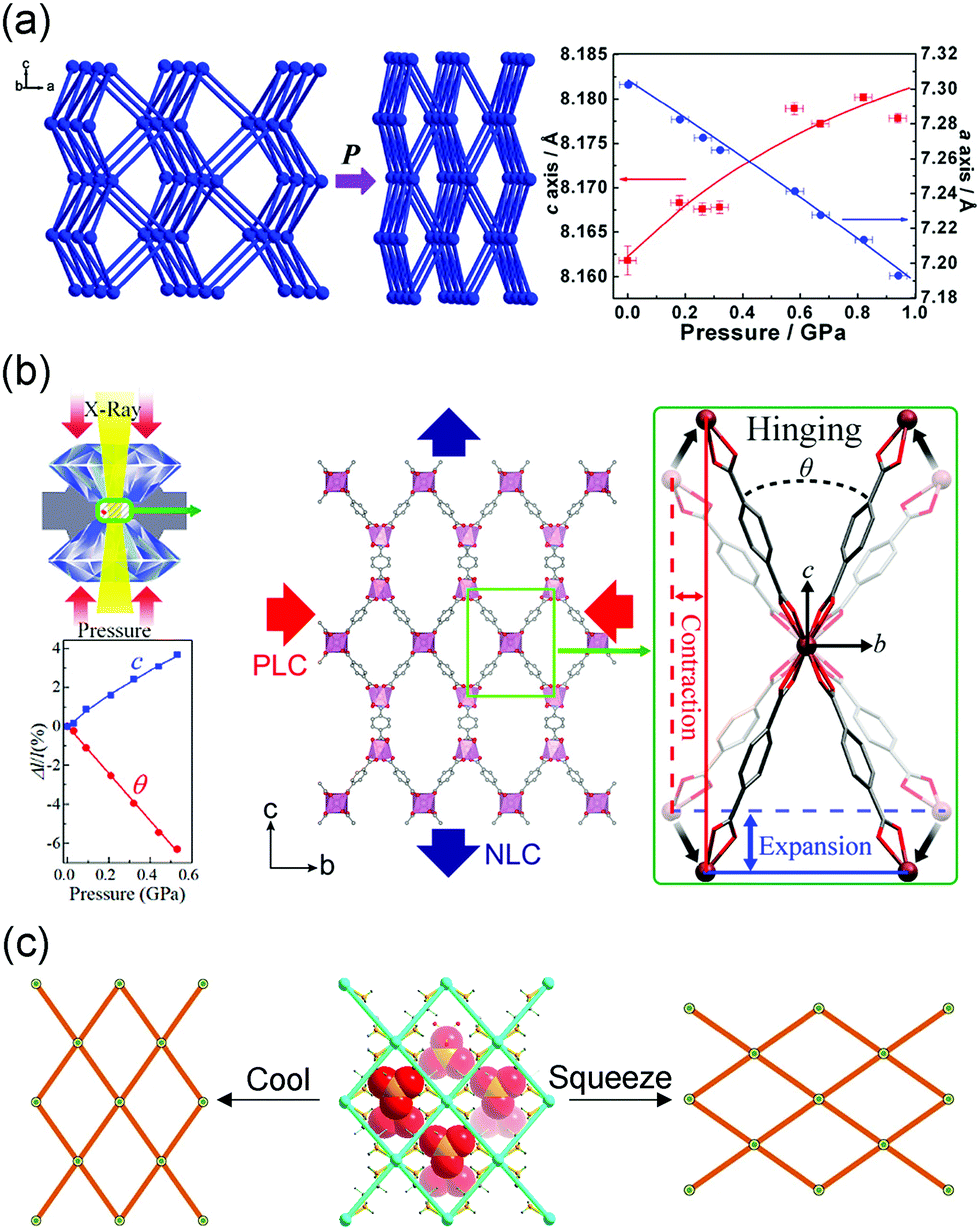
The NLC was initially discovered in inorganic compounds, such as LaNbO4 (Kl = −(∂lnl/∂p)T, Kl = −0.2 TPa−1),108 BAsO4 (Kl = −2 TPa−1),109 Se (Kl = −1.2 TPa−1).110 Cheetham et al. found that the MOF [NH4][Zn(HCOO)3] with the acs topology can exhibit NLC in the range of 0–1 GPa along its c axis with the coefficient Kc = −1.8 TPa−1.111In situ high pressure single crystal X-ray diffraction and DFT calculations revealed that the synergy effect of the contraction of Zn–O bonds and tilting of formate ligands caused the contraction of framework in the ab plane and NLC along the c axis (Fig. 16a). MFM-133, with a three-dimensional wine-rack motif, can exhibit NLC along its c axis with the coefficient −5.1 TPa−1 in the range 0–4.4 GPa.112 The NLC originates from an elongation of the octahedral cage. The substitution of Zr4+ with Hf4+ can slightly enhance the NLC to −7.9 TPa−1. Zou et al. found the β-quartz type MOF InH(BDC)2 can exhibit an extreme NLC with the coefficient −62.4 TPa−1 in the range 0–0.53 GPa (Fig. 16b).113 Some MOFs with the “wine-rack” geometries have been reported to show NLC and the details are listed in Table 2.
 | ||
| Fig. 16 Scheme of the wine-rack structural changes under hydrostatic pressure that exhibit NLC. (a) [NH4][Zn(HCOO)3]. (b) InH(BDC)2. (c) [Ag(ethylenediamine)]NO3. Partly taken from ref. 111, 113, and 116 with permission. | ||
| Name | Coefficient (TPa−1) | Pressure range (GPa) | Ref. |
|---|---|---|---|
| [NH4][Zn(HCOO)3] | β a = +16, βc = −1.8 | 0–1 | 111 |
| MFM-133(Zr) | β a = +6, βc = −5 | 0–4.4 | 112 |
| MFM-133(Hf) | β a = +10.7, βc = −7.9 | 0–4.9 | |
| InH(BDC)2 | β a = +102.4, βc = −62.4 | 0–0.53 | 113 |
| MIL-53(Al) | β a = +23, βb = −28, βc = +59 | 0–3 | 114 |
| NH2-MIL-53(Al) | β a = +99, βb = −27, βc = +76 | 0–2 | |
| Ag(mim) | β x1 = +55, βx2 = +26, βx3 = −4 | 0–6.4 | 74 |
| MCF-43 | β x1 = +120, βx2 = +57, βx3 = −47 | 0–0.77 | 115 |
It should be noted that, typically, there is an “inverse relationship” between temperature and pressure, so the NLC is usually accompanied by NTE in the same direction in the wine-rack structures. While Katrusiak et al. found that [Ag(ethylenediamine)]NO3 can exhibit NLC with the coefficient −28.4(18) TPa−1 coupled with large positive thermal expansion with the coefficient +149.2(12) mK−1 in the same direction, and the NTE is coupled with PLC in another direction (Fig. 16c).116 The unique phenomenon results from an interplay of the framework flexibility and the interactions between framework and NO3− anions. Increasing the temperature will increase the NO3− anions dynamics and the interactions with the framework, giving rise to a more rounded wine-rack. While under high pressure, the structural changes are mainly dominated by the scissor-like distortion.
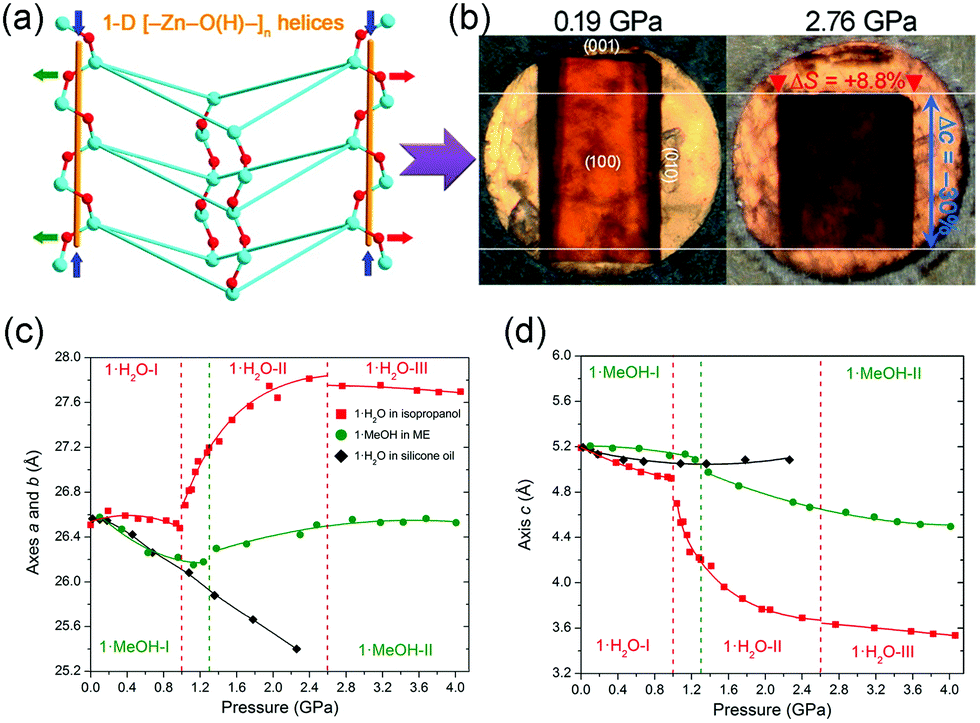
NAC means a material exhibit NLC along two directions, so it's rarer than NLC. As far as we know, to date, only two MOFs with NAC have been reported. Goodwin et al. found that Ag(tcm) (tcm = tricyanomethanide) exhibit NAC in the ac plane (βa = −3.5 TPa−1, βb = +66 TPa−1, and βc = −4.0 TPa−1), which arises from the flattening of the honeycomb-like layers under hydrostatic compression.117 Katrusiak et al. synthesized a MOF [Zn(L)2(OH)2]·H2O (L = 4-(1H-naphtho[2,3-d]imidazol-1-yl)benzoate),118 which crystallizes in the tetragonal phase with the space group I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) . The framework is composed of strongly puckered quadrangular nets which are interconnected through helical [–Zn–O(H)–]n chains along the c axis (Fig. 17a). Under hydrostatic pressure (1.0–2.6 GPa), the compression of helices and flattening of puckered rings combine to give an expansion in the ab plane (∼4.2%) and a substantial compression along the c axis (∼−21.5%) (Fig. 17c and d). The NAC coefficient for this compound in the ab plane can reach −72 TPa−1, which is the strongest ever reported. What's more, replacing the guest H2O molecules with MeOH will largely weaken the magnitude of NAC (∼15 times).
. The framework is composed of strongly puckered quadrangular nets which are interconnected through helical [–Zn–O(H)–]n chains along the c axis (Fig. 17a). Under hydrostatic pressure (1.0–2.6 GPa), the compression of helices and flattening of puckered rings combine to give an expansion in the ab plane (∼4.2%) and a substantial compression along the c axis (∼−21.5%) (Fig. 17c and d). The NAC coefficient for this compound in the ab plane can reach −72 TPa−1, which is the strongest ever reported. What's more, replacing the guest H2O molecules with MeOH will largely weaken the magnitude of NAC (∼15 times).
 | ||
| Fig. 17 (a) A perspective view of the quadrangular rings connected by the [–Zn–O(H)–]n helices to form a column along the c axis. (b) Photograph of a single crystal of [Zn(L)2(OH)2]·H2O compressed in isopropanol in the diamond-anvil cell (DAC) chamber. Lattice parameter changes along a axis (c) and c axis (d). Reprinted with permission from ref. 117. | ||
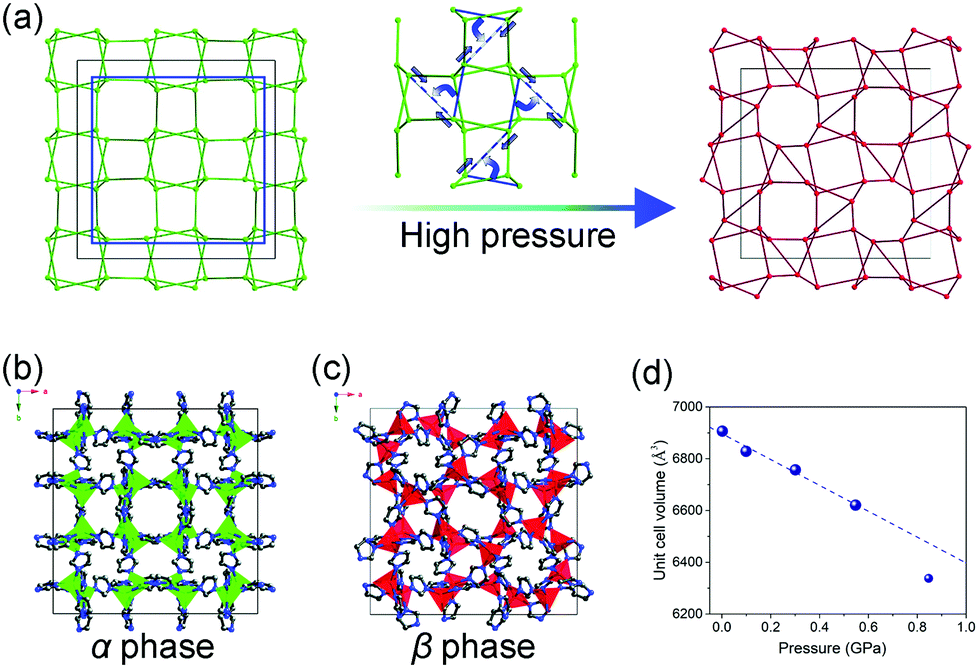
The inherent pores of MOFs make them easy to be compressed, when the applied pressure surpasses the limit that MOF can stand, the framework may collapse and form a dense phase. For example, ZIF-8 loses porosity and long range order upon compression at ∼2 GPa.119 The number coordination bonds between Zr(IV) ions and carboxylate groups in UiO-66 decreases by half at 1.9 GPa.120 This kind of volume change usually correspond to the type II (Fig. 15b), while the collapsed frameworks are usually amorphous state, which makes it difficult to directly obtain the unit cell volume. While in some cases, the pressure can induce structural changes without loss of crystallinity. The ZIF-zni (α-phase) used to be known as the most stable state among its polymorphs (ZIF-1, ZIF-2, ZIF-3, ZIF-4 etc.).121,122 While it can transform to a denser β-phase within 0.54–0.85 GPa pressure range (Fig. 18).123 A symmetry reduction can be observed as the crystal converts from the I41cd (α-phase) to its subgroup I41 (β-phase) (Fig. 18b and c). The mechanism of the transition involves cooperative bond rearrangements in the ab plane (Fig. 18a), which results in a contraction of distance between two neighboring Zn2+ from 7.1072(5) Å to 5.934(1) Å. This phase transition is accompanied by a contraction of unit cell volume by 2.1% (3.2% contraction of lattice parameter a and 4.5% expansion of lattice parameter c) (Fig. 18d). Additionally, the β-phase can be retained upon pressure release, indicating the irreversibility of this transition.
 | ||
| Fig. 18 (a) Scheme of the framework change under high pressure. Crystal structure of ZIF-zni in α phase (b) and β phase (c). (d) Unit cell volume of ZIF-zni at different temperatures. Partly taken from ref. 123 with permission. | ||
The hydrostatic pressure induced phase transitions can also be reversible. For these materials, the unit cell volume changes usually correspond to type III (Fig. 15c). For example, the compound [tmenH2][Er(HCOO)4]2 (tmenH22+ = N,N,N′,N′-tetramethylethylenediammonium) can undergo a reversible first-order phase transition at about 0.6 GPa (Fig. 19a), which is accompanied by a significant volume contraction of 10% (Fig. 19c and d).124 The transition arises from a striking pressure induced bond arrangement. The chelating formate groups that bound to the Er3+ will convert to bridging μ2-ligands connecting neighboring Er3+ under high pressure (Fig. 19a and b), which results in a reduction of distance between two neighboring Er3+ from ∼8.3 (ambient) to ∼6.6 Å (1.07 GPa).
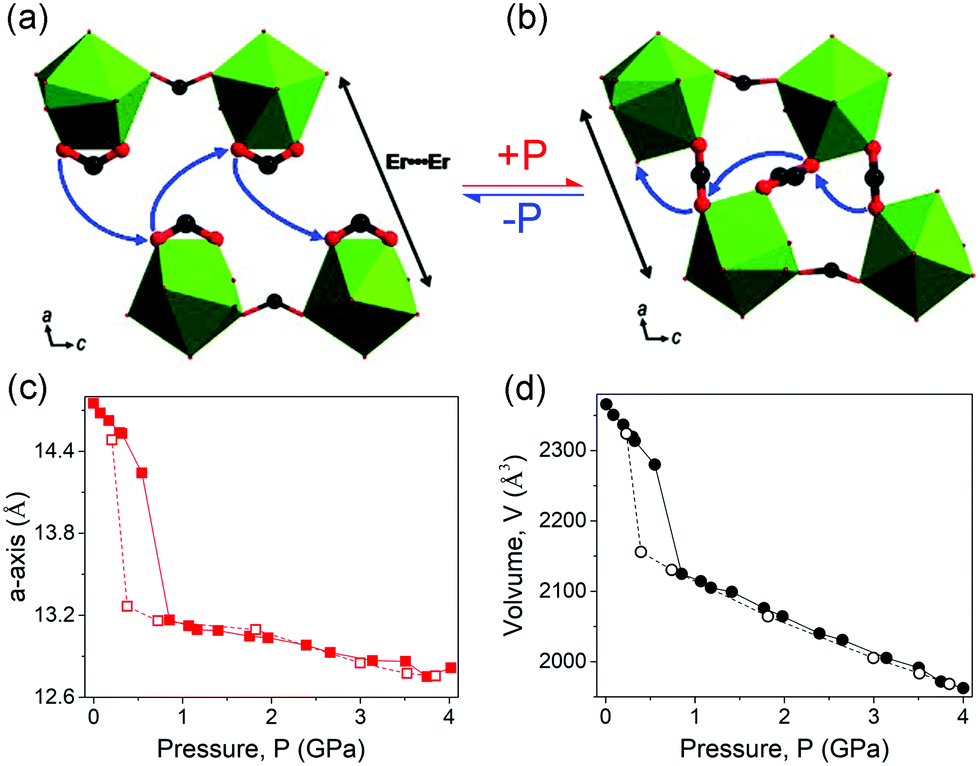 | ||
| Fig. 19 Partial crystal structure changes of the [tmenH2][Er(HCOO)4]2 before (a) and after (b) compression. Lattice parameter changes of a (c) and volume (d). The data collected with increasing and decreasing pressure are plotted with filled and empty spheres, respectively. Reprinted with permission from ref. 124. | ||
Intriguingly, Clegg et al. found that the 1D coordination polymer [(Cu2L2(1-methylpiperazine)2]n, where H2L is 1,1′-(1,3-phenylene)-bis(4,4-dimethylpentane-1,3-dione)), can underwent a reversible phase transition at a very low elevated pressure (∼0.05 GPa).125 At ambient pressure, the structure are constructed from alternating 5-coordinate and 6-coordinate dinuclear complex bridged by 1-methylpiperazine. With increasing pressure, part of the Cu–N bonds breaks and the bridging piperazine flips, resulting in a rare depolymerization process from the 1D coordination chain to the discrete complex.
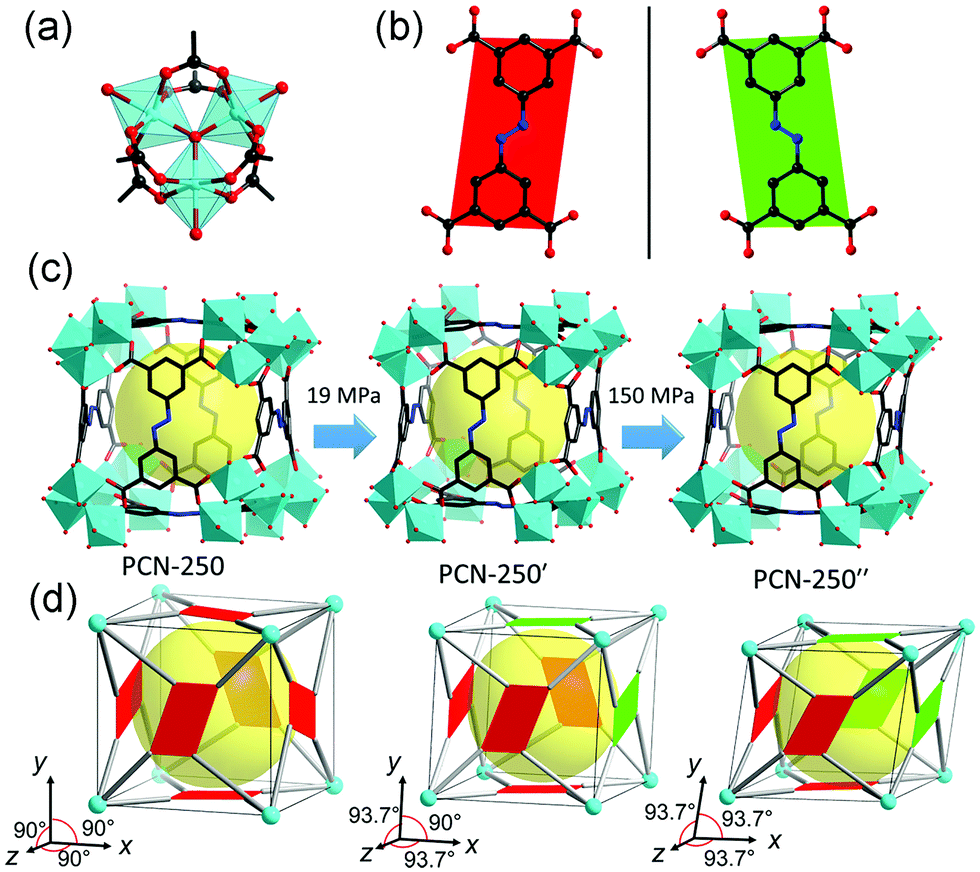 | ||
| Fig. 20 (a) Structure of the Fe3–oxo cluster. (b) ABTC ligand with two different kinds of configurations. (c) Crystal structure of PCN-250 at different pressures. (d) Scheme of the pressure induced structural transformation including the inclination of unit cell and flipping of ligands. Reprinted with permission from ref. 126. | ||
Electrical field
It has been known that the breathing behavior of MIL-53 can be triggered by guest adsorption, thermal and mechanical stimuli. Recently, by the mean of molecular dynamic simulation, Ghoufi et al. found that this transition can also be triggered by the application of external electric field. The MIL-53(Cr) can switch from the LP state to the CP state at E = 1.75 V nm−1 associated with 35% volume changes.127 The structural transformation is fully reversible but with a hysteresis of 1.00 V nm−1. This electrically switchable breathing behavior paves the way toward a variant of the electrical swing adsorption (ESA) process.The most representative materials toward electric field are piezoelectrics, which allow the inter-conversion between electric voltage and mechanical stress. In an electric field, the asymmetric displacement of ions will result in changes in the crystal dimension, which is proportional to the applied field. For example, the BaTiO3 single crystal can generate a recoverable strain of 0.75% at a field of 200 V mm−1.128
ZIF-8 crystallizes in the noncentrosymmetric space group I3m, and was found to show a large intrinsic second-order nonlinear optical response.129 This may make it responsive to electric field polarization. Caro et al. found that the separation factor of ZIF-8 membrane for the C3H6/C3H8 mixture can increase from 6 to 8 after applying the electrical field (Fig. 21e),130 while the influence of E-field to the permeance of HKUST-1 is negligible. In situ powder diffraction measurements reveal that the lattice of ZIF-8 can transform to polarized Cm and R3m polymorphs under E-field (Fig. 21a–c). The limiting pore diameter of the Cm phase (the most dominant phase) is 3.6 Å, a little larger than the pristine cubic ZIF-8 (3.4 Å). The DFT calculations also prove that the applying E-field can stiffen the network, inhibiting the linker rotation. The stiffen of the network and pore diameter changes are responsible for the enhanced C3H6/C3H8 separation. Additionally, the E-field can be used to modify the dipolar attractions between monodisperse ZIF-8 crystals, assembling the monodisperse ZIF-8 crystals to linear chains.131 Under electric field, the Cu(TCNQ) (TCNQ = 7,7,8,8-tetracyanoquinodimethane) can reversibly transform from a high-resistance phase to a conducting phase and a continuous lattice contraction was observed.132
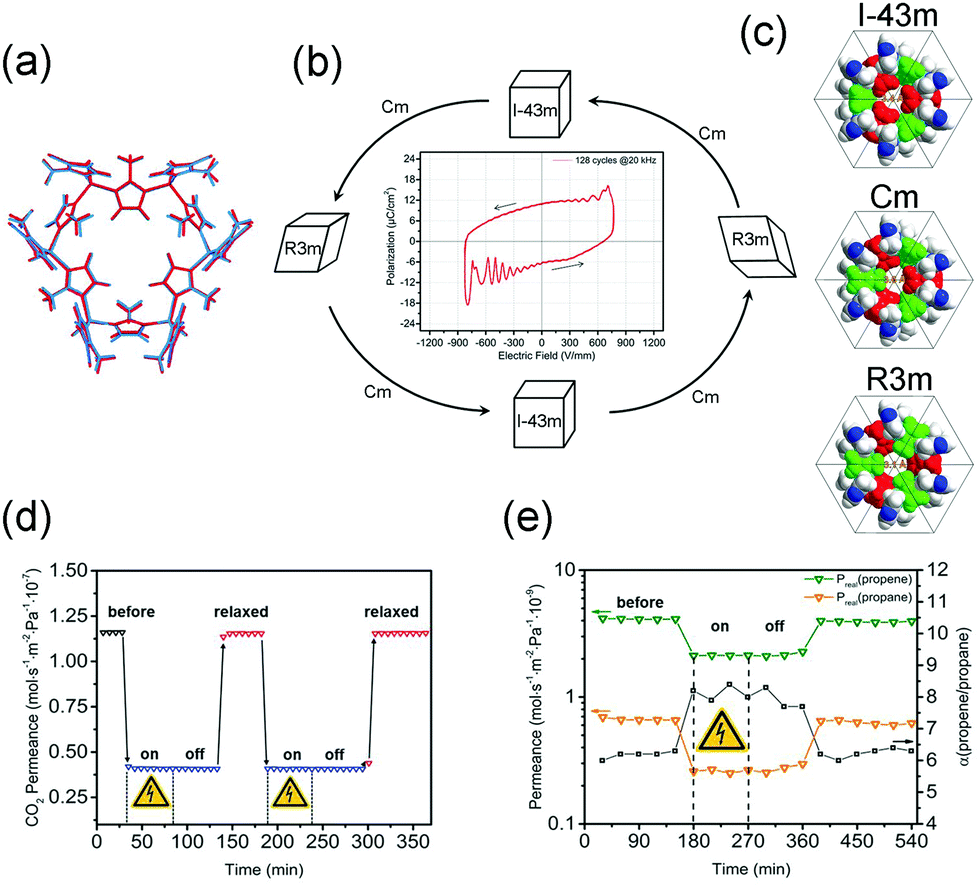 | ||
| Fig. 21 (a) Lattice overlap of the undistributed ZIF-8 gate with the same lattice distorted by external electric-field. (b) Scheme of the structural changes of ZIF-8 under electric field. (c) A representation of the primitive cells of ZIF-8 gate as a space-filling model of different simulated polymorphs. (d) A demonstration of the in situ switching of CO2 is shown in a plot of permeance versus time for the 1 μm membrane. (e) For C3H6/C3H8 mixture in ZIF-8, the real separation factor α increase upon E-field poling. Reprinted with permission from ref. 130. | ||
Light
The designability of MOFs makes it possible to introduce photosensitive functional groups, such as azobenzene, into the framework. By controlling the light source, the crystal structures and pore environments can be modified. Normally, two categories can be used to fabricate light-responsive MOFs. One is to fill the pores of MOFs with photosensitive guests and the other is to decorate the bridging ligands with photosensitive functional groups. The most representative photosensitive molecule is azobenzene, which can translate its conformation from trans- to cis-state under ultraviolet (UV) light (λ = 365 nm) irradiation (Fig. 22a). Kitagawa et al. introduced the azobenzene molecules into the pores of a flexible MOF. The light induced conformational changes of azobenzene would force the framework to underwent a phase transition from orthorhombic to tetragonal phase.133 This transition is accompanied with a significant enhancement of N2 uptake.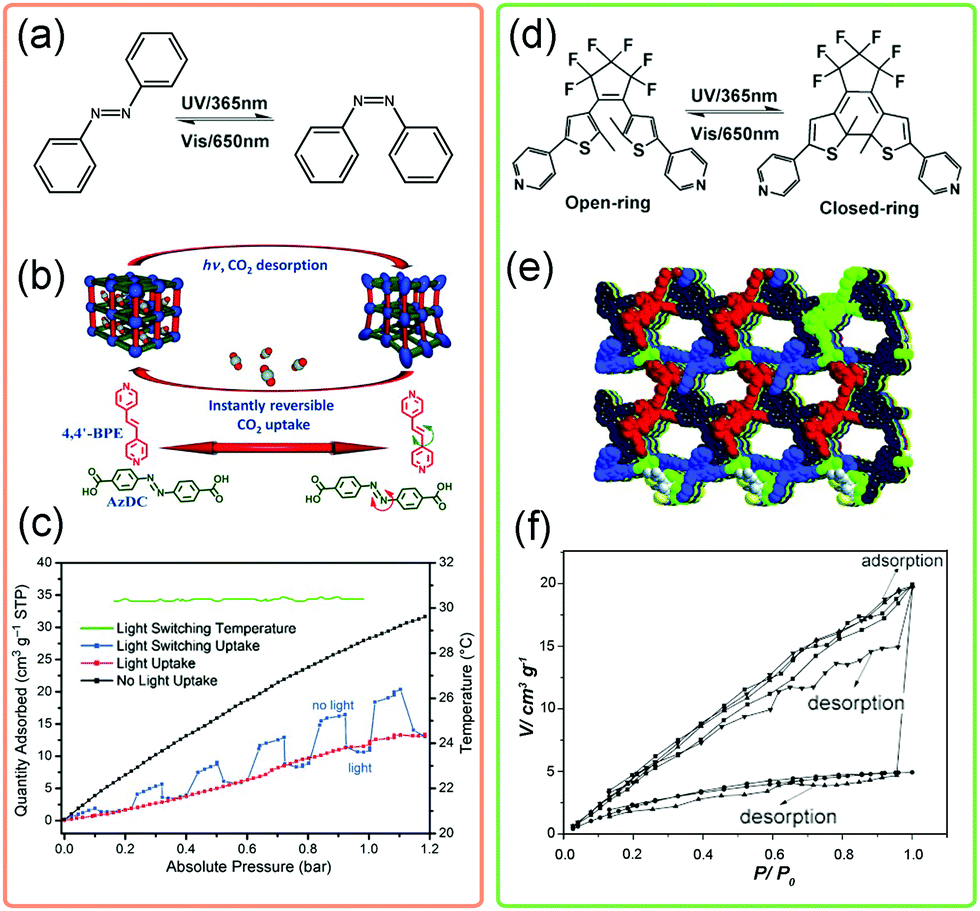 | ||
| Fig. 22 (a) Scheme of the conformational changes of the azobenzene molecule upon UV irradiation. (b) Illustration of the guest triggered framework changes of Zn(AzDC)(4,4′-BPE)0.5. (c) CO2 adsorption isotherms of Zn(AzDC)(4,4′-BPE)0.5 at 303 K in the presence of light (red), absence of light (black) and light switching environment (blue). (d) Scheme of the conformational changes of the diarylethene molecule upon UV irradiation. (e) Crystal structure of the diarylethene MOF. (f) The static and dynamic CO2 isotherms under different conditions. Partly taken from ref. 134 and 138 with permission. | ||
Alternatively, the photosensitive groups can be introduced on the bridging ligands of MOFs. Hill et al. constructed a MOF using azobenzene-4,4′-dicarboxylate (AzDC) as bridging ligands and trans-1,2-bis(4-pyridy)ethylene (4,4′-BPE) as pillars (Fig. 22b).134 Since the azobenzene derivatives acted as bridging units in this structure, tightly bounded with metal ions, the irradiation induced conformational changes would be largely suppressed. While the local bending about the azo group was allowed. Notably, this dynamic flexibility enables the framework to rapidly switch back to the native state, resulting in an instantaneous release of ∼64% of the adsorbed CO2 release under switching light (Fig. 22c). Similarly, the robust PCN-250, constructed from 3,3′,5,5′-azobenzene tetracarboxylic acid (ABTC), was also found to exhibit suppressed bending around the azo group under UV irradiation, realizing the light triggered regeneration of MOFs.135 What's more, the azobenzene can be introduced on the ligands as dangling groups, enabling finely control of the gate-opening and closing behaviors.136,137
Besides, the diarylethene can undergo a fast reversible ring-open to ring-closed transformation under UV irradiation (Fig. 22d). Guo et al. synthesized a diarylethene based MOF (Fig. 22e), with the dia topology.138 After UV irradiation, the CO2 adsorption amount can be significantly enhanced nearly 4 times as that of non-irradiated one and the desorption capacity can reach 75% under static irradiation. The light induced ring-open and ring-closed transformation was proposed to be responsible for the light responsive gas uptake properties. Additionally, the diarylethene unit can incarcerate gas molecules, such as C2H2, C2H4etc. So the conformational change of diarylethene unit will largely influence MOFs’ separation properties. For example, the diarylethene MOF shows ultrahigh C2H2/C2H4 selectivity of 47.1 at 195 K, while under UV irradiation the value is only 3.0 (∼15.7 times weakened).139
Summary
In this feature article, we have mainly summarized how the structure changes under external stimuli (e.g. guest adsorption/desorption, temperature, pressure, electric field and light). The flexibility has been known to widely exist in MOFs.140–143 Even some of the “rigid” MOFs, such as MOF-5, HKUST-1, MIL-68, and ZIFs, have been found to exhibit some degree of flexibility due to the strong dynamic vibration modes. Every coin has two sides. On the one hand, the flexibility endows MOFs with some fascinating physical or chemical properties, making them show potential applications in gas separation, drug delivery, catalysis and so on. On the other hand, such sensitivities may weaken their tolerance toward environmental fluctuations, including the atmosphere, temperature, pressure etc. A fundamental understanding of how the structure changes under external stimuli can not only deepen the knowledge of structure–property relationships but also provide guidance for the rational utilization of stimuli to enhance MOFs’ practical performances. Some successful examples have been reported. For example, by finely control of the temperature, the gas diffusion process in Cu(OPTZ) can be rationally regulated, facilitating kinetic-based gas separations.97 The luminescence property of tetraphenylethylene based MOFs can be tuned by mechanical pressure.144Although the diversity of organic ligands and SBUs allows infinite MOFs to be synthesized and decorated, the utilization of external stimuli opens up a new dimension for the modification of MOFs’ performances. There is wide scope for further development. For example, some of MOFs may respond to multiple stimuli, enabling further improving their performance via combined stimuli treatments.145 Notably, when multiple stimuli are applied, there may exist competition or synergy effects, which is a big challenge to elucidate the underling mechanism. Additionally, current characterization of structural changes of MOFs under stimuli are mainly dependent on the crystallographic analyses. Although measured in situ, the diffraction result is hard to distinguish the dynamic vibrations and steric disorders. While the flexibility of MOFs usually concerns dynamic motions. The combination of X-ray diffraction measurements with other characterization approaches (such as magic angle spinning nuclear magnetic resonance spectroscopy, terahertz spectroscopy, etc.) may give a deeper insight into the structural changes of MOFs under external stimuli. In summary, there is still a long way to go. We wish this feature article can be help to the development of smart materials whose structures and properties can be finely controlled by external stimuli.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation of China (Grant No. 21875285), the Fundamental Research Funds for the Central Universities (20CX06047A), Taishan Scholar Foundation (ts201511019) and Development Projects of Shandong Province (Grant No. 2019JZZY010331).Notes and references
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS.
- M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O'keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319–330 CrossRef CAS PubMed.
- S. Kitagawa, R. Kitaura and S. i. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed.
- J. Li, R. J. Kuppler and H. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- A. Schoedel, M. Li, D. Li, M. O’Keeffe and O. M. Yaghi, Chem. Rev., 2016, 116, 12466–12535 CrossRef CAS PubMed.
- Z. Chen, P. Li, R. Anderson, X. Wang, X. Zhang, L. Robison, L. R. Redfern, S. Moribe, T. Islamoglu and D. A. Gómez-Gualdrón, Science, 2020, 368, 297–303 CrossRef CAS.
- B. Chen, Y. Yang, F. Zapata, G. Lin, G. Qian and E. B. Lobkovsky, Adv. Mater., 2007, 19, 1693–1696 CrossRef CAS.
- Z. Lin, Z. M. Zhang, Y. S. Chen and W. Lin, Angew. Chem., Int. Ed., 2016, 55, 13739–13743 CrossRef CAS PubMed.
- X. Song, S. Y. Song, S. N. Zhao, Z. M. Hao, M. Zhu, X. Meng, L. L. Wu and H. J. Zhang, Adv. Funct. Mater., 2014, 24, 4034–4041 CrossRef CAS.
- G. Férey and C. Serre, Chem. Soc. Rev., 2009, 38, 1380–1399 RSC.
- S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695 CrossRef CAS PubMed.
- S. Kitagawa and K. Uemura, Chem. Soc. Rev., 2005, 34, 109–119 RSC.
- B. Hoskins and R. Robson, J. Am. Chem. Soc., 1990, 112, 1546–1554 CrossRef CAS.
- C. Serre, F. Millange, C. Thouvenot, M. Nogues, G. Marsolier, D. Louër and G. Férey, J. Am. Chem. Soc., 2002, 124, 13519–13526 CrossRef CAS.
- Z.-J. Lin, J. Lü, M. Hong and R. Cao, Chem. Soc. Rev., 2014, 43, 5867–5895 RSC.
- G. Alberti, S. Murcia-Mascaros and R. Vivani, J. Am. Chem. Soc., 1998, 120, 9291–9295 CrossRef CAS.
- J. H. Lee, S. Jeoung, Y. G. Chung and H. R. Moon, Coord. Chem. Rev., 2019, 389, 161–188 CrossRef CAS.
- C. Serre, C. Mellot-Draznieks, S. Surblé, N. Audebrand, Y. Filinchuk and G. Férey, Science, 2007, 315, 1828–1831 CrossRef CAS.
- P. L. Llewellyn, G. Maurin, T. Devic, S. Loera-Serna, N. Rosenbach, C. Serre, S. Bourrelly, P. Horcajada, Y. Filinchuk and G. Férey, J. Am. Chem. Soc., 2008, 130, 12808–12814 CrossRef CAS.
- C. Volkringer, D. Popov, T. Loiseau, N. Guillou, G. Ferey, M. Haouas, F. Taulelle, C. Mellot-Draznieks, M. Burghammer and C. Riekel, Nat. Mater., 2007, 6, 760–764 CrossRef CAS PubMed.
- R. Kitaura, S. Kitagawa, Y. Kubota, T. C. Kobayashi, K. Kindo, Y. Mita, A. Matsuo, M. Kobayashi, H.-C. Chang and T. C. Ozawa, Science, 2002, 298, 2358–2361 CrossRef CAS PubMed.
- Y. Kubota, M. Takata, T. C. Kobayashi and S. Kitagawa, Coord. Chem. Rev., 2007, 251, 2510–2521 CrossRef CAS.
- T. Yildirim and M. Hartman, Phys. Rev. Lett., 2005, 95, 215504 CrossRef CAS PubMed.
- J. A. Mason, J. Oktawiec, M. K. Taylor, M. R. Hudson, J. Rodriguez, J. E. Bachman, M. I. Gonzalez, A. Cervellino, A. Guagliardi, C. M. Brown, P. L. Llewellyn, N. Masciocchi and J. R. Long, Nature, 2015, 527, 357–361 CrossRef CAS PubMed.
- S. Couck, J. F. Denayer, G. V. Baron, T. Rémy, J. Gascon and F. Kapteijn, J. Am. Chem. Soc., 2009, 131, 6326–6327 CrossRef CAS.
- L. Hamon, P. L. Llewellyn, T. Devic, A. Ghoufi, G. Clet, V. Guillerm, G. D. Pirngruber, G. Maurin, C. Serre, G. Driver, W. V. Beek, E. Jolimaitre, A. Vimont, M. Daturi and G. Ferey, J. Am. Chem. Soc., 2009, 131, 17490–17499 CrossRef CAS PubMed.
- F. Salles, H. Jobic, A. Ghoufi, P. L. Llewellyn, C. Serre, S. Bourrelly, G. Férey and G. Maurin, Angew. Chem., Int. Ed., 2009, 48, 8335–8339 CrossRef CAS.
- J. P. Zhang and X. M. Chen, J. Am. Chem. Soc., 2009, 131, 5516–5521 CrossRef CAS PubMed.
- J. Jia, F. Xu, Z. Long, X. Hou and M. J. Sepaniak, Chem. Commun., 2013, 49, 4670–4672 RSC.
- A. Douvali, A. C. Tsipis, S. V. Eliseeva, S. Petoud, G. S. Papaefstathiou, C. D. Malliakas, I. Papadas, G. S. Armatas, I. Margiolaki, M. G. Kanatzidis, T. Lazarides and M. J. Manos, Angew. Chem., Int. Ed., 2015, 54, 1651–1656 CrossRef CAS PubMed.
- S. Yuan, L. Zou, H. Li, Y. P. Chen, J. Qin, Q. Zhang, W. Lu, M. B. Hall and H. C. Zhou, Angew. Chem., Int. Ed., 2016, 55, 10776–10780 CrossRef CAS PubMed.
- M. Mon, J. s. Ferrando-Soria, T. Grancha, F. R. Fortea-Pérez, J. Gascon, A. Leyva-Pérez, D. Armentano and E. Pardo, J. Am. Chem. Soc., 2016, 138, 7864–7867 CrossRef CAS PubMed.
- P. Horcajada, C. Serre, G. Maurin, N. A. Ramsahye, F. Balas, M. Vallet-Regi, M. Sebban, F. Taulelle and G. Férey, J. Am. Chem. Soc., 2008, 130, 6774–6780 CrossRef CAS.
- M. X. Wu and Y. W. Yang, Adv. Mater., 2017, 29, 1606134 CrossRef PubMed.
- T. M. McDonald, J. A. Mason, X. Kong, E. D. Bloch, D. Gygi, A. Dani, V. Crocella, F. Giordanino, S. O. Odoh and W. S. Drisdell, Nature, 2015, 519, 303–308 CrossRef CAS PubMed.
- W. Cheng, X. Zhao, H. Su, F. Tang, W. Che, H. Zhang and Q. Liu, Nat. Energy, 2019, 4, 115–122 CrossRef CAS.
- B. F. Hoskins and R. Robson, J. Am. Chem. Soc., 1989, 111, 5962–5964 CrossRef CAS.
- O. Yaghi and H. Li, J. Am. Chem. Soc., 1995, 117, 10401–10402 CrossRef CAS.
- M. Kandiah, M. H. Nilsen, S. Usseglio, S. Jakobsen, U. Olsbye, M. Tilset, C. Larabi, E. A. Quadrelli, F. Bonino and K. P. Lillerud, Chem. Mater., 2010, 22, 6632–6640 CrossRef CAS.
- Y. Bai, Y. Dou, L.-H. Xie, W. Rutledge, J.-R. Li and H. C. Zhou, Chem. Soc. Rev., 2016, 45, 2327–2367 RSC.
- S. Kitagawa and M. Kondo, Bull. Chem. Soc. Jpn., 1998, 71, 1739–1753 CrossRef CAS.
- K. Biradha and M. Fujita, Angew. Chem., Int. Ed., 2002, 41, 3392–3395 CrossRef CAS.
- P. Horcajada, F. Salles, S. Wuttke, T. Devic, D. Heurtaux, G. Maurin, A. Vimont, M. Daturi, O. David and E. Magnier, J. Am. Chem. Soc., 2011, 133, 17839–17847 CrossRef CAS PubMed.
- W. Morris, B. Volosskiy, S. Demir, F. Gándara, P. L. McGrier, H. Furukawa, D. Cascio, J. F. Stoddart and O. M. Yaghi, Inorg. Chem., 2012, 51, 6443–6445 CrossRef CAS.
- S. B. Kalidindi, S. Nayak, M. E. Briggs, S. Jansat, A. P. Katsoulidis, G. J. Miller, J. E. Warren, D. Antypov, F. Corà and B. Slater, Angew. Chem., Int. Ed., 2015, 54, 221–226 CrossRef CAS PubMed.
- O. V. Gutov, W. Bury, D. A. Gomez-Gualdron, V. Krungleviciute, D. Fairen-Jimenez, J. E. Mondloch, A. A. Sarjeant, S. S. Al-Juaid, R. Q. Snurr and J. T. Hupp, Chem. – Eur. J., 2014, 20, 12389–12393 CrossRef CAS PubMed.
- R. Zou, X. Ren, F. Huang, Y. Zhao, J. Liu, X. Jing, F. Liao, Y. Wang, J. Lin and R. Zou, J. Mater. Chem. A, 2015, 3, 23493–23500 RSC.
- P. Deria, D. A. Gómez-Gualdrón, W. Bury, H. T. Schaef, T. C. Wang, P. K. Thallapally, A. A. Sarjeant, R. Q. Snurr, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2015, 137, 13183–13190 CrossRef CAS.
- Y.-S. Wei, K.-J. Chen, P.-Q. Liao, B.-Y. Zhu, R.-B. Lin, H.-L. Zhou, B.-Y. Wang, W. Xue, J.-P. Zhang and X.-M. Chen, Chem. Sci., 2013, 4, 1539–1546 RSC.
- F. Millange, N. Guillou, R. I. Walton, J.-M. Grenèche, I. Margiolaki and G. Férey, Chem. Commun., 2008, 4732–4734 RSC.
- S. Biswas, T. Ahnfeldt and N. Stock, Inorg. Chem., 2011, 50, 9518–9526 CrossRef CAS PubMed.
- D. N. Dybtsev, H. Chun and K. Kim, Angew. Chem., Int. Ed., 2004, 43, 5033–5036 CrossRef CAS.
- K. S. Park, Z. Ni, A. P. Côté, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O’Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186–10191 CrossRef CAS PubMed.
- P. Zhao, G. I. Lampronti, G. O. Lloyd, M. T. Wharmby, S. b. Facq, A. K. Cheetham and S. A. Redfern, Chem. Mater., 2014, 26, 1767–1769 CrossRef CAS PubMed.
- Y. Du, K. Mao, B. Wooler, A. K. Sharma, D. Colmyer, M. Nines and S. C. Weston, J. Phys. Chem. C, 2017, 121, 28090–28095 CrossRef CAS.
- A. Boutin, M. A. Springuel-Huet, A. Nossov, A. Gedeon, T. Loiseau, C. Volkringer, G. Férey, F. X. Coudert and A. H. Fuchs, Angew. Chem., Int. Ed., 2009, 48, 8314–8317 CrossRef CAS.
- F. Salles, G. Maurin, C. Serre, P. L. Llewellyn, C. Knöfel, H. J. Choi, Y. Filinchuk, L. Oliviero, A. Vimont and J. R. Long, J. Am. Chem. Soc., 2010, 132, 13782–13788 CrossRef CAS PubMed.
- E. J. Carrington, C. A. McAnally, A. J. Fletcher, S. P. Thompson, M. Warren and L. Brammer, Nat. Chem., 2017, 9, 882 CrossRef CAS.
- H. Yang, T. X. Trieu, X. Zhao, Y. Wang, Y. Wang, P. Feng and X. Bu, Angew. Chem., Int. Ed., 2019, 58, 11757–11762 CrossRef CAS PubMed.
- M.-H. Yu, B. Space, D. Franz, W. Zhou, C. He, L. Li, R. Krishna, Z. Chang, W. Li and T.-L. Hu, J. Am. Chem. Soc., 2019, 141, 17703–17712 CrossRef CAS PubMed.
- S. Krause, V. Bon, I. Senkovska, U. Stoeck, D. Wallacher, D. M. Többens, S. Zander, R. S. Pillai, G. Maurin and F.-X. Coudert, Nature, 2016, 532, 348–352 CrossRef CAS.
- G. J. Halder, C. J. Kepert, B. Moubaraki, K. S. Murray and J. D. Cashion, Science, 2002, 298, 1762–1765 CrossRef CAS PubMed.
- D. A. Reed, B. K. Keitz, J. Oktawiec, J. A. Mason, T. Runčevski, D. J. Xiao, L. E. Darago, V. Crocellà, S. Bordiga and J. R. Long, Nature, 2017, 550, 96–100 CrossRef CAS.
- J. Chen, L. Hu, J. Deng and X. Xing, Chem. Soc. Rev., 2015, 44, 3522–3567 RSC.
- D. Dubbeldam, K. S. Walton, D. E. Ellis and R. Q. Snurr, Angew. Chem., Int. Ed., 2007, 46, 4496–4499 CrossRef CAS.
- A. L. Goodwin, M. Calleja, M. J. Conterio, M. T. Dove, J. S. Evans, D. A. Keen, L. Peters and M. G. Tucker, Science, 2008, 319, 794–797 CrossRef CAS PubMed.
- Y. Liu, J.-H. Her, A. Dailly, A. J. Ramirez-Cuesta, D. A. Neumann and C. M. Brown, J. Am. Chem. Soc., 2008, 130, 11813–11818 CrossRef CAS PubMed.
- C. Nanthamathee, S. Ling, B. Slater and M. P. Attfield, Chem. Mater., 2015, 27, 85–95 CrossRef CAS.
- S. J. Baxter, A. Schneemann, A. D. Ready, P. Wijeratne, A. P. Wilkinson and N. C. Burtch, J. Am. Chem. Soc., 2019, 141, 12849–12854 CrossRef CAS PubMed.
- S. Henke, A. Schneemann and R. A. Fischer, Adv. Funct. Mater., 2013, 23, 5990–5996 CrossRef CAS.
- Y. Kim, R. Haldar, H. Kim, J. Koo and K. Kim, Dalton Trans., 2016, 45, 4187–4192 RSC.
- L. D. DeVries, P. M. Barron, E. P. Hurley, C. Hu and W. Choe, J. Am. Chem. Soc., 2011, 133, 14848–14851 CrossRef CAS PubMed.
- J. M. Ogborn, I. E. Collings, S. A. Moggach, A. L. Thompson and A. L. Goodwin, Chem. Sci., 2012, 3, 3011–3017 RSC.
- H.-L. Zhou, R.-B. Lin, C.-T. He, Y.-B. Zhang, N. Feng, Q. Wang, F. Deng, J.-P. Zhang and X.-M. Chen, Nat. Commun., 2013, 4, 1–8 RSC.
- L. Zhang, X. Kuang, X. Wu, W. Yang and C. Lu, Dalton Trans., 2014, 43, 7146–7152 RSC.
- J. Pang, C. Liu, Y. Huang, M. Wu, F. Jiang, D. Yuan, F. Hu, K. Su, G. Liu and M. Hong, Angew. Chem., Int. Ed., 2016, 55, 7478–7482 CrossRef CAS PubMed.
- P. Lama, R. K. Das, V. J. Smith and L. J. Barbour, Chem. Commun., 2014, 50, 6464–6467 RSC.
- A. Gładysiak, S. M. Moosavi, L. Sarkisov, B. Smit and K. C. Stylianou, CrystEngComm, 2019, 21, 5292–5298 RSC.
- Z. Liu, C. Liu, Q. Li, J. Chen and X. Xing, Phys. Chem. Chem. Phys., 2017, 19, 24436–24439 RSC.
- Z. Liu, Q. Gao, J. Chen, J. Deng, K. Lin and X. Xing, Chem. Commun., 2018, 54, 5164–5176 RSC.
- Z. Liu, X. Jiang, C. Wang, C. Liu, Z. Lin, J. Deng, J. Chen and X. Xing, Inorg. Chem. Front., 2019, 6, 1675–1679 RSC.
- H.-L. Zhou, Y.-B. Zhang, J.-P. Zhang and X.-M. Chen, Nat. Commun., 2015, 6, 1–7 Search PubMed.
- Z. Liu, K. Lin, Y. Ren, K. Kato, Y. Cao, J. Deng, J. Chen and X. Xing, Chem. Commun., 2019, 55, 4107–4110 RSC.
- J. L. Rowsell, E. C. Spencer, J. Eckert, J. A. Howard and O. M. Yaghi, Science, 2005, 309, 1350–1354 CrossRef CAS PubMed.
- Y. Wu, A. Kobayashi, G. J. Halder, V. K. Peterson, K. W. Chapman, N. Lock, P. D. Southon and C. J. Kepert, Angew. Chem., Int. Ed., 2008, 47, 8929–8932 CrossRef CAS PubMed.
- V. K. Peterson, G. J. Kearley, Y. Wu, A. J. Ramirez-Cuesta, E. Kemner and C. J. Kepert, Angew. Chem., Int. Ed., 2010, 49, 585–588 CrossRef CAS PubMed.
- B. Chen, M. Eddaoudi, S. Hyde, M. O'Keeffe and O. Yaghi, Science, 2001, 291, 1021–1023 CrossRef CAS PubMed.
- Y. Wu, V. K. Peterson, E. Luks, T. A. Darwish and C. J. Kepert, Angew. Chem., Int. Ed., 2014, 126, 5275–5278 CrossRef.
- M. J. Cliffe, J. A. Hill, C. A. Murray, F.-X. Coudert and A. L. Goodwin, Phys. Chem. Chem. Phys., 2015, 17, 11586–11592 RSC.
- H. L. Zhou, J. Bai, J. W. Ye, Z. W. Mo, W. X. Zhang, J. P. Zhang and X. M. Chen, ChemPlusChem, 2016, 81, 817–821 CrossRef CAS PubMed.
- Z. Liu, Q. Li, H. Zhu, K. Lin, J. Deng, J. Chen and X. Xing, Chem. Commun., 2018, 54, 5712–5715 RSC.
- Z. Liu, R. Ma, J. Deng, J. Chen and X. Xing, Chem. Mater., 2020, 32, 2893–2898 CrossRef CAS.
- N. Lock, Y. Wu, M. Christensen, L. J. Cameron, V. K. Peterson, A. J. Bridgeman, C. J. Kepert and B. B. Iversen, J. Phys. Chem. C, 2010, 114, 16181–16186 CrossRef CAS.
- S. L. Gould, D. Tranchemontagne, O. M. Yaghi and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2008, 130, 3246–3247 CrossRef CAS PubMed.
- S. Nandi, P. De Luna, R. Maity, D. Chakraborty, T. Daff, T. Burns, T. K. Woo and R. Vaidhyanathan, Mater. Horiz., 2019, 6, 1883–1891 RSC.
- C. Gu, N. Hosono, J.-J. Zheng, Y. Sato, S. Kusaka, S. Sakaki and S. Kitagawa, Science, 2019, 363, 387–391 CrossRef CAS PubMed.
- Q. Gao, J. Xu, D. Cao, Z. Chang and X. H. Bu, Angew. Chem., Int. Ed., 2016, 55, 15027–15030 CrossRef CAS PubMed.
- M. C. Bernini, F. Gándara, M. Iglesias, N. Snejko, E. Gutiérrez-Puebla, E. V. Brusau, G. E. Narda and M. Á. Monge, Chem. – Eur. J., 2009, 15, 4896–4905 CrossRef CAS PubMed.
- M. Zhu, X. Z. Song, S. Y. Song, S. N. Zhao, X. Meng, L. L. Wu, C. Wang and H. J. Zhang, Adv. Sci., 2015, 2, 1500012 CrossRef PubMed.
- M. T. Wharmby, S. Henke, T. D. Bennett, S. R. Bajpe, I. Schwedler, S. P. Thompson, F. Gozzo, P. Simoncic, C. Mellot-Draznieks and H. Tao, Angew. Chem., Int. Ed., 2015, 54, 6447–6451 CrossRef CAS PubMed.
- T. D. Bennett, A. L. Goodwin, M. T. Dove, D. A. Keen, M. G. Tucker, E. R. Barney, A. K. Soper, E. G. Bithell, J.-C. Tan and A. K. Cheetham, Phys. Rev. Lett., 2010, 104, 115503 CrossRef PubMed.
- L. Vanduyfhuys, S. Rogge, J. Wieme, S. Vandenbrande, G. Maurin, M. Waroquier and V. Van Speybroeck, Nat. Commun., 2018, 9, 1–9 CrossRef CAS PubMed.
- R. H. Baughman, S. Stafström, C. Cui and S. O. Dantas, Science, 1998, 279, 1522–1524 CrossRef CAS PubMed.
- A. J. Graham, D. R. Allan, A. Muszkiewicz, C. A. Morrison and S. A. Moggach, Angew. Chem., Int. Ed., 2011, 50, 11138–11141 CrossRef CAS.
- S. A. Moggach, T. D. Bennett and A. K. Cheetham, Angew. Chem., Int. Ed., 2009, 121, 7221–7223 CrossRef.
- A. J. Graham, J.-C. Tan, D. R. Allan and S. A. Moggach, Chem. Commun., 2012, 48, 1535–1537 RSC.
- J. Mariathasan, L. Finger and R. Hazen, Acta Crystallogr., Sect. B: Struct. Sci., 1985, 41, 179–184 CrossRef.
- J. Haines, C. Chateau, J. Leger, C. Bogicevic, S. Hull, D. Klug and J. Tse, Phys. Rev. Lett., 2003, 91, 015503 CrossRef CAS PubMed.
- D. McCann, L. Cartz, R. Schmunk and Y. Harker, J. Appl. Phys., 1972, 43, 1432–1436 CrossRef CAS.
- W. Li, M. R. Probert, M. Kosa, T. D. Bennett, A. Thirumurugan, R. P. Burwood, M. Parinello, J. A. Howard and A. K. Cheetham, J. Am. Chem. Soc., 2012, 134, 11940–11943 CrossRef CAS PubMed.
- Y. Yan, A. E. O’Connor, G. Kanthasamy, G. Atkinson, D. R. Allan, A. J. Blake and M. Schröder, J. Am. Chem. Soc., 2018, 140, 3952–3958 CrossRef CAS.
- Q. Zeng, K. Wang and B. Zou, J. Am. Chem. Soc., 2017, 139, 15648–15651 CrossRef CAS PubMed.
- P. Serra-Crespo, A. Dikhtiarenko, E. Stavitski, J. Juan-Alcañiz, F. Kapteijn, F.-X. Coudert and J. Gascon, CrystEngComm, 2015, 17, 276–280 RSC.
- Q. Zeng, K. Wang and B. Zou, ACS Mater. Lett., 2020, 2, 291–295 CrossRef CAS.
- W. Cai and A. Katrusiak, Nat. Commun., 2014, 5, 1–8 Search PubMed.
- S. A. Hodgson, J. Adamson, S. J. Hunt, M. J. Cliffe, A. B. Cairns, A. L. Thompson, M. G. Tucker, N. P. Funnell and A. L. Goodwin, Chem. Commun., 2014, 50, 5264–5266 RSC.
- W. Cai, A. Gładysiak, M. Anioła, V. J. Smith, L. J. Barbour and A. Katrusiak, J. Am. Chem. Soc., 2015, 137, 9296–9301 CrossRef CAS PubMed.
- Z. Su, Y.-R. Miao, S.-M. Mao, G.-H. Zhang, S. Dillon, J. T. Miller and K. S. Suslick, J. Am. Chem. Soc., 2015, 137, 1750–1753 CrossRef CAS PubMed.
- Z. Su, Y.-R. Miao, G. Zhang, J. T. Miller and K. S. Suslick, Chem. Sci., 2017, 8, 8004–8011 RSC.
- C. A. Schröder, I. A. Baburin, L. Van Wüllen, M. Wiebcke and S. Leoni, CrystEngComm, 2013, 15, 4036–4040 RSC.
- D. W. Lewis, A. R. Ruiz-Salvador, A. Gómez, L. M. Rodriguez-Albelo, F.-X. Coudert, B. Slater, A. K. Cheetham and C. Mellot-Draznieks, CrystEngComm, 2009, 11, 2272–2276 RSC.
- E. C. Spencer, R. J. Angel, N. L. Ross, B. E. Hanson and J. A. Howard, J. Am. Chem. Soc., 2009, 131, 4022–4026 CrossRef CAS PubMed.
- E. C. Spencer, M. S. Kiran, W. Li, U. Ramamurty, N. L. Ross and A. K. Cheetham, Angew. Chem., Int. Ed., 2014, 53, 5583–5586 CrossRef CAS PubMed.
- J. K. Clegg, A. J. Brock, K. A. Jolliffe, L. F. Lindoy, S. Parsons, P. A. Tasker and F. J. White, Chem. – Eur. J., 2017, 23, 12480–12483 CrossRef CAS PubMed.
- S. Yuan, X. Sun, J. Pang, C. Lollar, J.-S. Qin, Z. Perry, E. Joseph, X. Wang, Y. Fang and M. Bosch, Joule, 2017, 1, 806–815 CrossRef CAS.
- A. Ghoufi, K. Benhamed, L. Boukli-Hacene and G. Maurin, ACS Cent. Sci., 2017, 3, 394–398 CrossRef CAS PubMed.
- X. Ren, Nat. Mater., 2004, 3, 91–94 CrossRef CAS PubMed.
- S. Van Cleuvenbergen, I. Stassen, E. Gobechiya, Y. Zhang, K. Markey, D. E. De Vos, C. Kirschhock, B. Champagne, T. Verbiest and M. A. Van Der Veen, Chem. Mater., 2016, 28, 3203–3209 CrossRef CAS.
- A. Knebel, B. Geppert, K. Volgmann, D. Kolokolov, A. Stepanov, J. Twiefel, P. Heitjans, D. Volkmer and J. Caro, Science, 2017, 358, 347–351 CrossRef CAS PubMed.
- N. Yanai, M. Sindoro, J. Yan and S. Granick, J. Am. Chem. Soc., 2013, 135, 34–37 CrossRef CAS PubMed.
- C. A. Fernandez, P. C. Martin, T. Schaef, M. E. Bowden, P. K. Thallapally, L. Dang, W. Xu, X. Chen and B. P. McGrail, Sci. Rep., 2014, 4, 1–6 Search PubMed.
- N. Yanai, T. Uemura, M. Inoue, R. Matsuda, T. Fukushima, M. Tsujimoto, S. Isoda and S. Kitagawa, J. Am. Chem. Soc., 2012, 134, 4501–4504 CrossRef CAS.
- R. Lyndon, K. Konstas, B. P. Ladewig, P. D. Southon, P. C. J. Kepert and M. R. Hill, Angew. Chem., Int. Ed., 2013, 52, 3695–3698 CrossRef CAS.
- H. Li, M. R. Martinez, Z. Perry, H. C. Zhou, P. Falcaro, C. Doblin, S. Lim, A. J. Hill, B. Halstead and M. R. Hill, Chem. – Eur. J., 2016, 22, 11176–11179 CrossRef CAS.
- J. Park, D. Yuan, K. T. Pham, J.-R. Li, A. Yakovenko and H.-C. Zhou, J. Am. Chem. Soc., 2012, 134, 99–102 CrossRef CAS PubMed.
- J. W. Brown, B. L. Henderson, M. D. Kiesz, A. C. Whalley, W. Morris, S. Grunder, H. Deng, H. Furukawa, J. I. Zink and J. F. Stoddart, Chem. Sci., 2013, 4, 2858–2864 RSC.
- F. Luo, C. B. Fan, M. B. Luo, X. L. Wu, Y. Zhu, S. Z. Pu, W. Y. Xu and G. C. Guo, Angew. Chem., Int. Ed., 2014, 53, 9298–9301 CrossRef CAS PubMed.
- C. B. Fan, L. Le Gong, L. Huang, F. Luo, R. Krishna, X. F. Yi, A. M. Zheng, L. Zhang, S. Z. Pu and X. F. Feng, Angew. Chem., Int. Ed., 2017, 56, 7900–7906 CrossRef CAS PubMed.
- A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Chem. Soc. Rev., 2014, 43, 6062–6096 RSC.
- J.-P. Zhang, H.-L. Zhou, D.-D. Zhou, P.-Q. Liao and X.-M. Chen, Natl. Sci. Rev., 2018, 5, 907–919 CrossRef CAS.
- Z. Chang, D. H. Yang, J. Xu, T. L. Hu and X. H. Bu, Adv. Mater., 2015, 27, 5432–5441 CrossRef CAS PubMed.
- F.-X. Coudert, Chem. Mater., 2015, 27, 1905–1916 CrossRef CAS.
- C.-X. Chen, Z.-W. Wei, Y.-N. Fan, P.-Y. Su, Y.-Y. Ai, Q.-F. Qiu, K. Wu, S.-Y. Yin, M. Pan and C.-Y. Su, Chem, 2018, 4, 2658–2669 CAS.
- H. Li, M. M. Sadiq, K. Suzuki, C. Doblin, S. Lim, P. Falcaro, A. J. Hill and M. R. Hill, J. Mater. Chem. A, 2016, 4, 18757–18762 RSC.
| This journal is © The Royal Society of Chemistry 2020 |