
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStability of blueberry anthocyanin, anthocyanidin and pyranoanthocyanidin pigments and their inhibitory effects and mechanisms in human cervical cancer HeLa cells†
Fengguang Pan,
Yanjun Liu ,
Jingbo Liu and
Erlei Wang*
,
Jingbo Liu and
Erlei Wang*
Lab of Nutrition and Functional Food, College of Food Science and Engineering, Jilin University, Changchun 130062, Jilin, China. E-mail: wel@jlu.edu.cn; panfg@jlu.edu.cn
First published on 8th April 2019
Abstract
Anthocyan-rich foods have attracted lots of attention because of their potential biological and pharmacological applications. Anthocyanins richly exist in blueberry fruits and have been proved to possess various bioactive properties. Despite the potential application of anthocyanins in the food, pharmaceutical and cosmetic industries, their use is limited because of their relative instability. Recently, much attention has been given to a particular family of anthocyanin derivatives, the pyranoanthocyanins that have been indicated to have higher stability than the original anthocyanins. However, the anti-cancer activity of pyranoanthocyanins is largely unknown. The objective of this study was to conduct a comparative analysis on the stability and anti-cancer activities of anthocyanins, anthocyanidins and pyranoanthocyanidins. Pyranoanthocyanidins exhibited the highest stability in the pH range 3.0–9.0, while anthocyanidins had the strongest inhibition on HeLa cells among the three anthocyan pigments. All the anthocyan pigments could effectively induce cell cycle arrest at the G2/M phase in conjunction with a marked increase in the expression of the p53 protein. Exposure of HeLa cells to three anthocyan pigments caused pronounced late apoptosis that might be involved in the activation of the p38 MAPK/p53 signaling pathway. These findings suggest that anthocyanidins and pyranoanthocyanidins might be more promising anti-cancer agents than anthocyanins and warrant further evaluation regarding the molecular mechanisms.
Introduction
Anthocyans are natural colorants belonging to the flavonoid family and are widely spread in all tissues of higher plants.1 Anthocyans are liable for colors ranging from red to blue of fruits, vegetables, flowers, and roots. The structural differences between individual anthocyans lie in the number of hydroxyl groups, the methylation degree of the –OH groups, the kinds of sugar attachments, or the number and position of aliphatic or aromatic acids fixed to sugars.2 The most common anthocyans are glycosylated or acyl-glycosylated forms (named anthocyanins), while sugar-free or deacyl-glycosylated forms of anthocyanins are called anthocyanin aglycones or anthocyanidins.3 The most common anthocyanidins in plants are delphinidin, cyanidin, petunidin, peonidin, pelargonidin and malvidin.4 Anthocyan-rich foods have attracted much attention because of their potential biological and pharmacological applications. Growing evidence suggests that anthocyans play an important role in the prevention of several types of human cancer, such as colon, breast, prostate, oral and stomach cancers,5,6 while showing no toxic effects on normal human cells.7 However, several factors have restricted the widespread applications of anthocyanins or anthocyanidins, especially their weak stability. Anthocyans are highly unstable and very susceptible to pH, light, temperature, oxygen, solvents, metallic ions and some enzymes. Recently, a new family of anthocyanin derivatives described as pyranoanthocyanins was found in aged red wines8 and fermented fruit juices.9 The pyranoanthocyanins formed during wine aging play an important role in wine color and contribute to the progressive shift of initial violet-red color of young wines to a more orangish hues.10 However, the main interest of pyranoanthocyanins is that they have a greater stability against pH changes and SO2 bleaching than that of the anthocyanins.11–13 This stability is believed to result from the substitutions on the C-4 position that includes an additional ring D formed between the hydroxyl group in C-5 and C-4 of the anthocyanidin pyran ring (Fig. 1A).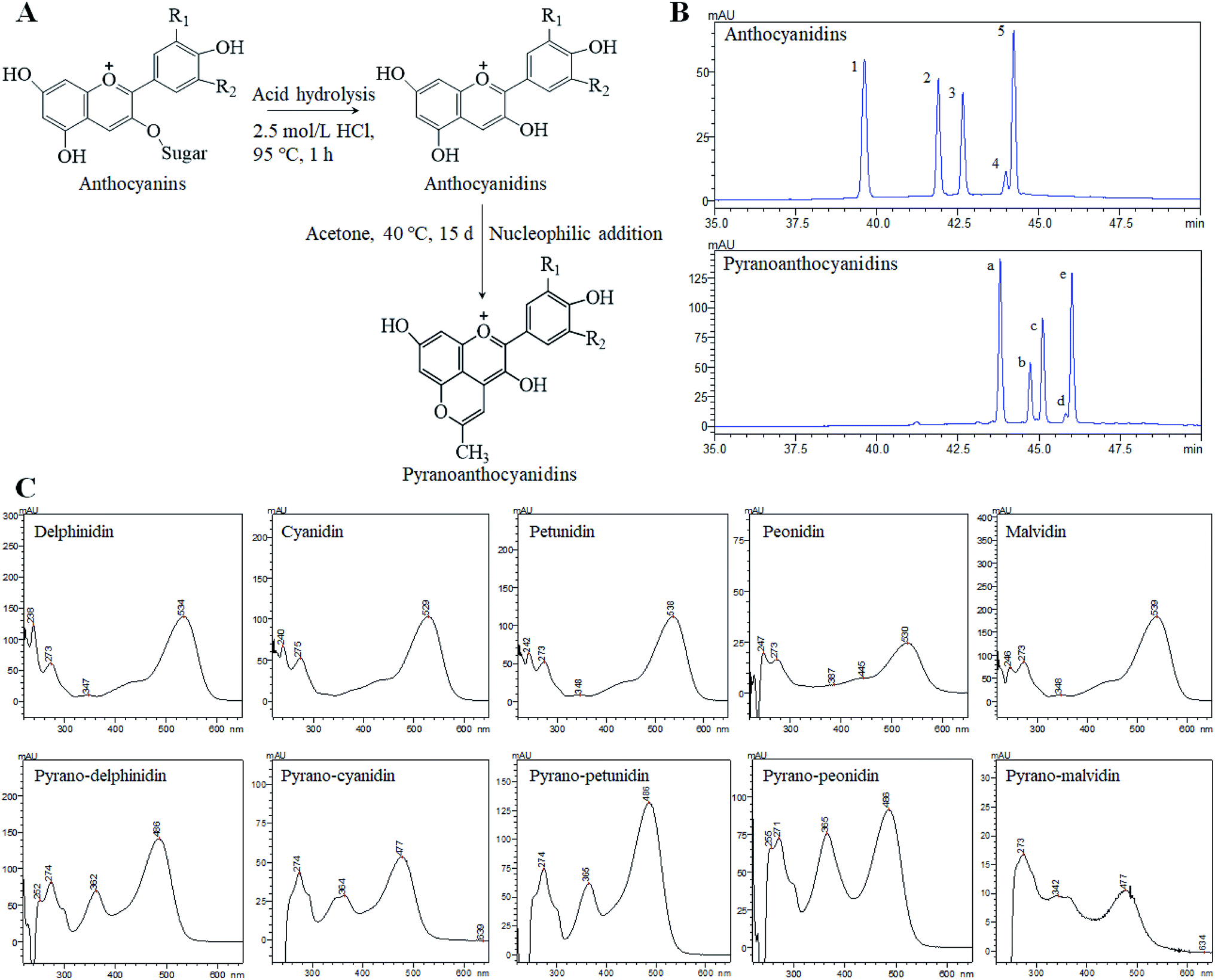 | ||
| Fig. 1 The formation process of pyranoanthocyanidins (A) and a comparative analysis of anthocyanidins and pyranoanthocyanidins using the HPLC-DAD method (B) and UV-Vis spectrum (C). Notes: R1 = OH, OCH3; R2 = OH, H, OCH3. The HPLC peak numbers 1–5 and a–e correspond to those in ESI Table S1.† | ||
To date, many studies have investigated the inhibitory effects and mechanisms of anthocyanins from different plant materials on the growth of several cancer cells. Delphinidin can suppress matrix metalloproteinase-induced MCF-7 human breast carcinoma cell invasion through the specific inhibition of NF-κB-dependent MMP-9 gene expression.14 Anthocyanin-rich extracts from mulberry could inhibit gastric cancer cell growth by inducing cell apoptosis via the activation of p38/p53 and p38/c-jun signaling pathways.15 The anthocyanin and anthocyanidin extracts from purple-shoot tea could inhibit the proliferation of HT-29 and COLO 320DM colorectal cancer cell lines through the cellular mechanism of cell cycle arrest in the G0/G1 phase, as well as apoptosis induction by Bax and concomitant cleavage of caspase-3 and PARP.16 Moreover, recent studies have proposed that the inhibitory mechanisms of anthocyanins on cancer chemoprevention can be grouped into three aspects: (1) the antioxidation, (2) the molecular mechanism related to anticarcinogenesis, and (3) the molecular mechanism involved in apoptosis induction.17 In recent years, several reports have explored the optimal preparation methods and conditions for the extraction and synthesis of different types of pyranoanthocyanins, such as methyl pyranoanthocyanins, carboxyl pyranoanthocyanins, portisins, oxovitisins and so on.18 However, there are currently rare studies that focus on the assessment of the anticancer activities of pyranoanthocyanins, especially the forms of pyranoanthocyanidins or pyranoanthocyanin aglycones. In view of the predominant anticancer activities of common anthocyanins, pyranoanthocyanidins might also possess similar anticancer effects, though the molecular remains unclear. Considering the predominant stability of pyranoanthocyanin compounds, it is necessary to investigate the potential anticancer activities of pyranoanthocyanidins on human cancer cell growth in the present study.
In the present research, we synthesized and characterized pyranoanthocyanidins using blueberry anthocyanidin extracts and conducted a comparative analysis on the stability and anticancer activities of three anthocyan forms (anthocyanins, anthocyanidins and pyranoanthocyanidins). The human cervical cancer HeLa cell lines were treated with three anthocyan pigments and assessed for viability, cell morphology, cell cycle distribution, apoptosis and changes in the expression levels of relevant proteins involved in the cell cycle and apoptosis.
Materials and methods
Reagents
Standards of cyanidin-3-O-glucoside chloride [molecular weight (MW) 484.84, purity ≥ 98%] and cyanidin chloride (MW 322.7, purity ≥ 97%) were purchased from Sigma-Aldrich Chemical Co. (St Louis, MO, USA). Cyclin D1 mouse monoclonal antibody, p53 mouse monoclonal antibody, p38 mitogen-activated protein kinases (p38 MAPK) mouse monoclonal antibody, caspase-3 rabbit polyclonal antibody, β-actin mouse monoclonal antibody, peroxidase-conjugated goat anti-mouse antibody, and HRP-labeled goat anti-rabbit IgG were all purchased from Beyotime Biotechnology (Shanghai, China). Ethanol, ethyl acetate, hydrochloric acid (HCl), acetone and acetic acid were obtained from Beijing Chemistry Factory (Beijing, China). Chromatographic grade methanol (MeOH) was purchased from Merck (Darmstadt, Germany).Preparation of three anthocyan pigments
The extraction methods and identification of blueberry anthocyanin and anthocyanidin extracts were described in our previous study.19 Briefly, anthocyanin and anthocyanidin extracts were prepared from the lyophilized fruits of low-bush wild blueberries (V. uliginosum L.) by combined column chromatography techniques, especially the employment of Amberlite XAD-7HP column and C18 Sep-Pak cartridge (Waters).Pyranoanthocyanidins were synthesized from the reaction between anthocyanidin extracts and acetone through the nucleophilic addition reactions.20 First, blueberry anthocyanidins were dissolved in acetone/ethanol/water (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2, v/v), resulting in an anthocyanidin concentration of 200 μg mL−1. The pH value of this solution was adjusted to 3.0 with a hydrochloric acid solution, and the reaction temperature was set at 40 °C. The reaction lasted for 15 days until the absorption peaks of the anthocyanidins were no longer detected by HPLC photodiode array (PDA) detector at 530 nm. Excess acetone and ethanol reagents were removed with a rotary evaporator below 60 °C. Then, pyranoanthocyanidin pigments were purified by solid phase extraction using C18 Sep-Pak cartridge (Waters, 500 mg). The cartridge was first activated with 3.0 mL of anhydrous ethanol and 3.0 mL of 0.01% aqueous HCl. Pyranoanthocyanidin concentrate was adsorbed on the cartridge. After washing the cartridge with 2.0 mL of 0.01% aqueous HCl, the pyranoanthocyanidin fraction was collected by elution with 60% aqueous ethanol containing 0.01% HCl (v/v). Then, ethanol was removed by a rotary evaporator below 60 °C and freeze-dried to powders. Finally, the anthocyan samples were analyzed by HPLC and HPLC-MS (ESI Method S1†).
:1:2, v/v), resulting in an anthocyanidin concentration of 200 μg mL−1. The pH value of this solution was adjusted to 3.0 with a hydrochloric acid solution, and the reaction temperature was set at 40 °C. The reaction lasted for 15 days until the absorption peaks of the anthocyanidins were no longer detected by HPLC photodiode array (PDA) detector at 530 nm. Excess acetone and ethanol reagents were removed with a rotary evaporator below 60 °C. Then, pyranoanthocyanidin pigments were purified by solid phase extraction using C18 Sep-Pak cartridge (Waters, 500 mg). The cartridge was first activated with 3.0 mL of anhydrous ethanol and 3.0 mL of 0.01% aqueous HCl. Pyranoanthocyanidin concentrate was adsorbed on the cartridge. After washing the cartridge with 2.0 mL of 0.01% aqueous HCl, the pyranoanthocyanidin fraction was collected by elution with 60% aqueous ethanol containing 0.01% HCl (v/v). Then, ethanol was removed by a rotary evaporator below 60 °C and freeze-dried to powders. Finally, the anthocyan samples were analyzed by HPLC and HPLC-MS (ESI Method S1†).
Stability of anthocyanin, anthocyanidin and pyranoanthocyanidin pigments
The effects of temperature and pH on the stability of anthocyanin, anthocyanidin and pyranoanthocyanidin pigments were studied. The pH stability of three anthocyan pigments was determined at seven different pH values. The three samples were dissolved in McIlvaine buffer at pH 0.0, 1.0, 2.0, 3.0, 5.0, 7.0, and 9.0 (200 μg mL−1). The pH value was verified by pH-meter measurement. The UV-visible spectra absorbance maxima of each sample were measured after 2 h of storage in the dark at room temperature (25 °C). The effects of temperature on the stability of three anthocyan pigments were assessed at three different temperatures (25 °C, 40 °C, and 80 °C). Each anthocyan buffer solution was divided into three portions, which were stored at 25 °C, 40 °C, and 80 °C. Each of the anthocyan sample solutions (200 μg mL−1) was sealed in a small flask tube with a screw cap. Due to the different degradation rates of each anthocyan pigments, each heating treatment has a different storage period. After heating treatment, the effects of temperature on the stability of three anthocyan pigments were evaluated by UV spectrophotometer (UV-2550, Shimadzu, Japan).Cell lines and cell culture
HeLa cells (human cervical cancer HeLa cells) were obtained from Shanghai Cell Bank (Shanghai, China). HeLa cells were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% heat-inactivated fetal bovine serum, 4.5 g L−1 glucose, 1 mmol L−1 glutamine, 100 U mL−1 penicillin and 100 μg mL−1 streptomycin at 37 °C in a humidified atmosphere with 5% CO2, and the medium was changed every day.Cell viability assay
Cell viability was measured using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) colorimetric assay, which is based on the conversion of MTT to formazan crystals by mitochondrial dehydrogenases. HeLa cells were seeded in 96-well plates at a density of 8000 cells per well for 24 h of incubation. Afterwards, cells were incubated in anthocyanin, anthocyanidin and pyranoanthocyanidin pigments with concentration gradients of 100, 200, 250, 300, 400, 500, 600, 800 and 1000 μg mL−1 for 24 h, respectively. Then, 20 μL of MTT (5 mg mL−1 solution in PBS) was added to the culture medium. After 4 h of incubation at 37 °C, the supernatant was aspirated, and the blue-produced formazan crystals were dissolved in 160 μL of DMSO. Absorbance was measured by a Bio-Rad 680 Microplate reader (Bio-Rad Laboratories, CA, USA) at 490 nm. Cell viability was expressed as a percentage of the control culture value, which was considered as 100% viable.Cell morphology observation by the Calcein-AM/PI double staining method
The cell morphology after treatments with anthocyanin, anthocyanidin and pyranoanthocyanidin pigments were investigated by the Calcein-AM (acetoxymethyl ester)/PI (propidium iodide) Double Staining Kit (Molecular probes, Eugene, OR, USA). Calcein-AM can remove the AM group by esterase action in living cells and produce Calcein, which emits strong green fluorescence. PI can enter dead cells and insert into the double helix of cell DNA to produce red fluorescence. HeLa cells were seeded in 6-well plates at a density of 1 × 106 cells per well in complete medium. After 24 h of incubation, each well was treated with different culturing media: anthocyanin, anthocyanidin and pyranoanthocyanidin pigments at various concentrations (0, 100, 200, 400, and 600 μg mL−1) for 24 h. Then, the medium was rinsed with PBS, and the cells were labeled with Calcein-AM/PI stain. The cells were examined under a fluorescence microscope combined with a digital camera.Cell cycle by flow cytometric analysis
Cell cycle analysis was performed using a Cell Cycle Detection Kit (KeyGen BioTech Co., Nanjing, China). In brief, HeLa cells were seeded in 6-well plates at a density of 1 × 106 cells per well and cultured overnight. After that, the cells were treated with different concentrations of anthocyanins (0, 100, 200, 400, and 600 μg mL−1), anthocyanidins (0, 50, 200, and 400 μg mL−1) or pyranoanthocyanidins (0, 100, 200, 400, and 600 μg mL−1) for 24 h. Then, the cells were collected with trypsin–EDTA, washed with cold phosphate-buffered saline (PBS) and fixed with cold 70% ethanol at 4 °C for 2 h. The cells were then washed twice with cold PBS to remove the ethanol. The cells were soaked in PI/RNase A (9/1, v/v) staining solution at room temperature for 30 min. The distribution of cell cycles was measured by flow cytometry based on the precursors in the DNA content. FACSCalibur (BD Biosciences; San Jose, CA, USA) was used for flow cell analysis, and FACSDiva Software (version 6.1.3; BD Biosciences) was used to analyze data.Cell apoptosis by flow cytometric analysis
Cell apoptosis analysis was detected by flow cytometry with an Annexin V-FITC/PI Apoptosis Detection Kit (KeyGen BioTech Co., Nanjing, China). In brief, HeLa cells were treated with various doses of anthocyanins (0, 200, 400, 600, and 800 μg mL−1), anthocyanidins (0, 200, 400, and 600 μg mL−1) or pyranoanthocyanidins (0, 200, 400, 600, and 800 μg mL−1) for 24 h. Then, the cells were harvested, washed twice with PBS and suspended in binding buffer. Then, the cells were stained with annexin V-FITC and PI in the dark for 10 min. Then, the cells were analyzed by flow cytometry at wavelengths of 488 and 530 nm. Data were analyzed using the Flow Jo 7.6 software (Tree Star, Inc., Ashland, OR, USA).Western blot analysis
The analysis of p53, cyclin D1, p38 MAPK and caspase-3 in HeLa cells was performed on whole cell lysates. To analyze the expression levels of proteins, anthocyanins (0, 100, 200 and 400 μg mL−1), anthocyanidins (0, 50, 150 and 300 μg mL−1) or pyranoanthocyanidins (0, 100, 200 and 400 μg mL−1) were added to the medium, incubated for 24 h, and then washed with ice-cold phosphate-buffered saline (PBS). Then, the total proteins were extracted by adding RIPA lysis buffer containing a 1% protease inhibitor cocktail and centrifuged at 12000 rpm for 25 min at 4 °C to remove cell debris. An enhanced BCA protein assay kit (Beyotime Biotechnology) was used to determine the protein concentration. Equal amounts of cytolytic proteins (60 μg) were mixed with a quarter of the electrophoresis loading buffer, boiled for 5 min, and then electrophoresed on a 10% SDS-polyacrylamide gel at 120 V for 90 min. Proteins were transferred to polyvinylidene fluoride (PVDF) membranes and incubated with primary antibodies (p53, cyclin D1, p38 MAPK, caspase-3 and β-actin). Then, the secondary antibody (peroxidase-conjugated goat anti-mouse antibody or HRP-labeled goat anti-rabbit IgG) was used to bind the corresponding primary antibodies. Finally, proteins were incubated with ECL plus (GE Healthcare Buckinghamshire, UK) and detected by exposure to X-ray films.
Statistical analysis
Each experiment was performed in triplicate and repeated at least three times. The results are presented as the mean ± standard deviation (SD). Differences between the groups were calculated by Student's t-test using SPSS Statistics 17.0.1 (Chicago, USA). A significant difference was set at p < 0.05.Results and discussion
Composition and contents in three blueberry anthocyan pigments
The composition of anthocyanin, anthocyanidin and pyranoanthocyanidin pigments was analyzed by ultraviolet-visible spectrum (UV-Vis), HPLC, and HPLC-PDA-ESI-MS/MS, as well as the relevant publications from earlier studies.21,22 The composition of monomers in blueberry anthocyanin and anthocyanidin extracts has been reported in our previous research.19 In general, at least 16 anthocyanin monomers were identified in blueberry anthocyanin extract, and delphinidin-3-O-glucoside, petunidin-3-O-glucoside and malvidin-3-O-glucoside are three dominant anthocyanin monomers, accounting for approximately 64% of the total anthocyanin content. The anthocyanidin extracts were composed of five anthocyanidin monomers, and three major anthocyanidin peaks (delphinidin, petunidin and malvidin) accounted for approximately 84% of the total anthocyanidin content compared with the minor peaks (cyanidin and peonidin). Pyranoanthocyanidin pigments could be obtained through the reaction of anthocyanidin extracts with acetone. As shown in Fig. 1B, five anthocyanidin monomers were completely transformed to methyl pyranoanthocyanidins. The chemical structures of five new components were further confirmed by UV-Vis spectroscopy (Fig. 1C) and HPLC-PDA-ESI-MS/MS (Table S1 in ESI†). The five new methyl pyranoanthocyanidins (λmax at ∼480 nm) exhibited a hypsochromic spectral shift of 50 nm from the maximum absorption (λmax at ∼530 nm) of anthocyanidins (Fig. 1C). In addition, the MS data of these peaks were further applied to confirm their identities (Fig. S1 in ESI†). The molecular ion of peak a gave a signal in the mass spectrum at m/z 341.16, 38 amu higher than the molecular ion of peak 1 (delphinidin) at m/z 303.0, which indicated cycloaddition reactions between the C-4 and 5-OH positions of anthocyanidins and acetone.23 It is likely that the other four peaks (peaks b, c, d and e) also possessed an additional 38 amu when compared to the relevant anthocyanidin peaks (peaks 2, 3, 4 and 5 respectively), respectively. Thus, the identities of five new methyl pyranoanthocyanidins in pyranoanthocyanidin pigments were confirmed (Table S1†). After purifying and freeze-drying, three anthocyan pigments were obtained with a purity of 63.5% for anthocyanin extracts, 70.9% for anthocyanidins, and 67.2% for pyranoanthocyanidin pigments based on the quantitative analysis of HPLC.Thermal stability and pH of three blueberry anthocyan pigments
It is well known that the stability of anthocyanins can be affected by a number of factors, such as temperature, oxygen, light, pH and some enzymes, thus, the instability of anthocyanins has largely limited its application. The present study focused on investigating the color stability with respect to the pH and temperature of purified anthocyanidin and pyranoanthocyanidin pigments in comparison with the parent anthocyanin extracts. Fig. 2A shows the absorbance variation in aqueous solutions of three anthocyan pigments at different pH values in the range of 0.0–9.0. The three anthocyan pigments had a considerably stable absorbance within the pH range of 0.0–2.0, at least 80% of the color was still intact after 2 h of storage at 25 °C. Above pH 3.0, the stability of anthocyanins and anthocyanidins decreased dramatically to reach the minimum at pH range 5.0–9.0. Surprisingly, the stability of pyranoanthocyanidins remained almost unchanged in the pH range of 3.0–9.0, and 75% of pyranoanthocyanidins remained intact after 2 h of storage at 25 °C (Fig. S2 in ESI†). These results indicated that pyranoanthocyanidins exhibited stronger stabilities compared with anthocyanins and anthocyanidins, and the stability might be related to the particular structure of pyranoanthocyanidins (pyranic rings or the planar aromatic moieties) that prevents the nucleophilic attack of water, thus hindering the formation of a carbinol base.24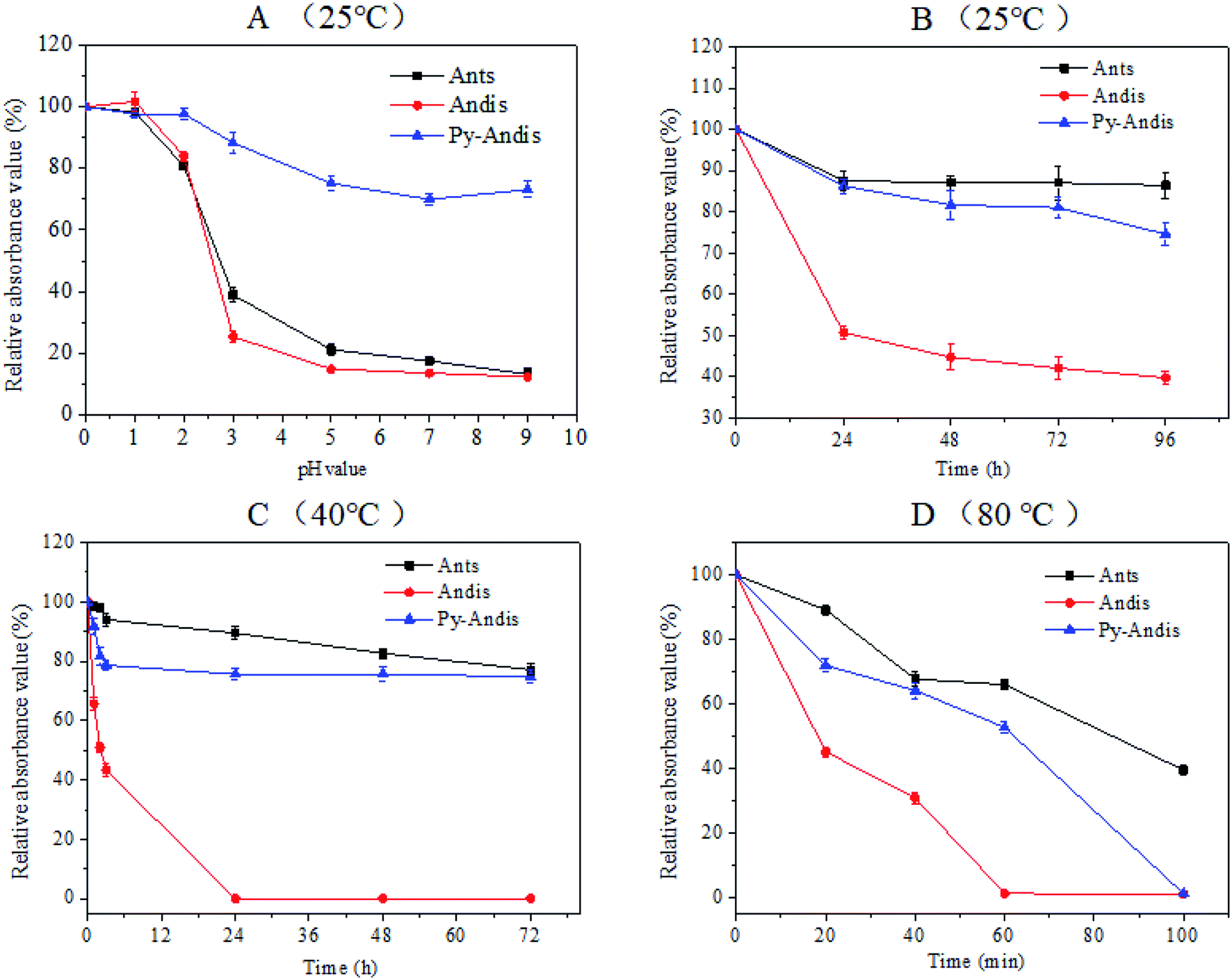 | ||
| Fig. 2 Absorbance changes at the λmax of each anthocyan aqueous solution at different pH values (A) and temperatures (B–D). Each absorbance value was expressed as a percentage relative to the absorbance at pH 0.0. Values are expressed as the means ± SD (n = 3). Abbreviations: Ants, anthocyanins; Andis, anthocyanidins; Py-Andis, pyranoanthocyanidins. | ||
The thermal stability of three anthocyan pigments was also studied at 25 °C, 40 °C and 80 °C (Fig. 2B–D). As shown in Fig. 2B and C, the absorbances of anthocyanin and pyranoanthocyanidin aqueous solutions decreased slightly with increasing time at 25 °C and 40 °C, respectively (Fig. S3 and S4 in ESI†). In contrast, the absorbances of anthocyanidins decreased significantly at 25 °C and 40 °C. The absorbance of the three anthocyan pigments decreased much faster at 80 °C than at the lower temperatures (Fig. 2D). After 60 min of heating at 80 °C, anthocyanins and pyranoanthocyanidins showed stronger thermal stability than anthocyanidins, displaying 70% and 55% relative absorbance values, respectively, while anthocyanidins were completely degraded (Fig. S5 in ESI†). These data suggest that the C-4 substituted structure confers higher thermal stability of pyranoanthocyanidin pigments in comparison with anthocyanidins.
MTT results
The cytotoxic effects of the three anthocyan samples in HeLa cancer cells were assessed using the MTT assay. Fig. 3A shows the cell viability of HeLa cells, which was expressed as % of the control after 24 h incubation in the presence of different concentrations of anthocyanin samples (100–1000 μg mL−1). All three anthocyan samples showed an obvious cytotoxic effect on the HeLa cells, and a dose-dependent decrease of cell viability was observed after treatment for 24 h. The most active sample among the three anthocyan samples was anthocyanidins, which showed highly significant effects (p < 0.01) on the HeLa cells at lower concentrations (100, 200, 250 and 300 μg mL−1) compared with the other samples. Pyranoanthocyanidins caused a significant reduction (p < 0.05) in cell viability at concentrations ranging between 250 and 1000 μg mL−1. Anthocyanins showed the lowest cytotoxic effects on HeLa cells among the three anthocyan samples. HeLa cells treated with anthocyanidins revealed the lowest IC50 values (275.33 ± 1.67 μg mL−1), followed by pyranoanthocyanidins (536.91 ± 2.03 μg mL−1), and both samples were about two-fold more active than anthocyanins (1095.28 ± 1.72 μg mL−1). According to the literature,5 similar observations on the effects of anthocyanin glycosides in HeLa cells have been reported, in which the anthocyanin fraction inhibited the viability of HeLa cells by 40% at 200 μg mL−1 after 48 h of treatment. The present results also showed that anthocyanidins and pyranoanthocyanidins displayed more potential antitumor activity than the anthocyanin glycosides.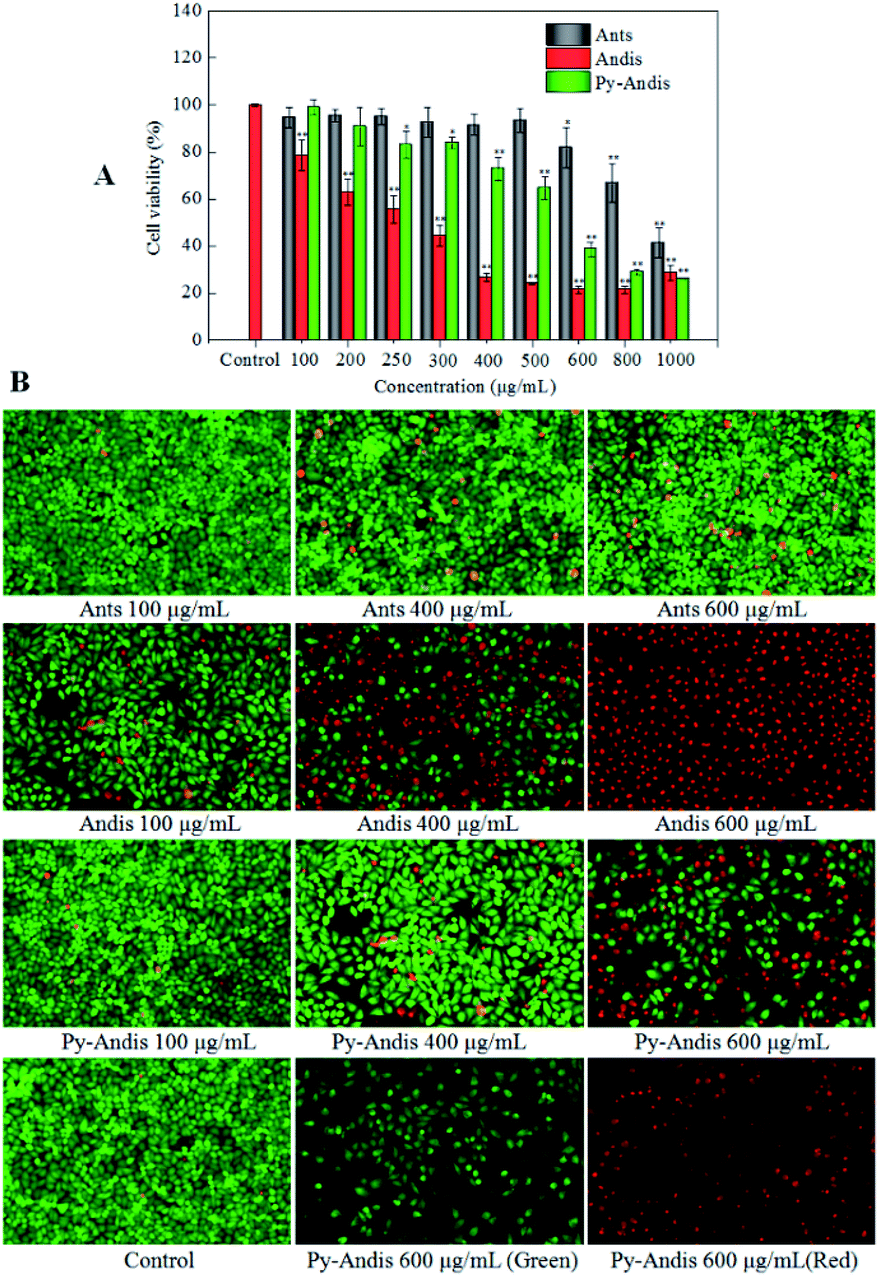 | ||
| Fig. 3 Cell viability and morphology after 24 h of treatment with different concentrations of anthocyanins (Ants), anthocyanidins (Andis), and pyranoanthocyanidins (Py-Andis). Cell viability was evaluated by the MTT assay. After the cells were Stained with Calcein-AM/PI, the live cells produce green fluorescence and the dead and dying cells produce red fluorescence. The results are expressed as the mean ± SD (n = 3). (**) p < 0.01, compared to the control group. (*) p < 0.05, compared to the control group. | ||
Based on early toxicological studies, anthocyanin-rich extracts had been proven to have very low toxicity or no adverse effects on several tested animals.25 Recently, the toxicological studies26 demonstrated that methyl pyranoanthocyanins showed cytoprotective effects on H2O2 induced MRC-5 cell damage, which suggested that pyranoanthocyanins are safe functional ingredients incorporated in food and beverages. Likewise, we preliminarily deduced that the aglycones (pyranoanthocyanidins) of methyl pyranoanthocyanins are regarded as safe and healthy functional ingredients used as food additives. However, the toxicological evaluation of pyranoanthocyanidins remains to be studied in future.
Cell morphology observation
To address how the anthocyan samples suppressed the proliferation of HeLa cancer cells, a live/dead cell viability kit (Calcein-AM/PI) was used to analyze the morphology of live and dead cells. HeLa cells without anthocyanin samples were used as controls. As shown in Fig. 3B, the live cells emit green fluorescence as a result of the conversion of calcein-AM to calcein in response to cytosolic esterase activity, while the dead and dying cells emit red fluorescence because of the interaction between the PI dye and nucleic acids. After 24 h of treatment with anthocyanin samples, no significant changes were seen in the number of live and dead HeLa cells between the anthocyanin and pyranoanthocyanidin samples at the concentrations of 100 and 400 μg mL−1 compared with the control group, however, anthocyanidins caused significant changes in the number of live and dead cells when its concentration ranged from 100 to 600 μg mL−1. In addition, HeLa cells in the control group showed spindle or polygonal morphology at 24 h of treatment, and the cells treated with the three anthocyan samples at low concentrations (100 and 400 μg mL−1) also showed similar morphology. However, some HeLa cells treated with anthocyanin samples at high concentrations (600 μg mL−1) showed irregularly polygonal morphology and produced dark green fluorescence. The morphological images of cells treated with anthocyanidins and pyranoanthocyanidins at 600 μg mL−1 suggested that the cell nuclei with chromatin condensation and apoptotic bodies were shaped, which appeared as a typical pattern of DNA fragmentation because of the internucleosomal cleavage connected to apoptosis.27 The present results were generally consistent with the cytotoxic evaluation of the three anthocyanin samples by MTT assay, where anthocyanidins showed the strongest inhibition on HeLa cells, followed by pyranoanthocyanidins, while anthocyanins ranked last among the three anthocyan samples.Cell cycle distribution
To explore the mechanism by which anthocyanins, anthocyanidins and pyranoanthocyanidins inhibit cell viability, we first investigated the distribution of cells in different phases of the cell cycle by flow cytometry analysis. Fig. 4A shows the cell cycle distribution percentages in the G0/G1, S and G2/M phases using the nuclei DNA content of HeLa cells after 24 h of treatment, and Fig. 4B shows typical histograms of the cell distribution in cells treated with or without the three anthocyan samples. The HeLa cells treated with anthocyanins in the concentration ranges of 100–600 μg mL−1 resulted in a highly significant (p < 0.01) accumulation of the cells at G2/M phase, and the cells at G0/G1 phase were not noticeably affected with an increasing concentration compared with the control group. The cell cycle distribution of HeLa cells treated with pyranoanthocyanidins at different concentrations also induced cell cycle G2/M arrest. The cells treated with anthocyanidins at 400 μg mL−1 caused an arrest of the cell cycle at both the S and G2/M phases. These results indicated that the three anthocyanin samples effectively inhibited the proliferation of HeLa cells by inducing cell cycle arrest at the G2/M phase. In particular, anthocyanins and pyranoanthocyanidins at concentrations of 100–600 μg mL−1 significantly (p < 0.01) induced cell cycle arrest at the G2/M phase, and anthocyanidins at 400 μg mL−1 effectively caused cell cycle arrest at both S and G2/M phases.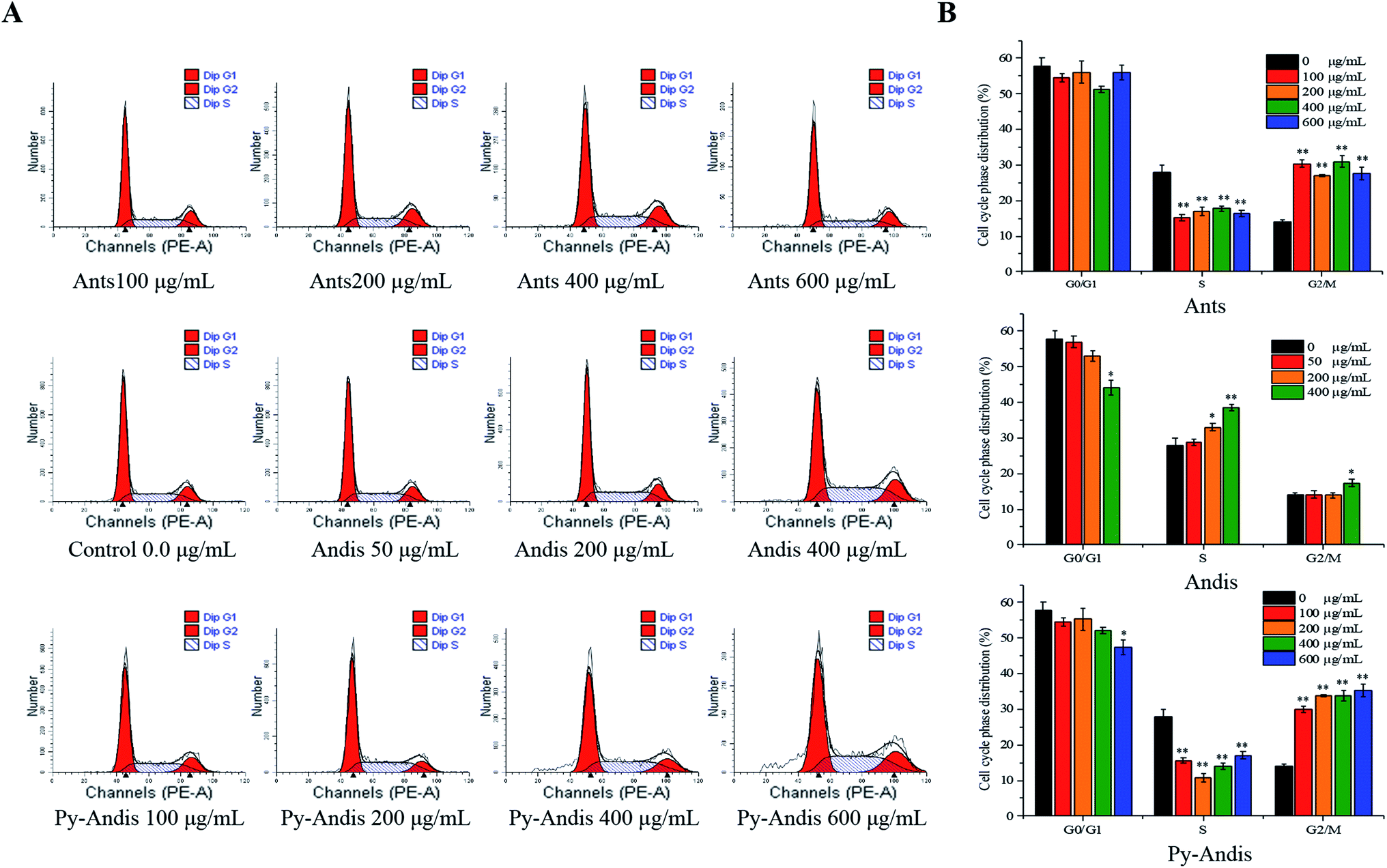 | ||
| Fig. 4 Cell cycle distribution G0/G1, S and G2/M phases were analyzed using a flow cytometry method. HeLa cells were treated with different concentrations of anthocyanins (Ants), anthocyanidins (Andis), and pyranoanthocyanidins (Py-Andis) for 24 h, and the cell cycle distribution was assessed by the nuclear DNA content of HeLa cells (A). The cell cycle distribution is shown by histograms (B). The percentage of cells in each phase is shown as the mean ± SD for three experiments. (**) p < 0.01, compared to the control group. (*) p < 0.05, compared to the control group. | ||
Cell apoptosis
The MTT results showed that HeLa cells treated with three anthocyan samples for 24 h decreased cell viability in a dose-dependent manner. However, the kinds of inhibitory mechanisms associated with the inhibitory effects of anthocyanin samples still need further experiments. During apoptosis, lipid asymmetry deviated and phosphatidylserine (PS) was exposed to the outer leaflet of the plasma membrane that can be assayed by annexin V.28 Therefore, we investigated the proapoptotic effects of three anthocyan samples by annexin V-FITC/PI staining and subsequent flow cytometry analyses. Treatment of HeLa cells with three anthocyan samples resulted in a marked increase in early and late apoptosis of cells compared with the control group, especially late apoptosis (Fig. 5A). Anthocyanidins (400 μg mL−1) treatment increased early (66.3%) and late (18.1%) apoptosis of cells up to approximately 84.4% compared to the control group. In contrast, the addition of anthocyanins and pyranoanthocyanidins at 400 μg mL−1 induced total apoptotic cells to approximately 31.95% and 39.6%, respectively. Anthocyanidins treatment at 600 μg mL−1 resulted in a more pronounced increase in cell apoptosis (95.8%) compared to anthocyanins (70.32%) and pyranoanthocyanidins (71.9%). Pyranoanthocyanidins at the highest dose of 800 μg mL−1 exhibited a higher level of apoptotic cells (83.83%) compared to anthocyanins (71.30%). As shown in Fig. 5B, exposure of HeLa cells to three anthocyan samples in the range of 0–600 μg mL−1 caused pronounced late apoptosis in a concentration-dependent manner. The present results indicated that anthocyanidins are the most potent agents in apoptosis induction among the three anthocyan samples, followed by treatments with pyranoanthocyanidins and anthocyanins.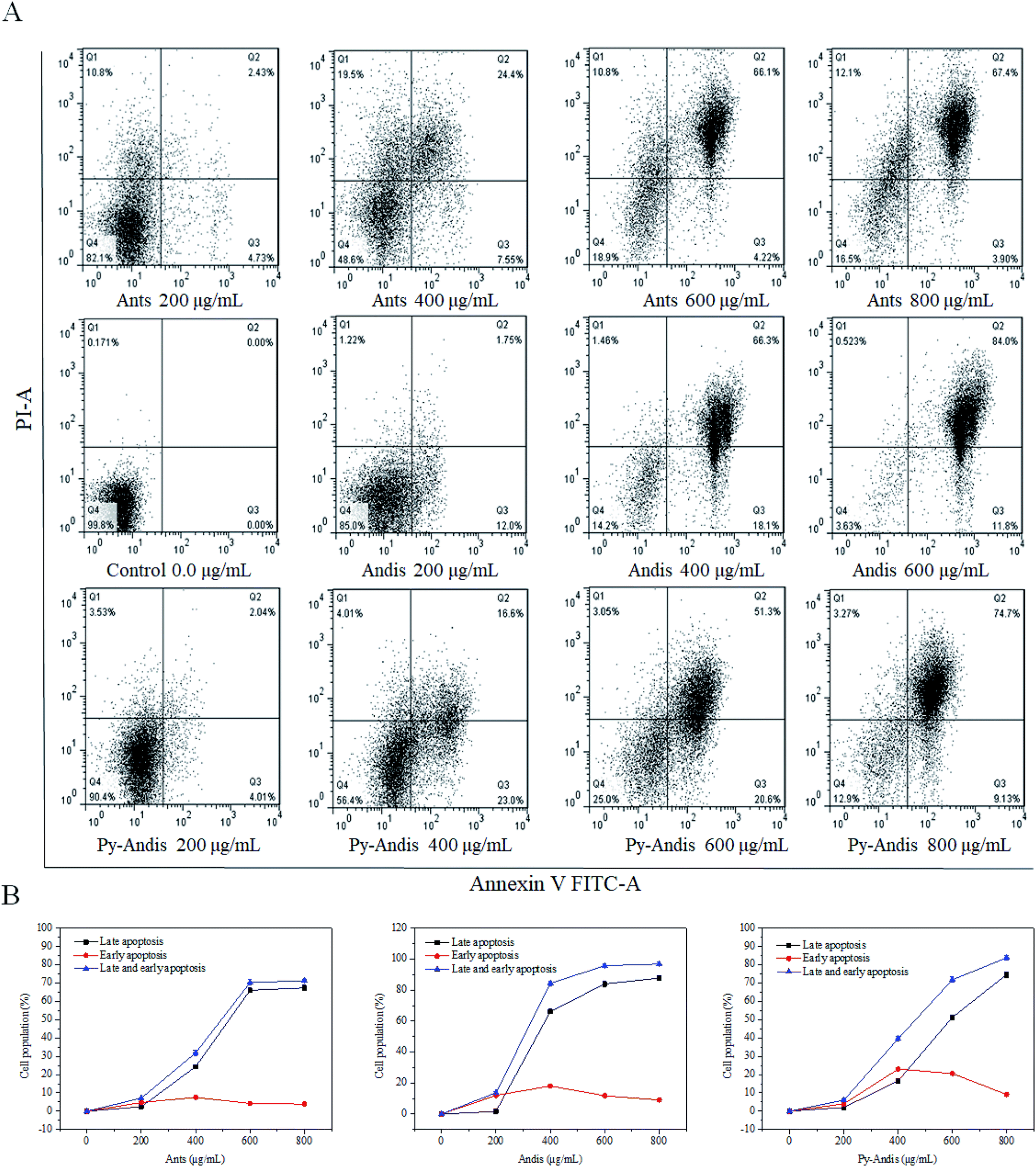 | ||
| Fig. 5 Induction of apoptosis in HeLa cells by anthocyanins (Antis), anthocyanidins (Andis), and pyranoanthocyanidins (Py-Andis). After treatment with three anthocyan samples for 24 h, the HeLa cells were double stained by Annexin V-FITC/PI and then subjected to flow cytometry. Data are expressed as mean ± SD of three independent experiments. Q1 denotes the necrotic cells; Q2, the cells of late apoptosis; Q3, the cells of early apoptosis; Q4, the viable cells. | ||
Western blot analysis
The above results showed that the inhibitory effects of three anthocyan samples on HeLa cell proliferation were accompanied by the cell cycle at different stages and were associated with cell apoptosis. To further elucidate the possible mechanisms underlying the cell cycle arrest or cell apoptosis of three anthocyan samples, we investigated the expression levels of several typical proteins, such as p53, cyclin D1, p38 MAPK, and caspase-3, in HeLa cells using western blotting.There is growing evidence of an association between p53 levels and cell cycle arrest as well as apoptosis induced by multiple phytochemicals from fruits or vegetables in cancer cells.15,29 Specially, the activation of p53 levels results in cell cycle arrest or apoptosis in response to DNA damage.30 To verify whether p53 was involved in anthocyanin-induced apoptosis or cell cycle arrest in HeLa cells, the levels of p53 in HeLa cells were determined using Western blot analysis. Exposure of HeLa cells to anthocyanins and pyranoanthocyanidins for 24 h at concentrations of (0–400 μg mL−1) and anthocyanidins (0–300 μg mL−1) resulted in a significant (p < 0.01) increase in levels of p53 in a dose-dependent manner (Fig. 6). The upregulation of p53 protein expression levels may be associated with the phosphatidylinositol 3-kinase (PI3K) signal pathway, the extracellular signal-regulated kinase (ERK) signal pathway, or the p38 mitogen-activated protein kinase (MAPK) signal pathway.31 As a kind of stress-activated protein kinase (SAPK), p38 MAPK plays an important role in the suppression effects of tumors because it can effectively regulate cell survival and proliferation.32 Several studies suggested that p38 MAPK and p53 were upregulated simultaneously in the apoptotic response to DNA damage.32 Treatment with 100, 200, and 400 μg mL−1 anthocyanins and pyranoanthocyanidins significantly (p < 0.01) increased the expression levels of p38 MAPK in HeLa cells in a dose-dependent manner compared with those in the control group (Fig. 6A and C, respectively). Anthocyanidins at 150 and 300 μg mL−1 also significantly (p < 0.05) increased the expression levels of p38 MAPK compared with the control group. The activation of p38 MAPK signal pathway has been reported to be helpful to the regulation of cell apoptosis or differentiation by the phosphorylation of transcription factors such as p53 or activator protein 1.33–35 These present findings suggest that the p38 MAPK/p53 signaling pathway might be involved in the apoptotic process of HeLa cancer cells induced by three anthocyan samples.
 | ||
| Fig. 6 Expression of cell cycle regulatory and apoptosis-induced proteins. HeLa cells were treated with different concentrations of anthocyanins (Ants), anthocyanidins (Andis), and pyranoanthocyanidins (Py-Andis) for 24 h, and then the expression levels of different proteins were analyzed by western blotting. β-Actin was used as loading control. Data are expressed as the mean ± SD of three independent experiments. | ||
The expression of the caspase-3 protein was measured to further clarify the hypothesis that the p38 MAPK/p53 signaling pathway is involved in the apoptotic induction process of three anthocyan sample-treated HeLa cells. Treatment with anthocyanins and pyranoanthocyanidins at 100, 200 and 400 μg mL−1 for 24 h and anthocyanidins at 50, 150, and 300 μg mL−1 for 24 h significantly (p < 0.05) upregulated the expression levels of the caspase-3 protein (Fig. 6) compared with the control group. The higher caspase-3 expression in HeLa cells following treatment indicated that the caspase family proteins were activated through the cellular apoptotic pathway, which is mainly composed of caspase-3 and cleaved-caspase-3, caspase-12 and cleaved-caspase-12. The upregulation of p53 and p38 MAPK as well as the activation of caspase-3 could be explained by the fact that anthocyanin samples triggered the intrinsic apoptotic pathway, where the Bcl-2 family proteins could be activated by p53. Subsequently, the expression of Bcl-2 is attenuated and the expression of Bax and t-Bid is increased, and then the cytochrome C is released from mitochondria into the cytoplasm.15 Cytosolic cytochrome C has been proven to bind apoptosis protease-activating factor-1 (Apaf-1) and then form the complex celled the apoptosome, which could mediate the activation of caspase-9 and caspase-3.36 Activated caspases cleave various target proteins, and then disable the normal cellular procedures, as well as breakdown the structural components of the cells, thus resulting in cell apoptosis.37 The above results demonstrated that three anthocyan samples induced cell apoptosis via the activation p38 MAPK/p53 signaling pathway, which supports our hypothesis.
The results of the cell cycle distribution indicated that three anthocyan samples could effectively induce cell cycle arrest at the G2/M phase. Cell cycle progression could be regulated by a large number of cell cycle regulatory proteins, such as p53, p21, p27, cyclin A, cyclin B, cyclin D1, and cyclin E.38,39 Thus, we performed Western blot analysis to detect the protein expression of several relevant regulators involved in G2/M phase arrest. As shown in Fig. 6, treatment of HeLa cells with three anthocyan samples led to the upregulation of p53 and the downregulation of cyclin D1 in a dose-dependent manner. The expression of p21 can be regulated by the tumor suppressor protein p53 to control the cell cycle.40 The expression levels of p21, p53 and CD25C are commonly used as markers for cell cycle arrest at the G2/M phase.31 The present results suggested that the anthocyanin sample-treated cell cycle arrest at the G2/M phase might be associated with the upregulation of p53 levels. Cyclin D1 is usually required for cells to process from the G0/G1 phase into the S phase, which will degrade when cells enter the S phase. However, the present results indicated that the HeLa cells treated with anthocyan samples were delayed at the G2/M phase, which suggests that downregulation of cyclin D1 is not directly associated with the cell cycle at the G2/M phase. Cyclin D1 might be one of the downstream effectors of p53, and the downregulation of cyclin D1 is associated closely with the status of p53.41 Thus, the present findings indicated that the anthocyan samples inhibited the proliferation of HeLa cells by causing an arrest of the cell cycle at the G2/M phase, with an increase in the levels of p53 and a concomitant decrease in the levels of cyclin D1.
Conclusions
Overall, our study clearly indicates that blueberry anthocyanin extracts and two derivatives, anthocyanidin and pyranoanthocyanidin pigments, were able to inhibit the proliferation of HeLa cancer cells through cell cycle arrest at the G2/M phase and the induction of late cell apoptosis. The inhibitory mechanisms of three anthocyan pigments on HeLa cells might be associated with the upregulation of the p53 protein and the activation of the p38 MAPK/p53 signaling pathway. In view of the stability and bioactivity of the three anthocyan samples, we concluded that both anthocyanidins and pyranoanthocyanidins are good candidates for cancer therapy, but further studies are necessary for investigating the inhibitory mechanisms and clinical applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by the National Natural Science Foundation of China (Grant No. 31271907 and No. 51503200) and the Thirteenth Five-Year Plan Science and Technology Research Project of Jilin Province Education Department (Grant No. JJKH20180090KJ).References
- S. Yıldırım, A. Kadıoğlu and A. Sağlam, et al., Fast determination of anthocyanins and free pelargonidin in fruits, fruit juices, and fruit wines by high-performance liquid chromatography using a core-shell column, J. Sep. Sci., 2016, 39(20), 3927–3935 CrossRef PubMed.
- A. Srovnalova, M. Svecarova and M. K. Zapletalova, et al., Effects of Anthocyanidins and Anthocyanins on the Expression and Catalytic Activities of CYP2A6, CYP2B6, CYP2C9, and CYP3A4 in Primary Human Hepatocytes and Human Liver Microsomes, J. Agric. Food Chem., 2014, 62(3), 789–797 CrossRef CAS PubMed.
- G. A. Macz-Pop, J. C. Rivas-Gonzalo and J. J. Pérez-Alonso, et al., Natural occurrence of free anthocyanin aglycones in beans (Phaseolus vulgaris L.), Food Chem., 2006, 94(3), 448–456 CrossRef CAS.
- X. Wu, S. Mckay and R. L. Prior, Aglycones and sugar moieties alter anthocyanin absorption and metabolism after berry consumption in weanling pigs, J. Nutr., 2005, 135(10), 2417–2424 CrossRef CAS PubMed.
- D. Rugină, Z. Sconţa and L. Leopold, et al., Antioxidant activities of chokeberry extracts and the cytotoxic action of their anthocyanin fraction on HeLa human cervical tumor cells, J. Med. Food, 2012, 15(8), 700 CrossRef PubMed.
- S. Li, J. Li and Y. Sun, et al., Transport of Flavanolic Monomers and Procyanidin Dimer A2 across Human Adenocarcinoma Stomach Cells (MKN-28), J. Agric. Food Chem., 2019, 67(12), 3354–3362 CrossRef CAS PubMed.
- M. E. Camire, S. Kubow and D. J. Donnelly, Potatoes and human health, Crit. Rev. Food Sci. Nutr., 2009, 49(10), 823–840 CrossRef CAS PubMed.
- A. Marquez, M. P. Serratosa and J. Merida, Pyranoanthocyanin Derived Pigments in Wine: Structure and Formation during Winemaking, J. Chem., 2013, 2013, 1–15 Search PubMed.
- S. Hillebrand, P. Quast and D. Bonerz, et al., 5-Carboxypyranocyanidin-3-(2(G)-glucosylrutinoside) a vitisine derivative from sour cherry juice: characterization and colour contribution, Dtsch. Lebensm.-Rundsch., 2008, 104(3), 120–126 CAS.
- M. García-Marino, M. L. Escudero-Gilete and M. T. Escribano-Bailón, et al., Colorimetric characteristics of the phenolic fractions obtained from Tempranillo and Graciano wines through the use of different instrumental techniques, Anal. Chim. Acta, 2012, 732(4), 153–161 CrossRef PubMed.
- K. Yoshida, N. Nagai and Y. Ichikawa, et al., Structure of two purple pigments, catechiopyranocyanidins A and B from the seed-coat of the small red bean, Vigna angularis, Sci. Rep., 2019, 9(1), 1484 CrossRef PubMed.
- H. E. Jingren, A. R. F. Carvalho and N. Mateus, Spectral Features and Stability of Oligomeric Pyranoanthocyanin-flavanol Pigments Isolated from Red Wines, J. Agric. Food Chem., 2010, 58(16), 9249–9258 CrossRef PubMed.
- M. Rentzsch, F. Weber and D. Durner, et al., Variation of pyranoanthocyanins in red wines of different varieties and vintages and the impact of pinotin A addition on their color parameters, Eur. Food Res. Technol., 2009, 229(4), 689–696 CrossRef CAS.
- N. K. Im, W. J. Jang and C. H. Jeong, et al., Delphinidin suppresses PMA-induced MMP-9 expression by blocking the NF-κB activation through MAPK signaling pathways in MCF-7 human breast carcinoma cells, J. Med. Food, 2014, 17(8), 855–861 CrossRef CAS PubMed.
- H. P. Huang, Y. C. Chang and C. H. Wu, et al., Anthocyanin-rich Mulberry extract inhibit the gastric cancer cell growth in vitro and xenograft mice by inducing signals of p38/p53 and c-jun, Food Chem., 2011, 129(4), 1703–1709 CrossRef CAS.
- C. P. Hsu, Y. T. Shih and B. R. Lin, et al., Inhibitory effect and mechanisms of an anthocyanins- and anthocyanidins-rich extract from purple-shoot tea on colorectal carcinoma cell proliferation, J. Agric. Food Chem., 2012, 60(14), 3686–3692 CrossRef CAS PubMed.
- Y. C. Chang, H. P. Huang and J. D. Hsu, et al., Hibiscus anthocyanins rich extract-induced apoptotic cell death in human promyelocytic leukemia cells, Toxicol. Appl. Pharmacol., 2005, 205(3), 201–212 CrossRef CAS PubMed.
- M. Rentzsch, M. Schwarz and P. Winterhalter, Pyranoanthocyanins – an overview on structures, occurrence, and pathways of formation, Trends Food Sci. Technol., 2007, 18(10), 526–534 CrossRef CAS.
- E. Wang, Y. Liu and C. Xu, et al., Antiproliferative and proapoptotic activities of anthocyanin and anthocyanidin extracts from blueberry fruits on B16-F10 melanoma cells, Food Nutr. Res., 2017, 61(1), 1325308 CrossRef PubMed.
- Y. Lu and L. Y. Foo, Unusual anthocyanin reaction with acetone leading to pyranoanthocyanin formation, Tetrahedron Lett., 2001, 42(7), 1371–1373 CrossRef CAS.
- J. S. Barnes, H. P. Nguyen and S. J. Shen, et al., General method for extraction of blueberry anthocyanins and identification using high performance liquid chromatography-electrospray ionization-ion trap-time of flight-mass spectrometry, J. Chromatogr. A, 2009, 1216(23), 4728–4735 CrossRef CAS PubMed.
- V. Gavrilova, M. Kajdzanoska and V. Gjamovski, Separation, Characterization and Quantification of Phenolic Compounds in Blueberries and Red and Black Currants by HPLC-DAD-ESI-MS, J. Agric. Food Chem., 2011, 59(8), 4009–4018 CrossRef CAS PubMed.
- V. D. Freitas and N. Mateus, Formation of pyranoanthocyanins in red wines: a new and diverse class of anthocyanin derivatives, Anal. Bioanal. Chem., 2011, 401(5), 1463–1473 CrossRef PubMed.
- J. He, A. R. F. Carvalho and N. Mateus, et al., Spectral Features and Stability of Oligomeric Pyranoanthocyanin-flavanol Pigments Isolated from Red Wines, J. Agric. Food Chem., 2010, 58(16), 9249–9258 CrossRef CAS PubMed.
- A. Smeriglio, D. Barreca and E. Bellocco, et al., Chemistry, Pharmacology and Health Benefits of Anthocyanins, Phytother Res., 2016, 30(8), 1265–1286 CrossRef CAS PubMed.
- Z. Zhu, N. Wu and M. Kuang, et al., Preparation and toxicological evaluation of methyl pyranoanthocyanin, Food Chem. Toxicol., 2015, 83, 125–132 CrossRef CAS PubMed.
- S. H. Lee, S. M. Park and S. M. Park, et al., Induction of apoptosis in human leukemia U937 cells by anthocyanins through down-regulation of Bcl-2 and activation of caspases, Int. J. Oncol., 2009, 34(4), 1077–1083 CAS.
- N. S. Namazi, H. Sepehri and L. Delphi, et al., Eupatorin and Salvigenin Potentiate Doxorubicin-Induced Apoptosis and Cell Cycle Arrest in HT-29 and SW948 Human Colon Cancer Cells, Asian Pac. J. Cancer Prev. APJCP, 2018, 19(1), 131–139 Search PubMed.
- J. A. Vaz, C. F. R. Ferreira, C. Tavares, G. M. Almeida, A. Martins and M. H. Vasconcelos, Suillus collinitus methanolic extract increases p53 expression and causes cell cycle arrest and apoptosis in a breast cancer cell line, Food Chem., 2012, 135(2), 596–602 CrossRef CAS PubMed.
- S. H. Park, M. Seong and H. Y. Lee, p38 MAPK-induced MDM2 degradation confers paclitaxel resistance through p53-mediated regulation of EGFR in human lung cancer cells, Oncotarget, 2016, 7(7), 8184–8199 Search PubMed.
- M. Chen, C. Chou and C. Shun, et al., Iron suppresses ovarian granulosa cell proliferation and arrests cell cycle through regulating p38 MAPK/p53/p21 pathway, Biol. Reprod., 2017, 106(3), e249 Search PubMed.
- L. R. Coulthard, D. E. White and D. L. Jones, et al., p38(MAPK): stress responses from molecular mechanisms to therapeutics, Trends Mol. Med., 2009, 15(8), 369–379 CrossRef CAS PubMed.
- J. Ouyang, Z. H. Zhang and Y. X. Zhou, et al., Up-regulation of Tight-Junction Proteins by p38 Mitogen-Activated Protein Kinase/p53 Inhibition Leads to a Reduction of Injury to the Intestinal Mucosal Barrier in Severe Acute Pancreatitis, Pancreas, 2016, 45(8), 1136 CrossRef CAS PubMed.
- I. Samuel, S. Zaheer and A. Zaheer, Bile-pancreatic juice exclusion increases p38 MAPK activation and TNF-α production in ligation-induced acute pancreatitis in rats, Pancreatology, 2005, 5(1), 20–26 CrossRef CAS PubMed.
- B. Li, J. Si and J. W. Dewille, Ultraviolet radiation (UVR) activates p38 MAP kinase and induces post-transcriptional stabilization of the C/EBPdelta mRNA in G0 growth arrested mammary epithelial cells, J. Cell. Biochem., 2010, 103(5), 1657–1669 CrossRef PubMed.
- D. S. Küpper, F. C. Valera and R. Malinsky, et al., Expression of apoptosis mediators p53 and caspase 3, 7, and 9 in chronic rhinosinusitis with nasal polyposis, Am. J. Rhinol. Allergy, 2014, 28(3), 187 CrossRef PubMed.
- J. H. Qiu, A. Asai and S. Chi, et al., Proteasome inhibitors induce cytochrome c-caspase-3-like protease-mediated apoptosis in cultured cortical neurons, J. Neurosci., 2000, 20(1), 259 CrossRef CAS PubMed.
- P. R. Vidya, M. R. Senthil and S. Maitreyi, et al., The flavonoid quercetin induces cell cycle arrest and mitochondria-mediated apoptosis in human cervical cancer (HeLa) cells through p53 induction and NF-κB inhibition, Eur. J. Pharmacol., 2010, 649(1), 84–91 CrossRef PubMed.
- M. L. Schoelch, J. A. Regezi and N. P. Dekker, et al., Cell cycle proteins and the development of oral squamous cell carcinoma, Oral Oncol., 1999, 35(3), 333–342 CrossRef CAS PubMed.
- W. Ahmed, M. Rahmani and P. Dent, et al., The cyclin-dependent kinase inhibitor p21(CIP1/WAF1) blocks paclitaxel-induced G2M arrest and attenuates mitochondrial injury and apoptosis in p53-null human leukemia cells, Cell Cycle, 2004, 3(10), 1305–1311 CrossRef CAS PubMed.
- R. P. Fathi, F. Shamileh and G. S. Mahmood, et al., Induction of G1 cell cycle arrest and cyclin D1 down-regulation in response to pericarp extract of Banehin human breast cancer T47D cells, Daru, J. Fac. Pharm., Tehran Univ. Med. Sci., 2012, 20(1), 101 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra01772k |
| This journal is © The Royal Society of Chemistry 2019 |