
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign of efficient bifunctional catalysts for direct conversion of syngas into lower olefins via methanol/dimethyl ether intermediates†
Xiaoliang
Liu
a,
Wei
Zhou
a,
Yudan
Yang
a,
Kang
Cheng
*a,
Jincan
Kang
a,
Lei
Zhang
b,
Guoquan
Zhang
b,
Xiaojian
Min
b,
Qinghong
Zhang
*a and
Ye
Wang
 *a
*a
aState Key Laboratory of Physical Chemistry of Solid Surfaces, Collaborative Innovation Center of Chemistry for Energy Materials, National Engineering Laboratory for Green Chemical Productions of Alcohols, Ethers and Esters, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, P. R. China. E-mail: kangcheng@xmu.edu.cn; zhangqh@xmu.edu.cn; wangye@xmu.edu.cn; Fax: +86-592-218-3047; Tel: +86-592-218-7470
bState Energy Key Lab of Clean Coal Grading Conversion, Shaanxi Coal and Chemical Technology Institute Co., Ltd., Xi'an 710070, P. R. China
First published on 30th April 2018
Abstract
The direct conversion of syngas into lower olefins is a highly attractive route for the synthesis of lower olefins. The selectivity of lower olefins via the conventional Fischer–Tropsch (FT) synthesis is restricted to ∼60% with high CH4 selectivity due to the limitation by the Anderson–Schulz–Flory (ASF) distribution. Here, we report the design of bifunctional catalysts for the direct conversion of syngas into lower olefins with selectivity significantly breaking the ASF distribution. The selectivity of C2–C4 olefins reached 87% at a CO conversion of 10% and was sustained at 77% by increasing CO conversion to 29% over a bifunctional catalyst composed of Zn-doped ZrO2 nanoparticles and zeolite SSZ-13 nanocrystals. The selectivity of CH4 was lower than 3% at the same time. It is demonstrated that the molar ratio of Zn/Zr, the density of Brønsted acid sites of SSZ-13 and the proximity of the two components play crucial roles in determining CO conversion and lower-olefin selectivity. Our kinetic studies indicate that methanol and dimethyl ether (DME) are key reaction intermediates, and the conversion of syngas to methanol/DME is the rate-determining step over the bifunctional catalyst. Formate and methoxide species have been observed on Zn-doped ZrO2 surfaces during the activation of CO in H2, and the formed methanol/DME are transformed into lower olefins in SSZ-13.
Introduction
Lower olefins (C2–C4 olefins), which are key building blocks for the manufacture of plastics, cosmetics and pharmaceuticals, are produced by cracking of petroleum-based naphtha in the current chemical industry.1 The growing demand for lower olefins has motivated a lot of recent research activities for synthesis of lower olefins from alternative carbon resources such as natural gas or shale gas, coal and renewable biomass via synthesis gas (syngas, CO/H2).2–4 Carbon dioxide can also be used as a renewable source of syngas.5Fischer–Tropsch (FT) synthesis is a classic route for the conversion of syngas to hydrocarbons.6,7 Many studies have been devoted to synthesizing lower olefins from syngas via the FT route on Fe-based or Co2C-based catalyst.2,4,7–14 However, the selectivity of C2–C4 olefins can hardly exceed 60% due to the limitation by the FT reaction mechanism. Generally, FT synthesis proceeds via the dissociation of CO, formation of surface CHx species (x = 1–3), coupling of CHx species to CnHm intermediates and the hydrogenation or dehydrogenation of CnHm intermediates to paraffin or olefin products (Fig. 1a).15–17 The coupling of CHx species is uncontrollable on surfaces of Fe- or Co-based catalysts, and the hydrocarbon products usually follow a statistical distribution known as the Anderson–Schulz–Flory (ASF) distribution.18 The maximum selectivity of C2–C4 hydrocarbons (including paraffins and olefins) is 58% according to the ideal ASF distribution.19
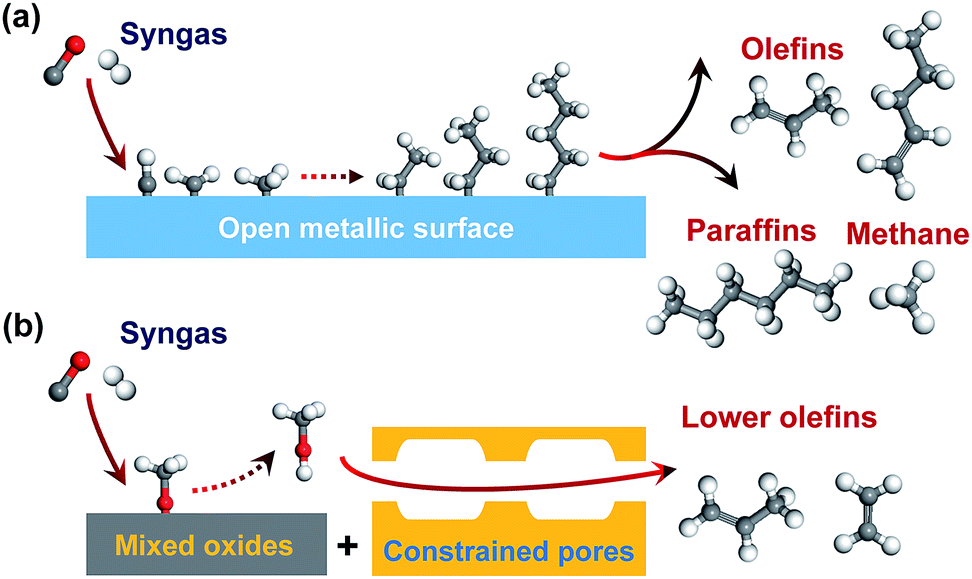 | ||
| Fig. 1 Illustration of two types of direct conversion of syngas into lower olefins. (a) FT-synthesis route on a Fe-based catalyst. (b) SMO (syngas via methanol intermediate to olefin) route on one bifunctional catalyst. | ||
The conversion of syngas into lower olefins involves a series of elementary steps (at least including CO activation and C–C coupling), and thus cannot be precisely controlled by using one kind of active sites as in FT synthesis. Thus, we have recently proposed a new reaction-coupling strategy for selectivity control in syngas conversion.20 The idea is to separate CO activation and C–C coupling on different active sites, which are integrated into one bifunctional catalyst. The reaction coupling may enable a better control of each step, in particular the C–C coupling by choosing an active component capable of performing selective C–C coupling. C2–C4 olefins can be produced from syngas via two consecutive processes, i.e., syngas to methanol (methanol synthesis) and methanol to olefins (MTO). As compared to the two-process route, a single-process using reaction coupling, which we define as the SMO process (Fig. 1b), i.e., the conversion of syngas via methanol intermediate into olefins, would be more energy- and cost-efficient. The product selectivity can be tuned by the shape selectivity of molecular sieves. Zeolites with chabasite (CHA) topology are known to be suitable for the selective formation of C2–C4 olefins.21,22
Actually, some early papers reported the conversion of syngas to hydrocarbons using mixed catalysts composed of a methanol-synthesis component (e.g., Pd/SiO2 or Cu–Zn oxide) and a zeolite, but the major products were C2–C4 paraffins.23–25 The formation of aromatics was also observed in a few early studies,26 and the selectivity of aromatics could be significantly enhanced recently through further improving the bifunctional catalyst.27,28 However, almost no C2–C4 olefins were formed in the early work.23–26 It is likely that C2–C4 olefins can easily undergo hydrogenation over the mixed catalysts, and thus the selective formation of C2–C4 olefins is a big challenge.
Recently, Jiao et al. reported an OX-ZEO process, i.e., the use of a composite catalyst containing a mixed metal oxide (ZnCrOx) and zeolite (MSAPO), for the direct conversion of syngas into lower olefins.3 The selectivity of C2–C4 olefins reached ∼80% at a CO conversion of 17% at 673 K under 2.5 MPa of syngas with a H2/CO ratio of 1.5. They excluded the possibility of methanol as a reaction intermediate and proposed that C2–C4 olefins were formed from syngas via a ketene (CH2CO) intermediate.3 At the same time, we communicated that a bifunctional catalyst composed of ZrO2–ZnO binary oxide and SAPO-34 could catalyse the conversion of syngas to lower olefins with a selectivity of 74% at 11% CO conversion at 673 K under 1.0 MPa of syngas with a H2/CO ratio of 2.29 The selectivity of C2–C4 olefins significantly breaks the upper limit of 60% for the conventional FT catalyst. These two pieces of work open a new avenue for the highly selective conversion of syngas beyond the classic FT process. However, the chemistry of this new process, in particular the reaction mechanism, is still unclear.
Here, we report our recent attempt to clarify the key factors that influence the catalytic behaviours of the bifunctional system. We adopt SSZ-13, an aluminosilicate zeolite with the same CHA-topology as SAPO-34, as the active component for the selective coupling of the intermediate to form C2–C4 olefins. Similar to SAPO-34, SSZ-13 is an efficient catalyst for the MTO reaction.30,31 Furthermore, SSZ-13 has attracted much attention as a commercial catalyst for the selective catalytic reduction of NOx with NH3 because of its high structural stability.32 In this work, we first utilize H–SSZ-13 in combination with metal oxides for the direct conversion of syngas into C2–C4 olefins. The present work also aims to provide insights into reaction mechanism. The reaction kinetics, reaction intermediates and possible routes for the formation of products (including lower olefins and major by-products) will also be discussed.
Results and discussion
Active components matching with each other for direct conversion of syngas to lower olefins
The use of one bifunctional catalyst to integrate the two reactions in tandem, i.e., syngas to methanol and subsequent MTO is quite challenging. The two reactions typically occur under different reaction conditions; methanol synthesis is operated at temperatures <573 K due to the thermodynamic requirement (Fig. S1a, ESI†), whereas the MTO reaction needs higher temperatures (typically 673–723 K) because of the high activation energy for C–C coupling and the removal of coke deposition.33 The continuous removal of methanol from methanol-synthesis catalyst by the tandem MTO reaction can overcome the thermodynamic limitation based on Le Chatelier's principle (Fig. S1b, ESI†).29,34 The equilibrium conversion of CO into lower olefins (C2H4 as an example) is ∼80% at 673 K and a syngas pressure of 3 MPa (Fig. S1b, ESI†). Thus, the catalyst being capable of selectively producing methanol at ∼673 K should be developed. We clarified that methane became a dominant product at 673 K over Cu–Zn–Al oxide, a typical methanol-synthesis catalyst (Fig. S2a, ESI†). Moreover, due to the strong hydrogenation ability of Cu–Zn–Al catalyst, the formed C2–C4 olefins were easy to be transformed to C2–C4 paraffins by hydrogenation. Almost no C2–C4 olefins were obtained on the bifunctional Cu–Zn–Al/SSZ-13–45H catalyst, which composed of Cu–Zn–Al oxide and H–Na–SSZ-13 with a 45% H+-exchanging degree (denoted as SSZ-13–45H) (Fig. S2b, ESI†). Here, SSZ-13–45H was used as the typical component for C–C coupling in the present work. As already mentioned, many studies by integrating a Cu- or Pd-based methanol-synthesis catalyst with a zeolite only led to the formation of saturated C2–C4 paraffins or gasoline-range hydrocarbons from syngas.23–26,35–37 Therefore, the development of a high-temperature methanol-synthesis catalyst with proper hydrogenation ability is the key to matching the MTO reaction for the synthesis of C2–C4 olefins.We discovered a Zn-doped ZrO2 (denoted as Zn–ZrO2) catalyst, which showed high selectivity of methanol and dimethyl ether (DME), a dehydrative product from methanol, from syngas in a wide-temperature range (Fig. 2a). The bifunctional catalyst composed of Zn–ZrO2 and SSZ-13–45H provided CH3OH/DME as the major products at <600 K (Fig. 2b), confirming that the zeolite-catalysed C–C coupling of CH3OH/DME can only take place at higher temperatures. The selectivity of C2–C4 olefins increased at the expense of that of CH3OH/DME upon increasing the reaction temperature. At the same time, the conversion of CO also increased significantly, becoming much higher than that on the Zn–ZrO2 catalyst at the corresponding temperature. These observations indicate the transformation of CH3OH/DME into C2–C4 olefins over the SSZ-13 component, which thermodynamically drives the conversion of syngas. The C2–C4 olefin selectivity and the CO conversion increased to 75% and 23%, respectively, by increasing the reaction temperature to 673 K (Fig. 2b). A further increase in temperature decreased the selectivity of C2–C4 olefins and increased that of C2–C4 paraffins due to the hydrogenation of lower olefins.
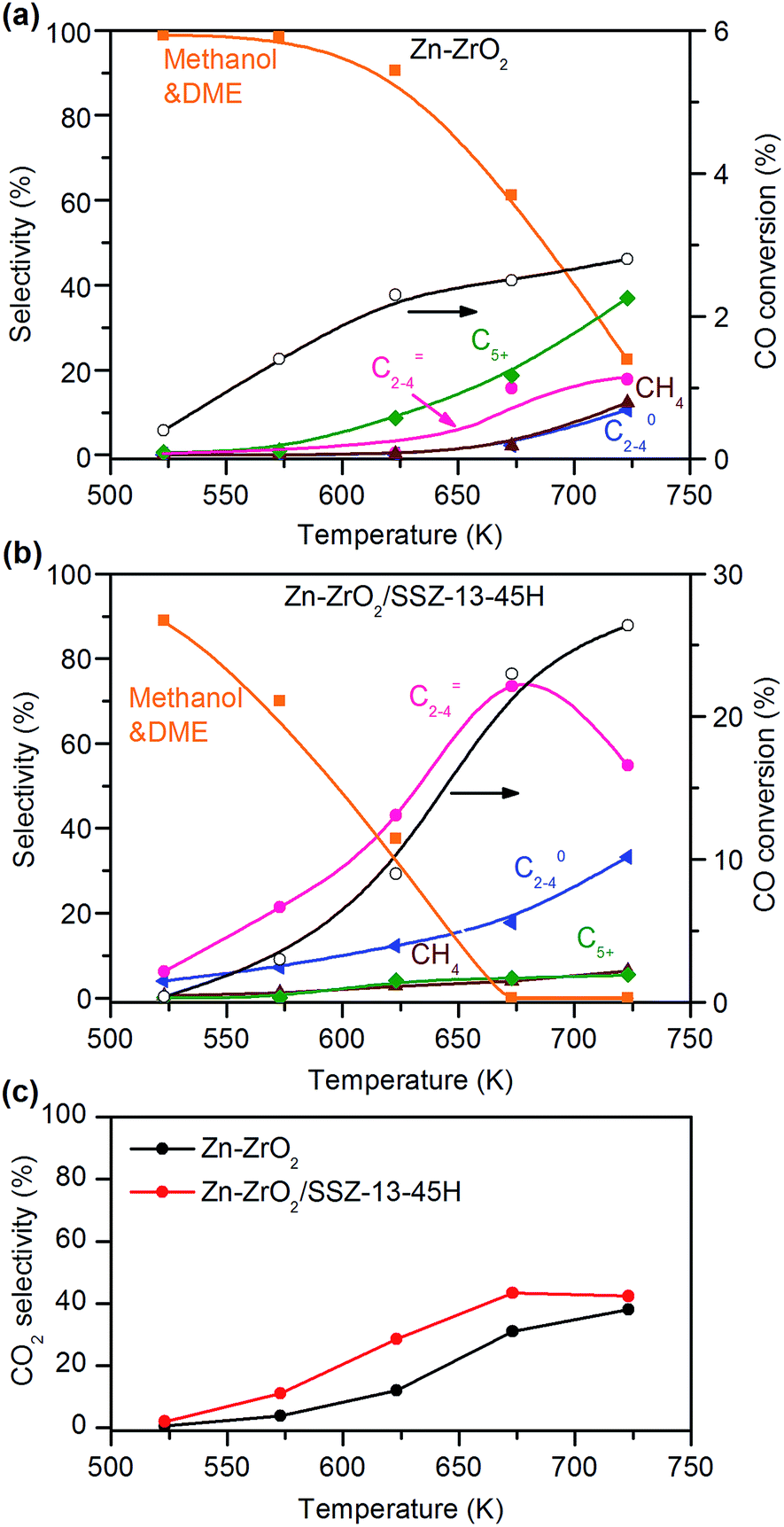 | ||
Fig. 2 Catalytic behaviours of Zn–ZrO2 and Zn–ZrO2/SSZ-13–45H catalysts for the conversion of syngas at different temperatures. (a) Zn–ZrO2 (Zn/Zr = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :16). (b) Zn–ZrO2/SSZ-13–45H. (c) CO2 selectivity. Reaction conditions: W(Zn–ZrO2) = 0.20 g; W(Zn–ZrO2/SSZ-13–45H) = 0.60 g; H2/CO = 2; P = 3 MPa; F = 30 mL min−1; time on stream, 10 h. :16). (b) Zn–ZrO2/SSZ-13–45H. (c) CO2 selectivity. Reaction conditions: W(Zn–ZrO2) = 0.20 g; W(Zn–ZrO2/SSZ-13–45H) = 0.60 g; H2/CO = 2; P = 3 MPa; F = 30 mL min−1; time on stream, 10 h. | ||
CO2 was also formed over both Zn–ZrO2 and Zn–ZrO2/SSZ-13–45H catalysts. The selectivity of CO2 was calculated separately from the hydrogenation of CO and the values depended on reaction temperature (Fig. 2c). The selectivity of CO2 was slightly higher over the Zn–ZrO2/SSZ-13–45H catalyst than over the Zn–ZrO2, but the degree of difference of CO2 selectivity between the two catalysts was much lower than that of CO conversion.
Key factors controlling the catalytic behaviour of bifunctional catalysts
:64) could increase the conversion of CO from 3.9% to 17% without a significant change in the selectivity of C2–C4 olefins. The selectivity of C2–C4 olefins could keep at 75% with an increase in CO conversion to 23% by raising the Zn/Zr ratio to 1:16. A further increase in the ratio of Zn/Zr to ≥1:4 considerably decreased the selectivity of C2–C4 olefins and increased that of C2–C4 paraffins, indicating that the hydrogenation of lower olefins became significant on catalysts with Zn/Zr ratios ≥1:4. When the Zn/Zr ratio exceeded 1:1, CO conversion also decreased. Thus, the control of Zn/Zr ratio is crucial to both CO conversion activity and product selectivity.
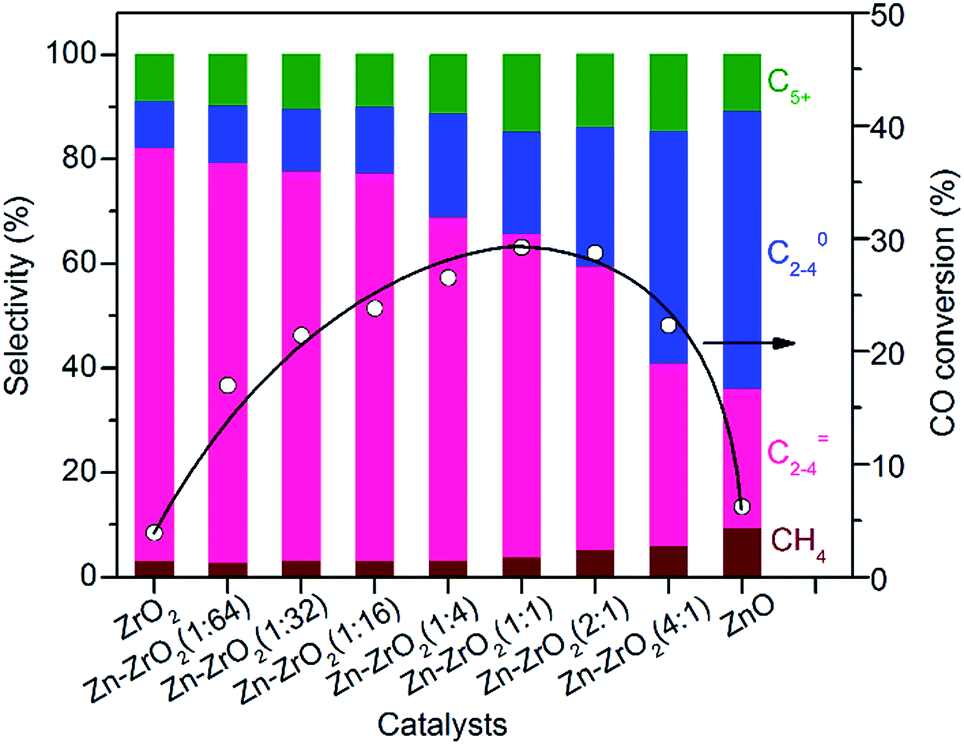 | ||
| Fig. 3 Effect of Zn/Zr molar ratio on catalytic behaviours of the bifunctional Zn–ZrO2/H–SSZ-13–45H catalyst. The selectivity of CO2 did not change with the ratio of Zn/Zr and was in the range of 41–45% for each catalyst. Reaction conditions: catalyst, 0.60 g; H2/CO = 2; P = 3 MPa; T = 673 K; F = 30 mL min−1; time on stream, 10 h. | ||
We have performed characterizations to gain insights into the effect of Zn/Zr ratio on structures of Zn–ZrO2 particles. The X-ray diffraction (XRD) measurements showed that ZrO2 particles were mainly in the tetragonal crystalline phase with an average size of 6.0 nm (Fig. S3 and Table S1, ESI†). The addition of Zn did not change the crystal structure of ZrO2 and no diffraction peaks belonging to ZnO were observed from XRD at Zn/Zr ratio of ≤1:16. Thus, such a small amount of ZnO may be incorporated into ZrO2 matrices, forming a solid solution, or may exist as small ZnO clusters on ZrO2. The formation of ZnO–ZrO2 solid solution was recently reported.38 At a higher Zn/Zr ratio (≥1:4), diffraction peaks ascribed to hexagonal ZnO became observable (Fig. S3, ESI†). The size of ZrO2 crystallites estimated by the Scherrer equation from XRD patterns did not change significantly with Zn addition (Table S1, ESI†). The transmission electron microscopy (TEM) measurements showed that the addition of Zn increased the mean size of Zn–ZrO2 particles (Fig. S4, ESI†). In particular, as the Zn/Zr ≥4:1, the mean size of Zn–ZrO2 particles increased steeply and the size distribution became significantly wide (Fig. S4 and Table S1, ESI†), possibly because of the segregation of large ZnO particles.
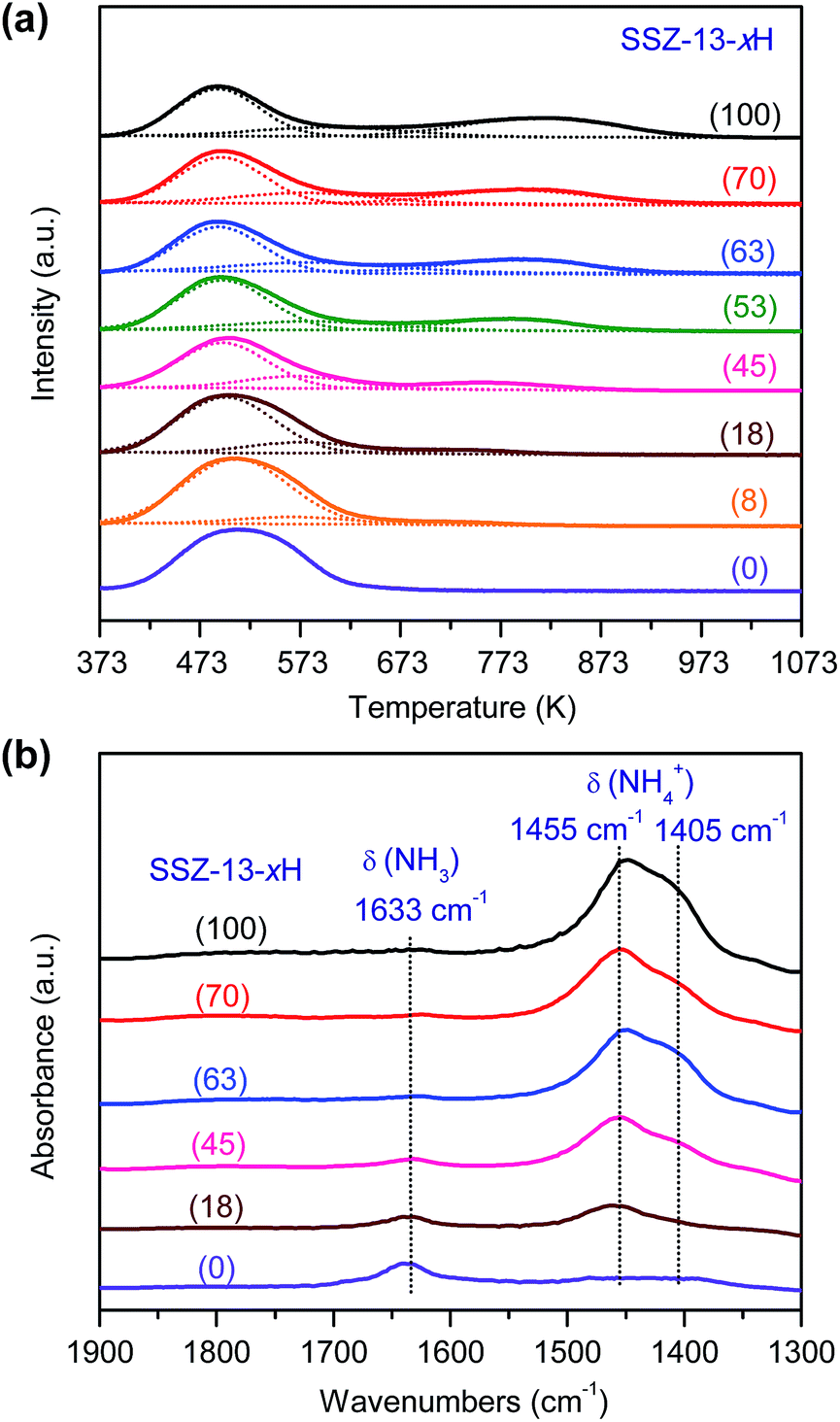 | ||
| Fig. 4 Characterization of acidity of SSZ-13 with different H+-exchanging degrees. (a) NH3-TPD profiles. (b) NH3-adsorbed FT-IR spectra. The number in the parentheses after each sample denotes the H+-exchanging degree. | ||
The bifunctional catalysts containing Zn–ZrO2 with a Zn/Zr ratio of 1:16 and SSZ-13 with different densities of Brønsted acid sites were investigated for the conversion of syngas. The CO conversion increased significantly from 5% to ∼25% with the density of Brønsted acid sites (Fig. 5a). The Brønsted acid site is known to catalyse the MTO reaction but is not responsible for CO activation. Thus, the significant increase in CO conversion should stem from the thermodynamic driving force because of the rapid removal of methanol/DME to form lower olefins by the Brønsted acid sites. This was confirmed by the change in product selectivity. CH3OH and DME were formed as the major products over the bifunctional catalyst with a lower density of Brønsted acid sites (Fig. 5b). The increase in the density of Brønsted acid sites decreased the selectivity of CH3OH/DME and increased that of lower olefins. The selectivity of C2–C4 olefins reached 75% when the density of Brønsted acid sites rose to 0.10 mmol g−1. However, a further increase in the density of Brønsted acid sites rather decreased the selectivity of C2–C4 olefins and increased that of C2–C4 paraffins. Thus, there is an optimum density of Brønsted acid sites for the selective formation of lower olefins. These results clearly demonstrate that the Brønsted acidity is a key factor in determining not only the conversion of CO but also the selectivity of lower olefins.
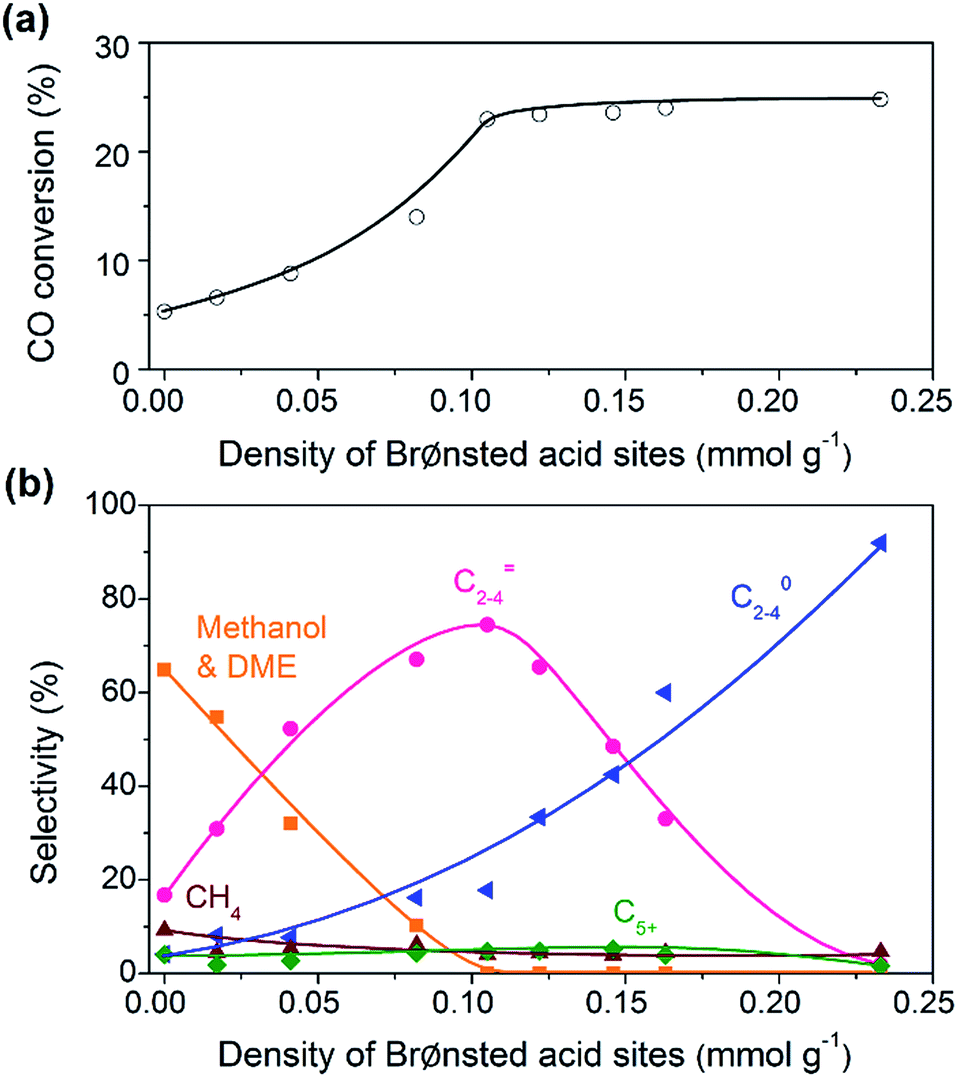 | ||
| Fig. 5 Effect of density of Brønsted acid sites on catalytic behaviours of the bifunctional Zn–ZrO2/SSZ-13 catalyst. (a) CO conversion. (b) Product selectivity. The selectivity of CO2 did not change with the density of Brønsted acid sites and was in the range of 41–45%. Reaction conditions: catalyst, 0.60 g; H2/CO = 2; P = 3 MPa; T = 673 K; F = 30 mL min−1; time on stream, 10 h. | ||
:16) and SSZ-13 in the bifunctional catalyst on catalytic behaviours. The separation of Zn–ZrO2 and SSZ-13 by quartz wool in one reactor, i.e., the dual-bed model, led to not only low activity but also decreased selectivity of C2–C4 olefins (Fig. 6a). We believe that the low CO conversion is due to the lack of the thermodynamic driving force when the zeolite SSZ-13 is far from the Zn–ZrO2 or the two components are separated in such a millimetre distance. The high selectivity of CH4 and C2–C4 paraffins probably results from the repeated contact of products with Zn–ZrO2 in the first bed and SSZ-13 in the second bed, leading to deep hydrogenation and the formation of saturated products instead of lower olefins. The saturated lower paraffins could not be converted on the subsequent zeolite catalyst. The use of mixture of Zn–ZrO2 granules and SSZ-13 granules with sizes 250–600 μm increases the proximity of the two components to micrometre scale, resulting in significant increases in both CO conversion and C2–C4 olefin selectivity (Fig. 6b). The grinding of micrometre-sized SSZ-13 crystallites (Fig. S5, ESI†) with Zn–ZrO2 powders with a mean particle size of 8.9 nm together, which was typically used in our work, caused the formation of Zn–ZrO2 nanoparticles dispersed on micrometre-sized SSZ-13 crystallites, and this further increased the conversion of CO and the selectivity of C2–C4 olefins (Fig. 6c). To further increase the proximity of the two components, we have synthesized SSZ-13 with a mean size of 230 nm (Fig. S6, ESI†) and Zn–ZrO2 nanoparticles with a smaller mean size of 4.8 nm by an alcogel method (Fig. S7, ESI†). The grinding of nano-sized SSZ-13 crystallites with powders of Zn–ZrO2 nanoparticles with mean sizes of 8.9 nm and 4.8 nm further increased the proximity between the two components into nano-scale (Fig. S8, ESI,†6d and e). The results in Fig. 6 display that the closer proximity between the two components results in higher CO conversion and higher C2–C4 olefin selectivity.
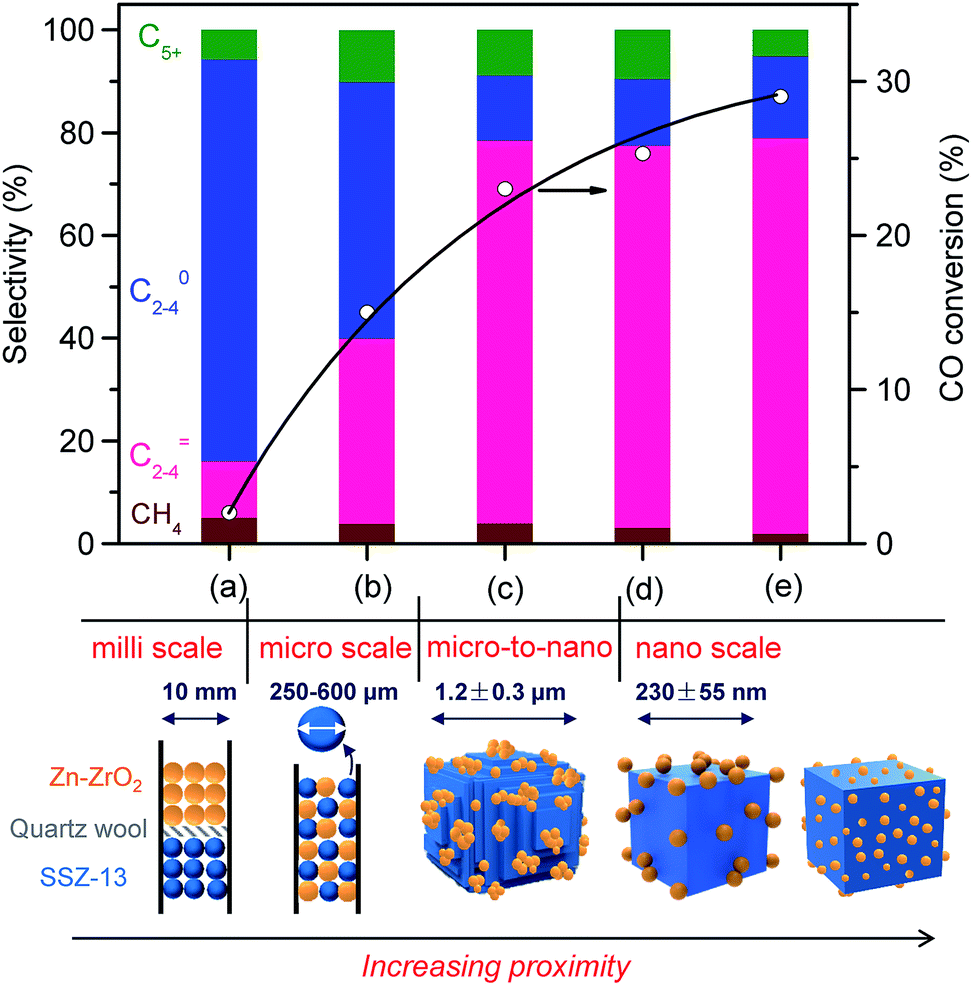 | ||
| Fig. 6 Effect of proximity of Zn-doped ZrO2 and SSZ-13 on catalytic behaviours of the bifunctional catalyst. (a) Dual bed (0.20 g Zn–ZrO2 + 0.40 g SSZ-13–45H); (b) mixing of granules of two components with sizes of 250–600 μm; (c) Zn–ZrO2 (8.9 nm)/micro-SSZ-13–45H; (d) Zn–ZrO2 (8.9 nm)/nano-SSZ-13; (e) Zn–ZrO2 (4.8 nm)/nano-SSZ-13. The selectivity of CO2 was in the range of 41–45% for each type of proximity. Reaction conditions: catalyst, 0.60 g; H2/CO = 2; P = 3 MPa; T = 673 K; F = 30 mL min−1; time on stream, 10 h. | ||
We obtained a CO conversion of 29% and a lower-olefin selectivity of 77% over the bifunctional catalyst containing Zn–ZrO2 nanoparticles with a mean size of 4.8 nm finely dispersed on nano-sized SSZ-13, denoted as Zn–ZrO2 (4.8 nm)/nano-SSZ-13. This combination of CO conversion and C2–C4 olefin selectivity is much better than that reported for our previous catalyst.29 The selectivity of C2–C4 olefins was 71% at 22% CO conversion over the ZnCrOx/MSAPO catalyst at H2/CO ratio of 2 and 673 K reported by Jiao et al.3 A recently reported MnO/MSAPO catalyst showed a C2–C4 olefin selectivity of 79% at CO conversion of 10.1% at 673 K and the increase in CO conversion to 15.4% decreased the C2–C4 olefin selectivity to 68.9%.42 Thus, our present catalyst is outstanding for the direct conversion of syngas into lower olefins. The major by-products in hydrocarbons were C2–C4 paraffins and the selectivity of C2–C4 paraffins was 18% over our catalyst. We further displayed the detailed selectivities of C2, C3 and C4 olefins and paraffins in Fig. 7a. It becomes clear that propylene with a selectivity of 49% is the most abundant olefin in the product, followed by ethylene (19%) and butenes (8.3%). Among C2–C4 paraffins, the selectivity of propane is the highest. The ratios of olefin to paraffin for C2, C3 and C4 hydrocarbons are 3.9, 4.4 and 3.3, respectively. This catalyst was also quite stable during a long-term reaction (Fig. 7b). The CO conversion and C2–C4 olefin selectivity were sustained at 25% and 75% after 160 h of reaction. The selectivity of undesired CH4 was around 3%. The changes in the selectivities of C2, C3 and C4 olefins were also insignificant after 160 h of reaction.
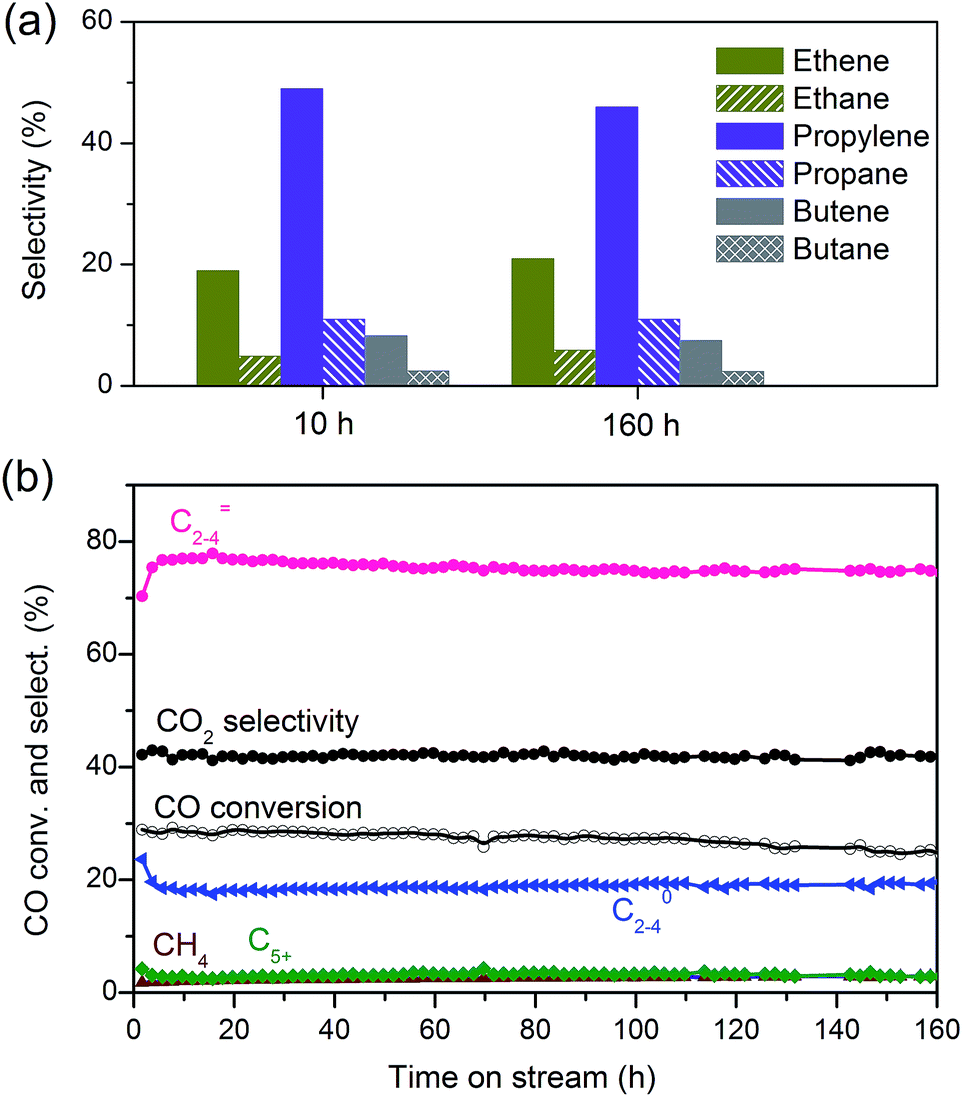 | ||
| Fig. 7 (a) Selectivities of C2, C3 and C4 olefins and paraffins over Zn–ZrO2 (4.8 nm)/nano-SSZ-13. (b) Stability of Zn–ZrO2 (4.8 nm)/nano-SSZ-13 catalyst. Reaction conditions: catalyst, 0.60 g; H2/CO = 2; P = 3 MPa; T = 673 K; F = 30 mL min−1. | ||
Structure–property relationship
The understanding of the roles of ZrO2 and ZnO in the bifunctional catalyst is crucial to future catalyst design for the conversion of syngas into lower olefins. ZrO2 is known to be capable of chemisorbing and activating CO.43 However, the activity of the bifunctional catalyst containing ZrO2 and SSZ-13 was very low (Fig. 3) probably due to its low capability of activating H2. An early report showed that the presence of Cu on ZrO2 could help H2 activation and enhance methanol synthesis.44 Nevertheless, the presence of Cu would accelerate the hydrogenation of lower olefins more significantly, leading to lower paraffins as the predominant products as in the case of Cu–Zn–Al oxide catalyst (Fig. S2, ESI†). One of our most important discoveries is that ZnO is an efficient component for the activation of H2, offering hydrogen species for the selective hydrogenation of CO with lower olefins surviving. The addition of small amount of Zn could significantly enhance CO conversion while keeping the high selectivity of lower olefins. It is well known that H2 can be activated via heterolytic dissociation on ZnO.45 The hydrogen species thus formed may be different from those formed via homolytic dissociation of H2 on metallic surfaces. Our result further suggests that the catalyst with –Zn–O– domains incorporated into ZrO2 lattice or with small ZnO clusters shows higher selectivity of lower olefins, whereas considerable paraffins are formed on catalysts containing larger ZnO particles. Therefore, the unique hydrogenation ability of heterolytically dissociated hydrogen species and their controlled surface concentration are the key to keeping lower olefins from hydrogenation.The CHA topology of zeolite in the bifunctional catalyst is certainly an important factor for selective formation of lower olefins. Our result reveals that the present SSZ-13 is more selective toward the formation of propylene (Fig. 7a), possibly because the size of pore windows of SSZ-13 matches better with that of propylene molecule. In addition to the topology, the Brønsted acidity is a key parameter. The Brønsted acid site catalyses the C–C coupling, transforming intermediates coming from Zn–ZrO2 nanoparticles into lower olefins. Although the Brønsted acid site does not directly participate in CO activation, CO conversion can be significantly enhanced by increasing the density of Brønsted acid sites because of the thermodynamic driving force. The product selectivity also depends on the density of Brønsted acid sites; there is an optimum density for the selective formation of lower olefins. Methanol and DME are the major products at a lower density of Brønsted acid sites, while the higher density of Brønsted acid sites results in considerable formation of paraffins. Although zeolites are not typical hydrogenation catalysts, the hydrogenation of olefins on transition-metal-free zeolites has been reported in a few early papers.46 The catalytic functions of Brønsted acid sites in zeolites for the dissociation of H2 and the hydrogenation of olefins have also been studied both experimentally and theoretically.47,48
How the proximity of the two functional sites affects the catalytic behaviour is a key issue for bifunctional catalysis but has not been well explored so far. A recent study uncovered that the too closer intimacy of Pt nanoparticles and the Brønsted acid sites in HY zeolite was detrimental to the selectivity of hydrocracking products.49 Our present study suggests that the closer proximity favours CO conversion and C2–C4 olefin selectivity. We propose that the closer proximity can enable a faster transfer of reaction intermediates formed on Zn–ZrO2 nanoparticles to SSZ-13, and thus is beneficial to the selective formation of lower olefins.
Kinetic studies
:16) catalysts (Fig. 8). This indicates that the reaction proceeds steadily over both catalysts. CH3OH and DME were formed as main products over the Zn–ZrO2 catalyst, and the total selectivity of methanol and DME exceeded 70% at shorter contact times. The prolonging of contact time decreased the selectivity of CH3OH/DME and increased that of C5+ hydrocarbons (Fig. 8a). The formation of C5+ hydrocarbons at longer contact times may be catalysed by the acid sites on mixed oxides.50 CH3OH and DME were also formed as the major products over the Zn–ZrO2/SSZ-13–45H catalyst at shorter contact time (Fig. 8b). The increase in contact time significantly increased the selectivity of C2–C4 olefins at expense of that of CH3OH/DME. When the contact time exceeded 1.2 s g mL−1, the selectivity of C2–C4 olefins decreased and that of C2–C4 paraffins increased more significantly due to the hydrogenation of olefins. These results are in agreement with those obtained by changing reaction temperature (Fig. 2b) and the density of Brønsted acid sites (Fig. 5b), providing further evidence that CH3OH and DME are the reaction intermediates on our bifunctional catalyst. The major reaction pathways for the conversion of CO on the bifunctional catalyst can be expressed as:| CO + H2 → CH3OH/DME → C2–C4 olefins → C2–C4 paraffins | (1) |
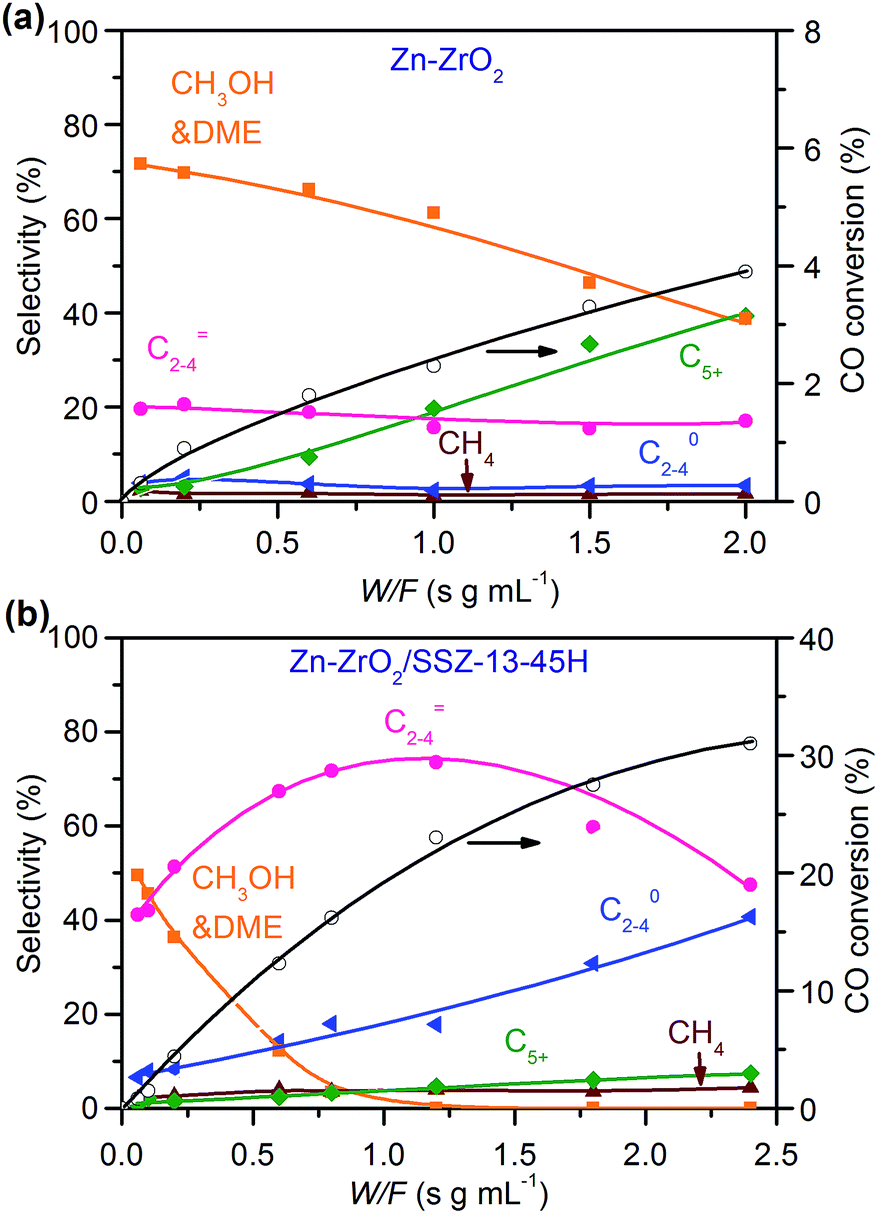 | ||
| Fig. 8 Effect of contact time on catalytic behaviours of Zn–ZrO2 and Zn–ZrO2/SSZ-13–45H catalysts. Reaction conditions: W(Zn–ZrO2) = 0.030–1.0 g; W(Zn–ZrO2/SSZ-13–45H) = 0.030–1.2 g; H2/CO = 2/1; P = 3 MPa; T = 673 K; F = 30 mL min−1; time on stream, 10 h. | ||
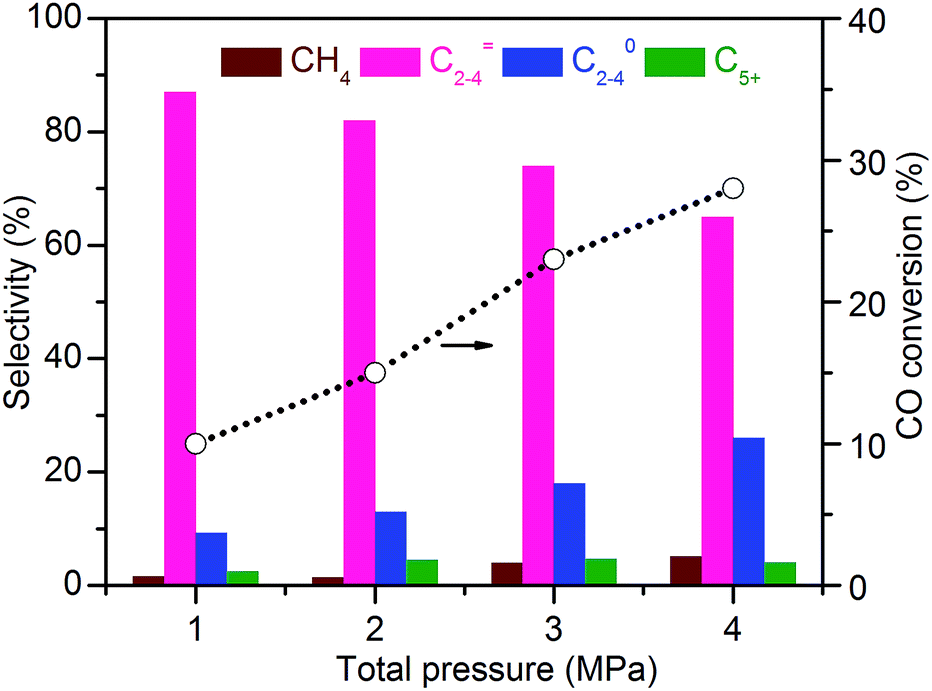 | ||
| Fig. 9 Effect of total pressure of syngas on catalytic behaviours of Zn–ZrO2/SSZ-13–45H catalyst. Reaction conditions: catalyst, 0.60 g; H2/CO = 2/1; T = 673 K; F = 30 mL min−1; time on stream, 10 h. | ||
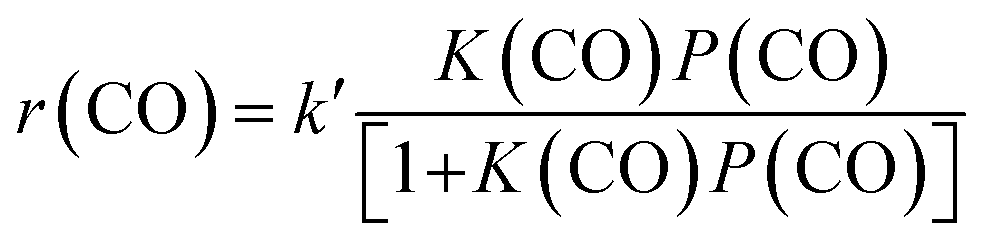 | (2) |
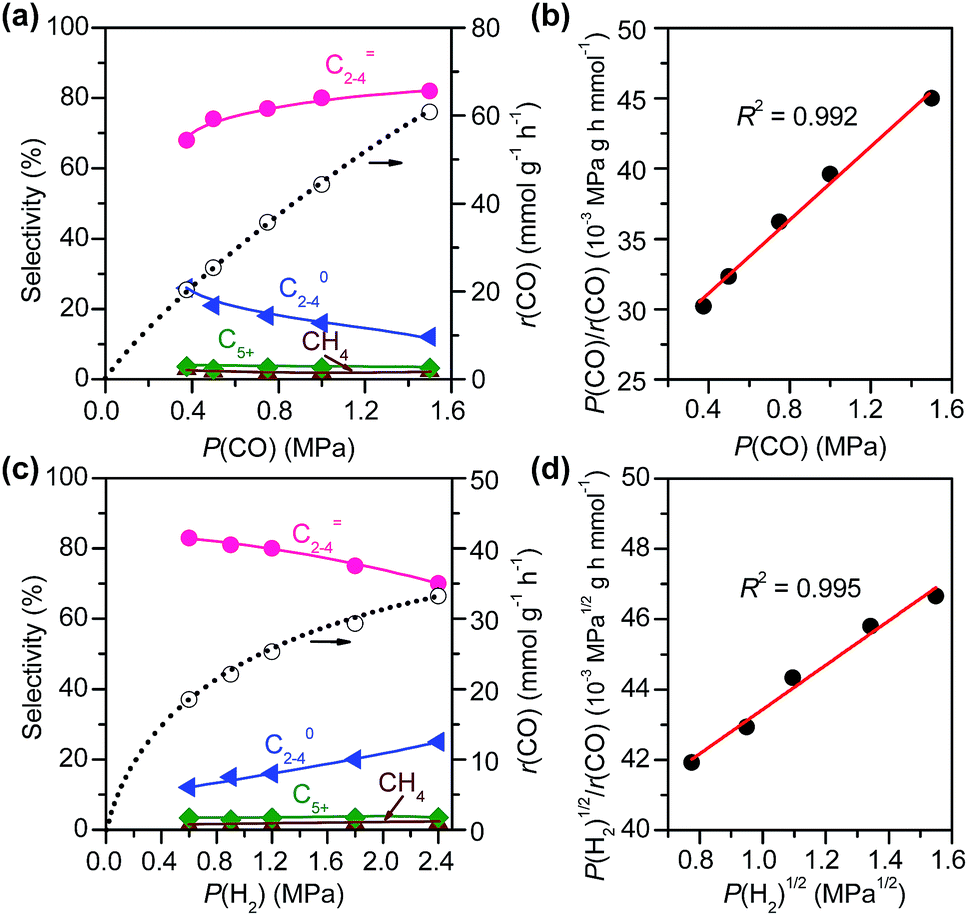 | ||
| Fig. 10 Effects of pressures of CO and H2 on catalytic behaviours of Zn–ZrO2/SSZ-13–45H catalyst. (a) Effect of CO pressure at a fixed H2 pressure. (b) Plot of P(CO)/r(CO) vs. P(CO). Reaction conditions: W = 0.40 g; P(H2) = 1.5 MPa; T = 673 K; F(total) = 50 mL min−1; P(total) = 3 MPa; time on stream, 10 h. (c) Effect of H2 partial pressure at a fixed CO pressure. (d) Plot of P(H2)1/2/r(CO) vs. P(H2)1/2. Reaction conditions: W = 0.40 g; P(CO) = 0.6 MPa; T = 673 K; F(total) = 50 mL min−1; P(total) = 3 MPa; time on stream, 10 h. | ||
The result on H2-pressure dependence shows that the rate of CO conversion is less than 0.5-order dependent on P(H2) (Fig. S10b, ESI†). Our analysis suggests a Langmuir-adsorption model with H2 dissociatively adsorbed on surface sites; the plot of P(H2)1/2/r(CO) versus P(H2)1/2 could be fitted to a good straight line with a R2 of 0.995. Thus, the rate of CO conversion at a fixed CO pressure can be expressed as:
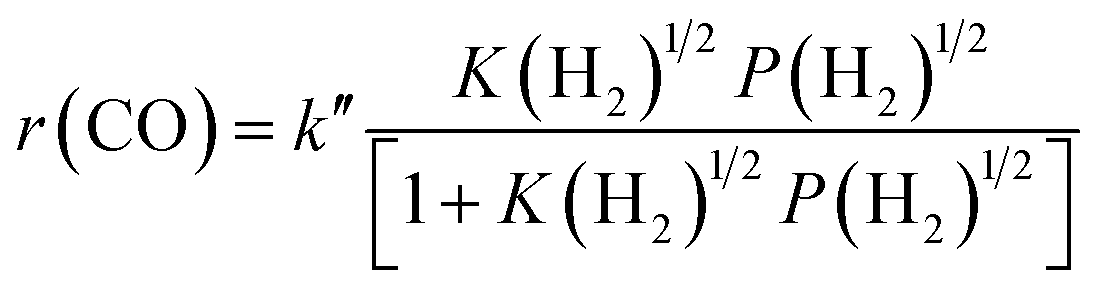 | (3) |
By combining the results described above, we can express the rate equation of CO conversion as:
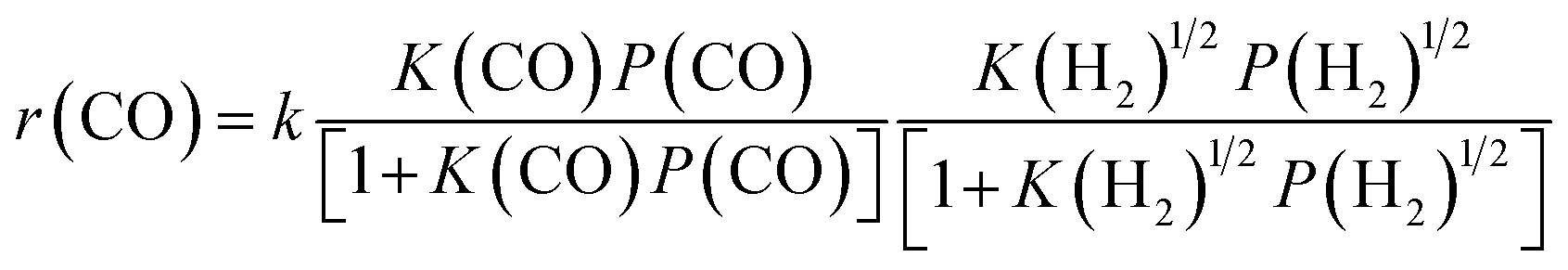 | (4) |
In addition to the CO conversion rate, the product selectivities are also dependent on the pressures of CO and H2. A higher pressure of CO favours the selectivity of C2–C4 olefins, whereas the increase in H2 pressure leads to higher selectivity of C2–C4 paraffins. This can be explained by the enhancement in hydrogenation of C2–C4 olefins at a higher H2/CO ratio.
Reaction mechanism
To further confirm that CH3OH is an intermediate, we have performed the conversion of CH3OH at 673 K under high-pressure H2 over zeolite SSZ-13 with different densities of Brønsted acid sites. Fig. 11 displays that the conversion of methanol increases with the density of Brønsted acid sites and reaches 97% at a density of Brønsted acid sites of 0.10 mmol g−1. The selectivity of C2–C4 olefins was kept at 65–70% when the density of Brønsted acid sites was lower than 0.10 mmol g−1. A further increase in the density of Brønsted acid sites caused the remarkable hydrogenation of C2–C4 olefins into C2–C4 paraffins. These trends agree well with those observed for the conversion of syngas; methanol/DME can be completely converted as the density of Brønsted acid sites increases to 0.10 mmol g−1 and a higher density of Brønsted acid sites would cause the hydrogenation of C2–C4 olefins into C2–C4 paraffins (Fig. 5). These results provide further evidence that methanol and DME are key reaction intermediates for the formation of lower olefins from syngas over our bifunctional catalysts. Furthermore, the rapid conversion of CH3OH over the catalyst with an adequate density of Brønsted acid sites agrees well with that the conversion of syngas into CH3OH/DME is the rate-determining step for the formation of lower olefins over the bifunctional catalyst.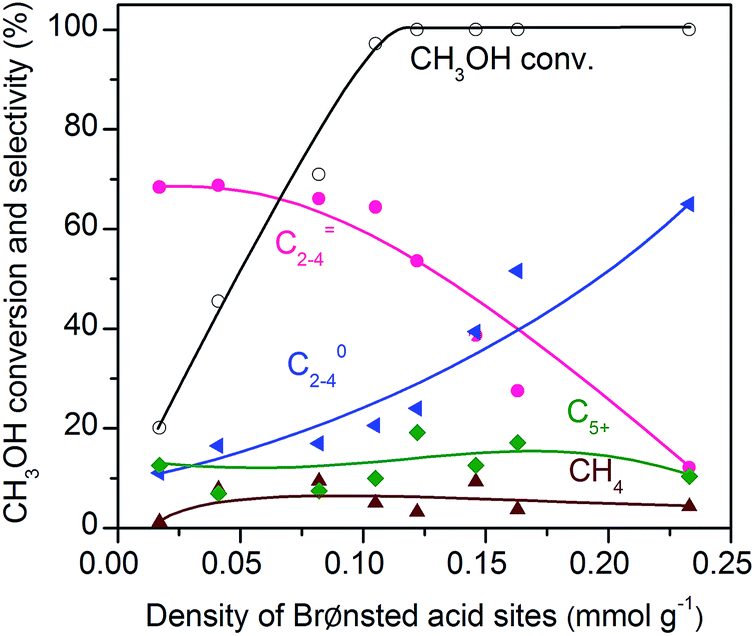 | ||
| Fig. 11 Effect of density of Brønsted acid sites for the conversion of methanol over SSZ-13 zeolites. Reaction conditions: catalyst, 0.4 g; F(H2) = 30 mL min−1; F(liquid CH3OH) = 0.01 mL min−1; P = 2 MPa; T = 673 K; time on stream, 3 h. | ||
To gain further insights into the adsorbed species on the surfaces of Zn–ZrO2 nanoparticles, we have performed in situ diffuse reflection infrared Fourier-transform spectroscopy (DRIFTS) studies. The adsorption of CO on the pre-reduced Zn–ZrO2 surface resulted in IR bands at 2974, 2870, 2733, 1594 and 1366 cm−1 at 553 K (Fig. 12). These IR bands can be ascribed to surface formate species.51,52 The intensity of formate species increased as the CO adsorption time increased from 1 to 30 min. The surface hydroxyl groups on metal oxide surfaces have been proposed to participate the formation of HCOO species.53 Our DRIFTS studies confirmed the existence of multi-coordinated hydroxyl species on fresh Zn–ZrO2 surfaces (Fig. S11, ESI†). The intensity of the hydroxyl group decreased after the adsorption of CO (Fig. S12, ESI†). This observation confirms the formation of HCOO species through the interaction of CO with surface hydroxyl groups. After switching from CO to CO/H2, we observed the generation of methoxide species located at 2930, 2820, 1152 and 1052 cm−1 together with formate species.51,52
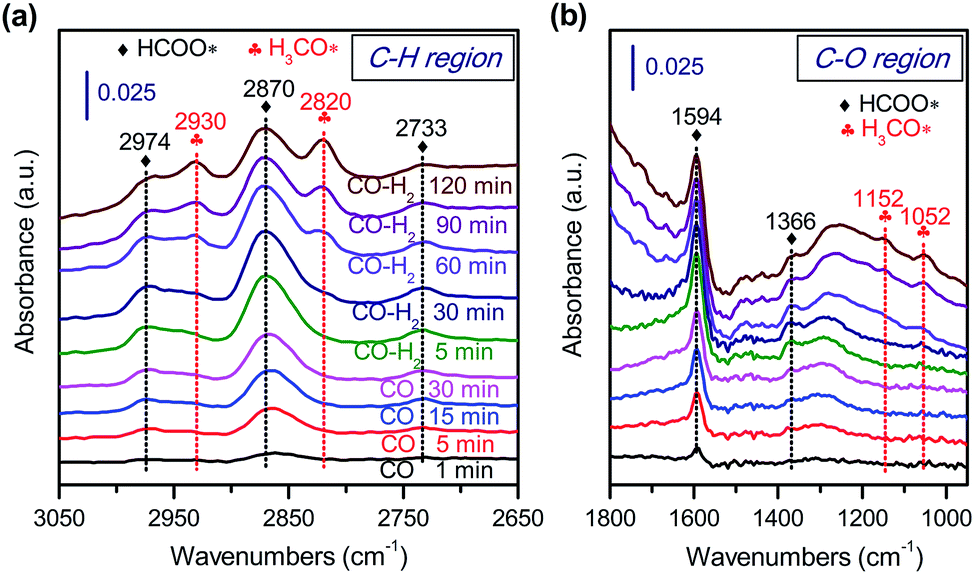 | ||
| Fig. 12 In situ DRIFT spectra for the conversion of CO on Zn–ZrO2 catalyst. (a) C–H region in 3050–2650 cm−1. (b) C–O region in 1800–1000 cm−1. | ||
The results described above and from literature allow us to speculate a possible reaction mechanism for the hydrogenation of CO on Zn–ZrO2 surfaces (Fig. 13). ZrO2 surfaces may activate CO, while H2 can be heterolytically dissociated on the –Zn–O– domains with H− bonded to Zn2+ and H+ bonded to O2−.45,53,54 The surface reaction of adsorbed CO with hydroxyl groups to form formate species should be a facile step as indicated by our in situ DRIFT studies (Fig. 12).55 The hydrogenation of formate species to form methoxide species needs the participation of H species from the heterolytic dissociation of H2. Based on previous studies,56,57 we speculate that the H− species might participate in the hydrogenation of formate, leading to the formation of methoxide species, whereas the H+ species might be involved in the formation of methanol and the recovery of surface hydroxyl groups (Fig. 13). Methanol can diffuse into the cages of SSZ-13, forming C2–C4 olefins catalysed by the Brønsted acid site. The rate equation (eqn (4)) could be interpreted by assuming that the surface reaction between adsorbed CO and dissociatively adsorbed H species on different active sites via a Langmuir–Hinshelwood mechanism is the rate-determining step (see details in ESI†). It is noteworthy that the hydrogen species formed by the heterolytic dissociation of H2 should play a very important role in the selective hydrogenation of CO while keeping lower olefins from hydrogenation. Further experimental and computational studies on the nature of such hydrogen species and their working mechanism are definitely needed in the future work.
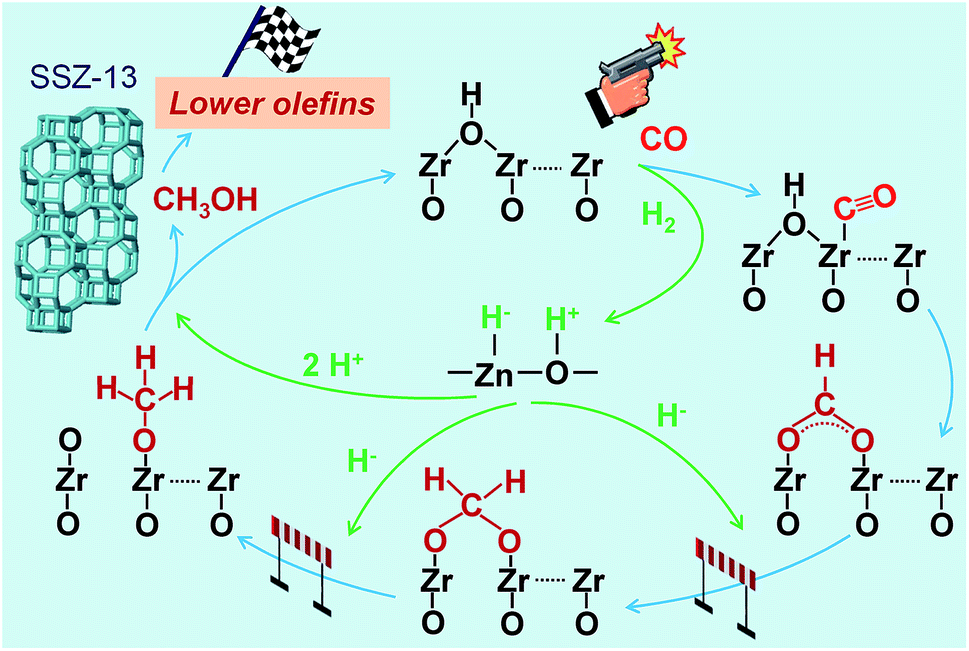 | ||
| Fig. 13 A possible reaction mechanism for the conversion of syngas into lower olefins over Zn–ZrO2/SSZ-13 catalyst. | ||
CO2 was formed as a major by-product over our bifunctional catalyst. We found that the selectivity of CO2 depended significantly on reaction temperature (Fig. 2c), but it did not change significantly with Zn/Zr ratio, the density of Brønsted acid sites and the proximity of the two components (Fig. 3, 5 and 6). The selectivity of CO2 was in a narrow range of 41–45% for the bifunctional catalyst at 673 K. Jiao et al. also observed the formation of CO2 with a selectivity of 45% at 673 K over the ZnCrOx/MSAPO catalyst.3 They proposed that CO2 was produced by the Boudouard reaction (2CO → CO2 + C*), and the C* was responsible for the formation of CH2 and CH2CO (ketene) species in gas phase, which was believed to be the key intermediate for the formation of C2–C4 olefins in their system.3 However, we observed the formation of methoxide species on our catalyst surfaces. Thus, we consider that the breakage of C–O bond does not take place on our Zn–ZrO2 surfaces but in the cage of zeolite SSZ-13. This is possibly because of the difference in metal oxide catalysts used for CO activation. It is quite interesting that the use of different metal oxides can lead to very different reaction mechanisms.
We speculate that the formation of CO2 arises from the water–gas shift (WGS) reaction (CO + H2O → CO2 + H2, ΔH0298 = −41 kJ mol−1), which is kinetically favourable at high temperatures. In fact, both ZnO and ZrO2 have been reported to be the active catalysts for the WGS reaction.58,59 We confirmed that the WGS reaction could easily occur on our Zn–ZrO2 catalyst. The H2O conversion was >90% at a H2O/CO molar ratio of ≤2.5/10 (Table S3, ESI†), which was the case on our catalyst by assuming a CO conversion <30%. This approaches the thermodynamic equilibrium for the WGS reaction. By assuming that the WGS reaction arrives at thermodynamic equilibrium over our catalyst, we have evaluated the selectivity of CO2 contributed solely from the WGS reaction. The result reveals that the selectivity of CO2 is around 45% at a CO conversion of 10–40% at 673 K (Table S4, ESI†). The selectivity of CO2 observed experimentally on our Zn–ZrO2/SSZ-13 catalyst was 43% at 673 K, which is very close to such an estimation. Thus, we believe that the generation of CO2 over our Zn–ZrO2-based catalysts is caused by the WGS reaction. The WGS reaction is helpful when the H2/CO ratio in syngas is low, but this reaction should be inhibited if syngas is produced from natural gas or shale gas. We expect that the design of methanol-synthesis catalyst with suppressed WGS activity may lead to lower CO2 selectivity.
Conclusions
We have demonstrated that the Zn–ZrO2/SSZ-13 catalyst efficiently bridges syngas to methanol/DME and methanol/DME to lower olefins. The selectivity of C2–C4 olefins reaches 87% at a 10% CO conversion and 77% at a 29% CO conversion at 673 K. Propylene with a selectivity of 49% is the major component in lower olefins. The bifunctional catalyst is stable during a 160 h of reaction. Our studies point out that the design of a high-temperature methanol-synthesis catalyst with controlled hydrogenation ability is one of the most important keys to realizing the selective formation of lower olefins. As compared to Cu-based catalysts, the Zn-doped ZrO2 is outstanding in keeping methanol at a high temperature (673 K) and lower olefins from hydrogenation on the bifunctional catalyst under high-pressure H2. There is an optimum molar ratio of Zn/Zr for the formation of lower olefins. Our work suggests that ZrO2 works for CO activation, while the –Zn–O– domains or small ZnO clusters are responsible for the activation of H2 possibly through heterolytic dissociation. The Brønsted acid site in the cages of zeolite SSZ-13 catalyses the C–C coupling to form lower olefins. The density of Brønsted acid sites affects both CO conversion and C2–C4 olefin selectivity. An appropriate density of Brønsted acid sites has been found for the selective formation of C2–C4 olefins because the hydrogenation of C2–C4 olefins also occurs on the Brønsted acid site. The proximity between Zn-doped ZrO2 and SSZ-13 plays a pivotal role in determining the catalytic behaviours. The closer proximity can facilitate the transfer of reaction intermediates, thus favouring both the activity and lower olefin selectivity. Our kinetic studies strongly indicate that methanol and DME are the key reaction intermediates on our bifunctional catalyst and the formation of methanol/DME is the rate-determining step. We have observed the formation of formate and methoxide species on Zn-doped ZrO2 surfaces, the precursors of methanol. Our studies also suggest that CO2, a major by-product, is formed by the water–gas shift reaction. It should be pointed out that although this work provides a reaction-coupling approach for the direct conversion of syngas into lower olefins with high selectivity breaking the ASF distribution, there are still limitations in both the CO conversion and the product selectivity. The formation of CO2 by the water–gas shift reaction decreases the final yield of C2–C4 olefins from CO. The control of hydrogenation ability of Zn-doped ZrO2 to suppress the hydrogenation of lower olefin products limits the conversion of CO at the same time. The CO conversion obtained in this work is much lower than the equilibrium conversion (∼80%). Future studies to improve the CO conversion and to inhibit the generation of CO2 are needed.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Key Research and Development Program of Ministry of Science and Technology (2017YFB0602201), the National Natural Science Foundation of China (Nos. 91545203, 21503174, 21433008, 21403177 and 21673188), and Shaanxi Coal and Chemical Technology Institute Co., Ltd.Notes and references
- J. J. H. B. Sattler, J. Ruiz-Martinez, E. Santillan-Jimenez and B. M. Weckhuysen, Chem. Rev., 2014, 114, 10613–10653 CrossRef CAS PubMed.
- H. M. Torres Galvis, J. H. Bitter, C. B. Khare, M. Ruitenbeek, A. I. Dugulan and K. P. de Jong, Science, 2012, 335, 835–838 CrossRef CAS PubMed.
- F. Jiao, J. Li, X. Pan, J. Xiao, H. Li, H. Ma, M. Wei, Y. Pan, Z. Zhou, M. Li, S. Miao, J. Li, Y. Zhu, D. Xiao, T. He, J. Yang, F. Qi, Q. Fu and X. Bao, Science, 2016, 351, 1065–1068 CrossRef CAS PubMed.
- L. Zhong, F. Yu, Y. An, Y. Zhao, Y. Sun, Z. Li, T. Lin, Y. Lin, X. Qi, Y. Dai, L. Gu, J. Hu, S. Jin, Q. Shen and H. Wang, Nature, 2016, 538, 84–87 CrossRef CAS PubMed.
- W. Sheng, S. Kattel, S. Yao, B. Yan, Z. Liang, C. J. Hawxhurst, Q. Wu and J. G. Chen, Energy Environ. Sci., 2017, 10, 1180–1185 CAS.
- Q. Zhang, J. Kang and Y. Wang, ChemCatChem, 2010, 2, 1030–1058 CrossRef CAS.
- K. Cheng, J. Kang, D. L. King, V. Subramanian, C. Zhou, Q. Zhang and Y. Wang, Adv. Catal., 2017, 60, 125–208 Search PubMed.
- H. M. Torres Galvis and K. P. de Jong, ACS Catal., 2013, 3, 2130–2149 CrossRef CAS.
- H. M. Torres Galvis, A. C. J. Koeken, J. H. Bitter, T. Davidian, M. Ruitenbeek, A. I. Dugulan and K. P. de Jong, J. Catal., 2013, 303, 22–30 CrossRef CAS.
- X. Zhou, J. Ji, D. Wang, X. Duan, G. Qian, D. E. Chen and X. Zhou, Chem. Commun., 2015, 51, 8853–8856 RSC.
- Y. Cheng, J. Lin, K. Xu, H. Wang, X. Yao, Y. Pei, S. Yan, M. Qiao and B. Zong, ACS Catal., 2016, 6, 389–399 CrossRef CAS.
- Y. Cheng, J. Lin, T. Wu, H. Wang, S. Xie, Y. Pei, S. Yan, M. Qiao and B. Zong, Appl. Catal., B, 2017, 204, 475–485 CrossRef CAS.
- P. Zhai, C. Xu, R. Guo, X. Liu, M. Li, W. Li, X. Fu, C. Jia, J. Xie, M. Zhao, X. Wang, Y. W. Li, Q. Zhang, X. Wen and D. Ma, Angew. Chem., Int. Ed., 2016, 55, 9902–9907 CrossRef CAS PubMed.
- Z. Li, L. Zhong, F. Yu, Y. An, Y. Dai, Y. Yang, T. J. Lin, S. Li, H. Wang, P. Gao, Y. Sun and M. He, ACS Catal., 2017, 7, 3622–3631 CrossRef CAS.
- P. Biloen and W. M. H. Sachtler, Adv. Catal., 1981, 30, 165–216 CAS.
- R. A. van Santen, I. M. Ciobîcă, E. van Steen and M. M. Ghouri, Adv. Catal., 2011, 54, 127–187 CAS.
- R. A. van Santen, A. J. Markvoort, I. A. W. Filot, M. M. Ghouri and E. J. M. Hensen, Phys. Chem. Chem. Phys., 2013, 15, 17038–17063 RSC.
- P. J. Flory, J. Am. Chem. Soc., 1936, 58, 1877–1885 CrossRef CAS.
- Q. Zhang, K. Cheng, J. Kang, W. Deng and Y. Wang, ChemSusChem, 2014, 7, 1251–1264 CrossRef CAS PubMed.
- K. Cheng, J. Kang, Q. Zhang and Y. Wang, Sci. China: Chem., 2017, 60, 1382–1385 CrossRef CAS.
- U. Olsbye, S. Svelle, M. Bjørgen, P. Beato, T. V. W. Janssens, F. Joensen, S. Bordiga and K. P. Lillerud, Angew. Chem., Int. Ed., 2012, 51, 5810–5831 CrossRef CAS PubMed.
- P. Tian, Y. Wei, M. Ye and Z. Liu, ACS Catal., 2015, 5, 1922–1938 CrossRef CAS.
- K. Fujimoto, H. Saima and H. Tominaga, J. Catal., 1985, 94, 16–23 CrossRef CAS.
- K. Fujimoto, H. Saima and H. Tominaga, Ind. Eng. Chem. Res., 1988, 27, 920–926 CrossRef CAS.
- Q. Zhang, X. Li, K. Asami, S. Asaoka and K. Fujimoto, Fuel Process. Technol., 2004, 85, 1139–1150 CrossRef CAS.
- K. Fujimoto, Y. Kudo and H. Tomonaga, J. Catal., 1984, 87, 136–143 CrossRef CAS.
- K. Cheng, W. Zhou, J. Kang, S. He, S. Shi, Q. Zhang, Y. Pan, W. Wen and Y. Wang, Chem, 2017, 3, 334–347 CAS.
- P. Zhang, L. Tan, G. Yang and N. Tsubaki, Chem. Sci., 2017, 8, 7941–7946 RSC.
- K. Cheng, B. Gu, X. Liu, J. Kang, Q. Zhang and Y. Wang, Angew. Chem., Int. Ed., 2016, 55, 4725–4728 CrossRef CAS PubMed.
- F. Bleken, M. Bjørgen, L. Palumbo, S. Bordiga, S. Svelle, K. P. Lillerud and U. Olsbye, Top. Catal., 2009, 52, 218–228 CrossRef CAS.
- M. A. Deimund, L. Harrison, J. D. Lunn, Y. Liu, A. Malek, R. Shayib and M. E. Davis, ACS Catal., 2016, 6, 542–550 CrossRef CAS.
- A. M. Beale, F. Gao, I. Lezcano-Gonzalez, C. H. F. Peden and J. Szanyi, Chem. Soc. Rev., 2015, 44, 7371–7405 RSC.
- Y. Wei, C. Yuan, J. Li, S. Xu, Y. Zhou, J. Chen, Q. Wang, L. Xu, Y. Qi, Q. Zhang and Z. Liu, ChemSusChem, 2012, 5, 906–912 CrossRef CAS PubMed.
- U. Olsbye, Angew. Chem., Int. Ed., 2016, 55, 7294–7295 CrossRef CAS PubMed.
- R. A. Dagle, J. A. Lizarazo-Adarme, V. Lebarbier Dagle, M. J. Gray, J. F. White, D. L. King and D. R. Palo, Fuel Process. Technol., 2014, 123, 65–74 CrossRef CAS.
- C. Wang, X. Ma, Q. Ge and H. Xu, Catal. Sci. Technol., 2015, 5, 1847–1853 CAS.
- D. L. S. Nieskens, A. Ciftci, P. E. Groenendijk, M. F. Wielemaker and A. Malek, Ind. Eng. Chem. Res., 2017, 56, 2722–2732 CrossRef CAS.
- J. Wang, G. Li, Z. Li, C. Tang, Z. Feng, H. An, H. Liu, T. Liu and C. Li, Sci. Adv., 2017, 3, e1701290 CrossRef PubMed.
- N. Katada, H. Igi, J. H. Kim and M. Niwa, J. Phys. Chem. B, 1997, 101, 5969–5977 CrossRef CAS.
- J. Datka, B. Gil and A. Kubacka, Zeolites, 1995, 15, 501–506 CrossRef CAS.
- F. Yin, A. L. Blumenfeld, V. Gruver and J. J. Fripiat, J. Phys. Chem. B, 1997, 101, 1824–1830 CrossRef CAS.
- Y. Zhu, X. Pan, F. Jiao, J. Li, J. Yang, M. Ding, Y. Han, Z. Liu and X. Bao, ACS Catal., 2017, 7, 2800–2804 CrossRef CAS.
- M. Y. He and J. G. Ekerdt, J. Catal., 1984, 90, 17–23 CrossRef CAS.
- K. D. Jung and A. T. Bell, J. Catal., 2000, 193, 207–223 CrossRef CAS.
- A. B. Anderson and J. A. Nichols, J. Am. Chem. Soc., 1986, 108, 4742–4746 CrossRef CAS.
- M. M. Khabib, S. K. Yu and V. S. Nakhshunov, Russ. Chem. Rev., 1976, 45, 142–154 CrossRef.
- J. Kanai, J. A. Martens and P. A. Jacobs, J. Catal., 1992, 133, 527–534 CrossRef CAS.
- S. Senger and L. Radom, J. Am. Chem. Soc., 2000, 122, 2613–2620 CrossRef CAS.
- J. Zecevic, G. Vanbutsele, K. P. de Jong and J. A. Martens, Nature, 2015, 528, 245–248 CrossRef CAS PubMed.
- Y. Li, D. He, Q. Zhu, X. Zhang and B. Xu, J. Catal., 2004, 221, 584–593 CrossRef CAS.
- F. Ouyang, J. N. Kondo, K. Maruya and K. Domen, J. Phys. Chem. B, 1997, 101, 4867–4869 CrossRef CAS.
- S. Kattel, B. Yan, Y. Yang, J. G. Chen and P. Liu, J. Am. Chem. Soc., 2016, 138, 12440–12450 CrossRef CAS PubMed.
- N. B. Jackson and J. G. Ekerdt, J. Catal., 1986, 101, 90–102 CrossRef CAS.
- S. Kouva, K. Honkala, L. Lefferts and J. Kanervo, Catal. Sci. Technol., 2015, 5, 3473–3490 CAS.
- K. Maruya, A. Takasawa, T. Haraoka, M. Aikawa, T. Arai, K. Domen and T. Onishi, Stud. Surf. Sci. Catal., 1993, 75, 2733–2736 CrossRef CAS.
- H. H. Kung, Catal. Rev., 1980, 22, 235–259 CAS.
- G. Rossmüller, V. Kleinschmidt, J. Kossmann and C. Hättig, J. Phys. Chem. C, 2009, 113, 1418–1425 Search PubMed.
- D. G. Rethwisch and J. A. Dumesic, J. Catal., 1986, 101, 35–42 CrossRef CAS.
- S. Kouva, J. Andersin, K. Honkala, J. Lehtonen, L. Lefferts and J. Kanervo, Phys. Chem. Chem. Phys., 2014, 16, 20650–20664 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, interpretation of rate equation from kinetic analysis, supplementary figures and tables. See DOI: 10.1039/c8sc01597j |
| This journal is © The Royal Society of Chemistry 2018 |