
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rational design of silicon-based composites for high-energy storage devices
Jung Kyoo
Lee
a,
Changil
Oh
a,
Nahyeon
Kim
a,
Jang-Yeon
Hwang
b and
Yang-Kook
Sun
*b
aDepartment of Chemical Engineering, Dong-A University, Busan 604-714, Republic of Korea
bDepartment of Energy Engineering, Hanyang University, Seoul 133-791, Republic of Korea. E-mail: yksun@hanynag.ac.kr; Fax: +82-2-2282-7329; Tel: +82-2-2200-0524
First published on 7th March 2016
Abstract
Silicon-based composites are very promising anode materials for boosting the energy density of lithium-ion batteries (LIBs). These silicon-based anodes can also replace the dendrite forming lithium metal anodes in lithium metal-free Li–O2 and Li–S batteries, which can offer energy content far beyond that of current LIBs. However, it is challenging to design silicon-based materials for use as anodes in real energy storage devices. In this review, we discuss how to boost the energy content of LIBs, the pros and cons of silicon-based anodes, and challenges associated with silicon-based anodes. A major focus of this review is on the rational design of silicon-based composite anodes to address the outstanding issues. In addition, high energy LIBs and Li–S batteries that employ silicon-based anodes are introduced and discussed.
1. Introduction
In our current society, energy and environmental issues have been recognized as top priorities among humanity's most pressing problems.1,2 Due to the current heavy dependence on the usage of non-renewable fossil fuel, our society is facing climate problems mainly caused by increasing levels of atmospheric CO2. Thus, we have to reduce the global atmospheric CO2 emission to a level (500 ± 50 ppm) that prevents climate change in the future,2 even though global energy demand continues to increase. An integral approach to addressing carbon and climate issues has been proposed using a portfolio of technologies based on solid scientific, technical and industrial legitimacy.2 Along these lines, solar and wind power have been used to replace fossil fuel (i.e., coal-based) power. In addition, energy efficient electric vehicles are replacing current internal combustion vehicles. The adoption of these technologies can significantly reduce global carbon emissions if powerful energy storage devices are available.Among the many candidates for energy storage, lithium-ion batteries (LIBs) offer the highest energy density among rechargeable batteries. LIBs also offer relatively good life cycle and power capability. Thus, LIBs dominate the market for portable electronics and drones, and they show great promise for electric vehicles and large-scale energy storage systems. At present, however, LIBs based on a combination of lithium metal oxides or phosphate (∼170 mA h g−1) cathodes and graphitic carbon anodes (theoretical capacity = 372 mA h g−1) offer a specific energy of ∼150 W h kg−1,3 which is far below the specific energy required for electric vehicles to match the performance of internal combustion vehicles. Therefore, a significant amount of research has been devoted to developing electrode materials that possess higher capacities than those currently available. On the cathode side, Ni-rich NCM cathodes delivering capacity levels exceeding 200 mA h g−1 have recently been reported4–6 and research is on-going worldwide to incorporate new cathodes with capacity level as high as 250–300 mA h g−1 in commercial LIBs.7,8 Even new electrochemical systems such as Li–S and Li–O2 have been intensively studied in devising energy storage systems offering energy density levels far exceeding those of current LIBs.9,10 On the anode side, the theoretical capacity of graphitic carbon has already been achieved in commercial LIBs, indicating that the anode technology in current LIBs has reached its limit in terms of energy density. Hence, new anode materials offering much higher capacity than graphitic carbon must be implemented to further increase LIB energy content.
2. How do we boost the energy content of LIBs?
In Fig. 1, the total specific capacity of the electrode material is calculated as a function of the specific capacities of the anode (CA) (Fig. 1a) and cathode (CC) (Fig. 1b) using eqn (1) below.11,12 For this purpose, only the specific capacities of the active materials (cathode + anode) are taken into account. This is because the other components in the LIB cells such as the separator, current collectors and electrolytes have lower mass fractions than the active material (about 60 wt% and 76 vol% in a 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 650 cell, see Table 1 below) and the mass fraction of the housing material is cell-size dependent.
650 cell, see Table 1 below) and the mass fraction of the housing material is cell-size dependent.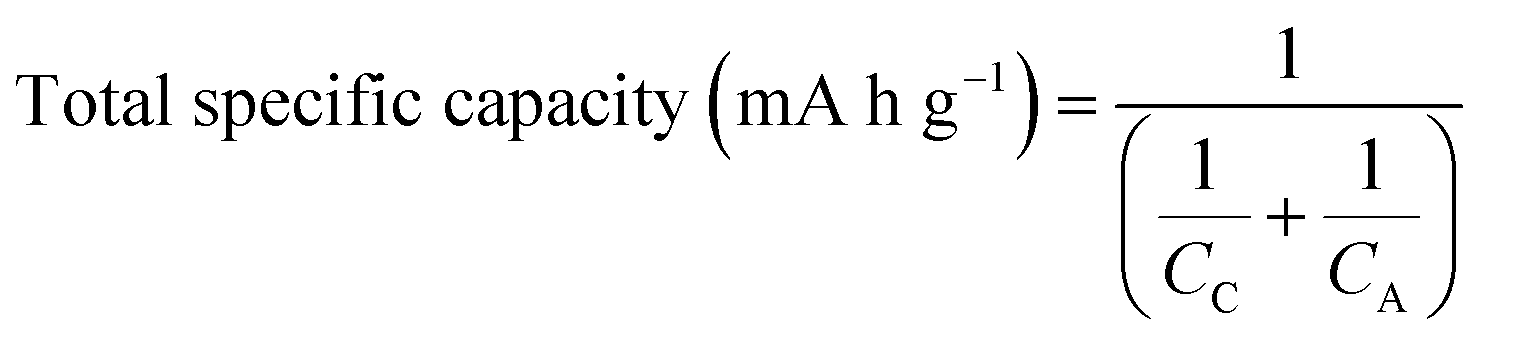 | (1) |
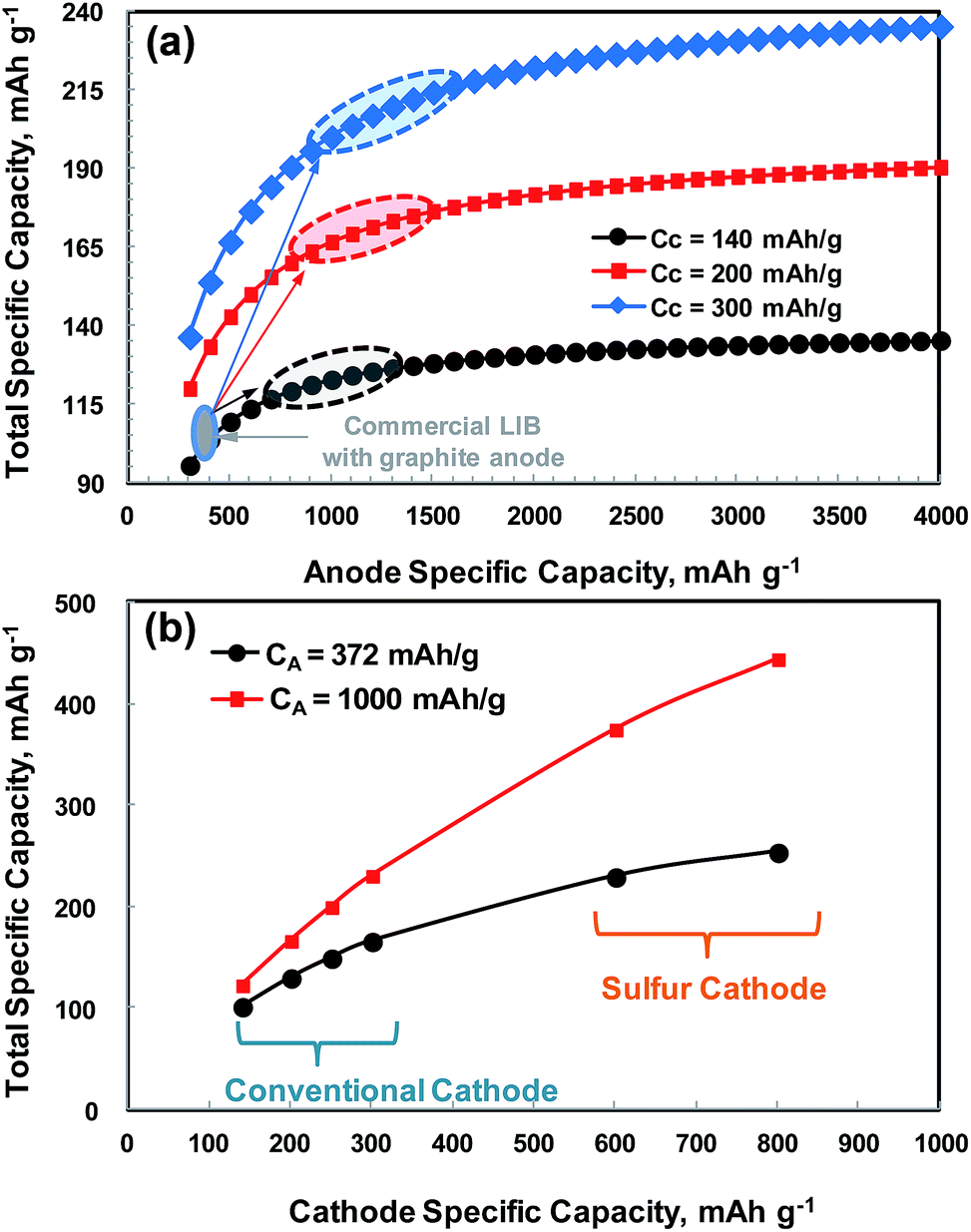 | ||
| Fig. 1 Total specific capacity of a LIB cell as a function of the specific capacity of (a) anode and (b) cathode. | ||
| Cell components | Weight | Volume | Thickness, μm | ||
|---|---|---|---|---|---|
| g | % | cm3 | % | ||
|
a Cathode material = LCO:LN1/2M1/4C1/4 = 2:1 wt ratio.
b Anode material = graphite.
|
|||||
| Separator | 1.2 | 2.7 | 1.8 | 10.9 | 19 |
| Cathode Al foil | 1.7 | 3.8 | 0.6 | 3.6 | 16 |
| Cathode active materiala | 18.3 | 41.3 | 6.5 | 39.4 | 91 |
| Anode Cu foil | 2.9 | 6.5 | 0.3 | 1.8 | 8 |
| Anode active materialb | 8.1 | 18.3 | 6.0 | 36.4 | 81 |
| Electrolyte | 4.6 | 10.4 | 1.3(?) | 7.9 | |
| Housing | 7.5 | 16.9 | |||
| Sum | 44.3 | 100.0 | 16.5 | 100.0 | |
As shown in Fig. 1a, the total specific capacity increases significantly with an increase in anode specific capacity up to about 800–1200 mA h g−1, and then reaches a saturation region, where the cathode specific capacity is fixed. The total specific capacity can also be significantly increased by using a high capacity cathode. The total specific capacity of current LIBs combined with a cathode of 140–170 mA h g−1 and a graphite anode (372 mA h g−1) is in the range of 101–117 mA h g−1. If the current cathodes providing 140–170 mA h g−1 are coupled with an anode whose capacity is 1000 mA h g−1, then the total specific capacity would be 123–145 mA h g−1, which corresponds to about a 21–25% increase in the total specific capacity. Similarly, when a cathode material of around 200 mA h g−1, which has been recently reported in the literature,5 is combined with a high capacity anode of 1000 mA h g−1, then ∼52% increase in the total specific capacity would be obtained over the current LIB (cathode of 155 mA h g−1 on average and graphite anode). With a more optimistic view, a ∼111% increase in the total specific capacity would even be possible when a cathode material of 250–300 mA h g−1, which has been intensively studied by many researchers,3,13 is available in the near future. To go beyond the horizon of LIBs in terms of energy content, Li–O2 and Li–S batteries could also be possible options.9 For these emerging technologies, alternative anodes to replace the dendrite-forming lithium metal are being intensively pursued. As compared in Fig. 1b, for example, if a sulfur cathode (in the form of S/C) whose composite capacity (based on sulfur + carbon weight) is in the range of 600–800 mA h g−1, which is a practicable range, is combined with an anode with a capacity of 1000 mA h g−1 (e.g., a lithiated-silicon/sulfur battery discussed below), then the total specific capacity would be in the range of 375–444 mA h g−1, which is far beyond those achievable with conventional LIBs (130 and 167 mA h g−1 when a cathode of 200 mA h g−1 is combined with an anode of 372 (graphite) and 1000 mA h g−1, respectively). However, considering the average working voltage of conventional LIBs (∼3.6 V) and Li metal-free Li–S (2.0 V or less), the specific energies would be 468–601 W h kg−1 and 750–858 W h kg−1, respectively. Thus, the specific energy gains of Li metal-free Li–S batteries over conventional to advanced LIBs would be in the range of 48–90% when the same amounts of cell components (other than active materials) are used in both battery systems. This could be a new milestone in LIB technology in terms of energy content, and it may provide electrical vehicles with the energy density required to reach a driving distance on the order of 400 km per charge.9
The shift of anode material from low capacity graphite to a high capacity material becomes more critical when one considers the volume of the LIBs. In electric vehicles and portable electronics, smaller LIBs are preferred and space is a critical factor in many cases. An example of the mass and volume fractions of the components of an 18650 cylindrical cell is given in Table 1.14 The mass content of the anode active material (graphite) in the cell is only 18.3 wt%, while the mass of the cathode active material (mixture of LCO:LN1/2M1/4C1/4 = 2:1 weight ratio) accounts for about 41 wt%. In terms of volume, however, graphite constitutes 36.4 vol% of the total, which is as much as that of the cathode active material (39.4 vol%). If a cathode material whose capacity is higher than that of the current cathodes (140–170 mA h g−1) is commercialized and is used in the same volume for manufacturing a higher energy LIB, a much larger amount of graphite must be used to match the cathode capacity. This will lead to a larger battery volume and the use of a larger amount of electrolyte due to increased anode porosity. The electrochemical performance of a thicker graphite anode would not be as good as that of a thinner one. One example of the commercial success in a LIB anode is a nanostructured SnCo anode used in SONY's Nexelion battery, which shows an increase in volumetric capacity by 50% over a conventional battery.15 The situation will become even worse when a much higher capacity cathode, i.e., S/C in Fig. 1b, is combined with a low capacity anode such as graphite. In this case, it may not be possible to design such an LIB cell due to the huge imbalance in the amount of active materials.
3. Why silicon-based anodes?
As discussed above, it is obvious that anode materials offering a specific capacity range of 800–1200 mA h g−1 have to be employed in replacement of a low capacity graphite anode to further increase both the specific and volumetric energies of current LIBs. Among the many candidates for anode materials,16 silicon-based anodes are the most promising because silicon has the highest lithium storage capacity among known elements; furthermore, its capacity is closest to that of metallic lithium, and silicon has a low voltage of about ∼0.4 V Li/Li+ for de-lithiation.17–25 As discussed above, silicon-based anodes can be used not only to boost the energy density of current LIBs but also to realize Li metal-free new battery systems such as Li–S and Li–O2 in the future.9,26 With these opportunities, silicon-based anodes have attracted increasing research interest in the past decade, and thus have been the subject of recent review topics.12,16,20,27–32 With the evolution of nanotechnology in material synthesis and characterization over the past decade, remarkable progress has been made recently in improving the electrochemical performances of silicon-based anodes for LIBs. Among the many synthesis strategies reported so far, nanoengineering of Si/C composites is believed to be the most promising approach for the massive commercialization of silicon-based anode materials. There is no doubt that advances in electrolyte additives33–37 and binders38–40 will provide other important contributions to enhancing the cycling stability of high capacity silicon-based anodes. Electrolyte additives enhance the stability of the solid electrolyte interface (SEI) layer, and new polymeric binders which have much stronger adhesive properties than the conventional polyvinylidene difluoride (PVDF) binder help maintain electrode integrity, leading to enhanced cycling stability of silicon-based anodes.41–44In this review, challenges related to silicon-based anode materials are briefly reviewed in comparison with state-of-the-art graphite anodes. The rest of this review will focus on the recent improvements in the electrochemical performances of Si/C composites through rational design. In addition, we provide representative results from full-cell LIBs and Li–S systems employing advanced silicon-based anodes, which are critical steps before bringing these systems to the market.
4. Challenges for silicon-based anodes
Table 2 compares the properties of lithium metal, graphite and silicon for LIB anodes. Graphite has a low theoretical capacity of 372 mA h g−1. Silicon has the largest specific capacity (3579 mA h g−1) among known elements that store lithium through an alloying reaction with lithium. Silicon has a lithium storage capacity that is even close to that of the lightest lithium metal (3862 mA h g−1). Li22Si5 was identified as the most Li-rich phase of the Li–Si system using equilibrium coulometric titration at a high temperature (415 °C).45 With a Li22Si5 (Li4.4Si) phase, the theoretical specific capacity of Si is generally calculated to be 4200 mA h g−1 with a net volume expansion of ∼310% compared to Si.19,45 However, Li15Si4 (Li3.75Si) has been proposed as the fully lithiated phase of silicon at room temperature, and this leads to a maximum capacity of 3579 mA h g−1 for Si.46,47 These values are commonly accepted in the scientific community and have shown agreement with the experimental results. Compared to Si, the Li15Si4 phase corresponds to a net volume expansion as high as 280%.47 The repeated huge volume expansion and contraction upon lithiation and delithiation, respectively, cause electrode pulverization, and thus loss of electrical contact between active materials and the active material–current collector interface. This leads to the common rapid capacity fading of silicon-based anodes. Another issue associated with silicon is its low electrical conductivity compared with graphite.| a The theoretical specific capacity of Si is based on Li15Si4. b Theoretical volumetric capacity is calculated by (theoretical specific capacity) × (density). | |||
|---|---|---|---|
| Anode materials | Li | C | Si |
| Density (g cm−3) | 0.53 | 2.25 | 2.33 |
| Lithiated phasea | Li | LiC6 | Li15Si4 |
| Theoretical specific capacity (mA h g−1) | 3862 | 372 | 3579a |
| Theoretical volumetric capacity (mA h cm−3)b | 2047 | 837 | 8339 |
| Volume change | 100 | 12 | 280a |
| Potential versus Li (V) | 0 | 0.05 | 0.4 |
| Conductivity, J cm−1 | Conductor | 0.61 × 103 | 2.52 × 10−6 |
Fig. 2 compares the typical voltage profiles of graphite and silicon particles having sizes on the order of micrometers. The average voltages for charging (Li intercalation) and discharging (Li de-intercalation) for graphite are 0.07 and 0.16 V vs. Li/Li+, respectively, and thus the voltage hysteresis is about 0.09 V vs. Li/Li+. Graphite allows very little room (0.07 V Li/Li+) for charging to avoid lithium plating during rapid charging. Silicon particles show a long voltage plateau at around ∼0.1 V vs. Li/Li+ in the first Li-alloying reaction (charging) to allow delivery of its theoretical capacity, and the voltage plateau increases to around 0.2 V vs. Li/Li+ in the subsequent cycles due to electrode polarization. The first de-alloying reaction occurs at around 0.4 V vs. Li/Li+ and it is slightly larger than that of graphite but much lower than those of metal oxides (typically 0.8–1.0 V vs. Li/Li+). Lithium plating during charging is less likely in silicon than in graphite but it will give a slightly lower discharge voltage (about 0.24 V vs. Li/Li+) than graphite when coupled with a specific cathode in a complete LIB cell. Considering the lithium storage capacity and operation voltage range, silicon is the best candidate for replacing graphite anodes. As shown in Fig. 2b, however, the micrometer-sized silicon particles lose capacity very quickly in subsequent cycles due to electrode pulverization caused by the large volume expansion discussed above.48 Hence, nanostructured silicon should mitigate the detrimental effects associated with volume expansion.
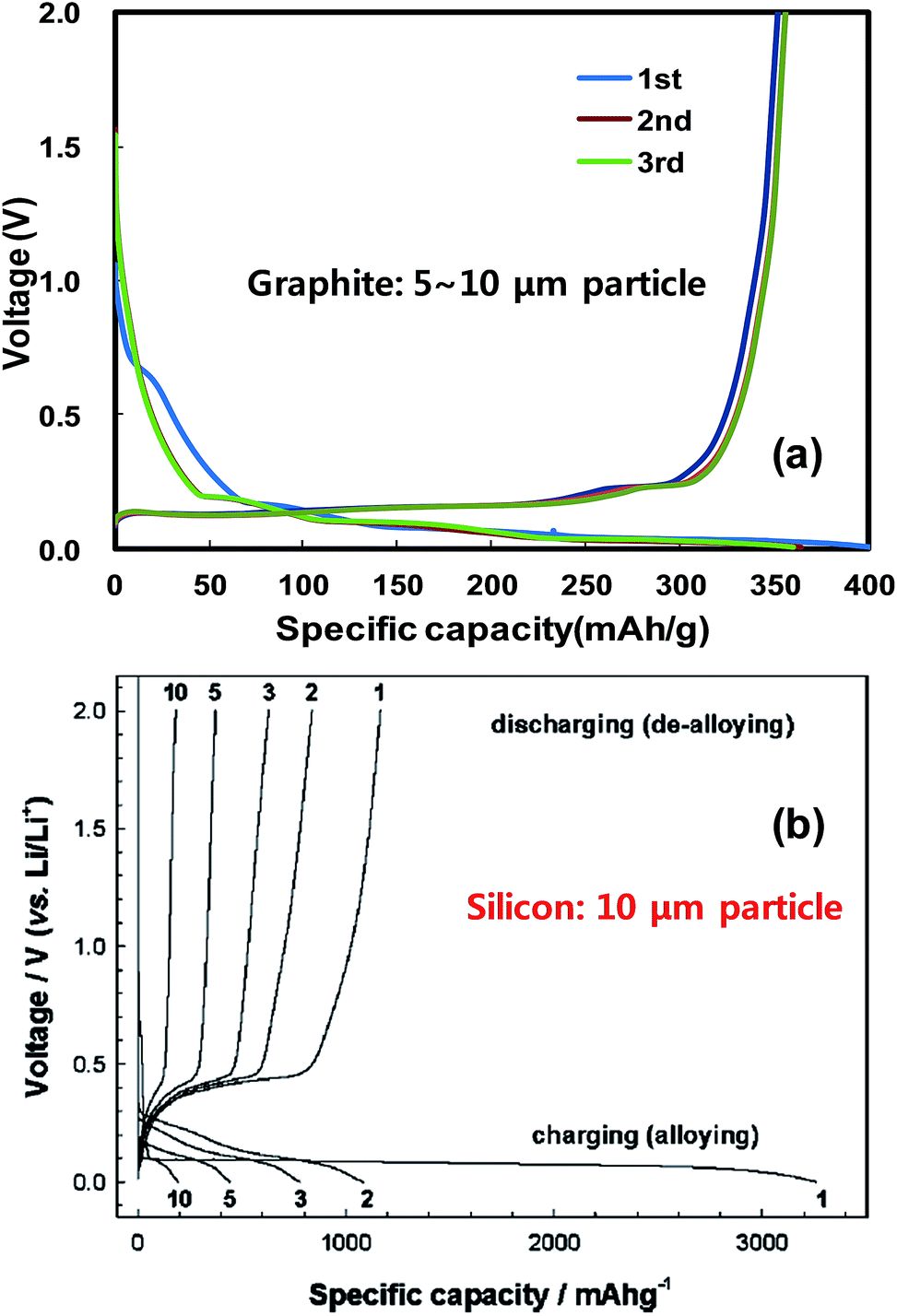 | ||
| Fig. 2 Typical galvanostatic charge–discharge voltage profiles obtained with anodes of (a) graphite powder and (b) silicon powder having sizes on the order of micrometers. (b) is reproduced with permission.48 Copyright 2004, The Electrochemical Society. | ||
Recently, Liu et al.49 investigated the lithiation of individual silicon nanoparticles using real time in situ transmission electron microscopy. Fig. 3 shows the size evolution of a single silicon nanoparticle as a function of lithiation time. For example, the diameter of a spherical silicon nanoparticle of 80 nm was increased to 130 nm after complete lithiation, corresponding to a net volume expansion of around 300%. Fig. 3b also clearly shows the time-dependent progressive lithiation of a silicon sphere from its shell (Li–Si alloy) to core (Si), indicating the co-existence of two-phase regions before completion of lithiation.47,49 Liu et al. also discovered a critical silicon particle diameter of ∼150 nm. Below this critical diameter, the particles did not fracture upon first lithiation, and above this diameter the particles initially formed surface cracks followed by fracture due to lithiation-induced swelling.49Fig. 3d shows the thickness changes for 60 μm thick electrodes prepared with graphite or ball-milled Si/graphite (1/2 by weight) in the first cycles.50 At full lithiation, graphite and Si/graphite showed about a 10 and 150% increase in electrode thickness, respectively. Thus, Si/graphite delivered only 71.1% of its initial capacity after 50 cycles. Furthermore, battery cell failure is highly probable because the current cell design cannot accommodate a large increase in electrode thickness.
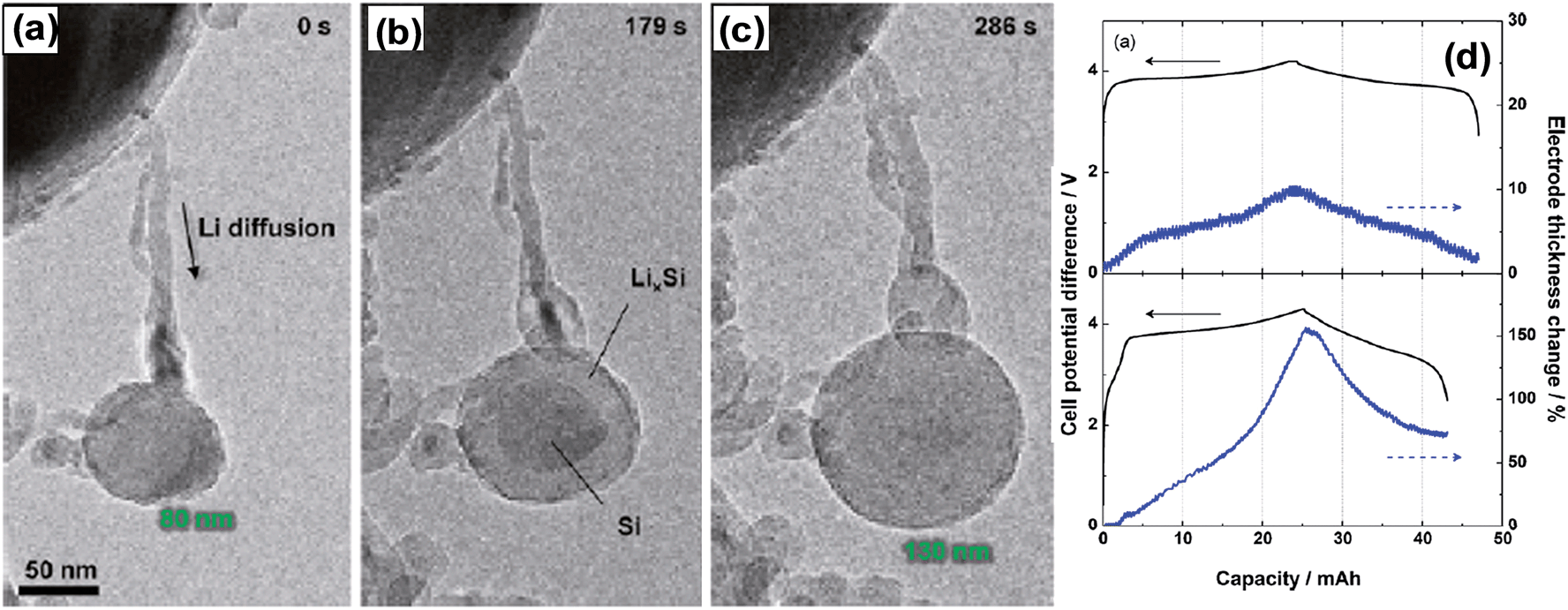 | ||
| Fig. 3 Chemical lithiation of Si nanoparticles without cracking: (a) pristine Si nanoparticle with D = 80 nm, and (b) core–shell structure during gradual lithiation from the surface to the center. The nanowire hanging the SiNPs provided the Li diffusion path. (c) Completion of lithiation without cracking. The diameter of the lithiated nanoparticles was 130 nm, corresponding to a volumetric expansion around 300%. Reproduced with permission.49 Copyright 2012, American Chemical Society. (d) The variations of the electrode thickness for the 1st cycle: (top) graphite electrode, and (bottom) Si (33.3 wt%)/graphite composite electrode. Reproduced with permission.50 Copyright 2010, Elsevier. | ||
Another issue arising from the huge volume expansion of silicon-based anode is the instability of the solid–electrolyte-interface (SEI) layer.17 At a potential below ∼1 V vs. Li/Li+, the organic electrolyte decomposes to form a passivating SEI layer on the surface of the electrode material during lithiation. The SEI layer is a lithium-ion conductor but an electronic insulator. Thus, the formation of a stable SEI layer on the electrode surface is critical for stable cycling of silicon-based anodes. Under the repetitive volume expansion and contraction of silicon, the SEI layer becomes unstable as shown in Fig. 4. The SEI layer initially formed during lithiation can be broken as silicon shrinks during delithiation. Then, a fresh silicon surface is exposed to the electrolyte and the SEI layer forms again, resulting in a very thick SEI layer after repeated cycles, which eventually leads to performance degradation in silicon-based anodes.
 | ||
| Fig. 4 Schematic of SEI formation on silicon surfaces. Reproduced with permission.17 Copyright 2012, Nature Publishing Group. | ||
As a form of silicon-based anode, SiOx (0 < x < 2), especially SiO, is also a very attractive material because of its high capacity (∼1600 mA h g−1), lower volume expansion than Si and low charge–discharge potential.51–54 However, an SiO anode shows intrinsically high irreversible capacity loss in the first cycle associated with Li–silicates and Li2O formation.52,53,55 The electrical conductivity and rate capability of SiO remain quite poor, and SiO undergoes large changes in volume during charge/discharge cycles. The intrinsic issues associated with the SiO anode resemble those described above for silicon, suggesting that the rational design strategies discussed below for silicon should be applicable to address the issues for SiO.56,57
5. Rational designs of silicon-based anodes
Important technical issues related to the electrochemical behavior of silicon anodes have been identified as described above, and these issues have been recently addressed through rational design to demonstrate silicon-based anodes capable of many hundreds to thousands of cycles. In this section, we highlight some selected strategies such as (1) nanostructured Si/C composites with empty spaces, (2) Si/C composites using graphene, and (3) the use of porous silicon structures. Finally, we introduce representative studies in high-energy LIBs and Li–S systems based on silicon-based anodes.5.1 Nanostructured Si/C composites with empty spaces
One of the most promising design approaches for silicon-based anodes is to form composites of insulating silicon with a conducting carbon matrix, which is believed to be a scalable option for mass production. Among the various nanostructured Si/C composites, the introduction of nano-scale empty spaces between silicon and the conducting carbon matrix22,58–61 that allow silicon to freely expand and contract was highly effective in mitigating the volume expansion problems associated with Si/C composites.Hertzberg et al.61 have proposed such a conceptual model design (Si-in-C tubes) and have successfully demonstrated the model in experiments with Si tubes attached to the pore wall of the carbon tube as schematically shown in Fig. 5. According to the computational modeling in Fig. 5a, the Si tube expands inward during lithiation since the rigid carbon wall restricts the outer expansion of the Si tube. Upon delithiation, the tube does not restore its initial geometry but rather contracts to a smaller size. During consecutive cycling, however, the Si tube freely expands and contracts inside the carbon tube. Thus, the change in the volume of the Si tube can be effectively accommodated inside the nanostructured Si/C composites. This model Si-in-C structure was built by using alumina membranes with ∼300 nm pores as a sacrificial template. Alumina membranes were subsequently coated with ∼40 nm C via chemical vapor deposition (CVD) of C3H6, Si of various thicknesses via CVD of SiH4, and ∼5 nm of CVD deposited C. After the CVD process, the alumina template was dissolved by HF etching to prepare the model Si-in-C tube structure shown in Fig. 5b. A transmission electron microscopy (TEM) image in Fig. 5c shows the contraction and partial delamination of the inner Si tube from the CNT surface after lithiation. As shown in Fig. 5d, one of the Si-in-C tubes (33 wt% Si) exhibited a capacity of over 1400 mA h g−1 at 80 mA g−1, and about 800 mA h g−1 at a high current of 1.7 A g−1 with an excellent cycling stability up to 250 cycles. The Si-in-C tubes also demonstrated a high coulombic efficiency (CE) of 99.6% for 2–50 cycles and >99.9% for cycles 50–250. Although the experimental processes might not be commercially viable for mass production, this model study provides guidance for the design of nanostructured Si/C composites offering excellent cycling stability at high capacity.
 | ||
| Fig. 5 (a) Results of the computational modeling of shape changes of a Si tube inside a rigid nanopore during reaction with Li, (b) SEM image of the composite Si-in-C tubes, (c) TEM image after Li extraction at the 10th cycle, and (d) deintercalation capacity retention and coulombic efficiency for the sample containing 33 wt% Si. The reported capacity is normalized by the total weight of C and Si. Reproduced with permission.61 Copyright 2010, American Chemical Society. | ||
Recently, Wu et al.59 also demonstrated a similar design concept to prepare SiNPs@CTs (silicon nanoparticles dispersed in carbon tubes) in which empty spaces were engineered between nanoparticles. The SiNPs@CT nanostructures were obtained by electrospinning a mixture of SiNPs and tetraethoxysilane (TEOS) into SiO2 nanofibers with embedded SiNPs. These nanofibers were coated with carbon followed by HF etching. The SiNPs@CTs delivered a capacity of around 1000 mA h g−1 at a current of 1 A g−1, and the capacity retention after 200 cycles was as high as 90%. The authors ascribed the excellent cycling stability to its structural properties in that (1) the empty space inside carbon tubes provides adequate space for Si expansion, and (2) a thin and uniform SEI layer is formed only on the outside of the carbon tubes since SiNPs are not directly in contact with the electrolyte. The latter suggests that the stability of the SEI layer is well controlled in a SiNPs@CT structure. More recently, Liu et al.62 fabricated Si/C nanocomposites inspired by the structure of a pomegranate, where single SiNPs are encapsulated by a conductive carbon layer that leaves sufficient void space for expansion and contraction following lithiation and delithiation. The resulting Si pomegranate is an advanced version of a previous yolk–shell design22 in terms of areal mass loading, material tap-density (related to volumetric capacity) and electrical properties.62 The Si pomegranate structure was prepared in a microemulsion of Si@SiO2 (SiO2 coated onto SiNPs) in a water-in-oil phase, followed by evaporation-driven self-assembly, subsequent carbon coating and removal of SiO2 layers by HF etching (see Fig. 6a). As shown in Fig. 6b, the Si pomegranate anode was cycled for 1000 galvanostatic cycles at a rate of C/2. Its capacity retention was more than 97% (0.003% decay per cycle) and over 1160 mA h g−1 capacity remained after 1000 cycles. The authors claim that the cycle stability is among the best of silicon-based anodes reported to date, but the mass loading of the active material on the electrode was only ∼0.2 mg cm−2, which is very low compared with the mass loading of graphite (in the range of 5–9 mg cm−2) in commercial LIBs. Fortunately, the authors also demonstrated excellent cycling performance with electrodes having much higher mass loading. For example, the authors prepared an electrode with a high mass loading of 3.12 mg cm−2 from a mixture of Si pomegranate and carbon nanotubes (CNTs) (7:3 ratio by mass, respectively). The uncalendered electrode, having a density of ∼0.4 g cm−3 and a thickness of ∼120 μm, showed a high areal capacity over 3 mA cm−2 at 0.7 mA cm−2. However, this electrode exhibited a volumetric capacity of only 310 mA h cm−3, which is much lower than that of graphite. The low volumetric capacity of the Si pomegranate is likely due to its low packing density on the electrode. With a conventional slurry coating and calendaring process, electrodes prepared with the Si pomegranate exhibited a volumetric capacity in the range of ∼900–1270 mA h cm−3, which is higher than the practical value for graphite anodes (<600 mA h cm−3). The above examples clearly demonstrate that the introduction of empty spaces between SiNPs in conductive carbon matrices can significantly enhance the cycling performances of Si-based anodes, which opens up new opportunities for the rational design of Si-based anodes. The examples discussed above have remaining issues related to the commercial viability of the synthesis processes.
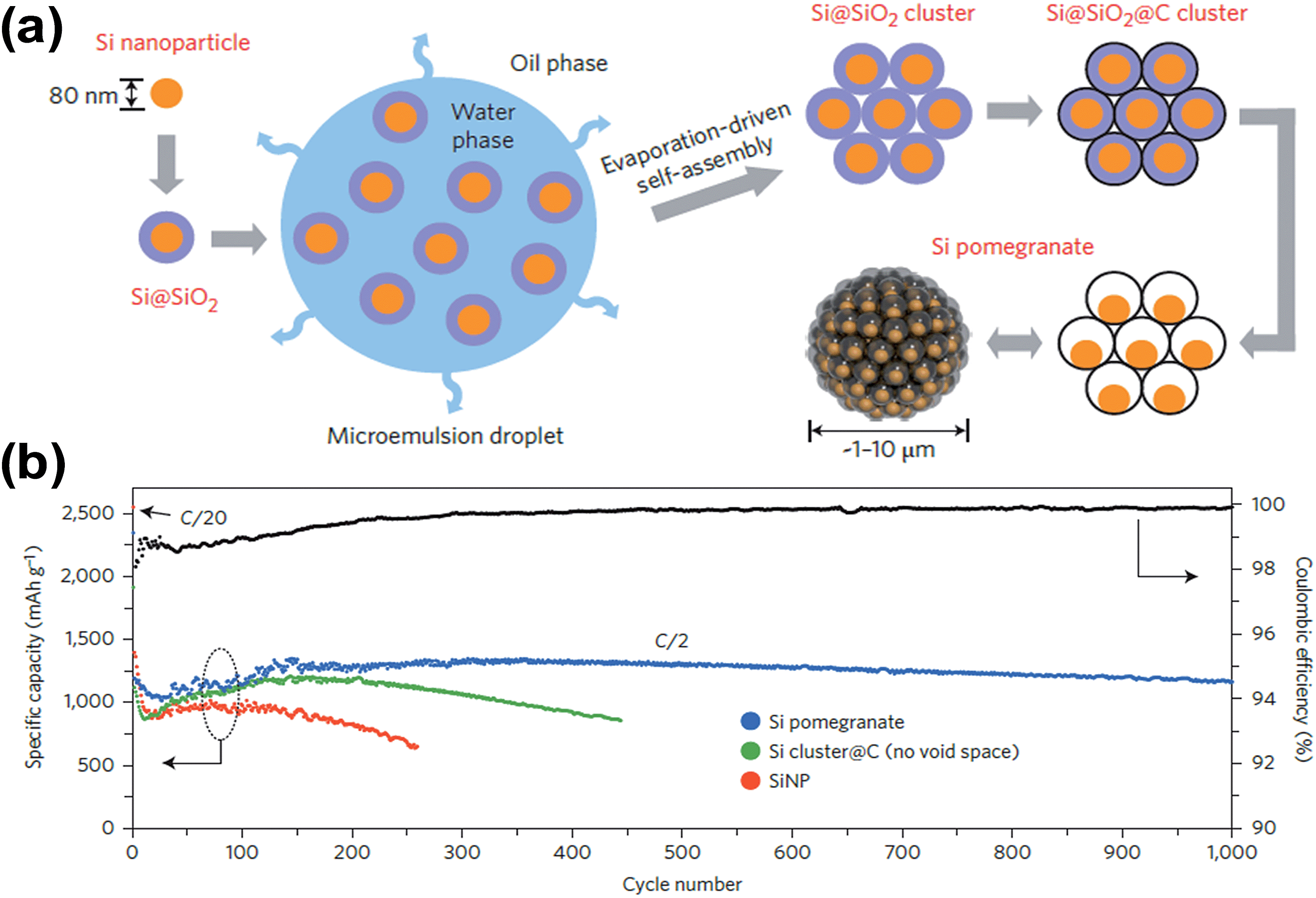 | ||
| Fig. 6 (a) Schematic of the fabrication process for silicon pomegranates, and (b) reversible delithiation capacity for the first 1000 galvanostatic cycles of the silicon pomegranate and other structures tested under the same conditions. Coulombic efficiency is plotted for the silicon pomegranate only. The mass loading of the active material was ∼0.2 mg cm−2. The rate was C/20 for the first cycle and C/2 for later cycles. (1C = 4.2 A g−1 active material.) Reproduced with permission.62 Copyright 2014, Nature Publishing Group. | ||
5.2 Si/C composites using graphene
Graphene, a single-atom-thick layer of sp2 hybridized carbon in the two-dimensional (2D) form, has been identified as an attractive supporting material that may address the huge volume expansion problems associated with Si anodes.63–72 Graphene's many appealing properties such as excellent electrical conductivity, superior mechanical and chemical properties, and extremely high aspect ratio have been successfully exploited in Si/C composites, which are very effective in achieving long cycle life. Lee et al.65 fabricated silicon nanoparticles–graphene paper composites by simple membrane filtering of a homogeneous mixture of SiNPs and graphene oxides (GO) in aqueous solution followed by thermal reduction in a H2/Ar gas flow. The paper composites are depicted in Fig. 7e. As shown, SiNPs are very well dispersed in the graphene composite, and a portion of the graphene sheets form a continuous, highly conducting 3D network that also serves as a structural scaffold to anchor the graphene sheets encapsulating the SiNPs. The Si–graphene paper composite delivered the theoretical capacity of silicon contained in the composites (>2200 mA h g−1 at 1000 mA g−1, Si content = 61 wt%). However, the paper composite showed moderate cycling performance and limited Li diffusion into the bulk structure for thicker paper at high currents. Nonetheless, the self-supporting paper composites are promising considering that they are flexible, very compact, have a high density and can be directly used as anodes in LIBs without the need for a binder, conductive additives or even a current collector (i.e., Cu foil). Zhao et al.67 reported similar self-supporting Si–graphene paper composites that improve upon the characteristic high resistance graphene material used for Li transport by forming diffusion channels with in-plane carbon vacancies. The composites provided high power capability. Therein, GO was treated in an aqueous HNO3 solution at different concentrations to generate in-plane defects on GO (denoted as deGO). The TEM image of a deGO stained with Pd clearly shows the porosity on the basal plane (Fig. 7a). The porous in-plane defects on thermally reduced deGO were <10 nm to >100 nm in size depending on the acid concentration. The SEM image for the paper cross-section in Fig. 7b reveals homogeneously dispersed SiNPs sandwiched between reassembled graphene sheets. As seen in Fig. 7c, the Si–graphene paper is flexible and remained integral when bent. This Si–graphene composite (Si content = 65–70 wt%) electrode exhibited an unprecedented reversible capacity of around 1100 mA h g−1 at a current of 8 A g−1, a rate equivalent to full discharge in 8 min (Fig. 7d). This Si–graphene composite also showed about 3200 mA h g−1 at a current of 1 A g−1, repeatable up to 99.9% for over 150 cycles. The XRD pattern of the Si–graphene sample also showed a sharp crystalline graphite diffraction peak indicating a composite structure as shown in Fig. 7e. Furthermore, the Si–graphene paper showed a specific capacity close to the theoretical value of silicon contained in the composite, and a high volumetric capacity on the order of 1400–4200 mA h cm−3 depending on calendaring. The authors ascribed the ultra-high power capability of the Si–graphene paper to the beneficial effects of in-plane defects and enhanced ion diffusivity throughout the composite paper. The authors claim that the preparation method employed can be easily scaled up for manufacturing electrodes using high-throughput processing protocols, such as spray-coating, inkjet printing, and roll-to-roll deposition. | ||
| Fig. 7 (a) TEM image of a Pd-stained graphene oxide sample, deGO-IV, (b) SEM image of the cross-section of a Si–deG-III paper, the inset shows Si nanoparticles embedded between graphene sheets uniformly. (c) Digital image of a Si–deGO-III paper, (d) specific delithiation capacity (solid) and coulombic efficiency (open) of Ar-reduced Si–deG-II between 0.02 and 1.5 V at 1 and 8 A g−1 (C/3 and 2.6C based on a theoretical capacity of 3052 mA h g−1), and between 0.1 and 0.55 V at 4 A g−1 (1.3C). Reproduced with permission.67 Copyright 2011, John Wiley and Sons. (e) Cross-sectional schematic drawing (not to scale) of a high-capacity, stable electrode, made of a continuous, conducting 3-D network of graphite (red) anchoring regions of the graphene–Si composite. Blue circles: Si nanoparticles, black lines: graphene sheets. Reproduced with permission.65 Copyright 2010, Royal Society of Chemistry. | ||
Luo et al.69 devised an aerosol-assisted capillary assembly method to prepare crumpled-graphene-wrapped SiNPs depicted in Fig. 8a. The aerosol synthesis route can serve as a rapid, scalable high throughput process capable of continuous operation. In the process, the colloidal mixture of SiNPs and GO was nebulized to form aerosol droplets, which were blown through a preheated tube furnace at 600 °C with N2 carrier gas. As seen in Fig. 8b, 50–100 nm diameter SiNPs were wrapped in the 5–10 nm thick crumpled graphene shell. The folds and wrinkles in the crumpled graphene capsule can accommodate the volume expansion of Si upon lithiation without losing electrical integrity. Compared to the bare SiNPs, the Si@crumpled graphene showed enhanced performance as lithium battery anodes in terms of cycling stability and coulombic efficiency. At the current of 1 A g−1, the composite delivered a capacity of 940 mA h g−1 in the 250th cycle with only 0.05% capacity loss per cycle after the initial stabilization cycles (Fig. 8c). In this work, the mass loading level of the electrode was very low (about 0.2 mg cm−2), and the composite exhibited a capacity (around 1200 mA h g−1 at 0.2 A g−1) far below the theoretical capacity of the composite containing 60 wt% Si. The material or electrode density and volumetric capacity of the composite are not available in the report. As the authors mentioned, further study of the synthesis conditions is required to address the above issues.
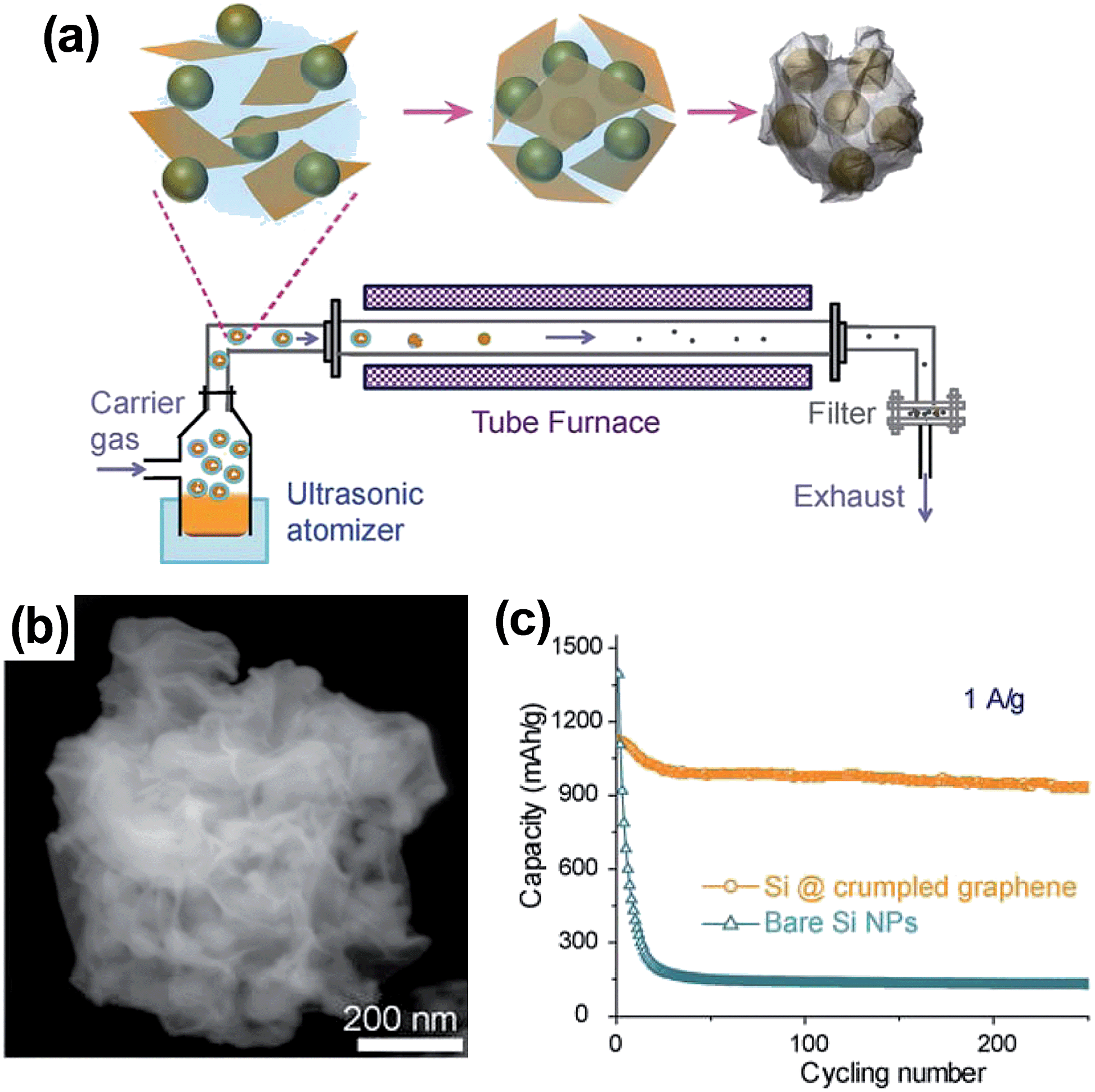 | ||
| Fig. 8 (a) Schematic drawings illustrating aerosol-assisted capillary assembly of crumpled-graphene-wrapped Si nanoparticles, (b) SEM images of a single capsule, and (c) charge/discharge cycling test of the composite capsules in comparison with the unwrapped Si nanoparticles at a constant current density of 1 A g−1. Reproduced with permission.69 Copyright 2012, American Chemical Society. | ||
Another interesting example of a Si–graphene composite was reported by Zhou et al.73 using self-assembly, which is an effective strategy for forming ideally mixed nanocomposites. Both SiNPs and GO have negative charges in aqueous solution due to the inherent surface oxide layer on SiNPs, and the ionization of the carboxylic and phenolic hydroxyl groups on GO. SiNPs were coated with a positively charged polyelectrolyte poly(diallydimethyl-ammonium) (PDDA) (Fig. 9a). The self-assembly was thermally reduced to yield the well-dispersed SiNPs encapsulated in graphene (Si-NP@G). The SEM and TEM images of Si-NP@G in Fig. 9b and c, respectively, show that the SiNPs are well wrapped by graphene sheets of about two layers to form a bulge-like micrometer-sized structure. When the Si-NP@G was cycled in the voltage range of 0.05–1.0 V vs. Li/Li+ at 100 mA g−1, it showed quite stable cycling performance and delivered a capacity of 1205 mA h g−1 after 150 cycles (Fig. 9d). In this work, the level of reversible capacity is much less than the theoretical capacity expected from the SiNPs in the Si-NP@G (80.1 wt% of SiNPs representing a theoretical capacity of Si = 3579 mA h g−1 × 0.801 = 2867 mA h g−1), possibly due to the rather shallow voltage window employed in the cycling test. Even though the cycling stability of Si-NP@G is rather promising, the material density and volumetric capacity for comparison with those of the state-of-the-art graphite anode are not provided in the report. However, this work successfully demonstrated an electrostatic attraction directed self-assembly approach for fabricating nanocomposites of SiNPs encapsulated in graphene.
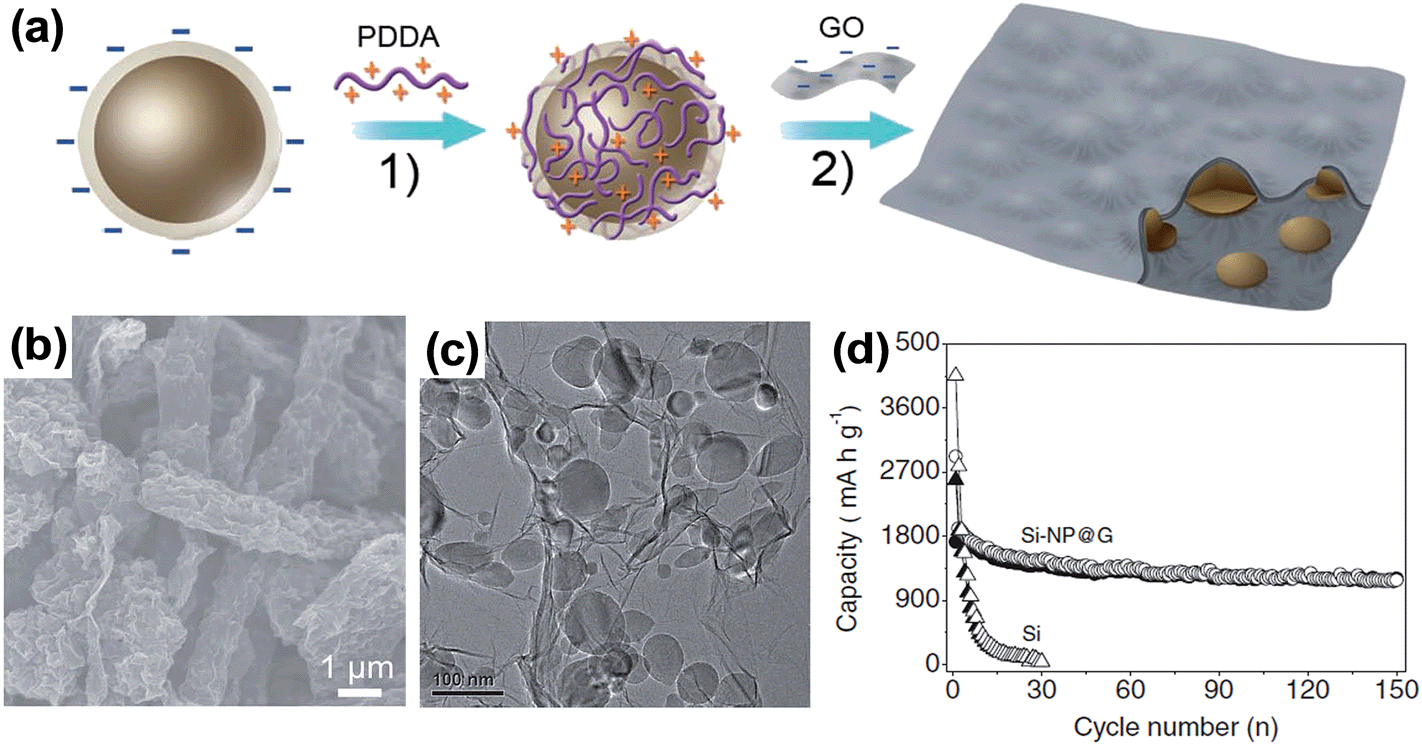 | ||
| Fig. 9 (a) Fabrication process of the Si-NP@G nanocomposite: (1) self-assembly between Si nanoparticles and PDDA to render the Si nanoparticle charged positively (hereafter abbreviated as Si-PDDA nanoparticles); and (2) self-assembly between positively charged Si–PDDA nanoparticles and negatively charged GO followed by freeze-drying, thermal reduction, and HF treatment. The golden balls and the black coatings represent Si nanoparticles and graphene sheets, respectively. (b) SEM image of the Si-NP@G nanocomposite, (c) TEM images of the Si-NP@G nanocomposite, and (d) cycling performance of the Si-NP@G nanocomposite and pure Si nanoparticles. Reproduced with permission.73 Copyright 2012, John Wiley and Sons. | ||
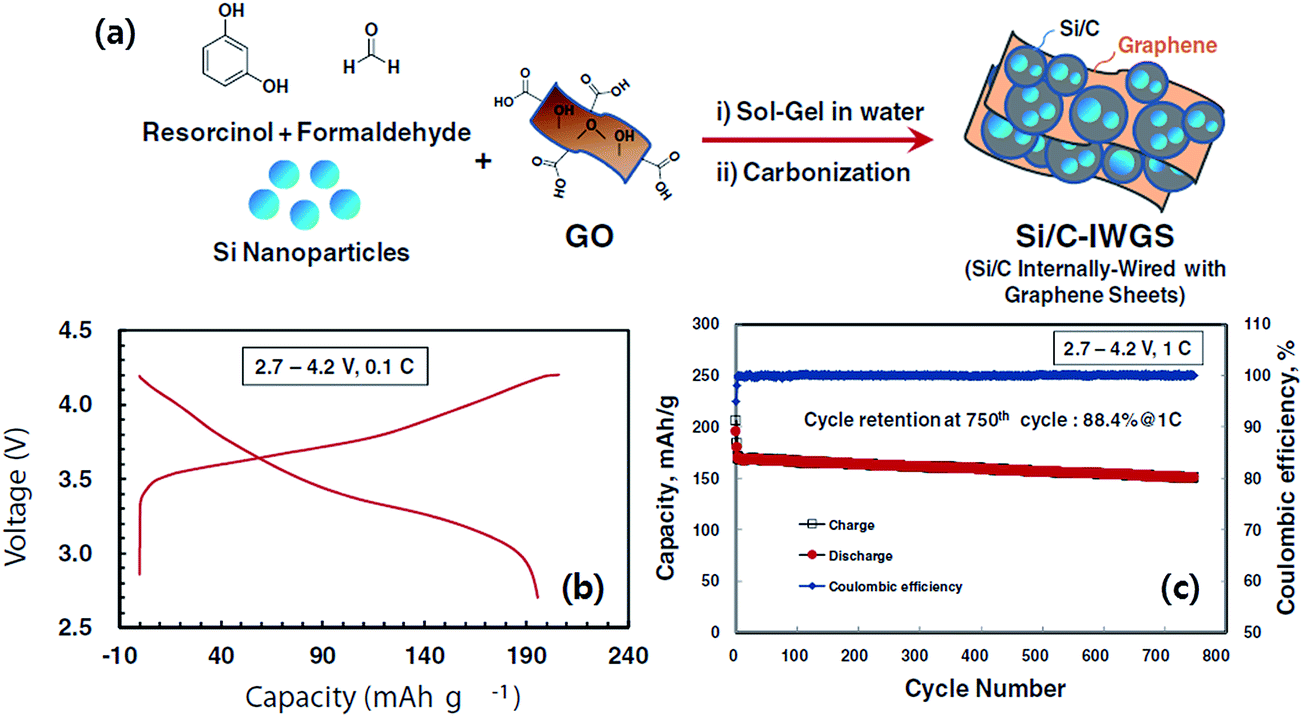
Recently, Kim et al.74 reported a simple sol–gel process in aqueous solution to synthesize Si/C-IWGN composites (Si/C internally wired with graphene networks) in which a small amount (1–10 wt%) of graphene sheets were dispersed to form electrical networks and to generate empty spaces. The Si/C-IWGN samples were prepared by using pre-formed SiNPs, GO and resorcinol-formaldehyde as the carbon source. Unlike the above strategies, the process provides dense particulate structures (Fig. 10a) with void spaces around the carbon-coated SiNPs connected by conducting graphene networks as seen in Fig. 10b and c. Compared to the conventional Si/C (SiNPs coated with carbon), the Si/C-IWGN samples showed enhanced cycling stability at high capacity (Fig. 10d). The changes in electrode thickness (see the inset of Fig. 10d) in the Si/C-IWGN samples were controlled in the range of 18–45% due to the void spaces generated by graphene networks in the composites. In contrast, the changes in electrode thickness in the Si/C samples were over 100%, which caused electrode pulverization that led to a short cycle life as seen in Fig. 10d. The authors also reported that the volumetric capacities of the Si/C-IWGN electrode were 1.4–1.7 times higher than that obtained from graphite.
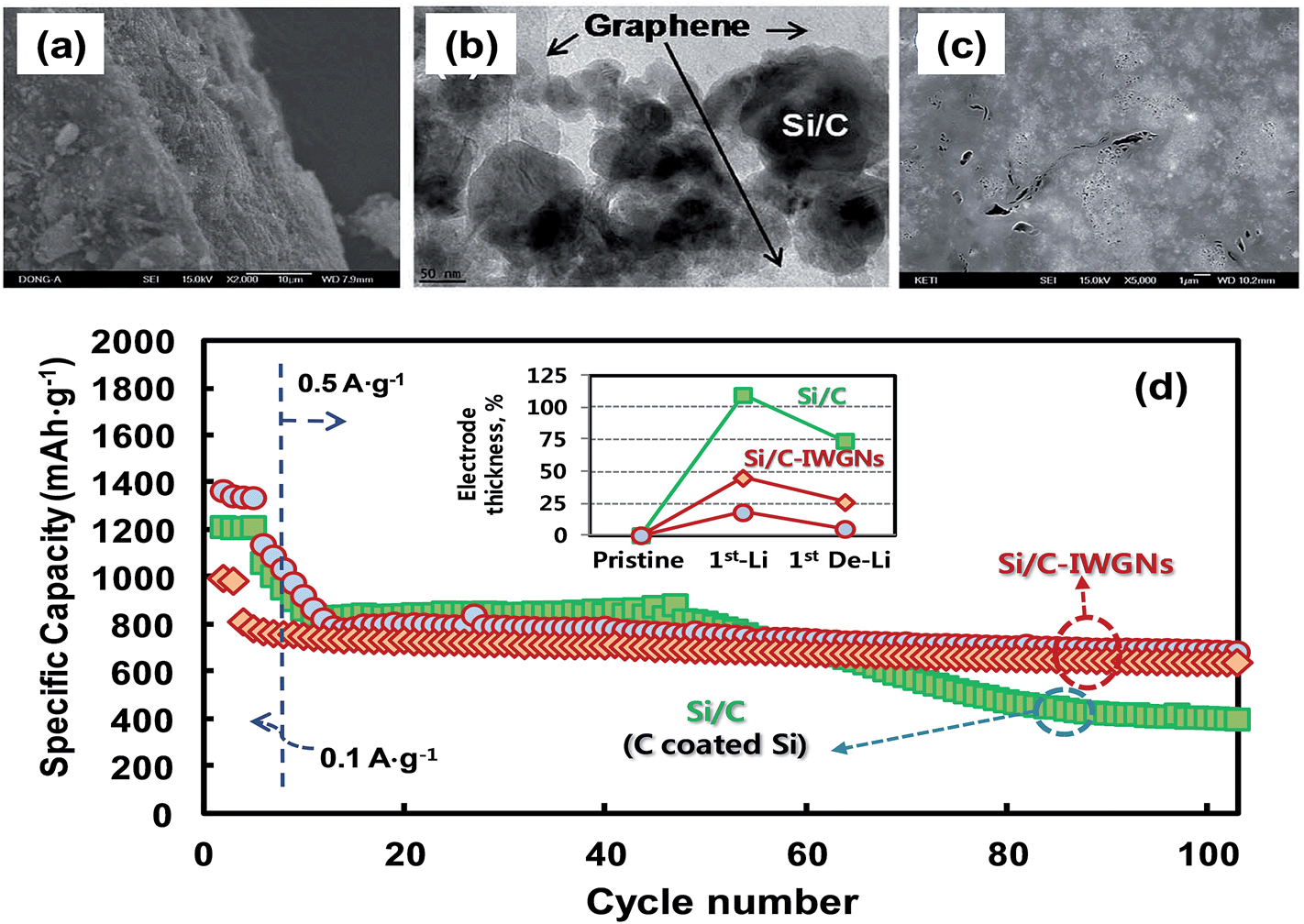 | ||
| Fig. 10 (a) SEM and (b) TEM images of a Si/C-IWGN composite, and (c) SEM image of a particle cross-section of a Si/C-IWGN composite, and (d) cycling performances of Si/C and Si/C-IWGN composites (the inset shows electrode thickness increases of Si/C and Si/C-IWGNs electrodes during the 1st charge–discharge cycles). Reproduced with permission.74 Copyright 2015, Royal Society of Chemistry. | ||
As demonstrated in the strategies introduced here and elsewhere,72,75 graphene can be used in the design of high performance silicon-based anodes since the production cost of graphene is expected to further decrease in the future.
5.3 Use of porous silicon structures
Researchers have used porous silicon structures as another approach to tackle the volume expansion problems associated with silicon-based anodes.76–80 Bao et al.81 published one of the pioneering studies on the preparation of porous silicon structures from 3D silica assemblies. In this work, a low-temperature (650 °C) magnesiothermic reduction process was demonstrated to convert three-dimensional nanostructured silica micro-assemblies into microporous nanocrystalline silicon replicas that retain their starting silica morphology. The magnesiothermic reduction of silica (SiO2) proceeds by the following reaction (2) with gaseous Mg.| 2Mg(g) + SiO2(s) → 2MgO(s) + Si(s) | (2) |
By following reaction (2), Du et al.82 prepared porous silicon hollow (PHSi) nanospheres replicated from mesoporous silica hollow nanospheres (MHSiO2). For this purpose, magnesium powder was mixed with preformed MHSiO2 at a 1:1 weight ratio, and the mixture was heated at 650 °C for 4 h, The MgO was then dissolved in a HCl solution and HF etching was conducted to remove the residual SiO2. Finally, polypyrrole was bound to PHSi to obtain the PPy@PHSi composite. The TEM image in Fig. 11a clearly shows that the PHSi is spherical and uniform with diameters of approximately 0.6 μm. The TEM image in Fig. 11b shows that an amorphous PPy layer (PPy content of 27.4 wt%) is uniformly coated on PHSi in PPy@PHSi. As shown in Fig. 11d, the PPy@PHSi composite provided an ultra-high capacity of over 2000 mA h g−1 that was well maintained with an 88% capacity retention after 250 cycles at a current of 1 A g−1. A capacity of 1161 mA h g−1 was maintained for 100 cycles even at a current of 4 A g−1. As seen in the TEM image of the PPy@PHSi after 250 cycles, the spherical morphology and hollow structure were well retained. The size of the nanocomposite increased from approximately 700 to over 900 nm in diameter. The authors ascribed the outstanding cycling stability of the PPy@PHSi to the synergistic action of the porous hollow structure and the surface PPy coating, which can accommodate huge changes in volume and can buffer the large mechanical stress due to the presence of the free volume in the hollow interior and the surface PPy coating. However, the authors did not provide the volumetric capacity of the highly macroporous hollow structure or the electrode parameters such as the loading amount of the active material (in mg cm−2) and coating thickness (in μm) to compare its electrochemical performances with other designs of silicon-based anodes.
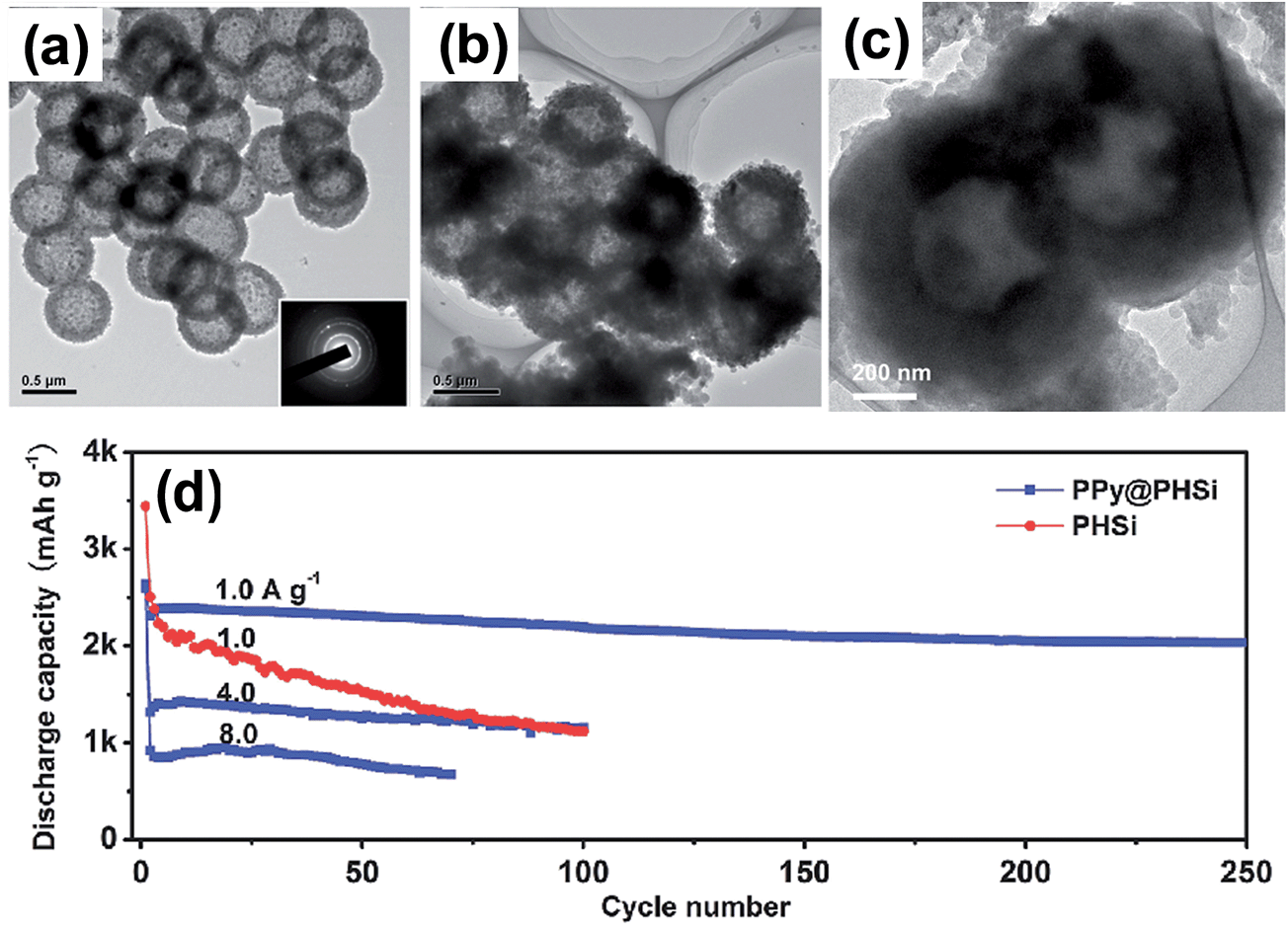 | ||
| Fig. 11 TEM images of the (a) PHSi nanospheres, (b) PPy@PHSi nanocomposite and (c) PPy@PHSi electrode after 250 cycles at a current density of 1.0 A g−1. (d) Cycling performance of the PHSi and PPy@PHSi nanocomposite at a variety of current densities. Reproduced with permission.82 Copyright 2014, John Wiley and Sons. | ||
Bang et al.77 reported a direct synthesis strategy for 3D macroporous bulk silicon structures from commercially available bulk silicon powders having an average particle size of 10 μm. For this purpose, they employed a combined process of electroless metal (Ag) deposition via a galvanic displacement reaction and subsequent metal-assisted chemical etching processes as shown in Fig. 12a. The bulk Ag-deposited Si in Fig. 12b was converted into the 3D macroporous bulk Si shown in Fig. 12c and d with a yield level of 30–40%. The surface of the bulk Si was coated with carbon by the pyrolytic process of acetylene gas to enhance the conductivity. The carbon coated 3D macroporous bulk Si exhibited a high first charge capacity of 2390 mA h g−1 at a rate of 0.1C with a remarkably high coulombic efficiency of 94.4% in the first cycle (Fig. 12e). The carbon-coated Si electrodes exhibited excellent rate capability by delivering 92% of the capacity of a 0.1C rate at a high rate of 5C. After 50 cycles, the electrode thickness increased from 18 to 25 μm, which corresponds to about 39% increase in thickness. The electrodes also showed an ultra-high volumetric capacity of approximately 2830 mA h cm−3, which is ∼5 times the value of ∼600 mA h cm−3 for practical graphite anodes.
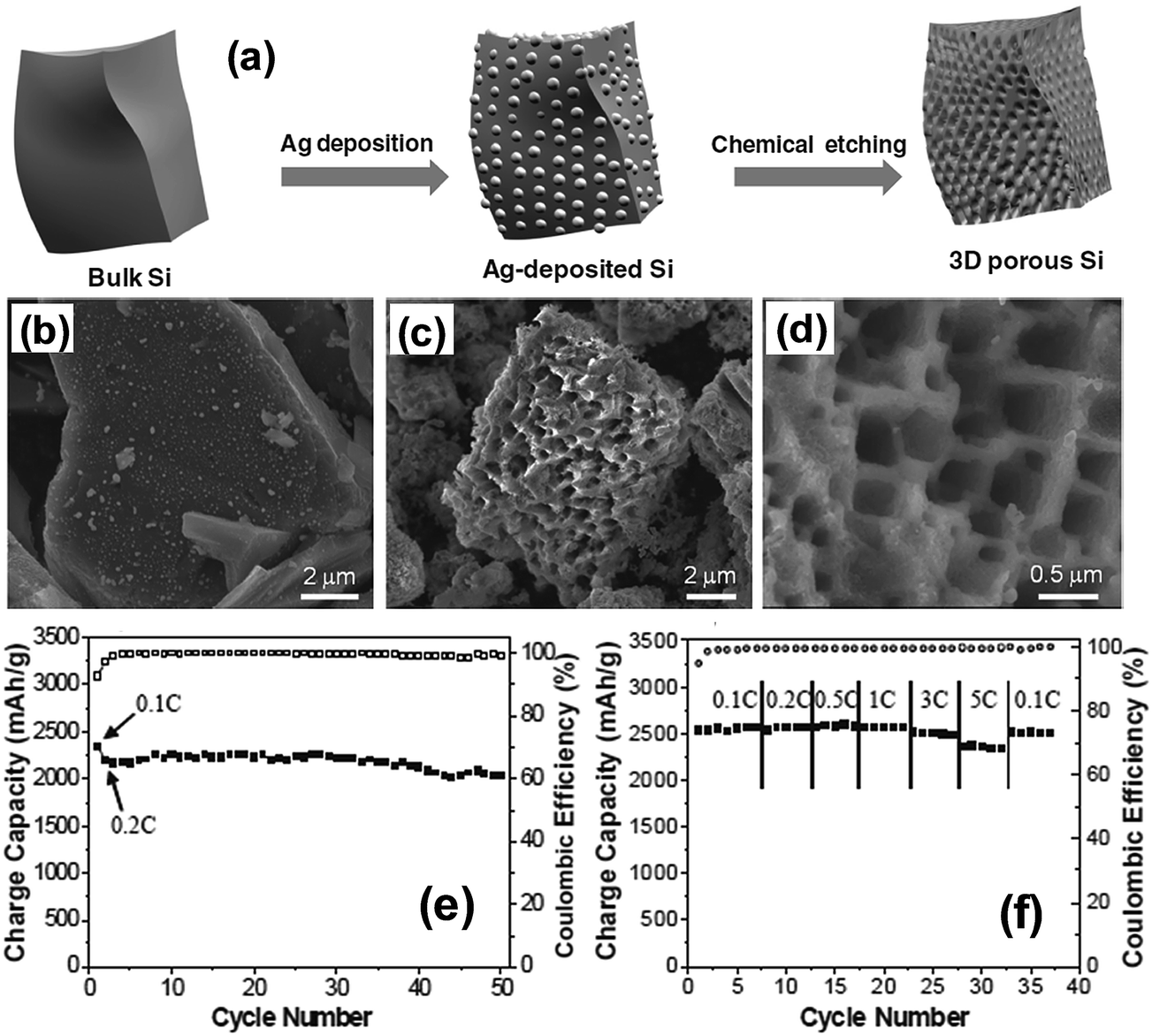 | ||
| Fig. 12 (a) Schematic illustration of the preparation of macroporous Si powders. Ag nanoparticles were deposited onto the surface of bulk silicon via a galvanic reaction, and subsequently, the Ag-deposited Si was chemically etched to make 3D porous Si particles. SEM image of (b) Ag deposited Si, (c) chemically etched Si and (d) magnified SEM image of samples seen in (b). (e) Plot of charge capacity vs. cycle number, and (f) rate capabilities of the carbon-coated porous Si anodes. Reproduced with permission.77 Copyright 2012, John Wiley and Sons. | ||
More recently, Lu et al.80 designed nonfilling carbon-coated porous silicon microparticles (nC-pSiMPs) in which the porous silicon microparticles (pSiMPs) consisted of many interconnected primary SiNPs, and only the outer surface of the pSiMPs was coated with carbon, leaving the interior pore structures unfilled, as depicted in Fig. 13a. The nC-pSiMP structure was obtained by the following procedure. Commercial SiO microparticles were first conformally coated with a layer of resorcinol-formaldehyde resin. Then, the resin-coated structures were heated to 950 °C under Ar for 5 h to carbonize the resin. This heat treatment also induced phase separation in SiO into interconnected SiNPs embedded in a SiO2 matrix due to the thermal disproportionation of SiO. The nC-pSiMP structure was obtained after removing the SiO2 matrix by HF etching. According to the thermal disproportionation reaction (3) below, the volume ratio of Si to void space was about 3:7 after removing SiO2. This volume ratio allowed for free volume expansion of Si materials without breaking the C shell.
| 2SiO → Si + SiO2 | (3) |
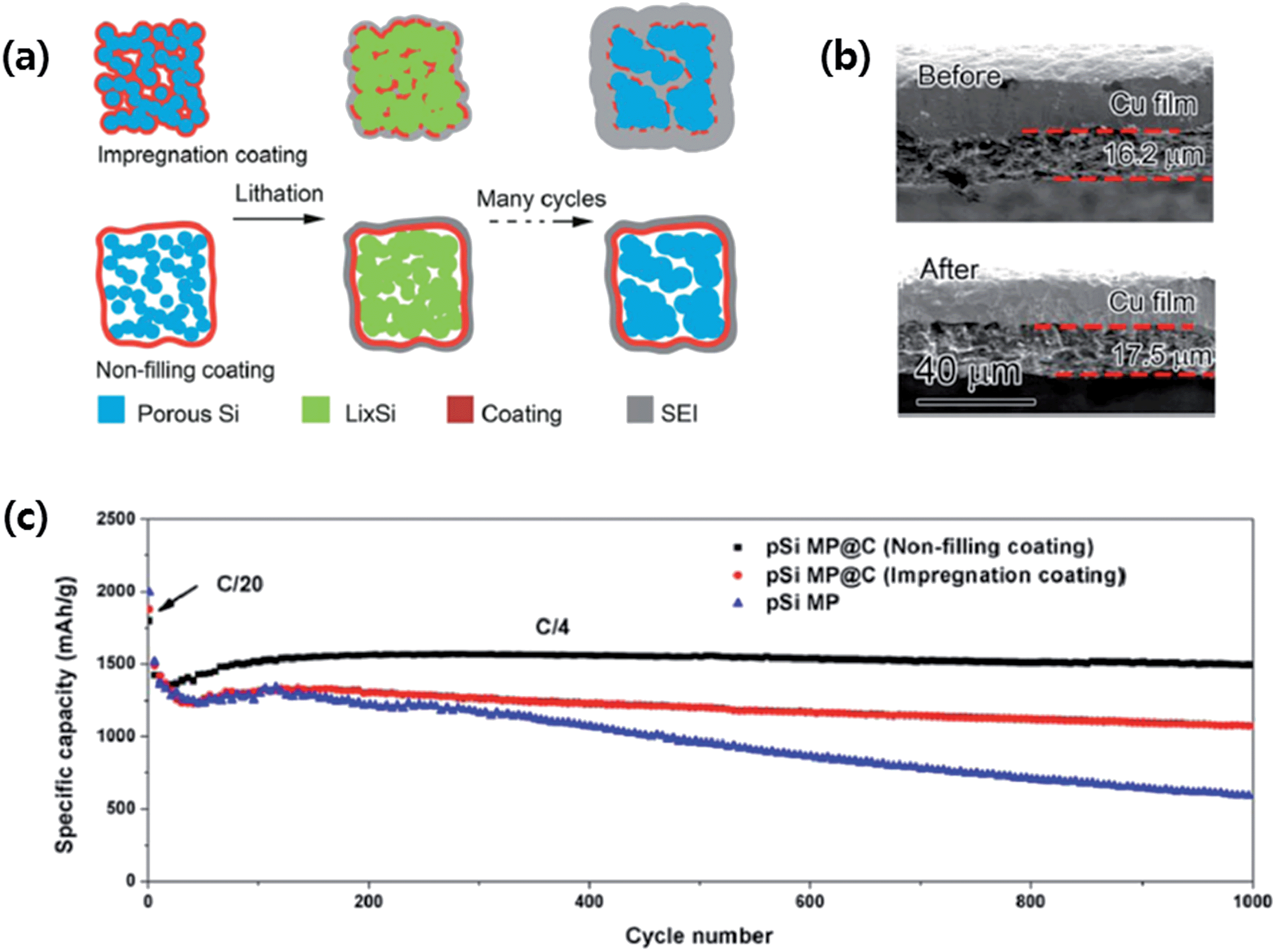 | ||
| Fig. 13 (a) Schematic of coating design on mesoporous Si microparticles (pSiMPs) and their structural evolution during cycling, (b) thickness of an nC-pSiMP electrode before cycling and after lithiation to 0.05 V at the 100th cycle, and (c) reversible delithiation capacity for the first 1000 galvanostatic cycles of the pSiMPs with different coatings. The active material mass loading was around 0.5 mg cm−2. The rate was C/20 for the first three cycles and then C/4 for later cycles. 1C = 4.2 A g−1. Reproduced with permission.80 Copyright 2015, American Chemical Society. | ||
As schematically depicted in Fig. 13a, the volume expansion of Si in the nC-pSiMPs is accommodated inside the C shell with a uniform SEI layer forming on the outer surface of the C shell. On the other hand, in the impregnated carbon coated structure, the carbon layer on each SiNPs is broken due to the huge volume expansion, which exposes the Si surface to the electrolyte, resulting in excessive SEI formation. As a result, the nC-pSiMP (the active material mass loading was around 0.5 mg cm−2) electrode showed excellent cycling stability up to 1000 cycles with a high reversible specific capacity of ∼1500 mA h g−1 at a rate of C/4 (1C = 4.2 A g−1 active materials). This value was much better than those of iC-pSiMP and pSiMP electrodes as compared in Fig. 13c. The thickness of the electrode slightly increased (only ∼7%) from 16.2 to 17.3 μm after 100 deep cycles (Fig. 13b), the volumetric capacity for this anode was determined to be 1003 mA h cm−3, which is much larger than the 600 mA h cm−3 obtained from graphite.83 The authors also demonstrated stable cycling of the nC-pSiMPs up to 100 cycles at an areal capacity of 2.84 mA h cm−2 with an active material mass loading of 2.01 mg cm−2.
The porous silicon structures highlighted here and elsewhere84 are a viable option in the design of high performance silicon-based anodes due to their unique structure, which can effectively accommodate the mechanical stress and strain caused by the huge volume expansion. In addition, the porous structure facilitates fast lithium diffusion, which leads to high-rate performance.
6. High-energy LIBs using silicon-based anodes
The rational designs of silicon-based anodes highlighted above have shown very promising performances in standard half-cell tests with Li foil as the reference electrode. Nonetheless, it is desirable to understand the electrochemical response of silicon-based anodes in full LIB cells coupled with specific cathodes as in a real battery. This is an essential step that must be taken before commercialization. Noticeable progress has been made recently in attempts to build full LIB cells with silicon-based anodes85 based on the accumulated knowledge of electrochemical behavior and advances in nanostructured design and synthesis of silicon-based anodes.Fridman et al. constructed full LIB cells in which amorphous silicon film anodes were combined with the integrated xLi2MnO3·(1 − x)LiNiyMnzCo1−y−zO286 and 5 V LiNi0.5Mn1.5O4 spinel87 cathodes. In this work, the silicon thin film (about 6 μm thick) electrodes were prepared by DC magnetron sputtering of n-type silicon onto roughened copper foil (Fig. 14a). The silicon film anodes showed very stable cycling for more than 1500 cycles using the “charge capacity limited (at 600 mA h g−1) procedure” with FEC-based electrolytes (Fig. 14b). The preconditioned Si electrodes were then combined with a high capacity Li-rich layered cathode materials xLi2MnO3·(1 − x)LiNiyMnzCo1−y−zO2 and the cycling results are shown in Fig. 14c and d. The cell delivered a capacity of 210–220 mA h g−1 at a current of C/8 during the first 100 cycles and about 195 mA h g−1 after 200 cycles with respect to the active cathode mass. Similar Si electrodes were combined with LiNi0.5Mn1.5O4 cathodes. Fig. 14e and f show the cycling results of a complete LiNi0.5Mn1.5O4/Si cell at 0.5C current rate. The capacity retention of the cell was 92.2% after 200 cycles, 88.5% after 300 cycles and 74.2% after 500 cycles. This performance suggests that amorphous film Si electrodes can be successfully combined with a 5 V LiNi0.5Mn1.5O4 spinel cathode in complete high voltage Li-ion cells with 1 M LiPF6 FEC/DMC electrolyte.
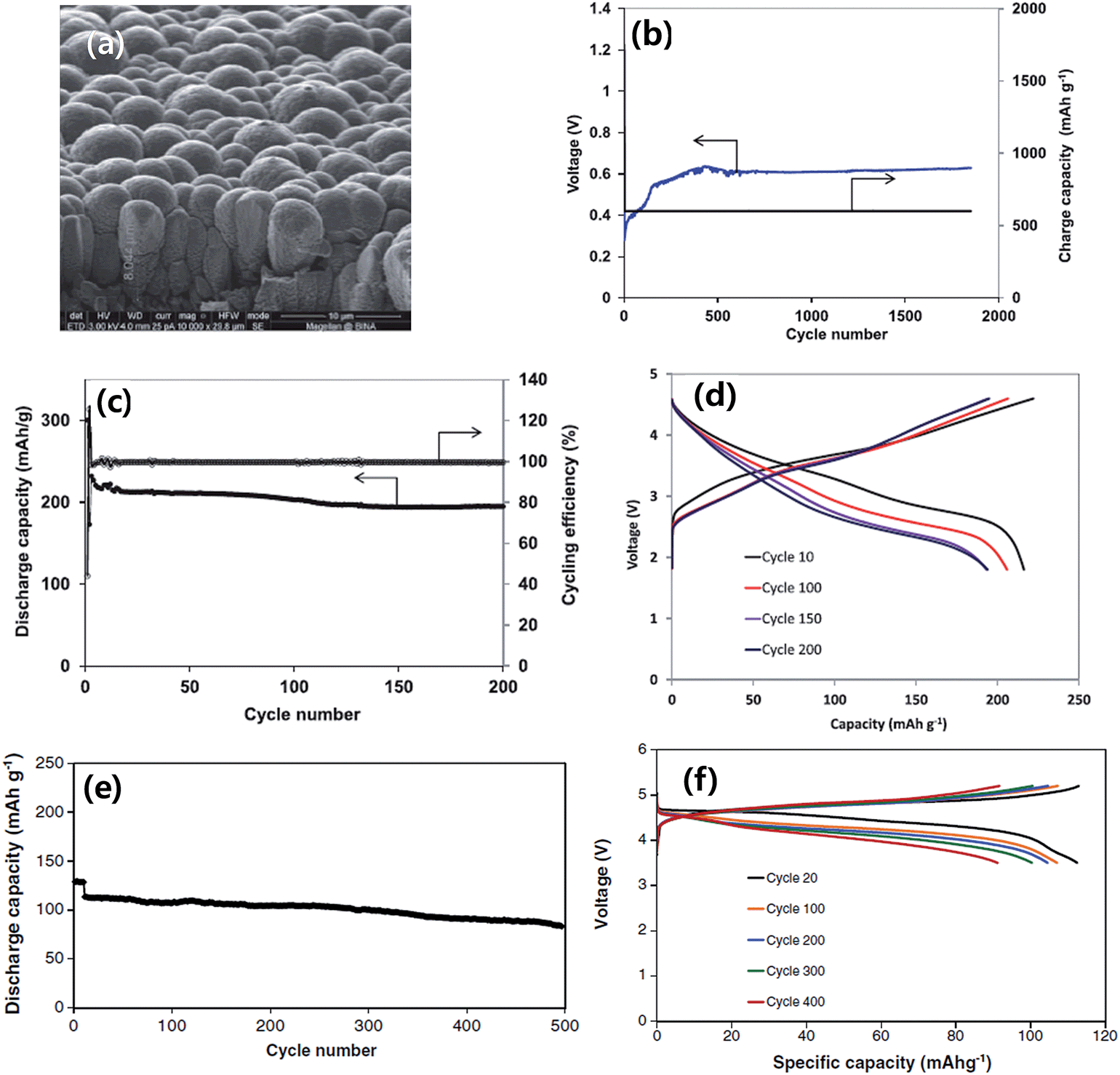 | ||
| Fig. 14 (a) SEM image of pristine silicon film (∼6 μm thick) deposited onto roughened copper foil, (b) specific charge capacity and voltage at the end of charge vs. cycle number with a cutoff discharge voltage of 10 mV vs. Li/Li+ and limiting charge capacity of 600 mA h g−1. (c and d) Results of the galvanostatic cycling of xLi2MnO3·(1 − x)LiNiyMnzCo1−y−zO2/Si cells. Current rate C/8, 30 °C; (c) specific discharge capacity and cycling efficiency vs. cycle number, and (d) voltage profile of the cell. Reproduced with permission.86 Copyright 2013, The Electrochemical Society. (e) Typical curve of discharge capacity vs. cycle number, and (f) voltage profile obtained upon galvanostatic cycling of LiNi0.5Mn1.5O4/Si cells at 0.5C rate (10 initial cycles at C/8 rate) in the FEC-based electrolyte solution. Reproduced with permission.87 Copyright 2013, Elsevier. | ||
Ko et al.88 combined an amorphous SiNPs backboned graphene nanocomposite (a-SBG) with a LiCoO2 cathode in a full LIB cell. The a-SBG was prepared by pyrolytic decomposition of SiH4 on reduced graphene, which was obtained by thermal reduction of freeze-dried porous graphene oxide. The a-SBG contained amorphous SiNPs (82 wt%) in sizes of 5–10 nm and showed a self-compacting behavior as the electrode thickness decreased by 31% after 100 cycles (Fig. 15a). The a-SBG also showed high power capability and an excellent cycling stability up to 1000 cycles at a high delithiation current of 14 A g−1 with a fixed lithiation current of 2.8 A g−1 (Fig. 15b). The full cell, a-SBG/LiCoO2, showed a highly reversible capacity of 162 mA h g−1 in the first cycle in the voltage range of 2.5–4.3 V (Fig. 15c). The full cell retained 83% and 82% of its initial capacity after 100 cycles at ultrafast delithiation rates of 5 and 7C, respectively, indicating good rate capability of the a-SBG/LiCoO2 full cell (Fig. 15d).
 | ||
| Fig. 15 (a) Schematic view of a-SBG nanocomposites before and after electrochemical cycling, (b) cycling performance of a-SBG at a charge current density of 14 A g−1 and a discharge density of 2.8 A g−1 over 1000 cycles, (c and d) electrochemical responses of a Li ion cell consisting of the a-SBG anode and a LiCoO2 cathode; (c) galvanostatic charge/discharge profile of the full cell obtained under constant current at a rate of 0.1C with constant voltage applied to 0.05C at the end of the process in the potential range of 2.5–4.3 V, and (d) cycling performance during fast discharging at rates of 5 and 7C for 100 cycles. Reproduced with permission.88 Copyright 2014, American Chemical Society. | ||
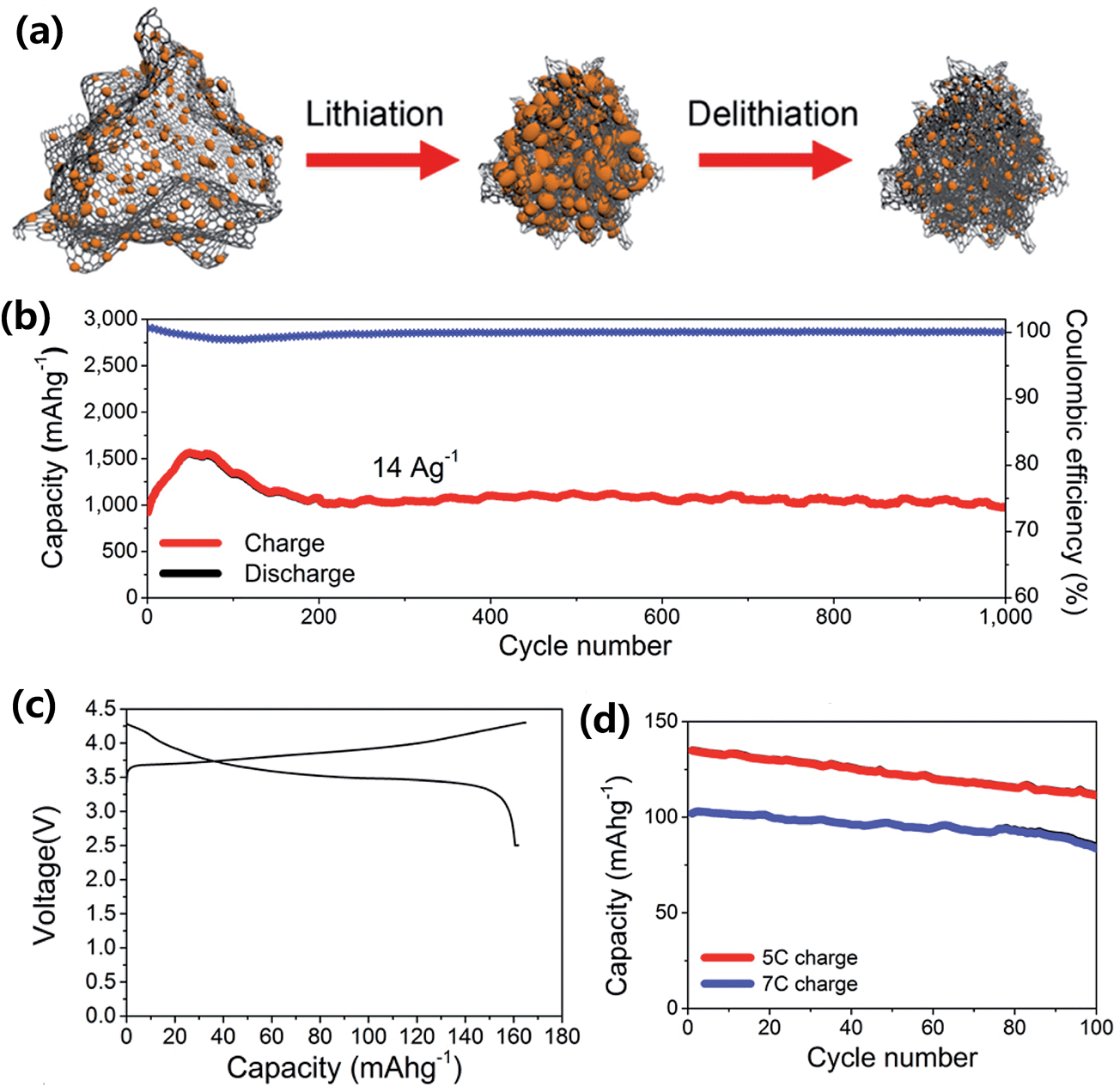
A silicon-based composite capable of very long cyclability in a full LIB cell was reported by Chae et al.71 They combined a Si/C composite internally wired with graphene sheets (Si/C-IWGS) with a novel Ni-rich layered full concentration gradient (FCG) cathode (Li[Ni0.75Co0.1MnO0.15O]2) to form a high energy LIB cell. The Si/C-IWGS was prepared by a one-pot sol–gel reaction of SiNPs, GO and a resorcinol-formaldehyde mixture in an aqueous solution, followed by carbonization in Ar (Fig. 16a). The Si/C-IWGS anode material is characterized by a simple synthesis process, highly dispersed and conducting graphene sheets with interconnecting Si/C particle aggregates, void spaces generated by graphene sheets,71,74 good cycling stability up to 200 cycles, and high capacity (880 mA h g−1 at 100 mA g−1). The Si/C-IWGS, however, showed rather low CE in the first cycle. Therefore, the authors pretreated the Si/C-IWGC anode to reduce the first irreversible capacity loss before the full cell assembly. The Ni-rich FCG cathode they used is characterized by high capacity (210 mA h g−1 at 0.1C), and high cycling stability (92.4% retention after 100 cycles at 0.5C in the range of 2.7–4.3 V). The Ni-rich FCG structure also ensures safety against thermal runaway with the potential of explosion. This is because the concentration of Ni, which has high reactivity with the electrolyte at elevated temperatures, decreases linearly from the center towards the particle surface while the Mn concentration increases gradually. Fig. 16b shows the charge/discharge voltage profile of the full cell cycled between 2.7 and 4.2 V at a 0.1C rate with respect to the cathode. The first charge and discharge capacities were 206 and 196 mA h g−1, respectively, giving a CE as high as 95%. The specific energy density of the full cell estimated from the voltage profile was 720 W h kg−1. This corresponds to a practical specific energy density of 240 W h kg−1 by assuming a 1/3 reduction factor (which is applicable for LIBs using a graphite anode) to account for the weight of the electrolyte, current collectors, and aluminum case. Thus, this battery can offer an energy density far beyond those offered by current LIBs. As shown in Fig. 16c, the full cell showed an unprecedented cycling stability up to 750 cycles at 1C in the voltage range of 2.7–4.2 V. The capacity retention after 750 cycles was 88.4%, which corresponds to a capacity fading of 0.016% per cycle.
 | ||
| Fig. 16 (a) Schematic diagram of the preparation of Si/C-IWGS, (b) voltage profile of the lithium ion battery based on the Si/CIWGS anode and FCG cathode cycled between 2.7 and 4.2 V at a 0.1C rate, and (c) cycling performance of the lithium ion battery based on the Si/C-IWGS anode and FCG cathode cycled between 2.7 and 4.2 V at a 1.0C rate. Reproduced with permission.71 Copyright 2014, John Wiley and Sons. | ||
As highlighted here and elsewhere,85 it is quite convincing that silicon-based anodes can be combined in LIBs offering high energy, high power and long cycle life.
7. High-energy Li–S batteries based on silicon anodes
Silicon-based anodes can also be employed in the development of new Li–S battery systems, where high capacity silicon-based anodes are employed to replace dendrite-forming Li metal anodes.9,23,25,26,89–91 This battery system may have a brighter future in terms of commercialization than the conventional Li–S system based on a cell configuration consisting of sulfur as the cathode, lithium metal as the anode, and a lithium salt in an aprotic organic solvent as the electrolyte.10A new battery system was developed by Hassoun et al.,21 who reported a lithium metal-free new battery version consisting of a “discharged” state cathode (Li2S/C composite), a lithium alloy anode (Sn/C composite), and a gel polymer electrolyte (CGPE, see ref. 21 for details). They successfully demonstrated that the new battery can provide specific energy on the order of 1100 W h kg−1, and the battery was cycled more than 90–110 times92 with rather good cycling stability and excellent charge–discharge efficiency.
Yang et al.26 also reported a lithium metal-free battery system based on a Li2S/CMK-3 cathode and a silicon nanowire (SiNW) anode as described in Fig. 17a. In Fig. 17b, the theoretical specific energy density of the Li2S/silicon battery is compared with different types of LIBs based on the theoretical capacities of the active materials in the electrodes and the average operating voltage of the battery. According to their calculation, the Li2S/silicon battery has a theoretical specific energy of 1550 W h kg−1, which is four times that of conventional batteries. More recently, Rosenman et al.10 calculated the specific energy densities for Li–sulfur/Si and Si–Li–sulfur in carbon systems considering the practical capacity values (700–1000 mA h g−1 for the sulfur cathode, and 1000 mA h g−1 for the Si anode) and the average operating voltages. They estimated that the specific energy densities for Li–sulfur/Si and Si–Li–sulfur in carbon systems were in the range of 740–900 W h kg−1, while those of conventional batteries based on graphite anodes were in the range of 375–670 W h kg−1. In Yang's work, the “discharged” state of the cathode, Li2S/CMK-3, was achieved by reacting the sulfur in S/CMK-3 with n-butyl-lithium. Fig. 17c and d present the discharge capacity at various cycles at C/3 and the discharge voltage profile at different rates, respectively, of the Li2S/silicon full cell. The full cell maintained about 60% of its initial capacity at C/3 after 20 cycles with continuous drops in capacity with cycle. The average discharge voltage of the full cell was ∼1.7 V. At a rate of C/8, the first discharge capacity increased to 482 mA h g−1 (Fig. 17d), which corresponded to an initial specific energy of 630 W h kg−1. Even though the electrochemical performance of the full cell was not satisfactory based on the preliminary results, the novel battery system will inspire the development of lithium metal free Li–S batteries based on high capacity silicon anodes.
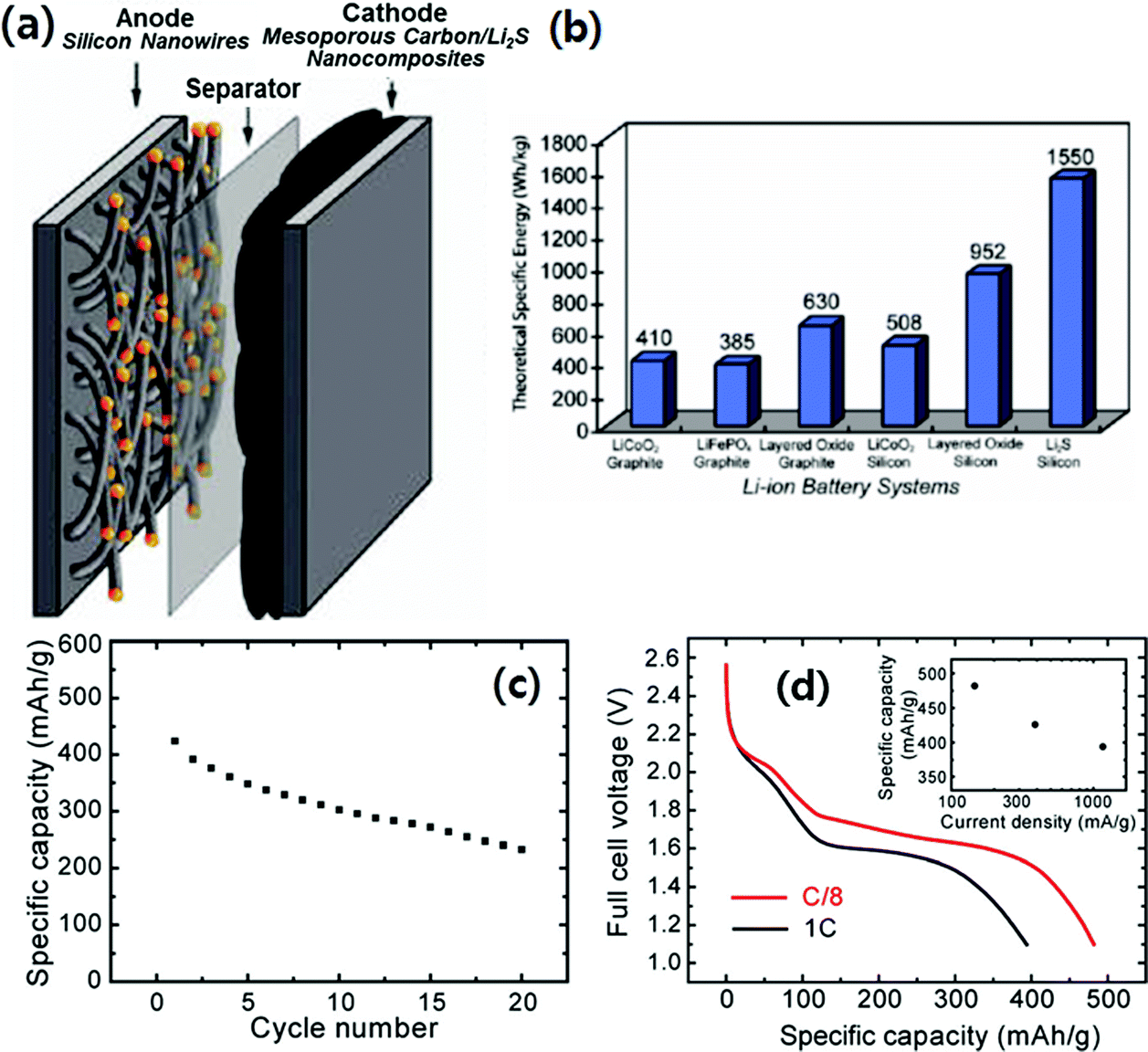 | ||
| Fig. 17 (a) Schematic diagram of battery structure; the cathode contains lithium sulfide (Li2S) encapsulated within ordered mesoporous carbon, and the anode consists of silicon nanowires grown by the VLS mechanism. (b) Comparison of theoretical specific energy for different types of Li-ion batteries. The theoretical specific energy is calculated based on the theoretical capacities of the active materials in the electrodes and the average operating voltage of the battery. (c) Specific discharge capacity with cycling of a full battery cell with a Li2S/CMK-3 mesoporous carbon nanocomposite cathode and a silicon nanowire anode. The current rate is C/3 (389 mA g−1) and the voltage range for the full cell is 1.2–2.6 V for the first cycle and 1.2–2.5 V for the following cycles. (d) First discharge voltage profiles of full battery cells with Li2S/CMK-3 mesoporous carbon nanocomposite cathodes and silicon nanowire anodes at rates of 1C (1166 mA g−1) and C/8 (146 mA g−1). The inset in (d) is a plot of the first discharge specific capacity of full cells operating at various current rates. Reproduced with permission.26 Copyright 2010, American Chemical Society. | ||
As opposed to the above lithium metal-free Li–S systems where the “discharged” state of cathodes, Li2S/C composites, was employed, a sulfur battery with a lithiated silicon (LixSi–S, LSS) system was reported by Pu et al.91 In the LSS battery, sulfur-impregnated on a mesoporous carbon (CMK-8), S/CMK-8 composite, was coupled with a pre-lithiated mesoporous Si synthesized by magnesiochemically reducing mesoporous silica (Fig. 18a). By employing mesoporous active materials for both lithiated silicon-based anodes and sulfur/carbon composite cathodes together with the CNT interlayer between the cathode and separator, the LSS full cell showed stable cycling with a capacity retention of 80% after 100 cycles at 0.5C (Fig. 18b). The LSS cell also showed high rate capability, delivering a discharge capacity of 465 mA h g−1 at 3C rate. Brückner et al.89 prepared an amorphous silicon layer (4 μm thick) sputter deposited onto a flexible carbon fiber (Fig. 18c) as the anode, which showed a stable cycling stability up to 300 cycles when cycled at a capacity limit of 1000 mA h g−1. The sulfur–carbon composite cathode was prepared by sulfur infiltration onto hollow carbon spheres, which delivered around 800 mA h g−1sulfur after 135 cycles with excellent cycling stability. The authors devised a balanced and stable Li–S full cell with a Si–C anode lithiated by short circuiting versus metallic lithium. As shown in Fig. 18d, the full cell showed good stability up to about 1400 cycles at a current of 836 mA g−1sulfur. The discharge capacity was 765 mA h g−1sulfur in the 5th cycle and faded extremely slowly (∼0.08%) over the following cycles at high coulombic efficiency.
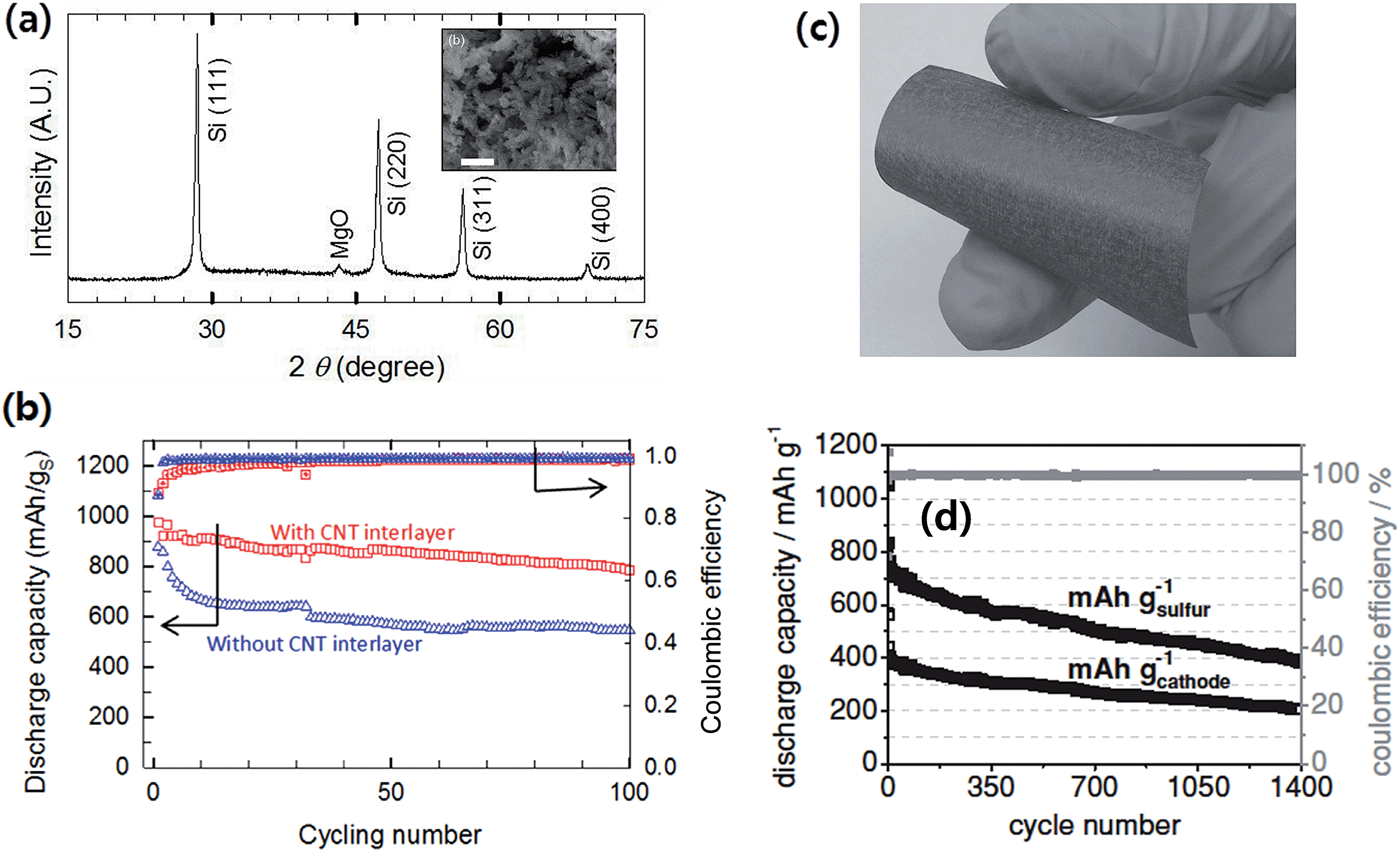 | ||
| Fig. 18 (a) X-ray diffraction (XRD) of the mesoporous Si after washing away MgO and SiO2 residues. The scale bars indicate 1 μm (the inset is the SEM image of mesoporous Si obtained by the magnesiothermic reduction). (b) Electrochemical performances of an LixSi–S (LSS) battery; discharge capacities and columbic efficiencies of the LSS battery with and without a CNT interlayer at 0.5C. Reproduced with permission.91 Copyright 2014, Elsevier. (c) Photograph of a-Si coated carbon non-woven (silicon layer thickness <1 μm, 0.95 mg silicon cm−2), and (d) discharge capacity (black) and coulombic efficiency (gray) of the full-cell setup. The discharge/charge current is 167 mA g−1 sulfur for three formation cycles and 836 mA g−1 sulfur (1.37 mA cm−2) in the subsequent cycles. The discharge capacity was 765 mA h g−1 sulfur in the 5th cycle and faded only 0.08% afterwards. The coulombic efficiency was 99.8% even in the 1390th cycle. Reproduced with permission.89 Copyright 2014, John Wiley and Sons. | ||
Very recently, Lee et al.90 demonstrated an ultrastable L-S full cell based on a lithiated Si–SiOx nanosphere anode and a dual-type (solid sulfur–polysulfide catholyte) sulfur cathode. In this work, the Si–SiOx nanosphere anode was prepared by the pyrolysis of hydrogen silsesquioxane. As seen in Fig. 19a, the lithiated Si/SiOx anode exhibited a reversible capacity of 830 mA h g−1 at 200 mA g−1 and maintained stable cycling up to 100 cycles. The anode also showed very low irreversible capacity loss in the first cycle because of the pre-lithiation. The sulfur cathode prepared by sulfur infiltration on activated carbon particles coated onto the surface of a gas-diffusion-layer (GDL) current collector showed a reversible capacity of ∼1000 mA h g−1 with excellent cycling stability for 100 cycles at a C/3 rate (Fig. 19b). As a result, the full cell delivered a specific capacity of 1100 mA h g−1 at 0.1C rate at an average voltage of about 1.8 V. Based on the cell capacity of 750 mA h g−1 at a 1C rate and the weight of the electrode materials, the energy density of the full cell was estimated to be 497 W h kg−1, which is more than double that of the commercially available LIBs. As seen in Fig. 19d, the full cell maintained about 85.5% of the initial capacity after 500 cycles, which is comparable to the cycle life of the commercially available LIBs. The full cell also exhibited very high CEs over 98.2% after the first cycle.
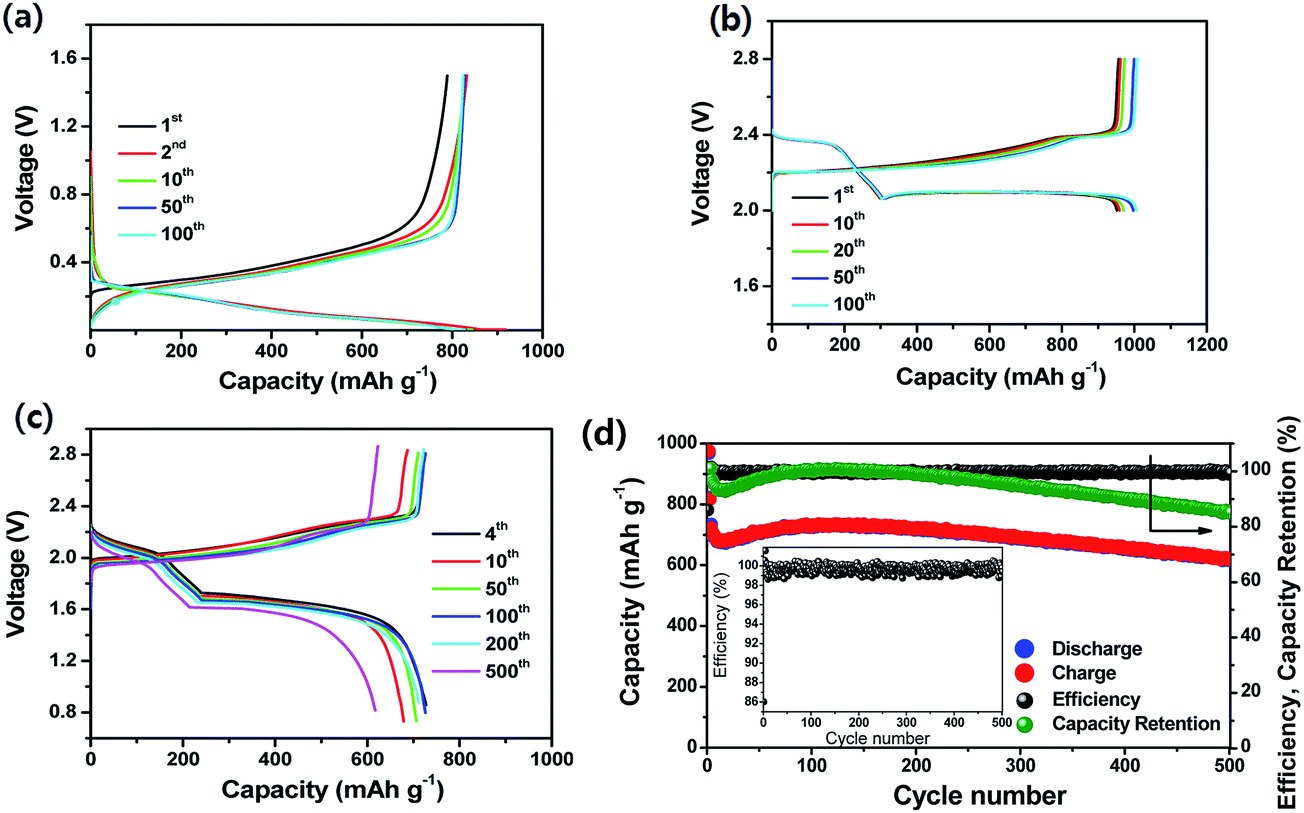 | ||
| Fig. 19 (a) Voltage profiles and (b) cycling performance of lithiated Si/SiOx nanosphere anode with a catholyte. Current, 200 mA g−1; voltage range, 1.5–0.005 V; temperature, 30 °C. (b) Voltage profiles of the dual-type sulfur cathode cycled at a rate of C/3. (c) Voltage profiles and (d) cyclic responses of the lithiated Si–SiOx/DME/DOL (1:1 v/v), Li2S8 (0.05 M), LiTFSI (1 M), LiNO3 (0.4 M)/AC-S full cell cycled at 1C rate. The upper and lower voltage limits are, respectively, 2.8 and 0.8 V. 1C = 1675 mA h g−1versus overall sulfur weight; temperature, 30 °C. Inset: magnification of coulombic efficiency. Reproduced with permission.90 Copyright 2015, American Chemical Society. | ||
As highlighted here and elsewhere,10 it is quite clear that silicon-based anodes can be used in the production of lithium metal-free Li–S batteries based on either the Si/Li2S or the lithiated Si/S system.
8. Summary and outlook
This contribution is focused on the development of advanced energy storage devices based on Li-ion batteries that can offer energy content beyond that is available with current LIB technologies. In order to boost the energy content of current LIBs to a level higher than is currently available with lower battery volume, graphite-based anodes have to be replaced with high capacity silicon-based anodes. As highlighted in this review, high capacity silicon-based anodes may also be able to work with high capacity cathodes (e.g., sulfur) in devising lithium metal-free Li–S batteries of various formats. These Li–S batteries could lead to an energy storage device offering higher energy densities than current LIBs by a factor of two.Over the last two decades, the technical issues associated with silicon-based anodes have been identified through multidisciplinary research efforts, and major issues have been addressed through rational designs to demonstrate stable cycling for several hundreds to thousands of cycles in the configuration of full-cells as well as half-cells. Nevertheless, there are still questions remaining to be answered before silicon-based anodes become common in high energy density devices. Silicon-based anode materials have to be improved much in the following areas: 1st cycle efficiency, volumetric capacity (related to material tap density), control of electrode volume expansion comparable to that of graphite, operation efficiency and safety over a wide-temperature range, and better control and understanding of the interfacial reactions between the silicon electrode and electrolyte. Along these lines, it would be beneficial to compare the electrochemical responses of silicon-based anodes with those of commercial graphite anodes. At the same time, the production cost of silicon-based anodes has to be low enough coupled with readily scalable processes in order to address larger applications such as electric vehicles. In addition to the materials designs and applications briefly discussed here, multidisciplinary studies of polymeric binders, electrolytes and electrolyte additives for enhancing the electrochemical performance will lead to the wide adoption of high-energy storage devices based on silicon-based anodes in the near future.
Acknowledgements
This work was supported by the Human Resource Training Program for Regional Innovation and Creativity through the Ministry of Education and the National Research Foundation of Korea (NRF-2014H1C1A1073093), by the National Research Foundation of Korea Grant funded by the Ministry of Education (NRF-2014R1A1A2057691) and by the Human Resources Development program (No. 20124010203310) of the Korea Institute of Energy Technology Evaluation and Planning (KETEP) grant funded by the Korea government Ministry of Trade, Industry and Energy.References
- R. E. Smalley, MRS Bulletin, 2005, 30, 412 CrossRef.
- S. Pacala and R. Socolow, Science, 2004, 305, 968 CrossRef CAS PubMed.
- M. M. Thackeray, C. Wolverton and E. D. Isaacs, Energy Environ. Sci., 2012, 5, 7854 CAS.
- Y. K. Sun, B. R. Lee, H. J. Noh, H. M. Wu, S. T. Myung and K. Amine, J. Mater. Chem., 2011, 21, 10108 RSC.
- Y. K. Sun, Z. H. Chen, H. J. Noh, D. J. Lee, H. G. Jung, Y. Ren, S. Wang, C. S. Yoon, S. T. Myung and K. Amine, Nat. Mater., 2012, 11, 942 CrossRef CAS PubMed.
- H. J. Noh, Z. Chen, C. S. Yoon, J. Lu, K. Amine and Y. K. Sun, Chem. Mater., 2013, 25, 2109 CrossRef CAS.
- H. J. Yu and H. S. Zhou, J. Phys. Chem. Lett., 2013, 4, 1268 CrossRef CAS PubMed.
- J. H. Yan, X. B. Liu and B. Y. Li, RSC Adv., 2014, 4, 63268 RSC.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J. M. Tarascon, Nat. Mater., 2012, 11, 19 CrossRef CAS PubMed.
- A. Rosenman, E. Markevich, G. Salitra, D. Aurbach, A. Garsuch and F. F. Chesneau, Adv. Energy Mater., 2015, 5, 21 Search PubMed.
- M. Yoshio, T. Tsumura and N. Dimov, J. Power Sources, 2005, 146, 10 CrossRef CAS.
- U. Kasavajjula, C. Wang and A. J. Appleby, J. Power Sources, 2007, 163, 1003 CrossRef CAS.
- M. Sathiya, G. Rousse, K. Ramesha, C. P. Laisa, H. Vezin, M. T. Sougrati, M. L. Doublet, D. Foix, D. Gonbeau, W. Walker, A. S. Prakash, M. Ben Hassine, L. Dupont and J. M. Tarascon, Nat. Mater., 2013, 12, 827 CrossRef CAS PubMed.
- A. W. Golubkov, D. Fuchs, J. Wagner, H. Wiltsche, C. Stangl, G. Fauler, G. Voitic, A. Thaler and V. Hacker, RSC Adv., 2014, 4, 3633 RSC.
- J. J. Chen, Materials, 2013, 6, 156 CrossRef.
- D. Larcher, S. Beattie, M. Morcrette, K. Edstroem, J. C. Jumas and J. M. Tarascon, J. Mater. Chem., 2007, 17, 3759 RSC.
- H. Wu, G. Chan, J. W. Choi, I. Ryu, Y. Yao, M. T. McDowell, S. W. Lee, A. Jackson, Y. Yang, L. B. Hu and Y. Cui, Nat. Nanotechnol., 2012, 7, 309 Search PubMed.
- R. A. Huggins, J. Power Sources, 1999, 82, 13 CrossRef.
- S. D. Beattie, D. Larcher, M. Morcrette, B. Simon and J. M. Tarascon, J. Electrochem. Soc., 2008, 155, A158 CrossRef CAS.
- H. Kim, E. J. Lee and Y. K. Sun, Mater. Today, 2014, 17, 285 CrossRef CAS.
- J. Hassoun and B. Scrosati, Angew. Chem., Int. Ed., 2010, 49, 2371 CrossRef CAS PubMed.
- N. Liu, H. Wu, M. T. McDowell, Y. Yao, C. Wang and Y. Cui, Nano Lett., 2012, 12, 3315 CrossRef CAS PubMed.
- R. Elazari, G. Salitra, G. Gershinsky, A. Garsuch, A. Panchenko and D. Aurbach, Electrochem. Commun., 2012, 14, 21 CrossRef CAS.
- J. Hassoun, J. Kim, D. J. Lee, H. G. Jung, S. M. Lee, Y. K. Sun and B. Scrosati, J. Power Sources, 2012, 202, 308 CrossRef CAS.
- M. Agostini, J. Hassoun, J. Liu, M. Jeong, H. Nara, T. Momma, T. Osaka, Y. K. Sun and B. Scrosati, ACS Appl. Mater. Interfaces, 2014, 6, 10924 CAS.
- Y. Yang, M. T. McDowell, A. Jackson, J. J. Cha, S. S. Hong and Y. Cui, Nano Lett., 2010, 10, 1486 CrossRef CAS PubMed.
- D. L. Ma, Z. Y. Cao and A. M. Hu, Nano-Micro Lett., 2014, 6, 347 CrossRef.
- H. K. Liu, Z. P. Guo, J. Z. Wang and K. Konstantinov, J. Mater. Chem., 2010, 20, 10055 RSC.
- H. Wu and Y. Cui, Nano Today, 2012, 7, 414 CrossRef CAS.
- M. T. McDowell, S. W. Lee, W. D. Nix and Y. Cui, Adv. Mater., 2013, 25, 4966 CrossRef CAS PubMed.
- B. Liang, Y. P. Liu and Y. H. Xu, J. Power Sources, 2014, 267, 469 CrossRef CAS.
- J. R. Szczech and S. Jin, Energy Environ. Sci., 2011, 4, 56 CAS.
- R. McMillan, H. Slegr, Z. X. Shu and W. D. Wang, J. Power Sources, 1999, 81, 20 CrossRef.
- D. Aurbach, K. Gamolsky, B. Markovsky, Y. Gofer, M. Schmidt and U. Heider, Electrochim. Acta, 2002, 47, 1423 CrossRef CAS.
- N. S. Choi, K. H. Yew, K. Y. Lee, M. Sung, H. Kim and S. S. Kim, J. Power Sources, 2006, 161, 1254 CrossRef CAS.
- H. Nakai, T. Kubota, A. Kita and A. Kawashima, J. Electrochem. Soc., 2011, 158, A798 CrossRef CAS.
- R. Elazari, G. Salitra, G. Gershinsky, A. Garsuch, A. Panchenko and D. Aurbach, J. Electrochem. Soc., 2012, 159, A1440 CrossRef CAS.
- H. K. Park, B. S. Kong and E. S. Oh, Electrochem. Commun., 2011, 13, 1051 CrossRef CAS.
- I. Kovalenko, B. Zdyrko, A. Magasinski, B. Hertzberg, Z. Milicev, R. Burtovyy, I. Luzinov and G. Yushin, Science, 2011, 334, 75 CrossRef CAS PubMed.
- H. Zhao, Z. H. Wang, P. Lu, M. Jiang, F. F. Shi, X. Y. Song, Z. Y. Zheng, X. Zhou, Y. B. Fu, G. Abdelbast, X. C. Xiao, Z. Liu, V. S. Battaglia, K. Zaghib and G. Liu, Nano Lett., 2014, 14, 6704 CrossRef CAS PubMed.
- J. Li, R. B. Lewis and J. R. Dahn, Electrochem. Solid-State Lett., 2007, 10, A17 CrossRef CAS.
- T. W. Kwon, Y. K. Jeong, I. Lee, T. S. Kim, J. W. Choi and A. Coskun, Adv. Mater., 2014, 26, 7979 CrossRef CAS PubMed.
- Y. K. Jeong, T. W. Kwon, I. Lee, T. S. Kim, A. Coskun and J. W. Choi, Energy Environ. Sci., 2015, 8, 1224 CAS.
- J. Liu, Q. Zhang, Z. Y. Wu, J. H. Wu, J. T. Li, L. Huang and S. G. Sun, Chem. Commun., 2014, 50, 6386 RSC.
- B. A. Boukamp, G. C. Lesh and R. A. Huggins, J. Electrochem. Soc., 1981, 128, 725 CrossRef CAS.
- M. N. Obrovac and L. Christensen, Electrochem. Solid-State Lett., 2004, 7, A93 CrossRef CAS.
- M. N. Obrovac and L. J. Krause, J. Electrochem. Soc., 2007, 154, A103 CrossRef CAS.
- J. H. Ryu, J. W. Kim, Y. E. Sung and S. M. Oh, Electrochem. Solid-State Lett., 2004, 7, A306 CrossRef CAS.
- X. H. Liu, L. Zhong, S. Huang, S. X. Mao, T. Zhu and J. Y. Huang, ACS Nano, 2012, 6, 1522 CrossRef CAS PubMed.
- S. S. Hwang, C. G. Cho and H. Kim, Electrochim. Acta, 2010, 55, 3236 CrossRef CAS.
- J. Yang, Y. Takeda, N. Imanishi, C. Capiglia, J. Y. Xie and O. Yamamoto, Solid State Ionics, 2002, 152, 125 CrossRef.
- M. Miyachi, H. Yamamoto, H. Kawai, T. Ohta and M. Shirakata, J. Electrochem. Soc., 2005, 152, A2089 CrossRef CAS.
- J. H. Kim, C. M. Park, H. Kim, Y. J. Kim and H. J. Sohn, J. Electroanal. Chem., 2011, 661, 245 CrossRef CAS.
- S. Goriparti, E. Miele, F. De Angelis, E. Di Fabrizio, R. P. Zaccaria and C. Capiglia, J. Power Sources, 2014, 257, 421 CrossRef CAS.
- A. Yamano, M. Morishita, M. Yanagida and T. Sakai, J. Electrochem. Soc., 2015, 162, A1730 CrossRef CAS.
- J. H. Kim, H. J. Sohn, H. Kim, G. Jeong and W. Choi, J. Power Sources, 2007, 170, 456 CrossRef CAS.
- J. I. Lee and S. Park, Nano Energy, 2013, 2, 146 CrossRef CAS.
- A. Magasinski, P. Dixon, B. Hertzberg, A. Kvit, J. Ayala and G. Yushin, Nat. Mater., 2010, 9, 461 CrossRef CAS.
- H. Wu, G. Y. Zheng, N. A. Liu, T. J. Carney, Y. Yang and Y. Cui, Nano Lett., 2012, 12, 904 CrossRef CAS PubMed.
- S. R. Chen, M. L. Gordin, R. Yi, G. Howlett, H. Sohn and D. H. Wang, Phys. Chem. Chem. Phys., 2012, 14, 12741 RSC.
- B. Hertzberg, A. Alexeev and G. Yushin, J. Am. Chem. Soc., 2010, 132, 8548 CrossRef CAS PubMed.
- N. Liu, Z. D. Lu, J. Zhao, M. T. McDowell, H. W. Lee, W. T. Zhao and Y. Cui, Nat. Nanotechnol., 2014, 9, 187 CrossRef CAS PubMed.
- B. Wang, X. L. Li, X. F. Zhang, B. Luo, M. H. Jin, M. H. Liang, S. A. Dayeh, S. T. Picraux and L. J. Zhi, ACS Nano, 2013, 7, 1437 CrossRef CAS PubMed.
- S. M. Paek, E. Yoo and I. Honma, Nano Lett., 2009, 9, 72 CrossRef CAS PubMed.
- J. K. Lee, K. B. Smith, C. M. Hayner and H. H. Kung, Chem. Commun., 2010, 46, 2025 RSC.
- T. Yoon, M. Cho, Y. W. Suh, E. S. Oh and J. K. Lee, J. Nanosci. Nanotechnol., 2011, 11, 10193 CrossRef CAS PubMed.
- X. Zhao, C. M. Hayner, M. C. Kung and H. H. Kung, Adv. Energy Mater., 2011, 1, 1079 CrossRef CAS.
- K. Evanoff, A. Magasinski, J. B. Yang and G. Yushin, Adv. Energy Mater., 2011, 1, 495 CrossRef CAS.
- J. Y. Luo, X. Zhao, J. S. Wu, H. D. Jang, H. H. Kung and J. X. Huang, J. Phys. Chem. Lett., 2012, 3, 1824 CrossRef CAS PubMed.
- J. G. Ren, Q. H. Wu, G. Hong, W. J. Zhang, H. Wu, K. Amine, J. Yang and S. T. Lee, Energy Technol., 2013, 1, 77 CrossRef CAS.
- C. Chae, H. J. Noh, J. K. Lee, B. Scrosati and Y. K. Sun, Adv. Funct. Mater., 2014, 24, 3036 CrossRef CAS.
- B. Luo and L. Zhi, Energy Environ. Sci., 2015, 8, 456 CAS.
- X. S. Zhou, Y. X. Yin, L. J. Wan and Y. G. Guo, Adv. Energy Mater., 2012, 2, 1086 CrossRef CAS.
- J. Kim, C. Oh, C. Chae, D. H. Yeom, J. Choi, N. Kim, E. S. Oh and J. K. Lee, J. Mater. Chem. A, 2015, 3, 18684 CAS.
- R. Raccichini, A. Varzi, S. Passerini and B. Scrosati, Nat. Mater., 2014, 14, 271 CrossRef PubMed.
- H. Kim, B. Han, J. Choo and J. Cho, Angew. Chem., Int. Ed., 2008, 47, 10151 CrossRef CAS PubMed.
- B. M. Bang, J. I. Lee, H. Kim, J. Cho and S. Park, Adv. Energy Mater., 2012, 2, 878 CrossRef CAS.
- R. Yi, F. Dai, M. L. Gordin, S. R. Chen and D. H. Wang, Adv. Energy Mater., 2013, 3, 295 CrossRef CAS.
- X. L. Li, M. Gu, S. Y. Hu, R. Kennard, P. F. Yan, X. L. Chen, C. M. Wang, M. J. Sailor, J. G. Zhang and J. Liu, Nat. Commun., 2014, 5, 7 Search PubMed.
- Z. D. Lu, N. Liu, H. W. Lee, J. Zhao, W. Y. Li, Y. Z. Li and Y. Cui, ACS Nano, 2015, 9, 2540 CrossRef CAS PubMed.
- Z. H. Bao, M. R. Weatherspoon, S. Shian, Y. Cai, P. D. Graham, S. M. Allan, G. Ahmad, M. B. Dickerson, B. C. Church, Z. T. Kang, H. W. Abernathy, C. J. Summers, M. L. Liu and K. H. Sandhage, Nature, 2007, 446, 172 CrossRef CAS PubMed.
- F. H. Du, B. Li, W. Fu, Y. J. Xiong, K. X. Wang and J. S. Chen, Adv. Mater., 2014, 26, 6145 CrossRef CAS PubMed.
- M. N. Obrovac, L. Christensen, D. B. Le and J. R. Dahn, J. Electrochem. Soc., 2007, 154, A849 CrossRef CAS.
- M. Ge, X. Fang, J. Rong and C. Zhou, Nanotechnol., 2013, 24, 10 CrossRef PubMed.
- V. Aravindan, Y. S. Lee and S. Madhavi, Adv. Energy Mater., 2015, 5, 43 Search PubMed.
- K. Fridman, R. Sharabi, E. Markevich, R. Elazari, G. Salitra, G. Gershinsky, D. Aurbach, J. Lampert and M. Schulz-Dobrick, ECS Electrochem. Lett., 2013, 2, A84 CrossRef CAS.
- K. Fridman, R. Sharabi, R. Elazari, G. Gershinsky, E. Markevich, G. Salitra, D. Aurbach, A. Garsuch and J. Lampert, Electrochem. Commun., 2013, 33, 31 CrossRef CAS.
- M. Ko, S. Chae, S. Jeong, P. Oh and J. Cho, ACS Nano, 2014, 8, 8591 CrossRef CAS PubMed.
- J. Brückner, S. Thieme, F. Böttger-Hiller, I. Bauer, H. T. Grossmann, P. Strubel, H. Althues, S. Spange and S. Kaskel, Adv. Funct. Mater., 2014, 24, 1284 CrossRef.
- S. K. Lee, S. M. Oh, E. Park, B. Scrosati, J. Hassoun, M. S. Park, Y. J. Kim, H. Kim, I. Belharouak and Y. K. Sun, Nano Lett., 2015, 15, 2863 CrossRef CAS PubMed.
- X. Pu, G. Yang and C. H. Yu, Nano Energy, 2014, 9, 318 CrossRef CAS.
- J. Hassoun, Y. K. Sun and B. Scrosati, J. Power Sources, 2011, 196, 343 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |