
Thermodynamic calculations of oxygen self-diffusion in mixed-oxide nuclear fuels†
D. C. Parfitt*a,
M. W. D. Cooperb,
M. J. D. Rushtonc,
S.-R. G. Christopoulosa,
M. E. Fitzpatricka and
A. Chroneos*a
aFaculty of Engineering, Environment and Computing, Coventry University, Priory Street, Coventry CV1 5FB, UK. E-mail: david.parfitt@coventry.ac.uk; ab8104@coventry.ac.uk
bMaterials Science and Technology Division, Los Alamos National Laboratory, P.O. Box 1663, Los Alamos, NM 87545, USA
cCentre for Nuclear Engineering (CNE) & Department of Materials, Imperial College London, South Kensington Campus, London SW7 2AZ, UK
First published on 29th July 2016
Abstract
Mixed-oxide fuels containing uranium with thorium and/or plutonium may play an important part in future nuclear fuel cycles. There are, however, significantly less data available for these materials than conventional uranium dioxide fuel. In the present study, we employ molecular dynamics calculations to simulate the elastic properties and thermal expansivity of a range of mixed oxide compositions. These are then used to support equations of state and oxygen self-diffusion models to provide a self-consistent prediction of the behaviour of these mixed oxide fuels at arbitrary compositions.
1. Introduction
Uranium, plutonium and thorium dioxides are all of considerable interest to the nuclear industry owing to their use as nuclear fuels1–4 either in isolation or in mixed-oxide fuels (MOx). These materials are important for the sustainability of the nuclear industry both in alternative fuel cycles for traditional light-water reactors or for advanced fuels in support of the Generation IV reactor designs. Mixed thorium and uranium based fuels benefit from higher melting points and thermal conductivity relative to pure UO2 fuels; the mixture also provides a source of fissile isotopes which are lacking from pure ThO2. Thorium is also greatly more abundant than uranium.1 The use of PuO2, directly or as a mixed fuel form, would enable a route to dispose of legacy stockpiles of plutonium-rich material.The three stoichiometric oxides, UO2, ThO2 and PuO2, and their solid solutions all share the same parent fluorite crystal structure. The diffusion of oxygen within this lattice is of fundamental importance in determining the physical properties of the fuel and is linked to the solubility and migration of fission products,5 and the recovery rate and relative tolerance of the oxides to radiation damage.6 The accretion of oxygen point defects into planar clusters may also drive the formation of high-burn-up microstructures during reactor operation.7
The shared symmetry and solid solubility that exists between UO2, PuO2 and ThO2 crystals allows a wide range of nominal fuel compositions to be fabricated. This flexibility presents a challenge for fuel performance codes as many of the important physical and mechanical parameters will vary with metal-ion content. On a microstructural level, the fuel is even more complex as significant compositional heterogeneities exist at start of life (owing to the fabrication route) and subsequently evolve during operation due to both temperature- and irradiation-induced segregation.8,9 Consequently, there is a need to provide a self-consistent estimate of the effect of metal-ion content on both the elastic and defect properties of these materials.
Thermodynamic models can correlate atomic defect properties with bulk properties.10–12 In the cBΩ model introduced by Varotsos and Alexopoulos11,12 the defect Gibbs energy (gi) is proportional to the isothermal bulk modulus (B) and the mean volume per atom (Ω) through a constant c. The cBΩ model has been previously employed to describe point defect processes including self-diffusion in a number of systems including oxides, semiconductors, and metals.13–21
Oxygen self-diffusion in stoichiometric fluorite oxides is vacancy-mediated, in which the oxygen atoms exchange positions with adjacent oxygen vacancies. As a single diffusion mechanism is dominant over a wide temperature range, the cBΩ model is well suited to understanding self-diffusion in these materials. We use the model in this case to separate out the contribution to diffusion from changes in the elastic properties of the lattice between the different materials from the intrinsic defect processes that are common to all of the considered compositions. This allows us to parameterise the models for better predictive capabilities, without the need to perform many separate simulations, and also to understand the effect of mixed metal compositions upon lattice diffusion. The combination of these should enable improved predictions of the behaviour of new fuel forms and a more complete understanding of the effect of compositional heterogeneities within current fuels.
2. Methodology
2.1 Molecular dynamics
Molecular dynamics (MD) simulations have often been used to provide data on the diffusion properties of energy related materials such as MOx.3,22,23 The recent Cooper–Rushton–Grimes (CRG) potential set24 has been used for numerous fluorite structured oxides and reproduces their thermomechanical and thermophysical properties and the Cauchy violation over a wide temperature range.22,25,26 The CRG potential's fidelity in describing these properties arises from the introduction of many-body interactions to the model's description of interatomic forces. This is achieved using the embedded atom method.27 The CRG model was successfully used in the calculation of diffusion properties in CeO2, U1−xThxO2 and Pu1−xUxO2.22,23,28MD calculations in this study were performed within the Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS) code.29 To produce equilibrium volume and diffusion constants, the following process was used:
(1) A 10 × 10 × 10 supercell of fluorite-structured MxN1−xO2 (M = U, N = Pu, Th) was generated. Metal ions were randomly allocated to each cation site. The starting lattice parameter is arbitrary provided it is sufficiently close to the final equilibrium value for each pressure, temperature and composition. It was set to be 5.47 Å.
(2) Before commencing the dynamic part of the simulation, each ion was slightly displaced (by around 0.1 Å) in a random direction from its lattice site. The initial velocities of all of the ions were set to a random value with the constraint that the overall velocity distribution conformed to a Maxwell Boltzmann distribution at an (arbitrary) temperature of 300 K. Statistically-independent random seeds were used in each simulation.
(3) A molecular dynamics simulation was performed in a constant pressure–temperature (NPT) ensemble. For 10 ps the temperature of the thermostat was ramped from the initial value of 300 K to the final value of temperature T. A subsequent 20 ps was calculated with the thermostat held at T. Control of the temperature and pressure in the simulation was through a Nosé–Hoover thermostat and barostat with relaxation times of 0.1 and 0.5 ps respectively.
(4) For calculation of the diffusion constant, the simulation box size was set to the average cell size over the previous 20 ps. The ensemble was set to constant volume and energy (NVE) (no thermostat or barostat) and the positions and displacements of the ions were recorded over a subsequent 1 ns.
This process allowed us to establish the equilibrium volume at a given composition, temperature and pressure of the system. We assume that the random selection of cation positions, and initiation of random velocities and displacements mean that these data are independent and statistically representative of the bulk crystal behaviour of the compositions at a given pressure and temperature.
To calculate the diffusivity D, we use a linear fit to the mean squared displacement:
| 〈(ri(t) − ri(0))2〉 = 6Dt | (1) |
In order to investigate the bulk and diffusion properties of this system, we generated data at specific compositions of MxN1−xO2 for M = U, N = Pu, Th and x = 0.0, 0.25, 0.5, 0.75 and 1.0. To generate the thermoelastic properties, 100 simulations per compositions were run at a randomly selected set of temperatures and pressures in the range T = {50, 2500 K} and P = {−7.5, 7.5 GPa}; in contrast to the diffusion calculations, only a single cation arrangement was used per P, T point, however we believe the large number of total simulations is sufficient to provide appropriate averaging over the different cation distributions.
2.2 Equation of state
The pressure–volume–temperature data were used to fit a Rose–Vinet equation of state30 in which a material of volume, V, at an equilibrium temperature, T, and pressure, P, are linked through an equation of the form:
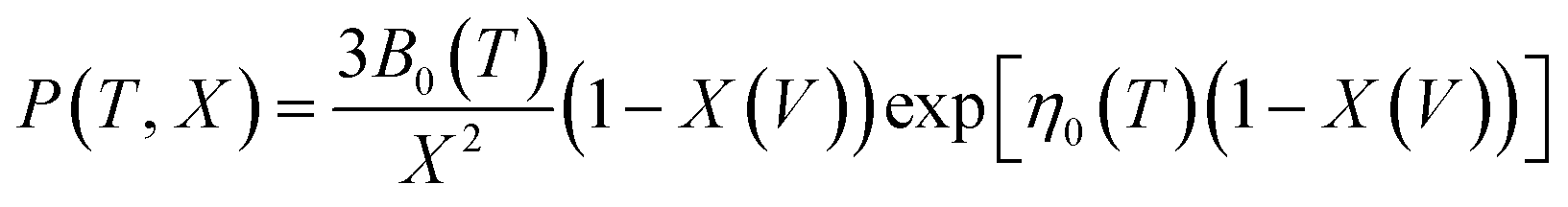 | (2) |
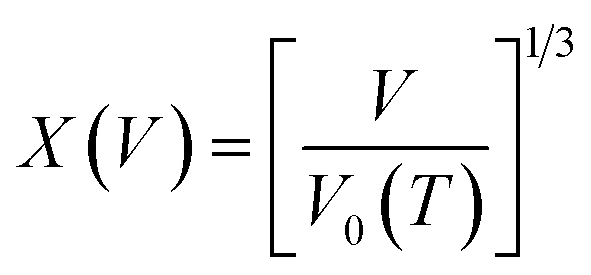 | (3) |
 | (4) |
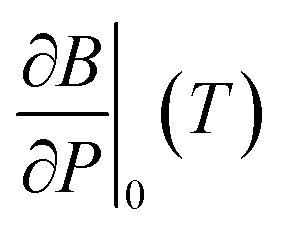 is the pressure derivative of the bulk modulus at zero pressure as a function of temperature.
is the pressure derivative of the bulk modulus at zero pressure as a function of temperature.
The physical origin of the Rose–Vinet equation of state is based on splitting the volume derivative of the Helmholtz free energy, F(V, T) into two terms:
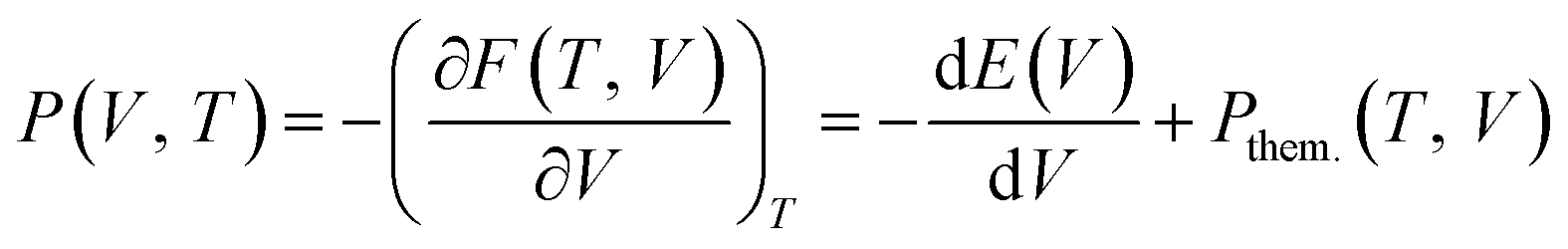 | (5) |
| P(T,V) = P(TR, V) + α0(TR)B0(TR)(T − TR) | (6) |
This expression is valid above a material's Debye temperature (300–400 K for the mixed oxides considered here34,35) and in the absence of any structural phase transformations that would alter E(V).
Vinet et al.36 have considered analytical functions that may represent the pressure isotherm P(TR, V) at a non-zero temperature and have noted that, for a wide range of solids, the following relationship holds:
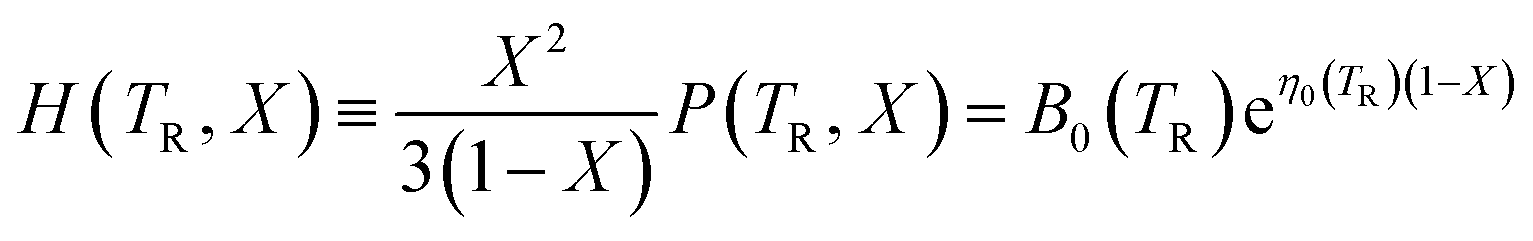 | (7) |
To provide parameters for the Rose–Vinet equation of state it is necessary to select an (arbitrary) reference temperature, TR, taken here to be 300 K. It follows that the pressure can then be calculation at a point (T, V) by substitution of the pressure isotherm P(TR, X) from eqn (7) into eqn (6) to obtain:
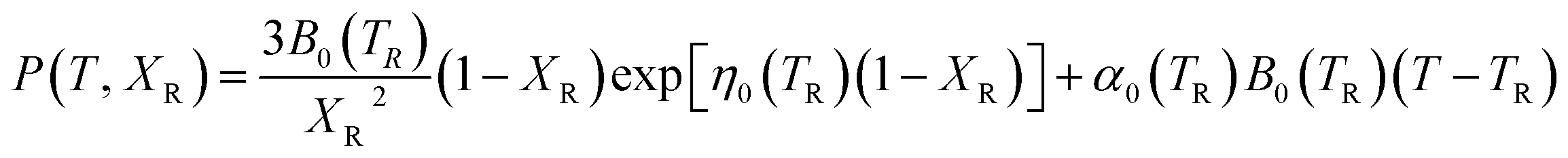 | (8) |
The isothermal bulk modulus then follows from:
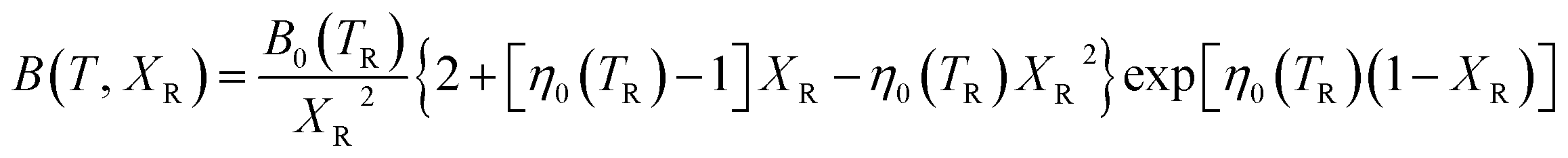 | (9) |
 | (10) |
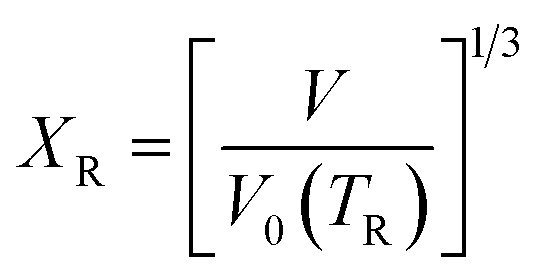 | (11) |
The functional form of this equation of state is more complex than the more widely used isothermal Birch–Murnaghan equation of state,37,38 however it has the significant advantage that it allows predictions of the volume of a material at an arbitrary temperature and pressure from only four parameters: the thermal expansion coefficient, volume, bulk modulus and pressure derivative of bulk modulus evaluated at zero pressure and a single reference temperature TR.
In order to predict the thermodynamic properties at an arbitrary pressure and temperature we use a Levenberg–Marquardt least-squares algorithm39,40 to fit the four parameters, V0(TR), B0(TR), 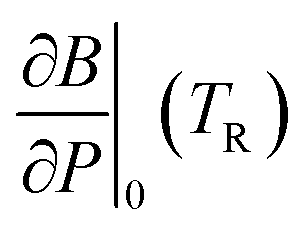 and α0(TR) to the sets of P, V, T data generated by the simulations. From these fitted values, we can use eqn (8)–(10) to predict volume, bulk modulus and the pressure derivative of the bulk modulus; thermal expansivity at temperature (instantaneous or averaged) may then be numerically calculated from the appropriate temperature derivatives of eqn (4).
and α0(TR) to the sets of P, V, T data generated by the simulations. From these fitted values, we can use eqn (8)–(10) to predict volume, bulk modulus and the pressure derivative of the bulk modulus; thermal expansivity at temperature (instantaneous or averaged) may then be numerically calculated from the appropriate temperature derivatives of eqn (4).
2.3 Diffusion model
The vacancy-mediated oxygen self-diffusion is controlled by the Gibbs activation energy, gact. Consequently, the diffusivity, D, is defined by the following relation:19
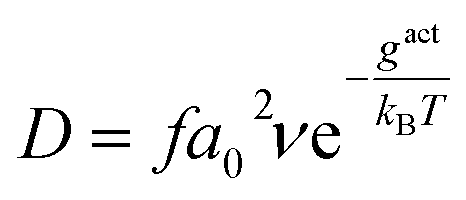 | (12) |
In order to link the bulk and defect properties of the lattice to the resultant oxygen diffusivity we have employed the cBΩ model. In this the Gibbs defect energy gi for oxygen defect formation is associated with the bulk properties of the material through:18,19
| gi = ciBΩ | (13) |
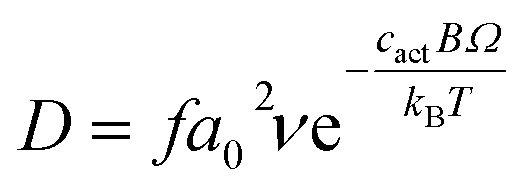 | (14) |
3. Results and discussion
3.1 Equation of state
The Rose–Vinet equation of state was fitted to the MD data generated at each of the three end members and three intermediate compositions UxTh1−xO2 and UxPu1−xO2 (x = 0.25, 0.5, 0.75). The fitted results for the bulk modulus, derivative of the bulk modulus and thermal expansivity are reported in Table 1. The use of these values, in association with the equations given in Section 2, allows the thermoelastic properties of each composition to be estimated at temperatures and pressures below the superionic transition. The empirical potentials used in the present study are designed to accurately reproduce the elastic properties of the matrix, so it is unsurprising that the data agree well with previous studies for the binary oxides. However the use of statistically-averaged ternary compositions (e.g. UxPu1−xO2), and the simultaneous treatment of volume and temperature in the Rose–Vinet equation of state, allow predictions of volume at an arbitrary composition, pressure and temperature.| Composition | Rose–Vinet parameters (values at TR = 300 K) | |||
|---|---|---|---|---|
| Volume per formula unit/Å3 | Bulk modulus/GPa | Derivative bulk modulus | Volume expansivity/10−5 K−1 | |
| ThO2 | 43.75 | 184.33 | 6.20 | 2.66 |
| U0.25Th0.75O2 | 42.99 | 188.91 | 6.69 | 2.77 |
| U0.50Th0.50O2 | 42.25 | 194.54 | 7.00 | 2.85 |
| U0.75Th0.25O2 | 41.56 | 198.85 | 7.17 | 2.90 |
| UO2 | 40.87 | 207.36 | 7.46 | 2.90 |
| Pu0.25U0.75O2 | 40.46 | 207.80 | 7.31 | 2.95 |
| Pu0.50U0.50O2 | 40.06 | 207.83 | 7.43 | 2.95 |
| Pu0.75U0.25O2 | 39.64 | 210.33 | 7.43 | 2.95 |
| PuO2 | 39.24 | 211.91 | 7.42 | 2.93 |
In Fig. 1, data are shown for the thermal expansion and compressibility of two example systems, UO2 and a mixture U0.5Th0.5O2. These data are not included in the original fit dataset but it can be seen that the equation of state does an excellent job of describing the variation in volume within the range of the fitted data. At high temperatures (greater than 2500 K) there is evidence in Fig. 1 of the onset of the superionic transition, which marks the beginning of significant anion disorder in the lattice. The equation of state is not fitted to data greater than 2500 K, and if it were, it is not capable of capturing the discontinuous change in thermal expansion that occurs at this transition.
 | ||
| Fig. 1 Plots of the predicted volume per formula unit against temperature (i) and pressure (ii) for (a) UO2 and (b) U0.5Th0.5O2. The dashed lines represent the Rose–Vinet fitted equation of state and the points are individual simulation results. | ||
The bulk modulus of a theoretical mixed oxide system MxN1−xO2 with complete solid solubility is given by,19
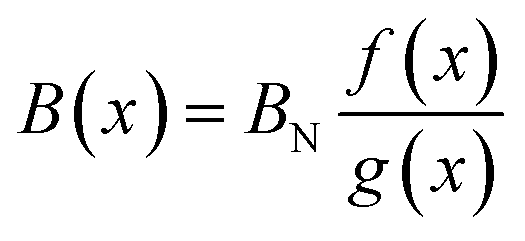 | (15) |
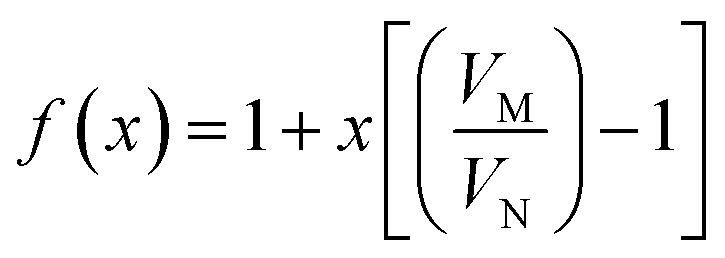 | (16) |
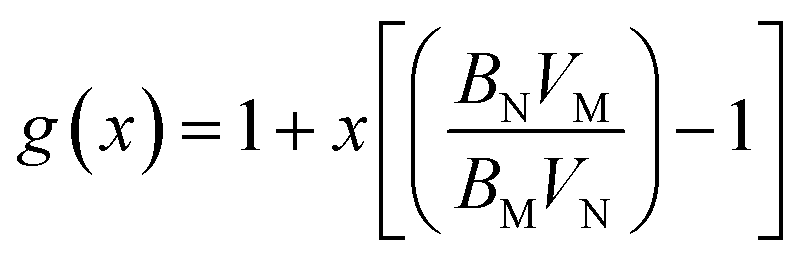 | (17) |
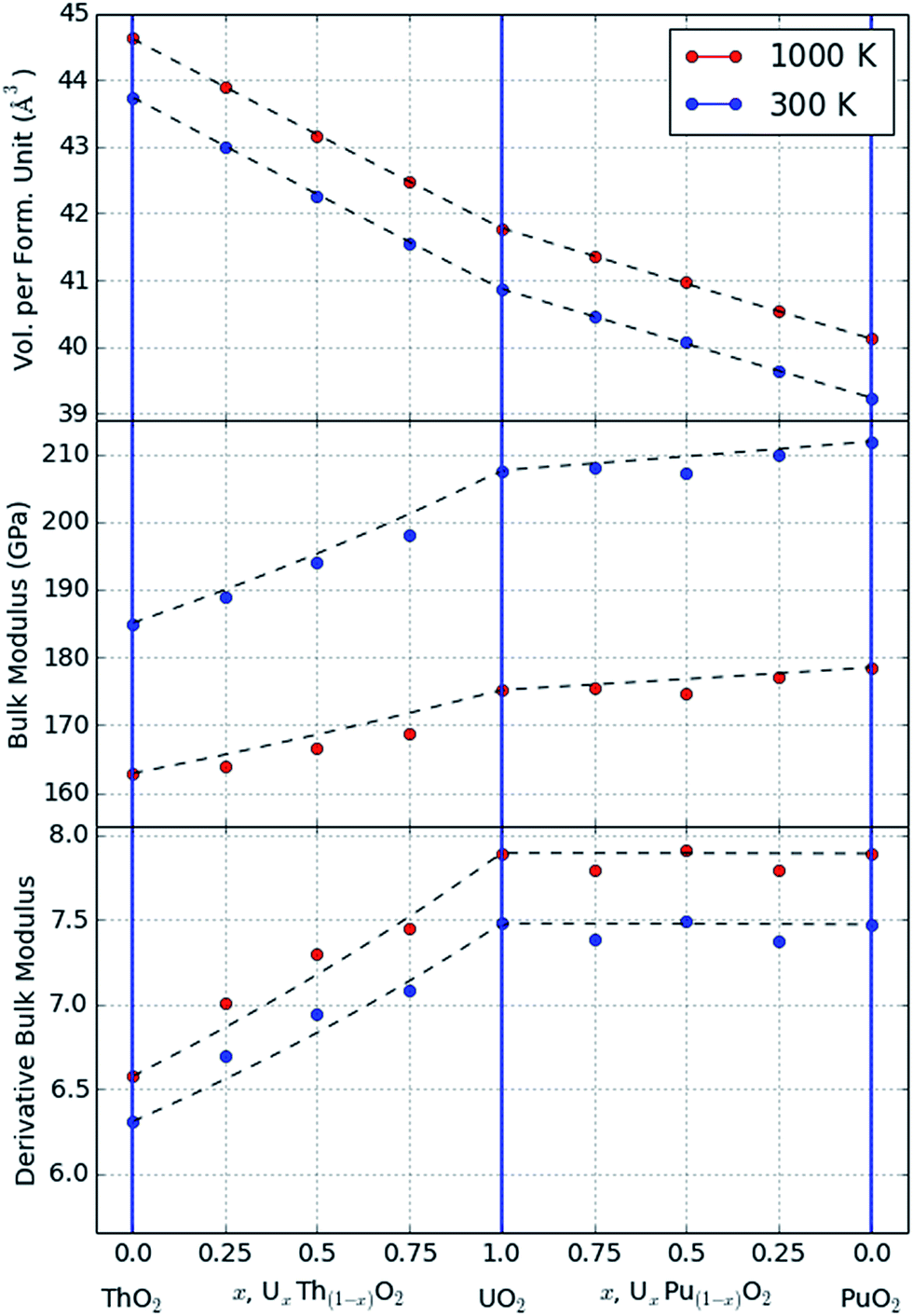 | ||
| Fig. 2 Plot of the value of bulk modulus calculated from the theoretical expression and fitting to the MD data. | ||
The pressure derivative of the bulk modulus as a function of composition can then be found from eqn (9) as:
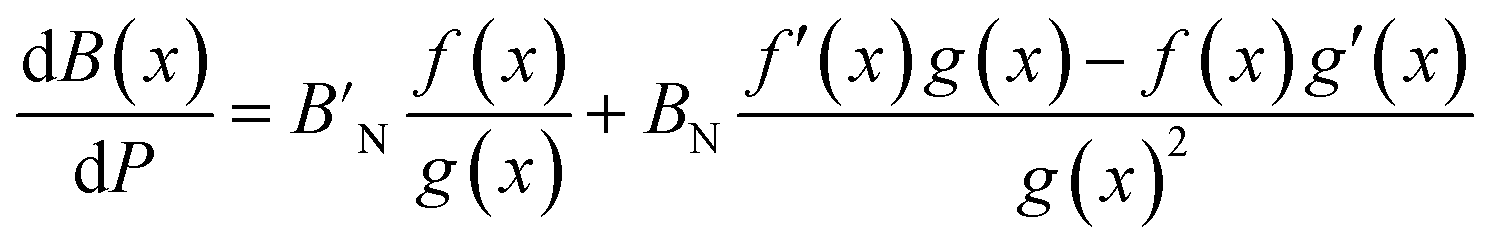 | (18) |
 | (19) |
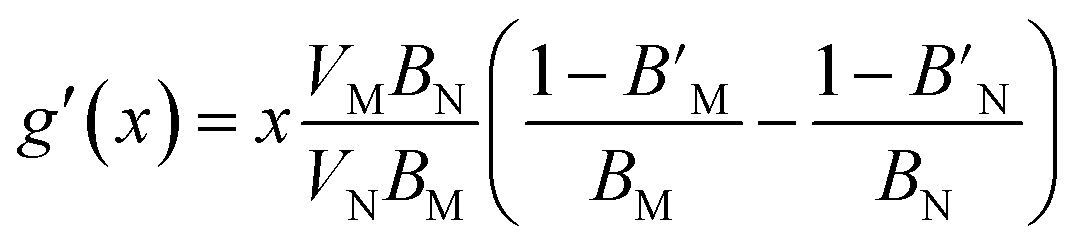 | (20) |
 given above. The agreement is reasonable and therefore we argue that the elastic properties at an arbitrary composition are well-represented by eqn (9) and (12).
given above. The agreement is reasonable and therefore we argue that the elastic properties at an arbitrary composition are well-represented by eqn (9) and (12).
The final term in the Rose–Vinet equation of state is the thermal expansivity. In Fig. 3 we show the variation in linear thermal expansion at zero pressure defined as:
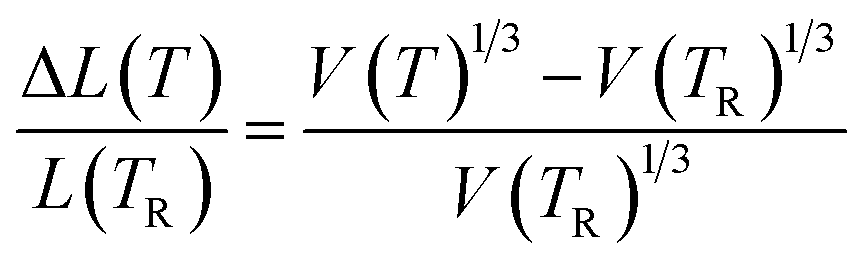 | (21) |
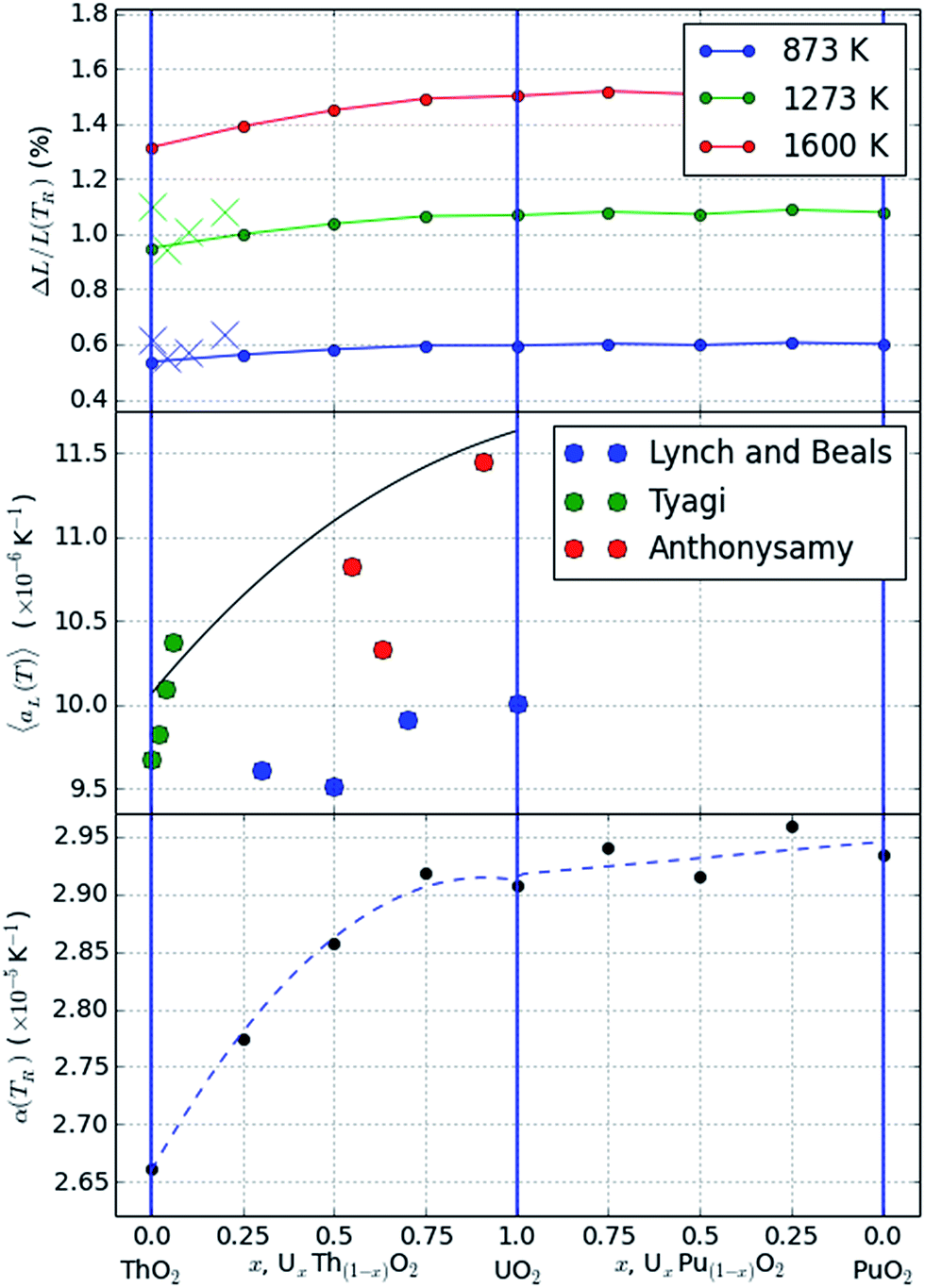 | ||
| Fig. 3 Plots showing the thermal expansion across a range of compositions. A common set of Rose–Vinet equations of state is used in all cases. The top plot shows the predictions at a range of temperatures and compared with experimental data41 at 873 K (blue crosses) and 1173 K (green crosses); the middle plot shows the average thermal expansion between 293 K and 1600 K compared with data from ref. 43–46. The bottom plot shows the value of α(TR) from eqn (4). | ||
This is shown across the different compositions indicating that the compositional variation is relatively minor for the PuO2–UO2 system but greater for ThO2–UO2. The difference also becomes progressively greater when considering the higher temperature data in agreement with experimental reports.41
In order to better compare with literature data on the thermal behaviour, we have also calculated the average change in linear thermal expansivity as:
 | (22) |
This is plotted for T = 1600 K in Fig. 3(b).
Experimental data showing the changes in thermal expansivity with composition are compared with the model predictions in Fig. 3(a) and (b). We have selected the data from ref. 41 for comparison with the change in thermal expansivity at 873 K and 1173 K and the data from ref. 42 and originally reported by Anthonysamy et al.43 Tyagi et al.44,45 and Lynch and Beals46 for comparison with the average linear thermal expansivity between 293 K and 1600 K.
The experimental data for thermal expansivity of the mixed ThO2–UO2 system is scattered, particularly when averaged over large temperature ranges as seen in Fig. 3(b); this is likely due to either heterogeneities in material microstructure or significant deviations from stoichiometry as a function of temperature. The absolute values of average thermal expansivity are however consistent with the models and the trends are similar within each dataset. In particular, both the models and experimental data support a non-linear increase in thermal expansivity with the addition of uranium into ThO2 which is not mirrored in the PuO2–UO2 system.
The fitted value of (volumetric) thermal expansivity at the reference temperature of 300 K is also shown in Fig. 3(c). The change mimics the trends of thermal expansion, although the differences in absolute values of expansion are only evident at high temperatures.
In the case of thermal expansivity, no general theory of variation with composition was apparent and so we fit these data using a polynomial function of the form:
 | (23) |
| Th1−xUxO2 | U1−xPuxO2 | |||
|---|---|---|---|---|
| α(x, TR) | cact(x) | α(x,TR) | cact(x) | |
| a0 | 2.6583 | 0.304 | 2.90715 | 0.292 |
| a1 | 0.5676 | −0.102 | 0.04159 | −0.0612 |
| a2 | −0.3188 | −0.0206 | — | 0.0327 |
| a3 | — | 0.245 | — | −0.0139 |
| a4 | — | −0.133 | — | — |
| D0 | — | 0.45108 | — | 0.45108 |
3.2 Oxygen self-diffusion
In previous studies, the link between atomic parameters and the diffusivity of UO2, PuO2 and ThO2 end-members was established13,14 using the mean value method.47–49 In principle, further equations could be determined for each U–Th–Pu composition. However, to avoid a proliferation of different terms, we fit the data using a universal value for the pre-exponential and making the activation coefficient compositionally dependent, i.e. fitting an equation of the form,
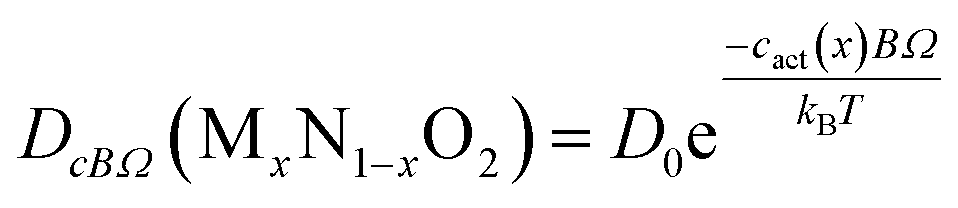 | (24) |
Similarly to the case of thermal expansion, there is no general expression for the variability of activation coefficient with composition and so we fit the data using a second set of polynomial expressions of the form:
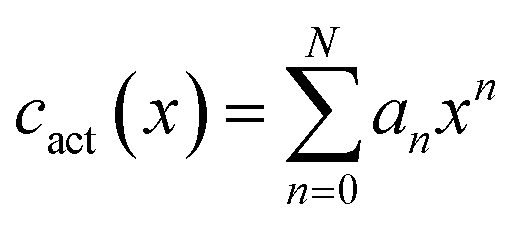 | (25) |
We apply the same logic to the fitting of the thermal expansivity data, however because of the greater variability of the cact parameter and the increased sensitivity of the final diffusion coefficient, a fourth-order polynomial (N = 4) is used for the ThO2–UO2 system and a third-order (N = 3) polynomial for the PuO2–UO2 system. Fitted values are reported in Table 2.
The results of these simulation data at each composition and the fitted polynomial functions are shown in Fig. 4. The activation coefficient varies smoothly and systematically with composition; this behaviour is monotonic in the PuO2–UO2 system representing an increase in oxygen diffusivity as Pu content is increased.22 However, in the case of the ThO2–UO2 system there is a pronounced minimum in the activation coefficient coincident with the maximum in oxygen ion diffusivity from molecular dynamics simulations. This curious feature was previously discussed by Cooper et al.23 where it was attributed to the existence of Frenkel pair formation enthalpies in the mixed system that lay below those of either of the end members. Fig. 4 of ref. 23 shows this splitting of the vacancy formation enthalpy demonstrating this effect.
 | ||
| Fig. 4 Plots showing the O-ion diffusivity and fitted values of the activation coefficient cact as a function of oxide composition. The dashed lines show a polynomial fit to the data which is fourth-order for the UxTh1−xO2 system and third-order for the UxPu1−xO2 system. | ||
3.3 Combined model
Using the results of the activation coefficient fitting and the Rose–Vinet equation of state, we now summarise the model coefficients which can be used to predict the oxygen ion self-diffusion and elastic properties at an arbitrary composition in the Th1–xUxO2 or Pu1–xUxO2 phase diagram. The Rose–Vinet equation of state and cBΩ diffusion model parameters are as follows:· Volume, V0(TR, x), linear interpolation between end members.
· Bulk modulus, B0(TR, x), eqn (15).
· Pressure derivative of bulk modulus,  , eqn (18).
, eqn (18).
· Thermal expansivity, α0(TR, x), polynomial fit using coefficients in Table 2.
· Activation coefficient, cact(x), polynomial fit using coefficients in Table 2.
These values can then be used in association with eqn (8)–(10) in order to predict the elastic properties and oxygen ion self-diffusivity at an arbitrary pressure, temperature and volume. We show an example of this use of this model in Fig. 5 which compares the volume and diffusivity values calculated from MD simulations for both the PuO2–UO2 and ThO2–UO2 systems with those predicted from the combined model.
 | ||
| Fig. 5 Predicted and calculated values of the (a) volume per formula unit and (b) oxygen ion diffusivity for a range of different compositions. | ||
3.4 Application to other systems
In addition to the properties at different temperatures, it is also of note that through the use of the cBΩ model the predictions of oxygen self-diffusion are equally applicable to a range of pressures. Therefore, these models should also account for the elastic properties and oxygen migration in mixed oxide regions subject to significant compressive or tensile stress. The stresses investigated in this paper are far above those encountered as a macroscopic stress in a nuclear fuel pin; however they would be useful in investigating diffusion around microstructural defects. For example, they may be help account for the enhanced diffusion close to dislocation cores (where elastic deformation in the surrounding lattice can lead to significant levels of strain).50,51 Equally, greatly enhanced diffusion has also been reported in similar fluorite-structured materials in which compositionally distinct regions lead to the generation of significant interface strains.52–55These results would also be applicable to modelling oxide corrosion layers formed on metallic uranium, plutonium or thorium. In these cases, the oxide layers are formed under significant compressive stresses due to the mismatch in lattice parameter between the oxide and the underlying metal substrate. Oxygen transport through these layers is one of the controlling factors that determine the overall oxidation rate.
The validation of these models is problematic due to the difficulty and expense of obtaining experimental data on these systems; the scatter in experimental values in Fig. 3(b) being a pertinent illustration. Whilst experimental data remains the sole test of scientific truth, the use of ab initio methods coupled with molecular dynamics may in the future enable these calculations to be replicated with density functional theory. These would reduce the number of fitted parameters in the empirical potentials through an appeal to the sounder theoretical basis of non-relativistic quantum mechanics. Calculation on LiGe2(PO4)3 (ref. 56) have shown the use of these types of simulations in obtaining self-diffusion coefficients similar to those obtained here and these have also successfully replicated diffusion coefficients and relatively subtle chemistry effects in uranium-containing molten salt reactions.57 These techniques are however computationally expensive, particularly for UO2 where subtle correlations between the uranium ion f-electrons can confound the identification of the true ground state electronic structure.58–60
4. Conclusions
In this paper we have examined the link between the composition of mixed oxide fuels, their bulk elastic properties and the value of oxygen self-diffusion. This has provided a predictive model of the elastic properties for the mixed oxides together with an estimate of the oxygen ion self-diffusion.The addition of UO2 into PuO2 produces a mixed oxide which exhibits thermoelastic properties and oxygen self-diffusion values which are in-line with the average properties. In particular, the changes in elastic properties and thermal expansion are small, and captured by simple interpolation between the two end members. There is a noticeable non-linearity in the oxygen self-diffusion coefficient at high temperature; however this is bounded by the maximum value in PuO2. The behaviour of the ThO2–UO2 system is more interesting in that there is a substantial difference in properties between the two end members and the mixed oxide exhibits properties that are poorly predicted by linear interpolation between them. This is most dramatic in the oxygen self-diffusion coefficients, but also evident in the thermal expansivity where a simple linear average between the two end-members may significantly under-predict the calculated values.
This combined model can be used to predict volume changes, elastic properties, thermal expansion and oxygen-ion self-diffusion as a function of stress, temperature and composition. As well as investigating the effect of heterogeneity in fuels, this may also offer insight into the behaviour of the material around microstructural features that impose significant lattice strain, for example edge dislocations, gas bubbles or metal-oxide interfaces.
Acknowledgements
MWDC is funded by the Department of Energy (DoE), Office for Nuclear Energy, Nuclear Energy Advanced Modelling and Simulation (NEAMS) program. SRGC, MEF and AC are grateful for funding from the Lloyd's Register Foundation, a charitable foundation helping to protect life and property by supporting engineering-related education, public engagement and the application of research.References
- M. Lung and O. Gremm, Perspectives of the thorium fuel cycle, Nucl. Eng. Des., 1998, 180, 133–146, DOI:10.1016/S0029-5493(97)00296-3.
- W. E. Lee, M. Gilbert, S. T. Murphy and R. W. Grimes, Opportunities for Advanced Ceramics and Composites in the Nuclear Sector, J. Am. Ceram. Soc., 2013, 96, 2005–2030, DOI:10.1111/jace.12406.
- M. Stan, Discovery and design of nuclear fuels, Mater. Today, 2009, 12, 20–28, DOI:10.1016/s1369-7021(09)70295-0.
- M. Kazimi, Thorium fuel for nuclear energy: An unconventional tactic might one day ease concerns that spent fuel could be used to make a bomb, Am. Sci., 2003, 91, 408–415, DOI:10.1511/2003.5.408.
- M. Bertolus, M. Freyss, B. Dorado, G. Martin, K. Hoang and S. Maillard, et al., Linking atomic and mesoscopic scales for the modelling of the transport properties of uranium dioxide under irradiation, J. Nucl. Mater., 2015, 462, 475–495, DOI:10.1016/j.jnucmat.2015.02.026.
- K. E. Sickafus, R. W. Grimes, J. A. Valdez, A. Cleave, M. Tang and M. Ishimaru, et al., Radiation-induced amorphization resistance and radiation tolerance in structurally related oxides, Nat. Mater., 2007, 6, 217–223, DOI:10.1038/nmat1842.
- T. Ichinomiya, B. P. Uberuaga, K. E. Sickafus, Y. Nishiura, M. Itakura and Y. Chen, et al., Temperature accelerated dynamics study of migration process of oxygen defects in UO2, J. Nucl. Mater., 2009, 384, 315–321, DOI:10.1016/j.jnucmat.2008.12.040.
- H. Kleykamp, Post-irradiation studies on LWR-MOX fuel fabricated by the optimized co-milling process, J. Nucl. Mater., 2004, 324, 198–202, DOI:10.1016/j.jnucmat.2003.10.004.
- G. Oudinet, I. Munoz-Viallard, L. Aufore, M.-J. Gotta, J. M. Becker and G. Chiarelli, et al., Characterization of plutonium distribution in MIMAS MOX by image analysis, J. Nucl. Mater., 2008, 375, 86–94, DOI:10.1016/j.jnucmat.2007.10.013.
- J. Philibert, Some Thoughts and/or Questions about Activation Energy and Pre-Exponential Factor, Defect Diffus. Forum, 2006, 249, 61–72, DOI:10.4028/www.scientific.net/DDF.249.61.
- P. Varotsos and K. Alexopoulos, Estimation of the migration enthalpy and entropy for cation vacancy motion in alkali halides with the NaCl-type structure, Phys. Rev. B: Condens. Matter Mater. Phys., 1977, 15, 2348–2351, DOI:10.1103/PhysRevB.15.2348.
- P. Varotsos and K. Alexopoulos, Calculation of the formation entropy of vacancies due to anharmonic effects, Phys. Rev. B: Condens. Matter Mater. Phys., 1977, 15, 4111–4114, DOI:10.1103/PhysRevB.15.4111.
- A. Chroneos and R. V. Vovk, Modeling self-diffusion in UO2 and ThO2 by connecting point defect parameters with bulk properties, Solid State Ionics, 2015, 274, 1–3, DOI:10.1016/j.ssi.2015.02.010.
- A. Chroneos, M. E. Fitzpatrick and L. H. Tsoukalas, Describing oxygen self-diffusion in PuO2 by connecting point defect parameters with bulk properties, J. Mater. Sci.: Mater. Electron., 2015, 26, 3287–3290, DOI:10.1007/s10854-015-2829-2.
- P. A. Varotsos, Calculation of point defect parameters in diamond, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 172107, DOI:10.1103/PhysRevB.75.172107.
- E. S. Skordas, Comment on “LiH as a Li+ and H− ion provider” by Khang Hoang, Chris G. Van de Walle, Solid State Ionics 253 (2013) 53, Solid State Ionics, 2014, 261, 26–27, DOI:10.1016/j.ssi.2014.04.001.
- F. Vallianatos and V. Saltas, Application of the cBΩ model to the calculation of diffusion parameters of He in olivine, Phys. Chem. Miner., 2013, 41, 181–188, DOI:10.1007/s00269-013-0636-y.
- P. Varotsos, Comparison of models that interconnect point defect parameters in solids with bulk properties, J. Appl. Phys., 2007, 101, 123503, DOI:10.1063/1.2745359.
- P. A. Varotsos and K. D. Alexopoulos, Thermodynamics of point defects and their relation with bulk properties, North-Holland, 1986 Search PubMed.
- V. Saltas and F. Vallianatos, Thermodynamic calculations of self- and hetero-diffusion parameters in germanium, Mater. Chem. Phys., 2015, 163, 507 CrossRef CAS.
- V. Saltas, A. Chroneos and F. Vallianatos, A thermodynamic approach of self- and hetero-diffusion in GaAs: connecting point defect parameters with bulk properties, RSC Adv., 2016, 6, 53324 RSC.
- M. W. D. Cooper, S. T. Murphy, M. J. D. Rushton and R. W. Grimes, Thermophysical properties and oxygen transport in the (Ux,Pu1−x)O2 lattice, J. Nucl. Mater., 2015, 461, 206–214, DOI:10.1016/j.jnucmat.2015.03.024.
- M. W. D. Cooper, S. T. Murphy, P. C. M. Fossati, M. J. D. Rushton and R. W. Grimes, Thermophysical and anion diffusion properties of (Ux,Th1−x)O2, Proc. R. Soc. A, 2014, 470, 20140427, DOI:10.1098/rspa.2014.0427.
- M. W. D. Cooper, M. J. D. Rushton and R. W. Grimes, A many-body potential approach to modelling the thermomechanical properties of actinide oxides, J. Phys.: Condens. Matter, 2014, 26, 105401, DOI:10.1088/0953-8984/26/10/105401.
- M. W. D. Cooper, R. W. Grimes, M. E. Fitzpatrick and A. Chroneos, Modeling oxygen self-diffusion in UO2 under pressure, Solid State Ionics, 2015, 282, 26–30, DOI:10.1016/j.ssi.2015.09.006.
- M. W. D. Cooper, S. C. Middleburgh and R. W. Grimes, Modelling the thermal conductivity of (UxTh1−x)O2 and (UxPu1−x)O2, J. Nucl. Mater., 2015, 466, 29–35, DOI:10.1016/j.jnucmat.2015.07.022.
- M. S. Daw and M. I. Baskes, Embedded-atom method: derivation and application to impurities, surfaces, and other defects in metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1984, 29, 6443–6453, DOI:10.1103/PhysRevB.29.6443.
- M. J. D. Rushton and A. Chroneos, Impact of uniaxial strain and doping on oxygen diffusion in CeO2, Sci. Rep., 2014, 4, 6068, DOI:10.1038/srep06068.
- S. Plimpton, Fast Parallel Algorithms for Short – Range Molecular Dynamics, J. Comput. Phys., 1995, 117, 1–19, DOI:10.1006/jcph.1995.1039.
- P. Vinet, J. R. Smith, J. Ferrante and J. H. Rose, Temperature effects on the universal equation of state, Phys. Rev. B: Condens. Matter Mater. Phys., 1987, 35, 1945–1953, DOI:10.1103/PhysRevB.35.1945.
- J. H. Rose, J. R. Smith, F. Guinea and J. Ferrante, Universal features of the equation of state of metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1984, 29, 2963–2969, DOI:10.1103/PhysRevB.29.2963.
- O. L. Anderson, A universal thermal equation-of-state, J. Geodyn., 1984, 1, 185–214, DOI:10.1016/0264-3707(84)90027-9.
- M. S. Anderson and C. A. Swenson, Experimental equations of state for cesium and lithium metals to 20 kbar and the high-pressure behavior of the alkali metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 31, 668–680 CrossRef CAS.
- G. Dolling, R. A. Cowley and A. D. B. Woods, The Crystal Dynamics of Uranium Dioxide, 2011, DOI:10.1139/p65-135.
- I. J. Fritz, Elastic properties of UO2 at high pressure, J. Appl. Phys., 1976, 47, 4353, DOI:10.1063/1.322438.
- P. Vinet, J. R. Smith, J. Ferrante and J. H. Rose, Temperature effects on the universal equation of state, Phys. Rev. B: Condens. Matter Mater. Phys., 1987, 35, 1945–1953 CrossRef CAS.
- F. Birch, Finite Elastic Strain of Cubic Crystals, Phys. Rev., 1947, 71, 809–824, DOI:10.1103/PhysRev.71.809.
- F. D. Murnaghan, The Compressibility of Media under Extreme Pressures, Proc. Natl. Acad. Sci. U. S. A., 1944, 30, 244–247, DOI:10.1073/pnas.30.9.244.
- K. Levenberg, A Method for the Solution of Certain Non-Linear Problems in Least Squares, Q. Appl. Math., 1944, 2, 164–168 Search PubMed.
- D. Marquardt, An Algorithm for Least-Squares Estimation of Nonlinear Parameters, SIAM J. Appl. Math., 1963, 11, 431 CrossRef.
- A. K. Sengupta, J. Banerjee, R. K. Bhagat, R. Ramachandran, S. Majumdar and R. Division, Thermal Expansion Data of (Th,U)O2 Fuels, BARC/2000/E/008, 2000.
- K. Bakker, E. H. P. Cordfunke, R. J. M. Konings and R. P. C. Schram, Critical evaluation of the thermal properties of ThO2 and Thl−yUyO2 and a survey of the literature data on Th1−yPuyO2, J. Nucl. Mater., 1997, 250, 1–12 CrossRef CAS.
- S. Anthonysamy, G. Panneerselvam, S. Bera, S. V. Narasimhan and P. R. Vasudeva Rao, Studies on thermal expansion and XPS of urania+thoria solid solutions, J. Nucl. Mater., 2000, 281, 15–21 CrossRef CAS.
- A. K. Tyagi, M. D. Mathews, B. R. Ambekar and R. Ramachandran, Thermal expansion of ThO2 4 and 6 wt% UO2 by HT-XRD, Thermochim. Acta, 2004, 421, 69–71 CrossRef CAS.
- A. K. Tyagi and M. D. Mathews, Thermal expansion of ThO2 wt% UO2 by HT-XRD, J. Nucl. Mater., 2000, 278, 123–125 CrossRef CAS.
- E. D. Lynch and R. J. Beals, Annual Report ANL-6677, Argonne National Laboratory, 1962.
- B.-H. Zhang and X.-P. Wu, Diffusion of aluminum in MgO: a thermodynamic approach study, Chin. Phys. B, 2013, 22, 056601, DOI:10.1088/1674-1056/22/5/056601.
- B. Zhang and X. Wu, Calculation of self-diffusion coefficients in diamond, Appl. Phys. Lett., 2012, 100, 051901, DOI:10.1063/1.3680600.
- B. Zhang, Calculation of self-diffusion coefficients in iron, AIP Adv., 2014, 4, 017128, DOI:10.1063/1.4863462.
- S. T. Murphy, E. E. Jay and R. W. Grimes, Pipe diffusion at dislocations in UO2, J. Nucl. Mater., 2014, 447, 143–149, DOI:10.1016/j.jnucmat.2013.12.029.
- D. C. Parfitt, C. L. Bishop, M. R. Wenman and R. W. Grimes, Strain fields and line energies of dislocations in uranium dioxide, J. Phys.: Condens. Matter, 2010, 22, 175004, DOI:10.1088/0953-8984/22/17/175004.
- N. Sata, K. Eberman, K. Eberl and J. Maier, Mesoscopic fast ion conduction in nanometre-scale planar heterostructures, Nature, 2000, 408, 946–949, DOI:10.1038/35050047.
- X. X. Guo, I. Matei, J.-S. Lee and J. Maier, Ion conduction across nanosized CaF2/BaF2 multilayer heterostructures, Appl. Phys. Lett., 2007, 91, 103102, DOI:10.1063/1.2779254.
- J. Garcia-Barriocanal, A. Rivera-Calzada, M. Varela, Z. Sefrioui, E. Iborra and C. Leon, et al., Colossal ionic conductivity at interfaces of epitaxial ZrO2:Y2O3/SrTiO3 heterostructures, Science, 2008, 321, 676–680, DOI:10.1126/science.1156393.
- M. J. D. Rushton and A. Chroneos, Impact of uniaxial strain and doping on oxygen diffusion in CeO2, Sci. Rep., 2014, 4, 2–7, DOI:10.1038/srep06068.
- J. Kang, H. Chung, C. Doh, B. Kang and B. Han, Integrated study of first principles calculations and experimental measurements for Li-ionic conductivity in Al-doped solid-state LiGe2(PO4)3 electrolyte, J. Power Sources, 2015, 293, 11–16 CrossRef CAS.
- C. Kwon, J. Kang, W. Kang, D. Kwak and B. Han, First principles study of the thermodynamic and kinetic properties of U in an electrorefining system using molybdenum cathode and LiCl-KCl eutectic molten salt, Electrochim. Acta, 2016, 195, 216–222 CrossRef CAS.
- E. Vathonne, J. Wiktor, M. Freyss, G. Jomard and M. Bertolus, DFT+U investigation of charged point defects and clusters in UO2, J. Phys.: Condens. Matter, 2014, 26, 325501 CrossRef PubMed.
- B. Dorado, M. Freyss, B. Amadon, M. Bertolus, G. Jomard and P. Garcia, Advances in first-principles modelling of point defects in UO2: f electron correlations and the issue of local energy minima, J. Phys.: Condens. Matter, 2013, 25, 333201 CrossRef CAS PubMed.
- B. Dorado, B. Amadon, M. Freyss and M. Bertolus, DFT+U calculations of the ground state and metastable states of uranium dioxide, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 235125 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra14424a |
| This journal is © The Royal Society of Chemistry 2016 |